一些肿瘤是按孟德尔方式遗传的,亦即由单个基因的异常决定的。它们通常以常染色体显性方式遗传,并有不同程度的恶变倾向,故也称为遗传性癌前改变。现举例如下。
1.家族性结肠息肉家族性结肠息肉(familialpolyposis coli,FPC)又称为家族性腺瘤样息肉症,在人群中的发病率为1:100000。表现为青少年时结肠和直肠已有多发性息肉,其中一些早晚将恶变。90%未经治疗的患者将死于结肠癌。FPC的基因现定位于5q21.
2.Ⅰ型神经纤维瘤Ⅰ型神经纤维瘤(neurofibromatosis,NF1)患者沿躯干的外周神经有多发的神经纤维瘤,皮肤上则可见多个浅棕色的“牛奶咖啡斑”,腋窝有广泛的雀斑,在少数患者肿瘤还有恶变倾向。现知与NF1发生密切有关的是一个肿瘤抑制基因,称为NF1基因,它定位于17q11.2,并已分离克隆。
此外,基底细胞痣综合征(basalcell nevus syndrome)、恶性黑素瘤(malignant melanoma)等属于遗传性肿瘤。
还有一些肿瘤既有遗传的,也有散发的。前者临床上按常染色体显性方式遗传,属遗传型,常为双侧性或多发性,发病早于散发型病例。这些肿瘤大多来源于神经或胚胎组织,虽然比较罕见,但在肿瘤病因研究中具有重要意义,故择要介绍如下。
1.视网膜母细胞瘤 视网膜母细胞瘤(retinoblastoma),为眼球视网膜的恶性肿瘤,多见于幼儿,大部分患者(70%)2岁前就诊,发病率为1:15000-28000。肿瘤的恶性程度很高,可随血循环转移,也能直接侵入颅内(图9-2)。
视网膜母细胞瘤可分为遗传型和散发型。大约40%的病例属遗传型,即由于父母患病或携带有突变基因,或父母的生殖细胞发生突变,在患儿出生时全身细胞已有一次视网膜母细胞瘤基因(Rb1)的突变。另约60%则是患者本人Rb1基因两次体细胞突变的结果,属非遗传型。遗传型患者常为双侧或多发肿瘤,平均发病年龄也较散发型者为早(15个月:30个月)。双侧性患者中还有少数患者可见一条13号染色体异常,主要是其长臂1区4带的缺失。13号长臂的这一区带正是视网膜母细胞瘤基因所在之处。
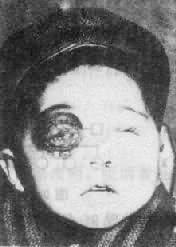
图9-2 双侧视网膜母细胞瘤右眼已萎缩
2.神经母细胞瘤 神经母细胞瘤(neuroblastoma)也是一种常见于儿童的恶性胚胎瘤,起源于神经嵴,活婴中的发病率为1:10000。神经母细胞瘤为常染色体显性遗传性肿瘤。有的神经母细胞瘤还合并有来源于神经嵴的其他肿瘤,如多发性神经纤维瘤、节神经瘤、嗜铬细胞瘤等。
3.Wilms瘤 Wilms瘤即肾母细胞瘤(nephr oblastoma),是一种婴幼儿肾的恶性胚胎性肿瘤,约占全部肾肿瘤6%,活婴中的发病率约为1:10000,3/4的肿瘤均在4岁以前发病。也可分为遗传型(38%)和非遗传型(62%),前者双侧性肿瘤较多,发病年龄较早,呈常染色体显性遗传,有明显的家族聚集现象。患者可伴有无虹膜症、半侧肥大、假两性畸形以及智力低下等。
一些易患Wilms瘤的无虹膜症患者有11号染色体短臂1区(11p13)缺失,而在Wilms瘤细胞中也曾发现11p13的缺失。现在认为11p13和11p15上有2个与肿瘤有关的基因,它们的异常都可能与Wilms瘤的发生有关。