第一章 绪论
第一节 临床生物化学的领域和性质
临床生物化学是化学、生物化学与临床医学的结合,目前已经发展成为一门成熟的独立学科。临床生物化学有其独特的研究领域、性质和作用,是一门理论和实践性较强的、边缘性的应用学科,以化学和医学知识为主要基础。广义上讲,临床生物化学是研究器官、组织人体体液的化学组成和进行着的生物化学过程,以及疾病、药物对这些过程的影响,为疾病诊断、病情监测、药物疗效、预后判断和疾病预防等各个方面提供信息和理论依据。临床生物化学除了要求应用化学与医学方面的理论知识和技术外,还应与生物学、物理学、教学、电子学等各方面的知识密切联系,广泛地应用这些学科领域的新成就。
在日常实践中,临床生物化学的主要作用有两个方面:
第一,阐述有关疾病的生物化学基础和疾病发生发展过程中的生物化学变化。这些生物化学改变可以是原发性的,也可能是某种原因引起器官病损或并发症导致体液生化组成发生的一系列继发性的改变。这部分内容又称之为化学病理学(chemical pathology)。
第二,开发应用临床生物化学检验方法和技术,对检验结果的数据及其临床意义作出评价,用以帮助临床诊断以及采取适宜的治疗。这部分内容有两方面的侧重点:在阐明疾病生化诊断的原理方面,侧重于论述疾病的生化机制,比较接近化学病理学的范畴;而在技术方法的开发应用方面,偏重于临床生物化学实验室的应用,有人称之为临床化学(clinical chemistry),其中一部分内容又称之为诊断生物化学(diagnosticclinical chemistry)。
由于社会和经济的发展,其它相应学科的进展以及新技术的应用,临床生物化学这门学科及其实验室技术在近二三十年中获得迅速发展和完善。它在临床医学中所起的作用和地位已日益受到重视,并已成为任何医院及有关研究部门建设中不可缺少的重要组成部分。它是检验医学中的主干学科之一,它的服务质量直接关系到整个医疗水平的提高和疾病防治的效果。
第二节 临床生物化学发展的简要回顾和现状
临床生物化学成为一门独立的学科还只是近四五十年的事,因此它是相当年轻的学科。追溯其发展过程,它是与许多相关学科(包括化学、生理学、药物学、病理学、临床医学等等)相互联系、相互渗透的结果。
在临床生物化学学科发展史上,有几次技术上和概念方面的重大突破,促使了本学科的进步和发展。
(一)“临床化学”名词的由来
“临床化学”一词是在第二次世界大战后、本世纪50年代开始较广泛地使用的。19世纪以前只是有一些化学家、生理学家和临床医生研究人体在健康与疾病时的化学组分的变化,包括血液及尿中蛋白质、糖及无机物等物质。1918年,Lichtwitz首先采用“临床化学”作为教科书名公开出版。1931年,Peter及Van Slyke又出版了两卷以《临床化学》为名的专著,第一次概括了这一领域的主要内容,它标志着这一学科的初步形成。
(二)体液生物化学组分的分析应用及“细胞内环境相对稳定”概念
19世纪以来就有一系列关于健康与疾病时体液生物化学组成的研究。它包括Berzelius、Liebig、Simon、BenceJones、Folin以及我国早期生物化学家吴宪等人的杰出工作。1926年,Waiter Gannon 使用了“homeostasis”(内环境相对稳定)一词,取代和发展了ClaudeBernard的细胞内环境恒定的概念,这对推动临床生物化学的发展起着深远的影响,在过去50年中成为实验性研究的指导思想。至今临床生物化学中相当部分的工作就是细胞外液(即Bernard提出的内环境)的临床生化。由Van Slyke等人开创的体液水、电解质与酸碱平衡这一领域中的理论与实践在临床诊断和治疗中所起作用就是一个具有代表性范例。
(三)比色法和光度法在临床生物化学实验室中的应用
比色法和光度法对促进这一领域中工作的质和量方面的变化起了根本性推动作用。19世纪和20世纪初,血液及尿中成分多采用传统的重量分析和容量分析法(滴定法),其灵敏度不高,标本用量多,耗费时间长,方法繁琐,限制了它在临床上的广泛应用。20世纪初,特别是从1904年Folin用比色法测定肌酐开始,建立了一系列血液生物化学成分测定的比色分析法。Duboseq第一个设计了目测比色法。值得提到的是,1924年我国北京协和医学院建立了由吴宪教授主持的生物化学系,成为当时我国医学生物化学教学与研究的中心。该系除了讲授基础生物化学外,还开设了血尿分析法、酶学、血液分析等进修课程,培养了我国第一批生物化学家和临床生物化学工作者;在血液分析、血滤液制备以及改建和发展新的比色分析法等方面作了一系列工作,并报告了我国正常成人血液化学成分的正常参考值。本世纪30年代后,由于光电比色计的应用,临床生物化学实验室的分析才发生了根本性的改观。至今,光度计和分光光度法在现代临床生物化学分析中仍占有突出的地位。
(四)血清酶活力测定作为细胞与组织损伤的重要指标
本世纪50年代后,应用血清酶活力测定作为监测细胞、器官损害及肿瘤生长的指标,使临床生物化学的工作又增加了新的内容。近30年来它已发展成诊断酶学这一分支。1908年Wohlgemuth首先提出,以检测尿中淀粉活力作为急性胰腺炎的诊断指标。以后又有血清碱性磷酸酶和脂酶的测定,但由于当时方法学存在的困难,应用进展缓慢。1954年Ladue、Worblewski、Karmen等人先后发现血清乳酸脱氢酶及转氨酶在不少疾病时增高,此后血清酶在诊断上的应用和研究非常活跃。目前方法学上又有了很大发展,同工酶的概念和检测以及酶谱分析,都大大地增加了诊断的特异性和灵敏度。
(五)治疗性药物监测成为临床生物化学的一个重要分支
由于病人对治疗药物的反应和代谢存在着个体差异,随着新的、有效的微量检测药物血浓度技术的发展,以及药代动力学知识的进展,治疗性药物监测工作在现代化医院中占有的比重日益增加。在有些大医院中,它的工作量已达整个临床生物化学工作的1/3左右。在我国,治疗性药物监测的工作也正在开展,并已受到重视。这对促使临床医生更有效、合理地使用药物,提高疗效,减少药物的副作用,了解药物在体内的转化与代谢规律等方面都具有重要意义。
(六)超微量的仪器分析、免疫学、分子生物学、放射性同位素等技术在生物化学实验室中的应用
这些新技术的应用使临床生物化学工作内容有可能日益扩大深入。近10多年来,对于体内一些微量蛋白质、多肽等生物活性物质的测定,基因(核酸片段)的分析,微量元素的分析,以及它们在多种疾病中的变化,为临床医学提供了极有价值的数据。
(七)自动化装置与电子计算机数据处理系统
近20年来,由于临床生物化学工作内容迅速扩大,促进了分析仪器的机械化和自动化,1957年Skeggs等首先在临床生物化学实验室中引用了连续流动式分析装置(continuous flow analysis),1964年后使用多通道分析仪(multichannel analyzer)和离心式分析仪(centrifugalanalyzer, 1969),加上微处理机的使用,使临床化学工作大大改进了分析的质和量,提供了检测大批标本的工作程序,改进了对结果的处理和作用,设计出各种组合报告(profile reporting)。例如将蛋白质、血清酶、电解质和血气等多种项目配套分析结果,经过处理(分析、结合),使数据转化为更高层次的报告。为了解某一器官的功能概貌,可组合一系列相关试验,经综合、分析作出评价。目前在肝功能、肾功能、心肌损害、肿瘤标志、血脂分析以及内分泌功能检测方面的成套试验(profile tests)已被广泛地使用。
(八)临床生物化学的国际性、全国性及地方性学会和出版刊物
由于临床生物化学已发展为一个得到确认的学科和专业,在不少国家都已成立了专门的全国性学会,并有它自己的十分活跃的、有成果的国际性学会。国际纯化学与应用化学协会(IUPAC)设有临床化学专业委员会(Commission of ClinicalChemistry,Division of Biological Chemistry, International Union of Pure andApplied Chemistry),成立于1952年。此外,国际性的临床化学协会(International Federation of Clinical Chemistry, IFCC)亦组织大量学术活动,并设有教育委员会,制定一系列有关培训人才和政策性的文件。在某些国家由于历史的和现实的条件,学会的活动可能与其它临床实验室学科和生物化学学会合并进行。我国目前临床生物化学的学术活动有两个主要方面,一是属于中华医学会下设的临床检验学会的临床生物化学专业委员会,一是属于中国生物化学学会下属的医学的生物化学专业委员会。国际性的专业出版刊物和杂志有:《临床化学杂志》(Clinical Chemistry,美国)、《临床化学学报》(Clinical Chemistry Acta,荷兰)、《临床生物化学年鉴》(Annuals of Clinical Biochemistry,英国)以及《临床生物化学评论》(Clinical Biochemistry Reviews,加拿大)等有较大的影响。我国出版的《国外医学-临床生物化学与检验学分册》(始于1980年)是全国性的情报刊物。其它有关临床生物化学为主要内容的国内外文献杂志刊物种类日益增多,可供参考。
第三节 临床生物化学的现状及其作用
(一)临床生物化学在医疗保健工作、疾病诊断与治疗中的作用
根据国内外近10多年来的统计,临床生化的检测项目不断扩大,工作量也以每年近10%-20%的速度增加。不少项目广泛地应用于肝、胆、胰腺等消化系统疾病以及肾疾病的诊断与治疗,在外科手术及创伤后患者体液电解质与酸碱平衡紊乱的监测有着重要意义。体液中酶与同工酶的活力测定为临床医生判断病情提供了十分有价值的信息,特别在心肌梗死、肝细胞损害、肿瘤及神经肌肉病损等方面近年来有很大新进展。在多种内分泌疾病与先天性代谢障碍疾病的确诊和病情随访中,专一的生物化学检测项目起着决定性的作用。此外,在糖尿病昏迷、肺性脑病的治疗中,由于应用临床生物化学指标紧密监护治疗进程,病死率已大大降低。在一些肿瘤化疗、强心甙及抗心律不齐、抗癫痫、抗抑郁等治疗性药物的控制使用中,能监测血药浓度,根据个体差异合理调整剂量及给药间隔,以提高疗效、减少毒副作用,有着十分重要的指导意义。
(二)临床生物化学理论与技术在医学教育中的作用与地位
由于临床生物化学在医学理论与实践方面的重要性,医学生在学习基础课和临床课中都应充分结合有关的临床生物化学知识。目前,多数院校是从讲授基础生化开始的。一般说,基础生物化学大部分是取材于动物和正常人的,在深度上也可以包括病理的材料。临床生物化学的教学计划在各个国家则有很大的不同,就是在一个国家内,由于本课程的组织者的认识深浅不同,也存在着较大的差异。但是,从最近几年来各国的医学教育改革及内容来看,在所有国家,在整个医学教育过程中,接触临床生物化学知识的比重正在不断增长。近年来,不少大学在后期教育中开设了有关临床实验室科学的课程或相应的讲座,或开展床边专题讨论,有不少课程是为研究生开设的。应当积极地为医学生、医学研究及临床医生提供能有效地利用实验室来进行日常医疗活动的条件。对于非实验室临床专家来说,要求他们通晓详尽的方法学是不现实的,但他们对于检测方法的原理及所得分析结果的限度和评价有所理解是十分必要的。现在,不少教学医院在为医学生和研究生开设临床生化课程的同时,亦为临床医生们提供相应的实验室活动中心,提供一定的经费、投资设备,以促进临床与实验室的合作,这对于现代医学的进步起着十分有益的作用。
(三)临床生物化学实验室面临的任务
临床生物化学室工作内容的不断增长,急需培养专门人才和建立工作质量控制程序、我国有关调查资料表明,如何切实加强和提高实验室的分析质量是刻不容缓的。因此制定提高各级检验人员素质的相应的教育培训计划和考核法规,实行质量控制等制度都是很关键的措施。具体内容应包括培养具有领导和监管临床生物化学实验室工作能力的各级领导人才和熟练的技术干部;及时引进和开发可靠性更高的新方法;协助对分析数据的处理,积极参与临床有关咨询;与临床医生密切合作,对临床生物化学的理论原理和技术应用不断进行总结和研讨。
分析手段的现代化、自动化以及微处理机的使用,是现代生化实验室的重要组成。能否合理地选用仪器,取决于日常工作量、使用人员素质以及对使用效益和经济水平等有关因素的充分了解。制定各项测定项目最适用的分析方法,是一个实验室工作的极其重要的环节,它要求充分考虑到方法的精密度、准确性以及实用性。为保证这一目的,在较大医院的临床生物化学检验部门,有必要组织一定力量进行有关方法学的开发工作,经常研讨新的方法学及自动化设置,经过试用,逐步推广于常规工作。
人们越来越意识到,对疾病本质和过程的透彻理解,在很大程度上需要有关生物化学分析的确切信息。临床医生正面临着应付实验室带来大量分析数据的新课题。因此临床生物化学工作者有必要在这方面和医生合作,进行更多的“翻译”、“加工”,将生物化学分析结果的信息转化为更高层次的医学语言,从而为医学科学和临床诊疗的提高服务。
第四节 本书的主要内容与使用
本书主要供高等医学检验专业开设的临床生物化学理论课程使用。学习本课程前,学生应已具备有关化学基础、生物化学、生理学及临床医学的相关知识。本课程将主要给予学生有关临床生物化学的理论知识,着重于对疾病本质的生化机制、休液中生物化学组分变化的病理生理学基础以及生物化学诊断原理的阐述,包括有关方法学应用的基本原理及其临床意义的判断;阐述临床生物化学实验室的检测项目,检测结果数据与临床的联系(即正确有效地将临床实验室数据转化为临床诊断信息)。至于详尽的实验技术程序和操作要点,将在《临床生物化学实验指导手册》中介绍,这部分特别是为训练临床检验专业的学生所必需。
本书的主要内容包括以下5个部分:
1.绪论包括临床生物化学的学科领域和性质,学科发展的简要回顾与现状、作用、地位以及面临的主要任务,有关质量保证制度的建立以及临床生化实验室的有效使用。
2.以物质分类介绍疾病时的临床生物化学包括糖尿病及其它糖代谢紊乱,蛋白质与临床,血浆脂蛋白系统及其代谢紊乱,体液的平衡失调及生化诊断,钙、磷、镁的代谢障碍,微量元素与疾病等。
3.以器官或组织病损为主介绍有关疾病时的临床生物化学包括内分泌腺、肾、肝胆疾病时的代谢紊乱及其生化诊断,神经与精神系统疾病的临床生化以及遗传疾病的生化诊断。
4.诊断生物化学中的某些专题包括诊断酶学、肿瘤生化标志物、妊娠及其并发症的生化诊断、治疗性药物监测等。
治疗性药物监测在发达国家已经发展成为相当成熟独立的分支学科,并为临床实验室日常检测的主项目内容。在我国医学检验专业中,多数亦已不同程序地开设本课程,在本教材第二次修改中经反复研究,结合我国具体学科发展情况,将此项内容列入本教材作为专章介绍。
5.诊断生物化学中常用的某些技术包括仪器分析技术、免疫化学技术、分子生物学技术、酶学分析技术以及同位素应用分析技术等。从学科发展的现状及各类应用技术的重要地位来说,它们应是临床生物化学的重要组成部分,但由于这些技术在不同学校根据各校的具体条件在课程组合时作了不同的安排,不少内容与基础知识在不同程度上于相关的基础或边缘学科中讲解,或打下了一定基础,或作为选修课程给予一定的发展,因此本书仅从这一主干学科培训内容的总体要求出发,作一提纲式的紧密结合检验应用的概述,反映了有关知识在学科发展中的系统性,避免了不必要的课程之间的重复。
第二章 蛋白质与临床诊断
本章着重介绍几类在临床医学中很受重视的蛋白质。在疾病产生发展过程中,组织和细胞内蛋白质的表达合成可增加或受一定程度的抑制,或从细胞内释放出进入血循环的量增加,这些变化或对发病机制的阐明和理解有重要意义,或对疾病损害的部位和程度提供有价值的信息,可供临床医师和实验室医师参考。
第一节 健康与疾病时的血浆蛋白质
一、概念
血浆蛋白质是血浆固体成分中含量很多、组成极为复杂、功能广泛的一类化合物。目前已经研究的血浆不下500种,其中已分离出接近纯品者有200种。近10多年来,出现和使用了不少新技术,用于分析血浆内较微量的个别蛋白质,并研究其在疾病时的变化,这些资料有助于疾病的诊断并提供有价值的病理生理信息。研究有关个别蛋白质的结构、功能、代谢的知识正在迅速积累,并成为很活跃的研究领域。
血浆蛋白基础医学、临床医学及生物化学家的广泛重视,正在进展的领域有以下几个方面:
1.生物化学家分离纯化各种血浆蛋白质组分,研究它们的物理性质、氨基酸的组成及顺序,以及某些蛋白质中结合的糖、脂类、金属化合物、活性多肽、类固醇激素和其它各种化合物。许多工作是对这些蛋白、降解、转换更新与代谢调节的研究。
2.生理学家与病理生理学家长期以来对血浆蛋白质的生理功能感兴趣研究它的胶体性质、缓冲性质和生理作用,在运输脂类、多种金属和微生时、元素中的作用,在结合和调节活性激素外外源性药物体内过程中的作用,以及血浆蛋白质在反蝒肾小球滤过、肾小管回收功能和肝细胞功能方面的意义。
血浆蛋白质亦广泛地应用于研究营养学问题,特别是蛋白质的营养不良。
3.血浆蛋白质具有遗传的变异如结合珠蛋白和转铁蛋白等在不同人群中常见有结构上的差异。还有一些虽然是少见的遗传变异(如缺乏某一种脂蛋白或免疫球蛋白),可表现一定的临床症状,亦具有临床医学上的意义。由于血液是人体组成中最易获得的标本,遗传学家常利用血浆蛋白质结构上的差异作为研究人群与家族遗传特征的标志。
4.在临床生物化学实验室中血浆蛋白质的分析一直是最主要的常规工作之一。最早就用于有关肝及肾疾病和血液恶性肿瘤的诊断与预后的监测。近年来由于个别蛋白质微量和特异的分析检测技术的进展,为不少病理过程和疾病的诊断又提供了新的信息。
血浆蛋白质中不少特殊成分的研究,如血液凝固因子、免疫球蛋白组分及补体系统组分的检测,在血液学与免疫学中都是基本的理论和实践。
5.在进化与个体发育的生物化学研究中,已发现有不少正常胎儿时期的蛋白质可以在恶性癌肿病人中重新出现。血浆蛋白质合成的调控,如急性时相反应蛋白的表达与释放,在临床医学中是长期受注意而又尚未完全解决的课题,它与限制炎症过程密切相关。此外,尚有不少蛋白质水解酶的特异抑制物在血浆中循环,它们具有十分重要的代谢调控作用,虽然其体内过程尚未被完全阐明。
6.血浆蛋白质在实际工作中还广泛地用于组织与细胞培养。血浆中含有各种细胞刺激因子,它们对细胞的活力、增殖、分化、胸内酶的合成及细胞特殊功能起着特殊的作用。必须指出,各种血浆蛋白质组分对不同类型的细胞起的作用有特异性,已发展成为细胞生物学研究的重要分支。
7.在人类不少疾病,包括常见的两种疾病——动脉粥样硬化及肿瘤的发病学研究,以及最常见的糖尿病及其并发症的发病机制中,血浆蛋白质均有广泛的涉及。
一般教科书将血浆蛋白质多方面的功能概括为:①营养;②缓冲与胶体渗透压;③运载(包括类固醇、甲状腺激素、维生素与、脂类、金属与微量元素、药物等);④免疫与防御功能(包括免疫球蛋白与补体);⑤凝血与纤维蛋白溶解;⑥各种醇的特殊功能;⑦代谢调控等几大方面。
血浆蛋白质的分类是一个较为复杂的问题,随着分离方法和对血浆蛋白质功能了解的进展,显然可以从不同角度来进行归纳分类。早先通过盐析法将血浆蛋白质分为白蛋白和球蛋白两大类,目前看来最实际的还是通过醋酸纤维薄膜电泳或琼脂糖凝胶电泳获得有关血浆蛋白质全貌的图谱,即将血浆蛋白质分为白蛋白和α12β、γ球蛋白5个主要区带,根据不同的电泳条件还可将各个区带进一步分离。在琼脂糖凝胶电泳中常可分出个区带。如果采用聚丙烯酰胺凝胶电泳在适当条件下可以分出30多个区带。应当理解,这咱分离区带并非截然。近年免疫化学技术分析法的进展提供了对个别蛋白质测量的新方法,二者结合可以为血浆蛋白质的分析和临床意义积累很好的有用资料。
目前许多学者试图按功能进行分类,如营养、修补、运输、载体、补体系统和凝血因子等等。
本章首先将血浆蛋白质按电泳和功能为主,试行分类归纳于表、表2-2供参考,以便对血浆蛋白质种类有一总的概念。然后再依次介绍一些具有代表性的个别血浆蛋白质的理化特性、功能与临床意义、常见疾病时血浆蛋白质的变化图谱以及血浆蛋白质的检测及其临床应用。
表2-1 血浆蛋白质的分类与特征(以区带电泳为主要技术分类)
| 蛋白质 | 成人参考值 | 半寿期(天) | 分子量(万) | 等电点 | 含糖量 | 简注 |
| 前白蛋白 | 200-400 | 5.4 | 4.7 | 营养指标 | ||
| 白蛋白 | 35000-50000 | 15-19 | 6.64 | 4-5.8 | 在许多疾病时降低,有较广泛的载体功能 | |
| α1区带球蛋白 | ||||||
| α1-抗胰蛋酶 | 780-2000 | 4 | 5.5 | 4.8 | 12 | APR,抗胰蛋白水解酶先天缺陷易导致肺气肿,肝硬化 |
| α1酸性糖蛋白 | 500-1500 | 5 | 4.0 | 2.7-4 | 45 | APR |
| α1脂蛋白 | 1700-3250 | 20 | 脂类运输 | |||
| α1胎儿球蛋白 | 0.03 | 6.9 | 胎儿期蛋白 | |||
| α2区带球蛋白 | ||||||
| 结合珠蛋白 | 300-2150 | 2 | 8.5-40 | 4.1 | 12 | APR,结合,溶血时减少 |
| α>2巨球蛋白 | 1250-4100 | 5 | 80 | 5.4 | 8 | 肾病期增加,抗蛋白水解酶 |
| 铜蓝蛋白 | 200-500 | 4.5 | 16 | 4.4 | APR,含铜 | |
| β1区带蛋白 | ||||||
| 转铁蛋白 | 2000-3500 | 7 | 7.7 | 5.7 | 6 | 负性APR,转运Fe在低色素贫血时增加 |
| 血红素结合蛋白 | 500-1150 | 5.7 | 结合血红素 | |||
| β脂蛋白 | 600-1550 | 300 | 脂类运输 | |||
| C4 | 20.6 | 7 | 补体系统 | |||
| β2区带蛋白 | ||||||
| 纤维蛋白原 | 2000-4000 | 2.5 | 34 | 5.5 | 3 | APR,纤维蛋白前身参与血凝 |
| C3 | 700-1500 | 18 | 2 | APR,补体系统 | ||
| β2-微球蛋白 | 1-2 | 1.18 | ||||
| γ区带球蛋白 | ||||||
| IgG | 5250-16500 | 24 | 16 | 6-7.3 | 3 | 抗体组分 |
| IgA | 400-3900 | 6 | 17 | 8 | ||
| IgM | 250-3100 | 5 | 90 | 12 | ||
| C-反应蛋白 | <8 | 12 | 6.2 | APR,防御蛋白 |
1.表2-1中所列仅为部分主要的血浆蛋白质组分;
2.正常参考值随选用检测方法和年龄有所不同;
血浆蛋白质的功能分类
| 功能分类 | 功能特征 |
| 运输载体 | |
| 血浆脂蛋白系统 | 包括乳糜微粒、VLDL、LDL、HDL等,运输胆固醇、磷脂、甘油酯及脂肪酸(详见脂蛋白代谢) |
| 前白蛋白与白蛋白 | 运输游离脂酸、甲状腺素、多种药物(如阿司匹林、巴比妥类、青霉素等) |
| 甲状腺素结合球蛋白 | 特异高亲和力结合甲状腺激素 |
| 皮质素结合球蛋白 | 特异高亲和力结合皮质醇 |
| 类固醇激素结合球蛋白 | 特异高亲和力结合类固醇激素 |
| 视黄醛结合蛋白 | 结合视黄醛 |
| 转铁蛋白 | 运输铁 |
| 结合珠蛋白 | 结合血红蛋白 |
| 血色素结合蛋白 | 结合血红素 |
| 铜蓝蛋白 | 运输铜 |
| 补体系统 | 至少有13种具有酶激活性的蛋白质,C1q、C1r、C1s、C2、C3、C4、C5、C6、C7、C8、C9、C1脂酶抑制物和备解素等 |
| 凝血系统 | 包括纤维蛋白原在内的10种以上蛋白质 |
| 激肽系统 | 包括激肽原及激肽酶,释放激肽 |
| 免疫功能 | 包括IgG、M、D、E、A,C反应蛋白 |
| 蛋白酶的抑制物 | 至少有6种以上具有蛋白酶抑制作用的血浆蛋白质,包括α1-抗胰蛋白酶、α1-抗糜蛋白酶、α2-巨球蛋白等 |
| 免疫抑制作用 | αFP等 |
| 其它具有酶活性的蛋白质 | 来自组织细胞的可溶性蛋白质或由于细胞破裂而进入血循环的细胞内酶,但也有一些血浆中的酶具有重要的调节代谢作用,如LCAT |
| 具有激素活性的蛋白质 | 胰岛素等 |
二、血浆蛋白质的理化性质、功能与临床意义
(一)前白蛋白
前白蛋白(prealbumin,PA),分子量5.4万,由肝细胞合成,在电泳分离时,常显示在白蛋白的前方,其半寿期很短,仅约12小时。因此,测定其在血浆中的浓度对于了解蛋白质在营养不良和肝功能不全,比之白蛋白和转铁蛋白具有更高的敏感性。PA除了作为组织修补的材料外,还可视作一种运载蛋白,可结合T4与T3,而对T3的亲和力更大。PA与视黄醇结合蛋白形成复合物,具有运载维生素A的作用。在急性炎症、恶性肿瘤、肝硬化或肾炎时其血浓度下降。
(二)白蛋白
白蛋白(albumin,Alb)系由肝实质细胞合成,在血浆中的半寿期约为15-19天,是血浆中含量最多的蛋白质,占血浆总蛋白的40%-60%。其合成率虽然受食物中蛋白质含量的影响,但主要受血浆中白蛋白水平调节,在肝细胞中没有储存,在所有细胞外液中都含有微量的白蛋白。关于白蛋白在肾小球中的滤过情况,一般认为在正常情况下其量甚微,约为血浆中白蛋白的0.04%,按此计算每天从肾小球滤过液中排出的白蛋白即可达3.6g,为终尿中蛋白质排出量的30-40倍,可见滤过液中多数白蛋白是可被肾小管重新吸收的。有实验证实白蛋白在近曲小管中吸收,在小管细胞中被溶酶体中的水解酶降解为小分子片段而进入血循环。白蛋白可以在不同组织中被细胞内吞而摄取,其氨基酸可被用为组织修补。
白蛋白的分子结构已于1975年阐明,为含585个氨基酸残基的单链多肽,分子量为66458,分子中含17个二硫键,不含有糖的组分。在体液pH7.4的环境中,白蛋白为负离子,每分子可以带有200个以上负电荷。它是血浆中很主要的载体,许多水溶性差的物质可以通过与白蛋白的结合而被运输。这些物质包括胆红素、长链脂肪酸(每分子可以结合4-6个分子)、胆汁酸盐、前列腺素、类固醇激素、金属离子(如Cu2+、Ni2+、Ca2+)药物(如阿司匹林、青霉素等)。
具有活性的激素或药物当与白蛋白结合时,可以不表现其活性,而视为其储存形式,由于这种结合的可逆性和处于动态平衡,因此在调节这些激素和药物的代谢上,具有重要意义。
血浆白蛋白另一重要功能是纤维血浆的胶体渗透压,并具有相当的缓冲酸与碱的能力。
临床意义:
1.血浆白蛋白浓度可以受饮食中蛋白质摄入量影响,在一定程度上可以作为个体营养状态的评价指标。
2.在血浆白蛋白浓度明显下降的情况下,可以影响许多配体在血循环中的存在形式,包括内源性的代谢物(Ca2+、脂肪酸)、激素和外源性的药物。在同样血浓度下,由于白蛋白的含量降低,其结合部分减少,而游离部分相对增加,这些游离状态的配体一方面更易作用于细胞受体而发挥其活性作用,一方面也更易被代谢分解,或由于其分子小而经肾排泄。
3.血浆白蛋白的增高较少见,在严重失水时,对监测血浓缩有诊断意义。
4.低白蛋白血症在不少疾病时常见,可有以下几方面的原因:
(1)由于白蛋白的合成降低:常见于急性或慢性肝疾病,但由于白蛋白的半寿期较长,因此,在部分急性肝病患者,血浆白蛋白的浓度降低可以表现不明显。
(2)由于营养不良或吸收不良。
(3)遗传性缺陷:无白蛋白血症是极少见的一种代谢性缺损,血浆白蛋白含量常低于1g/L。但可以没有症状(如水肿),可能部分由于血管中球蛋白含量代偿性升高。
(4)由于组织损伤(外科手术或创伤)或炎症(感染性疾病)引起的白蛋白分解代谢增加。
(5)白蛋白的异常丢失:由于肾病综合征、慢性肾小球肾炎、糖尿病、系统性红斑狼疮等而有白蛋白由尿中损失,有时每天可以由尿中排出蛋白达5g以上,超过肝的代偿能力。在溃疡性结肠炎及其它肠管炎症或肿瘤时也可由肠管损失一定量的蛋白质。在烧伤及渗出性皮炎可从皮肤丧失大量蛋白质。
(6)白蛋白的分布异常:如门静脉高压引起的腹水中有大量蛋白质,是从血管内渗漏入腹腔。
5.已发现有20多种以上白蛋白的遗传性变异。这些个体可以不表现病症,在电泳分析时血浆蛋白质的白蛋白区带可以出现2条或1条宽带,有人称之为双蛋白血症。当某些药物大量应用(如青霉素大剂量注射使血浓度增高时)而与白蛋白结合时,也可使白蛋白出现异常区带。
目前关于血浆或血清白蛋白的测定,最常使用的方法是利用其与某些染料如溴甲酚绿(bromcresolgreen,BCG)或溴甲酚紫)(bromcresolpurple,BCP)特异性的结合能力而加以定量。在pH4.2的条件下,BCG可与白蛋白定量地、特异地结合,而不受血浆中其它球蛋白的干扰。结合后的复合物在628nm有特殊吸收峰,而可与游离的染料相区别,这一吸收峰一般不受血浆中可能存在的其它化合物(如胆红素、血红素等)的影响,测定时应控制染料的浓度、反应的pH和时间。这是很实用的方法。一般用血浆量为20μl,在白蛋白10-60g/L浓度范围内呈良好线性关系、批内C·V值<3%,正常成人参考值为35-50g/L,在直立姿势采血,由于血浓缩其值可略高3g/L。
(三)α1-抗胰蛋白酶
α-抗胰蛋白酶(α1-antitrypsin,α1AT或AAT),是具有蛋白酶抑制作用的一种急性时相反应蛋白,分子量为5.5万,P1值4.8,含有10%-12%糖。在醋酸纤维薄膜或琼脂糖电泳中泳动于α1区带,是这一区带的主要组分。区带中的另2个主要组分;α1-酸性糖蛋白含糖量特别高,α1-脂蛋白含脂类特别高,因此蛋白质的染色都很浅。作为蛋白酶的抑制物,它不仅作用于胰蛋白酶,同时也作用于糜蛋白酶、尿激酶、肾素、胶原酶、弹性蛋白酶、纤溶酶和凝血酶等。AAT占血清中抑制蛋白酶活力的90%左右。AAT的抑制作用有明显的pH依赖性,最大活力处于中性和弱碱性,当pH4.5时活性基本丧失,这一特点具有重要的生理意义。
一般认为AAT的主要功能是对抗由多形核白细胞吞噬作用时释放的溶酶体蛋白水解酶。由于AAT的分子量较小,它可透过毛细血管进入组织液与蛋白水解酶结合而又回到血管内,AAT结合的蛋白酶复合物并有可能转移到α2-巨球蛋白分子上,经血循环转运而在单核吞噬细胞系统中被降低、消失。
AAT具有多种遗传分型,利用不同pH的缓冲剂和电泳支持物,迄今已分离鉴定有33种等位基因(allotypes),其中最多见的是PiMM型(为M型蛋白抑制物的纯合子体)占人群的90%以上,另外还有两种蛋白称为Z型和S型,可表现为以下遗传分型:PiZZ、PiSS、PiSZ、PiMZ、PiMS,S型蛋白与M蛋白之间的氨基酸残基仅有一个差异。对蛋白酶的抑制作用主要限于血循环中M型蛋白的浓度。以MM型的蛋白酶抑制能力为100%相比,ZZ型的相对活力仅为15%、SS为60%、MZ为57%、MS为80%,其它则无活性。
临床意义:低血浆AAT可以发现于胎儿呼吸窘迫综合征。AAT缺陷(ZZ型、SS型甚至MS表现型)常伴有早年(20-30岁)出现的肺气肿,由于吸入尘埃和细菌引起肺部多形核白细胞的吞噬活跃,引起溶酶体弹性蛋白酶释放,当M型AAT蛋白缺乏时,蛋白水解酶过度地作用于肺泡壁的弹性纤维而导致肺气肿的发生。AAT的缺陷,特别是ZZ表现型可引起肝细胞的损害而致肝硬化,机制未明。常用测定方法,一种是基于胰蛋白酶的抑制能力(trypsininhibitorycapacity),但目前已有免疫化学方法,供应M蛋白AAT的试剂盒来测定。正常参考值为新生儿1450-2700mg/L、成人780-2000mg/L。如果排除急性时相反应的存在,正常人血浆浓度<500mg/L提示可能存在变异的表现型,可进一步通过等电聚焦或淀粉胶电泳证实。
(四)α1-酸性糖蛋白
α-酸性糖蛋白(α1-acidglycoproteinTimes New Roman,AAG,早期称之为乳清类粘蛋白)分子量近4万,含糖约45%,pI为2.7-3.5,包括等分子的已糖、已糖胺和唾液酸。
AAG是主要的急性时相反应蛋白,在急性炎症时增高,显然与免疫防御功能有关,但详细机制尚待阐明。早期工作认为肝是合成α1-糖蛋白的唯一器官,近年有证据认为某些肿瘤组织亦可以合成。分解代谢首先经过唾液酸的分子降解而后蛋白质部分很快在肝中消失。AAG可以结合利多卡因和普萘洛尔(心得安),在急性心肌梗死时AAG作为一种急性时相反应蛋白可以升高,而干扰药物剂量的有效浓度。
临床意义:AAG的测定目前主要作为急性时相反应的指标,在风湿病、恶性肿瘤及心肌梗死患者亦常增高,在营养不良、严重肝损害等情况下降低。测定方法:使用AAG的抗体制成免疫化学试剂盒,可设计成免疫扩散或浊度法检测。正常参考值为500-1500mg/L,亦可采用过氯酸和磷钨酸分级沉淀AAG后,测定蛋白质或含糖量来计算之。
(五)甲胎蛋白
正常情况下甲胎蛋白(α-fetoprotein,αFP或AFP)主要在胎儿肝中合成,分子量6.9万,在胎儿13周AFP占血浆蛋白总量的1/3。在妊娠30周达最高峰,以后逐渐下降,出生时血浆中浓度为高峰期的1%左右,约40mg/L,在周岁时接近成人水平(低于30μmg/L)。
临床意义:在产妇羊水或母体血浆中AFP可用于胎儿产前监测。如在神经管缺损、脊柱裂、无脑儿等时,AFP可由开放的神经管进入羊水而导致其在羊水中含量显著升高。胎儿在宫腔内死亡、畸胎瘤等先天缺陷亦可有羊水中AFP增高。AFP可经羊水部分进入母体血循环。在85%脊柱裂及无脑儿的母体,血浆AFP在妊娠16-18周可见升高而有诊断价值,但必须与临床经验结合,以免出现假阳性的错误。
在成人,AFP可以在大约80%的肝癌患者血清中升高,在生殖细胞肿瘤出现AFP阳性率为50%。在其它肠胃管肿瘤如胰腺癌或肺癌及肝硬化等患者亦可出现不同程度的升高。
测定方法:根据不同标本可选用不同方法。羊水可采用免疫扩散或火箭电泳法。一般放射免疫测定标本需先加以稀释。注意避免胎儿血(AFP浓度比羊水高200倍)的污染。血浆标本可采用放射免疫或酶标免疫法测定。反向免疫电泳亦用于对肝病患者的筛选试验。在乙型肝炎流行区,AFP的普查可用以早期筛选肝癌。血清正常参考值,健康成人<30μg/L(或30ng/ml),新生儿<50mg/L,妊娠母体20周20-100μg/L,羊水(20周妊娠)5-25mg/L。
(六)结合珠蛋白
结合珠蛋白(haptoglobin,Hp)在血浆中与游离的血红蛋白结合,是一种急性时相反应蛋白。在CAM电泳及琼脂糖凝胶电泳中位于α2区带。分子中有两对肽链(α与β链)形成α2β2四聚体。α链有α1及α2两种。而α1又发现有α1F及α1S两种遗传变异体(F表示电泳迁移率相对为fast,S表示slow,两种变异体的多肽链只有一个氨基酸的残基组成不同),由于α1F、α1S、α2三种等位基因编码形成αβ聚合体,因此个体之间可有多种遗传表现型(表2-3)。不同个体,由遗传获得的特征基因型决定了血浆中Hp的性质,这就是所谓基因多形性(polymorphism)的表现。还有一些血浆蛋白质也表现有相似的遗传变异,如β脂蛋白、α1AT、IgG等,Hp在遗传研究上是颇为引起兴趣的课题。
表2-3 结合珠蛋白的几种遗传表现型
| 表现型 | 亚单位的结构 | 备注 |
| Hp1-1 | (α1F)2β2α1Fα1Sβ2(α1S)2β2 | 分子量约为8万,α链含氨基酸残基83个,β链含氨基酸残基245个 |
| Hp2-1 | (α1Sα2β2)n(α1Fα2β2)n | 分子量为12万-20万的聚合体,由于n不同,可以在电泳中出现多条带 |
| Hp2-2 | (α2β)nn=3-8 | 分子量为16万-40万,由于n不同,可在电泳中出现多条带 |
Hp的主要功能是能与红细胞中释出的自由形式存在的血红蛋白结合,每分子Hp可以结合两分子的Hp。结合是不可逆的,一旦结合后,复合物在几分钟之内转运到肝,肝细胞上有特异受体,可十分有效地结合Hp-Hb复合物进入肝细胞而被降解,氨基酸和铁可被机体再利用。因此Hp可以防止Hb从肾丢失而为机体有效地保留铁。在一次急性血管内溶血时血循环中的Hp可以结合3g以上的Hb。Hp在溶血后含量急剧降低,Hp与Hb结合后不能重新被利用,但急性溶血后其在血浆中的浓度一般在一周内即可由再生而恢复。
临床意义:正常参考值范围较宽,因此一次测定的价值不大,连续观察可用于监测急性时相反应和溶血是否处于进行状态。
急性时相反应中血浆Hp增加,当烧伤和肾病综合征引起大量白蛋白丢失的情况下亦可增加,血管内溶血如溶血性贫血、输血反应、疟疾时Hp含量明显下降。此外,在严重肝病患者Hp的合成降低。在新生儿期只有成人的10%-20%(50-480mg/L),6个月后肝成熟,血浆Hp即达成人水平(300-2150mg/L)。
(七)α2-巨球蛋白
α2巨球蛋白(α2-macroglobulin,α2MG或AMG)是血浆中分子量最大的蛋白质。分子量约为65.2万-80万,含糖量约8%,由4个亚单位组成。它与淋巴网状系统细胞的发育和功能有密切联系(虽然确切的机制尚未明确)。
α2MG最突出的特性是能与多种分子和离子结合。特别是它能与不少蛋白水解酶结合而影响这些酶的活性。如与许多肽链内切酶(包括丝氨酸、巯基、羧基蛋白水解酶和一些金属蛋白水解酶)的结合。这些蛋白水解酶有纤维蛋白溶酶、胃蛋白酶、糜蛋白酶、胰蛋白酶及组织蛋白酶D等。研究表明,α2MG与蛋白水解酶相互作用可使α2MG的分子构象发生变化,当酶处于复合物状态时,酶的活性部位没有失活,但不容易作用于大分子底物,若底物为分子量小的蛋白质,即使有其它抗蛋白酶的存在,也能被α2MG-蛋白酶复合物所催化而水解。这样,α2MG起到有选择地保护某些蛋白酶活性的作用,这在免疫反应中可能具有重要意义。
α2MG是由肝细胞与单核吞噬细胞系统中合成,半寿期约5天,但当与蛋白水解酶结合成为复合物后其清除率加速。
在低白蛋白血症时,α2MG含量可增高,可能系一种代偿机制以保持血浆胶体渗透压。妊娠期及口服避孕药时血浓度增高。机制不明。可采用免疫化学法测定,正常成人参考值为1500-3500μg/L。
(八)铜蓝蛋白
铜蓝蛋白(ceruloplasmin,CER)是一种含铜的α2糖蛋白,分子量约为12万-16万,不易纯化。目前所知为一个单链多肽,每分子含6-7个铜原子,由于含铜而呈蓝色,含糖约10%,末端唾液酸与多肽链连接,具有遗传上的基因多形性。
CER具有氧化酶的活性,对多酚及多胺类底物有催化其氧化的能力。最近研究认为CER可催化Fe2+氧化为Fe3+。对于CER是否是铜的载体存在不同看法。血清中铜的含量虽有95%以非扩散状态处于CER,而有5%呈可透析状态由肠管吸收而运输到肝的,在肝中渗入CER载体蛋白(apoprotein)后又经唾液酸结合,最后释入血循环。在血循环中CER可视为铜的没有毒性的代谢库。细胞可以利用CER分子中的铜来合成含铜的酶蛋白,例如单胺氧化酶、抗坏血酸氧化酶等。
近年来另一研究结果认为CER起着抗氧化剂的作用。在血循环中CER的抗氧化活力可以防止组织中脂质过氧化物和自由基的生成,特别在炎症时具有重要意义。
CER也属于一种急性时相反应蛋白。
临床意义:血浆CER在感染、创伤和肿瘤时增加。其最特殊的作用在于协助Wilson病的诊断,即患者血浆CER含量明显下降,而伴有血浆可透析的铜含量增加。大部分患者可有肝功能损害并伴有神经系统的症状,如不及时治疗,此病是进行性和致命的,因此宜及时诊断,并可用铜螯合剂-青霉胺治疗。血浆CER在营养不良、严重肝病及肾病综合征时亦往往下降。妇女妊娠期、口服避孕药时其含量有明显增加。
(九)转铁蛋白
转铁蛋白(transferrin,TRF,siderophilin)是血浆中主要的含铁蛋白质,负责运载由消化管吸收的铁和由红细胞降解释放的铁。以TRF-Fe3+的复合物形式进入骨髓中,供成熟红细胞的生成。
TRF分子量约7.7万,为单链糖蛋白,含糖量约6%。TRF可逆地结合多价离子,包括铁、铜、锌、钴等。每一分子TRF可结合两个三价铁原子。TRF主要由肝细胞合成,半寿期为7天。血浆中TRF的浓度受铁供应的调节,在缺铁状态时,血浆TRF浓度上升,经铁有效治疗后恢复到正常水平。
临床意义:血浆中TRF水平可用于贫血的诊断和对治疗的监测。在缺铁性的低血色素贫血中TRF的水平增高(由于其合成增加),但其铁的饱和度很低(正常值在30%-38%)。相反,如果贫血是由于红细胞对铁的利用障碍(如再生障碍性贫血),则血浆中TRF正常或低下,但铁的饱和度增高。在铁负荷过量时,TRF水平正常,但饱和度可超过50%,甚至达90%。
TRF在急性时相反应中往往降低。因此在炎症、恶性病变时常随着白蛋白、前白蛋白同时下降。在慢性肝疾病及营养不良时亦下降,因此可以作为营养状态的一项指标。
妊娠及口服避孕药或雌激素注射可使血浆TRF升高。
有免疫试剂盒供应抗体级标准品。用免疫扩散或浊度法检测。正常成人参考值为2200-4000mg/L。新生儿为1300-2750mg/L。临床评价时常同时测定血清铁含量及TRF的铁结合容量(TIBC),并可计算出的TRF铁饱和度(%)。TRF亦可通过测定而间接计算估得,其计算方程式如下:
TRF(mg/L)=TIBC(μg/L)×0.70
(十)血红素结合蛋白
血红素结合蛋白(hemopexin,Hpx)分子量5.7万,单链多肽,含糖量约22%。正常血浆中含量为500-1000μg/L,和游离血红素有特异结合能力。它可配合结合珠蛋白对血红蛋白进行处理。当广泛溶血时,血浆结合珠蛋白耗竭,循环中游离的血红蛋白可降解为珠蛋白和血红素两部分。血红素不溶于水,可与Hpx结合成复合物而运输到肝,分子中的铁可被机体重新利用,卟啉环降解为胆红素而由胆管排出。Hpx并不能与血红蛋白结合,仅可与血红素可逆地结合,而在血循环中反复利用,这是机体有效地保存铁的又一种方式,而避免血红蛋白和血红素从肾排出体外。
(十一)β2-微球蛋白
β2-微球蛋白(β2-microglobulin,BMG)分子量为11800,存在于所有有核细胞的表面,特别是淋巴细胞和肿瘤细胞,并由此释放入血循环。它是细胞表面人类淋巴细胞抗原(HLA)的β链(轻链)部分(为一条单链多肽),分子内含一对二硫键,不含糖。半寿期约107分钟,可透过肾小球,但尿仅有滤过量的1%,几乎完全可由肾小管回收。
临床意义:在肾功能衰竭、炎症及肿瘤时,血浆中浓度可升高。主要的临床应用在于监测肾小管功能。特别用于肾移植后,如有排斥反应影响肾小管功能时,可出现尿中BMG排出量增加。在急性白血病和淋巴瘤有神经系统浸润时,脑脊液中BMG可增高。因含量微,常用放射免疫方法测定,正常血浆BMG参考值为1.0-2.6μg/L,尿中0.03-0.37mg/d。
(十二)C-反应蛋白
在急性炎症病人血清中出现的可以结合肺炎球菌细胞壁C-多糖的蛋白质(1941年发现),命名为C-反应蛋白(C-reactiveprotein,CRP)。最早采用半定量的沉淀试验,现在制备优质的抗血清,可以建立高灵敏度、高特异性、重复性好的定量测定方法。CRP是第一个被认为是急性时相反应蛋白的,在急性创伤和感染时其血浓度急剧升高。CRP由肝细胞所合成。
CRP含5个多肽链亚单位,非共价地结合为盘形多聚体。分子量为11.5万-14万。电泳分布在慢γ区带,有时可以延伸到β区带。其电泳迁移率易受一些因素影响,如钙离子及缓冲液的成分。
CRP不仅结合多种细胞、真菌及原虫等体内的多糖物质,在钙离子存在下,还可以结合卵磷脂和核酸。结合后的复合体具有对补体系统的激活作用,作用于C1q。CRP可以引发对侵入细胞的免疫调理作用和吞噬作用,而表现炎症反应。
临床意义:作为急性时相反应的一个极灵敏的指标,血浆中CRP浓度在急性心肌梗死、创伤、感染、炎症、外科手术、肿癌浸润时迅速显著地增高,可达正常水平的2000倍。结合临床病史,有助于随访病程。特别在炎症过程中,随访风湿病、系统性红斑狼疮、白血病等。
采用何种免疫化学检测法,取决于各实验室条件和对灵敏度、特异性的要求。免疫扩散、放射免疫、浊度法,以及酶标免疫测定方法均有实用价值。正常值:800-8000μg/L(免疫扩散或浊度法)。
(十三)其他血浆蛋白质
血浆脂蛋白系统将于第四章详细介绍,免疫球蛋白及补体系统由临床免疫学详细介绍,凝血因子由临床血液学详细介绍,此处略去。此外,在血浆中还有一些蛋白质仅择其特点,简介如下:
1.α1-抗糜蛋白酶、间α胰蛋白酶抑制物处于α1、α2区带间。前者分子量6.8万,为急性时相反应蛋白之一;后者分子量16万,可分裂为碎片,具有抑制蛋白酶的作用。
2.一些来源于胎盘的血浆蛋白质除具有激素作用的人类绒毛膜促性腺激素(分子量约4万)及胎盘催乳素(lactogen)外,尚有妊娠相关血浆蛋白质(pregnancy-associatep lasmaprotein,PAPP-A分子量为75万,PAPP-B分子量100万左右)。妊娠特异β-球蛋白(SP)分子量约9万,妊娠期升高,可作为妊娠指标及监测胎儿胎盘功能。
3.溶菌酶分子量约1.5万。正常存在于细胞内的溶酶体及外分泌液(如唾液)中,有天然杀菌作用。由颗粒白细胞及单核细胞中产生,而不存在于淋巴细胞。因此在结核和单核细胞白血病中增高,电泳中可出现于γ区带之后,此溶菌酶可从肾小球滤过,但多数被肾小管重吸收而在小管上皮细胞内分解。可用于肾小管功能的检查。正常血清参考值为3.6-7.8mg/L。
4.癌胚抗原分子量近20万的糖蛋白。在结肠、肺、胰腺、胃及乳腺恶性肿瘤时血浆中浓度可升高。特异性不高,但可用于手术后随访监测手术是否清除彻底及复发,亦可用于监测化疗的进展情况。正常血浆浓度<2.5μg/L。
三、关于血浆蛋白质的正常参考值
上节中列举了一些血浆蛋白的正常参考范围,但必须指出这些数字是相对的。根据多个实验室选用的方法和蛋白质标准品有差异。
特别应提及,近年来许多评论家对“健康”与“疾病”个体正常值的界限提出了更有实用价值的新概念(参阅第二十二章)。除此之外,由于在蛋白质的测定中采用的标准品(基准物质)存在的问题更为复杂,使得各个实验室裼之间的参考值范围不易取得一致。卫生部检验中心和世界卫生组织曾推行供应和采用公认的血浆蛋白质标准品,但尚未能普遍实现。混合血清标准或各个商品化的标准品也很难统一。因此,各家文献中列出的参考值有较大的变异。此点在建立方法学与质量控制中应予妥善处理。关于不同年龄、性别与个体间的差异,作以下几点归纳,可供参考。
(一)年龄组的变异
1.Gitlin等1975年发表了一个很详尽的新生儿和胎儿血浆蛋白成分的数值。以新生儿血浆蛋白浓度/成人血浆蛋白浓度相对比值来看,AFP、α2MG、α1AT浓度在新生儿期显著高于成人。Alb、纤维蛋白原、IgG与正常成人接近,其他各成分特别是IgM、IgA及C3、C4补体成分均偏低。
2.对8-95岁的年龄组分布调查有以下几点特征:
(1)Alb在50岁前保持稳定,50岁以后有下降趋势。
(2)α1酸性糖蛋白在男30岁、女40岁后有上升趋势。
(3)α1脂蛋白、α1AT40岁后有上升趋势。
(4)Hp随年龄增加而增加。
(5)α2MG在40岁前随年龄增加而下降,到老年时又略有上升。
(6)转铁蛋白在男性40岁后有随年龄增加而逐步下降的趋势,女性则在30岁左右达高峰,以后亦逐步下降。
(7)IgA在出生前逐步上升,中年期达高峰。
(二)关于性别的差异
1.男性成人略高于女性的有Alb、α1-酸性糖蛋白、IgA等。
2.女性略高于男性成人的有α1脂蛋白、铜蓝蛋白、α2MG。在妊娠期明显增高的有铜蓝蛋白、转铁蛋白等。
(三)个体不同时期的差异及个体间的差异
Statland等1976年曾用同一方法测定个体24小时内及不同天内的血前浆蛋白水平的变异,并与人群间的变异相比较,获得的概念是个人不同时期的变异大大地小于人群间不同个体间的变异(表2-4)。因此提出用正常健康状态下本身的血浆蛋白质数值作为正常参考值更有效和合理。这一概念是否能通过建立个人健康档案而普遍地用于今后实际工作中去,尚待努力。
表2-4 五种血浆蛋白在个体内与人群间的差异
| 血浆蛋白 | 个体内不同时间的变异(CV%) | 不同个体间的变异(CV%) |
| 结合珠蛋白 | 9.5 | 71 |
| α1-酸性糖蛋白 | 11 | 42 |
| 转铁蛋白 | 2.5 | 9 |
| α1-抗胰蛋白酶 | 3 | 16 |
| α2-巨球蛋白 | 3 | 17 |
四、疾病时血浆蛋白质变化的图谱特征
(一)关于急性时相反应蛋白
急性时相反应蛋白(acutephasereactants,APR)包括AAT、AAG、Hp、CER、C4、C3、纤维蛋白原、C-反应球蛋白等等。其血浆浓度在炎症、创伤、心肌梗死、感染、肿瘤等情况下显着上升。另外有3种蛋白质:前白蛋白、白蛋白及转铁蛋白则出现相应的低下。以上这类蛋白质统称为急性时相反应蛋白,这一现象可称为急性时相反应。这是机体防御机制的一个部分,其详尽机制尚未十分清楚。
当机体处于炎症或损伤状态时,由于组织坏死及组织更新的增加,血浆蛋白质相继出现一系列特征性变化,这些变化与炎症创伤的时间进程相关,可用于鉴别急性、亚急性与慢性病理状态。在一定程度上与病理损伤的性质和范围也有相关。
例如单纯的手术创伤,C-反应蛋白及α1抗糜蛋白酶在6-8小时内即上升。继之在12小时内α1AG上升。在严重病例继之可见到AAT、Hp、C4及纤维蛋白原的增加,最后C3及CER增加,2-5天内达到主峰,同时伴有PA、Alb及TRF的相应下降。如无并发感染,则免疫球蛋白可以没有特殊变化,α2MG亦可无变化。因此结合后几项可以作为监测患者有否伴随失水及血容量变化的指标。以上变化可用表2-5简示。
表2-5 手术创伤后APR的变化
| 血浆蛋白质 | 6-8小时 | 12小时 | 24小时 | 2-3天 | 1周 |
| 前白蛋白 | ↓ | ↓↓ | ↓ | ||
| 白蛋白 | ↓ | ↓ | ↓ | ||
| α-脂蛋白 | ↓ | ↓ | |||
| α1-酸性糖蛋白 | ↑ | ↑↑ | ↑↑↑ | ↑↑ | |
| α1抗胰蛋白酶 | ↑↑↑ | ↑ | |||
| α1-抗糜蛋白酶 | ↑ | ↑ | ↑↑ | ↑↑↑ | ↑↑ |
| α2巨球蛋白 | |||||
| 铜蓝蛋白 | |||||
| 结合珠蛋白 | ↑ | ↑↑ | ↑ | ||
| 血红素结合蛋白 | ↑ | ||||
| 转铁蛋白 | ↓ | ↓↓ | ↓ | ||
| C3 | ↑ | ||||
| 纤维蛋白原 | ↑ | ↑↑ | ↑ | ||
| C-反应蛋白 | ↑ | ↑ | ↑↑ | ↑↑↑ | ↑ |
空格表示无变化
急性心肌梗死后的APR变化常与时间进程及损伤程度相关。一般也可分为3个时期:
1.损伤早期C-反应蛋白、αAG、α1AT,α1AC、Hp及纤维蛋白原均很快上升,3周左右逐步恢复正常。
2.PA、Alb、TRF、α-脂蛋白、IgG5天内明显下降,3周左右逐步恢复。
3.C3、CER中等度增加,2周达高峰,C4、α2MG、IgM变化较小。
其中C-反应蛋白、α-糖蛋白及结合珠蛋白3项与梗死区大小和血清酶的变化呈一定相关。以上现象目前的解释是:在损伤和炎症细胞释放某引起生物活性介质,有证据提示是一些小分子的蛋白质,如白细胞内源性介质(leukocyticeudogenousmediators,LEM)等参与,目前知道有白细胞介质(interleukin)IL-6,肿瘤坏死因子α及β,干扰素以及血小板活化因子等。可导致肝细胞中上述蛋白质的合成增加,以及前白蛋白、白蛋白及转铁蛋白在肝细胞中的合成减少。
(二)风湿病
风湿病可表现急性或慢性炎症过程,包括多方面的变化。炎症主要累及结缔组织,但可伴有多系统的损害。患者血浆蛋白的异常改变主要包括急性炎症反应和由于抗原刺激引起的免疫系统增强的反应,其特征为:①免疫球蛋白升高,特别是IgA,并可有IgG及IgM的升高;②炎症活动期可有α1AG、Hp及C3成分升高。
(三)肝疾病
肝是合成大多数血浆蛋白质的主要器官,肝的枯否细胞可参与免疫细胞的生成调节,因此肝疾病中可以影响到很多血浆蛋白质的变化。在急性肝炎时,可以出现非典型的急性时相反应,如乙型肝炎活动期AAT增高,α1AG大致正常,而Hp常偏低,IgM起病时即可上升,PA、Alb往往下降,特别PA是肝功能损害的敏感指标。肝硬化时可有以下特征:①IgG出现弥散性的增高,以及IgA的明显升高;②α1AT是肝细胞损害的一个敏感指标,升高显著;③C-反应蛋白、CER及纤维蛋白原轻度升高;④α1AG、Hp、C3可由于肝细胞损害而偏低;⑤PA、Alb、α1脂蛋白及TRF明显降低;⑥α2MG则可出现明显地增高。其全貌可参见表2-6。
(四)选择性蛋白质的丢失
肾病患者或某些肠道疾病患者,常可导致血浆蛋白质丢失。其量常与蛋白质的分子量相关。小分子量的白蛋白丢失最为明显,而大分子量的蛋白则可有绝对含量的增加(由于肝细胞补偿性的合成增加)。其特征往往表现为:①Alb明显低下,同时PA、α1AG、αAT及TRF下降;②α2MG、β-脂蛋白及Hp多聚体的增加(Hp2-1、Hp2-2);③免疫球蛋白中Igg 降低,而IgM可有增加。
至于严重肾病致肾小球失去分子筛作用,或严重肠道炎症导致非选择性的蛋白丢失,以及全血丧失均可表现为广泛的低血浆蛋白质血症。这类全低血浆蛋白质图谱也可以在充血性心力衰竭、肝功能衰竭、全血稀释及营养不良时见到。
(五)妊娠期及高雌激素血症
正常妊娠时表现为:①PA、Alb、α1AG及IgG略有降低;②α1AT、CER、TRF及纤维蛋白原有显著增高。α1-脂蛋白可有中度增加。
在用雌激素治疗的个体以及口服避孕药者,可有类似上述血浆蛋白质图谱。
表2-6 几种疾病时血浆蛋白质的变化图谱
| 乙型肝炎 | 肝硬化 | 选择性蛋白丢失 | 妊娠与高雌激素血症 | |
| 前白蛋白 | ↓ | ↓ | ↓ | ↓ |
| 白蛋白 | N或↓ | ↓ | ↓ | ↓ |
| α-脂蛋白 | ↓ | ↑ | ||
| α1-酸性糖蛋白 | ↓ | ↓ | ↓ | |
| α1-抗胰蛋白酶 | ↓ | ↑↑ | ↓ | ↑↑ |
| α2-巨球蛋白 | ↑ | ↑↑ | ↑ | |
| 铜蓝蛋白 | N↑ | ↑↑ | ||
| 结合珠蛋白 | ↓ | N↓ | N | |
| 转铁蛋白 | ↓ | ↑↑ | ||
| β-脂蛋白 | ||||
| C3 | N↓ | N | ||
| 纤维蛋白原 | N | ↑↑ | ||
| IgG | ↑ | ↓ | ||
| IgA | ↑↑ | N | ||
| IgM | ↑ | N↑ | N | |
| C-反应蛋白 | N | |||
| 电泳图谱特征 | PA带↓Alb略↓αβ不规则↑宽γ带(有时可与β融成一片 | PA明显↓Alb明显↓ 宽γ带 | Alb明显↓α2↑β↑γ↓ | Alb略↓α2↑β↑ |
(六)遗传性缺陷
血浆蛋白质的遗传性缺陷,包括个别蛋白质发生变异或其量的完全缺乏与基本缺乏。这一现象多数是由于编码的相应蛋白质基因发生遗传上的突变或缺失。举例如下:
1.α1抗胰蛋白酶缺乏病,患者血浆中α1AT可仅为正常的10%,是一种常染色体的隐性遗传。杂合子患者血清中αAT含量也低于正常。由于α1AT占α1区带中蛋白质的大部分,这种异常在血清电泳中可以初步识别。进一步作免疫化学检查可以确诊。
2.结合珠蛋白缺乏病。
3.转铁蛋白缺乏病,为常染色体显性遗传。
4.铜蓝蛋白缺乏病,为常染色体隐性遗传。
5.补体成分缺失,此病少见。患者可完全缺乏某种补体成分,对感染的易感性增加。
6.免疫球蛋白缺乏,可表现为反复感染,可有一种或多种免疫球蛋白的缺陷。如无γ球蛋白血症或低γ球蛋白血症,全部免疫球蛋白组分均可降低。
7.无白蛋白血症,为极罕见的遗传病,完全缺乏时患者可以不发生严重症状,这是由于球蛋白代偿性的增加。
五、血浆蛋白质的检测及其临床应用
血浆蛋白质的检测及其临床应用可以概括为以下几方面:
1.定量地用化学方法测定血浆总蛋白质以及白蛋白。
2.通过电泳将血浆(或血清)蛋白质初步分离,可以半定量地检测主要蛋白质的组分及其图谱,如Alb、α1、α2、β1、β2、γ等区带,并以相对百分比表示之。
3 .特异的定量测定个别蛋白质,多采用免疫化学的技术,通过制备特异的抗血清(或抗体)测定抗原-抗体复合物。依据抗原抗体结合及其复合物的检测手段可有浊度亮度法、沉淀法、免疫扩散法、免疫电泳法等。如果含量很微的蛋白质则采用放射免疫测定法(RIA)及酶免疫测定法(EIA)。
此处仅从临床的需要,对方法学的临床应用及其进展作一简介。
(一)血清总蛋白质的测定
新鲜全血采取后经自然凝固,析出血清,除去含量约为2-4μg/L的纤维蛋白原,剩下的即为血清蛋白质。健康成人在活动状态采血,其含量为63-83g/L,平卧休息时为60-78g/L。血浆总蛋白质含量的变化不外两大原因;一是血容量的改变(浓缩或稀释);二是个别蛋白质组分的明显增加或减少。血浓缩时的高血浆蛋白血症,各个组分成比例的增加(病史中有失水史)。血稀释时的低血浆蛋白血症亦是相对的,各组分蛋白质仍保持正常的比例。
由于个别蛋白质的变化所致的低蛋白血症,最多见的原因是低血浆白蛋白。轻度的高蛋白血症可由于慢性感染性疾病引起的多克隆,弥散性的γ球蛋白增多症是由于多发性骨髓瘤或异常蛋白血症时单克隆免疫球蛋白增多。
应当指出,在进行化学定量测定血浆蛋白质时,我们作了如下假定:①所有血浆蛋白是纯的多肽链(糖脂类和金属有机物等均不计在内),其含氮量平均为16%;②几百种血浆蛋白其理化性质虽不同,但与化学试剂作用产生的反应(如呈色、沉淀)是一致的。显然,这是过于理想化了的,事实上前一种情况是不存在的,后一种情况在不同蛋白质之间也有很大的差别,因此采用任何一种化学方法作血浆蛋白质的测定,严格来讲都是从实用出发的,是相对的定量。
至今,凯氏定氮法仍然是建立各个具体方法时采用的参考标准方法。
双缩脲比色法是目前首先推荐的蛋白质定量方法。方法操作简便,虽然双缩脲试剂有大同不异。其中酒石酸钾纳可以稳定在碱性溶液中的铜离子,含有碘化物作为抗氧化剂。双缩脲反应生成的复合物其吸收峰为540nm。可采用公认的标准牛血清白蛋白作为标准品,经精确称量,必要时用凯氏定氮法标定。各地质控中心提供的混合标准血清可作为第二参考,血清用量100μl,在10-120g/L浓度范围内呈良好线性关系,批内CV值<2%,其它常用的方法还有:
1.基于蛋白分子中含有酪氨酸和色氨酸而使用的酚试剂比色法 由于各种蛋白质分子中上述两种氨基酸的组成比例不同,特别是白蛋白含色氨酸为0.2%,而γ-球蛋白中含量达2%-3%,导致较大的差异。Lowry的改良法在酚试剂中加入Cu2+,集中原法和双缩脲反应两者的作用,使呈色灵敏度提高。其中75%的呈色依赖于Cu2+。反应产物最佳吸收峰在650-750nm,方法灵敏度为双缩脲方法的100倍左右。有利于检测较微量的蛋白质。但试剂反应仍易受多种化合物的干扰。
2.采用280nm和215/225紫外吸收值,计算蛋白质含量 280nm 是由于蛋白质分子中存在芳香族氨基酸所致。方法的特异性和准确性受蛋白分子中该种氨基酸的含量比例影响甚大。尿酸和肝红素在280nm附近有干扰。紫外区200-225nm是肽健的强吸收峰。在此区域其吸收值为280nm的10-30倍,将血清稀释1000-2000倍可以消除干扰物质的影响。
3.采用沉淀反应进行散射比浊法 用磺柳酸、三氯醋酸等配方,此方法甚为简便,不需特殊仪器,技术关键在于:①选择最佳试剂浓度及温度;②混匀技术;③选用的标准;④待测标本中的蛋白浓度。
4.染料结合法 蛋白质可与某些染料特异结合,如氨基黑(amino black)与考马亮蓝(comassive brilliant blue )。这一性质除了可以用于电泳后的蛋白质区带染色,亦可用于总蛋白质的定量。缺点是多种蛋白质与染料的结合力不一致。考马亮蓝在与蛋白质结合后的吸收峰从465nm移向595nm,这一性质可用分光光度法来定量检测。
关于用化学方法测定白蛋白,现多采用特异性的染料(BCG或BCP)结合法,已于第一节中介绍。
(二)血清蛋白质的电泳分析
醋酸纤维薄膜(ACM)和琼脂糖凝胶是目前最广泛采用的两类介质。巴比妥缓冲液pH8.6,离子强度0.05,标本用量3-5μl,标准电泳条件为CAM每厘米宽电流0.75mA,琼脂糖约为每厘米宽10mA,电泳时间40-60分钟,电泳前沿达6cm左右。虽然目前已开展和应用不少个别蛋白质的测定方法,但血浆蛋白质电泳图谱至今仍然是了解血浆蛋白质全貌的有价值的方法,可用为初筛试验,以提供较全面的信息。正常血清电泳后可以很好地分为5条区带(Alb、α1、α2、β1、β2),新鲜标本可以分出β带(以C3成分为主)。由于各条区带中各个蛋白质组分的重叠、覆盖(如CER常被α2MG及Hp所掩盖),以及某些蛋白质染色带很浅(如脂蛋白和α1糖蛋白),可以用其它染色方法辅助。目前除了常使用的氨基黑和丽春红染料外,还采用灵敏度更高的考马亮蓝。
用血清蛋白质电泳测定各组分的含量,通常可采用各区带的浓度百分比(%)或绝对浓度(g/L)表示之。
用醋酸纤维薄膜电泳测得正常小儿及成人血清蛋白质的参考值可参见表2-7。
在疾病情况下血清蛋白质可以出现多种变化。根据它们在血清蛋白质电泳图谱上的异常特征,不少学者曾试将其分为以下类型,参见表2-8及图2-1。
表2-7 各年龄组血清蛋白质电泳正常值(X±SD)范围
| 年龄 | 总蛋白质(g/L) | 蛋白质各组分的浓度比(%) | ||||
| 白蛋白 | α1 | α2 | β | γ | ||
| 脐带血 | 57 | 69.4 | 2.5 | 5.4 | 7.0 | 15.4 |
| ±12 | ±5.6 | ±1.0 | ±1.6 | ±2.4 | ±4.4 | |
| 新生儿 | 60 | 64.2 | 3.5 | 14.1 | 9.2 | 6.2 |
| ±12 | ±13.2 | ±3.6 | ±2.1 | ±2.4 | ±4.8 | |
| 1-5岁 | 68 | 65.2 | 3.1 | 11.2 | 10.2 | 10.4 |
| ±11 | ±7.8 | ±1.3 | ±2.7 | ±2.6 | ±5.5 | |
| 6-12岁 | 69 | 61.7 | 3.0 | 10.5 | 10.2 | 14.7 |
| ±14 | ±6.1 | ±1.8 | ±3.4 | ±2.0 | ±5.8 | |
| 成人△1 | 64 | 2.8 | 6.6 | 7.2 | 14.4 | |
| 45-78 | 0.8-6.2 | 3.4-12 | 4.8-14 | 7.0-25 | ||
| 2 | 64 | 2.8 | 5.9 | 9.8 | 17.5 | |
| 55-71 | 0.9-4.8 | 2.6-9.2 | 6.3-13.3 | 12.2-22.3 | ||
| 3 | 68.7 | 3.1 | 6.0 | 8.2 | 13.8 | |
| 53-76.6 | 1.3-5.0 | 2.7-9.9 | 5.5-14.9 | 8.8-21.3 | ||
| 4 | 62.2 | 4.2 | 6.6 | 10.2 | 17.33 | |
| 47-77 | 0.7-7.6 | 2.4-10.8 | 4-16.4 | 8.8-25.7 | ||
| 5 | 65.7 | 2.28 | 5.7 | 8.8 | 17.5 | |
| 58.8-72.7 | 0.7-3.8 | 3.6-7.8 | 6.3-11.3 | 12.3-22.7 | ||
△资料取自不同作者:①上海医科大学n=50;②重庆医科大学n=123;③同济医科大学n=132;④苏州医学院n=100;⑤湖南医科大学n=100
表2-8 异常血清蛋白质电泳图谱的分型及其特征
| 血清蛋白质的图谱类型 | 总蛋白质 | Alb | α1 | α2 | β | γ | ||
| 1.低蛋白血症 | ↓↓ | ↓↓ | N↑ | N | ↓ | N↑ | ||
| 2.肾病型 | ↓↓ | ↓↓ | ↑ | ↑↑ | 不定 | |||
| 3.肝硬化型 | ↓N↑ | ↓↓ | N↓ | N↓ | β-γ↑(融合) | |||
| 4.急性炎症或急性时相反应症 | N | ↓N | ↑ | ↑ | N | |||
| 5.慢性炎症型 | ↓ | ↑ | ↑ | ↑ | ||||
| 6.弥漫性肝损害型 | ↓N | ↓↓ | ↑↓ | ↑ | ||||
| 7.弥漫宽γ球蛋白血症型 | ↑ | ↓N | ↑↑ | |||||
| 8.M蛋白血症型 | 在α-γ区带中出现M蛋白峰-M区带峰 | |||||||
| 9.高α2(β)-球蛋白血症 | ↓ | ↑↑ | ↑ | |||||
| 10.妊娠型(高α型) | ↓N | ↓ | ↑ | ↑ | N | |||
| 11.蛋白质缺陷型 | 个别区带出现特征性缺乏 | |||||||
上述电泳图谱分型有助于临床疾病判断的参考。在某些蛋白质异常增多的情况下,可出现异常区带。如高浓度的αFP可以在Alb与α1区带间出现一条清晰的新带(有人称之为肝癌型);CRP异常增高可出现特殊界限的γ区带;单核细胞白血病可出现由于溶菌酶异常增多的γ后区带等;单克隆免疫球蛋白异常症(M蛋白血症)则在α-γ区带中出现一条很深的界限截然的M区带。
在大剂量使用青霉素或水杨酸等药物时,由于药物与白蛋白的结合,可导致这部分白蛋白电泳迁移率的加快而出现区带状的改变。
急性时相反应型常以α1及α2区带加深为特征;妊娠型以α1区带增高为特征,伴有β区带的增高;以α2区带增高为特征的图谱常见于风湿病等免疫反应性疾病。其它慢性炎症则同时有α1、α2及γ-球蛋白的增加。在肝硬化及慢性肝炎伴肝硬化及慢性肝炎伴肝硬化可以出现β、γ区带融合弥散的宽γ带。慢性迁延型肝炎、慢性活动型肝炎及慢性反复感染可以出现多条γ区带的加深。
(三)免疫化学法测定个别蛋白质
散射比浊法和透射比浊法由于测定方法简便、快速而被广泛使用。许多试剂盒供应抗血清及标准蛋白质,即可建立此测定法。此技术可以测定抗原-抗体复合物(沉淀颗粒)形成的量(终点法),亦可采用测定复合物形成的速率(动力学方法,即通过散射浊度计测定抗原-抗体混合反应复合物颗粒形成的时间,即反应速率。一定条件下,反应速率与反应体系中抗原的含量直线相关,可以通过制备标准曲线而计算)。
现已有设计完善的带微电脑进行数据处理的散射浊度计和透射浊度计,以免疫化学系统(immuno-chemicalsystem,ICs )可供应。免疫扩散法不需昂贵设备,放射免疫法则需要液体闪烁计数器及应用放射性同位素。
现将几种常用的免疫化学测定法的特点列表总结比较于表2-9、10中。
表2-9 电泳与免疫化学检测血清蛋白列表比较
| 电泳法(CAM法) | 各种免疫化学分析法 |
| 前白蛋白 | 前白蛋白 |
| 白蛋白 | 白蛋白 |
| α1球蛋白(区带) | α1抗胰蛋白酶 |
| α1酸性糖蛋白 | |
| α1脂球蛋白 | |
| α1脂蛋白 | |
| 甲状腺激素结合球蛋白,皮质醇结合蛋白 | |
| α2球蛋白(区带) | 结合珠蛋白 |
| α2巨球蛋白 | |
| 铜蓝蛋白 | |
| 前β脂蛋白 | |
| β球蛋白(区带) | 转铁蛋白 |
| 血红素结合蛋白 | |
| 补体C4,C3 | |
| β2微球蛋白 | |
| γ球蛋白(区带) | IgG,A,M,D,E |
| C反应球蛋白 |
表2-10 几种免疫化学测定方法的比较
| 灵敏度 | 准确性 | 检测时间 |
| 散射比浊法 10mg/L | 批内CV值<5% | 几分钟(动力学法) |
| 1小时 (终点法) | ||
| 透射比浊法 20-30mg/L | 批内CV值5%-10% | 1小时内 |
| 免疫扩散法 >20mg/L | 批间CV值5%-15% | 1-2天 |
| 放射免疫法 μg/L | 批内CV值5%-10% | 几小时 |
关于免疫化学测定方法中的标准品和方法的标准化问题:含有准确含量的纯抗原蛋白不易买到。制备的抗血清由于其来源不同,其特异性和灵敏度效价有很大差异,一个纯蛋白制剂来制备高特异性的抗血清亦非轻易之举。因此抗血清的制备和方法的标准化是方法推广和使用的关键。据美国病理学会的研究报告,测α1AT用同一标本,使用5种不同的方法在510个实验室报告的结果从1.6-2.34g/L。同一研究中C3补体的测定采用了8种不同方法,测定结果为149-282mg/L。
世界卫生组织目前提供以下参考标准品:IgG 、IgA、IgM、IgD、IgE、αFP、CEA、Alb、C3、CER及TRP。使用国际单位(IU),没有使用绝对的质量单位。美国疾病控制中心(CDC)供应的国家参考标准品人血清蛋白质含13种蛋白质,亦使用WHO的国际单位标明含量。
由于不易获得稳定的抗血清和标准化的参考蛋白质,各个实验室还不得不根据自己的条件建立自己的正常参考值。
第二节 细胞骨架蛋白——组织特异性蛋白的鉴定及其意义
人类基因组(genome)含有3万-5万个可单拷贝的结构基因,但在一个细胞生长的特定条件下,往往只有少数基因表达。有些基因几乎普遍地在所有细胞活跃地处于表达状态,并保证其表达产物的功能。如多数酵解酶类蛋白、钙调节蛋白。此外,多数细胞均有其细胞骨架的特定组分,或产生特定代谢的酶。这些蛋白在各细胞中表现出它的组织特异性(tissue-speificprotein),当有关细胞损害过程中,它们在血循环中可以出现,可以反应该细胞的特异损害。这一事实被利用于监测某组织是否进行性损害的一种非损伤性的手段,并正在发展中。
有几种方法可以检测出特殊蛋白的组织特异性,如果该蛋白本身就是酶,可以直接测定酶的活性。例如肌酸激酶在骨骼肌中含量很高,但它们并不存在于肝内。血清中该酶的活性可以反应肌肉的损害,特别是骨骼肌的病变——肌萎缩症,或心肌损害。如果该蛋白并非具有酶活性,但可以表现一定的抗原性,可用于制备相应的抗体,用相应的免疫学方法来检测。新的组织特异性蛋白质也可以通过高分辨率的双相电泳色谱而定位,可以比较不同的组织,而检出特殊器官中特异性蛋白的存在。
一个真核细胞的胞质部分(往往指细胞在去除其亚细胞的结构组分——包括线粒体和内质网及核微粒等成分后,残留下的可溶性部分),往往尚含有20%-30%高浓度蛋白质溶液,各个蛋白质之间具有弱的相互作用力。这些蛋白质可导致细胞内水形成两个部分,即水化的结合水分子与蛋白分子表面结合以及自由水。这些细胞质中的由蛋白丝组成的非膜相结构统称为细胞骨架(cytoskeleton),根据目前的研究,按纤维直径的大小又可将其分为微管(microtube,直径约24nm)、微丝(microfilament,直径约5-8nm)、中间纤维或称中间丝(intermediatefilament,直径约7-12nm)以及比微丝更细且不规则的纤维网,称为微梁格(microtrabecularlatticesystem,直径<6nm)。
细胞骨架蛋白在细胞运动、分裂、信息传递、能量转换、代谢调控以及纤维细胞形态方面具有重要作用。
一、微管
微管可在所有哺乳类动物细胞中存在,除了红细胞外,所有微管均由约55ku的α及β微管蛋白(tubulin)组成。它们正常时以αβ二聚体形式存在(110ku)并以头尾相连的方式聚合,形成微管蛋白原纤维(protofilament),由13根这样的原纤维构成一个中空的微管。
从各种组织中提纯微管蛋白可以发现还存在一些其他蛋白成分(5%-20%),称之谓微管相关蛋白(microtubeassociatedproteinsorMAPS’)。这些蛋白具有组织特异性,表现出从相同αβ二聚体聚合形成的微管具有独特的性质,已从人类不同组织中发现了多种α及β微管的等点异质体(variants),并追踪微管基因表现出部分基因家族,某些基因被认为是编码独特的微管蛋白。
在人类至少发现两种明显区别的α-微管蛋白及三种明显区别的β-微管基因,空们产生具有特定功能的微管蛋白mRNA,由于这些编码在结构组分上十分近似蛋白质分子,在不同组织存在多少特异性的具有差异表达的微管蛋白亚型,尚待深入研究。
二、微丝
微丝(microfilament)也普遍存在于所有真核细胞中,是一个实心状的纤维,一般细胞中含量约占细胞内总蛋白质的1%-2%,但在活动较强的细胞中可占20%-30%。
微丝的主要化学成分是肌动蛋白(actin)和肌球蛋白(myosin),如同微管蛋白,肌动蛋白的基因组成一个超家族并有多种结构极为相似的组成。在肌细胞中至少存在4种不同的肌动蛋白:①骨骼肌的条纹纤维;②心肌的条纹纤维;③血管壁的平滑肌;④胃肠道壁的平滑肌。它们在氨基酸组分上有微小的差异(大约在400个氨基酸残基序列中有4-6个变异),在肌肉与非肌细胞中都还存在β及γ肌动蛋白,它们与具有横纹的α肌动蛋白可有25个氨基酸的差异。
单体的或G-肌动蛋白可聚合为呈纤维状的F-肌动蛋白,它们可由Mg2+及高浓度的K+或Na+诱导而聚合,聚合后ATP水解为ADP及C-肌动蛋白ADP单体,而从组装成F-肌动蛋白的多聚体上游离下来。在骨骼肌肌动蛋白的细丝与肌球蛋白的粗丝相互作用产生肌收缩(肌球蛋白可以起作肌动蛋白激活的ATPase的作用)。肌球蛋白也存在于哺乳动物的非肌细胞中(但以非聚合状态存在)。
总之,微丝具有多种功能,在不同细胞的表现不同,在肌细胞组成粗肌丝、细肌丝,可以收缩(收缩蛋白),在非肌细胞中主要起支撑作用、非肌性运动和信息传导作用。
三、中间纤维
细胞骨架的第三种纤维结构称中等纤维或中间纤维(intermediatefilment,IF),又称中丝,为中空的骨状结构,直径介于微管和微丝之间,其化学组成比较复杂,在不同细胞中,成分变化较大。
免疫学试验与生物化学方法已确认动物细胞中有5种中间纤维,可见于表2-11。
表2-11 中间纤维丝蛋白的分类
| 中间纤维的类型 | 分子量(ku) | 多肽数 | 细胞类型 |
| 波形蛋白纤维(vimentin) | 54 | 1 | 成纤维细胞 |
| 上皮细胞 | |||
| 软骨细胞 | |||
| 淋巴细胞 | |||
| 结蛋白纤维(desmin) | 53 | 1 | 骨骼肌,心肌 |
| 平滑肌 | |||
| 角质蛋白纤维(keratins) | 40-68 | 19 | 上皮 |
| 包括各种角蛋白丝 | |||
| (头皮,指甲等) | |||
| 神经胶质蛋白纤维(glialfibrillaryacidicprotein,GFAP) | 51 | 1 | 神经胶质细胞 |
| 经中等纤维 | 63,160,200 | 3 | 只存在于中枢神经系统、外周神经细胞的树突与轴突及核周围 |
以上5种中间纤维有共同的基本结构,即由311-314个氨基酸构建成一个中央α螺旋杆状区,两侧则是大小和化学组成不同的端区。端区的多样性决定了中间纤维外形和性质的差异和特异性。
表2-11中见人类角蛋白可有19种不同型别,可由双相凝胶电泳行为及氨基酸序列而区别,它们又可分为I型(1-9)、II型(10-19),它们表现组织定位的特异性。例如4、13号与食管和舌的上皮相关,3、12则和角膜相关。这5种中间纤维蛋白存在于原始的脊椎动物,可见在进化过程中具有较强的保守性。多种中间纤维蛋白的抗体目前可用于免疫组织化学方法来检测人类的癌肿,并诊断哪些细胞已转化为恶性,例如抗细胞角蛋白抗体可用于鉴别多种类型皮肤和肺部的肿瘤等。
第三节 细胞调节因子
一、概述
细胞调节因子是一组小分子或中等分子量的可溶性蛋白质(多肽)与糖蛋白,具有强大的和多方面的生物效应。它们均作用于特异的靶细胞表面受体,通过细胞内信号传导和第二信使介导,调节细胞的增殖、分化、生长、出血、骨发生、免疫过程、创伤的愈合、炎症反应等。较大分子量的调节因子前身物质经蛋白酶切水解为较小的活性分子(成熟分子),糖蛋白分子中的含糖结构对其药理动力学有显着影响。已知不少疾病过程与细胞因子生成的失平衡有密切联系,它们异常过度的分泌可诱发和延长病理过程,有些疾病可恶化,或受到一些起始因子(如病毒与细胞的感染等)的作用后,造成细胞分泌异常而发病。细胞因子的生成异常亦与一些免疫介导的疾病有关,如过敏、哮喘、类风湿性关节炎、系统性红斑狼疮、自身免疫性全细胞缺乏症、牛皮癣等。细胞因子广泛地介入恶性肿瘤的增殖分化、转移,如多发性骨髓瘤,急性白血病,慢性淋巴细胞白血病,霍奇金病。细胞因子更广泛地从多层次以多种形式介导一系列炎症发病进程,如胰腺炎、毛细管渗出性综合征、骨质疏松症、创伤愈合、肾衰等。因此,近年来,细胞因子的分泌表达及其作用引起多方面专家的关注和研究,细胞因子的检测也迅速发展起来。
本节从生物化学与分子生物学基础对其发展中的几个基本问题作一简介。
细胞因子的发现近半个世纪,由于其生物学作用的多效性,细胞来源的多样性,曾经赋予过不少名称。自80年代基因工作和蛋白质的优化技术发展,细胞因子基因被克隆至今,虽然多种细胞因子的蛋白质一级结构被阐明,其相应的mRNA及DNA序列分析及其染色体定位被确认,但由于一种细胞因子可由多种细胞产生,不同细胞来源的细胞因子又有相似的生物学作用(多效性),加上生物学作用的环境依赖性,因此命名至今不够满意,更不甚统一,分类上的意义也是相对的,有的侧重考虑其细胞来源,有的着重于参照其生物学作用,见表2-12。
表2-12 细胞因子的分类
| 1.促红细胞生成素(EPO) | 5.肿瘤坏死因子(TNF) |
| 2.集落刺激因子(CSF) | 肿瘤坏死因子α(TNF-α) |
| 粒细胞巨噬细胞集落刺激因子(GM-CSF) | 肿瘤坏死因子β(TNF-β) |
| 粒细胞集落刺激因子(G-CSF) | 6.干扰素(IFN) |
| 巨噬细胞集落刺激因子(M-CSF或CSF-1) | 干扰素α-1(IFNα-1) |
| 干细胞因子(CSF) | 干扰素α-2(IFNα-2) |
| 白介素-3或多集落刺激因子(IL-3或multi-F) | 干扰素β(IFNβ) |
| 白血病抑制因子(LIF) | 干扰素γ(IFNγ) |
| 胰岛素样生长因子(IGF) | 7.转化生成因子(TGF) |
| 3.血小板生成素(TPO) | 转化生长因子β-1(TGFβ-1) |
| 4.白介素(IL) | 转化生成因子β-2(TGFβ-2) |
| 白介素-1α(IL-1α) | 转化生长因子β-3(TGFβ-3) |
| 白介素-1β(IL-1β) | 8.巨噬细胞炎性蛋白(MIP) |
| 白介素-2(IL-2) | 巨噬细胞炎性蛋白1α(MIP-1α) |
| 白介素-3(IL-3或multi-CSF) | 巨噬细胞炎性蛋白1β(MIP-1β) |
| 白介素-4到白介素-13(IL-4,IL-5,IL-6,IL-7,IL-8,IL-9,IL-10,IL-12,IL-13) | 巨噬细胞炎性蛋白2(MIP-2) |
| 9.其他 | |
| 单核细胞化学吸附蛋白-1(MCP-1) | |
| 血小板因子4(PF4) | |
| β-凝集球蛋白 | |
| 黑色瘤生长刺激活性(GROimgSA) |
有些研究者按其主要特征,归纳为以下几类:
1.天然免疫的效应分子(如α/β干扰素,TNF,IL-1,IL-6等)。
2.炎症反应的激活因子(IL-1,IL-5,IFN-1等)。
3.淋巴细胞活化生成与分化的调节因子(IL-2,IL-4,TGFβ等)。
4.未成熟免疫细胞生长分化的刺激因子(IL-3,IL-7,GM-CSF,M-CSF,G-CSF)。
也有一些研究者依据其生物学特征从临床病理出发分为以下6大类:
1.炎症因子(介导炎症发生发展)又称为炎症反应因子(inflamatoryfactor)(如IL-1,IL-6,IL-8,TNF等)。
2.杀伤因子(cytotoxicfactor)(如TNF-γ,TNF-β)。
3.干扰素(有抗病毒作用的IFN-γ,β,α)。
4.白介素类,促细胞活化、增殖、分化、调节其功能。
5.成血因子(hematopoieticfactor)(SCF,IL-3,GM-CSF,G-CSF,M-CSF等)
6.转化生长因子(transforminggrowthfactor,TGF)。
最近由于分子生物学研究的进展,将已发现的细胞因子以分子结构为基础,结合其基因组合受体类型,将它们归纳为以下6个不同的家族,列表2-13下可供参考。
表2-13 细胞因子的结构型家族
| 家族(family) | 细胞因子(cytokines) | 受体类型(receptortype) |
| Haematopoietins(4α-helicalbundles) | IL-2,IL-3,IL-4,IL-5,IL-6,IL-7,IL-8,IL-9,IL-14,G-CSF,GM-CSF,CNTF,OSM,LIF,Epo | CytokinereceptorclassI |
| IL-10,IFNαIFNβIFNγ | CytokinereceptorclassII | |
| EGF(β-sheet) | EGF,TGFα | Tyrosinekinase |
| FGFα,FGFβ | Splittyrosinekinase | |
| IL-1α,IL-1β,IL-1Rα | IL-1receptor | |
| TNF(jellyrollmotif) | TNFα,TNFβ,LTβ | |
| Cysteineknot | NGF | |
| TGFβ1,TGFβ2,TGFβ3 | Serine/threoninekinase | |
| PDGF,VEGF | Tyrosinekinase | |
| Chemokines | ||
| (triple-strandedantiparallelβ-sheetinGredkkeymotif) | IL-8,MIP-1α,MIP-1β,MIP-2,PF-4,PBP,I-309/TCA-3,MCP-1,MCP-2,MCP-3,γIP-10 | Rhodopsinsuperfamily |
各个细胞因子的生物化学及其分子生物学正在积累大量的资料,学者可从以下几方面了解和收集有关材料加以深入系统的研究。
1.使用过的其它名称,多数细胞因子在发现它们存在的早期,曾基于其生物效应的某一侧面或其发现的历史、细胞来源以及生物检测方法的使用而使用过多种名称,这些名称不一定是反映该因子最本质的方面,并易混淆,宜予注意。
2.分子大小、分子特征、分子量。
3.氨基酸序列和三维空间结构(包括来自X线及NMR获得的数据)
4.交叉反应性(crossreactivity),比较不同种属(如人和鼠)细胞因子氨基酸序列的一致性及其差异。
5.已知可以分泌该细胞因子的细胞类型及细胞因子的表达调控。
6.主要物理化学性质,包括前体的分子量和去除信号肽后成熟分子的分子量及其酶切点。
7.基因结构及其染色体定位,包括有关外显子和内含子的基因结构及相应的氨基酸序列。
8.受体的描述、类型,包括蛋白质的主区域细胞膜结合的模式和糖基化的程度,受体细胞分布,受体的物理化学性质,氨基酸序列,染色体定位等。
9.受体后信号传递体制和类型-细胞内信号传递途径的了解情况。
二、细胞调节因子实验室检测的简介
研究细胞因子在各种病理生理情况下的活动、分泌以及在治疗中使用细胞因子制剂的效应,都要求能有效地检测体内细胞因子的含量(浓度水平),以及检测细胞因子的功能(生物效应)。
从前以细胞因子的生物学特征作为其含量分析的基础,免疫分析及其它免疫化学或生物化学技术亦可用于此细胞因子的测定和部分鉴定,表2-14总结了已用于细胞因子功能测定和浓度(含量)测定的方法。
表2-14 细胞因子分析
| 生物分析 | 酶联免疫吸附分析(ELISA) |
| 动物分析 | 其它 |
| 增殖分析 | 受体结合分析 |
| 骨髓集落形成分析 | 免疫化学程序 |
| 细胞毒性或细胞稳定性分析 | 细胞或细胞中的测定 |
| 抗病毒分析 | 匀浆生物分析或免疫分析 |
| 次级分子分泌的诱导 | 免疫组化或免疫细胞分析 |
| 趋化作用或化学促进作用分析 | 流式细胞计数 |
| 细胞因子分泌活性的抑制 | 酶联免疫印迹(ELISPOT) |
| 免疫分析 | 反相溶血斑分析 |
| 放射免疫分析(RIA) | 细胞因子mRNA原位杂交 |
| 免疫放射光谱分析(IRMA) |
人细胞因子实验测定的标准化:人细胞因子的实验测定中仍存在诸多问题,选择分析方法不恰当则可能导致混乱甚至错误的结论。有时有必要同时选择几种方法分析某些细胞因子。生物分析方法特异性差,那些使用细胞株的生物分析可能与不同细胞因子反应,而且可受非细胞因子分子的影响。用于维持细胞生物的细胞因子能强烈地影响某些依赖细胞对一些细胞因子的反应程度。有的细胞在连续传代后可能失去对某些细胞因子的反应性,此外,由于抑制剂如受体拮抗剂、可溶性细胞因子受体或其他拮抗剂分子的存在,测得细胞因子活性结果可能会比实际活性偏低。
由于蛋白酶的存在或与抑制剂如可溶性受体形成复合物,免疫分析能测得失去生物活性或具部分生物活性的细胞因子分子,并可受到基质效应的影响。此外,特异性问题影响了细胞因子mRNA测定,而mRNA水平亦不一定与具生物活性的细胞因子水平相一致。其它影响自动免疫分析方法准确性的问题还包括抗体的选择、分析的不均匀、分子糖基化的不同、寡聚体的形成及循环受体的影响,虽然免疫分析的结果并不一定反映细胞因子的生物活性,但一般而言,免疫分析及其它配体结合分析都更容易标准化。
目前,英国已能提供下列细胞因子的标准化参考试剂:IL-1α,IL-1β,IL-2-9,GM-SCF,M-CSF,TGF-α,TGF-β,TGF-γ,SCF,LIF(白血病抑制因子),IFN-α1,IFN-α2b,IFNγ,还能供应IFN-α2a,IFN-β及IFN-γ的国际标准。但于细胞因子分子的不均匀,其分析的标准化仍是问题。因此,要求各实验室的细胞因子分析应参考国际标准。某些细胞因子具有易被破坏的特性,从而要求非常细致和标准化的取样。
随着细胞因子在治疗上的不断应用,其测定方法亦得到了广泛的发展,临床检师必须选用正常的分析方法支持临床医生,为临床诊断提供相对简易的实验程序和准确的操作技术。
第三章 糖代谢紊乱
糖类的营养价值主要是供给能量,此外糖也是人体的重要组成成分之一。正常人体内糖代谢的中心问题之一是维持血糖浓度的相对恒定。临床上重要的糖代谢紊乱也主要是血糖浓度过高(高血糖症)和过低(低血糖症)。本章重点讨论高血糖症,对低血糖症及部分先天性糖代谢异常仅作简要阐述。
第一节 概述
一、糖的重要生理作用
糖类(主要是淀粉)是食物的主要成分。食物中的淀粉、糖原、蔗糖和乳糖等,在肠道经消化成为单糖后再被吸收,然后由血液运送到全身各组器官,供细胞利用或合成糖原贮存。糖的生理作用主要体现在两方面:
1.氧化供能糖的主要生理功能是氧化供能,每g糖完全氧化可释放16750J(4kcal)能量。我国一般膳食中,糖类所供给的能量约为总能量的75%左右,糖也是最易被消化吸收的能源物质。要避免酮症的发生必须保证每100g的膳食中至少要含5g糖类物质,要阻止因肌饿或高脂膳食引起的酮症,每日膳食中须有50~100g糖类。
2.人体的主要组成成分之一糖和蛋白质结合形成的糖蛋白,是某些激素、酶、血液中凝血因子和抗体的成分,细胞膜上某些激素受体、离子通道和血型物质等也是糖蛋白。结缔组织基质的主要成分-蛋白多糖,是由氨基多糖和蛋白质所结合组成的。糖和脂类结合则形成糖脂,糖脂是神经组织和生物膜的重要组份。糖在体内可以转化成为脂肪、非必需氨基酸,并以核糖形式参与核酸的组成。
在整个人体重中,糖占人体干重的2%。
所以,糖既是人体重要的人供能物质,又是人体重要的组成成分之一。糖代谢障碍,首先导致机体能量供给障碍,由此可以产生一系列代谢变化,最终造成多方位的代谢紊乱,重者将危及生命。
二、血糖及其来源与去路
血糖是指血液中糖,由于正常人血液中糖主要是葡萄糖,且测定血糖的方法也主要是检测葡萄糖,所以一般认为,血糖是指血液中的葡萄糖。正常人空腹血糖浓度为4.4~6.7mmol/L(80~120mg/100ml),它是糖在体内的运输形式。全身各组织都从血液中摄取葡萄糖以氧化供能,特别是脑、肾、红细胞、视网膜等组织合成糖原能力极低,几乎没有糖原贮存,必须不断由血液供应葡萄糖。当血糖下降到一定程度时,就会严重妨碍脑等组织的能量代射,从而影响它们的功能。所以维持血糖浓度的相对恒定有着重要的临床意义。
正常人血糖浓度虽有波动,但可保持相对恒定在4.4~6.7mmol/L范围内。这些神经、肝脏等组织和激素对血糖的调节作用,使血糖的来源和去路达到动态平衡的结果。
血糖的来源有:①食物中的糖类物质经消化吸收进入血中,这是血糖的主要来源;②肝贮存的糖原分解成葡萄糖入血,这是空腹时血糖的直接来源;③在禁食情况下,以甘油、某些有机酸及生糖氨基酸为主的非糖物质,通过糖异生作用转变成葡萄糖,以补充血糖。
血糖的去路有:①葡萄糖在各组织细胞中氧化分解供能,这是血糖的主要去路;②餐后肝、肌肉等组织可将葡萄糖合成糖原,糖原是糖的贮存形式;③转变为非糖物质,如脂肪、非必需基酸等;④转变成其它糖及糖衍生物,如核糖、脱氧核糖、氨基多糖、糖醛酸等;⑤当血糖浓度高于8.9mmol/L(160mg/100ml)时,则随尿排出,形成糖尿。正常人血糖虽然经肾小球滤过,但全部都被肾小管吸收,故尿中糖极微量,常规检查为阴性。只有在血糖浓度高于8.9mmol/L,即超过肾小管重吸收能力时,尿糖检查才为阳性。糖尿多见于某些病理情况,如糖尿病等。
血糖的来源与去路总结为图3-1。
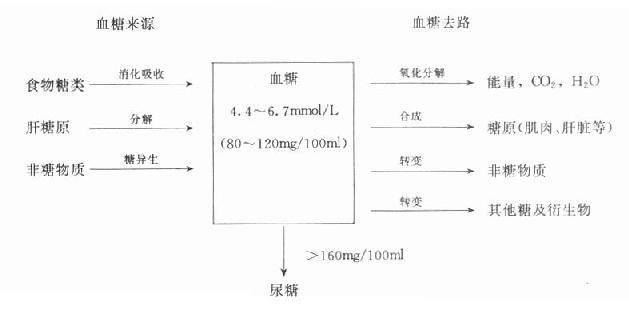
图3-1 血糖的来源与去路
三、血糖浓度的调节
血液浓度能维持相对恒定是由于机体内存在一整套高效率的调节机制,精细地控制着血糖的来源与去路,使之达到动态平衡。
(一)神经系统的调节作用
神经系统对血糖浓度的调节作用主要通过以下丘脑和自主神经系统对所控制激素的分泌,后者再通过影响血糖来源与去路关键酶的活性来实现。神经系统的调节最终通过细胞水平的调节来达到目的。
下丘脑一方面通过内脏神经作用于肾上腺髓质,刺激肾上腺素的分泌;另一方面也作用于胰岛α-细胞,使其分泌胰高血糖素;同时还可以直接作用于肝。三方面共同作用的结果是使肝细胞的磷酸化酶活化,使糖原分解加速;糖异生关键酶的活性增加,糖异生作用增加,从而使血糖浓度升高。
下丘脑了可通过迷走神经兴奋,使胰腺β-细胞分泌胰岛素,同时还可直接作用于肝,使肝细胞内糖原合成酶活化,促进肝糖原的合成;此外还抑制糖异生途径,促进糖的氧化和转化,总体上使血糖的去路增加,来源减少,最终达到使血糖浓度降低的目的。
(二)激素的调节作用
调节血糖浓度的激素可分为两大类,即降低血糖浓度的激素和升高血糖浓度的激素。各类激素调节糖代谢反应从而影响血糖浓度的机制在表3-1中简要说明。
表3-1 激素对血糖浓度的调节作用
| 降低血糖的激素 | 升高血糖的激素 | ||||
| 激素 | 对糖代谢影响 | 促进释放的主要因素 | 激素 | 对糖代谢影响 | 促进释放的主要因素 |
| 胰岛素 | 1.促进肌肉、脂肪组织细胞膜对葡萄糖通透性,使血糖容易进入细胞内(肝、脑例外) | 高血糖、高氨基酸、迷走神经兴奋、胰泌素、胰高血糖素 | 肾上腺素 | 1.促进肝糖分解为血糖 | 交感神经兴奋,低血糖 |
| 2.促进肝葡萄糖激酶活性,使血糖易进入肝细胞内合成肝糖原 | 2.促进肌糖原酵解 | ||||
| 3.促进糖氧化分解 | 3.促进糖异生 | ||||
| 4.促进糖转变成脂肪 | 胰糖高血素 | 1.促进肝糖原分解成血糖 | 低血糖、低氨基酸、促胰酶素(肝囊收缩素) | ||
| 5.抑制糖异生 | 2.促进糖异生 | ||||
| 糖皮质激素 | 1.促进肝外组织蛋白质分解生成氨基酸 | 应激 | |||
| 2.促进肝内糖异生 | |||||
| 生长素 | 早期:有胰岛素样作用(时间很短) | 低血糖,运动,应激 | |||
| 晚期:有抗胰岛素作用(主要作用) | |||||
1.胰岛素胰岛素是胰岛β细胞分泌的一种蛋白类激素,由51个氨基酸组成。血中葡萄糖或氨基酸浓度高时,可促进胰岛素的分泌。
胰岛素对血糖的调节机制,首先是使肌肉和脂肪组织细胞膜对葡萄糖的通透性增加,利于血糖进入这些组织进行代谢。胰岛素还能诱导葡萄糖激酶、磷酸果糖激酶和丙酮酸激酶的合成,加速细胞内葡萄糖的分解利用。胰岛素通过使细胞内cAMP含量减少,激活糖原合成酶和丙酮酸脱氢酶系,抑制磷酸化酶和糖异生关键酶等,使糖原合成增加,糖的氧化利用、糖转变为脂肪的反应增加,血糖去路增快;使糖原分解和糖异生减少或受抑制,使血糖来源减少,最终使血糖浓度降低。
近年来从人血清中分离出的类胰岛素生长因子(insulin-likegrowthfactor,IGF,也称somatomedins)其化学结构和生物学特性类似胰岛素,但IGF的免疫学性质与胰岛素完全不同。IGF通过IGF受体和胰岛素受体而发挥作用。但IGF促进血糖降低的快速效应仅相当于胰岛素的一部分,例如:①促进脂肪细胞转变、摄取和氧化葡萄糖,并合成脂肪的强度仅为胰岛素的1/50或1/100;②对心肌细胞摄取葡萄糖的作用为胰岛素的1/2或1/5;对骨骼肌摄取、氧化葡萄糖及合成糖原的作用只有胰岛素的1/20。IGF的长期效应是促进生长。
2.胰高血糖素是胰岛α细胞合成和分泌的由29个氨基酸组成的肽类激素,分子量为3500。其一级结构和一些胃肠道活性肽如胰泌素、肠抑制胃肽(GIP)等类似。血糖降时胰高血糖素分泌增加,高糖饮食后其分泌则减少。
胰高血糖素主要通过提高靶细胞内cAMP含量达到调节血糖浓度的目的。细胞内的cAMP可激活依赖cAMP的蛋白激酶,后者通过酶蛋白的共价修饰改变细胞内酶的活性,即激活糖原分解和糖异生的关键酶,抑制糖原合成和糖氧化的关键酶,使血糖升高。该蛋白激酶还激活脂肪组织的激素敏感性脂肪酶,加速脂肪的动员和氧化供能,减少组织对糖的利用,从而加重血糖升高。目前认为,胰高血糖素是使血糖浓度升高的最重要的激素。
胰高血糖素的前体为无活性的胰高血糖素原。由肠道上皮细胞生成和分泌的类似胰高血糖素的物质叫肠高血糖素。所以,用一般免疫法测得的高血糖素由胰高血糖素、胰高血糖素原、肠高血糖素3种形式组成,正常血浆中的基础浓度为50-100ng/L。
在激素发挥调节血浆浓度的作用中,最重要的是胰岛素和胰高血糖素。肾上腺素在应激时发挥作用,而肾上腺皮质激素、生长激素等都可影响血糖水平,但在生理性调节中仅居次要地位。
综上所述,胰岛素和胰高血糖素是调节血糖浓度的主要激素。而血糖水平保持恒定则是糖、脂肪、氨基酸代谢协调的结果。
⒊肝在糖代谢调节中的作用肝是调节血糖浓度的主要器官,这不仅仅是因为肝内糖代谢的途径很多,而关键还在于有些代谢途径为肝所特有。
餐后食物中糖类经消化吸收,以葡萄糖形式大量进入血液,使血糖浓度暂时轻度升高。此时葡萄糖直接促进肝等组织摄取葡萄糖,使肝细胞内糖原合成明显增加,同时也抑制肝糖原的分解,减少其向血中释放葡萄糖,同时还使糖转为脂肪,结果是餐后血糖浓度仅轻度升高,并很快恢复至正常范围。饥饿时肝通过自己特有的葡萄糖-6-磷酸酶,将贮存的肝糖原分解成葡萄糖以提供血糖,而肌糖原则不能转为葡萄糖。
肝还是糖异生的主要器官(表3-2),在生理情况下,甘油、氨基酸等非糖物质主要在肝细胞骨转变成葡萄糖,以补充血糖因空腹所致血糖来源不足。这是因为糖异生途径的关键酶:丙酮酸羧化酶、磷酸烯醇式丙铜酸羧激酶的活性似肝最高。饥饿或剧烈运动时,肝脏利用非糖物质转变成糖的作用尤为显著。此外,肝所具有的果糖二磷酸酶、葡萄糖-6-磷酸酶在其他单糖转化为葡萄糖的方面也起着重要作用。
表3-2 空腹和长期饥饿时的糖异生作用
| 葡萄糖的异生作用 | ||||
| 条件 | 异生器官 | |||
| 速率(克/天) | 肝 | 肾 | ||
| 空腹 | 150-300 | >90% | <10% | |
| 饥饿5-6周后 | 86 | 50% | <45% | |
由此可见,肝在血糖的来源与去路方面所发挥的作用较其他器官全面,所以它是维持血糖恒定的关键器官。当机体需要时,通过神经-激素的作用,使肝细胞内各种糖代谢途径的酶活性改变,实现肝维持血糖浓度恒定的目的。当肝功能严重受损时,进食糖类或输注葡萄糖液都可发生一时性高血糖甚至糖尿,而饥饿时则可出现低血糖症状。
现将血糖三个水平的调节简要总结于图3-2。
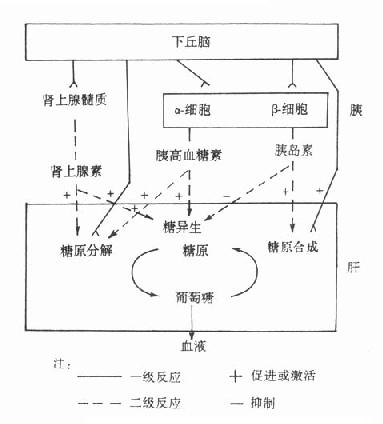
图3-2 血糖调节的主要机制
第二节 高血糖症与糖尿病
高血糖症(hyperglycemia)是指空腹血糖高于正常上限7.3mmol/L(130mg/dl),血糖高于肾糖阈值9.0mmol/L(160mg/dl),则出现尿糖。在某些生理情况下,如情绪激动致交感神经系统兴奋,促使肾上腺素等分泌增加,使血糖浓度升高,出现尿糖,称为情感性糖尿;一次性食入大量糖,血糖急剧升高,出现糖尿,称为饮食性尿糖。上述两种暂时性高血糖及尿糖均为生理性高血糖及尿糖,受试者空腹血糖浓度均在正常水平,且无临床症状和意义。
临床上最常见的病理性高血糖症是糖尿病。糖尿病是一种以糖代谢为主要表现的慢性、复杂的代谢性疾病,系胰岛素相对或绝对不足,或利用缺陷而引起。虽然糖尿病的病因和生化缺陷尚未被彻底阐明,但目前较一致的认识是:该病是一种家族性疾病,其易感性有很大的遗传因素。糖尿病的临床特征是血糖浓度持续升高,甚至出现糖尿。重症病人常伴有脂类、蛋白质代谢紊乱和水、电解质、酸碱平衡紊乱,甚至出现一系列并发症,重者可致死亡。糖尿病是临床常见病之一,我国发病率为1%以下。
一、糖尿病的分型
糖尿病的分型曾经较混乱,过去临床上习惯按发病年龄将其分为儿童或青少年型和成年型两类,现在一般分为三型。
⒈胰岛素依赖型糖尿病(insulindependentdiabetesmellitus,IDDM也称为Ⅰ型糖尿病)此型好发于青春期(20岁以下),对胰岛素治疗敏感。其发病和遗传因素密切相关,大多数(80%以上)病人属于自身免疫性疾病,患者血清中发现有胰岛素抗体,使胰岛素不能发挥其正常生物学活性。病人胰岛素分泌菜单现为逐渐减退,以致完全丧失,需依赖补给胰岛素才能生存。由于患者的胰岛β细胞有缺陷,胰岛素生成和分泌不足,血中胰岛素水平低下,胰岛素/胰高血糖素比值降低,即使在葡萄糖从肠道吸收期间,此比值也不会增高。病人临床症状典型,病情较重,易出现酮症酸中毒,甚至昏迷。Ⅰ型糖尿病的突出特点是:从肠道进入体内的食物不论过多还是不足,各组织持续处于分解代谢状态,如同饥饿一样,其结果将直接威胁生命。
⒉非胰岛素依赖型糖尿病(noninsulindependentdiabetesmellitus,NIDDM,也称为Ⅱ型糖尿病)常见于中壮年肥胖个体。患者血中胰岛素水平不低,甚至有所升高(但低于同等肥胖而无糖尿病的个体)。此型的病变在于靶细胞膜上胰岛素受体数目减少或缺陷,表现为激素-受体亲和力减弱,有的则为正常结合,但结合后反应异常。此种胰岛素受体缺陷所致靶细胞对胰岛素的反应差,不能将胰岛素信息转换为生物效应的现象,称为胰岛素抗性。
Ⅱ型糖尿病人临床症状一般较轻,不发生酮症酸中毒,且对胰岛素治疗不敏感,也不适于用胰岛素治疗。病人可通过控制饮食使症状减轻,通过减肥阻止病情发展。但是,若不能有效控制病情的发展,病情可发展成与Ⅰ型一样,还将伴发神经、眼、肾和心血管系统的疾病。
⒊其它型糖尿病即继发性糖尿病。凡是由于其它已知原因的疾病引起的胰岛素分泌受损,或胰岛素不能正常发挥作用而引起的高血糖症均属此型。如胰腺疾病(胰腺炎等)、胰岛素拮抗激素分泌异常(肢端肥大症、甲状腺功能亢进、皮质醇增多症等)、肝脏疾患(肝硬化等)和某些药物(糖皮质激素、雌激素等)等。
在临床上,各型糖尿病糖代谢异常的共同特征是胰岛素缺乏样表现,病人血糖升高的程度与糖尿病病情的轻重程度密切相关,而NIDDM型约占总病例数的80%-90%(与Ⅰ型比较)。
二、糖尿病的主要代谢紊乱
糖尿病病人代谢异常主要表现在以下四方面:①糖代谢紊乱-高血糖和糖尿;②脂类代谢紊乱-高脂血症、酮症酸中毒;③体重减轻和生长迟缓;④微血管病变、神经病变等并发症。
在人类糖尿病患者中,除少数IDDM型糖尿病人血浆胰岛素减少外,大多数NIDDM型糖尿病人血浆胰岛素的含量正常或升高,这表明糖尿病代谢异常除因胰岛素不足外,还有其它因素存在,其中特别是对胰岛素有拮抗作用的激素如胰高血糖素、生长素、糖皮质激素、儿茶酚胺类激素的分泌过多。临床上可以见到,糖尿病人不论男女,其昼夜生长素水平均高于正常人,这些病人有的在糖尿病前期就已出现生长素分泌增高的现象。此外,病人对一些能引起生长素分泌的刺激(如低血糖、运动等)也反应过高,在运动后儿茶酚胺的分泌也高于正常。所以,糖尿病虽然与胰岛素缺乏的关系最大,但把糖尿病代谢异常产生的原因完全归咎于胰岛素缺乏显然是不够全面的。对胰岛素有拮抗作用的激素,它们对上述四方面代谢变化也有明显的影响。
(一)糖代谢紊乱
糖尿病时常见血糖升高,这是因为胰岛素/胰高血糖素比值降低,肝的糖酵解、糖原合成及生脂作用等途径不易启动,使血糖的去路受阻,而肝糖原分解和糖异生加强,血糖来源增加。在IDDM型糖尿病人,由于胰岛素缺乏,许多组织不能摄取并利用葡萄糖,使血糖进一步上升。糖尿病病人空腹时出现高血糖,主要是因为糖异生作用增强。一般糖异生的速度主要依赖于胰岛素与胰高血糖素、皮质醇、儿茶酚胺等激素之间的平衡,其中胰高血糖素的作用最重要。有人认为糖尿病病人空腹高血糖有25%以上是由于胰高血糖素分泌过多引起,因此主张给病人生长素抑制胰高血糖素的分泌。由于胰岛素/胰高血糖素比值降低,血糖的去路受阻,而糖异生作用却不断进行,肝失去了缓冲血糖水平的能力,因此饱食时造成高血糠。病人体内蛋白质降解为糖异生提供了大量原料,进一步促进糖异生作用,结果是病人在饥饿状态下,血中葡萄糖浓度仍持续升高。
血糖过高可经肾脏排出,引起糖尿,并产生渗透性利尿。糖尿病病人在肾功能正常的情况下,血糖浓度一般不会超过28mmol/L(500mg/dl)。有些老年患者,不但血糖升高,同时伴有肾功能障碍,其血糖含量可极度升高超过33.6mmol/L(>600mg/dl),使细胞外液的渗透压急剧上升,引起脑细胞脱水,出现高渗性高血糖昏迷。在糖尿病患者中,高渗性高血糖性昏迷的死亡率高于糖尿病酮症酸中毒。
(二)脂类代谢紊乱
糖尿病时,由于胰岛素/胰高血糖素比值降低,脂肪分解加速,使大量脂肪酸和甘油进入肝脏。过多的脂肪酸再酯化成甘油三酯,并以VLDL的形式释放入血,造成高VLDL血症(Ⅳ型高脂血症)。此外,LPL(脂蛋白脂肪酶)活性依赖胰岛素/胰高血糖素的高比值,糖尿病时此比值低下,LPL活性降低,VLDL和CM难以从血浆清除,因此除VLDL进一步升高外,还可以出现高CM血症。糖尿病人由于存在高脂血症,所以容易伴发动脉粥样硬化。
糖尿病病人血浆胆固醇常常升高,可能是由于生长素、肾上腺素、去甲肾上腺素增多,这些激素使胆固醇合成的限速酶-HMG-CoA还原酶增加,进而使胆固醇合成增加。
糖尿病时,肝合成甘油三酯的速度增加,如果合成的速度大于释放的速度时,则甘油三酯可以在肝内堆积,形成脂肪肝。
糖尿病时,脂类代谢紊乱除能发生高脂血症外,还会造成酮血症。IDDM型糖尿病人较NIDDM型容易发生酮症。这是因为胰岛素/胰高血糖素比值降低,脂肪酸合成明显减少,而脂肪组织的脂解速度却大大加速,血中脂肪酸升高,肝内脂肪酸氧化增强,酮体大量生成。当酮体生成量超过肝外组织氧化利用它的能力时,就发生酮体堆积,出现酮血症和酮尿症,严重时可发展为酮症酸中毒。
(三)体重减轻和生长迟缓
胰岛素是一种以促进合成代谢为主的储存激素。当胰岛素不足时,体内蛋白质和脂肪的合成均下降,而分解则加速,这是病人体重减轻的重要原因。另一方面,葡萄糖由肾排出造成的渗透性利尿,大量失水,使体重进一步减轻。病人同时还可伴有水、电解质和酸碱平衡失衡。
胰岛素和生长素对促进蛋白质合成具有协同作用,而且生长素促进合成代谢所需要的能量也依赖于胰岛素促进物质的氧化。缺乏胰岛素的糖尿病患儿,即使体内生长素水平较高,仍可见到生长迟缓的现象。
(四)微血管、神经病变和白内障的发生
微血管病变是糖尿病人的严重并发病,其病变主要是肌肉和肾小球等组织的毛细血管基底膜增厚(膜上有大量糖蛋白沉着)以及视网膜血管异常。产生这种病变的原因还不清楚,多数人认为与生长素升高有关。因为糖尿病人血浆生长素水平的高低常与微血管病变有一致的关系,而且生长素介质有促进粘多糖合成的作用。由于高血糖时,许多蛋白质可发生糖基化作用,所以也有人提出,蛋白质的糖基化作用增强,可以促进糖尿病患者发生如冠心病、视网膜病变、肾病及神经病变等一系列并发症。蛋白质糖基化作用增强,是糖尿病患者血管损伤的原因。
糖尿病时,脑细胞中的葡萄糖含量随血糖浓度上升而增加。葡萄糖在脑细胞中经醛糖还原酶和山梨醇脱氢酶催化、转化为山梨醇和果糖。山梨醇和果糖不能被脑细胞利用,又不容易逸出脑细胞,从而造成脑细胞内高渗。
葡萄糖+NADPH+H+醛糖还原酶山梨醇+NADP+
山梨醇+NAD+山梨醇脱氢酶果糖+NADH+H+
当用胰岛素使血糖突然下降时,细胞外液水分可因脑细胞内高渗而向细胞内转移,使治疗中的糖尿病酮症酸中毒病人发生脑水肿。此外,山梨醇可使神经纤维内的渗透压升高,吸水而引起髓鞘损害,从而影响神经传导。出现糖尿病周围神经炎。同样,过高的葡萄糖进入晶状体后,形成的山梨醇和果糖不能再逸出晶状体,致使晶状体内晶体渗透压升高,水进入晶状体的纤维中导致纤维积水、液化而断裂。再加上代谢紊乱、晶状体中的ATP和还原型谷胱甘肽等化合物含量降低、α-晶体蛋白的糖基化等,最终使晶状体肿胀,出现空泡,其中某些透明蛋白质变性,聚合或沉淀,导致白内障。
综上所述,糖尿病可引起体内一系列的代谢紊乱,临床上病人出现三多一少症状,即多尿、多饮、多食和体重减少。高血糖引起的高渗性利尿是多尿的根本原因;而多尿所致的脱水刺激机体产生口渴感又导致多饮;体内糖利用障碍,能量代谢紊乱所致的饥饿感使得病人多食;大量蛋白质和脂肪的分解及脱水使病人体重减轻。在IDDM人群中,三多一少症状比较明显,重型病人更加突出。而在NIDDM病人中,往往没有典型的这些症状。
三、糖尿病的生物化学检测
(一)血糖的测定
血糖的测定是糖尿病生物化学检测中最常见的方法之一,糖尿病患者均有不同程度的血糖升高。
测定血糖的标本以血浆最为方便,测得结果最可靠。一般情况下全血葡萄糖浓度比血浆低10%-15%,毛细血管血样与静脉血样二者的测定值在空腹时无区别,但餐后1小时血样,二者血浆血糖水平可相差2.27±0.66mmol/L。测血糖的血浆中取空腹、进食一小时或随机取血,一般采用空腹血样本。抗凝剂用草酸钾氧化钠(2mg/ml可在24小时内阻止葡萄糖酵解)。正常人空腹血浆葡萄糖浓度的参考范围为3.9-6.7mmol/L。若空腹静脉血糖浓度大于8mmol/L,且有临床症状,可诊断为糖尿病。若小于6mmol/L,则可除外糖尿病;若在6.0-7.0mmol/L之间,应复查做进一步检查。进餐后1小时,血糖浓度可一时性升高,并伴有胰岛素分泌增多。若餐后1小时血糖明显增高,而血浆胰岛素为低水平,则可论断为糖尿病;若餐后2小时,血浆葡萄糖浓度大于7mmol/L,可怀疑为糖尿病。因为正常人餐后,葡萄糖的来源增加,血中葡萄糖浓度会反应性的一时升高,但多不超过肾糖阈,故尿病试验为阴性。高糖的刺激使胰岛素分泌增加,后者作用的结果,使餐后2小时内血糖浓度恢复到空腹水平。只有在胰岛素不足时,餐后血糖才持续升高,且不恢复到空腹水平。如果随机血样浓度大于11mmol/L,也可论断为糖尿病。
测定血糖的方法主要有两种,即化学法和酶法。化学法具有操作简便、快速、试剂价廉等优点,但由于干扰较大,所以仍属不理想的方法。已糖激酶法以其高度特异性,方法灵敏、准确,干扰因素少等优点被公认为参考方法。我国临床检验中心推荐的葡萄糖氧化酶-过氧化物酶比色法(GOD-POD法)目前已广泛用于临床。本法分析性能与已糖激酶法相近,且试剂稳定、价格便宜、操作简便。酶法测血糖还可以标准化、程序化、宜用于自动分析仪。用酶制剂制成的试纸条或胶片,直接用毛细血管进行快速分析,方便了患者根据血糖浓度自行调节胰岛素的剂量,以监控糖尿病的胰岛素治疗。固相酶试片法还提高了酶制剂的稳定性,所以是大有发展前途的。
(二)尿糖的测定
正常人24小时由尿排出的葡萄糖少于0.5g,在常规尿葡萄糖检测时为阴性。只有当血糖浓度高于8.9-9.9mmol/L(160-180mg/dl),超过肾小管重吸收能力时,尿糖试验才为阳性。所以将肾对葡萄糖的吸收能力用血糖浓度8.9-9.9mmol/L(160-180mg/dl)表示,即此值为正常肾糖阈。临床上有些糖尿病是由于受试者肾糖阈值低于正常人,如妊娠妇女由于肾糖阈值降低,可出现暂时性糖尿。而长期患糖尿病的患者其肾糖阈值可高于正常人。
尿糖测定已广泛用于对糖尿病的初判断,通常作为过筛程序的一部分。尿糖测定一般不需要准确定量,当尿糖浓度为5.55-11.1mmol/L时,应考虑糖尿病。尿标本以膀胱排空再饮水后30分钟为宜,这样更能准确地反映病人的代谢情况。
肾性尿糖是由于慢性肾炎、肾病综合征等疾病引起肾脏对糖的重吸收障碍而出现的尿糖,但病人血糖及糖耐量曲线基本正常,这与糖尿病性尿糖有根本的区别。
(三)口服葡萄糖耐量试验(oralglucosetolerancetest,OGTT)
OGTT是一种葡萄糖负荷试验,用以了解机体对葡萄糖的调节能力。当空腹血浆葡萄糖浓度在6-7mmol/L之间而又怀疑为糖尿病时,作此试验可以帮助明确诊断。
WHO标准化的OGTT是:试验前3日,每日食物中糖含量应不低于150g,且维持正常活动。影响试验的药物(表3-3)应在3日前停用。试验前病人应10-16小时不进食。坐位取血后5分钟内饮入250ml含75g无水葡萄糖的糖水,以后每隔30分钟取血1次,共4次,历时2小时。整个试验中不可吸烟、喝咖啡、喝茶或进食。儿童给予葡萄糖量为0.75g/kg体重。于采血的同时,每隔1小时留取尿液做尿糖半定量试验。必要时可适当延长血标本的收集时间,可长达口服葡萄糖后6小时。
表3-3影响葡萄糖耐量的常用药物
| 可引起血糖升高的药物 | 可引起血糖降低的药物 |
| 咖啡因、儿茶酚胺、皮质醇类、阿密替林、呋喃苯胺酸(速尿)、氯噻酮、噻嗪类、氯压定、吲哚美辛(消炎痛)、口服避孕药、氟哌啶醇、碳酸锂 | 磺胺、磺酰脲、乙醇、水杨酸盐 |
一般根据5次葡萄糖水平,以测定血糖的时间为横座标(空腹时为0时),血糖浓度为纵座标,绘制耐糖曲线。临床上常用的方法是清晨抽空腹血后,口服100g葡萄糖(或按1.5-1.75g/kg体重),再于给糖后0.5、1、2、3小时各取血1次,将测得的血糖按上述方法做耐糖曲线。本试验常用于协助诊断糖代谢紊乱的疾病。
⒈正常糖耐量正常人由于存在精细的代谢调节机制,服糖后0.5-1小时血糖浓度暂时略有升高,耐糖曲线显示峰值<10mmol/L,但尿糖阴性。1小时后血糖逐渐降低,一般2小时左右恢复至空腹3.9-6.7mmol/L水平。此种糖耐量曲线说明机体处理糖负荷的能力良好(图3-3)。
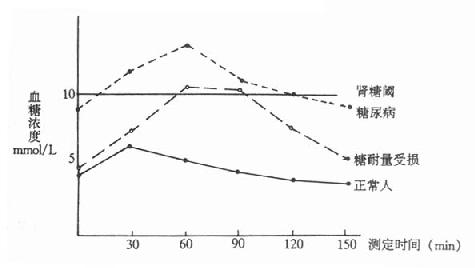
图3-3 葡萄糖耐量曲线
⒉糖尿病性糖耐量典型的糖尿病人糖耐量试验为:患者空腹血糖≥8.0mmol/L,高于正常值;服糖后血糖急剧升高,血糖增高的时间仍为0.5~1小时,但峰值超过10mmol/L,并出现尿糖;以后血糖浓度恢复缓慢,常常2小时以后仍高于空腹水平。说明病人处理摄入糖的能力降低。
此时重要的判断指标是服糖后2小时血糖浓度仍然高于空腹水平。对于早期糖尿病人,可只表现为OGTT后2小时血糖浓度仍高于8mmol/L。若空腹血糖正常而OGTT后2小时血糖大于11mmol/L,以及空腹血糖>8mmol/L而OGTT2小时的血糖水平在8-10.9mmol/L者,均应诊断为糖尿病。
⒊糖耐量受损如果非妊娠的成年人OGTT呈现空腹葡萄糖水平<8.0mmol/L,服糖后60、90分钟的血糖≥11mmol/L(有人30分钟也可达此值)而2小时血糖值在8-11mmol/L之间则为轻度耐糖能力下降,称为亚临床或无症状的糖尿病。这些病人几年后可能有1/3恢复正常,1/3仍为糖耐量受损,1/3则转为糖尿病(每年约1%~5%)。近来发现,这些病人容易发生小血管合并症,如冠心病、脑血管病,而不会发生微血管合并症,如视网膜病、肾病。
耐糖试验受许多因素影响,如年龄、饮食、劳动、应激、药物、胃肠功能、标本采集和葡萄糖测定方法等。所以临床上要具体情况具体分析。
对于OGTT正常而有糖尿病家族史的病人,可以进行可的松OGTT,即在应激的同时再给予糖负荷,通过加强机体对胰岛素分泌的要求来提高耐糖试验的敏感性,用以观察机体有无潜在的耐糖缺陷。具体做法是,在第1次OGTT后给病人口服50mg醋酸可的松,6小时再服1次,两小时后进行第2次OGTT。年龄在50岁以下的受试者,若血糖浓度在60分钟时高于10.2mmol/L,90分钟高于9.4mmol/L,120分钟高于9.0mmol/L,表示有潜伏性糖尿病。若两次OGTT的血糖浓度相差2.9mmol/L以上,说明病人对皮质类固醇的影响很敏感。
50岁以上的老年人对葡萄糖的耐受能力有下降趋势,所以不宜作此类试验。对于老年人的OGTT结果的判断,要注意区别是正常老年伴有的变化,还是糖尿病所致。儿童糖尿病的诊断标准与成人相同,但儿童空腹血浆葡萄糖的正常值比成人高0.83mmol/L。
由于妊娠性糖尿病致先天畸形及胎儿死亡增多,所以对孕妇进行有否糖尿病的检测应予以重视,特别对肥胖、直系亲属有糖尿病、有流产、畸胎或滞胎历史的孕妇,更应进行OGTT,以便发现糖尿病,并及时进行治疗、控制。
糖耐量试验虽然可以反映机体近期糖代谢的情况,但由于采血次数较多给病人带来一定的痛苦。临床上对于血糖持续增高并伴有糖尿,再结合病史及体征能够确诊的病人,毋须再做OGTT。
⒋糖化蛋白的测定成人红细胞的血红蛋白(Hb)主要是HbA1,占90%以上,HbA2占2.5%,HbF占0.2%,其余为HbA3。HbA3为连接有已糖的HbA1,其中HbA3a占Hb总量的0.8%,HbA3b占1.6%,HbA3c占4%。凡连接有已糖的HbA1,统称为糖化血红蛋白(glycoseylated,hemoglobin,GHb)。HbA3a、HbA3b、HbA3c也可分别表示为HbA1a、HbA1b、HbA1c。HbA1a还可分为HbA1a1和HbA1a2。HbA1c是Hbβ链的氨基末端缬氨酸残基与葡萄糖醛基通过非酶促反应缩合而成。HbA1a1是与1,6-二磷酸果糖结合,HbA1a2则是与6-磷酸葡萄糖结合形成的GHB。HbA1b的结构还不清楚。在四种GHb中HbA1c最多,占GHb总量的80%。有报道,正常人血液中HbA1c约占血红蛋白总量的5%-8%,而糖尿病时可达8%-30%。
GHb是在红细胞生存期间,HbA1与血中已糖(主要为葡萄糖)缓慢、连续的非酶促反应产物,为HbA1合成化学修饰的结果。GHb的形成取决于血糖浓度和作用时间,生成量与血中葡萄糖浓度成正比。红细胞平均寿命为120天,因此GHb的浓度反映测定日前2-3个月内受试者血糖的平均水平,而与血糖的短期波动无关。所以目前测定糖化血红蛋白,只作为糖尿病病人6-10周前血糖水平的定量指标。在新发生的糖尿病病人,临床检测只有血糖水平增高,而GHb正常;而未控制的糖尿病病人,则既有高血糖,也有GHb增多;在糖尿病已被控制的人群中,可见到血糖正常,GHb水平仍较高。这是因为GHb的形成与消失均需要数周时间。所以GHb水平不能反映近期的血糖水平,不能提供近期的治疗效果。但它是糖尿病长期监控的良好指标,尤其对IDDM和妊娠期性糖尿病的治疗监控有用。
血清白蛋白亦可糖基化,而且白蛋白的半寿期仅为19天。因此测定糖化白蛋白可了解糖尿病近二周的血糖水平,反映糖尿病治疗的较近期效果。
GHb的测定方法为:首先将红细胞样品在等渗盐溶液中放置一定时间以除去细胞中游离的葡萄糖,然后将细胞溶解并离心取上清液,进行离子交换层析。洗脱液用分光光度法在410nm处测吸收光度。在所测得的峰值中,HbA1a、HbA1b和HbA1c均为GHb。在血红蛋白的电泳实验中,GHb为快动组分。
正常人GHb为6.5%±1.5%。临床上随机测GHb,若<8%,多不考虑糖尿病;当所测的GHb>9%,预报糖尿病的准确度约78%,灵敏度为68%,特异性94%;若测得GHb>10%,则有89%为糖尿病,灵敏度43%,特异性99%,有效率86%。GHb的测定还可协助判断预后。据报道糖尿病合并视网膜病的病人,如果GHb为8%-10%,表示病变为中等程度,可以用激光治疗;若GHb>10%,则为严重损害,预后较差。
糖化白蛋白在糖尿病组为6.1%-22.3%,非糖尿病组为1.9%-5.8%,与糖化血红蛋白相关良好(γ=0.91)。
总之,目前测定糖化蛋白主要测GHb,其在临床上对糖尿病人治疗效果、监测病人对治疗的适应性方面应用较广,且是一个很好的参数,而对糖尿病的诊断作用不如血糖和OGTT灵敏。
⒌胰岛素、胰岛素原和C肽的测定胰岛素是由胰岛β细胞合成和分泌的一种蛋白质。和其他蛋白质一样,有粗面内质网的核糖核蛋白体上新合成的胰岛素是由102个氨基酸组成的前体,称为前胰岛素原(preproinsulin)。前胰岛素原穿过内质网膜进入腔内,随即切去前面由16个氨基酸组成的信号肽,生成胰岛素原(proinsulin,86肽),并输送到高尔基体贮存。胰岛素原是一条直链多肽,两端分别是胰岛素A链(21肽)和B链(30肽)的肽段,中间是一条由35个氨基酸组成的肽段,与A链的N端和B链的C端相连(图3-4)。当胰岛素分泌时,在蛋白水解酶的作用下,将这条连接肽两端分别切下2个碱性氨基酸(精-精、精-赖),生成胰岛素和C肽(connectingpeptide,即连接肽),二者一起分泌入血。所以了解胰岛素合成及分泌功能时,可以测定胰岛素原、胰岛素和C肽。
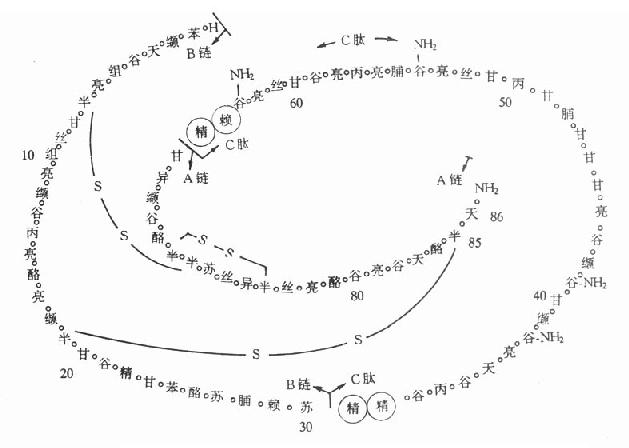
图3-4 人类胰岛素原的一级结构
用胰岛素或C肽的抗体建立起来的RIA方法,都可用于胰岛素分泌功能的测定。分泌入血的胰岛素流经肝时,50%以上将被肝细胞摄取,继而降解,其在血循环中的半寿期约为5分钟。胰岛素每天仅不足1%由尿排出,而被肾小管重吸收的胰岛素60%在肾实质降解。胰岛素的基础分泌量为每小时0.5-1.0单位,进食后分泌量可增加3-5倍。在胰岛素依赖型糖尿病病人血清中常常发现有胰岛素抗体,后者使胰岛素不能发挥其正常生物活性,而且还使胰岛素分泌功能逐渐减退,以至完全丧失。虽然检查胰岛素分泌功能对诊断IDDM有重要意义,但因血中本身存在胰岛素抗体,患者又使用了外源性胰岛素治疗,故用RIA测定血中胰岛素浓度,或口服葡萄糖耐量试验都不能得到准确的结果。若改为测定C肽则可克服这些干扰。
由于胰岛β细胞分泌胰岛素的同时也等摩尔地释放了C肽,所以测定C肽可以反映β细胞生成和分泌胰岛素的能力。特别在用胰岛素治疗了的病人,测血浆中C肽水平更能准确反映胰岛素功能,况且C肽在循环中很少被肝代谢,C肽的清除率也小于胰岛素。测定不同时间外周血浆中C肽和胰岛素量,可估计肝摄取处理胰岛素的能力。近年来用测定基础C肽及其对某些刺激因子的反应来估计糖尿病病人胰岛素的依赖性。目前认为糖尿病病人空腹血浆C肽≥1.9μg/L,口服甲苯磺丁脲后5分钟,C肽增至20.4μg/L,此类病人90%可通过限制食物和用降低血糖的药物控制病情,治疗中不需用胰岛素。若病人空腹C肽<1.9μg/L,则需要用胰岛素治疗。正常人空腹血浆C肽为2.2μg/L。
正常人每天C肽分泌总量的4%出现于尿中,所以尿C肽的测定也可作为β细胞分泌功能的指标。
目前C肽的测定已用于糖尿病的分型,但由于尚缺乏标准方法,空腹血浆C肽的参考范围变化相当大,故需进一步改进。
由于糖尿病和糖耐量受损的病人血浆中都可见到低胰岛素或高胰岛素水平,所以血浆胰岛素测定对诊断糖尿病价值不大。如果病人已用胰岛素治疗了6周,血中已产生了抗胰岛素抗体,此时用一般的放免法检测胰岛素更无意义。NIDDM病人多数与靶细胞受体数目减少有关。口服葡萄糖后血液胰岛素增高的程度显著高于非肥胖正常人,说明其胰岛素分泌功能正常,病因则是靶细胞对胰岛素的敏感性降低。病人血中胰岛素增高,即可引起受体的减数调节,进一步降低靶细胞的敏感性,以致病情逐渐加重。显然这类患者不适于用胰岛素治疗。对于这些病人,用RIA法测定血中胰岛素含量可以反映胰岛分泌功能,不必改用C肽测定。临床上长期大量应用胰岛素,常常发现胰岛素的疗效逐渐下降,甚至完全丧失应答能力。已经证明,这是靶细胞对激素作用的一种自我调节功能,称为激素受体的减数调节。减数调节的结果是靶细胞表面受体数目逐渐减少。
胰岛β细胞分泌胰岛素时也分泌有少量胰岛素原,约占血浆总胰岛素的5%。由于胰岛素原的生物学活性仅为胰岛素的3%-6%,且血浆中含量更微量,检测时需大量样本,所以临床并不常用。当血循环中胰岛素水平太高且有病理情况时,需测定胰岛素原,方法是灵敏度和特异性均高于RIA的双点放免法。临床上发现,胰岛瘤病人有胰岛素原增高。患糖尿病的儿童也表现有胰岛素原分泌增加。
由于糖尿病是一个复杂的代谢紊乱性疾病,病人除了有上述代谢紊乱的表现外,还将出现多系统、多方面的代谢紊乱,故实验室检查也应多方面辅助检查。如糖尿病合并有酮症或酮症酸中毒时,可做血、尿酮体及血气分析;如病人合并有高脂血症,则应做血脂或血浆脂蛋白检测,以便正确治疗;糖尿病病人无酮症酸中毒,但却出现昏迷时,可能为血糖过高所引起的高渗性昏迷,检测其血、尿渗透压可见均升高;对于有脱水症状的病人,则应检测血清电解质。
第三节 低血糖症
低血糖症(hypoglycaemia)是指空腹血糖浓度低于某一极限,临床出现一系列因血糖浓度太低引起的症候群。由于临床上出现低血糖症状时的血浆葡萄糖浓度极不一致,而且血糖浓度下降的速度比其绝对值对机体的影响更大,所以究竟血糖浓度降低到什么程度方可诊断为低血糖症一直存着不同的看法。一般认为成人血浆葡萄糖浓度低于2.8mmol/L(50mg/dl),全血葡萄糖浓度低于2.2mmol/L(40mg/dl),或空腹血浆葡萄糖浓度低于3.3mmol/L(60mg/dl)称为低血糖。也有人提出,以葡萄糖氧化酶法测定血浆葡萄糖浓度,低于2.2mmol/L(40mg/dl)作为低血糖的指标。因为在进餐前静脉血浆葡萄糖浓度也很少下降到2.2mmol/L以下。
低血糖症是由于血糖的来源小于去路,即食入糖减少,肝糖原分解少,肝将非糖物质转化为葡萄糖少,也可以是组织消耗利用葡萄增多和加速所致。
引起低血糖的原因很多,较常见的原因有:①胰岛β细胞增生和肿瘤等病变使胰岛素分泌过多,致血糖来源减少,去路增加,造成血糖降低;②使用胰岛素或降血糖药物过多;③垂体前叶或肾上腺皮质功能减退,使对抗胰岛素或肾上皮质激素分泌减少,结果同胰岛素分泌过多;④肝严重损害时不能有效地调节血糖,当糖摄入不足时很易发生低血糖;⑤长期饥饿、剧烈运动或高烧病人因代谢率增加,血糖消耗过多。
低血糖时可出现饥饿感,四肢无力以及交感神经兴奋而发生的面色苍白、心慌、出冷汗等症状。脑组织主要以葡萄糖作为能源,对低血糖比较敏感,即使轻度低血糖就可以发生头昏、倦怠。低血糖影响脑的正常功能还可发现为肢体与口周麻木,记忆减退和运动不协调,严重时出现意识丧失,昏迷(血糖降至40mg/dl以下可出现低血糖昏迷),如没有及时纠正可导致死亡。对于低血糖病人,若能及时静脉输注葡萄糖或口服补糖,以上症状可迅速纠正和缓解。
正常人脑组织储存糖极少(约0.5mg/g组织),仅可维持数分钟脑功能的需要,正常脑功能的维持要靠血循环不断供给葡萄糖和氧气。血糖浓度一旦降低,下丘脑中枢就会发出信号使交感神经活动增强:①通过肝交感神经末梢直接促进肝糖原分解;②使肾上腺髓质迅速分泌肾上腺素,增强糖原分解,增强糖异生作用,并降低组织摄取葡萄糖和抑制胰岛素的分泌,以便提高血糖浓度,缓解低血糖;③刺激胰高血糖素分泌,促进肝糖原分解和糖异生。此外,低血糖还刺激下丘脑释放促肾上腺皮质激素(ACTH)和生长素释放因子,促使垂体前叶分泌ACTH和生长素。ACTH又刺激肾上腺皮质分泌糖皮质激素,促进糖异生,抑制外周组织利用葡萄糖。以上总的效应是代偿性的对抗低血糖。如果低血糖是缓慢发生,数小时血糖浓度才低达2.2mmol/L,此时则可以无上述激素反应。
低血糖症的诊断通过测定血中葡萄糖浓度即可确定,但要根本性治疗则需要进一步找出低血糖的发病原因。
低血糖症的分类常用临床分类法,即将低血糖症分为空腹性低血糖和刺激性低血糖症两类,见表3-4。
一、空腹型低血糖症
正常人一般不会因为饥饿而发生低血糖症,这是因为正常的调节机制可维持血糖浓度>2.8mmol/L(50mg/dl)。成年人空腹时发生低血糖症往往由于葡萄糖利用过多或生成不足。如果怀疑病人有此型低血糖症,可以按图3-5所示检查程序找出原因。
表3-4 低血糖的临床分类
| ⒈空腹性低血糖症 | |
| ①高胰岛素血症:良性、恶性和多发性胰岛瘤,小腺瘤病胰岛细胞增殖症; | |
| ②非胰性肿瘤;③肝和肾疾病;④内分泌疾病:垂体、肾上腺、下丘脑等;⑤ | |
| 先天性代谢病:糖原贮积病等;⑥各型新生儿低血糖症;⑦自身免疫性疾病; | |
| ⑧饥饿 | |
| ⒉刺激性低血糖症 | |
| ①外源性低血糖因子:药物、毒物;②反应性低血糖症;③遗传性果糖失耐; | |
| ④半乳糖血症;⑤酒精性低血糖症 |
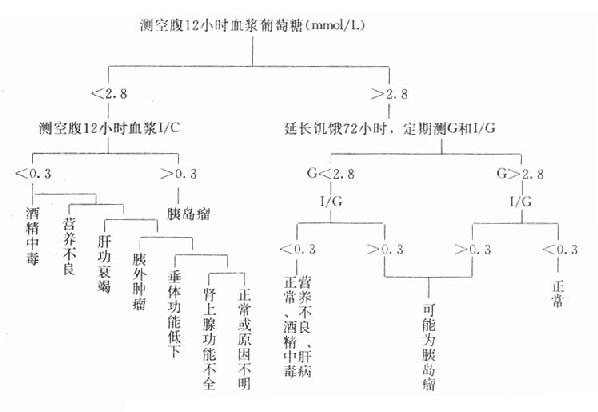
图3-5 空腹型低血糖症的诊断程序
若测得12小时空腹血浆葡萄糖浓度<2.8mmol/L,可诊断为低血糖症。进一步检测胰岛素(I)与葡萄糖(G)的比值(I/G),如I/G>0.3,提示为胰岛素瘤,如测其C肽和胰岛素原也会增高。若I/G>0.3的低血糖者,还可做以下检测以进行鉴别诊断(见力图3-6)。胰外肿瘤病人血清中可以有类胰岛素活性物质,后者具有降低血糖的作用。空腹性低血糖预后比刺激性低血糖症差,且难以用食物控制。
二、刺激性低血糖症
刺激性低血糖症空腹时血糖并无明显降低,其往往是给予适当的刺激,如进食才诱发,表现为反应性低血糖,临床上用胰岛素治疗糖尿病时最常见,也可由干扰糖异生或促进糖利用的药物、毒物或糖(半乳糖、果糖)引起,餐后低血糖症为刺激性低血糖症的一大类,低血糖发生于进餐后1-5小时,且可用OGTT诊断(图3-7)。
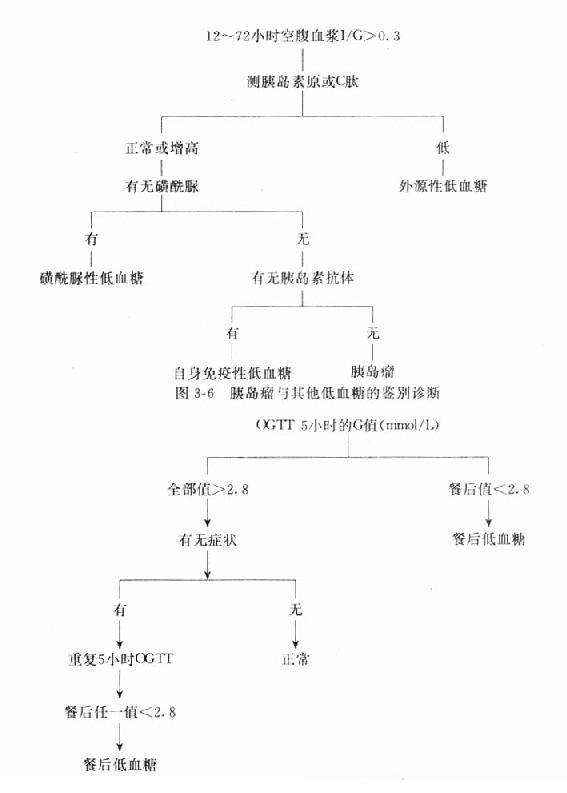
图3-7 餐后低血糖的诊断程序
餐后低血糖按病史与OGTT反应类型可分为三型(见图3-8):
⒈功能性低血糖症(反应性低血糖症)为发生于餐后或OGTT2-5小时的暂时性低血糖。多发于心理动力学异常的妇女。病人有交感神经兴奋的症状,病人耐糖曲线的前部正常或接近空腹水平,有的病人可见胰岛素延迟分泌。病人血浆胰岛素不高,甲状腺素、肾上腺皮质激素缺乏的病人可出现此型低血糖症。
⒉Ⅱ型糖尿病或糖耐量受损伴有的低血糖症病人空腹血糖正常,OGTT前2小时似糖耐量受损或Ⅱ型糖尿病,但食糖后3-5小时,血浆葡萄糖浓度迅速降低达最低点。产生的原因可能是持续高血糖引起胰岛素延迟分泌,且表现出后期胰岛素高,致使血糖后期迅速下降。
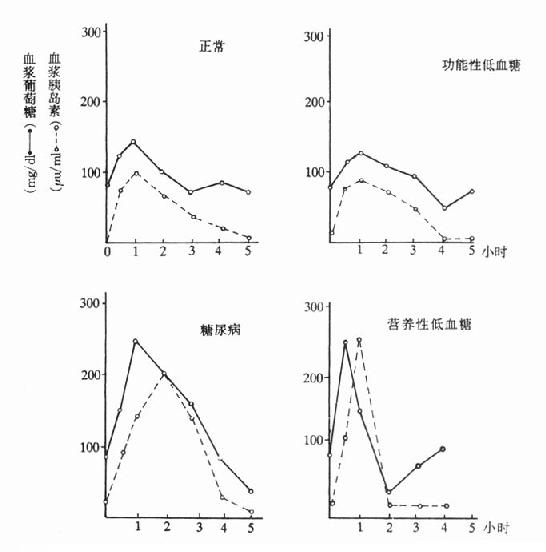
图3-8 餐后低血糖症血浆葡萄糖与胰岛素的比较
⒊营养性低血糖症低血糖常发生于餐后1-3小时。病人大多有上消化道手术或迷走神经切除,由于胃迅速排空,使葡萄糖吸收增快,血浆葡萄糖明显快速增高,刺激胰岛素一时性过多分泌,致使血糖浓度迅速降低,出现低血糖症。
餐后低血糖症血标本一般采集服糖后5小时内或病人低血糖症状时的血液。对高度怀疑者,虽一次OGTT为正常,还是应该再次检测。
临床上同一病人既可发生空腹性低血糖,也可发生餐后低血糖。对于这类病人,治疗首先在于纠正空腹性低血糖症。
早产儿比足月新生儿对低血糖更为敏感,且儿童对低血糖的敏感性也高于成年人。原因是儿童大脑占体重的比例比成人高;新生儿酮体生成能力低,很难以酮体作为大脑的有效能源;新生儿糖异生能力还未达到足以防止饥饿性低血糖的水平;早产儿肝糖原储量少于足月新生儿,空腹时糖原很快就消耗掉等。
第四节 糖代谢的先天性异常
糖代谢障碍并不都出现血糖浓度异常,本节简要阐述糖代谢酶的先天性、遗传性缺损所致糖代谢异常。
一、糖原代谢先天性异常
最常见的是糖原贮积病(glycogenstoragediseases),这是由于糖原生成和分解的酶系统先天性缺陷所引起的一组糖原合成或分解异常、使糖原在细胞中过多贮积、或糖原分子异常的遗传性疾病。肝是糖原贮积病的主要受累器官,其次是心脏和肌肉(见表3-5)。
表3-5 各糖原贮积病及其临床体征
| 分型 | 酶的缺陷 | 受累器官 | 临床表现 |
| Ⅰ(von-Gierke病) | 葡萄糖-6-磷酸酶 | 肝、肾 | 肝明显肿大、发育受阻、严重低血糖、酮症、高尿酸血症伴有痛风性关节炎、高脂血症 |
| Ⅱ(Pompe病) | 1,4-α-D葡萄糖苷酶(溶酶体) | 肝、心、肌等 | 常常在2岁前心力、呼吸衰竭致死 |
| Ⅱ(Cori病) | 脱支酶 | 肌肉、肝 | 类似Ⅰ型,但程度较轻 |
| Ⅳ(Andersen病) | 分支酶 | 肝、脾 | 进行性肝硬化,常在2岁前因肝功能衰竭死亡 |
| Ⅴ(McArdle病) | 磷酸化酶 | 肌肉 | 由于疼痛,肌肉剧烈运动受限,否则病人可以正常发育 |
| Ⅵ(Hers病) | 磷酸化酶 | 肝脏 | 类似Ⅰ型,但程度较轻 |
| Ⅶ | 磷酸果糖激酶 | 肌肉 | 与Ⅴ型类似 |
| Ⅷ | 磷酸化酶激酶 | 肝脏 | 轻度肝肿大和轻度低血糖 |
| Ⅸ※ | 糖原合酶 | 肝 |
注:※糖原缺乏
由于酶缺陷不同,故糖原贮积病分为许多型。Ⅰ型糖原贮积病是1929年由VonGierke命名,所以也叫VonGierke病,为最常见的糖原贮积病。本型是因肝、小肠粘膜和肾脏的葡萄糖-6-磷酸酶或6-磷酸葡萄糖转位酶缺乏所致。前一种酶缺乏为Ⅰa型,后一种酶缺乏为Ⅰb型。该病为常染色体隐性遗传性疾病,发病率占人群比例为1/200000。
各型糖原贮积病的预后及治疗各不相同,故有必要对其进行鉴别(见表3-6)。在检测糖原贮积病时最可靠的方法是用肝或肌肉的活检标本测定特异酶活性。临床上有的病人还可见到一种以上缺陷的酶。
二、糖分解代谢途径的先天异常
糖分解代谢途径先天代谢异常可有丙酮酸激酶缺乏病,丙酮酸脱氢酶缺乏症和磷酸果糖代谢异常所致恶性发烧。
(一)丙酮酸激酶(PK)缺乏病
在糖酵解过程中,丙酮酸激酶催化磷酸烯醇式丙酮酸生成烯醇式丙酮酸,同时产生ATP,是酵解途径产生ATP的反应之一,PK缺乏将导致成熟红细胞缺乏ATP,进而发生溶血。
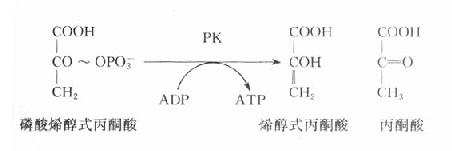
表3-6 各型糖原贮积病的特征
| 类型 | Ⅰ | Ⅱ | Ⅲ | Ⅳ | Ⅴ | Ⅵ | Ⅶ | Ⅷ | Ⅸa | Ⅸb | Ⅹ | ||
| 受害器官 | 肝肾 | 所有组积 | 肝、肌、心 | 肝单核吞噬细包系统 | 肌肉 | 肝 | 肌肉 | 脑 | 肝 | 肝 | 肝、肌肉 | 肝 | |
| 糖原结构 | N | N | 短枝异常 | 长枝异常 | N | N | N | 巨大颗粒 | N | N | |||
| 酶缺陷 | G-6-P酶 | α葡萄糖 苷酶 | 脱枝酶 | 分枝酶 | 肌磷酸化酶 | 肝磷酸化酶 | 磷酸果粒糠酶 | 肝磷酸化酶 | 同左 | 糖原合成酶 | |||
| 空腹低血糖 | ++ | + | ± | ± | ++ | ||||||||
| 对胰高血 糖素反应 |
血糖 | ↑N | 0↑ | ↑ | ↑ | 0± | ↑ | ± | N进餐0空腹 | ||||
| 血乳酸 | ↑↑ | ↑ | ↑ | ||||||||||
| 半乳糖 果糖试验 |
血糖 | ↑ | ↑ | ↑ | ↑ | ↑ | ↑ | ↑ | |||||
| 血乳酸 | ↑↑ | ↑ | ↑ | ||||||||||
| 剧烈运动后血乳酸 | ↑ | ↑ | ↑ | ↑ | ↑ | ||||||||
| 脂质代谢 | ↑↑ | N | 饿后 FFa ↑ |
运动后肌摄取 FA↑ |
TG↑ Ch↑ |
TG↑ Ch↑ |
TG↑ Ch↑ |
||||||
| 其它诊断性试验 | 肝活检 | 红细胞抗原 ↑ | 红细胞糖原结构异常 | 红细胞磷酸化酶↓ | |||||||||
| 临床特征 | 生长停滞,肝大 | 心衰 | 似Ⅰ型但不明显 | 肝硬化腹水 | 肌无力 | 肝大 | 肌无力 | 意识障碍 | 肝大,常染色体隐性遗传 | 肝大伴性遗传 | 肝大 | 轻度肝大 | |
注:N-正常;O-阴性;↑-升高;↓降低;±-可疑
网织红细胞中含有线粒体,故可通过糖有氧氧化产生足量的ATP。而成熟红细胞中不含线粒体,完全依赖糖酵解供能。红细胞内生成的ATP主要用于维持细胞内外的离子梯度,特别是通过Na+-K+-ATP酶维持细胞内外Na+、K+浓度梯度。这对于维持红细胞双凹形状十分重要。若缺乏ATP,红细胞将发生肿胀,易发生溶血,实验室检查可以见到自身溶血试验阳性。PK的遗传缺陷是糖酵解途径中遗传性缺陷导致溶血性贫血的最多见原因。PK缺陷时,细胞中PK活性仅为正常细胞的5%-25%,故虽然PK缺陷少见,但其造成的溶血性贫血却对机体危害甚大。
(二)丙酮酸脱氢酶复合物缺乏症
丙酮酸脱氢酶复合物由丙铜酸脱氢酶、二氢硫辛酸转乙酰基酶、二氢硫辛酸脱氢酶及NAD+、FAD、CoASH、焦磷酸硫胺素、硫辛酸三个酶、五个辅助因子组成,其氧化的丙酮酸氧化脱羧生成乙酰辅酶A的反应是糖进入三羧酸循环、彻底氧化成CO2和水、产生大量ATP的关键。
在儿童中发现有多种丙酮酸代谢异常的疾病,其中有些是由于丙酮酸脱氢酶复合物中某些组份先天性缺陷所致。该酶复合物中各种亚基(催化亚基和调节亚基)都可能发生先天性缺陷。这些缺陷都可使丙酮酸不能继续氧化产生ATP,使脑组织不能有效地利用葡萄糖供能,进而影响了儿童大脑的发育和功能,严重者可导致死亡。
丙酮酸不能进一步氧化,致使患儿血液中乳酸、丙酮酸和丙氨酸的浓度显著升高,出现慢性乳酸酸中毒。丙酮酸脱氢酶的缺陷可以通过皮肤成纤维细胞培养并进行酶学测定予以测定。此类病人在一定程度上可通过进食生酮食物和限制糖的摄入使病情缓解或得到控制。
(三)磷酸果糖代谢异常
磷酸果糖激酶与果糖-1,6-二磷酸酶是作用相反的一对酶,它们所催化的化学反应是糖代谢途径中的一处无效循环(又称底物循环)。

由于酶的遗传性缺陷,以上无效循环得不到控制,造成ATP大量分解产热:ATP+H2O→ADP+Pi+热。临床上可因服用氟烷而诱发恶性发烧。
恶性发烧是一种罕见的遗传缺陷性疾病,其发病率占儿童的1/15000,成人的1/50000-1/100000。病人常因服用某种药物,如吸入氟烷而在几分钟内突然发病,表现为体温骤然升高、代谢性和呼吸性酸中毒,以及高血钾症和肌肉强直。人们认为,氟烷可以促进肌肉中上述两个酶所催化的耗能无效循环,诱发恶性发烧的产生。
三、其它糖代谢异常
(一)红细胞中6-磷酸葡萄糖脱氢酶遗传缺陷或变异
6-磷酸葡萄糖脱氢酶(G-6-PD)催化6-磷酸葡萄糖脱氢生成6-磷酸葡萄内脂,脱下的氢由NADP接受。此步反应是磷酸戊糖途径的关键部位,所产生的NADPH在维持红细胞的正常形态与功能方面起了重要作用。
G-6-PD缺乏病是伴性遗传性疾病,临床并不罕见。女性杂合子含有两族红细胞:一族酶活性正常,另一族缺乏G-6-PD。轻型G-6-PD缺乏病人红细胞中G-6-PD活性比正常人低10倍。在一般情况下磷酸戊糖途径提供的NADPH还能维持还原型谷胱甘肽的水平,保证红细胞的正常形态与功能。当红细胞中NADPH的需要量增加,如服用抗疟疾药扑疟喹啉时,正常人不会有什么危害,而G-6-PD缺乏病人红细胞中磷酸戊糖途径的代谢速度则不能相应增加,提供的NADPH不能保证维持还原型谷胱甘肽所应有的水平,可引起严重的溶血性贫血,俗称蚕豆黄。
(二)先天性家族性非溶血性黄疸
人类先天性家族性非溶血性黄疸(Grigler-Najjar综合征)是由于缺乏UDP-葡萄糖醛酸基转移酶,使胆红素不能与葡萄糖醛酸结合,形成结合胆红素,使胆红素以不易运输和排泄的游离形在体内堆积所致的先天性疾病。
正常人通过糖醛酸途径产生葡萄糖醛酸,后者在UDP-葡萄糖醛酸基转移酶的催化下,可与内源性如类固醇、胆红素和外源性如药物等物质结合,生成相应的葡萄糖苷酸化合物。结合型的葡萄糖苷酸化合物具有较强的酸性,在生理pH下有较高的溶解度,易于运输排泄。这在体内类固醇激素的灭活和胆红素的代谢,以及许多生物转化作用中具有重要意义。而病人因先天缺乏此酶,使其不但对胆红素代谢造成异常,同时也缺乏结合外源性物质生成葡萄糖苷酸化合物的能力。
(三)果糖代谢异常
果糖是食物中糖的一部分,主要来自蔗糖。从肠道吸收的果糖大部分在肝内通过1-磷酸果糖(F-1-P)途径代谢。代谢途径见图3-9。果糖代谢障碍是因与果糖代谢有关的酶缺乏所致。
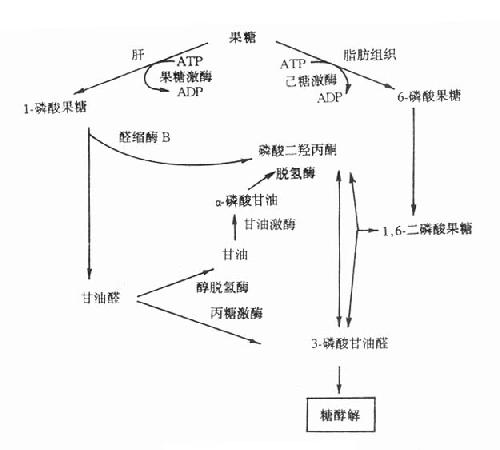
图3-9 果糖代谢途径
1.实质性果糖尿(essentialfructosuria)又称为原发性果糖尿症,它是由于果糖激酶缺乏所引起的常染色体隐性遗传疾病。正常人血中果糖比葡萄糖代谢快,其半寿期果糖为20分钟,葡萄糖为45分钟一次服用50g果糖后,通常在2小时之内血中果糖浓度就降至空腹水平(0-0.44mmol/L)。果糖激酶缺乏者一次服用50g果糖后,病人血中果糖浓度异常高,2小时内果糖仍未消失,并出现果糖尿。此型果糖尿又称为Ⅰ型果糖尿。病人无低血糖表现,这是因为病人机体内葡萄糖与乳糖代谢均正常。
2.果糖不耐受(fructoseintolerance)此病为常染色体隐性遗传性疾病,杂合子无症状。多数病人在断奶后给予蔗糖饮食时才发病,严重病例可致死亡。有些病例可由于手术前后给予果糖或静脉注射山梨醇引起严重肝、肾损伤时才发现。
果糖不耐受症的临术表现可明显不同。在婴儿可表现为呕吐、进食少、肝大、精神淡漠、生长停滞等。病人尿分析有果糖、葡萄糖等还原物质,多数病人有蛋白尿、非特异的氨基酸尿以及血浆转酶活性增高。有的病儿伴有其它肝、肾损害指标。在大龄儿童或成人则无症状,只在进甜食后,可有腹部不适、呕吐或腹泻。果糖不耐受的一个重要特征是服用果糖后出现严重的低血糖,病人即使肝糖原储备丰富也会在此时发生低血糖,这是由于过量的1-磷酸果糖抑制了肝磷酸化酶所致。
此症是由于1-磷酸果糖醛缩酶(醛缩酶B)缺陷引起,病人肝内1-磷酸果糖醛缩酶活性几乎完全缺失,而1,6-二磷酸果糖醛缩酶活性降低50%以上,造成肝内1-磷酸果糖的堆积及Pi和ATP的消耗。由于Pi大量消耗,肝线粒体氧化磷酸化减少,造成ATP缺乏。后者缺乏使肝细胞ATP依赖性离子泵功能障碍,膜内外离子梯度不能维持,细胞肿胀,细胞内容物外溢。

3.1,6-二磷酸果糖酶缺乏症此症为常染色体隐性遗传病,多在婴儿时发病。病儿表现为肌无力、呕吐、嗜睡、生长停滞和肝肿大等,感染可促使急性发作。若不治疗,在婴儿期就可死亡。
实验室检查可见空腹血糖低,即空腹性低血糖、酮血症、乳酸血症和血浆丙氨酸水平增高。诊断依据低血糖症,确诊需用肝、肾、肠活检标本测定该酶活性。
治疗主要通过食物疗法,食含果糖少的食物,少吃多餐,避免饥饿,一般疗效还可以,预后尚可。
5.半乳糖血症半乳糖在体内的正常代谢途径如图3-10所示。
影响半乳糖利用的各种因素均可引起半乳糖血症(galactosemia),但遗传性半乳糖血症主要有两种。
1-磷酸半乳糖尿苷转移酶所致的半乳糖症是最多见的遗传性半乳糖血症,其为常染色体隐性遗传疾病。患者半乳糖代谢终止于1-磷酸半乳糖阶段。杂合子病人此酶仅活性低下,但如果不持续服用半乳糖饮食,酶活性还可以维持健康。而纯合子病人此酶完全缺乏,如食用富含半乳糖的食物,病人会出现严重病变,甚至死亡。如果及时停用含半乳糖的食物,病人除智力障碍外,其他的各种症状均可消失。此型半乳糖血症临床表现为生长停滞,喂奶后呕吐和腹泻,继而出现黄疸、溶血、肝大、智力障碍,检查中可见患儿血清半乳糖水平明显升高,尿中出现半乳糖,红细胞中1-磷酸半乳糖尿苷酰转移酶缺乏。患儿进食半乳糖或乳糖后,常伴有低血糖和高半乳糖血症。
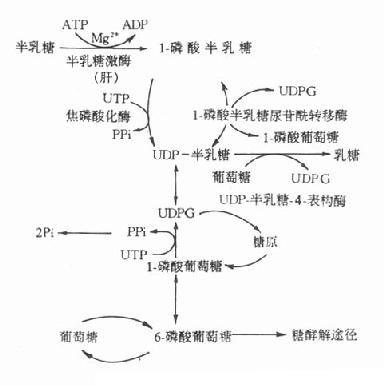
图3-10 半乳糖代谢途径
另一种遗传性半乳糖血症是由于半乳糖激酶缺乏所致,此型症状较轻,新生儿期不表现症状,往往发生白内障后才被确诊。
半乳糖血症的危害不是由于缺乏某种必需物质,而是由于产生毒性物质所致。半乳糖还原产物-半乳糖醇,在细胞内高浓度,如贮积于晶体,将吸收水进入晶体,造成晶体肿胀、混浊,引起白内障。此外1-磷酸半乳糖可能是某些毒性物质的前体,或者高浓度时本身就具有毒性作用。体外实验已证实,1-磷酸半乳糖可抑制磷酸葡萄糖变位酶和G-6-P酶的活性。
(四)粘多糖沉积症
因蛋白聚糖降解酶先天性缺陷所引起的蛋白聚糖分解代谢障碍,将导致产生各种类型的粘多糖沉积症(mucopo-lysaccharidoses)。其特征是过多的寡聚糖堆积与排泄。由表3-7可知,各型粘多糖沉积症的代谢基础相似,但遗传类型和临床表现各不相同。
表3-7 粘多糖沉积症的酶缺陷
| 名称 | 代号 | 酶缺陷 | 酶学测定样品 | 生化改变 | 遗传特性 |
| Hurler,Scheie综合征 | MPSⅠ | α-L-艾杜糖酸苷酶 | 成纤维细胞、白细胞、组织、羊水细胞 | 尿和组织中DS、HS增多,成纤维细胞中DS增加 | 常染色体隐 性 |
| Hunter综合征 | MPSⅡ | 艾杜糖醛酸硫酯酶 | 血清、成纤维细胞、白细胞、组织、羊水、羊水细胞 | 同上 | X连隐性 |
| Sanfilippo综合征A | MPSⅢA | HS-N-硫酸酯酶(硫酰胺酶) | 成纤维细胞、白细胞、组织、羊水细胞 | HS在尿中和组织中增多、DS在成纤维细胞中增多 | 常染色体隐性 |
| Sanfilippo综合征B | MPSⅢB | α-N乙酰葡萄胺苷 | 血清、成纤维细胞、白细胞、组织、羊水细胞 | HS出现于尿中 | 同上 |
| Sanfilippo综合征C | MPSⅢC | 乙酰基转移酶 | 成纤维细胞 | HS出现于尿中 | 同上 |
| Morquio综合征 | MPSⅣ | N-乙酰半乳糖胺-6-硫酸酯酶 | 成纤维细胞 | KS和CS出现于尿中 | 同上 |
| Morquio综合征 | β-半乳糖甘酶 | 成纤维细胞 | KS出现于尿中 | ||
| Maroteaux-Lamy综合征 | MPSⅥ | N-乙酰半乳糖胺4-硫酸酯酶(芳香硫酸酯酶B) | 成纤维细胞、白细胞、组织、羊水细胞 | DS出现于尿中 | 同上 |
| β-葡糖醛酸苷酶缺乏症无名疾病 | MPSⅦ | β-葡糖醛酸苷酶 | 血清、成纤维细胞、白细胞、羊水细胞 | DS、HS(±)出现于尿中 | 同上 |
| 无名疾病 | MPSⅧ | N-乙酰葡糖胺6-硫酸酯酶 | 成纤维细胞 | KS和HS(±)出现于尿中 | 同上 |
注:MPS-粘多糖沉积症DS-磷酸皮肤素
HS-硫酸乙酰肝素CD-硫酸软骨素
KS-硫酸角质素
遗传性粘多糖沉积症约占出生婴儿的1/30000。
第四章 血浆蛋白及其代谢紊乱
血浆脂类包括游离胆固醇(freecholesterol,FC)、胆固醇酯(cholesterolester,CE)、磷脂(phospholipid,PL)、甘油三酯(triacylglycerol/triglyceride,TG)、糖酯、游离脂肪酸(freefattyacid,FFA)等。血浆中最多的脂质有胆固醇(总胆固醇totalcholesterol,TC)、PL和LG,血浆脂质总量为4.0-7.0g/L。
血浆脂类简称血脂,其含量与全身相比只占其小部分,然而其代谢却非常活跃。肠道吸收的外源性食物酯类、肝合成的内源性脂类及脂肪组织贮存的脂肪动员都必须先经血液再到其他组织,因此,血脂水平可反映全身脂类代谢的状态。由于血脂的不断降解和重新合成在正常地进行,并保持动态平衡,血脂含量的变动也就稳定在一定的范围内。测定血浆脂类可及时地反映体内脂类代谢状况。就测定方法而言,从历史上看,测定血浆胆固醇是最古老的先行方法,其后陆续进行PL、TG和FFA的定量测定,尔后又增加脂蛋白及其载脂蛋白的测定,这些项目,已是目前临床上用于了解人体脂类代谢状况的系列指标。
第一节 血浆脂蛋白
一、血浆脂蛋白的分类
脂蛋白属于一类物质,因结构及组成的差异,有多种形式存在,尽管如此,仍有许多共同之处,一般都是以不溶于水的TG和CE为核心,表面覆盖有少量蛋白质和极性的PL、FFA,它们的亲水基因暴露在表面突入周围水相,从而使脂蛋白颗粒能稳定地分散在水相血浆中,如图4-1所示。
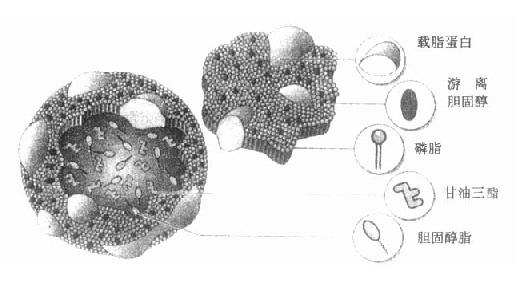
图4-1 脂蛋白结构图
血浆脂蛋白的分类方法主要有电泳法和超速离心法
(一)超速离心法
超速离心法是根据各种脂蛋白在一定密度的介质中进行离心时,因漂浮速率不同而进行分离的方法。脂蛋白中有两种比重不同的蛋白质和脂质,蛋白质含量高者,比重大;相反脂类含量高者,比重小。从低到高调整介质密度后超速离心,可依次将不同密度的脂蛋白分开。通常可将血浆脂蛋白分为乳糜微粒(chylomicron,CM)、极低密度脂蛋白(verylowdensitylipoprotein,VLDL)、低密度脂蛋白(lowdensitylipoprotein,LDL)和高密度脂蛋白(highdensitylipoprotein,HDL)等四大类。
(二)电泳法
由于血浆脂蛋白表面电荷量大小不同,在电场中,其迁移速率也不同,从而将血浆脂蛋白分为乳糜微粒、β-脂蛋白、前β-脂蛋白和α-脂蛋白等四种。α-脂蛋白中蛋白质含量最高,在电场作用下,电荷量大,分子量小,电泳速度最快,电泳在相当于α1球蛋白的位置。CM的蛋白质含量很低,98%是不带电荷的脂类,特别是甘油三酯含量最高。在电场中几乎不移动,所以停留在原点。为了取样方便,多以血清代替血浆。正常人空腹血清在一般电泳谱上无乳糜微粒。电泳分类法的脂蛋白种类与超速离心法的脂蛋白分类相应关系如图4-2所示。
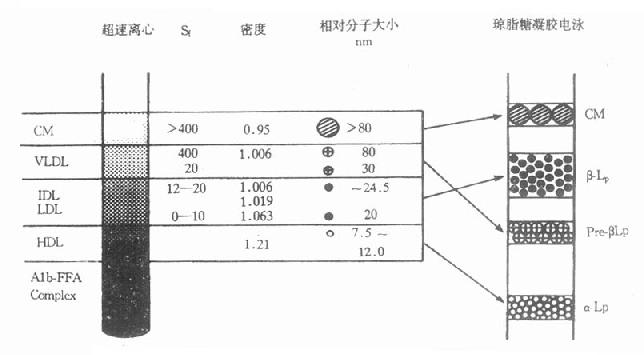
图4-2 超速离心法与电泳法分离血浆脂蛋白的相应名称
二、脂蛋白组成与结构
一般认为血浆脂蛋白都具有类似的结构,呈球状,在颗粒表面是极性分子,如蛋白质,磷脂,故具有亲水性;非极性分子如甘油三酯、胆固醇酯则藏于其内部。磷脂的极性部分可与蛋白质结合,非极性部分可与其它脂类结合,作为连接蛋白质和脂类的桥梁,使非水溶性的脂类固系在脂蛋白中。磷脂和胆固醇对维系脂蛋白的构型均具有重要作用。
1.乳糜微粒CM颗粒最大,约为500nm大小,脂类含量高达98%,蛋白质含量少于2%,因此密度极低。CM由小肠粘膜细胞在吸收食物脂类(主要是甘油三酯)时合成,经乳糜导管,胸导管到血液。主要功能为运输外源性甘油三酯。
2.极低密度脂蛋白VLDL中TG主要在肝脏利用脂肪酸和葡萄糖合成。若食物摄取过量糖或体内脂肪动用过多,均可导致血VLDL增高。VLDL中脂类占85%-90%,其中TG占55%,其密度也很低。VLDL是运输内源性TG的主要形式。
3.低密度脂蛋白LDL的结构大致可分为三层:内层,占15%的蛋白质构成核心,被一圈磷脂分子包围;中层,非极性脂类居中,并插入内外层,与非极性部分结合;外层,85%的蛋白质构成框架,磷脂的非极性部分镶嵌在框架中,其极性部分与水溶性的蛋白质等亲水基团突入周围水相,使其脂蛋白稳定地分散于水溶液中;游离胆固醇分布于三层之中。
4.高密度脂蛋白HDL是一组不均一的脂蛋白,经超速离心和等电聚焦电泳,可把HDL分成若干亚族。各亚族具有不同的密度,颗粒大小及分子量不尽相同,脂质和载脂蛋白比例不同,经X射线衍射研究证实为三维形态结构。现有资料提示,HDL是对称的准球形颗粒,具有一低电子密度的核心的外壳。低电子密度的中心由非极性脂质所占据,高电子密度是部分由磷脂极性头和蛋白质组成的颗粒外壳。经园二色分析证实,HDL的蛋白部分有2/3是α-螺旋结构,其余为无规则结构。带电荷的极性氨基酸残基构成α-螺旋的极性面,而疏水侧链则占据另一面。氨基酸按顺序排列在螺旋区域形成两性结构。目前认为,HDL的结构是α螺旋区平行于脂蛋白颗粒表面,非极性氨基酸残基伸展到颗粒的非极性核心区域;磷脂的脂肪酰链则垂直于脂蛋白颗粒表面的螺旋形载脂蛋白;胆固醇酯深埋在HDL颗粒的亲脂核心内;而游离的胆固醇可能与颗粒表面在磷脂极性头和载脂蛋白结合。
HDL主要由肝合成,小肠也可合成。HDL按密度大小又可分为HDL1、HDL2和HDL3。HDL1又称为HDLc,仅在摄取高胆固醇膳食后才在血中出现,健康人血浆中主要含HDL2和HDL3。HDL主要是将胆固醇从肝外组织转运到肝进行代谢。
5.脂蛋白(a)Berg于1963年在血浆脂蛋白电泳时发现β-脂蛋白部分有一种新的抗原成分,并与LDL结合,将此抗原成分命名为脂蛋白(a)[lipoprotein(a),LP(a)]。其后证实,LP(a)核心部分由甘油三酯、磷脂、胆固醇、胆固醇酯等脂质和载脂蛋白B100组成,结构类似LDL,并含有LDL中没有的载脂蛋白(a)[apolipoprotein(a),Apo(a)]。Apo(a)与纤溶酸原具有高度同源性,在纤溶系统多个环节发挥作用,从而影响动脉粥样硬化性疾病的发生和发展。有足够证据表明,Lp(a)是动脉粥样硬化性疾病的一项独立危险因子。Lp(a)含有两类载脂蛋白,即ApoB100和Apo(a),两者通过1至2个二硫键共价相连,若用还原剂巯基乙醇处理Lp(a)时,Apo(a)可从Lp(a)的分子上脱落下来,成为不含脂质的一类糖蛋白。剩下不含Apo(a)仅含ApoB100的颗粒,称为Lp(a-)。
第二节 载脂蛋白
脂蛋白中的蛋白部分称为载脂蛋白(apolipoprotein/apoprotein,Apo)。载脂蛋白在脂蛋白代谢中具有重要的生理功能。Apo构成并稳定脂蛋白的结构,修饰并影响与脂蛋白代谢有关的酶的活性。作为脂蛋白受体的配体,参与脂蛋白与细胞表面脂蛋白受体的结合及其代谢过程。
一、载脂蛋白组成与结构特点及生理功用
Apo种类很多,一般分为5-7类,其氨基酸序列大多数已阐明,Apo种类的命名是按1972年Alaupovic建议的命名方法,用英文字母顺序编码,即ABC顺,每一类还有亚类。
(一)载脂蛋白A族
ApoA可分为ApoAⅠ,AⅡ,AⅣ。ApoAⅠ和AⅡ大部分分布在HDL中,是HDL的主要载脂蛋白。
1.ApoAⅠApoAⅠ是ApoA族最多的一种组份,先后从人HDL中分离纯化得到ApoAⅠ,并阐明了ApoAⅠ的氨基酸序列,也预测了其二级结构的要点。人成熟的ApoAⅠ由243个氨基酸残基组成,是单一多肽链,分子量为28.3ku。人及大鼠、猴、兔、牛、鸭、树鼷等动物的ApoAⅠ已分离纯化。人和其他种属的ApoAⅠ的氨基末端为Asp,羧基末端为Gln,其分子中不含半胱氨酸和异亮氨酸。经等电点聚焦电泳证实,人和动物的ApoAⅠ都是不均一的,有10种不同的亚组份,至少有6种多态性。
目前所知,ApoAⅠ的氨基酸残基的排列有其自身的特征:①极性氨基酸残基含量较多,并以1,2或1,4相反离子对的形式排列,即Glu-Arg、Glu-Lys或Asp=lys、Asp-Arg。②疏水氨基酸残基一对对地出现在1,2或1,4反离子对的附近,因此很容易形成特有的双性螺旋二级结构。极性与非极性氨基酸残基排列的方式是载脂蛋白的一个共性。由疏水氨基酸残基组成螺旋的非极性面,由带电荷亲水的氨基酸残基组成螺旋极性面,故称为双性螺旋。与一般蛋白质的α-螺旋不同,这种双性螺旋既有亲脂的一面又有亲水的一面。ApoAⅠ富含双性螺旋结构,对于维持其正常的生理功能是非常重要的。
ApoAⅠ主要存在于HDL中,在HDL3中ApoAⅠ占载脂蛋白的65%,在HDL2中ApoAⅠ占载脂蛋白的62%,在CM、VLDL和LDL中也有少量存在。血浆中呈现β迁移率的一种β-HDL,其内80%为ApoAⅠ。
Ⅰ的生理功能有:①组成载脂蛋白并维持其结构的稳定性与完整性。实验表明,纯化的ApoAⅠ在水溶液中可以自发地和脂类结合。用CNBr法将ApoAⅠ裂解成四个肽段,发现仅有羧基末端的肽段可自发和磷脂结合。后来进一步确认这一段是ApoAⅠ224-242段,这一段既可维持双性螺旋的结构,又可以维持和脂质结合所具备的疏水性。②ApoAⅠ可以激活卵磷脂胆固醇酰基转移酶(LCAT)的活性。已经证实,ApoAⅠ是通过激活LCAT,再催化胆固醇酯化。ApoAⅠ肽段Ⅲ(肽段116-151)是激活作用的中心。③ApoAⅠ可作为HDL受体的配体,含ApoAⅠ脂蛋白可以和转铁蛋白及铜蓝蛋白形成大分子复合物以运输铁和铜离子。
Ⅰ由肝和小肠合成,血浆中生物半寿期为45天。
2.ApoAⅡApoAⅡ是HDL中第二种含量多的载脂蛋白,在HDL2中占载脂蛋白的15%,在HDL3中占载脂蛋白25%,在CM中占载脂蛋白的7%-10%,VLDL中也存在少量。到1985年,ApoAⅡ蛋白质的氨基酸序列,cDNA序列及基因序列均已阐明。ApoAⅡ是由两条多肽链的77个氨基酸残基组成。ApoAⅡ在不加还原剂的SDS-PAGE中测出分子量是17ku,在人血浆中以二聚体形式存在。ApoAⅡ的单体分子量为8.7ku。ApoAⅡ蛋白的C端氨基酸残基为谷氨酸,N端为吡咯烷酮酸,缺乏组氨酸、精氨酸及色氨酸。ApoAⅡ有多态性存在。
ApoAⅡ生理功能是:①维持HDL结构,ApoAⅡ肽段12-31和肽段50-77具有与磷脂结合的能力。经二级结构分析认为,残基17-30和51-62形成的双性螺旋结构是人ApoAⅡ与脂质结合的分子基础。②激活肝脂酶,用以水解CM和VLDL中的TG和PL。还有报道,ApoAⅡ可抑制LCAT活性。
ApoAⅡ由肝和小肠合成。人血浆中的ApoAⅡ生物半寿期为4.4天。
3.ApoAⅣ最先从大鼠HDL和CM中发现载脂蛋白AⅣ,以后证实人血浆中也有ApoAⅣ存在,主要分布于密度大于1.211g/ml部分。成熟ApoAⅣ由376个氨基酸残基组成。经SDS-PAGE确认大鼠和人ApoAⅣ分子量为44-46ku。人和大鼠氨基酸组成相似,是一种糖蛋白,含有6%的碳水化合物,其中甘露醇占1.8%,半乳糖占1.55%,N-乙酰葡萄糖胺占1.55%,唾液酸占1.1%。ApoAⅣ有多态性存在,生物半寿期为10小时。
ApoAⅣ生理功能目前尚不完全清楚,据推测ApoAⅣ在胆固醇逆向转运过程中起着重要作用。体外的ApoAⅣ可以促进LCAT的胆固醇酯化反应,并认为是LCAT的激活剂。ApoAⅣ由肝和小肠合成,并有五种多态型。
(二)Apob族
体外实验表明,ApoB是难溶于水的蛋白质。目前所知,ApoB族可分为两个亚类,即ApoB48和ApoB100。ApoB主要成分是B100,其次为B48,其它形式的如ApoB75、ApoB41、ApoB36等均为ApoB100的不同降解产物。
1.ApoB100ApoB是单链糖蛋白,分子量为51ku,主要在肝,少数在小肠合成。ApoB100由4536个氨基酸残基组成。包括27个(或24)氨基酸信号肽和4536个氨基酸残基的成熟单体蛋白。1986年ApoB100的全部氨基酸残基排列顺序及结构已经阐明。当年已测出ApoB100的cDNA序列,ApoB100分子中含有25个Cys残基。其中有11个Cys残基集中分布在前面500氨基酸组成区域,形成链内二硫键,所以N端高度交联成典型球形结构。Cys残基通过硫酯键与软脂酸、硬脂酸相结合,使ApoB牢固地连接着脂质成分。ApoB100中,对脂类结合十分必要的区域结构在203-2506和4002-4527氨基酸残基之间。两个结构区域重复出现两性亲脂α-螺旋区段;另有一种结合脂质的重要结构是含疏水和亲水性氨基酸交替排列的两性亲脂β-折叠结构。这种结构分布在整个分子序列中,但集中于四个富含脯氨酸区,这种富含脯氨酸的重复序列是ApoB所特有的,使ApoB能够将磷脂侧链深埋其间并使之紧密结合。由于ApoB的两性α-螺旋和富含脯氨酸的疏水肽以及可被脂酰化的Cys残基形成的特殊结构,在VLDL和LDL从分泌到被清除的整个过程中,使α-螺旋能够与单层极性脂牢固地结合,从而使其不在脂蛋白分子间转换,这是与其他载脂蛋白不同之处。
ApoB100的生理功能有:①合成装配和分泌富含甘油三酯的VLDL;②是LDL的结构蛋白。③LDL受体的配体,并可调节LDL从血浆中的清除速率。
2.ApoB48ApoB48因分子量是ApoB100的48%而得名。存在于CM中,不与其他脂蛋白分子交换。ApoB48在小肠合成,是组装CM所必需的载脂蛋白。小肠细胞分泌CM后进入淋巴液,并通过胸导管再进入血液循环,再分布到毛细血管的内皮细胞,主要是骨骼肌体和脂肪组织的内皮细胞,脂肪酶可水解CM中甘油三酯的80%-90%,剩下的脂蛋白颗粒则称为CM残粒,尓后送到肝,被肝脂酶进一步代谢,最后被能够识别ApoE的残粒受体摄取。该残粒受体是LDL受体,还是与LDL受体相关的蛋白或其他蛋白质,目前尚不清楚。
人血浆中ApoB48生物半寿期仅5-10分钟,分解速度很快,血浆中的浓度很低,约相当于ApoB100的0.1%。经SDS-PAGE电泳染色在VLDL组分中可检出痕量的ApoB48。进食丰富的脂肪后,ApoB48/ApoB100比值明显增加。
(三)ApoC族
ApoC是目前所知载脂蛋白中分子量最小的一类。最先从VLDL中分离出一种含有少量磷脂的低分子量载脂蛋白,并命名为载脂蛋白C。此后双在HDL中发现有ApoC,并进一步确认ApoC有三种亚型,即ApocⅠ、Ⅱ、Ⅲ。
Apoc是由57个氨基酸残基组成的单一多肽链,其序列已测出,不含半胱氨酸、组氨酸和酪氨酸。分子量为6625u。人ApoCⅠ二级结构中有55%α-螺旋结构,极易与磷脂结合,它是LCAT的激活剂。
ApoCⅡ是由79个氨基酸残基组成单一多肽链,氨基酸顺序已测出,分子量为9110u,有两种多态型,pⅠ分别为4.86和4.69。不含半胱氨酸和丝氨酸,其二级结构的α-螺旋约占23%。ApoCⅡ可激活多种来源的脂蛋白脂肪酶(LPL),其结构中第55-78位氨基酸残基是维持其对LPL激活作用的最短的必须区域。羧基端43-50位氨基酸残基为α-螺旋结构的脂质结合区。
ApoCⅢ由79个氨基酸残基组成单一多肽链,由于第74位苏氨酸残基所带唾液酸个数不同,又可分为ApoCⅢ、CⅢ1、CⅢ2三个亚类,也是其多态性,等电点分别为5.02,4.82和4.62。ApoCⅢ氨基酸序列已测出,分子量为8764u,其二级结构在不同状态下,α-螺旋约占22%-54%不等。ApoCⅢ的α-螺旋结构极易与磷脂结合。
ApoC族生理功能有:①同磷脂相互作用,维持脂蛋白结构:在溶液中呈特殊的立体双性离子,带负电荷的酸性氨基酸与磷脂带正电荷的基团作用,具有很强的磷脂结合活性。由于与磷脂的相互作用,使ApoC族的α-螺旋结构增加,而磷脂的单个酯酰链的运动则受到限制,从而影响磷脂从凝胶态到液晶态的转变,两者作用的结果,从而固系了脂蛋白的结构;②对酯酶有激活作用,HDL的磷脂在流动性增加时,ApoCⅠ通过HDL脂层表面后促进了LCAT的催化作用;③ApoCⅡ可以激活LPL,其激活机制可能是:LPL通常与外周循环肝素样分子结合并附着于血管内皮上,当LPL接触CM或VLDL时,LPL便同脂蛋白颗粒表面的磷脂发生作用,进而结合于脂蛋白颗粒上,其内的ApoCⅡ与LPL发生作用,改变LPL的空间结构,进而催化水解甘油三酯。
Apoc主要由肝合成,小肠也合成少量。
(四)ApoE
ApoE是一种富含精氨酸的碱性蛋白,人AppE由299个氨基酸残基组成,分子量为34.145ku,含32个Arg和12个Lys,存在于血浆CM、VLDL及其残粒中,β-VLDL中含ApoE量高于VLDL,一部分ApoE在血液中与ApoAⅡ形成复合体。已测出ApoE的蛋白质一级结构,建立ApoE和cDNA序列,并确认ApoE有3个等位基因异构体以及基因在染色体上的定位。据推算和测定,在溶液中ApoE有62%的α-螺旋、9%的β-片层、11%的β-转角和18%的无规则线团。ApoE分子可以被凝血酶水解为N-端和C-端两个区域,N-端区(1-191)为22ku的可溶性球蛋白,此区域较稳定;C-端区(216-299)分子量为10ku,螺旋程度很高,不稳定,是与脂蛋白的结合区,ApoE主要由肝脏合成,近年来发现脑、肾、骨骼、肾上腺及巨噬细胞也能合成ApoE。
ApoE生理功能有:①是LDL受体的配体,也是肝细胞CM残粒受体的配体,它与脂蛋白代谢密切相关;②ApoE具有多态性,多态性与个体血脂水平及动脉粥样硬化发生发展密切相关。
(五)Apo(a)
早期测定脂蛋白(a)[LP(a)]在人群分布率为30%。目前采用更灵敏的方法发现几乎存在于所有人群中,仅是血浆深度差异很大,波动在0-1000mg/L的范围。1987年克隆了人Apo(a)的基因序列,并推导出氨基酸序列,提示Apo(a)的分子结构与纤溶酶原极为相似。Apo(a)含有一个疏水信号序列,37个Kringle-4拷贝、1个Kringle-5及1个胰蛋白酶样区。第36个Kringle-4含有一个额外未配对半胱氨酸,推测此处可能是Apo(a)以二硫键与ApoB结合的部位。经胰蛋白酶限制性水解Apo(a)发现,Apo(a)中的Krtingle-4有75%-85%的氨基酸与纤溶酶原的第391-472个氨基相同,有共同的抗原簇,两者表现有交叉反应。纤溶酶原(PG)是一种丝氨酸蛋白酶原,含有791个氨酸残基,结构中含有5个富含半胱氨酸的“Kringle”样结构,即Kringle1-5,在Kringle-5的后面为一丝氨酸蛋白酶区。PG与Apo(a)结构相似,如图4-3所示。
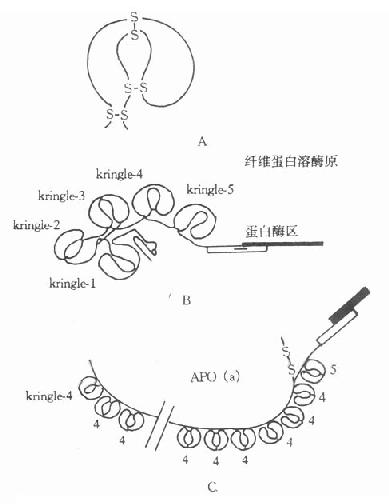
图4-3 载脂蛋白(a)结构示意图
A:KringleB:纤维蛋白溶酶原C:Apo(a)
Kringle结构是三对二硫键组成的三套环形结构,含有78-82个氨基酸残基,因其序列的书写形式酷似一种丹麦面糕而得名。在其第1与第6、第2与第4、第3与第5半胱氨酸上,连成三个二硫键。这种结构也出现在前凝血酶、尿激酶、链激酶和纤溶酶原激活剂(t-PA)的组份中。由于Apo(a)分子中的Kringle-4数目可在15-27之间变化,从而导致Apo(a)有多种不同的异构体。
Apo(a)结构中有一蛋白酶区,推测其功能可能是一种酶。在分子中相当于PG蛋白酶的丝氨酸被精氨酸代替,可使其丧失酶的功能。由于Kringle结构与PG相似,推测Apo(a)可能结合到象PG受体或纤维蛋白那样的大分子上,再加上LP(a)颗粒携带的胆固醇结合到血管损伤部位,因此它不仅促进动脉粥样硬化形成,也阻碍血管内凝血块的溶解。
二、载脂蛋白的基因结构及表型
(一)基因多态性概念
各种生物都能通过生殖产生子代,子代和亲代之间,不论在形态构造或生理功能的特点上都很相似,这种现象称为遗传(heredity)。但是,亲代和子代之间,子代的各个体之间不会完全相同,总会有所差异,这种现象叫变异(variation)。遗传和变异是生命的特征。遗传和变异的现象是多样而复杂的,正因为如此,才导致生物界的多种多样性,生物体所具有的遗传性状称为表型或表现型(phernotype)。生物体所具有的特异基因成分称为基因型(genotype)。表型是基因型与环境因素相互作用的结果。遗传物质是相对稳定的,但是又是可变的,遗传物质的变化以及由其所引起表型的改变,称为突变(mutation)。遗传物质突变包括染色体畸变和基因突变。基因突变是染色体中某一点上发生化学改变,所以又称为点突变(pointmutation)。基因结构和遗传表型的研究是深入了解脂蛋白代谢缺陷症的分子生物学基础,逆向遗传学方法(reversegeneticapproach)则使其有可能在蛋白质水平系统地分析结构和功能的关系。现已采用一个特定的cDNA探针从基因文库中筛选所需要的基因进行cDNA克隆,测定其核苷酸序列,然后从核苷酸序列推断蛋白质氨基酸序列。目前,已分离出许多与动脉粥样硬化有关的脂蛋白的cDNA克隆,并将其蛋白质一级结构的氨基酸排列顺序和基因的核苷酸顺序测出。现已查明,ApoAⅠ、AⅣ、E、B、CⅡ和(a)都存在着异构体,也就是说存在着各种不同的表型或基因型,并可分别从蛋白质水平和核酸水平进行分型。现分别介绍几种主要载脂蛋白的基因结构。
(二)载脂蛋白基因结构特点
人血浆中载脂蛋白的结构及功能,经过近十年的深入研究,已了解得较为清楚。大部分载脂蛋白的基因和cDNA都已得到分离和确定,其核苷酸顺序也进行了测定。除ApoAⅣ,B、(a)外,它们的共同特点是含有三个内含子(intron)和四个外显子(exon),其内含子插入外显子的位置大致相同,基本上按照生理功能的不同,将其加以分隔。第一个内含子把5′-末端的非翻译区和翻译区分开;第二个内含子把信号肽编码(singnalpeptide)和功能蛋白编码区分开;第三个内含子则把原肽编码区和成熟肽编码区分开。这些基因的第一、二、三外显子的核苷酸数量也相差无几,第四个外显子核苷酸数量不同而导致各种载脂蛋白基因长度不同。从生物进化角度考虑,上述载脂蛋白基因结构相似性,提示可能来源于一个共同的祖先,即ApoCⅠ基因。ApoAⅣ与其他载脂蛋白基因结构不同,它只含有三个外显子。载脂蛋白基因结构的另一特点是几个基因相接很近,定位于同一染色体的一个位点上或附近,呈紧密连锁状态。如ApoAⅠ、CⅢ和AⅣ基因位于第11号染色体长臂2区,形成一个约15kb的基因簇。还有一个紧密连锁的基因簇是ApoE、CⅠ和CⅡ基因,同位于第19号染色体长臂3区,见图4-4。
ApoA-Ⅱ基因定位于第1号染色体长臂2区,ApoB基因定位于第2号染色体短臂2区,Apo(a)基因定位于第6号染色体长臂2区。
(三)载脂蛋白基因结构
1.ApoAⅠApoAⅠ基因长1863bp,含有三个内含子,第一个内含子位于5′端非翻译区;第二个内含子位于翻译区的AⅠ前肽区内;第三个内含子插入翻译成熟AⅠ第43氨基酸残基处。ApoAⅠ基因含有四个外显子,分布于ApoAⅠ基因的不同区域,ApoAⅠ基因与ApoCⅢ、AⅣ基因相连成簇,CⅢ基因居中,转录方向与AⅠ和AⅣ基因相反。位于AⅠ和CⅢ基因共同3′区的DNA序列,可能参与对AⅠ基因的转录调控。
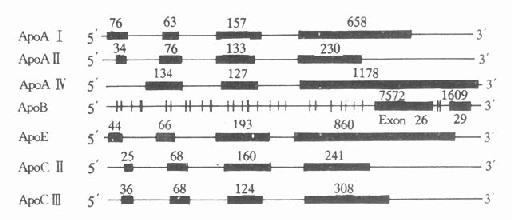
图4-4 人载脂蛋白AⅠ、AⅡ、AⅣ、B100、
CⅡ、CⅢ和E基因结构示意图
粗线代表外显子,粗线之间的细线代表内含子,粗线上缘数字
代表该段核苷酸数目
2.ApoBApoB族位于2号染色体P23→Pter区,是由非翻译区、编码区、TAA终止密码子和一个3′端的非翻译区组成。ApoB100基因全长43kb,含29个外显子和28个内含子见图4-4,其中第26和第29两个外显子特别长,分别含有7552和1905bp,外显子2最短,仅39bp(从211-249)。内含子则以第27个为最短(107bp)。人群中至少有14种不同的3′端高变异等位基因区,75%的人群在此区是不均一的。
ApoB48和ApoB100除了在结构上有关外,ApoB48的形成机制目前尚无完全一致的看法,主要认为有合成ApoB48的基因存在。1987年被发现ApoB48是由ApoB100通过一种新的机制涉及到mRNA的编辑而产生的。在测定从人小肠基因库分离的ApoBcDNA的序列时发现,小肠ApoBcDNA的第6666个核苷酸为T,而从肝分离的ApoBcDNA克隆在此位置为C。将T替换C则6666处产生一终止编码(TAA),TAA替换CAA编码使ApoB100的2153位氨基酸应为Gln,预示血浆中存在的ApoB48应是相当于ApoB100的2153氨基末端为Gln。这一预测后来得到实验证实,并发现核苷酸上6666的替换C→T只发生在小肠的mRNA上,而不发生在小肠基因组(genomic)DNA上,因此这是转录以后的一种特殊形成的编辑小肠mRNA的结果.
3.ApoE人ApoE基因位于19号染色体长臂3区,含有四个外显子和三个内含子。
1975年首先观察到ApoE的多态性,利用等电聚焦电泳和SDS-PAGE可以确认ApoE的多态性。实验表明,ApoE有三种异构体(isoform)即E2、E3和E4。有的人只含有一种主要异构体即纯合子,有的人可含二种主要异构体为杂合子。由此可见,人群中可有六种不同的表现。根据ApoE表型提出ApoE基因模型认为,ApoE的合成是由位于一个基因位点上的三个等位基因所控制,即E2、E3和E4,每一个等位基因对应于一个主要异构体,产生三种纯合子(E2/2,E3/3,E4/4)和三种杂合子(E2/3,E2/4,E3/4)共六种常见表型,另外,还有极少见的异构体。一般认为,次要异构体是由主要异构体翻译后,经唾液酸糖化修饰后转变而来。ApoE3/3型又称野生型。ApoE的基因序列的112位和158位两种氨基酸残基即精氨酸(Arg)和半胱氨酸(Cys)的交换决定了异构体的种类。ApoE4在这两个位置上都是Arg;E2都是Cys;112和158位是Arg者为ApoE3异构体。自然人群中,基因频率(3)分布最高,ApoE3/3表型分布约70%,见图4-5。
4.ApoC族ApoCⅡ基因有3347bp,含有4个外显子和3个内含子。ApoCⅡ的羧基末端氨基酸序列是激活脂蛋白脂肪酶的活性功能区域。ApoCⅢ基因含有3133bp,有4个外显子和3个内含子。

图4-5 人ApoE三种主要异构体的氨基酸残基及基因密码的改变位置
5.Apo(a)运用cDNA探针进行染色体定位研究时发现,Apo(a)的基因位点在人第6号染色体长臂2区6-7带间,与血纤溶酶原(PLG)的基因位点有部分重叠。测定PLG基因跨距为525kb,由18个内含子与19个外显子组成,5个Kringle结构由各自两个外显子编码。Apo(a)cDNA分析表明,Apo(a)与PLG的基因有很多相似之处。
通过家系研究,目前已发现Apo(a)基因位点中至少有26个等位基因与多态性有关。这些等位基因至少表达有34种Apo(a)异构体。
第三节 脂蛋白受体
脂类在血液中以脂蛋白形式进行运送,并可与细胞膜上存在的特异受体相结合,被摄取进入细胞内进行代谢。迄今为止报道的受体已有很多种,研究最详尽的是LDL受体,其次是清道夫受体,再就是VLDL受体。这三种受体的氨基酸序列、构象及与配体的结合部位都已阐明,并且已成功地得到其cDNA。Brown和Goldstein于1974年研究家族性高胆固醇血症(familialhypercholesterolemia,FH)患者代谢缺陷时,在成纤维细胞膜上发现了LDL受体(LDLreceptor,LDLR)的存在。以后相继发现有VLDL受体和清道夫受体。脂蛋白受体在决定脂类代谢途径、参与脂类代谢、调节血浆脂蛋白水平等方面起重要的作用。脂蛋白受体的发现是脂类代谢研究的里程碑,推动了脂蛋白、载脂蛋白的深入研究。
一、LDL受体
最先从牛肾上腺分离出LDL受体,以后又分离了编码牛LDL受体羟基末端1/3氨基酸的cDNA,并初步阐明了牛LDL受体的cDNA,并且推导出人LDL受体的氨基酸序列。
(一)LDL受体结构
LDL受体是一种多功能蛋白,由836个氨基酸残基组成36面体结构蛋白,分子量约115ku,由五种不同的区域构成,各区域有其独特的功能,见图4-6。
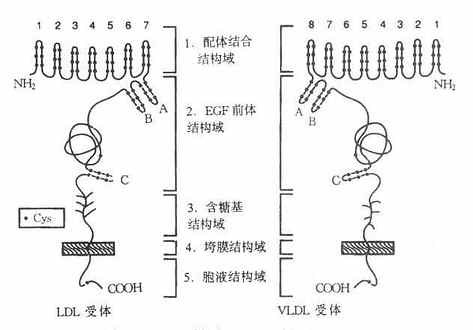
图4-6 LDL受体与VLDL受体结构示意图
1.配体结合结构域配体结合结构域由292个氨基酸残基组成,其中共有47个半胱氨酸(Cys)。含有七个由40个残基组成的与补体Cb和Cq类似的重复序列,每个重复系列中有6个半胱氨酸残基,所有42个半胱氨酸残基均已构成二硫键,重复序列2,3,6,7是结合LDL所必需,其中任何一种发生突变,均使受体丧失结合LDL的能力。重复序列5则与结合β-VLDL有关,若该序列突变时,受体结合β-VLDL的能力丧失60%。该受体不仅能结合LDL,还能结合VLDL、β-VLDL和VLDL残粒,它不仅能识别ApoB100,也可识别含ApoE的脂蛋白。ApoE、B100为LDL受体的配体,因此,LDL受体又称为ApoB100E受体。
2.EGF前体结构域该域约由400个氨基酸残基组成的肽段,有五个重复序列,每个重复序列包括25个氨基酸残基。EGF前体结构域与小鼠上皮细胞生长因子(epidermalgrowthfactor,EGF)前体有同源性,这一区域因此而得名。体外实验证实,这个区域的肽段,属于细胞膜外结构蛋白,起着支撑作用。
3.糖基结构域由58个氨基酸残基组成,是紧靠细胞膜面的肽段,有18个丝氨酸或苏氨酸,构成O-连接糖链,对LDL受体也有支撑作用。
4.跨膜结构由22个氨基酸残基组成,富含疏水氨基酸残基,属于跨膜蛋白,起着固系于细胞膜中的“抛锚”作用。这个区域若有缺陷则影响受体的细胞外分泌。
5.胞液结构域位于细胞膜的胞质侧,由50个氨基酸残基组成,C-末端位于胞质并“深埋”于胞质之中。
(二)LDL受体基因结构及功能
1.受体亲和性含ApoB100的脂蛋白可以与LDL受体以高亲和力结合,肠道分泌的ApoB48不是LDL受体的配体,所以肝脏不能清除完整的CM。
2.基因结构人LDL受体基因长度45ku,由18个外显子和17个内含子组成。
3.LDL受体途径LDL受体广泛分布于肝、动脉壁平滑肌细胞、肾上腺皮质细胞、血管内皮细胞、淋巴细胞、单核细胞和巨噬细胞,各组织或细胞分布的LDL受体活性差别很大。
LDL或其他含ApoB100、E的脂蛋白如VLDL、β-VLDL均可与LDL受体结合,内吞入细胞使其获得脂类,主要是胆固醇,这种代谢过程称为LDL受体途径(LDLreceptorpathway)。该途径依赖于LDL受体介导的细胞膜吞饮作用完成,如图4-7所示。当血浆中LDL与细胞膜上有被区域(coatedregion)的LDL受体结合(第1步),使其出现有被小窝(coatedpit)(第2步),并从膜上分离形成有被小泡(coatedvesicles)(第3步),其上的网格蛋白(clathrin)解聚脱落,再结合到膜上(第4步),其内的pH值降低,使受体与LDL解离(第5步),LDL受体重新回到膜上进行下一次循环(第6、7步)。有被小泡与溶酶体融合后,LDL经溶酶作用,胆固醇酯水解成游离胆固醇和脂肪酸,甘油三酯水解成脂肪酸,载脂蛋白B100水解成氨基酸。LDL被溶酶体水解形成的游离胆固醇再进入胞质的代谢库,供细胞膜等膜结构利用。胞内游离胆固醇在调节细胞胆固醇代谢上具有重要作用;若胞内浓度升高,可能出现下述种情况:①抑制HMGCoA还原酶,以减少自身的胆固醇合成;②抑制LDL受体基因的表达,减少LDL受体的合成,从而减少LDL的摄取,这种LDL受体减少的调节过程称为下调(downregulation);③激活内质网脂酰基CoA胆固醇酰转移酶(Acyl-CoAcholesterolacyltransferase,ACAT),使游离胆固醇在胞质内酯化成胆固醇酯贮存,以供细胞的需要。经上述三方面的变化,用以控制细胞内胆固醇含量处于正常动态平衡状态。血浆中胆固醇主要存在于LDL中,而65%-70%的LDL是依赖肝细胞的LDL受体清除。肝的LDL受体还影响LDL的合成速率及VLDL代谢。曾经认为人VLDL几乎全部在血循环中转变为LDL,LDL再被肝外组织摄取。现在经大鼠和兔实验研究表明,仅有15%以下转变为LDL,人则是小于50%的VLDL转变为LDL,大部分VLDL是以VLDL或VLDL残粒的形成被肝摄取。VLDL残粒与肝受体的亲和力比VLDL大很多。所以VLDL残粒被肝清除的速率比VLDL快。VLDL残粒大部分被肝清除,一小部分在肝脂酶作用下水解除去甘油三酯而转变成LDL。LDL受体还在乳糜微粒代谢中起一定作用。乳糜微粒中的ApoB48不能识别ApoB100E受体,所以肝不能清除完整的乳糜微粒。CM中虽有少量ApoE,因含有丰富的ApoC,可掩盖ApoE,而阻碍其与肝的ApoB、E受体结合,血液中乳糜微粒被脂蛋白脂肪酶水解去除其大部分甘油三酯核心后,同时丧失部分ApoC、A,生成乳糜微粒残粒后除去了阻碍ApoE与受体结合的因素,其残粒可迅速被肝清除,约有一半是通过LDL受体,另一半通过LDL受体相关蛋白代谢,其半寿期短。
总之,LDL受体主要功能是通过摄取Ch进入细胞内,用于细胞增殖和固醇类激素及胆汁酸盐的合成等。
二、VLDL受体
在ApoE100存在下,LDL受体可以结合LDL;有ApoE存在时,LDL受体既可结合LDL,又可结合VLDL、β-VLDL。与LDL受体不同,还有一种仅与含ApoE脂蛋白结合的特异受体存在,有以下临床现象及实验结果让人不得不推测还有另一种受体的存在:①纯合子FH患者血中乳糜微粒残粒并不增加;②LDL受体缺陷的WHHL兔乳糜微粒残粒仍正常地被肝摄取;③LDL受体下调状态下,乳糜微粒残粒可以在肝内异化,FH的LDL受体缺陷者或WHHL兔巨噬细胞不能利用LDL使之泡沫化,但可利用含ApoE脂蛋白的乳糜微粒残粒β-VLDL使其泡沫化,所以推测有对ApoE特异结合的另一种受体存在。
利用cDNA单克隆证明存在VLDL受体,其结构与LDL受体类似,如图4-6所示。由与LDL受体相同的五部分组成,即配体结合结构域、EGF前体结构域、含糖基结构域、跨膜结构域和胞液结构域。然而并非完全相同,配体域结构有32%的相同性,EGF前体结构域有52%的相同性;含糖基结构域仅有19%的相同性,跨膜域有32%相同性,胞质域有46%的相同性。LDL受体对含ApoB100的LDL,含ApoE的VLDL、β-VLDL、VLDL残粒有高亲和性。VLDL受体仅对含ApoE的脂蛋白VLDL、β-VLDL和VLDL残粒有高亲和性结合,并摄入细胞内,对LDL则为显著的低亲和性。VLDL受体在肝内几乎未发现,但是广泛分布在代谢活跃的心肌,骨骼肌、脂肪等组织细胞。
LDL受体受细胞内Ch(胆固醇,cholesterol)负反馈抑制,VLDL受体则不受其负反馈抑制。当VLDL受体的mRNA量成倍增加时,不受LDL乃至β-VLDL的影响。这是因为VLDL的配体关系使β-VLDL的摄取不受限制。这一点,对由单核细胞而来的巨噬细胞的泡沫化在早期动脉粥样硬化的斑块形成中有重要意义。
VLDL受体在脂肪细胞中多见,可能与肥胖成因有关。
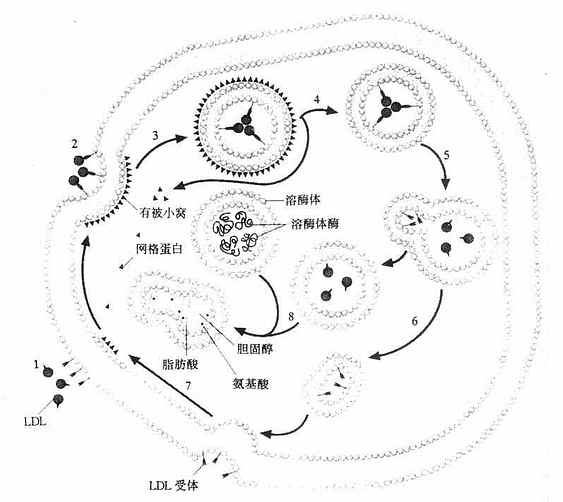
图4-7 LDL受体胞吞作用示意图
三、清道夫受体
遗传性的LDL受体缺陷的杂合子是不能摄取LDL的,但动脉粥样硬化斑块的巨噬细胞有从LDL来的胆固醇大量蓄积并泡沫化,其原因用LDL受体途径无法解释,因为从这条途径不可能摄取过多的脂质。Brown与Goldstein等使LDL乙酰化,从而导致不受细胞内胆固醇调节的过剩脂质也摄入,并出现异常蓄积,进而推测存在一种LDL受体途径以外的脂质摄取途径,使巨噬细胞摄取乙酰化LDL。Brown等人提出这种设想并定名为清道夫受体(scarengereceptor),以后许多实验证明了这种推测。其后,在细胞培养液中添加氧化剂使LDL氧化修饰,其结果使巨噬细胞摄取了这种变性LDL。现在认为,人体内脂质过氧化反应导致的变性LDL,可被巨噬细胞无限制地摄取入细胞内,这是因为变性LDL上带有各种分子的负电荷可与清道夫受体结合。
(一)清道夫受体结构
1990年用配体亲和层析和免疫亲和层析,将牛肺巨噬细胞清道夫受体纯化,并由其部分氨基酸序列克隆得到Ⅰ型、Ⅱ型清道夫受体cDNA。以后相继将人、兔和小鼠的清道夫受体cDNA克隆成功。该受体C-末端为半胱氨酸的为Ⅰ型,具有短肽结构的为Ⅱ型,清道夫受体共有两种亚基,以三聚体形式存在,是分子量为22万的膜糖蛋白;N末端在细胞膜内侧,C末端在膜外侧存在,是内翻外“inside-out”型的受体。该受体的Ⅰ、Ⅱ型均由六个区域部分组成,如图4-8所示。
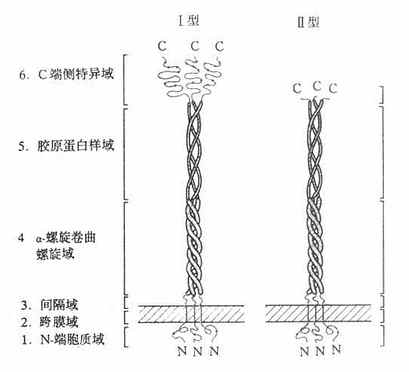
图4-8 清道夫受体结构示意图
1.N-端胞质域由50个氨基酸残基组成,可能与包涵素结合,类似LDL受体结构。其中央部分是磷酸化区域,是摄取配体的最重要的部位。
2.跨膜域(transmembrane)由第51-76氨基酸残基构成。为疏水性氨基酸组成的单一结构,“抛锚”固定于细胞膜上。
3.间隔域由第77-150氨基酸残基构成。
4.α-螺旋卷曲螺旋域(α-hericalcoiled-coil)由第151到271共121个氨基酸残基组成,此肽段常常先折叠成右手α-螺旋,每圈含3.5个氨基酸残基。这些α-螺旋又相互缠绕,构成平行的三股索状结构。这种右手螺旋索靠α-螺旋之间由脂肪族氨基酸的疏水核心来维持。
5.胶原蛋白样域属第273至343个氨基酸残基肽段,这种序列与胶原蛋白非常相似,推测这段肽链为右手胶原蛋白样三联体螺旋。
6.C-端侧特异域属第344至543个氨基酸残基肽段,为羧基末端,该段富含半胱氨酸。清道夫受体的8个半胱氨酸有6个在此范围,所以称为清道夫受体富含半胱氨酸域(scavengerreceptorcysteinrichdomainlike,srcR)。半胱氨酸的二硫键交联而成的区域非常紧密、牢固,形成球状,足以经受细胞外环境的影响,属于细胞外区域。
srcR域长约430nm,犹如三朵郁金香的“花苞”,由间隔域到α-螺旋卷曲螺旋域构成的“花茎”为支撑,这一“花茎”约占总长度的52%或胞外部分的62%。Ⅱ型清道夫受体没有srcR域,代之以6个氨基酸残基,所以是“截短”的清道夫受体,但Ⅱ型清道夫受体比Ⅰ型清道夫受体具有高亲和力结合和介导内移修饰LDL作用,配体谱很广。
(二)清道夫受体配体
清道夫受体配体谱广泛,有:①乙酰化或氧化等修饰的LDL;②多聚次黄嘌吟核苷酸,多聚鸟嘌吟核苷酸;③多糖如硫酸右旋糖酐;④某些磷脂,如丝氨酸磷脂,但卵磷脂不是配体;⑤细菌脂多糖,如内毒素等。这样广泛的配体谱的共同特点是多阴离子化合物。Ⅱ型清道夫受体没有srcR域,但仍具有与Ⅰ型相同的功能,显然配体结合域不在srcR域,推测其结合域在胶原蛋白样域,C末端的22个氨基酸残基作为配体识别位点,是结合多阴离子配体所必须的位点。
(三)清道夫受体功能
目前对于清道夫受体的功能还不十分清楚,是人们在研究巨噬细胞转变成泡沫细胞的机制时发现的。近年来大量实验证明LDL可被巨噬细胞、血管内皮细胞和平滑肌细胞氧化成氧化LDL,可通过清道夫受体被巨噬细胞摄骤,形成泡沫细胞。氧化LDL还能吸引单核细胞粘附于血管壁,对内皮细胞有毒性作用,从而促进粥样斑块形成。这些研究无疑阐明了巨噬细胞的清道夫受体在粥样斑块形成机制中起重要作用;另一方面,也推测巨噬细胞通过清道夫受体清除细胞外液中的修饰LDL,尤其是氧化LDL,是机体的一种防御功能。还有清除血管过多脂质,清除病菌毒素,摄取内毒素及其他多方面的功能。
清道夫受体分布于胎盘、肝、脾等单核吞噬细胞系统。
清道夫受体不仅在组织巨噬细胞内存在,在单核细胞分化由来的巨噬细胞侵入内皮下的过程中也见有该受体。兔、大鼠高脂肪膳食模型制作过程中,喂饲高胆固醇开始的几天见到LDL样粒子附着于血管壁,其后有单核细胞附着于内膜,巨噬细胞导致脂肪线条病巢形成,并以成百成千巨噬细胞簇出现,此时发现有大量的清道夫受体。当病灶逐步进入平滑肌细胞内膜后,其深部巨噬细胞仅有少量残存,受体量逐渐减少。若变性LDL显著增加时,清道夫受体摄取脂质的过程不受制约,可能这是脂质沉积的重要原因,也是动脉粥样硬化发病的重要机制。使LDL变性的主要因素是脂质的过氧化,然而是何种原因引起胆质过氧化的,尚待进一步研究。
第四节 脂代谢有关酶类与特殊蛋白质
参与脂质代谢的酶有LPL,HTGL,LCAT,ACAT,HMG-CoA还原酶,HMGCoA合成酶。脂质代谢过程中还有几种特殊蛋白质如CETP等。
一、脂蛋白脂肪酶
脂蛋白脂肪酶(lipoprteinlipase,LPL)是脂肪细胞、心肌细胞、骨骼肌细胞、乳腺细胞以及巨噬细胞等实质细胞合成和分泌的一种糖蛋白,分子量为60ku,含3%-8%碳水化合物。活性LPL以同源二聚体形式存在,通过静电引力与毛细血管内皮细胞表面的多聚糖结合,肝素可以促进此结合形式的LPL释放入血,并可提高其活性。LPL生理功能是催化CM和VLDL核心的TG分解为脂肪酸和单酸甘油酯,以供组织氧化供能和贮存。LPL还参与VLDL和HDL之间的载脂蛋白和磷脂的转换,ApoCⅡ为LPL必备的辅因子,其中的C端第61-79位氨基酸具有激活LPL的作用。在哺乳类动物如牛、鼠和猪等LPL的酶蛋白质一级结构有87%-94%的同源性,事实表明,LPL在进化过程中具有高度保守性,人类LPL、肝脂酶(hepatictriglyceridelipase,HTGL)及胰脂酶具有高度相似的氨基酸序列,推测三者可能起源于同一个基因家族,有共同的作用机制。
LPL基因位于第8染色体短臂8p22,长约35kb,由10个外显子和9个内含子组成,编码475个氨基酸残基的蛋白质,LPL基因位点存在多态性,主要分布在LPL基因内含子和侧翼序列中,其中内含子6中PVUⅡ多态位点和内含子8中HindⅢ多态位点与高脂血症有关,并为高脂血症的家系连锁分析提供了遗传标记。
LPL在实质细胞的粗面内质网合成,新合成的LPL留在核周围内质网,属于无活性酶,由mRNA翻译合成的无活性LPL,称为酶前体,再经糖基化后,才转化成活性LPL。从细胞中如何分泌,目前认为有两种机制,其一是细胞合成LPL后直接分泌,不贮存于细胞内,即称为基本型分泌;其二是调节型分泌,某些细胞新合成的LPL贮存在分泌管内,一旦细胞受到一个合适的促分泌刺激,LPL即分泌,此时分泌往往大于合成。所有细胞都具有基本型分泌,只有少部分细胞兼有两种分泌形式。存在于细胞膜外表面的硫酸肝素糖蛋白(heparinsulphateproteoglycans,HSPG)使酶保持一种无活力的浓缩状态,然后通过一个尚未阐明的机制由肝素促使分泌,即肝素后刺激血浆中得到活化的LPL,分布在含甘油三酯的脂蛋白中,主要是分解CM和VLDL的甘油三酯,并结合和附着在这些脂蛋白残粒中,可能作为肝摄取这些颗粒的信号。
LPL生理功能,目前认为是分解脂蛋白核成分的甘油三酯,也分解磷脂如卵磷脂、磷脂酰乙醇胺,并促使脂蛋白之间转移胆固醇、磷脂及载脂蛋白,其代谢产物游离脂肪酸为组织提供能量,或再酯化为TG,储存在脂肪组织中。另外,LPL还具有增加CM残粒结合到LPL受体上的能力,促进CM残粒摄取。
测定血浆LPL活性时,一定要静脉注射肝素,因为LPL对肝素亲和性很高。静脉注射肝素,使LPL从内皮细胞表面释放入血,这是测定血中LPL活性的一种必备操作。通常按每公斤体重10单位的量静脉注射,10分钟后采静脉血得到血浆再测LPL活性。一般静脉注射肝素后血浆总脂酶活性的1/3为LPL,剩余的几乎都是肝脂酶(HTGL)。目前还可用高浓度盐酸或鱼精蛋白选择性抑制LPL活性的方法测定其活性。最近报道,还可用LPL或HTGT抗体进行活性检测。
二、肝脂酶
肝脂酶(hepaticlipase或hepatiltriglyceridase或hepaticendothelaillipase,HL或HTGL)属于与血液循环中内源性TG代谢有关的酶之一,与LPL在功能上有相似之处,然而却是两种不同性质的酶。其特点是:①HL活性不需要ApoCⅡ作为激活剂;②SDS可抑制HL活性,而不受高盐浓度及鱼精蛋白的抑制;③主要作用于小颗粒脂蛋白,如VLDL、残粒残余CM及HDL,同时又调节胆固醇从周围组织转运到肝,使肝内的VLDL转化为LDL。经人及鼠cDNA克隆的DNA序列表明,HL是共有2个N连接多聚糖链的糖蛋白,含有499个氨基酸残基,分子量53ku,基因位于第15号染色体上。与分解代谢有关的丝氨酸位于145位。LPL和HL的基因同属一组基因族,在进化上较为保守。
HL在肝实质细胞中合成,在合成过程中,酶蛋白的糖化及紧随着的低聚糖化修饰过程是分泌HL的必要条件。免疫电镜研究表明,HL位于肝窦状隙内皮细胞表面,在肝素化后,HL可释放到血浆。激素可调节HL的释放,主要是类固醇激素,如雄性激素可升高HL酶活性,而雌性激素则相反。录怀孕或泌乳时,肝素化后血浆中HL活力与血浆的游离胆固醇或类固醇也呈负相关,肾上腺素抑制HL酶活性。另外胰岛素和甲状腺素在控制HL活力中有作用。
HL主要作用于VLDL、β-VLDL及VLDL残粒中的TG。HDL中积累的未酯化胆固醇在HL作用下由肝摄取,在HDL3转化为HDL2的过程中可防止肝外组织过量胆固醇的积累,其中HL起重要作用。
三、卵磷脂胆固醇脂酰转移酶
卵磷脂胆固醇脂酰转移酶(lecithin-cholesterolacyltransferase,LCAT)由肝合成释放入血液,以游离或与脂蛋白结合的形式存在,是一种在血浆中起催化作用的酶,其作用是将HDL的卵磷脂的C2位不饱和脂肪酸转移给游离胆固醇,生成溶血卵磷脂和胆固醇酯。血浆胆固醇几乎70%-80%是胆固醇酯,均是LCAT催化生成所致。LCAT常与HDL结合在一起,在HDL颗粒表面活性很高并起催化作用,对VLDL和LDL的颗粒几乎不起作用。LCAT在磷脂代谢中有重要的作用。
LCAT由416个氨基酸残基组成,分子量为6.3ku。属于糖蛋白,糖链约占24%,是维持其活性必不可少的组分,富含Glu、Asp、Gly、Pro、Leu。每一酶分子含4个Cys,其中两个连成二硫键。根据与胰脂酶序列的同源性比较,推测六肽Ⅰ178-G-H-S-L-G183可能是酶的活性中心。酶蛋白的α螺旋、β-折叠和其他结构比例分别为21%、24%和55%。LCATmRNA约为1400bp组成,其信号肽是440个氨基酸组成的密码子。
LCAT选择性底物是HDL,特别是新生盘状或小球形HDL3。HDL核心是LCAT酶反应产物胆固醇酯的贮存库,并通过胆固醇酯转移蛋白将CE转移至其他脂蛋白和细胞膜,并与其交换。
LCAT除肝细胞合成外,在小肠、脾、胰、胎盘、肾上腺等组织细胞发现有LCAT的mRNA,推测也可合成LCAT。
四、HMGCoA还原酶
HMGCoA还原酶(HMGCoAreductase)是合成胆固醇的限速酶,存在于小胞体膜,催化合成甲基二羟戊酸(mevalonicacid),并生成体内多种代谢产物,称之为甲基二羟戊酸途径。细胞内胆固醇水平调节主要依赖于内因性胆固醇合成途径和LDL受体摄取细胞外胆固醇的外因途径两条。Goldstein和Brown阐明其抑制机制认为,细胞内Ch可作为HMGCoA还原酶抑制剂使其活性降低,肝细胞膜上的LDL受体增加,从血中摄取Ch也增加,使血中胆固醇水平降低。设想使HMGCoA还原酶活性降低的药物可使血在胆固醇水平下降,尤其是对FH的杂合子患者,凡能使LDL受体数锐减的药物均可起治疗作用。
以Merinolin的HMGCoA还原酶抑制剂投入,使狗血中LDL消失速度上升,LDL产生速度下降;肝移植的小儿FH纯合子患者,用梅维诺林治疗可使LDL胆固醇降低40%,而LDL产生速度下降35%。这种抑制剂的投入使LDL合成减少的机制,有两种可能,一是胆固醇合成减少使VLDL生成量降低;第二是HMGCoA还原酶抑制剂使VLDL残粒或β-VLDL异化增加,转变成LDL减少。体外抑制实验也证实,从VLDL残粒转变到LDL的速度比正常状态下小20倍,与此同时LDL受体的亲和力也增加。
五、胆固醇酯转移蛋白
血浆胆固醇酯转移蛋白(cholesterolestertransferprotein,CETP)又称为脂质转运蛋白(lipidtransferprotein,LTP),从血浆d>1.21g/ml组份中精制得到,CETP的非极性氨基酸残基高达45%,是一种疏水性蛋白质,很容易被氧化而失活。CETP由肝、小肠、肾上腺、脾、脂肪组织及巨噬细胞合成的476个氨基酸残基组成的多肽,细胞内成熟蛋白分子量为740ku。最近已阐明其基因结构,存在于第16染色体,与LCAT的基因靠近。
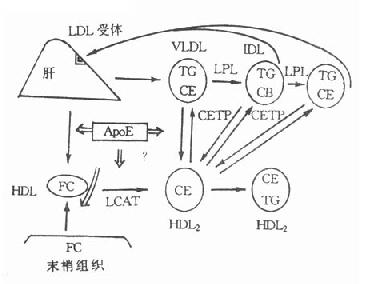
图4-9 胆固醇逆转运系统
CETP促进各脂蛋白之间脂质的交换和转运。CETP在完成和促进胆固醇逆转过程中充当着重要的角色。周围组织细胞膜的游离胆固醇与HDL结合后,被LCAT酯化成胆固醇酯,移入HDL核心,并可通过CETP转移给VLDL、LDL,再被肝的LDL及VLDL受体摄取入肝细胞,至此,完成了胆固醇从周围末梢组织细胞经HDL转运到肝细胞的过程,称之为胆固醇的逆转运(reversecholesteroltransport,RCT)如图4-9所示。
目前认为,血浆中各脂蛋白的胆固醇酯主要通过LCAT和CETP的共同作用生成。血浆中CE90%以上来自HDL,其中约70%的CE在CETP作用下由HDL转移至VLDL及LDL后被清除。CETP与LCAT一样也能与HDL结合在一起。
当血浆中CETP缺乏时,HDL中CE蓄积、TG降低,无法转运给VLDL及LDL,出现高HDL血症,而VLDL、LDL中的CE减少,TG增加。这是因为从HDL将CE转运到含ApoB脂蛋白上发生障碍所致。利用酶联免疫方法测血浆中CETP活性,此时其活性降低。
第五节 脂蛋白代谢
脂蛋白是血液中脂质的运输形式,并与细胞膜受体结合被摄入细胞内进行代谢。
一、乳糜微粒
CM是饮食高脂肪食物后,由肠壁细胞合成的富含TG的巨大脂蛋白,80-100nm。血中半寿期为10~15分钟,食后12小时,正常人血中几乎无CM。它在肠上皮细胞合成,并分泌入淋巴管。CM含有ApoAⅠ,AⅡ,AⅣ和B48。ApoB48含量多少与摄取食物的TG含量有关。ApoB48是合成CM所必须的蛋白质,CM从胸导管移行入血液过程中,其载脂蛋白的组份迅速改变。CM获得ApoC和E后,将ApoAⅠ移行到HDl,脱去ApoAⅣ,使进入血中的CM被末梢血管内皮细胞表面的LPL经ApoAⅡ激活,并作用于其内的TG,分解变成脂肪酸和单甘油脂肪酸,再进入肌肉、脂肪组织及心肌组织贮存或利用。CM表面的磷脂和Apo往HDL3移行,颗粒变小,结果转变成CM残粒,分别被肝脏LDL受体和清道夫受体识别并摄取。
CM生理功能是转运外源性脂类,主要是甘油三酯。其中TG在毛细血管中被水解成游离脂肪酸后进入各组织贮存或利用,而外源性胆固醇则全部进入肝。
二、极低密度脂蛋白
VLDL大小为30-80nm,含有甘油三酯、胆固醇、胆固醇酯和磷脂,TG占50%左右,蛋白质部分为ApoAⅠ、AⅣ、B100、C、E等。VLDL在肝脏合成,利用来自脂库的脂肪酸作为合成材料,其中胆固醇来自CM残粒及肝自身合成的部分。ApoB100全部由肝合成,肝合成的VLDL分泌后经静脉进入血液,再由VLDL内ApoCⅡ激活LPL,并水解其内的TG。由HDL的LCAT作用生成的胆固醇酯经CETP转送给VLDL进行交换,而VLDL中余下的磷脂、ApoE、C转移给HDL,VLDL转变成VLDL残粒(remnant),而后大部分通过VLDL受体摄入肝,小部分则转变成LDL继续进行代谢。
三、低密度脂蛋白
LDL是富含胆固醇的脂蛋白,其胆固醇主要来自从CE转运的高密度脂蛋白中的胆固醇。目前认为血浆中LDL的来源有两条途径:①主要途径是由VLDL异化代谢转变而来;②次要途径是肝合成后直接分泌到血液中。
LDL的降解是经LDL受体途径进行代谢,细胞膜表面的被覆陷窝是LDL受体存在部位,即LDL中的ApoB100被受体识别,将LDL结合到受体上陷窝内,其后再与膜分离形成内吞泡,在内吞泡内经膜H+-ATPase作用,pH降低变酸,LDL与受体分离并与溶酶体融合后,再经酶水解产生胆固醇进入运输小泡体,或者又经ACAT作用再酯化而蓄积。血浆中65%-70%的LDL是依赖LDL受体清除,少部分(约1/3)被周围组织(包括血管壁)摄取异化。一旦LDL受体缺陷,VLDL残粒由正常时大部分经肝LDL受体识别,而改为大部分转变成LDL,使血浆中LDL浓度增加。
四、高密度脂蛋白
HDL主要由肝和小肠合成。肝合成的新生HDL以磷脂和ApoAⅠ为主。在LCAT作用下,游离胆固醇变成胆固醇酯,脂蛋白则变成成熟球形HDL3,再经LPL作用转变成HDL2。
HDL可将蓄积于末梢组织的游离胆固醇与血液循环中脂蛋白或与某些大分子结合而运送到各组织细胞,主要是肝脏。实际上是胆固醇逆转(RCR),RCT促进组织细胞内胆固醇的清除,维持细胞内胆固醇量的相对衡定,从而限制动脉粥样硬化的发生发展,起到抗动脉粥样硬化作用。Golmset指出,LCAT通过转酯化反应完成新生盘状HDL向HDL3、HDL2的转化,减少血浆HDL中游离胆固醇的浓度,构成胆固醇从细胞膜流向血浆脂蛋白的浓度梯度,降低组织胆固醇的沉积。
综上所述,脂蛋白代谢主要途径如图4-10所示,作为食物中的脂质有甘油三酯,胆固醇等,以TG为主。TG经胰脂肪酶水解后,经肠道吸收于肠粘膜细胞内再合成TG(外源性TG),同时还有Ch、PL、ApoAⅣ、B48与之结合形成CM,进入淋巴管由胸导管再送到血液(图中①)。流入血液中的CM,从HDL接受ApoCⅡ、E及CE(图中②),各脏器毛细血管内皮细胞表面的LPL被ApoCⅡ活化水解CM(图中③),使之转变成仅有ApoB48和ApoE为主的CM残粒,被肝细胞的ApoB、E受体识别并摄取入肝细胞(图中④)。CM在血液中半寿期为5-15分钟,空腹时,血中一般无CM存在。
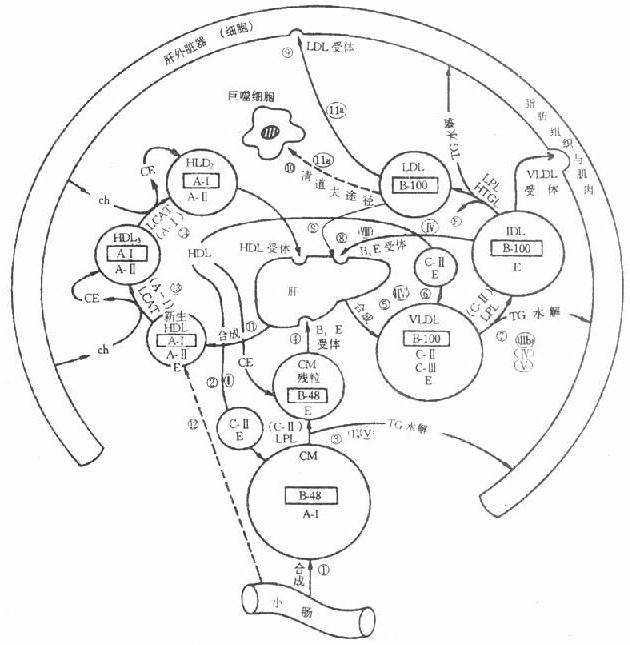
图4-10 脂蛋白主要代谢途径
另外,体内肝将糖及游离脂肪酸合成TG(内源性TG),再与Ch、ApoB100、ApoCⅡ及ApoE等结合形成VLDL释放入血中(图中⑤),血中VLDL再接受HDL上转移的ApoCⅡ、E(图中⑥)又在各组织毛细血管壁的LPL作用下,使其核心的TG不断水解(图中⑦),而后转变成LDL或者VLDL残粒,大部分被肝摄取,也可能被肝外摄取一小部分。血浆中VLDL很不均一,大颗粒的含TG高者可迅速清除,仅10%左右转变成LDL;小颗粒含TG少的VLDL清除较慢,40%转变成VLDL残粒。经肝细胞的LDL受体摄取(图中⑧),一部分又经LPL和HL作用水解TG成脂肪酸,供组织细胞利用,使其表面积缩小,并变成ApoB100为主的LDL。LDL一部分经肝及肝外LDL受体识别摄取入肝内(图中⑨);一部分氧化后经肝及肝外组织清道夫受体识别入肝外组织细胞(图中⑩)。
HDL是肝(图中⑾)和小肠(图中⑿)合成的一种脂蛋白,有从末梢组织运送Ch到肝内处理的生理功能。LCAT使卵磷脂的FFA转移到Ch上催化进行胆固醇酯化反应,HDL是其最适底物。刚合成分泌的HDL为新生HDL,呈圆状,含ApoE多,经LCAT作用转变成球状HDL3。图中⒀再转变成HDL2(图中⒁)。粘膜细胞也可合成一部分新生HDL(图中⑩),HDL还可将ApoCⅡ和ApoE转运给CM及VLDL。
脂蛋白代谢是血中脂质、脂蛋白、载脂蛋白及其受体和酶相互作用并密切相关的代谢过程。在脂蛋白代谢过程中多种环节受到障碍,有可能导致脂蛋白代谢紊乱。
第六节 脂蛋白代谢紊乱
脂蛋白代谢紊乱的常见现象是血中Ch或TG升高,或者是各种脂蛋白水平异常增高。
高脂蛋白血症(hyperlipoproteinemia)是指血浆中CM、VLDL、LDL、HDL等脂蛋白有一类或几类浓度过高的现象。一般根据血浆(血清)外观、血总胆固醇、甘油三酯浓度以及血清脂蛋白含量将高脂蛋白血症进行分型。1967年Fredrickson将高脂血症分为六型,从生化及遗传角度考虑,有其不合理的一面,因为同一型高脂血症可有几种不同的基因型,临床实践表明,这种分型仍有一定的应用价值,因此,一直沿用至今。确认各种类型的高脂蛋白血症时,一定要用血清总胆固醇及甘油三酯、脂蛋白水平交叉检验,以求准确定型。因为任何一种高脂蛋白血症都有Ch、TG一项或两项指标同时升高即高脂血症(hyperlipidemia)出现。高脂血症与高脂蛋白血症实际上是同义词,而后者说明脂质代谢的病理变化更为确切。
从脂蛋白代谢紊乱的原因分类可分为原发性和继发性两大类。原发性是遗传缺陷所致,如家族性高胆固醇血症。继发性是继发于许多疾病所致,如糖尿病、肾病等疾患可继发引起高脂血症。下面着重介绍原发性高脂蛋白血症。
从遗传基因角度考虑,原发性高脂蛋白血症一定由遗传基因突变引起。从生化角度考虑是基因突变所致的基因表达的产物蛋白质水平上的缺陷,如Apo、酶和受体蛋白的异常。Apo异常多见于ApoE变异,典型例子是Ⅲ型高脂蛋白血症,ApoB变异可引起无β-脂蛋白血症,表现为脂肪吸收障碍;ApoAⅠ异常,则有Tangier病出现。导致血清HDL和ApoAⅠ水平降低;LCAT和LPL缺陷以及受体缺陷同样导致脂蛋白代谢异常,如家族性高Ch血症。
一、高脂蛋白血症
1967年Fredrickson等用改进的纸上电泳法分离血浆脂蛋白,将高脂蛋白症分为五型,即Ⅰ、Ⅱ、Ⅲ、Ⅳ和Ⅴ型。1970年世界卫生组织(WHO)以临床检验表现型为基础分为六型,将原来的Ⅱ型又分为Ⅱa和Ⅱb两型,如表4-1所示。
(一)Ⅰ型高脂蛋白血症
患者血浆TG升高,Ch正常,CM、VLDL含量升高,LDL及HDL均降低。约有2/3的病人在10岁前发病。
患者新鲜血清外观呈乳白色混浊,4℃过夜,血浆上层出现“奶油样”上层。大部分患者伴有视网膜脂血症、急性胰腺炎及肝脾肿大。
表4-1 人高脂蛋白血症分型及其特征
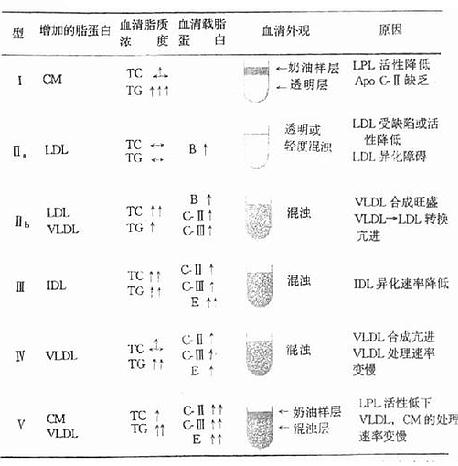
本症为常染色体隐性遗传,家族性遗传性纯合子型患者除血脂改变外,临床症状明显,而杂合子除TG高外,其症状不明显。Ⅰ型高脂血症又称为家族性高CM血症。发病原因主要是LPL的ApoCⅡ的遗传性缺陷,使LPL缺乏或者不能激活,CM中TG不能水解转变成CM残粒,无法被LDL受体识别进行代谢,从而造成CM在血浆中堆积。
(二)Ⅱa型高脂蛋白血症
Ⅱa型又称为家族性高胆固醇血症,血浆LDL和Ch明显升高。血清脂蛋白电泳呈现浓染的β-脂蛋白带,提示β-脂蛋白含量升高,故又称为高β-脂蛋白血症。纯合子病人在青春期即因动脉粥样硬化而死亡,这类病人除冠状动脉硬化外,还会出现黄色瘤和角膜弓状云等。
Ⅱa型有明显家族史,由LDL受体缺陷引起。纯合子型患者LDL受体完全缺陷,杂合子型者LDL受体只为正常的1/2。细胞不能通过膜上LDL受体从血中摄取LDL,使血浆中LDL升高。近年,Goldstein和Brown用纤维母细胞对遗传性高Ch血症遗传分析发现,LDL受体缺陷有三种不同的细胞表型:①无LDL受体型;②LDL受体缺陷型,细胞表面受体活性为正常的5%-20%;③入胞缺陷(内吞缺陷)型,即LDL受体可以与LDL结合,但是不能以正常速度内吞,从而导致LDL堆积于血浆中。目前所知,Ⅱa型是因为编码LDL受体的基因突变所致。
(三)Ⅱb型高脂蛋白血症
Ⅱb型又称为高β-脂蛋白及高前β-脂蛋白血症。血浆电泳图谱中,除β-脂蛋白增高外,前β-脂蛋白含量也升高,但二者并不融合。血浆中除LDL和Ch升高外,VLDL也升高,当然TG也升高,所以又称为混合型高脂蛋白血症。Ⅱb型与Ⅱa型的主要区别是前者LDL受体活性正常,患者多合并肥胖,糖代谢及胰岛素分泌异常,易伴发黄色瘤及动脉粥样硬化症。
Ⅱb型为显性遗传性疾患,体内VLDL合成量过多,ApoB100合成量比正常高两倍,LDL也增高。另外VLDL合成增加的同时,VLDL代谢分解速度并未增强,从而使过量合成的VLDL不能加速分解,造成血浆中VLDL蓄积,同时LDL代谢速度也减慢。
(四)Ⅲ型高脂蛋白血症
Ⅲ型属于家族遗传性高脂蛋白血症,血清电泳图谱上β-脂蛋白带与前β-脂蛋白带融合,呈现一个宽而浓染的色带,称为“阔β带”,因而出现β-移动度的VLDL,故也称为高β-VLDL血症。病因为ApoE异常,属显性遗传。正常人ApoE约65%-75%为E3/E3型,患者亦多见ApoE2/E2型,后者与受体结合力仅为前者的20%-40%。临床症状常见冠状动脉粥样硬化、外周血管病和黄色瘤等。
Ⅲ型患者代谢性改变是:①β-VLDL经肝的ApoE受体清除受阻;②肝从CM残粒获得的外源性胆固醇减少,自身合成Ch并分泌VLDL增多,使VLDL过度生成而堆积于血浆中;③LPL活性降低,VLDL在体外不能转变成LDL。Ⅲ型患者除β-VLDL出现外,Ch和TG均升高。Ⅲ型比较罕见。
(五)Ⅳ型高脂蛋白血症
Ⅳ型又称为家族性高甘油三酯血症或高VLDL血症,仅血浆TG升高,LDL正常,HDL降低。Ⅳ型高TG症的患者属于常染色体显性遗传。Ⅳ型发生动脉粥样硬化的危险性也增加,但不如Ⅱ型、Ⅲ型严重。发病原因尚不清楚。
(六)Ⅴ型高脂蛋白血症
Ⅴ型患者血清脂蛋白电泳图谱呈现CM及前β-脂蛋白深染,属于高CM和高前β-脂蛋白血症都存在的混合型高脂蛋白血症,故又称为高CM与高前β-脂蛋白血症。将该型血浆置于4℃冷藏10小时,可见上层为“奶油样”,下层为混浊状,属于罕见的血清外观。血清TG远远超出正常,LDC-C和HDL-C低于正常值,Ch在正常范围,VLDL-C超出正常范围,VLDL-C/VVLDL-TG低于0.3。若给予患者注入肝素后,血清CM消失。与Ⅲ型不同点是,Ⅴ型人血清VLDL-C/VLDL-TG为0.3以上。该型患者,若仅是LPL活性降低属显性遗传;如既有LPL活性降低,又有ApoCⅡ减少,则属于隐性遗传。常于20岁前发病,家族史明显的Ⅴ型者,丘疹状黄色瘤的发病率可高达30%-50%,并伴有急性胰腺炎及肝脾肿大。ApoCⅡ缺乏、LPL活性降低、VLDL生成过多而代谢率降低等原因导致VLDL在血内堆积。
各类型的高脂蛋白血症表型的诊断指标的变化值如表4-1所示
(七)高HDL血症
血浆HDL含量过高导致高HDL血症,也属于病理状态。HDL具有抗动脉粥样硬化作用,是人们公认的,然而并非血浆HDL含量越高越好。血浆HDL-胆固醇(HDL-C)含量超过1g/L,定义为高HDL血症。现已查明,高HDL血症是因为有CETP和HTGL等活性异常所致。高HDL血症分为原发性和继发性。原发性高HDL血症的病因有以下几种可能:①CETP缺损;②HTGL活性降低;③其他不明原因。继发性高HDL血症病因有:①运动失调;②饮酒过量;③原发性胆汁性肝硬化;④治疗高脂血症的药物引起;⑤其他原因。总之,CETP及HTGL活性降低是引起高HDL血症的主要原因。若CETP缺陷,HDL上的CE蓄积,使HDL增多;若HTGL活性降低,HTGL与HDL被肝细胞摄取减少并使HDL2→HDL3转换过程减慢而停留在血液中,并使其浓度增加,出现高HDL血症。血清中总胆固醇轻度或中度升高,HDL-C高达正常人3-5倍,血清ApoAⅠ、CⅢ、E明显增加,ApoB呈低值。因为CETP活性低,从HDL转运到含ApoB的脂蛋白的CE量减少,即运输障碍使CE量增加。高HDL血症多见于CETP缺乏者,易出现多分散LDL,而HDL颗粒变大。有实验表明,CETP活性低的动物作胆固醇负荷实验很容易形成动脉粥样硬化,对人而言,高HDL血症与动脉粥样硬化的关系有待进一步研究。
HDL按超速离心法可分为HDL2和HDL3。用聚丙烯酰胺梯度凝胶电泳可将HDL分为HDl1、2a、2b、3a、3b、3c六种亚组份颗粒,如图4-10所示。有报道冠心病患者大型颗粒HDL2b减少,小颗粒HDL3b增加,认为HDL2b的上升有抗动脉粥样硬化的作用,HDL3b颗粒的增加则可以引起脂质代谢障碍,动脉粥样硬化形成。
高脂蛋白血症分为六型,在临床诊治疾病过程中有一定的意义。从临床实验室诊断方法学考虑,作脂蛋白检测有一定难度,电泳分离法欠准确,按超速离心法,操作时间过长,难以快速定量。另外,高脂蛋白血症多数与遗传有关。目前可采用载脂蛋白基因分型以弥补按脂蛋白进行分型的不足之处。载脂蛋白的基因分型是目前研究脂质代谢及其探讨动脉粥样硬化发病机制的热门话题。
二、遗传性脂蛋白代谢异常
(一)ApoAⅠ异常症
Assmann分析近两万人,发现每500人中有1例ApoAⅠ结构基因杂合子出现,比野生型多一个或少一个正电荷或负电荷。大多数变异无明显血脂的变化。仅有ApoAⅠMarburg病在107位上的Lys缺失,引起轻度的TG升高。ApoAⅠ的Milano变异体(173Arg→Cys)血浆中HDL有所降低,然而冠心病发病率未见增加。ApoAⅠ和ApoCⅢ基因重排导致的变异可引起家族性ApoAⅠ和ApoCⅢ缺乏症,用EcroⅠ限制性内切酶分析ApoAⅠ基因,发现家族性早发性冠心病患者都出现6.5kb片段纯合子,正常人为13kb纯合子,其杂合子为13kb/6.5kb,推测纯合子6.5kb与动脉粥样硬化发病有关。
ApoAⅠ与ApoCⅢ缺陷者表现为血HDL水平降低,易出现早期动脉粥样硬化。有报道ApoAⅠ减少会导致LCAT活性降低,使含ApoCⅠ、ApoAⅢ的脂蛋白如CM置换发生障碍,从而在体内蓄积。
(二)ApoB异常症
ApoB缺陷将出现无β-脂蛋白血症或低β-脂蛋白血症。无β-脂蛋白血症是纯合子隐性遗传病,称为Bassen-Kornzeig综合征,有脂肪吸收障碍(脂肪泻)、红细胞变形(棘状红细胞症)和运动失调等症状。
低β-脂蛋白血症为显性遗传病,杂合子者血中LDL浓度低,与无β-脂蛋白血症有区别。经三个家族分析,患者肠粘膜细胞的ApoB48合成正常而不能合成ApoB100,即ApoB48外显子以外的ApoB100外显子领区异常,由于LDL受体领域附近的点突变(Arg3500→Glu),使LDL受体结合能力降低。
ApoB100的羧基端是其与LDL受体的结合区,有下列依据证明这一推测:①观察到30种单克隆抗体能与LDL受体结合的肽链区域结合,即ApoB100的2980-3780一段可被阻止,其他单克隆抗体无阻止作用;②缩短的ApoB异构体缺乏羧基端,则不能与LDL受体结合;③羧基端有三个区域可与肝素结合,该区域含有几簇带正电荷的氨基酸残基,其中之一的序列与ApoE区域的序列有同源性,这一区域称为ApoE区,已证实这一区域可与LDL受体结合,其序列为3359-3367氨基酸残基位置,带正电荷的氨基酸簇相当保守;④经化学修饰的带正电荷的ApoB100不能与HDL受体结合;⑤若ApoB100分子中Arg→Gln3500,LDL分子不能与LDL受体结合,导致高脂蛋白血症,如Innerarity的家族性高脂蛋白血症。
ApoB100的cDNA中某一核苷酸的变异或缺失,均可引起家族性低β-脂蛋白血症,迄今已发现有25种之多,血浆LDL浓度降低,杂合子患者血浆中ApoB和LDL浓度为正常的1/4-1/2。一般无症状,纯合子则更为严重,包括脂肪吸收不良、棘型红细胞、视网膜色素沉着和神经性肌肉退变。
ApoB100在血浆脂蛋白中分子量最大,氨基酸链最长,因此在合成蛋白质和形成脂蛋白的过程中,任何部位或环节均可能发生变异,可想而知,今后发现的ApoB100的变异会更多。
(三)ApoCⅡ异常症(遗传变异)
ApoCⅡ缺陷导致LPL活性降低。因为ApoCⅡ是LPL发挥催化作用不可缺少的辅因子。ApoCⅡ异常会出现高TG血症,高CM血症,发病率约1/10万。现已有ApoCⅡ有多种变异体的报道。
(四)ApoE异常症
ApoE是LDL受体的配体,其表型不同,与LDL受体结合的能力也不同,E4和E3几乎相同,E2几乎无结合功能。E2纯合子因为第158氨基酸残基突变,CM残粒或β-VLDL滞留导致高Ch、TG血症,高脂蛋白血症,易出现早期动脉粥样硬化。典型例子是家族性Ⅲ型高脂血症,ε2基因纯合子人群分布频率为1%,家族性Ⅲ型高脂血症发病率为10000人有2-3人。究其病因,ApoE2纯合子遗传缺陷因素是主要的,然而还有环境及生理性因素等的影响,如甲状腺功能亢进、肿瘤以及家族性复合型高脂蛋白血症等。
(五)LDL受体异常
LDL受体异常导致FH发生,属显性遗传,遗传频率约1/500。杂合子的高LDL血症易导致动脉粥样硬化。FH的LDL受体基因变异和LDL受体合成的过程中均可出现异常,将其分为四类:①单元1异常是因为mRNA转录障碍导致总体蛋白性质改变,生物学活性降低;②单元2异常是分子量为12万的受体前驱体异常,从小胞体到高尔基复合体运送障碍,ER潴留,与单元2相同,富含Cys领域阅读框的缺失(in-framedeletion)存在;③单元3变异,细胞表面的分子量160ku的成熟受体数量显著减少,使LDL受体结合能力下降;④单元4变异为受体不能局部化,使LDL无法结合而进入细胞内。这几种变化均与lDL受体结构有关。
(六)LPL与HTGL异常症
LPL与ApoCⅡ异常同样都是出现高CM血症,而血中VLDL并不升高,常伴有胰腺炎产生。
HTGL缺乏,有与Ⅲ型高脂血症类似的症状,CM残粒滞留。
(七)LCAT异常症
LCAT缺乏者,HDL中CE比例增加,使HDL处于新生未成熟圆盘状态,相反LDL的CE减少,TG增多。有角膜混浊、肾损害、溶血性贫血等症状,鱼眼病就是LCAT基因突变,使Cys替代Arg引起LCAT活性降低,致使HDL结构变化,并使血浆中ApoAⅠ、Ⅱ和HDL浓度只有正常人的20%。
(八)CETP异常症
CETP缺陷者或者活性受到强烈抑制则呈现高HDL血症,血浆LDL浓度降低,同时还有可能出现动脉粥样硬化症。
三、继发性高脂蛋白血症
某些原发性疾病在发病过程中导致脂质代谢紊乱,进而出现高脂蛋白血症,称为继发性高脂蛋白血症,引起继发性高脂血症或高脂蛋白血症的病因是多方面的,如糖尿病、肾病及某些内分泌紊乱等疾患。
第七节 脂蛋白代谢紊乱与动脉粥样硬化
正常的动脉壁内有内、中和外三层结构,内膜由单层内皮细胞及狭窄的内皮下层构成。内皮下层含胶原纤维、弹性纤维和基质等成分,幼年时期,内皮下层内几乎无平滑肌细胞,随年龄增长,平滑肌细胞以缓慢恒定的速度在内膜下堆积;中膜主要是平滑肌细胞;外膜有纤维母细胞和平滑肌细胞。
动脉粥样硬化(atherosclerosis,AS)主要损伤动脉内壁膜,严重累及中膜是动脉管壁胆固醇酯大量堆积成粥样硬化斑块,使血管壁纤维化增厚和狭窄的一种病理改变。主要侵犯大动脉和中等动脉,如主动脉、冠状动脉和脑动脉,导致某些脏器的局部组织供血不足,常出现心脑血管疾患,甚至有致命性损害。凡能增加动脉壁内Ch内流和沉积的脂蛋白如LDL、β-VLDL、OXLDL等,是致动脉粥样硬化的因素;凡能促进胆固醇从血管壁外运的脂蛋白如HDL、X-HDL,则具有抗动脉粥样硬化性作用,称之为抗动脉粥样硬化性因素。
有关动脉粥样硬化的发病机制,人们已研究了几个世纪。虽已从细胞水平深入到分子水平,取得一定的进展,尚还有许多问题没能解决,难题成堆。因为这是一种多因素导致的疾病,其发病机制极为复杂。诸如血管内皮细胞的功能变化、损害、剥离、外加血浆成分以巨噬细胞浸润、内膜层平滑肌细胞增殖过程等,这一连串的血管内皮细胞的损害与功能障碍,均与动脉粥样硬化症的发生有密切关系。目前认为其基本过程如图4-11所示。多种原因使血管内皮细胞损害,单核细胞粘附其上侵入内膜,并分化成巨噬细胞,与此同时,血小板也粘附着并分泌多种因子,使血管壁中膜平滑肌细胞游走进入内膜,巨噬细胞泡沫化,进一步使其游走进入内膜的平滑肌细胞增殖,形成粥样硬化斑块,见图4-11。
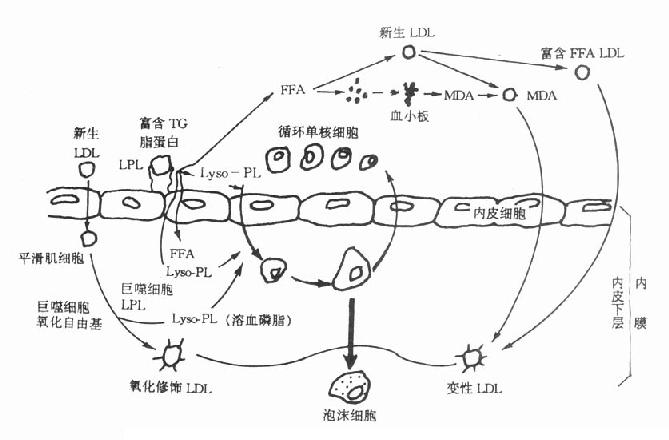
图4-11 动脉粥样硬化发生机制假说
动脉粥样硬化的动脉壁细胞内和细胞之间有大量的胆固醇酯堆积,即粥样硬化斑块中堆积有大量胆固醇。斑块中胆固醇主要来自血浆脂蛋白,在探讨AS的发病机制中必须探讨脂蛋白如何将胆固醇送入到动脉壁的细胞内,何种类型细胞与胆固醇在动脉壁内堆积有关。目前所知,动脉粥样硬化形成过程中有诸多因素参与,主要有三方面:第一是细胞因素,有血管内皮细胞、血管平滑肌细胞、血液中单核细胞、巨噬细胞和淋巴细胞等;第二是代谢物因素,有作用于平滑肌的增殖因子、游走因子、脂蛋白受体、凝血纤溶因子、血小板因子等;第三是物理学因素,如剪切应力(shearstres)等,与AS有关的脂蛋白因素有HDL、VLDL、CM、LDL、OX-LDL、LDLR、LPL、LP(a)等。
一、低密度脂蛋白与动脉粥样硬化
LDL经化学修饰成氧化LDL或被醋酸乙酰化成乙酰LDL,在代谢过程中,损伤血管壁内皮细胞,使管壁通透性增加,并刺激单核细胞游走进入管壁,形成巨噬细胞并泡沫化。化学修饰的LDL使其内ApoB100蛋白变性,经清道夫受体无限制地被巨噬细胞摄取而形成泡沫细胞,并停留在血管壁内,沉积大量的Ch,特别是ChE,致使动脉壁粥样硬化斑块形成。
二、脂蛋白脂肪酶与动脉粥样硬化
LPL与AS有关,常见有LPL活性增加者可预防动脉硬化性血管变化。但是LPL活性加强还有不利的一面,从CM和VLDL看是致动脉粥样硬化因素的立场考虑,血管壁的LPL使含TG的CM和VLDL的水解后,自身变成残粒,不仅被血管壁摄取,其水解产物脂肪酸又增加了Ch的溶解度,加快Ch进入血管壁的速度,促进动脉管壁的粥样斑块形成。
三、过氧化脂质与动脉粥样硬化
循环血液中过氧化脂质导致管壁损害引起AS是目前较为肯定的事实。利用TBA(thiobarbituricacid)法测定到血浆中有过氧化脂质存在,加入铜离子后,VLDL特别是LDL易被氧化,实验也表明AS的血管壁有氧化LDL存在。利用高效液相色谱法(HPLC)法与化学发光法测定出AS者,血浆有3nmol/L左右的过氧化脂肪酸及胆固醇酯羟自由基。也有学者认为,氧化LDL在血流中作用于血管壁的可能性很小,因为氧化LDL代谢速度非常快,很易被肝脏处理,几乎很少在血管内停留。
四、脂蛋白(a)与动脉粥样硬化
LP(a)与AS的心脏疾患有关,是促进AS的独立危险因子。Apo(a)与plasminogen(血纤维蛋白溶酶原)有相同的性质,二者竞争性抑制与内皮细胞表面结合,有助于血栓的形成。心肌梗死患者用血栓溶解疗法治疗有一定效果,溶解能力强弱与血浆LP(a)含量有关,若LP(a)与管壁表面t-PA结合,其结果是plasminogen活化受到抑制。
引起AS的起始因子(initiatingtactor)是什么?Ross,Glomseer提出的损害反应学说认为血小板凝集能力亢进,引起血管壁损害是其起始因子。其后又有学者提出起始因子是使血管内皮细胞层下发生完整的剥脱,并以泡沫细胞为主,外加平滑肌细胞的浸入,使单核细胞在循环血液中发生。Goldstoin提出变性LDL经巨噬细胞清道夫受体结合形成AS,为AS早期起始因子。最近又提出氧化LDL是早期AS的起始,即OXLDL学说;又认为LDL的磷脂经氧化产生溶血卵磷脂,成为循环中单核细胞的趋化物(chemoat-tratant),起主要作用。
第八节 脂蛋白和脂质测定方法学评价
血浆脂蛋白和脂质测定是临床生化检验的常规测定项目,其临床意义主要是:早期发现与诊断高脂蛋白血症;协助诊断动脉粥样硬化症;评价动脉粥样硬化疾患如冠心病和脑梗塞等危险度;监测评价饮食与药物治疗效果。
一、血浆脂蛋白测定
⒈超速离心分离纯化法超速离心法是根据血浆中各种脂蛋白的比重(密度)的差异,在强大离心力作用下进行分离纯化的一种方法。
血浆在不同密度盐溶液中经过超速离心,各种脂蛋白可按密度大小漂浮于不同盐溶液介质中。脂蛋白漂浮的衡量单位是漂浮率(Sf)。Gofman等于1950年确定,血浆脂蛋白在比重为1.063的NaCl溶液中(26℃),每秒每达因(ldyn=10-5N)克离心力的作用下的漂浮速度,称为1个Svedbery单位(10-13s)。Sf越大,脂蛋白密度越低。
一般操作方法是将血浆置于已准备好的不同密度梯度的盐溶媒介质中,在强大离心作用下,各脂蛋白依其自身的Sf不同,分散于离心管中的密度梯度溶媒层,达到分离纯化的目的。
超速离心法是分离纯化脂蛋白的有效技术,目前广泛应用于脂蛋白、载脂蛋白代谢的研究中。
⒉电泳分离法不同脂蛋白因蛋白质含量不同、其电荷量不同,故可用电泳方法进行分离,并根据血浆脂蛋白电泳迁移率不同予以判断确认。电泳支持物一般常用醋酸纤维薄膜、琼脂糖凝胶或聚丙酰胺凝胶。由于醋酸纤维薄膜要预处理,很繁杂,外加电泳时间过长,目前已很少使用。临床检验中主要采用琼脂糖凝胶电泳进行分离,快而较为准确,仪器设备要求不高,被广泛采用。聚丙烯酰胺凝胶作为支持物分离脂蛋白,值得推荐给临床使用。
电泳分离法不论用何种支持物,血浆脂蛋白需用亲脂染料如苏丹黑B等进行预染再电泳,电泳完毕,脂蛋白根据电荷量不同,移动在不同的位置,再置于光密度计内进行扫描,计算出各种脂蛋白的百分比,该数值乘以血浆总脂量,即可求出α-脂蛋白、β-脂蛋白和前β-脂蛋白含量,乳糜微粒停留在原点,无法测出其含量,正常空腹12h后,血浆中无CM存在。也可将电泳毕的琼脂糖凝胶脂蛋白区带切割置于试管中,加水溶解,进行比色,测出各自百分比,这一手工半定量法无法测准,属淘汰之列。
⒊沉淀分离法由于脂蛋白的组成及理化性质不同,在不同的聚阴离子和2价不同金属离子(Mn2+、Mg2+、Ca2+、Ni2+、Co2+)以及不同pH值条件下,使脂蛋白与聚阴离子结合形成复合物沉淀,以达到分离定量各种脂蛋白的目的。
如肝素锰沉淀血清中VLDL和LDL,离心沉淀,HDL则留在上清液,再定量HDL量,即为血浆HDL的量。也可采用聚乙烯硫酸盐沉淀LDL,离心去上清液留沉淀,再定量沉淀其中的LDL,即血浆的LDL量。脂蛋白是一种既有蛋白质又有胆固醇,还有磷脂的复合体,如何定量,尚无一种较为理想的方法。目前仅以测定脂蛋白中胆固醇总量的方法作为脂蛋白的定量依据,即测定HDL、LDL或VLDL中的胆固醇,并分别称为高密度脂蛋白-胆固醇(HDL-C)、低密度脂蛋白-胆固醇(LDL-C)或极低密度脂蛋白-胆固醇(VLDL-C)。这类测定方法是目前临床广泛使用的方法,快速并较为准确。
⒋遮蔽直接测定法利用脂蛋白中某种特异抗体等物质,先将血浆中LDL和VLDL包裹保护,不受测定胆固醇试剂的影响,而直接测定未被包裹的HDL中的胆固醇,在不离心分离条件下,测定出HDL的胆固醇含量,快速准确,是近年来发展的新技术,值得推广应用。
二、血清总脂质测定
血清总脂质主要包括FC、CE、PL和TG等。血清总胆质测定除作为脂质代谢紊乱及有关疾患的协助诊断外,还可用于血总脂增加的原发性胆汁酸肝硬化、肾病综合征或急慢性肝炎以及血总脂减少的重症肝炎、肝硬化等的严重肝实质性损害、恶液质、甲状腺功能亢进和吸收不良综合征等疾患的协助诊断。
血总脂一般随年龄增加而升高,40岁以上者显着增加,65-70岁者反而降低。测定方法不同,正常参考值有一定的差异。
测定方法分两大类:一类是抽提法,将血清脂质通过脂质抽提剂提入某一介质中,再进行定量;另一类是直接测定法,即无需抽提。
⒈脂质抽提法脂质存在于血清脂蛋白中,利用甲醇或乙醇使其与蛋白结合的脂质分离,再利用甲醇或乙醇的非极性有机溶媒使脂质溶于其中。Bloor溶剂(醚:醇为1:3,V/V)或FovCh溶剂(氯仿:甲醇为2:1,v/V)的醚氯仿等非极性溶剂的混合液,可提高切断脂质与蛋白质的结合的能力,达到抽提的目的。血清脂质抽提入有机溶液后,蒸发干固,除去有机溶液,通过加热氧化,再显色定量。
⒉脂质直接测定法如Sulfo-phospho-Vanillin法是加浓硫酸入血清加热,冷却后,加试剂显色(即SPV反应)直接测定出血清总脂质。
三、胆固醇测定
血清中胆固醇包括CE和FC,酯型的CE占70%,游离型FC占30%。FC中的C3的-OH在LACT作用下,可分别与亚油酸(43%)、油酸(24%)、软脂酸(10%)、亚麻油酸(6%)、花生四烯酸(65)、硬脂酸(3%)等脂肪酸结合而成。血清中胆固醇在LDL中最多,其次是HDL和VLDL,CM最少。血清总胆固醇测定方法分为化学法和酶法两大类。
⒈化学法化学法一般包括:①抽提;②皂化;③毛地黄皂苷沉淀纯化;④显色比色四个阶段。代表性的方法有Sperry-Webb法,包括①到④步骤操作过程,常规操作中多省去了②、③。或者用省去②、③步骤的Zak-Henly法及省去③步骤的Abell-Kendall法,现将此法作为标准参考方法予以利用。
⒉酶法测定CE在胆固醇酯酶(cholesterolesterase,CHE)作用下水解成FC和FFA,FC再经胆固醇氧化酶(cholesteroloxidase,COD)氧化成△4胆甾烯酮和H2O2,再分别定量O2的消耗或者H2O2的生成量,或者△4胆甾烯酮生成量,以作为FC的定量依据。该法是目前常规应用方法,快速准确,标本用量少,便于自动生化分析器作批量测定。
四、甘油三酯测定
甘油三酯又称中性脂肪,由于其甘油骨架上分别结合了3分子脂肪酸、2分子脂肪酸或1分子脂肪酸,所以分别存在有甘油三酯(TG)、甘油二酯(DG)和甘油一酯(MG)。血清中90%~95%是TG,TG中结合的脂肪酸分别为油酸(44%)、软脂酸(26%),亚油酸(16%)和棕榈油酸(7%)。
血清TG测定方法一般分为物理化学法、化学法及酶法三大类。
血清中TG的化学组成并不单一,准确求其分子量较为困难。因标准不同,测定结果存在差异。化学法多采用三棕榈精(软脂精)(分子量807.3)、三油精(分子量895.4),按摩尔浓度计算。酶法测定以三油精为标准物进行换算。
⒈化学法化学测定法包括:①TG的抽提分离;②皂化;③甘油糖的氧化;④氧化生成甲撑显色定量等四个阶段。操作较为繁杂,影响测定因素太多,共法准确性差,一般很少用。
⒉酶法测定酶法测定包括:①TG的抽提与皂化;②加水分解生成甘油糖定量两个阶段。目前常规检测应用的方法有甘油激酶(glycerolkinase,GK)法和甘油氧化酶(glyceroloxidase,glyod,GOD)法。操作简便,快速准确,并能在自动化生化分析仪上进行批量测定。
五、磷脂测定
磷脂(PL)并非单一的化合物,而是含有磷酸基和多种脂质的一类物质的总称。
血清中PL包括:①卵磷脂(60%)和溶血卵磷脂(2%-10%);②磷脂酰乙醇胺等(2%);③鞘磷脂(20%)。
血清PL定量方法包括测定无机磷化学法和酶法两大类。
⒈化学测定法包括①抽提分离;②灰化;③显色、比色三个阶段。
⒉酶测定法可分别利用磷脂酶A、B、C、D等4种酶作用,加水分解,测定其产物,对PL进行定量,一般多采用磷脂酶D(PL-D)。PL-D可作用于含有卵磷脂、溶血卵磷脂和鞘磷脂以及胆碱的磷脂,这三种磷脂约占血清总磷脂的95%。该法快速准确,便于自动化仪器进行批量检测。
六、游离脂肪酸测定
临床上将C10以上的脂肪酸称为游离脂肪酸(FFA)。正常血清中含有油酸(C18:1)占54%,软脂酸(C16:1)占34%,硬脂酸(C18:1)占6%,是其主要的FFA。另外还有月桂酸(C12:0)、肉豆蔻酸(C14:0)和花生四烯酸(C20:1)等含量很少的脂肪酸。与其他脂质比较,FFA在血中浓度很低,其含量水平极易受脂代谢、糖代谢和内分泌功能等因素影响,血中FFA半寿期为1-2min,极短。血清中的FFA是与清蛋白结合进行运输,属于一种极简单的脂蛋白。
测定血清FFA法有滴定法、比色法、原子分光光度法、高效液相层极法和酶法等。一般多以酶法测定。作FFA测定的标本一定要注意在4℃条件下分离血清并进行测定。因为血中有各种脂肪酶存在,极易也极快速使血中TG和PL的酯型脂肪酸分解成非酯化的FFA,使血中FFA值上升。贮存的标本仅限于24h内,若保存三天,其值约升高30%,使结果不准确。
⒈非酶法确定非酶法测定包括滴定法、比色法、原子吸收分光光度法和高效液相层析法。前三种方法准确性差,高效液相层析法仪器太昂贵,不便于批量操作。
⒉酶测定法血清中主要用脂肪酶测定。可分别测定产物乙酰CoA、AMP或辅酶A(CoA),进行定量。酶法测定结果准确可靠快速,易于批量检测。
七、载脂蛋白测定
血清载脂蛋白(Apo)包括ApoAⅠ、AⅡ、B100、CⅡ、CⅢ、E和(a),已属常规检测项目。血清中载脂蛋白均结合于脂蛋白中,测定时要加用解链剂,使脂蛋白中Apo暴露再进行测定。
目前测定血清中Apo的含量的方法是利用相应特异抗体试剂进行测定。现有羊抗人ApoAⅠ、AⅡ、B100、CⅡ、CⅢ、E和(a)等抗体试剂。测定原理是将某一特异抗体加到待测人血清中,即与血清中相应抗原形成抗原抗体复合物,根据复合物的量,即可测出血清中某一Apo含量。例如在人血清中加入抗人ApoAⅠ抗体,即与血清中ApoAⅠ(抗原)结合形成复合物,再定量即可测出血清中ApoAⅠ含量。
⒈免疫扩散法先制备含Apo抗体的琼脂糖凝胶,间隔等距离打孔,加入待测人血清,水平置于温箱(37℃)保温24或48h,抗原从孔中间向周围扩散,因凝胶板中有相应抗体,经一定时间扩散后,最后在凝胶孔周围形成一圆型沉淀圈,测量其圆直径,以圆的面积大小计算血清中Apo含量。该法要求在测量圆周大小时,一定要精确到0.1mm。这一测定方法可适用于以抗体为试剂贩所有微量蛋白的测定,但易受多种因素的影响,难以测准,有淘汰的趋势。
⒉免疫火箭电泳先制备含某一Apo抗体的琼脂糖凝胶,靠近阴极端打孔,加入待测血清,进行电泳。血清中Apo抗原往正极移动,一定时间(约3h)后,即可形成类似火箭的沉淀峰,根据火箭峰高度或面积的大小计算血清中Apo含量。在严格掌握电泳条件下,本法是一种较为简便、试剂用量少的好方法。
⒊免疫比浊法免疫比浊法分为免疫透射比浊法和免疫散射比浊法。
Apo的特异抗体与血清中相应的抗原结合形成抗原抗体免疫复合物,并形成微细颗粒,混悬于溶液介质中,光线通过这种浑浊介质溶液时被吸收一部分,吸收的多少与混浊颗粒的量成正比。在抗体量一定的条件下,抗原越多,抗原抗体复合物越多,溶液越浑浊,吸收光越多,以此对抗原进行定量,这种通过测定光吸收量的方法称为透射比浊法。该法是目前使用最为广泛的方法,快速准确,可在自动生化分析仪上作批量检测。
当光线通过含抗原体复合物的浑浊介质溶液的混悬颗粒时,出现光散射作用,散射光强度与溶液中混悬颗粒的量成正比,这种测定光散射强度的方法称为免疫散射比浊法。该法的进行,需要有具备测定散射光的仪器如散射比浊仪等。
免疫比浊时,由于抗原与抗体结合的过程中,吸收度与浓度之间不呈线性关系,一般是三次方程曲线关系。若要将抗原与抗体两个变量之间的变动特征恰当地反映出来,需要经过三次方程拟合成近似直线化的曲线方程,再进行运算。免疫比浊的实际操作过程中,可采用终点法或速率法,用五点或七点不同浓度进行定标,经三次曲线方程拟合求出一条能反映真实情况的浓度与吸光度的关系曲线方程,作为定量的工作曲线。如果以单一标准浓度定标,以一次方程直线回归运算,所测结果仅在标准浓度上下波动,真正的过低和过高值无法测准。
免疫比浊法所用抗体应是单价、特异并且高效价的,尤其是抗体若非单一而混有少量其他蛋白抗体,在血清中形成的复合物将是一种大杂烩,非单一蛋白的复合物,测出结果偏高而不准确。
第五章 体液平衡紊乱
人体内存在的液体称为体液(bodyfluid)。体液中有无机物和有机物,无机物与部分以离子形式存在的有机物统称为电解质。葡萄糖、尿素等不能解离的有机物称为非电解质。体液以细胞膜为界,可分为两大部分,即细胞内液(intracellulerfluid,ICF)和细胞外液(extracellulerfluid,ECF)。细胞外液因其存在部分不同,又可分为血浆和细胞间液,后者包括淋巴液。各部位体液之间处于动态平衡,其内的水与电解质也处于动态平衡,这种平衡状态,很易受外界或机体内部因素的影响,导致破坏出现代谢紊乱,即水电解质平衡紊乱和酸碱平衡紊乱。
第一节 体液平衡及调节
一、水平衡
人体内含水量的变动与年龄(图5-1)和疾病密切相关。
图5-1 人体水含量与年龄的关系
小儿每日水的摄入量(含体内氧化产生的水)略大于排出量,其中约有摄入量的0.5%-3.0%保留在体内用以生长。成年人每日通过饮水(约1200ml),摄食含的水(约1000ml)和体内氧化时释放出的水(约300ml)以维持水的需要量,平均总进水量约2500ml,摄入量等于排出量。小儿水交换速度比成人快,如体重7.0kg婴儿每日排出水约700ml,占细胞外液的1/3,但70kg体重的成人水交换率只占细胞外液的1/7,这就是小儿易引起脱水的原因。
机体对水的需要量与代谢热量成正比,每天散热所需水量约占体内总量的1/4,经体表面无形蒸发散失以维持体温,每散发0.58cal热需蒸发水量约1ml,如成人每日按代谢产热2000cal计算,无形蒸发失水量约为:
2000×1/4×1/0.58=826ml
(1cal=4.18J)
蒸发途径是由皮肤以所谓不显性出汗方式及呼出的水蒸气的形式排出,亦称为非显性失水。非显性失水量,加上每天最低排出尿量500ml,其总量约为1500ml,这就是每天需要补充水的数量,即临床的“生理需水量”。小儿需水量一般按每代谢100cal热需消耗120-150ml计,在禁食情况下,按基础代谢计算即60-90ml(kg·d)。
室温30℃时,每增加1℃,机体应增加当日需水量10%-13%。体温每增加1℃代谢热卡增加12%,生理需水量也增加。疾病过程中,如气管切开、严重烧伤、腹膜炎等代谢率增加,需水量也相应增加,相反,人工冬眠及手术后,应减少需水量。
二、钠、氯平衡
氯和钠是细胞外液的主要阴阳离子。体内Na+约有50%分布于细胞外液,约40%存在于骨骼,约10%存在于细胞内。机体通过膳食及食盐形式摄入氯和钠,成人每日需要量5-9g,一般是摄入体内NaCL的量大于其需要量,所以,一般情况下,人体不会缺钠缺氯。
Na+、CL-主要从肾排出,肾排钠量与食入量保持平衡。肾对保持体内钠的含量起有很重要的作用。当无钠摄入时,肾排钠减少,甚至可以不排钠,而让钠保留于体内,以维持体内钠的平衡。钠的摄入与排出往往伴随有氯的出入,钠与氯还有少部分以出汗形式丢失。
体液中Na+主要分布在细胞外液,K+主要分布在细胞内液。Na+有65%-71%是可交换的,其中85%存在于细胞外液,15%在细胞内液。用同位素标记Na+的实验证明,Na+既能透过细胞膜,还可通过主动转运机制,即细胞上存在的钠钾泵将Na+从细胞内泵出到细胞外,使细胞内的Na+保持在低水平。人体中K+约90%是可交换的,大部分存在于细胞内液。体液中细胞外液的Na+浓度高于细胞内液,K+是细胞内液高于细胞外液。
三、体液中的电解质
(一)体液电解质分布及平衡
血浆中主要电解质有Na+、Cl-、K+。细胞间液是血浆的超滤液,其电解质成分和浓度与血浆极为相似,不同之处是血浆含有较多的蛋白质,而细胞间液不含或仅含少量的蛋白质,由于蛋白质是大分子量物质,不易通过细胞膜,故血浆蛋白含量高于细胞间液。
由于测定细胞内电解质含量很困难,所以临床都以细胞外液的血浆或血清的电解质含量作为诊疗的参考依据。
细胞内液的电解质浓度是从肌肉活检或红细胞标本中测得,或以同位素示踪方法计算。细胞种类不同,其内电解质的种类及含量是有区别的。细胞内液主要阳离子是K+和Mg2+,主要阴离子是蛋白质和有机磷酸盐,而Na+、Cl-、HCO3-则很少。细胞内高K+和低Na+的维持,不是依赖细胞膜对这些离子的不同渗透性,而是依赖膜上的钠钾泵的主动转运。钠钾泵除了维持细胞内外电解质浓度外,还有助于肾的Na+和K+的转动,并在调节细胞内电解质的浓度方面起有重要的作用。
按Donnan平衡论,体液中阴离子总数应与阳离子总数相等,并保持电中性,往往是阴离子随阳离子总量的改变而变化,升高或降低阴离子以适应阳离子的改变。
血浆中Cl-、HCO3-总和与阳离子Na+浓度之间保持有一定比例关系,即:
Na+=HCO3-+Cl-+12(或10)mmol/L
若已知血浆Hco3-和Cl-浓度,Na+浓度可以从上式求得。
各体液渗透压均处于同一水平,即渗量摩尔为294-296mOsm/L,理论渗透压为756-760kPa。
(二)阴离子隙
阴离子隙(AG)是指细胞外液中所测的阳离子总数和阴离子总数之差,即正常人血清Na+、K+之和与HCO3-、Cl-之和的差值为AG值,用mmol/L表示,计算公式为:AG=(Na++K+))-(Cl-+HCO3-)。一般是利用血清中的电解质含量进行运算。血清K+浓度较低。且相当恒定,对AG正常参考值为8-16mmol/L,平均为12mmol/L。临床上利用血清主要阴阳离子的测定值即可算出AG值,它对代谢性酸中毒的病因及类型的鉴别诊断有一定的价值。在疾病过程中,因代谢紊乱,酸性产物增加,导致代谢性酸中毒症状最为多见。缺氧使糖酵解中乳酸产生过多,病人不能进食或糖尿病等脂代谢紊乱导致酮体产生增加,菌血症、烧伤等组织细胞大量破坏,蛋白质分解使含硫的产物(So42-)增多等一系列酸性代谢产物在血浆酸碱缓冲过程中,消耗了血浆HCO3-量,并使乳酸根、酮体的乙酰乙酸根及硫酸根等阴离子增加。机体为了保持体液阴阳离子平衡呈电中性,在Na+、K+离子浓度变动不大而阴离子中的这些酸性产物又增多的状况下,势必造成极易透过细胞膜的CL-转移,使血浆HCO3-与CL-之和减少,(Na++K+)-(HCO3-+CL-)之差值变大,AG值升高(图5-2)。
AG值的异常分为升高及降低两种。临床以AG升高多见,并以AG升高的临床意义较大。AG升高多见于代谢性酸中毒发生的全过程:①肾功能不全导致氮质血症或尿毒症时,引起磷酸盐和硫酸盐的潴留;②严重低氧血症、休克、组织缺氧等引起乳酸堆积;③饥饿时缺糖,脂肪动用分解加强,酮体堆积,糖尿病患者也同样使酮体过剩,形成酮血症、酮尿症。从AG考虑,可将代谢性酸中毒分为高AG代谢性酸中毒及AG正常的代谢性酸中毒,如高血氯性代谢性酸中毒。根据AG水平高低,判断代谢性酸中毒的病因,并可作为治疗的参考依据。
(三)体液的交换
正常状态下,人体内由外界摄入的水分及电解质与体内氧化产生的水(内生水),不断与各区间的体液进行交换。体液交换包括血浆与细胞间液、细胞间液与细胞内液之间的交换,前者交换的动力是血浆胶体渗透压与静水压(血压)之差,其中胶体渗透压在血浆与细胞间液的交换中起有主要作用,同时还可影响细胞外液的总量。细胞间液与细胞内液之间的交换主要决定于细胞内外液的渗透压,因为细胞膜对水与各种电解质的通透性不同,水总是向渗透压高的一侧移动。正常体液的分布、组成及容量三方面均在神经体液等因素调节下保持动态平衡,以保证机体各种生理活动的正常进行。
图5-2 代谢性酸中毒与阴离子隙
四、体液平衡的调节
体液平衡是维持机体生命活动的必不可缺少的条件。机体在生命活动的过程中,通过神经-体液因素调节体液的正常平衡。
(一)口渴感觉调节
口渴感觉是机体对水需要的一种极为重要的保护性生理机制。当机体缺水时,血浆和细胞间液的渗透压升高,下丘脑视前区渗透压感受器受到刺激,兴奋传到大脑皮质,引起口渴反射而思饮水。有人认为可能有渴感中枢,位于下丘脑,在调节渗透压感受器的附近。主要有效刺激是血浆晶体渗透压,用高渗盐水灌注山羊的渴感中枢部位,即可引起极度烦渴,大量喝水,直至产生严重水中毒;若破坏这一部分,渴感消失,则出现高渗性脱水。同时,渴感刺激也可引起抗利尿激素的释放,促使肾脏保留水分;反之,抑制渴感随即抑制分泌,引起水利尿。通常总体液缩减1%-2%即可引起渴感。严重的水丢失超过钠的丧失,如沙漠中水源断绝、炎症影响水的摄入、尿崩症失水过多等均可引起明显的细胞脱水和渴感。当血容量降低到5%-10%,有效循环量明显减少,如出血、腹泻也可引起口渴感。口渴感觉调节机制如图5-3所示。
(二)激素调节
⒈抗利尿激素(antidiuretichormone,ADH)ADH为下丘脑视上核神经细胞分泌,沿下丘脑-垂体束进入神经垂体贮存。ADH又称为血管加压素,是由9个氨基酸残基组成的9肽物,由于第1、6两个半胱氨酸间以二硫键相连成胱氨酸(H-半胱-酪-苯-谷胺-天胺-半胱-脯-精-甘-NH2),故也可称为8肽。ADH主要作用是增加肾远曲小管及集合管对水的重吸收作用,因此对肾脏浓缩功能有很大的影响。体液渗透压、血容量和血压等因素的改变,都可以影响ADH的分泌。主要通过血浆渗透压及有效循环血容量来调节,如图5-4所示。
图5-4 肾素-血管紧张素-醛固酮系统
⒉醛固酮(aldosterone)醛固酮是一种类固醇激素,由肾上腺皮质的球状带分泌,主要以游离形式存在,半寿期仅20-30分钟,在肝灭活,肾能灭活少部分。醛固酮的生理功能是促进肾远曲小管上皮细胞的排H+保Na+作用,使Na+重吸收,保留Na+(同时保留水)并促进K+的排出。
目前认为还有其他几种调节体液平衡的因素,如利尿钠激素(natriuretinhormone)可减少肾小管对钠的重吸收,尤其是对于慢性肾功能衰竭者具有重要作用。心房肌细胞产生的一种循环激素即心钠素(cardionatrin),又称为心房利钠因子或心房肽(arterialna-triureticpolypeptide,AMP),它可以增加肾小球滤过压,产生排钠利尿作用,又可增加肾小球旁器细胞的兴奋性,减少肾素的合成与分泌。
第二节 血气分析
生命的基本特征是不断地从环境中摄入营养物、水、无机盐和氧气,同时又不断地排出废物、呼出二氧化碳。机体需要氧气,用于体内的氧化过程,并主要用于能量代谢。追索到上亿年前,生物在进化过程中,逐渐地适应了有氧的环境,高等生物在有氧环境下,只能让其体内代谢物释放出大量能量,以维持生命活动。有无氧或少氧状态下,能量释放不完全,O2被机体利用的过程中,产生了CO2并排出体外,这种消耗O2产生CO2的过程中,均有赖于机体的气体交换系统,血液在气体交换中起有重要的作用。
一般而言,血气是指血液中所含的O2和CO2气体。血气分析是评价病人呼吸、氧化及酸碱平衡状态的必要指标。它包括血液的pH、PO2、PCO2的测定值,还包括经计算求得如TCO2、AB、BE、SatO2、ContO2等参数。血气分析的有关数据对临床疾病的诊断和治疗发挥着重要的作用。
一、血液气体运输
(一)氧的运输
⒈氧的运输与Hbo2解离曲线氧气随空气一道经呼吸作用而进入肺部,目前认为大气中的氧进入肺泡及其毛细血管的过程为:①大气与肺泡间的压力差使大气中的氧通过呼吸道流入肺泡;②肺泡与肺毛细血管之间的氧分压差又命名氧穿过肺泡呼吸表面而弥散进入肺毛细血管,再进入血液,其O2的大部分与Hb结合成氧合血红蛋白(HbO2)的形式存在,并进行运送,少部分以物理溶解形式存在,均随血流送往全身各组织器官。
血液中O2和CO2只有极少量以物理溶解形式存在,大部分O2以Hb为载体在肺部和组织之间往返运送。
Hb是运输O2和Co2的主要物质,将O2由肺运送到组织,又将CO2从组织运到肺部,在O2和Co2运输的整个过程中,均有赖于Hb载体对O2和CO2亲和力的反比关系:当PO2升高时,促进O2与Hb结合,PO2降低时O2与Hb解离。
![]()
肺部PO2(13.3kPa)高,Hb与O2结合而释放CO2;相反,组织中PCO2高,PO2(2.66-7.32kPa)低,CO2与Hb作用使O2从HbO2中释放到组织细胞供利用。
1L血浆仅能溶解O22.3ml,而97%-98%的O2是与Hb分子可逆性结合而运输,每gHb能结合O21.34ml,若1L血液含140gHb,则能携带O2188ml,其携带O2能力要比血浆溶解的量高81倍。若不是依赖Hb运送氧,单靠血浆溶解状态的氧运输,血液就得循环81次才能达到与Hb载体同等的运输O2的能力,这是不现实的。
测定动脉血和静脉血中存在的这种形式的O2含量及其差值,可以说明血液的O2运输状况。
血液中Hb并未全部与O2结合,如将血液与大气接触,因为大气PO2为21.147kPa(159mmHg),远高于肺泡气的PO213.566kPa(102mmHg),此时血液中所含的O2总量称为氧容量,其中与Hb结合的部分称为氧结合量,氧结合量的多少决定于Hb量的多少。
Hb与O2可逆结合的本质及解离程度主要取决于血液的PO2。血液与不同的PO2的气体接触,待平衡时,其中与O2结合成为HbO2的量也不同,PO2越高,变成HbO2量就越多,反之亦然。血液中HbO2量与Hb总量(包括Hb和HbO2)之比称为血氧饱和度:
血氧饱和度=HbO2/(Hb+HbO2)
若以PO2值为横座标,血氧饱和度为纵座标作图,求得血液中HbO2的O2解离曲线,称为HbO2解离曲线。血氧饱和度达到50%时相应的PO2称为P50,如图5-5所示。
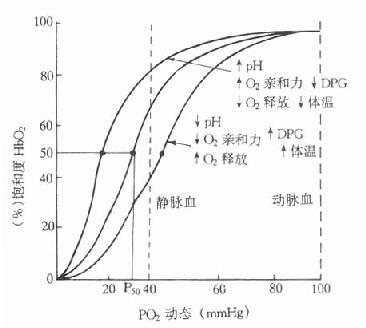
图5-5 正常人血红蛋白氧解离曲线
P50是表明Hb对O2亲和力大小或对O2较敏感的氧解离曲线的位置。P50正常参考值为3.54kPa。
⒉影响O2运输的因素
⑴pH值:当血液pH值由正常的7.40降至7.20时,Hb与O2的亲和力降低,氧解离曲线右移,释放O2增加。pH上升至7.6时,Hb对O2亲和力增加,曲线左移,这种因pH值改变而影响Hb携带O2能力的现象称为Bohr效应。反应式如下:
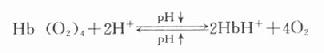
⑵PCO2:PCO2对O2运输的影响与pH作用相同,一方面是CO2可直接与Hb分子的某些基团结合并解离出H+:
![]()
也可以是CO2与H2O结合形成H2CO3并解离出H+:
![]()
上述两方面因素增加了H+浓度,产生Bohr效应,影响Hb对O2的亲和力,并通过影响HbO2的生成与解离,来影响O2的运输。
⑶温度:当温度升高时,Hb与O2亲和力变低,解离曲线右移,释放出O2;当温度降低时,Hb与O2结合更牢固,氧解曲线左移。
⑷2,3二磷酸甘油酸(2,3-DPG):2,3-DPG是红细胞糖酵解中2,3-DPG侧支循环的产物。2,3-DPG浓度高低直接导致H的构象变化,从而影响Hb对O2亲和性。因为脱氧hb中各亚基间存在8个盐键,使Hb分子呈紧密型(taut或tenseform,Tform,)即T型,当氧合时(HbO2),这些盐键可相继断裂,使HbO2呈松驰型(relaxedform,Rform)即R型,这种转变使O2与Hb的结合表现为协同作用(coordination)。Hb与O2的结合过程称为正协同作用(positivecooperation),当第一个O2与脱氧Hb结合后,可促进第二O2与第二个亚基相结合,依次类推直到形成Hb(O2)4为止。第四个O2与Hb的结合速度比第一个O2的结合速度快百倍之多。同样,O2与Hb的解离也现出负协同作用,反应式如下:
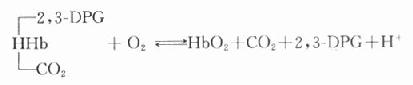
上式表明,H+、2,3DPG或CO2等物质浓度的变化对Hb氧合作用有相同的影响,其中任一物质浓度的变化都将影响Hb的R型与T型之间的平衡,从而改变Hb与O2的亲和力,反应式如下:
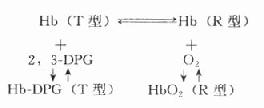
(二)CO2的运输
血液中CO2的存在形式有三种,即:①物理溶解;②HCO3-结合;③与Hb结合成氨基甲酸血红蛋白(HbNHCOO3-)。CO2在血液中的这三种存在形式,实际上也是其三种运输方式。动脉血中CO2含量比静脉血低,二者之差为2.17mmol/L,与O2恰好相反。因为组织细胞代谢过程中产生的CO2自细胞进入血液的静脉端毛细血管,使血浆中PCO2升高,其大部分CO2又扩散入红细胞,在红细胞内碳酸酐酶(carbonicanhydrase,C.A)的作用下,生成H2CO3,再解离成H+和HCO3-形式随循环进入肺部。因肺部PCO2低,PO2高,红细胞中HCO3-+H+→H2CO3→CO2+H2O的方向生成CO2,并通过呼吸排出CO2到体外。红细胞中一部分CO2以R-NHCOO-形式运送,约占CO2运输总量的13%-15%,溶解状态运送的CO2仅占8.8%。
组织缺O2时,糖酵解加强,致使红细胞中2,3-DPG增加,降低了Hb与O2的亲和力,使HbO2在组织中释放出更多的O2,以适应机体的需要。CO2可以通过H+参与Bohr效应,还直接与Hb结合形成HbNHCOO-,有助于稳定T型构象,并在运输CO2中起有一定作用。
(三)PO2、PCO2、pH、2,3-DPG对Hb运输气体的影响
血红蛋白除作为O2及CO2的运载体外,还控制CO2运输过程中H+量的多少,作为缓冲CO2产生的H2CO3中起有重要的作用。H2CO3的60%是在Hb运载O2及CO2过程中释放出H+,进而成为弱碱以完成缓冲H2CO3的作用,即:
![]()
上式表明,Hb与O2或CO2发生的反应互相协调,并通过Bohr效应恰当地处理了来自CO2的H+,使pH值衡定在很狭小的范围。这一过程称为CO2的等氢(isohydric)运输,如图(5-6)所示。
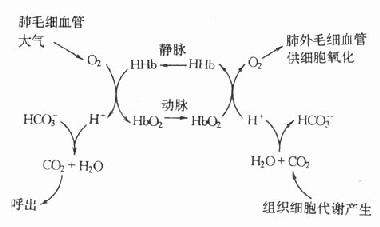
图5-6 O2和CO2的等氢运输
二、血液pH值及其运算
(一)溶液pH值
人体内的化学反应都是在体液中进行,不少化学反应受体液酸碱度的影响。任何溶液都有酸碱度,即使纯水也是一种微弱的电解质,因为纯水中亦有一小部分的水分子电离成H+和OH-保持电离平衡,不管H+浓度与OH-浓度如何改变,[H+]与[OH-]的乘积仍等于水的离子积常数Kw。也就是说,向纯水中加酸时,H+浓度增加多少倍,则OH-浓度就降低多少倍;反之,向纯水中加碱时,H+浓度降低多少倍,则OH-浓度就增加多少倍。所以,对某种水溶液,只要知道H+浓度就必然可以求出OH-浓度。习惯上采用H+浓度来表示溶液的酸碱度,纯水H+浓度为1×10-7mol/L,血液H+为3.98×10-8mmol/L。由于H+浓度太低,丹麦索楞逊于1909年首先使用H+浓度的负对数来表示溶液的酸碱度,称为pH值;
pH=-lg[H+]
用pH值表示溶液或血液酸碱度使用方便。
溶液pH值可利用比色法(如pH试纸)和电位法(如酸度汁)进行测定,前者一般可准确到0.2-0.3pH,后者精确度一般可达0.01-0.02pH单位。
(二)血液pH值及运算
血液pH之所以能恒定在较狭窄的正常范围内,主要是体内有一整套调节酸碱平衡的措施。首当其冲的是血液的缓冲作用。血液缓冲体系很多,以血浆中[HCO3-]/[H2CO3]体系最为重要,因为:①HCO3-的含量较其它缓冲体系高;②HCO3-浓度与H2CO3浓度比值为20:1,缓冲酸的能力远远比缓冲碱的能力大,这是血浆中其它缓冲对无法比拟的;③HCO3-与H2CO3的浓度易于调节。
血液pH主要是由[HCO3-]/[H2CO3]缓冲对所决定,据H-H公式运算:
pH=pKa+lg[HCO3-]/[H2CO3]
式中pKa值为6.1(37℃)
当血浆HCO3-为27.0mmol/L,H2CO3为1.35mmol/L时,血浆pH值是:
pH=6.1+lg27/1.35
=6.1+lg20/1
=6.1+1.3
=7.40
另外,血浆中H2CO3可通过PCO2进行运算即:
pH=pKa+lg[HCO3-]/[αPCO2]
式中α为CO2溶解常数,37℃时α为0.03mmol/L。已知上式中pH、HCO3-、PCO2的任两个数值亦可算出第三个数值。
三、血气分析仪测定原理及仪器结构
早年进行血氧和二氧化碳的测定是采用经典的VanSlyke量气法。此法原理是利用皂素破坏红细胞,再用铁氢化钾破坏血红蛋白以释放O2,加辛酸去泡剂等混合液,使这一反应过程在真空密闭条件下进行,让血液中所含CO2、O2和N2全部释放进密封真空管的液面之上,然后测量所释气体的压力。而后又用CO2吸收剂及O2吸收剂分别将两种气体吸收,根据压力的改变,再计算出CO2和O2的含量。该法准确可靠,然而操作繁锁,又使用大量水银,极易污染环境,现在较少使用。另外还使用化学法测定血浆HCO3-含量,此法操作要求严格的隔绝空气采血,否则很难测定准确。血气分析仪问世后,该法基本属于淘汰的方法。
当初丹麦Radiometer公司根据Astrup等学者的研究,提出血液pH与lgPCO2成线性关系,设计了最早的血气分析仪。最具代表性的产品是该公司70年代出品的BME系列。其后许多国家不同的厂家陆续改进,经过几代人的努力而使血气分析仪更加完善,更加自动化。目前血气分析仪型号虽然很多,然而都是测定血液pH、PCO2和PO2三项基本数据,再参考Hb及体温的数据计算出其他诊断参数。
国产仪器的研制也紧跟时代的步伐不断更新产品,如南京最早的产品DH-100型血气酸碱分析仪,以及后来的改进型,已陆续供应国内医院使用。
测定血气的仪器主要由专门的气敏电极分别测出O2、CO2和pH三个数据,并推算出一系列参数。血气分析仪生产厂家的型号很多,自动化程度也不尽相同,但其结构组成基本一致,一般包括电极(pH、PO2、PCO2)、进样室、CO2空气混合器、放大器元件、数字运算显示屏和打印机等部件,进行自动化分析,其所需样品少,检测速度快而准确。
(一)电极系统
⒈pH测定系统pH测定系统包括pH测定电极即玻璃电极、参化电极及两种电极间的液体介质。
pH电极是利用电位法原理测量溶液的H+浓度,其电极是一个对H+敏感的玻璃电极,同时必须用另一电位值已知的参比电极配套,通常与甘汞电极保持电接触。血样中的H+与玻璃电极膜中的金属离子进行交换,产生电位差,并与血样的H+浓度成正比,二者之间存在着对数关系。在电极内部有pH恒定的溶液,与玻璃膜接触。玻璃电极内部还有Ag/Agcl参比电极,浸在pH恒定液中,电极线连接伏特计,测量血样[H+]所产生的电位差,即中测pH值,并以数字显示再打印结果。参比电极里的KCl溶液通过它逸出与标本接触而形成接触面。因为Kcl浓度很大,所以血标本中离子组成的差异不会改变参比电极上的恒定电位(图5-7)。PH电极要求pH测定范围在6.8-8.0间,并能读出小数点以下三位,精密度达0.002pH单位,准确性达到±0.09pH单位。pH电极稳定性好,计数不漂移。
⒉PCO2电极PCO2电极属于CO2气敏电极。主要由特殊玻璃电极和Ag/Agcl参比电极及电极缓冲液组成,如图5-8所示。这种特殊的玻璃电极是对pH敏感的玻璃膜外包围着一层碳酸氢钠溶液(NaHCO35mmol/L、NaCl20mmol/L,并以Agcl溶液饱和),溶液的外侧再包一层气体可透膜。此膜是以聚四氟乙烯或硅胶为材料,可选择性让电中性能CO2通过,带电荷的H+及带负电荷的HCO3-不能通过。CO2则扩散入电极内,与电极里的碳酸氢钠溶液发生下列变化;见图5-7。使其内的NaHCO3、NaCl溶液的pH值发生改变,产生电位差,由电极套内的pH电极检测。pH值的改变与PCO2数值呈线性关系(△pH/logPCO2),根据这一关系即可测出PCO2值。
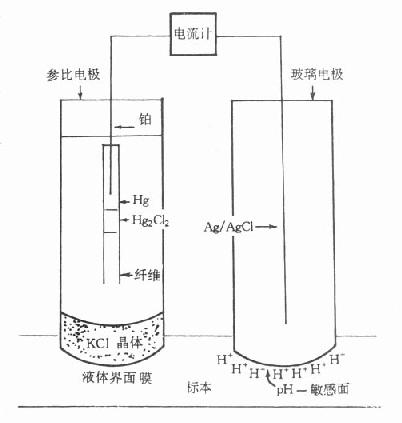
图5-7 pH电极结构示意图
PCO2电极灵敏度以-pH/lgPCO2=1.0为准,即PCO2上升1.33kPa,pH值下降1pH单位。测定范围为0.6-33.3kPa(37℃)。
Co2+H2O→H2CO3→H++HCO3-
⒊PO2电极PO2电极是一种对O2敏感的电极,属于电位法,电极结构如图5-8所示。以白金丝(Pt)为阴极,Ag/AgCl参比电极为阳极,以阴极与阳极之间的一层磷酸盐缓冲液藉以沟通,其外包裹一层聚丙烯膜,膜外接触血样品。此膜不能透过离子,仅O2可透过。当样品中的O2透过聚丙烯膜到达Pt阴极表面时,O2不断地被还原,产生如下化学变化:
阴极反应O2+2H2O+4e-→4OH-
电解质反应NaCl+OH-→NaOH+Cl-
阳极反应Ag++Cl-→Agcl+e-
氧的还原反应导致阴阳极之间产生电流,其强度与氧的扩散量或PO2成正比,以此测出PO2值(图5-9)。PO2电极可测定范围为0-106kPa。
(二)管道系统
主要由测量室、转换盘(有或无)系统、气路系统、溶液系统及泵体等组成。测量室有一套自动控制温度稳定于37℃的装置,转换盘是让样品进入并将有关溶液及气体送入测量室的装置,由计算机程序自动控制.气路系统由空气压缩机、CO2气瓶、气体混合器、湿化器、泵、阀门及有关管道组成。气体混合器将空气压缩机送来的空气(4-6个大气压,latm=101.3kPa)和CO2(纯度要求99.5%)气瓶送来的气体进行混合,混合后得到两种浓度不同的气体。由气体混合器中部出来的“气体1”含19.8%的O2和5.5%的CO2;“气体2”含9%-11%的CO2,从混合器的下部送出,需要时再进入测量室。液体管道系统使缓冲液进入测量室定标,保证样品吸入和废液排出的冲洗过程,管道系统中为保证仪器正常运转还没有一系列的自动检测装置。
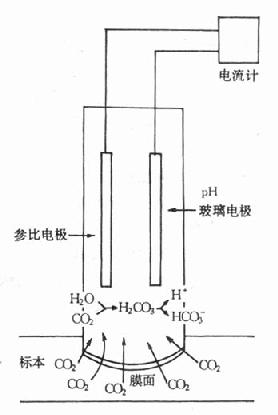
图5-8 PCO2电极结构示意图
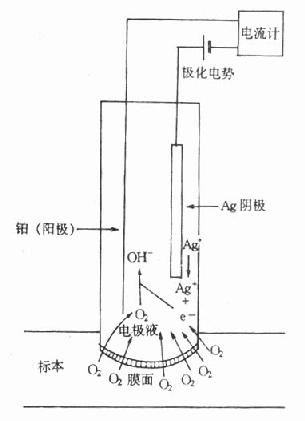
图5-9 PO2电极结构示意图
目前,血气分析仪种类很多,各有其特色。一般都具备有所需样品量少(25-100μl)、检测时间短(1-2)分钟、自动显示数据、打印结果等优点。
四、血气分析仪分析方法
(一)血标本采集
血气分析标本的收集是极为重要的,若处理不当,将产生很大的误差,甚至比仪器分析的误差还大,因此必须引起足够的重视。
血气标本以采动脉血或动脉化毛细血管血为主,静脉血也可供作血气测定。只有动脉血才能真实反映体内代谢氧化作用和酸碱平衡的状况,对O2检测的有关指标必须采集进入细胞之前的动脉血,也就是血液中从肺部运氧到组织细胞之间的动脉血,才能真正反映体内氧的运输状态。动脉血液的气体含量几乎无部位差异,从主动脉到末梢循环都是均一的。
对PCO2和pH的检测也以采集动脉血为好。血液循环无障碍的病人,静脉血的这两项指标基本也可反映体液酸碱状况。
⒈标本采取方法
⑴动脉血:肱动脉、股动脉、前臂动脉以及其他任何部位的动脉都可以进行采血。使用玻璃注射器采血,抗凝剂为肝素钠。每支肝素钠每亳升含12500U,相当于100mg,用20ml生理盐水稀释,分装成40支,消毒备用(4℃贮存)。临用时,注射器吸取肝素钠溶液一支,而后将肝素液来回抽动,使针筒局部湿润,多余肝素液全部排出弃之,注射器内死腔残留的肝素液即可抗凝。针刺动脉血管,让注射器内芯随动脉血进入注射器而自动上升,取1-2ml全血即可。拔针后,注射器不能回吸,只能稍外推,使血液充满针尖空隙,并排出第一滴血弃之,让空气排尽,将塑料嘴或橡皮泥封住针头,隔绝空气,再把注射器来回搓滚,混匀抗凝血,立即送检。或者采用微量取样器采集血标本。
⑵动脉化毛细血管血:所谓动脉化的毛细血管血就是指局部组织末梢经45℃温水热敷,使循环加速,血管扩张,局部毛细血管血液中PO2和PCO2值与毛细血管动脉端血液中的数值相近,此过程称为毛细血管动脉化。采血部位以手指、耳垂或婴儿的手足跟及拇趾为宜。用45℃热水敷局部,5-15min后或直至皮肤发红,而后穿刺,穿刺要深,使血液快速自动流出,弃去第一滴血。不能挤压,挤出的血液的测定结果不可信。未充分动脉化的毛细血管血的PO2测定值偏低,对pH、PCO2和HCO3-的测定结果影响不明显。
用肝素锂抗凝比肝素钠好,因为锂含量(3.5%-4.5%)比钠(9.5%-12.5%)少,可减少血中微纤维形成的可能;同时可排除了同一样本测定钠时出现错误的危险,特别是现在一些仪器将血气与电解质测定配套进行,即一份全血既测定血气又测定钠钾氯等电解质。
⑶静脉血:静脉血所测结果不适用于了解体内O2的运输状态,故PO2及有关推算数据仅供参考,对pH及PCO2等酸碱平衡指标是适用的。采静脉血尽可能不使用止血带。
⒉注意事项
⑴让病人处于安定舒适状态,卧床5分钟后采血。
⑵在病人进行治疗过程中采血要特别注意:①若进行辅助或人工呼吸时,采血前至少要等20分钟,让其在完全控制自如的人工呼吸状态下采血。②若病人进行氧气吸入时,作血气测定,应注意氧气流量,以备计算出该病人每分钟吸入的氧含量。例如病人吸氧速度为6L/min,吸氧器的呼吸循环纯氧气为3L/min,其余3L为周围空气(PCO2=0.209),因此病人每分钟得到3L纯氧及3×0.21=0.63L来自周围空气的氧气,因此6L总体积中含有3.6L氧气,含量为60%,此时PO2=0.6。③若是体外循环病人,应在血液得到混匀后再进行采血。
⑶抗凝剂以肝素锂为好。对于同时作血气、血钙或血锂的标本,则不能用肝素锂抗凝,因为肝素可与部分钙结合造成误差,此时就要用钙缓冲液肝素试剂抗凝。使用液体肝素抗凝剂浓度为500-1000U/ml为宜,含量过低,抗凝剂体积过大,易造成稀释误差;若含量过高也易引起误差。最好使肝素锂以均匀分布于毛细玻管周边壁上为宜,对标本既无稀释作用又有利于样品的抗凝。
⑷注意防止血标本与空气接触,应处于隔绝空气的状态。因为:①空气中PO2高(21.17kPa或150mmHg)于血液,PCO2低(0.040kPa或0.3mmHg)于血液,一旦血液与空气接触,大气中O2会从高压的空气中进入血液,造成血液PO2高的误差;CO2又会从高压的血液弥散到大气中,使血液PCO2测出结果偏低。大于标本10%的空气气泡会明显影响PO2值。②与空气接触,易造成空气污染血标本。
⑸标本放置时间:采出的全血中有活性红细胞,其代谢仍在继续进行,O2不断地被消耗,CO2不断地产生。有报道标本于体外37℃保存,每10分钟PCO2约增加1mmHg,pH值降低约0.01单位。血样于4℃保存1小时内,其中pH、PCO2值没有明显变化,PO2值则有改变。按要求,采取的血标本应在30分钟内检测完毕,如30分钟后不能检测,应将标本置于冰水中保存,最多不超过2小时,在30分钟到2小时之间,血PO2值是个怀疑值,仅供参考。
⑹采末梢血须是动脉化的毛细血管血,只有高灌注局部组织的代谢变化,其静脉血pH、PCO2、PO2与动脉血所测值才非常接近。
(二)仪器操作简介
目前使用的血气分析仪生产厂家多,型号各异,但性能和操作大同小异。现以AVL995血气酸碱分析仪为例,简要介绍该仪器的使用方法。
AVL945、995装有PO2、PCO2和pH电极,直接测定全血,实际上是测定血浆PO2、PCO2和pH,因为这些电极直接接触的标本是血浆,而未能伸入到红细胞内。测出这三个指标后,再通过仪器运算出其他指标。
⒈启动按仪器要求分别接通主机和空气压缩机电源,使空气压缩机压力到达额定的要求。再开启二氧化碳气瓶,使CO2气流量达到额定要求。分别检查洗涤液、参比液、标准缓冲液1和2等液体是否按要求装备。
⒉定标该机定标分两种形式,即两点定标和一点定标,与其他型号仪器一样可进行总两点自动定标。总两点定标是先用两种缓冲液对pH电极系统进行定标,再用混合后的两种不同含量的气体对PCO2和PO2电极进行定标。两点定标是让仪器建立合适的工作曲线。一点定标是每隔一定的时间检查一下电极偏离工作曲线的情况。开机后,两点定标自动进行是必须做的工作,并且不能中断。进行过两点定标后,仪器每隔12小时左右再自动进行下一次两点定标,必要时可根据情况任意选用定标程序再定标。两点定标后,每隔0.5-3小时,仪器用缓冲液1对pH电极系统进行一点定标。仪器还进行气体定标,先用气体2(CO2)对PCO2电极进行定标,最后用气体1(混合气)对PCO2和PO2电极进行定标。
⒊测量从开机到两点定标完成后,仪器屏幕上显示“READY”,即已准备好,此时可进行测量。一般测量用注射器进样或毛细管进样两种方式进行。
⑴注射器进样:按Syring(注射器)键,转换盘转到进样位置,用注射器慢慢注入血样,直到仪器屏幕显示Measure(测量),下行显示:拔出注射器,按START键。蠕动泵开始转动,将血样吸入测量室。当血样到达pH参比电极时,蠕动泵停转,血样停留在测量室中,仪器自动进行测量和计算。与此同时,输入病人Hb量及体温数、测出的pH、PO2值及其计算值在屏幕显示,并打印结果。由于AVL995Hb能自动测出病人血红蛋白值,这种型号的仪器就可不必另输Hb值,仅输入体温值即可。
测量一结束,仪器自动进行冲洗将血样冲走,干燥后,进行一点定标,然后返回READY状态,又可接着进行第二个样品的测量。
⑵毛细管进样:在仪器处于RDADY状态时,按Capillary键,转换盘转到进样位置。在进样口插入装有血样的毛细管,仪器便自动把血样吸入测量室,并停留在测量室自动进行检测,以下各种步骤与注射器进样法相同。
⑶微量样品测量法:当采集的血量不足40μl而又多于25μl时,仪器自动进行微量样品测量。进样后,仪器屏幕显示“微量样品”,下行显示“只测pH按1,其余按2”;如果还测pH、PCO2和PO2三个参数,需按“2”键。根据测量室血样进入的位置交替按“START键”和“1”键,直至pH测量完。仪器经运算后,即可打印结果。进行微量样品检测时,一定要按血样流动顺序进行,认真操作,其所测值与全量血样检测结果基本一致。
⑷维护和保养:按说明书要求,对仪器要定期保养和维护。特别是对电极的定期保养极为重要。
使用操作人员,一定要熟悉仪器的测定原理、各部件的工作性能,并熟读说明书,一般的故障应该学会处理,既大胆又心细。仪器一旦开机后,应该24小时连续开机使用,充分发挥仪器的效用,做到物尽其用。
五、血气检测的质量控制
(一)血气检测数据的校验
血气酸碱分析仪检测的数据是否准确是判断酸碱紊乱的先决条件,为此,首先应核实血气分析报告单上的数据是否可靠。可以用H-H公式进行核实;pH=6.1+lg([HCO3-]/PCO2×0.03),已知其中任意两个数据,即可计算出另一个数据。
(二)质量控制
目前使用的血气分析的参考试剂按基质不同分为水剂缓冲液、全血、血液基质、人造血氟碳化合物四种,使用最多的是水剂缓冲液,该质控物用安瓿封存,具有稳定、使用方便等优点。
水剂质控物是用Na2HPO4、KH2PO4及NaHCO3配成不同的pH缓冲液,再与不同浓度的CO2和O2平衡,以提供pH、PCO2和PO2及其换算的参考值。加入防腐剂贮存,有高、正常、低三种水平规格,以三种颜色予以标记:酸血症、低氧血症为红色标记,碱血症、高氧血症为蓝色标记,正常酸碱水平为黄色标记。质控物数据由多种型号仪器的几个实验室反复多次测定,再取X±S而确定一个参考范围。每种或每批质物均附有规定参考数据,不能通用。
制控物用安瓿装,液体并未充满整支安瓿,因安瓿中一定会出现一个液相,一个气相。液相为水及缓冲物质,气相则由O2、N2、CO2及汽水组成。根据物质运动规律,气相与液相之间不停地作分子交换,任一物质特别是气体不断地由气相进入液相,又从液相进入气相,这些物质处于两相间的平衡→不平衡→平衡的不断变化状态。这种平衡受温度因素影响很大。因此在使用血气质控物时应注意:①质控物在室温平衡后,再用力振摇2-3分钟,使气相与液相重新平衡;②开启安瓿后,应立即注入仪器中检测,再观察所测结果是否落在质控物范围内,如在范围内,表明该仪器处于正常运转状态,可以用于标本检测;③如果检测数据偏离参考范围,应检查原因,分别检查CO2纯度、标准缓冲液是否被污染、电极套是否要更换,电极是否过期等故障,在待一一排除,以确保仪器检测结果的准确性;④过期的质控物不能使用,无参考范围说明书的质控物也不能用,因为每一个批号的质控物的参考范围存在一定的差异。
第三节 体液平衡紊乱
一、水平衡紊乱
水平衡紊乱可表现为总体水过少或过多或总体水变化不大,但水分布有明显差异,即细胞内水增多而细胞外水减少,或细胞内减少而细胞外水增多。水平衡紊乱往往伴随有体液中电解质的改变及渗透压的变化。
(一)脱水
人体体液丢失造成细胞外液的减少,称为脱水。脱水因血浆钠浓度变化与否,又可将脱水分为高渗性、等渗性和低惨性脱水。
⒈高渗性脱水脱水以水丧失为主,与水比较电解质丢失较少,即每丧失1L体液的同时,丢失约300mOsm以下的电解质,使细胞外渗透压升高,多见于饮水不足,如高温作业大量出汗,或病人的非显性失水仍在进行,从而使水排出量增多。高渗透性脱水的特点是:①体液电解质浓度增加,血浆Na+浓度大于150mOsm/L或CL-与HCO3-浓度之和大于140mmol/L;②细胞外液量减少;③细胞内液水向细胞外液转移,造成细胞内液明显减少。临床症状表现为口渴、体温上升及各种神经症状出现,同时还有尿量减少,进而体重明显下降等症状。
⒉等渗性脱水主要是导致细胞外液的丢失。由于丢失的水和电解质基本平衡,即每丧失1L体液的同时约丢失300mOsm/L电解质,因而细胞外液渗透压保持正常,故称为等渗性脱水。常见于呕吐和腹泻等丧失消化液,此时患者体液电解质浓度无改变。正常时,血浆Na+浓度为130-150mmol/L或Cl-与HCO3-浓度之和为120-140mmol/L;但细胞外液量减少,细胞内液量正常。等渗性脱水对机体损害在于细胞外液量减少而导致血容量不足,血压下降、外周血液循环障碍等。
⒊低渗性脱水以电解质丢失为主,与水相比,电解质的丢失较多,即每丧失1L体液,同时丢失约300mOsm以上的电解质,因而细胞外液的渗透压较正常低,所以叫低渗性脱水。病因多见于丢失体液时,只补充水而不补充电解质所引起,如胃肠道消化液的丧失(腹泻,呕吐等)以及大量出汗情况下,仅补充水分而不补充从消化液和汗液中所丧失的电解质,从而导致低渗性脱水。此时,血浆Na+浓度小于130mmol/L或Cl-和HCO3-浓度之和小于120mmol/L。细胞外液量减少,细胞内液量增多,体重稍有减轻。如表5-1所示。
(二)水肿
当机体摄入水过多或排出减少,使体液中水增多、体重增加、血容量增多以及组织器官水肿,称为水肿或水中毒。一般是水增加致使体液超过体重的10%以上时,可出现水肿症状。引起水肿常见的原因有血浆蛋白浓度降低,或充血性心力衰竭,或水和电解质排泄障碍等,使体液增多出现水肿。水肿后,由于血浆渗透压出现不同的变化,又可分为高渗性、等渗性和低渗性水肿。
表5-1 脱水、水过多与钠的分布变化
| 总水量 | 细胞外液 | 细胞内液 | 总钠量 | 血浆Na+ | |
| 脱水 | |||||
| 高渗性 | ↓ | 轻微↓ | ↓ | 正常或轻微↓ | ↑ |
| 等渗性 | ↓ | ↓ | 正常 | ↑ | 正常 |
| 低渗性 | ↓ | ↓↓ | ↑ | ↓↓ | ↓ |
| 水过多 | |||||
| 水中毒 | ↑ | ↑ | ↑ | 正常 | ↓ |
| 细胞外液容量扩张 | |||||
| 等渗性 | ↑ | ↑ | 正常 | ↑ | 正常 |
| 低渗性 | ↑ | ↑ | ↑ | 轻微↑ | ↓ |
二、钠平衡紊乱
Na+离子是细胞外液最多的阳离子,对保持细胞外液容量、调节酸碱平衡、维持正常渗透压和细胞生理功能有重要意义。体内可交换的钠总量是细胞外液渗透压的主要决定因素,通过渗透压作用可影响细胞内液。细胞外液钠浓度的改变可由水、钠任一含量的变化而引起,故钠平衡紊乱常伴有水平衡紊乱。水与钠的正常代谢及平衡是维持人体内环境稳定的重要因素。
(一)低钠血症
机体摄入Na+过少造成血浆Na+浓度降低,小于130mmol/L,称为低钠血症。血浆Na+浓度是决定血浆渗透浓度(Posm)的主要决定因素,所以低钠血症通常是低渗透浓度的反映,故又称为低钠性低渗综合征。Psom降低导致水向细胞内转移,使细胞内水量过多,这是低钠血症产生症状和威胁生命的主要原因。
血浆钠浓度并不能说明钠在体内的总量和钠在体内的分布情况。低血钠可见于缺钠、多水或水与钠潴留等不同情况,是一个复杂的水与电解质紊乱。
引起低钠血症原因很多,可分为肾性和非肾性原因两大类。
⒈肾性原因肾功能正常情况下,机体很少是因为摄钠过少引起低钠血症的,因为肾脏有较强的保健能力。肾功能损害引起低钠血症有渗透性利尿、肾上腺功能低下、肾素生成障碍以及急、慢性肾功能衰竭等疾患。
⒉非肾性原因如呕吐、腹泻、肠瘘、大量出汗和烧伤等疾病过程,除钠丢失外还伴有不同比例水的丢失。低钠血症使细胞外液渗透压降低,引起水分向细胞内转移,进而出现细胞水肿,严重者有可能出现脑水肿和消化道紊乱。
(二)高钠血症
因进钠过多或水丢失过多所致,临床较少见。水的丢失大于钠的丢失的疾患有尿崩症、水样泻、出汗过多等以及糖尿病人,由于水随糖以糖尿形式排出体外等造成高钠血症。
高钠血症使细胞外液渗透压升高,细胞内水向细胞外转移,病人出现口渴等细胞内脱水症状。
三、钾平衡紊乱
(一)钾代谢
人体全身总钾量约为50mmol/kg。女性由于脂肪较多,体钾总量相对较少。约占总量98%的钾分布在细胞内。钾是维持细胞新陈代谢、调节体液渗透压、维持酸碱平衡和保持细胞应激功能的重要电解质之一。
人体钾的来源完全从外界摄入,每日摄入量50-75mmol/L,一般膳食每日可供钾50-100mmol/L,足够维持生理上的需求。钾90%由肠道吸收,80%-90%经肾脏排泄,还有10%左右经粪便排出。皮肤通常排出少量的钾,约5mmol/L,大量出汗时可排出较多的钾。机体有完整调节血K+水平的机制(图5-10)。
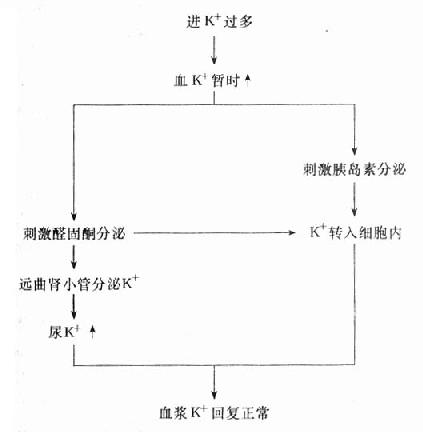
图5-10 血浆K=含量的调节
体内钾的排出主要途径是经肾以钾形式排出。肾排钾对维持钾的平衡起主要作用。肾对钾的排泄受到多种因素的影响,如醛固酮能促使近曲小管、髓袢升支、远曲小管和集合管各段肾小管细胞代谢过程,产生能量,加强对钠的重吸收和钾的排出。也可通过增强肾小管细胞膜对钾的渗透性,促进钾的排泌。醛固酮的分泌除受肾素-血管紧张素系统调节外,还受到血K+、Na+浓度的影响,当血K+升高血Na+降低时,醛固酮合成分泌增多,反之则分泌减少。体液酸碱平衡的改变也影响肾脏对钾的排泌,酸中毒时,尿钾增多;碱中毒时,尿钾减少。长期以来认为,酸碱平衡对肾排钾能力的影响主要通过远曲小管中Na+-H+与Na+-K+之间竞争性交换进行。近期研究认为,远曲小管的H+与K+的排泌呈平行而不是拮抗的关系,从而使人们对这传统观点提出了质疑。有人认为,酸碱平衡主要是通过增加或减少肾小管细胞内钾转运池对肾脏排K+功能产生影响。调节钾进入或逸出细胞膜的枢纽是血浆HCO3-,并非H+浓度。不论血钾水平如何,钾的浓度与细胞外液HCO3-的浓度直接有关,而与血pH的变化关系不大。
(二)钾平衡紊乱
钾平衡紊乱与否,要考虑钾总量和血钾浓度,二者既有区别又有联系。钾总量是指体内钾的总含量,由于钾主要分布在细胞内(约占总量的98%),此时血钾浓度并不能准确地反映体内总量状况。血钾浓度是指血清钾含量,血浆钾浓度要比血清钾浓度低0.5mmol/L左右。因为血液凝固成血块时,血小板及其它血细胞中钾释放少量入血清之故,临床以测血清钾为准。影响血钾浓度的因素有:①某种原因引起K+自细胞内移出到细胞外液时,则血钾浓度会增高,相反,细胞外液的钾进入细胞是时则血钾浓度会降低;②细胞外液受到稀释时,则血钾浓度降低,反之,细胞外液浓缩时,血钾浓度会增高;③钾总量是影响钾浓度的主要因素,如钾总量过多,往往血钾过高,缺钾则伴有低血钾。当细胞外液的钾大量进入细胞内或血浆受到过分稀释时,钾总量即使正常,甚至过多时,也可能出现低血钾,若细胞内钾向细胞外大量释放或血浆明显浓缩的情况下,钾总量即使正常甚至缺钾时也可能出现高血钾;④体液酸碱平衡紊乱,必定会影响到钾在细胞内外液的分布以及肾排钾量的变化。
临床观察拟为钾平衡失调的情况时,除了测定血清钾浓度外,还得分别从影响钾代谢以及钾平衡失调后代谢变化的多方面检查,如肾功能指标、血浆醛固酮及肾素水平、酸碱平衡指标以及尿pH、K+、Na+和Cl-的浓度。以便综合分析钾平衡紊乱的原因及其对机体代谢失调的影响程度。
⒈低钾血症血清钾低于3.5mmol/L以下,称为低血钾症。临床常见原因有:
⑴钾摄入不足的慢性消耗性疾病,术后较长时间未进食。因为人体钾来源全靠食物提供,长期进食不足或者禁食3-4天,由于钾来源不足,而肾仍照常排钾,很易引起体内缺钾造成低血钾症。
⑵钾排出增多,常见于严重腹泻、呕吐、胃肠减压和肠瘘。因为消化液丢失,消化液本身含有一定量钾,外加消化功能障碍,吸收减少,从而导致机体缺钾。肾上腺皮质激素有促进钾排泄及钠潴留作用,当长期应用肾上腺皮质激素时,均能引起低血钾。如心力衰竭,肝硬化患者,在长期使用汞利尿剂时,因大量排尿增加钾的丢失。
⑶细胞外钾进入细胞内,如静脉输入过多葡萄糖,尤其是加用胰岛素时,促进葡萄糖的利用,进而合成糖原,都有钾进入细胞内,很易造成低血钾,代谢性碱中毒或输入过多的碱性药物,形成急性碱血症,H+从细胞内进入细胞外,取而代之,细胞外K+进入细胞内,造成低血钾症。
⑷血浆稀释,也可能造成低血钾症。
低血钾改变了细胞内外K+含量的比例而影响了神经肌肉的兴奋性,也影响细胞膜的功能,使患者出现了低血钾的临床症状。最重要的是影响到心肌功能,表现为室上性心动过速、心传导阻滞、室性期外收缩和室性心动过速,严重者心跳停止于收缩期。血钾低于3.0mmol/L时可能有明显的神经症状,表现为胃肠管蠕动减少所致的便秘、腹胀、骨骼肌功能出现肌无力易激若的麻痹。心电图变化为:当血钾开始降低时,T波降低并增宽,Q-T时间延长,随着血钾进一步降低,伴有U波并呈双峰,再严重者,出现T波倒置,ST段下移等低血钾心电图的特殊表现。
⒉高血钾症血清钾高于5.5mmol/L者,称为高血钾症,临床引起高血钾症的原因有:
⑴钾输入过多,多见于钾溶液输入速度过快或量过大,特别是肾功能不全、尿量减少时,又输入钾溶液,尤其容易引起高血钾症。
⑵钾排泄障碍,各种原因的少尿或无尿,如急性肾功能衰竭的肾排钾障碍;
⑶细胞内的K+向细胞外转移,如大面积烧伤,组织细胞大量破坏,细胞内钾大量释放入血。代谢性酸中毒,血浆的H+往细胞内转移,取而代之,细胞内的钾转移到细胞外液;与此同时,肾小管上皮细胞泌H+增加,而泌K+减少,使钾潴留于体内。
高血钾症状主要是神经肌肉症状,如肌肉酸痛、苍白和肢体湿冷等一系列类似缺血现象。神经及神经肌肉联接处的兴奋性抑制,可发生心内传导阻滞,出现心跳变慢及心律不整,引起循环机能衰竭,甚至引起纤维性颤动,最后,心脏停跳于舒张期。
高血钾时,当其他症状不明显时,心电图呈现显著的改变,血钾高于6-7mmol/L时,T波高价而基底较窄,Q-T间期延长;当血钾升高达10mmol/L时,会出现异常增宽的QRS波群。有时心电图变化与血钾浓度不一定完全一致,甚至血K+高达8mmol/L,而心电图仍无明显变化者也有之。
第四节 酸碱平衡紊乱
正常人血液的酸碱度即pH值始终保持在一定的水平,其变动范围很小。血液酸碱度的相对恒定是机体进行正常生理活动的基本条件之一。机体每天在代谢过程中,均会产生一定量的酸性或碱性物质并不断地进入血液,都可能影响到血液的酸碱度,尽管如此,血液酸碱度仍恒定在pH7.35-7.45之间。健康机体是如此,在疾病过程中,人体仍是极力要使血液pH恒定在这狭小的范围内。之所以能使血液酸碱度如此稳定,是因为人体有一整套调节酸碱平衡的机制,首先依赖于血液内一些酸性或碱性物质并以一定的比例的缓冲体系来完成,而这种比例的恒定,却又有赖于肺和肾等脏器的调节作用,把过剩的酸或碱给予消除,使体内酸碱度保持相对平衡状态。机体这种调节酸碱物质含量及其比例,维持血液pH值在正常范围内的过程,称为酸碱平衡。
体内酸性或碱性物质过多,超出机体的调节能力,或者肺和肾功能障碍使调节酸碱平衡的功能障碍,均可使血浆中HCO3-与H2CO3浓度及其比值的变化超出正常范围而导致酸碱平衡紊乱如酸中毒或碱中毒。酸碱平衡紊乱是临床常见的一种症状,各种疾患均有可能出现。
一、酸碱平衡紊乱的诊断指标
近20年来由于酸碱平衡理论的认识不断深入,检测技术的不断提高以及血气分析仪器的广泛使用,使有关酸碱测定不仅准确快速而且微量化,现已成为临床日常重要的诊疗手段。为了正确诊断酸碱中毒的需要,对有关的酸碱中毒诊断指标的理论及临床意义列于表5-2。
二、酸碱平衡紊乱
正常状态下,机体有一套调节酸碱平衡的机制。疾病过程中,尽管有酸碱物质的增减变化,一般不易发生酸碱平衡紊乱,只有在严重情况下,机体内产生或丢失的酸碱过多而超过机体调节能力,或机体对酸碱调节机制出现障碍时,进而导致酸碱平衡失调。
表5-2 成人血气分析参数值及临床意义
| 指标 | 参考值 | 临床意义 |
| 酸碱度(pH) | 7.35-7.45(A)*7.33-7.43(V)* | <7.35酸中毒,>7.45碱中毒,在7.35-7.45之间,机体可能是①正常或②代偿型酸碱中毒 |
| 二氧化碳分压(pappen-heimer CO2,PCO2) | 4.66-6.0kPa(A)5.05-6.65kPa(V) | 反映肺泡PCO2值,PCO2增高示肺通气不足(原发或继发),CO2潴留;PCO2降低示肺通气过度(原发或继发),CO2排出过多 |
| 氧分压(pappenheimerO2,PO2)血红蛋白(hemoglobin | 9.98-13.30kPa (A)3.99-6.65kPa(V)120-160g/L(男) | 判断缺氧程度及呼吸功能,<7.3kPa示呼吸功能衰竭与氧含量、氧容量有密切关系的一项参数,Hb |
| Hb | 110-150g/L(女) | 降低携O2减少,缓冲酸碱能力降低,氧含量也降低,参与BE、SB及SatO2的运算 |
| P50 | 3.19-3.72kPa (A) | P50增高,氧解离曲线右移,Hb与O2亲和力降低;P50降低,氧解离曲线左移,Hb与O2亲和力增高 |
| 标准碳酸氢盐(standardbicarbonate,SB)实际碳酸氢盐(actualbicarbonate,AB) | 22.0-27.0mmol/L22.0-27.0mmol/L(AB=SB) | 表示血液HCO3-的储备量,SB增高示代谢性碱中毒;SB降低为代谢性酸中毒血液实测HCO3-量,AB>SB为呼吸性酸中毒,AB<SB为呼吸性碱中毒,AB增高和SB增高为代偿型碱中毒;AB降低和SB降低为代偿型酸中毒 |
| 缓冲碱(buffer base,BB | Bbb 45-54mmol/L(全血)BBP 41-43mmol/L(血浆) | BB降低为代谢性酸中毒或呼吸性碱中毒,BB增高为代谢性碱中毒或呼吸性酸中毒,BB降低而AB正常则提示Hb或血浆蛋白含量降低; |
| 碱剩余(碱超)(base xce-ss,BE) | -3-+3mmol/L | BE为正值,表示BB增高,为代谢性碱中毒;BE为负值,表示BB降低,为代谢性酸中毒或呼吸性酸碱中毒,因代偿关系,BE也可能升高或降低 |
| 总CO2(total CO2,TCO2) | 23-27mmol/L(A)24-28mmol/L(V) | 与代谢因素及呼吸因素有关,主要说明代谢因素影响酸碱平衡,因TCO2的95%为HCO3-量 |
| 阴离子隙(anion gap,AG) | AG=[Na+]+[K+]-[Cl-]为18±4mmol/L(V)AG=[Na+]-[Cl-][HCO3-]为12±4mmol/L(V) | AG增高是代谢性酸中毒AG正常的酸中毒可见于高Cl性代谢性酸中毒 |
| 氧含量(oxygen content,O2Cont) | 7.6-10.3mmol/L(A) | 判断缺氧程度和呼吸功能的指标 |
| 氧和度(O2saturation,O2 | 95%-98%(A) | 判断Hb与氧亲和力的指标;H+,2,3-DPG, |
| Sat | 60%~85%(V) | PCO2和PO2均影响O2Sat值 |
*:(A)为动脉,(V)为静脉
如果血浆HCO3-/H2CO3比值<20/1,pH值有低于正常下限(7.35)的倾向或<7.35,称为酸中毒(Acidosis)。由于HCO3/H2CO3比值>20/1pH高于正常上限(7.45)或>7.45,称为碱中毒(Alkalosis)。根据酸碱紊乱产生的原因,又可进一步分类,因血浆HCO3-水平下降造成的酸中毒,称为代谢性酸中毒(Metabolicacidosis),HCO3-增多产生的碱中毒,称为代谢性碱中毒。如因H2CO3增多使血浆pH值下降者,称为呼吸性酸中毒(respiratoryacidosis)。因H2CO3减少所造成的碱中毒称为呼吸性碱中毒。在发生酸碱紊乱后,机体的调节机制势必加强,以恢复HCO3-/H2CO3比值到正常水平,此为代偿过程。经过代偿后,如果HCO3-/H2CO3比值恢复到20/1,血浆pH值仍可维持在正常范围,称为代偿型酸碱中毒,属于临床认为的轻型酸碱中毒。如果经过代偿仍不能恢复到正常比值,血浆pH值必将发生明显变化。并超出正常值范围,称为失代偿型酸碱中毒。
血液酸碱度是用[H+]或pH来表示,其高低值取决于血液中H2CO3-与H2CO3的多少,三者的数值密切相关,可用Henderson-Hasselbalch(H-H)方程来表示,即:
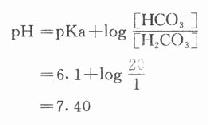
上式中pKa是常数,只有[HCO3-]/[/H2CO3]为20/1时,其pH值才能保持7.40±0.05,如果[HCO3-]量减少或者增加,机体将通过多种途径进行调节,使[HCO3-]量恢复到正常范围。机体酸碱中毒代偿程度如表5-3所示:
表5-3 酸碱中毒代偿程度
| 类型 | [HCO3-]/[H2CO3]比值 | [HCO3-]与[H2CO3]值 |
| 正常型 | 正常 | 正常 |
| 代偿型 | 恢复正常 | 异常 |
| 失代偿型 | 异常 | 异常 |
(一)酸中毒
根据引起酸中毒原因可分为代谢性酸中毒和呼吸性酸中毒。
⒈代谢性酸中毒常见原因有:①酸性代谢产物如乳糖酮体等产物增加;②酸性物质排障碍,如肾功能不全,尿液酸化不够;③碱丢物过多,如腹泻或重吸收HCO3-障碍。
过多代谢产物如乳酸、酮体进入血液后,机体通过多种途径进行调节,首先是血浆缓冲对HCO3-/H2CO3的缓冲作用:
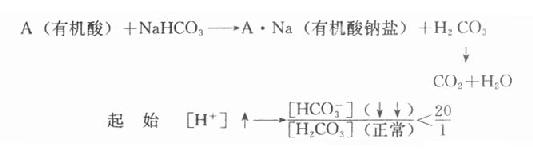
缓冲结果使血浆中HCO3-含量减少、CO2增多、PCO2升高,经肺调节,刺激呼吸中枢,加快呼吸,排出过多的CO2。与此同时,肾也进行调节,排酸保碱,以增加HCO3-的重吸收,其结果是:
代偿:
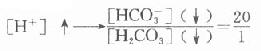
血浆中HCO3-降低,H2CO3也随之降低,在低水平保持[HCO3-]/[H2CO3]=20/1,血[HCO3-]低于正常水平,pH值仍在正常范围,即为代偿型代谢性酸中毒.
如果酸性产物继续增加,并超过肺和肾的调节能力,其结果是:
失代偿:
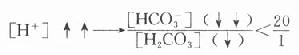
此时,血浆pH值下降至7.35以下者,称为失代偿型代谢性酸中毒。
肾调节酸碱平衡发挥的作用较晚,然而是极为重要并且是较为彻底的调节措施。肾主要通过H=-Na交换,K+-Na=交换以及排出过多的酸,达到调节的目的。
⒉呼吸性酸中毒由于肺部病变,使排出的CO2减少,使CO2潴留于体内,PCO2升高,H2CO3浓度增加,即:
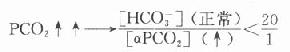
血液pH值有降低趋势,严重时,pH<7.35,这种因呼吸原因引起的酸中毒称为呼吸性酸中毒。呼吸性酸中毒患者,由于呼吸功能障碍,此时依赖于肺部再进行调节是不太可能,因此必须依赖于肾脏,排H+保Na+作用加强,肾小管回吸收Na+增加,而NaHCO3也随之吸收加强,补充血中NaHCO3,从而使血中NaHCO3浓度有一定程度的升高,有可能使血pH恢复正常范围,即
代偿:
![]()
若pH值仍在正常范围,仅PCO2和TCO2升高,此时称为代偿型呼吸性酸中毒。如病情继续发展严重,H2CO3浓度增加,血中PCO2、TCO2、H2CO3增加,经过代偿,HCO3-浓度也在增加,但H2CO3浓度增加速度高于HCO3-浓度的增长,使血液pH值小于7.35,所以称为代偿型呼吸酸中毒,即:
失代偿:
![]()
呼吸性酸中毒患者,由于肾脏排H+保Na+的作用加强,重吸收NaHCO3入血增加,此时,在血HCO3-浓度正常情况下使HCO3-浓度再升高,并高出正常值。这就是呼吸性酸中毒患者血生化指标的特点即呼吸性酸中毒患者,血HCO3-浓度是升高而不是降低的。
⒊代谢性碱中毒由于碱性物质进入体内过多或生成过多,或酸性物质产生过少而排出过多引起血浆HCO3-浓度升高,使血浆pH有升高的趋势,称为代谢性碱中毒。临床多见于:
⑴呕吐:使酸性胃液大量丢失,肠液的HCO3-重吸收增多。因为正常健康人胃粘膜的壁细胞,藉助碳酸酐酶的催化产生H2CO3,再解离为H+为HCO3-。H+从壁细胞中分泌入胃液与来自血浆中的Cl-结合成盐酸(HCl),而壁细胞中的HCO3-则重吸收入血浆与Na+结合成NaHCO3。向胃液中每排泌1分子HCl,则血中同时就多吸收1分子的NaHCO3,所以正常人在饭后血中出现暂时性的NaHCO3增高,这种现象称为“碱潮”。肠粘膜上皮细胞同样生成H2CO3,并解离成H+和HCO3-,HCO3-进入血液,当消化液丢失过多,NaHCO3未被HCl中和就重吸收,故血浆中NaHCO3含量增加。
⑵低钾低氯血症:使红细胞和肾小管上皮细胞内HCO3-进入血浆增多,又由于排K+保Na+减弱,排H+保Na+加强,从而由肾重吸收入血的NaHCO3增多,导致碱中毒。
⑶输入碱性药物过多:血浆中[HCO3-]增加,[HCO3]/[H2CO3]>20/1,pH有升高的趋势,甚至>7.45,导致如下变化:
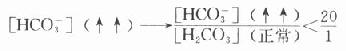
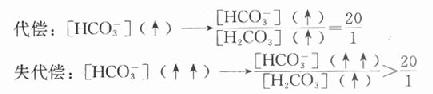
血液生化指标:pH≥7.45,SB明显升高,TCO2显著增加,BE往正值加大,PCO2升高,Cl-和K+减少。由于酸排出减少,NaHCO3排出增多,尿为碱性,尿NH4+也减少。但是当K+缺乏时H+-Na+交换加强,则有反向酸性尿。
⒋呼吸性碱中毒由于过度换气,CO2排出过多,使血浆PCO2降低,血浆[HCO3-]/[H2CO3]>20/1,pH有升高的趋势,这一现象即为呼吸性碱中毒。
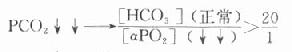
因血液CO2减少,使H2CO3浓度降低,CO2弥散入肾小管细胞量减少,分泌H+离子入肾小管腔也减少,H+-Na+交换减弱,HCO3-回吸收量减少,导致血浆中[HCO3-]水平也降低,血浆[HCO3-]/[H2CO3]在低水平正常水平下保持20/1,pH值仍在正常范围,此时属于代偿型呼吸性碱中毒,即:
![]()
如果,呼吸仍处于过度换气,CO2排出过多,PCO2降低,血浆HCO3-浓度无法与PCO2降低相平衡超过肾脏的代偿能力,其结果造成失代偿型呼吸型呼吸性碱中毒,此时pH>7.45,即:
失代偿:
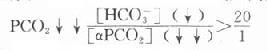
呼吸性碱中毒血液生化指标为血浆pH≥7.45,PCO2明显降低,TCO2减少,Cl-增高。K+轻度降低,AG轻度增高。
⒌混合性酸碱平衡紊乱临床上代谢性与呼吸性酸碱中毒,往往可以同时或相继出现,形成混合型酸碱平衡失调。不仅呼吸性与代谢性酸碱中毒可同时存在,甚至酸中毒与碱中毒也可能同时存在(表5-4)。
表5-4 血气变化值一览表
| 类型 | pH | PCO2 | BE | HCO3- | |||||
| pH值 | nmol/L | mmHg | kPa | mmol/L | mmol/L | ||||
| 正常 | 7.35-7.45 | 44.7-35.5 | 36-14 | 4.8-5.8 | +3.0-3.0 | 22-31 | |||
| 酸中毒 | 代谢性 | 代偿 | 不变 | ↓或不变 | ―→ | ↓ | |||
| 失代偿 | <7.35或[H+]>44.7 | ||||||||
| 呼吸性 | 代偿 | ↓ | ―→ | ↓↓ | |||||
| 失代偿 | |||||||||
| 不变 | ↑↑ | +→ | ↑ | ||||||
| <7.35或[H+]>44.7 | ↑↑ | +→ | ↑↑ | ||||||
| 代 | 代偿 | 不变 | ↑或不变 | +→ | ↑ | ||||
| 碱 | 谢 | ||||||||
| 性 | 失代偿 | >7.45或[H+]<33.5 | ↑ | +→ | ↑↑ | ||||
| 中 | |||||||||
| 呼 | 代 偿 | 不变 | ↓↓ | ―→ | ↓ | ||||
| 毒 | 吸 | ||||||||
| 性 | 失代偿 | >7.45或[H+]<33.5 | ↓↓ | ―→ | ↓↓ | ||||
“↓”示下降;“↑”示上升;双箭头示病情严重,“―→”示往负值降低;“+→”示往正值增加。
对于酸碱紊乱的实验诊断,主要依赖于血气分析仪检测系列酸碱指标。除测出血pH、PCO2和TCO2指标外,还可能推算出多项指标,一般有12-16项之多。根据这一系列参数,结合病人临床症状,对其酸碱中毒的类型,代偿程度以及治疗经过的观察,几乎有决定性的诊断价值。
如果没有合适的血气分析仪,也可通过有关参数运算出下述指标:
⑴pH=pKa+log[c]/αPCO2(mmHg)
⑵[HCO3-]=[αPCO2]antilog[pH-pKa]
⑶AG=AG=[Na+]+[K+]-[Cl-]-[HCO3-]或
AG=[Na+]-[Cl-]-[HCO3-]
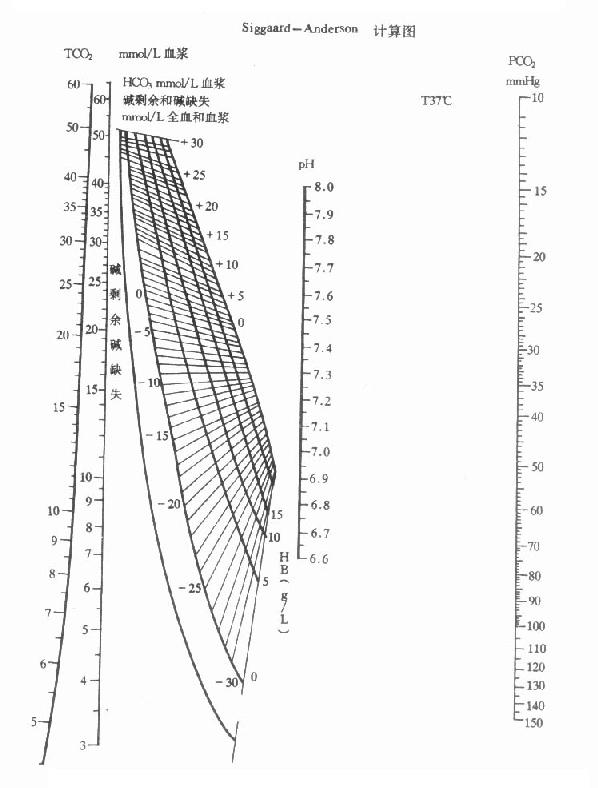
图5-11 PCO2pHBEHCO3和TCO2之间关系及其计算图
⑷TCO2=AB+[HCO3-]或者
=AB+0.03·PCO2(mmHg)
⑸PCO2(mmHg)(TCO2-AB)/0.03
另外,血pH、PCO2和[HCO3-]有关数据也可以从Siggaard-Andersen图(图5-11)直接查出。
第五节 酸碱平衡紊乱典型病例检验结果分析
病例一 代谢性酸中毒
一位有10年糖尿病史的45岁男性,因昏迷状态入院,体检血压12/5.3kPa,脉搏101次/min,呼吸28次/min。检验结果:
血球分析:MCV75fL、HCT0.65L/L,其他未见异常
生化检验:血糖10.1mmol/l、β-羟丁酸1.0mmol/L、尿素8.0mmol/L、K+5.0mmol/L、Na+160mmol/L、Cl-104mmol/L;pH7.136、PCO24.06kPa、PO29.91kPa、BE-18.0mmol/L、HCO3-9.9mmol/L、AG35mmol/L;尿:酮体(+++),糖(+++),酸性;脑脊液常规检查未见异常。
根据检验结果报告及有关临床症状体征诊断为糖尿病昏迷和代谢性酸中毒。经静脉滴注等渗盐水,以低渗盐水灌胃,静脉滴注胰岛素等抢救措施,6小时后,病人呼吸平稳,神志清醒,重复上述检验项目测定,除血K+为3.4mmol/L偏低外,其它项目均接近正常,临床以慎重地补钾,并适当减用胰岛素继续治疗。数月后,病人病情得到控制。
病案分析
患者因患糖尿病所致脂代谢障碍,酮体大量堆积或酮血症、酮尿症,血中大量乙酰乙酸及β-羟丁酸经血中HCO3-/H2CO3缓冲,使HCO3-减少致使HCO3-/H2CO3比值为<20/1,血pH<7.35(7.111)。机体通过肺加快呼吸,多排出缓冲酮体酸所产生的CO2,肾脏加快排出酮体酸盐,增加HCO3-的重吸收,尽管如此,病人仍出现失代偿型代谢性酸中毒,并因血糖未能及时进入细胞而堆积于血中,形成细胞外液的高渗状态,引起细胞内脱水尤以脑细胞脱水为重,外加PO2偏低,从而造成神经症状乃致昏迷。经抢救治疗,补充液体(含低渗液)及胰岛素,促使糖进入细胞代谢,减少脂肪动员,酮体产生减少,加上血、肺及肾的调节缓冲作用,使病人酸碱平衡紊乱得以恢复。
病例二 脱水
女性,62岁,因进食即呕吐10天而入院。近20天尿少色深,明显消瘦,卧床不起。体检:发育正常,营养差,精神恍惚,嗜睡,皮肤干燥松弛,眼窝深陷;呈重度脱水征。呼吸17次/min,血压16/9.3kPa,诊断为幽门梗阻。检验结果:
血球分析:MCV72fL、HCT0.56L/L,其余正常。
血液生化检验:血糖5.0mmol/L、尿素7.6mmol/L,K+3.61mmol/L、Na+158mmol/L、Cl-90mmol/L、pH7.50、PCO27.92kPa、PCO26.7kPa、BE+8.0/mmol/L、HCO3-45mmol/L、AG26.4mmol/L。
病案分析
患者系慢性十二指肠球部溃疡所致的幽门梗阻。呕吐未及一周,即出现严重的缺盐性脱水和低血氨性碱中毒。患者是幽门梗阻,频繁呕吐,丢失胃液并丧失胃酸,十二指肠液的HCO3-得不到中和即被吸收入血,使血中HCO3-量增加,造成代谢性碱中毒,又因血pH值高出正常范围,因此可以诊断为失代偿型代谢性碱中毒。由于长时间不能进食进水,还有胃液的丢失,从而导致严重脱水。血渗量摩尔高于正常,呈高渗性脱水,进一步使细胞脱水(MCV变小),尤其是脑细胞脱水,从而出现精神症状。
病例三 代谢性碱中毒
男性,46岁,因满腹疼痛以急性腹膜炎入院。入院后作血液分析、尿液分析粪常规、血气分析、肾功能检查确诊为急性弥漫性腹膜炎。急诊开腹探查,术中发现弥漫性腹膜炎是阑尾脓肿破裂所致,手术中切除阑尾,并作腹腔引流。术后病人胃肠减压五天后,又出现手麻、神志不清楚、血压下降、呼吸28次/分钟。化验检查血pH7.54、PCO26.44kPa、BE+10.6mmol/L、HCO3-40mmol/L、K+3.2mmol/L、Na+142mmol/L、Cl-105mmol/L,尿液pH呈酸性。诊断为低血钾性酸中毒。经补钾、生理盐水及多次补充新鲜血浆,症状明显好转,再次检验:血pH7.44、PCO2为5.7kPa、BE+3.0HCO3-28mmol/LK+4.2mmol/L、Na+148mmol/L、Cl-105mmol/L。
病案分析
该病人因较长时间减压,胃肠液丢失,尤其是K+的丢失过多,肾小管分泌K+减少,即K+-Na+交换减弱,而H+-Na+交换占优势,排H+过多,使血pH升高。同时又造成NaHCO3在血中增加,另外,该病人也有HCO3-的丢失,而K+的丢失更为严重,泌K+减少而泌H+增加,造成低血K+碱中毒,尿液呈酸性。胃肠减压停止,由于K+的补充,使血K+得到补充,肾脏的K+-Na+交换和K+-Na+交换恢复到正常,从而使病人酸碱平衡恢复到正常状态,病情逐渐好转。
病例四 呼吸性酸中毒
男性,65岁,因呼吸困难处于昏迷状态入院。病人有30年抽烟史,有慢性支气管炎,近五年病情逐渐加剧,实验室检验结果为:血生化检查,pH7.24、PCO28.6kPa、PO26.0kPa、BE+3.0mmol/L、HCO3-38mmol/L、AG18mmol/L、K+、Na+和Cl-分别为3.8、138和85mmol/L。血乳酸8.5mmol/L。肾功能正常,尿液偏碱性。
病案分析
病人因为慢性支气管炎,近月病情加重,呼吸困难,表现为通气不足,O2进量减少,CO2无法排出,积压于体内,即PCO2升高。肺部疾患引起的血TCO2及PCO2升高,使血中CO2不断扩散入肾小管细胞,导致CO2+H2O→H2CO3→HCO3-+H+,使H+升高进行H+-Na+交换,消除CO2的同时肾小管回吸收的HCO3-增加,从而导致呼吸性酸中毒的血HCO3-增加。因为代偿作用,血HCO3-高于正常值,使血pH超出正常范围。该病人因呼吸困难,PO2低,O2Sat降低,以及因缺氧使糖酵解加强,血乳酸增多,因此还可能伴有代谢性酸中毒。
病例五呼吸性碱中毒
男性,56岁,因小肠克隆病入院。后于硬膜外麻醉作肠切除术。术中病人紧张、呼吸加快,出现手足轻度发麻现象,临床诊断为呼吸性碱中毒。血液有关项目检验结果是:pH7.52、PCO24.0kPa、PO27.6kPa、BE-1.2mmol/L、HCO3-23.3mmol/L,K+4.5mmol/L、Na+134mmol/L、Cl-96mmol/L、AG19.3mmol/L,尿素及肾的肌酐清除率均在正常范围。
手术毕,经输液及维持能量需要,眼镇痛药,解除病人顾虑,症状逐渐好转,呼吸功能正常,伤口达一期愈合,病人逐渐好转,血液酸平衡指标,几乎都恢复正常。
病案分析
病人因呼吸过快,排出过多的CO2,使血中CO2减少。此时肾的代偿性调节起重要作用。肾小管产生H+减少,H+-Na+交换减弱,HCO3-的回吸收减少而排出增多,肾保留较多的Cl-,以填充较少的HCO3-在阴离子平衡中的位置;氨排泌减少,尿酸度降低,代偿结果使CO2保留于体内,血HCO3-相应减少,使HCO3-/H2CO3-的比值尽量接近保持在20/1,使血pH值也接近正常。尽管如此,患者血pH值仍是7.51,表明其为失代偿型代谢性碱中毒,血HCO3-降低属继发性的代偿的结果。手术完毕,病人情绪稳定,呼吸功能恢复到正常,有关血气分析指标已完全恢复正常。神志清楚,手脚麻木的感觉消失。
第六节 体液钾钠氯测定及方法学评价
占体重约5%的无机盐主要有Ca、Mg、K、Na、P和Cl,这六种无机元素约占体内无机盐总量的60%0-80%,主要存在于体液。
一、标本处理及含量变动的影响因素
体内所含元素约60种之多,作为定量检测无机元素主要是Na、K、Cl、Mg、Ca、P、Cu、Zn和Fe等。测定这些元素一般以各种体液为标本,主要是血清(浆)、尿、脑脊液和胸腹水。标本应注意及时处理及时检测。在贮存过程中,Na会进入红细胞内,而K会从红细胞进入血浆。与此同时Ca也会移入细胞内,Cl会在Cl转移过程中伴随血清CO2量的变化而改变。作为上述元素的测定标本要求采血后20min内分离出血清,在室温下可贮存数日,冰室内贮存数周,低温保存数月之久。
生理因素可影响其正常值的波动:如无机磷于餐后显着升高;血清铁早晨空腹呈高值,夜间呈低值,男性高于女性,不饱和铁结合力恰是女性高于男性。
血清中所含色素对测定结果也有影响,一般高胆红素、乳糜血及溶血等均影响测定结果,尤其是影响酶法测定值。
抗凝剂选择很重要,一般不用抗凝剂,以血清进行测定为宜。紧急情况下,应采用肝素锂抗凝。
二、无机元素存在形式及测定方法分类
(一)测定方法分类
体液或组织中无机盐的测定一般可用以下几类方法进行。
⒈原子吸收分光亮度法 测定Ca、Fe、Cu、Zn、Mg、P、Pb、Cd、Hg、Al和As可采用原子分光亮度法。以组织块为标本的检测更为适用。
⒉火焰亮度法 测定Na、K、Ca、Li、Cs和Zn可采用火焰亮度法。该方法属于经典的标准参考法,优点是结果准确可靠。国内检测Na、K的火焰亮度计种类较多。
⒊吸光亮度法 测定Ca、Fe、Cu、Mg、Zn和P可采用一般的比色分析法,此方法广为临床采用。
⒋螯合滴定法 测定Ca、Mg和Cl可采用特定的螯合剂滴定法进行,滴定过程中易受多种因素影响,难以得到准确的结果,属于淘汰之列。
⒌离子选择电极(iron selective electrode,ISE)法ISE法可用于Na、K、Cl、Ca、Li和H的测定,结果准确可靠,目前临床多采用ISE法。
⒍离子色谱法 离子色谱法需特殊条件,可用于Na、K、Cl、Br和F的测定。临床应用很少。
⒎电量滴定法 Cl、Fe也可采用电量滴定法,非医学方面应用较多。
(二)无机元素在体内存在形式
⒈Na、K和Cl 在体液及组织中,Na、K和Cl几乎全以离子形式存在。
⒉Ca和Mg 在体液中Ca和Mg约有一半与蛋白质结合,另一半以离子形式存在。
⒊Fe和Cu 体液中Fe与Cu几乎全部与蛋白质结合。
在体内以离子形式存在的状态下,可直接采用火焰光度法或离子选择电极法。与蛋白质结合状态的无机元素,为了精确测定,尚需考虑除去蛋白质再进行定量。故Ca、Mg、Fe和Cu等金属元素在严格控制蛋白质干扰的条件下,才能用直接法进行测定。
三、钾、钠测定方法学评价
⒈火焰亮度法 1950年开始使用并一直沿用至今的火焰亮度法检测血清、尿液、脑脊液及胸腹水的Na+和K+,是一种发射光谱分析法,准确可靠的好方法,广为临床采用。
测定方法分为内标准法和外标准法两种。外标准法操作误差较大,一般不采用。现在主要使用内部标准法,即标本及标准液采用加进相同浓度的内部标准元素锂或铯进行测定。操作时,将含锂的溶液作为稀释液,同时测定K、Na和Li的浓度,以标本与标准液的Na/Li与K/Li比值,计算Na、K浓度。由于血清稀释倍数大,血清蛋白质粘性的影响几乎可忽略不计。国产的火焰光度计,广为临床使用。
⒉化学测定法 主要利用复环王冠化合物如穴冠醚或球冠醚,亦称为冠醚,均为离子载体进行测定,由于大环结构内有空穴,分子内部氧原子有未共用电子对可与金属离子结合,根据空穴大小,可选择性结合不同直径的金属离子,从而可达到测出离子浓度的目的。测定血清K,一般采用普通冠醚阴离子染料进行比色定量。
⒊离子选择电极法 ISE法是采用灵敏的特定专用电极,在专用仪器上进行血清和尿等体液的K+、Na+的测定,因标本用量少,快速准确,几乎有取代其他方法的趋势。其仪器测定原理是离子选择电极与参比电极组合浸泡在待测标本溶液中,进行检测。目前已有的电极种类为:①玻璃膜电极,感应材料为玻璃薄膜的有pH电极、K+电极和Na+电极;②固相膜电极,由难溶性金属物质加压成型,以固体膜或单日膜作为感应膜的电极有Cl-电极和F-电极;③液态膜电极,将环氧树脂或内装聚氯乙稀为感应膜的Ca2+电极;④用缬氨霉素膜制成的K+电极。上述电极均有一定的寿命,因为电极使用一段时间就会自动老化,有效期长短不一。
目前有K、Na或K、Na、Cl或K、Na、Ca、Cl或K、Na、Cl、TCO2、pH组合的各种类型的电解质分析仪。标本用量少(100μl全血),1min出结果,快而准确。国产的同类仪器价格较便宜,操作简便。ISE法是目前所有方法中最为简便准确的方法。
⒋原子分光光度法 原子分光光度法可用于检测血清K+、Na+,操作繁杂,误差较大,不及火焰光度法简便。
四、氯的测定
体液中Cl以离子形式存在,几乎全与Na+平衡增减。测定Cl-的方法有多种。
⒈滴定法
⑴Molrr法:以K2CrO4为指示剂,用AgNO3滴定血清中Cl-,以过量的Ag+与CrO42-结合出现Ag2CrO4红色沉淀为终点,根据AgNO3消耗量计算出标本中Cl-的量。
⑵Sehales法:以二苯卡巴腙作指示剂,用Hg(NO3)2滴定血清中Cl-,以过量的Hg2+与二苯卡巴腙结合成不溶的紫蓝色化合物为终点,根据Hg(NO3)2消耗量计算血清Cl-的量。
滴定法需要熟练的操作,终点要准确,尽可能排除主观因素的干扰,否则误差很大。血清中过多的胆红素.血脂及血红蛋白(溶血)对结果干扰很大。滴定法有淘汰的趋势。
⒉化学测定法:在Fe+存在下,采用Hg(SCN)2与Cl-反应生成与Cl-等当量的SCN3-,再与Fe3+结合成Fe(SCN)3的红色化合物,进行比色,定量标本中Cl-的含量。
该法测定时,血清中其他因素如F.Br和I也可以起反应,其量少于1mmol/L,故可忽略不计。某些药物及胆红素均对其有影响。这种比色法消除主观因素的影响,并可在自动生化分析仪进行批量测定,属临床常用的一种方法。
⒊电量分析法:属于物理学方法。在恒定的电流下,以银电极置于标本中,从电极释放的Ag+与Cl-反应开始生成不解离的AgCl沉淀开始计时,当Cl-全部与Ag+作用完毕,游离的Ag+出现,其溶液电导明显增加,使仪器传感器和计时器立即切断电流并计算滴定所需的时间作为完成测定过程。氯的浓度用法拉第常数(9648库仑/摩尔氯化物)运算,库仑与滴定时间和电流的乘积成正比,以此计算出标本中Cl-的含量。
标本中Br-和I-对其有一定干扰,其量很少,可忽略不计。该法简便,快速,在控制好电流条件下,是个较好的方法。
⒋离子选择电极法 ISE法是目前测定Cl-的最好方法,因为测定Cl-的电极均与K+、Na+电极配套,仅需100μl全血即可测出标本中K+、Na+和Cl-的含量而快速、准确,操作简便,是目前广为临床使用的方法。
ISE法需用Cl-电极,该电极由氯化银.氯化铁-硫化汞为膜性材料作成固体膜电极,与Na+.K+电极组装于同一台仪器上,使用简便。标本中Br-和I-,虽有干扰,其量甚微,可忽略不计。该电极寿命较短,一般有效期仅4-6个月。Na+电极有效期8-10个月,K+电极6-8个月。近几年已出现长有效期(2年)的K+、Na+、Cl-电极,为临床的使用提供了方便。
第六章 钙磷镁与微量元素的临床生物化学
钙、磷、镁是人体的重要组成物质,具有广泛的生理功能,其代谢异常在临床上亦较多见。微量元素(traceelements)在体内具有广泛的生物学作用和临床意义,已引起医学界的重视。本章将扼要介绍钙磷镁及某些微量元素的生理作用、体内分布、代谢和代谢异常以及测定这些物质的临床意义。
第一节 钙、磷代谢及其异常
一、钙、磷的生理功用
钙盐和磷酸盐是人体内含量最高的无机盐,成人体内钙总量约为400-800g,约99%的钙和86%以上的磷存在于骨骼和牙齿中。
(一)体内Ca2+的生理功能
⒈血浆Ca2+可降低毛细血管和细胞膜的通透性,降低神经、肌肉的兴奋性当血浆Ca2+的浓度降低时,神经、肌肉的兴奋性增高,可引起抽搐。
⒉血浆Ca2+作为血浆凝血因子Ⅳ参与凝血过程它是因子Ⅸ、因子Ⅹ、凝血酶原、因子ⅩⅢ等的激活作用中不可缺少的辅因子。
⒊骨骼肌中的Ca2+可引起肌肉收缩当肌细胞内储存Ca2+受神经冲动而释放,Ca2+浓度增大到10-7-10-5mol/L时,Ca2+可迅速地与钙蛋白的钙结合亚基结合,引起一系列构象改变后导致肌肉收缩。
⒋Ca2+是重要的调节物质一方面作用于质膜,影响膜的通透性及膜的转运。一方面,在细胞内Ca2+作为第二信使起着重要的代谢调节作用。此外,Ca2+还是许多酶(脂肪酶、ATP酶)的激活剂,Ca2+还能抑制维生素D3-1α-羟化酶的活性,从而影响代谢。
(二)磷的生理功能
⒈血中磷酸盐(HPO42-/H2PO4-)是血液缓冲体系的重要组成成分。
⒉细胞内的磷酸盐参与许多酶促反应如磷酸基转移反应、加磷酸分解反应等。
⒊构成核苷酸辅酶类(如NAD+、NADP+、FMN、FAD、CoA等)和含磷酸根的辅酶(如TPP、磷酸吡哆醛等),还构成多种重要的核苷酸(如ATP、GTP、UTP、CTP、cAMP、cGMP等)。
⒋细胞膜磷脂在构成生物膜结构、维持膜的功能以及代谢调控上均发挥重要作用。酶蛋白及多种功能性蛋白质的磷酸与脱磷酸化则是代谢调节中化学修饰调节的最为普遍、最为重要的调节方式,与细胞的分化、增殖的调控有密切的关系。
二、钙、磷代谢及其调节
(一)血钙与血磷
⒈血钙血钙几乎全部存在于血浆中,正常人血钙波动甚小,保持于2.25-2.75mmol/L(10±1mg/dl或4.5-5.5mEq/L)。血浆钙分为可扩散钙和非扩散钙两大类。非扩散钙是指与蛋白质(主要是白蛋白)结合的钙,约占血浆总钙的40%,它们不通透毛细血管壁,也不具有前述生理功用。
血浆(清)钙的60%是可扩散钙,其中一部分(占血浆总钙的15%)是复合钙,即是与柠檬酸、重碳酸根等形成不解离的钙。发挥血钙生理作用的部分是离子钙,占总钙的45%,非扩散钙与离子钙之间可以互相转化。临床上常测定血清总钙量以观察血清离子钙的变化情况,方法简便。但由于非扩散钙部分可随血清白蛋白浓度的增减而改变,即同时引起血清总钙含量的增减,但不影响血清离子钙的浓度。目前已可应用离子选择电极等方法直接测定血清中离子钙的浓度,其正常参考值为0.94-1.26mmol/L。
血清pH值对血钙浓度有显著影响,酸中毒时蛋白结合钙向离子钙转化;碱中毒时,血浆离子钙浓度降低,此时虽血浆总钙含量无改变,亦可出现抽搐现象。
⒉血磷血液中的磷以有机磷和无机磷两种形式存在。有机磷酸酯和磷脂存在于血细胞和血浆中,含量甚大,血磷通常是指血浆中的无机磷,正常人血浆无机磷含量为成人1.1-1.3mmol/L(3.5-4.0mg/dl),婴儿为1.3-2.3mmol/L(4-7mg/dl),血浆无机磷酸盐的80%-85%以HPO42-的形式存在,其余为H2PO4-,PO43-仅含微量。
血浆磷的浓度不如血浆钙浓度稳定,新生儿血磷约为1.8mmol/L(5.5mg/dl),6个月婴儿可升高至2.1mmol/L(6.5mg/dl)。此后随年龄增长又逐步下降,15岁时达成人水平。儿童时期血磷高是由于儿童处于成骨旺盛期,碱性磷酸酶活性较高所致。成年人血磷也有一定的生理变动,进食、摄糖、注射胰岛素和肾上腺素等情况下,由于细胞内磷的利用增加,也可引起血磷降低。血钙与血磷之间也有一定的浓度关系,正常人钙、磷浓度(mg/dl)的乘积在36-40之间,病理条件下此值可高于40或低于36。
(二)钙磷代谢及其调节
⒈钙、磷的吸收与排泄正常成人日摄入钙量在0.6-1.0g之间。发育期儿童、少年、孕妇及授乳妇女需较多的钙。食物钙主要含于牛奶、乳制品及果菜中。钙主要在十二指肠吸收,成人每日可吸收0.1-0.4g,钙吸收主要是在活性维生素D3调节下的主动吸收。肠管pH明显地影响钙的吸收,偏碱时可以促进Ca3(PO4)2的生成,因而能减少钙的吸收。乳酸、氢基酸及胃酸等酸性物质有利于Ca3(H2PO4)2的形成,因此能促进钙的吸收。食物中的草酸和植酸可与钙形成不溶性盐,影响钙的吸收。食物中钙磷比例对吸收也有一定影响,Ca:P=2:1时吸收最佳。
钙通过肠管及肾排泄。由消化道排出的钙一部分是未吸收的食物钙,另一部分是肠管分泌的钙(每日可达600mg),分泌的钙量可因摄入高钙膳食而增加,严重腹泻排钙过多可导致缺钙。经肾排泄的钙占体内总排钙量的20%。每日由肾小球滤出约10g钙,其中约一半在近曲小管被重吸收,1/5在髓袢升段被吸收,其余在远曲小管和集合管被吸收,尿中排钙量只占滤过量的1.5%(约150mg)。尿钙的排出量受血钙浓度的直接影响,血钙低于2.4mmol/L(7.5mg/dl)时尿中无钙排出。
成人每日进食磷约1.0-1.5g,以有机磷酸酯和磷脂为主,在肠管内磷酸酶的作用下分解为无机磷酸盐。磷在空肠吸收最快,吸收率达70%,低磷膳食时吸收率可达90%。由于磷的吸收不良而引起的缺磷现象较少见,但长期口服氢氧化铝凝胶以及食物中钙、镁、铁离子过多,均可由于形成不溶性磷酸盐而影响磷的吸收。
肾是排泄磷的主要器官,肾排出的磷占总磷排出量的70%,30%由粪便排出。每天经肾小管滤过的磷可达5g,约85%-95%被肾小管(主要是近曲小管)重吸收。
⒉钙磷代谢的调节钙、磷的吸收与排泄、血钙与血磷的水平、机体各组织对钙磷的摄取利用和储存等都是在活性维生素D、甲状旁腺激素及降钙素这三种激素的调节下进行的。
⑴甲状旁腺素:甲状旁腺素(parathyroidhormone,PTH)是由甲状旁腺的主细胞合成并分泌的一种单链多肽。初合成的是含115个氨基酸残基的前甲状旁腺素原,再在粗面内质网去掉N端25个氨基酸残基形成甲状旁腺素原,后者在高尔基复合体内从N端去掉一个六肽,形成84个氨基酸残基的PTH,分子量9500。
PTH的合成与分泌受细胞外液Ca3+浓度的调节,血钙浓度低(如降至1.3mmol/L)时可明显促进PTH的合成与分泌;血钙浓度高(如达3.9mmol/L)则抑制PTH的合成与分泌。血钙在1.3-3.9mmol/L范围内,血钙浓度与PTH分泌呈负相关关系。
在肝枯否细胞及肾小管细胞,PTH均可被分解为N端两个片段,N片段具有PTH活性,可被肝细胞、肾及骨组织摄取,C片段无PTH活性(表)6-1。
表6-1 血中PTH各片段的活性比较
| 片段 | 分子量 | 产生器官 | 激素活性 | 靶器官 | 百分比 | 血中 | 代谢 |
| 完整PTH | 9500 | 甲状旁腺 | + | 肾 | <10 | <10分钟 | 肝、肾 |
| N片段 | 3000-4000 | 肝、肾 | + | 肾、骨 | <10 | <10分钟 | 肾、骨 |
| C片段 | 6000-7000 | 肝、肾 | - | / | <80 | >1小时 | 肾 |
| 甲状旁腺 |
PTH是维持血钙正常水平的最重要的调节因素,它有升高血钙、降低血磷和酸化血液等作用,其主要靶器官是骨、肾小管,其次是小肠粘膜等。PTH作用于靶细胞膜。活性腺苷酸环化酶系统,增加胞质内cAMP及焦磷酸盐浓度。cAMP能促进线粒体Ca2+转入胞质;焦磷酸盐则作用细胞膜外侧,使膜外侧Ca2+进入细胞,结果可引起胞质内Ca2+浓度增加,并激活细胞膜上的“钙泵”,将Ca2+主动转运至细胞外液,导致血钙升高。
PTH对骨的作用:骨是最大的钙储存库,PTH总的作用是促进溶骨,提高血钙。PTH可在数分钟到数小时内引起骨钙动员,使密质骨中的钙释放入血,此种作用迅速但不持久。数小时至数日内,PTH的作用是将前破骨细胞和间质细胞转化为破骨细胞,使破骨细胞数目增多,引起溶骨作用和骨钙的大量释放。PTH对破骨细胞的作用是使细胞内Ca2+浓度增加,进而促使溶酶体释放各种水解酶;另一方面抑制异柠檬酸脱氢酶等酶活性,使细胞内异柠檬酸、柠檬酸、乳酸、碳酸及透明质酸等浓度增高,促进溶骨。此外,胶原酶活性也显著升高,这均有利于溶骨作用。
PTH对肾的作用:主要是促进磷的排出及钙的重吸收,进而降低血磷,升高血钙。它作用于肾远曲小管和髓袢上升段以促进钙的重吸收;抑制近曲小管及远曲小管对磷的重吸收,使尿磷增加。
此外,PTH促进肾活性维生素D的形成,它能促进肾25-(OH)-D3lα-羟化酶的活性增高,从而促进25-(OH)-D3lα-羟化作用。
PTH对小肠的作用是促进肠管对钙的重吸收,这一作用是通过活性维生素D来实现的。PTH刺激肾25-(OH)-D3lα-羟化酶,促进lα,25-(OH)-D3的生成,后者作用于小肠,促进小肠对钙和磷的吸收。
⑵降钙素:降钙素(calcitonin,CT)是由甲状腺滤泡旁细胞(parafollicularcell,C细胞)合成、分泌的一种单链多肽激素,由32个氨基酸残基组成,分子量3500。CT在初合成时是由136个氨基酸残基组成的分子量为15000的前体物。此前体物中还含有一个称为下钙素(katacalicin)的21肽肽段。当血钙增高时,降钙素及下钙素等分子分泌,下钙素能增强降钙素降低血钙的作用,血钙低于正常时CT分泌减少。CT作用的主要靶器官是骨、肾和小肠。CT对骨的作用是抑制破骨细胞活性,从而抑制骨基质的分解和骨盐溶解,同时抑制破骨细胞的生成,还有使间质细胞转变为成骨细胞的作用,结果促进骨盐沉淀,降低血钙。此外,它还抑制肾小管对磷的重吸收,以增加尿磷,降低血磷。
⑶维生素D:天然存在的维生素D有两种,即维生素D2(麦角钙化醇,erqocalciferol)及维生素D3(胆钙化醇,cholecalciferol)。维生素D2及D3具有相同的生理作用,且都必须在体内进行一定的代谢转变,成为活化型后才能发挥其生物学作用,肝和肾是维生素D活化的主要器官。
肝细胞微粒体中有维生素D3-25-羟化酶系,可在NADPH、O2和Mg2+参与下将维生素D3羟化生成25-(OH)-D3。在肝生成的25-(OH)-D3与血浆中特异的α2-球蛋白(D结合蛋白)结合,运输至肾,在肾近曲小管上皮细胞线粒体中的25-(OH)2-D3lα-羟化酶系(包括黄素酶.铁硫蛋白和细胞色素P450)的催化下,羟化生成lα,25-(OH)2-D3。后者具有较强的生理活化,其活性比维生素D3高10-15倍,被视为维生素D的活化型,并被当作激素。
1α,25-(OH)2-D3能反馈地抑制25-OH-D3-1α-羟化酶的活性,但可诱导肾25-OH-D3-24-羟化酶的合成。故当体内1α,25(OH)2-D3减少时,25-(OH)-D3倾向于合成高活性的1α,25(OH)2-D3;而当1α,25(OH)2-D3过多时,可形成低活性的1,24,25-(OH)2-D3,这对于防止体内活性维生素D产生过多,控制维生素D中毒有重要意义。
无机磷可抑制25(OH)-D3-1α-羟化酶系的活性,故当血磷降低时可促进1,25-(OH)2-D3的生成,血磷正常或增高时,25-(OH)-D3-1α-羟化酶系活性降低。甲状旁腺激素亦可促进1α,25-(OH)2-D3的生成;而降钙素则抑制此过程。
活性维生素D3作用的靶器官主要是小肠、骨和肾。
对小肠的作用:1α,25-(OH)2-D3具有促进小肠对钙、磷的吸收和转运的双重作用,即促进肠粘膜细胞膜对钙的通透、细胞内的结合及转运。钙主要在十二指肠吸收,肠粘膜上皮细胞刷状缘存在着可控制Ca2+通透的孔道,而在基底膜一侧,Ca2+向血液的转运是在Ca2+-ATP酶作用下的主动耗能过程。
1α,25-(OH)2-D3进入肠粘膜上皮细胞后,首先与细胞液中特异受体结合,然后起到下述生理作用:①与受体结合的1α,25-(OH)2-D3直接作用于刷状缘,改变膜磷脂的结构与组成(增加磷脂酰胆碱和不饱和脂肪酸含量),从而增加钙的通透性;②与受体结合的1α,25-(OH)2-D3进入细胞核,加快DNA转录mRNA,促进与Ca2+转运有关的蛋白质(钙结合蛋白,Ca2+-ATP酶)的生物合成;③与受体结合的1α,25-(OH)2-D3还刺激基底膜腺苷酸环化酶的活化。这样,进入细胞的Ca2+和cAMP都作为第二信使,发挥其调节作用。
在1α,25-(OH)2-D3的作用下,细胞内钙浓度升高,一部分Ca2+进入线粒体,钙结合蛋白多位于粘膜细胞的基底膜侧,它可从线粒体接受Ca2+,再将Ca2+转运到基底膜的钙泵上,将Ca2+输送至血液中。小肠粘膜还可通过Na+-Ca2+交换体系(需与Na+,K+-ATP酶相偶联)将Ca2+转运至血液。
1α,25-(OH)2-D3还能促进小肠粘膜细胞对磷的吸收。
对骨的作用:1α,25-(OH)2-D3对骨的直接作用是促进溶骨,用佝偻病动物实验证明:骨钙沉积于骨端软骨的作用也以1α,25-(OH)2-D3为最高。1α,25-(OH)2-D3与PTH协同作用下,加速破骨细胞的形成,增强破骨细胞活性,通过促进肠管钙、磷的吸收及促进溶骨,使血钙、血磷水平增高以利于骨的钙化。也就是说,1α,25-(OH)2-D3能维持骨盐溶解与沉积的对立统一过程,有利于骨的更新与生长。
对肾的作用:1α,25-(OH)2-D3对肾小管上皮细胞的作用是促进对钙、磷的重吸收,其机制也是增加细胞内钙结合蛋白的生物合成。
在正常人体内,通过PTH、CT、1α,25-(OH)2-D3三者的相互制约,相互协调,以适应环境变化,保持血钙浓度的相对恒定。现将上述三种激素对钙磷代谢的影响列于表6-2。
表6-2 三种激素对钙磷代谢的影响
| 激素 | 肠钙吸收 | 溶骨作用 | 成骨作用 | 肾排钙 | 肾排磷 | 血钙 | 血磷 |
| PTH | ↑ | ↑↑ | ↓ | ↓ | ↑ | ↑ | ↓ |
| CT | ↓(生理剂量) | ↓ | ↑ | ↑ | ↑ | ↓ | ↓ |
| 1α,25-(OH)2-D3 | |||||||
| ↑↑ | ↑ | ↑ | ↓ | ↓ | ↑ | ↑ |
↑升高:↑↑显著升高;↓降低
三、钙、磷代谢异常
钙磷代谢的异常包括高钙血症、低钙血症、高钙尿症、高磷血症以及低橉血症。现分别叙述如下。
(一)高钙血症
按病因学分类(表6-3)引起高钙血症(hypercalcemia)的原因包括溶骨作用增强、小肠钙吸收增加以及肾对钙的重吸收增加等。高钙血症是由于过多的钙进入细胞外液,超过了细胞外液钙浓度调节系统的调节能力或钙浓度调节系统的异常所致。较多见的是恶性肿瘤,其次是原发性甲状旁腺功能亢进症。
上述引起高钙血症的原因中,最多见的是由于溶骨作用而引起的高钙血症。PTH、前列腺素、破骨细胞激活因子(osteoclastactivationfactor,OAF)、甲状腺素、1α,25-(OH)2-D3等都可促进溶骨作用。现将几种常见的高钙血症略加说明。
⒈原发性甲状腺功能亢进和PTH异位分泌这两种情况都造成PTH过多,促进溶骨作用,又促进了维生素D的活化,间接地促进了肠管对钙的吸收,引起高钙血症。
⒉恶性肿瘤恶性肿瘤骨转移是引起血钙升高的最常见的原因。65%的乳腺癌病人有骨转移,多发性骨髓瘤和Burkitt淋巴肉瘤亦多有骨转移。这些肿瘤细胞可分泌破骨细胞激活因子,这种多肽因子能激活破骨细胞。肾癌、胰腺癌、肺癌等即使未发生骨转移亦可引起高钙血症,这与前列腺素(尤其是PGE2)的增多导致溶骨作用有关。
表6-3 高钙血症的病因学分类
| ⒈溶骨作用增强 | 原发性甲状旁腺功能亢进症 |
| PTH的异位分泌 | |
| 甲状腺功能亢进 | |
| 恶性肿瘤(白血病、多发性骨髓瘤等) | |
| 废用性骨萎缩 | |
| ⒉小肠钙吸收增加 | 维生素D摄入过量 |
| 维生素A摄入过量 | |
| 类肉瘤病 | |
| ⒊肾对钙重吸收增多 | 应用噻嗪类药物 |
| ⒋其他 | 乳-碱综合征 |
| 肾上腺功能不全(如艾迪生病) | |
| 急性肾功能不全 | |
| 婴儿特发性高钙血症 |
⒊维生素D中毒治疗甲状旁腺功能低下或预防佝偻病而长期服用大量维生素D可造成维生素D中毒,维生素D在体内的半寿期长,中毒可持续数月,高钙高磷血症引起头痛恶心等一系列症状及软组织和肾的钙化。
⒋甲状腺功能亢进甲状腺素具有溶骨作用,中度甲亢病人约15%-20%伴有高钙血症。
(二)低钙血症
低钙血症(hypocalcemia)的病因学分类见表6-4。
表6-4 低钙血症的病因学分类
| ⒈溶骨作用减弱,成骨作用增强 | 甲状旁腺功能低下(原发性,手术) |
| 假性甲状旁腺功能低下 | |
| 甲状腺功能亢进病人手术后 | |
| 低镁血症(酒精中毒,吸收不良综合症) | |
| 恶性肿瘤骨转移(前列腺癌) | |
| ⒉肠管钙吸收的抑制 | 维生素D缺乏 |
| 维生素D摄入不足或紫外线照射不足 | |
| 吸收不全综合征 | |
| 维生素D活化受阻 | |
| ⒊其他 | 低白蛋白血症(肾病综合征) |
| 急性胰腺炎 | |
| 妊娠、肾功能不全 | |
| 大量输血 |
甲状旁腺功能低下可因甲状旁腺或甲状腺手术的失误所引起。由于PTH的分泌减少,骨中破骨细胞减少,成骨细胞增加,造成一时性低钙血症。假性甲状旁腺功能低下主要是由于PTH的靶器官受体异常,对PTH应答不良所引起。低镁血症时PTH的分泌减少,也可由于引起PTH靶器官对PTH反应低下而造成低钙血症。食物中维生素D缺少或紫外线照射不足,或消化系统疾病造成维生素D吸收障碍都可引起维生素D缺乏性佝偻病。此时血循环中的活性维生素D减少,引起肠钙吸收减少及血钙降低。血钙降低又刺激PTH的分泌,促进骨钙动员和增加肾小管对钙的重吸收,通过此种代偿机制维持血钙正常水平。
急性胰腺炎时机体对PTH的反应性降低,CT和胰高血糖素分泌亢进。急性胰腺炎时的低血钙与溶骨作用的抑制可能有关。
肾功能不全时由于肾功能低下而造成活性维生素D产生不足,结果PTH对溶骨的促进作用降低,造成低钙血症。
低血钙时神经、肌肉兴奋性增加,外界刺激可引起肌肉痉挛、手足搐搦。维生素D缺乏引起的佝偻病可表现方头、O形或X形腿、鸡胸及念珠胸,血清碱性磷酸酶可因软骨细胞增加而活性增高,可高达50-60布氏单位(正常值为5-15布氏单位)。
(三)高磷血症
高磷血症(hyperphosphoremia)主要是由于肾排出减少,溶骨作用亢进,磷摄入过多,磷向细胞外移出及细胞破坏等原因所造成。临床上常伴有血钙降低的各种症状和软组织的钙化现象。高血磷的病因可见表6-5。
表6-5 高磷血症
| ⒈摄入磷过多 | 摄取磷过多 |
| 小肠磷吸收亢进(维生素D过量) | |
| 使用含磷缓泻剂 | |
| 磷酸盐静注 | |
| ⒉磷向细胞外移出或组织破坏 | 呼吸性酸中毒 |
| 糖尿病性酮症、酸中毒 | |
| 乳酸性酸中毒 | |
| 骨骼肌破坏 | |
| 高热 | |
| 恶性肿瘤(化疗) | |
| 淋巴性白血病 | |
| ⒊肾排磷减少 | 肾功能不全(急性、慢性) |
| 甲状旁腺功能低下 | |
| 原发性甲状旁腺功能低下 | |
| 继发性甲状旁腺功能低下 | |
| 假性甲状旁腺功能低下 | |
| 甲状腺功能亢进症 | |
| 肢端肥大症 | |
| ⒋其他 | 镁缺乏 |
| 细胞外液量减少 | |
| 家族性间歇性高磷血症 |
⒈急、慢性肾功能不全肾小球滤过率在20-30ml/min以下时,肾排磷减少,血磷上升,血钙降低,PTH的分泌增多。肾的病变使1α,25-(OH)2-D3的生成减少,血浆钙浓度降低,也有利于高磷血症的发生。
⒉甲状旁腺甲状旁腺功能低下,尿排磷减少,导致血磷增高。
⒊维生素D中毒由于维生素D的活性型促进溶骨,并促进小肠对钙、磷的吸收以及肾对磷的重吸收,因而维生素D中毒时伴有高磷血症。
⒋甲状腺功能亢进同时出现高钙血症与高磷血症,这与溶骨作用亢进有关。
⒌肢端肥大症活动期可出现高磷血症。生长激素可促进小肠对钙的重吸收,增加尿钙排出,减少尿磷排泄,导致血磷增高。
(四)低磷血症
血清无机磷的降低即低磷血症(hypophosphoremia)可由于小肠磷吸收减低、尿磷排泄增加、磷向细胞内转移等所引起,引起低磷血症的病因学分类见表6-6。
表6-6 低磷血症
| ⒈小肠磷吸收减低 | 对磷摄取不足,饥饿、呕吐 |
| 1α,25-(OH)2-D3不足〔维生素D缺乏、维生素D活化障 | |
| 碍,对1α,25-(OH)2-D3感受性低下〕 | |
| 吸收不良综合征 | |
| 结合磷酸的制酸剂(氢氧化铝凝胶、碳酸铝、氢氧化镁等) | |
| ⒉尿磷排泄增加 | 急性乙醇中毒 |
| 甲状旁腺功能亢进症(原发性、继发性) | |
| 肾小管性酸中毒 | |
| Fanconi综合征 | |
| 维生素D抵抗性佝偻病 | |
| 代谢性酸中毒 | |
| 糖尿病 | |
| 糖皮质类固醇投予 | |
| 利尿剂投予 | |
| ⒊磷向细胞内转移 | 摄取糖类 |
| 静注葡萄糖、果糖、甘油 | |
| 高热量输液 | |
| 营养恢复综合征 | |
| 过度换气综合征(呼吸性碱中毒) | |
| 应用胰岛素、乳酸钠 | |
| 投予水杨酸 | |
| 应用雄性激素 | |
| 严重烫伤恢复期投予葡萄糖 |
⒈甲状旁腺功能亢进症无论原发性或继发性甲状旁腺功能亢进均可使PTH过多,无机磷随尿排出增多,造成低磷血症。
⒉维生素D缺乏维生素D摄入减少或肾1α-羟化酶受抑制,1α,25-(OH)2-D3合成不足,均可引起小肠磷吸收降低,尿排磷增加,导致低磷血症。
⒊肾小管性酸中毒与Fanconi综合征肾小管性酸中毒时由于H+排出受阻导致钙磷代谢障碍,出现骨软化症、肾钙化、肾结石等。酸中毒可抑制肾小管对钙的重吸收,出现低钙血症、高PTH和低磷血症。Fanconi综合征表现为高钙血症、低磷血症和高碱性磷酸酶血症。低磷血症是由于肾小管对磷的重吸收显著减低所致。病人尿中大量排磷,即使血磷很低,尿磷仍很高。
四、尿路结石症的生物化学
尿路结石症是地区性常见病、多发病。我国尿石症的多发地区是广东、广西、贵州、云南、四川、福建、台湾、浙江、江西、安徽、湖南、湖北、陕西、河南东部、山东、苏北、吉林双城堡等地。
尿石症大体可分为上尿路结石症(肾结石、输尿管结石)与下尿路结石症(膀胱结石、尿道结石等)。随着工农业生产的发展和生活水平的提高,上尿路结石就诊率明显升高,下尿路结石明显下降。
(一)尿路结石的化学组成及结构特点
尿石一般由三部分组成:①中心部或核;②沉积的晶体物质;③基质。
尿石的核心通常位于尿石的中心,是尿石生成中最早出现的部分。核心的组成常能反映当时体内的代谢变化。对尿石核心的分析对探讨尿石的成因也有一定价值。有的尿石核心由晶体物质及基质组成,也有的尿石核心是凝血块、细胞碎屑、管型、菌落、异物等。
尿石的基质是各种尿石共有的成分,是由粘蛋白类构成,基质在含钙尿石中占结石总量的2.5%左右,在胱氨酸结石中基质约占9%-11%,而在罕见的基质结石中基质可达结石总重量的60%左右。基质作为结石的网架,晶体沉积于其上。
尿石的晶体物质种类较多,其性质及来源各不相同,尿石的晶体成分见表6-7。
表6-7 尿石的晶体成分
| 成分 | 分子式 |
| 一水草酸钙 | CaC2O4·H2O |
| 二水草酸钙 | CaC2O4·2H2O |
| 磷酸镁铵 | MgNH4PO4·6H2O |
| 碳酸磷灰石 | Ca10(PO4,CO3OH)6(OH)2 |
| 羟基磷灰石 | Ca10(PO4)6(OH)2 |
| 二水磷酸氢钙 | CaHPO4·2H2O |
| 尿酸 | C5H4N4O3 |
| 尿酸钠 | NaHC5H2N4O3·H2O |
| 尿酸铵 | NH4HC5H2N4O3·H2O |
| 磷酸三钙 | Ca3(PO4)2 |
| 胱氨酸 | [-S.CH2CH(NH2)·COOH]2 |
| 黄嘌呤 | C5H4N4O2 |
一种尿石可含一种或数种晶体成分,据国内有关报告初步统计,我国国内尿路结石以含草酸钙的混合结石最为多见,个别地区尿酸结石较其他地区为高。尿石分析可用化学分析方法、微量重结晶分析法、X线粉晶分析、红外线光谱分析、偏光显微镜分析等。
尿石的结构及其意义见表6-8。
(二)尿路结石的成因及形成机制
尿路结石的成因问题颇为复杂,它涉及结石成分由尿中排泄增加、尿液pH的变化、环境及某些生活习惯、营养因素、神经因素及尿液淤滞等。
表6-8 尿石的结构特点及其意义
| 结构 | 组成成分 | 意义 |
| 核心 | 基质与晶体成分(草酸盐、磷酸 | 为尿石生成中最早出现的部分,肾小管中形成 |
| 盐)、血块、细胞碎屑管型、菌 | 的微石进入肾盏,长大成为尿石 | |
| 落、异物等,核心一个或多个 | ||
| 同心层 | 由晶体及基质组成年轮样结构,基质也 | 在草酸钙结石及尿酸结石中同心层结构明 |
| 具有同心层结构,形成结石网架 | 显 | |
| 放射条纹 | 与同心层相垂直,长度相当于1个同心 | 只存在于一水草酸钙、尿酸或磷酸氢钙等单一 |
| 层的宽度,由纤维状基质组成 | 成分区域,与同心层并存 | |
| 小球体 | 每个尿石中皆有不同数目的1mm左右 | 小球体即微小结石,尿石可为多个小球体的聚 |
| 的小球体存在,由基质及晶体组成 | 合体,说明微小结石不断从肾乳头脱落,聚集 | |
| 成尿石 |
⒈高钙尿症尿钙的增高与含钙尿石的形成有密切关系,但是高钙尿症并非形成尿石的唯一决定性因素。许多尿石患者尿钙含量正常,高钙尿症也不一定都合并尿石症。只是在原发性甲状旁腺功能亢进症、肾小管性酸中毒、特发性高钙尿症等情况下较多见尿石的发生。
⒉草酸代谢紊乱草酸钙是尿路结石的主要成分。尿中排泄的草酸约有40%来自富含草酸的食物,还有一半是来自体内氨基酸(甘氨酸、丝氨酸、羟脯氨酸)和维生素C的代谢转变。原发性高草酸症是先天性的酶缺陷所致,包括Ⅰ型(α-酮戊二酸-乙醛酸裂合酶缺陷)和Ⅱ型(D-甘油酸脱氢酶缺陷)。原发性高草酸尿症Ⅰ型由前述三种氨基酸代谢生成的乙醛酸难于进一步转变成α-羟基β-酮基已二酸,因而造成乙醛酸堆积,由乙醛酸生成草酸增加。原发性高草酸尿症Ⅱ型则表现为由丝氨酸生成的羟基丙酮酸堆积,导致L-甘油酸和草酸的生成增加。
维生素B6及维生素B1构成上述氨基酸代谢酶的辅酶,因而当维生素B6与B1缺乏时亦可导致草酸代谢紊乱,即乙醛酸向氨基酸的转化发生障碍而转向草酸的生成,因而促进草酸钙结石的生成。草酸代谢紊乱与镁的缺乏也有密切的关系,动物实验表明,饲以缺镁食饵即可形成草酸钙结石,而且这种结石形成过程可因投予镁而被阻止。据认为镁的吸收可以抑制钙的吸收,镁离子可使尿中草酸钙的溶解度显著增高,有人曾主张测定尿中镁/磷比值,若镁/磷比值在0.1以下即可认为是“镁失调尿”,他们认为“镁失调尿”是草酸钙结石形成的重要条件。
内生性草酸的35%-50%是由维生素C转变而来。维素C(L-抗坏血酸)在体内首先被氧化成L-脱氢抗坏血酸(在细胞色素、分子氧等参加下),再经内酯酶作用而生成2,3-二酮古洛糖酸,后者再分解成草酸和L-苏阿糖酸。大量服用维生素C可引起内生性草酸的大量生成而有发生草酸钙结石的可能,动物实验也表明了这一点。
⒊尿酸代谢紊乱尿酸是体内嘌呤代谢的产物。发生高尿酸血症的原因在于组织中核酸分解破坏的增加以及摄取富含嘌呤及核酸食物的增加。约25%的痛风病人有尿酸结石,尿酸结石病人的25%合并痛风。高尿酸血症可同时出现高尿酸尿症。
随着鱼肉类食品及乙醇饮料摄取量的增加,痛风及无症状性高尿酸血症有增加的趋势。痛风已被看做是以原发性高尿酸血症为基础、以关节炎发作为主要症状,合并肾损害(痛风肾)、高血压、脂类及糖代谢异常、肥胖等的全身性代谢异常的疾病。其尿液pH有强烈的偏酸的倾向,因而易形成尿酸结晶和尿酸结石。血清及尿中尿酸浓度越高,肾结石发生率也越高。尿pH越低,肾结石发生率亦越高。尿酸在偏酸(pH6以下)条件下易处于过饱和状态,而利于尿酸结石的形成。
黄嘌呤是嘌呤代谢的中间产物,溶解度低。黄嘌呤结石是由于黄嘌呤氧化酶的缺陷而致嘌呤代谢障碍,尿中黄嘌呤沉积而成为尿石。
⒋胱氨酸代谢障碍胱氨酸尿症是肾及肠道中氨基酸转运的先天性异常。尿中胱氨酸、鸟氨酸、赖氨酸等排出增加,可出现复发性尿路结石。
⒌尿pH与结石形成的关系(表6-9)。
表6-9 成石物质的溶解度与尿pH的关系
| 成石物质 | 酸性尿中情况 | 碱性尿中情况 |
| 尿 酸 | (pH5.0附近)沉淀 | (pH6.5以上)溶解 |
| 胱氨酸 | (pH4.5-7.0)沉淀 | (pH7.5-7.8)溶解 |
| 草酸钙 | (pH6.0以下)沉淀 | (pH>7)沉淀 |
| 磷酸钙 | (pH6.0以下)溶解 | (pH>7)沉淀 |
| 磷酸镁铵 | (pH6.0以下)溶解 | (pH>7)沉淀 |
由表6-9可以看出不同成分尿石的形成是在不同的pH下进行的,在偏酸性的尿中易形成尿酸及胱氨酸结石;在偏碱性尿中易形成磷酸钙及磷酸镁铵结石。
⒍尿中结晶抑制物的缺乏尿中成石物质的浓度一般都大于其溶解度,因而都处于过饱和状态。在正常尿中存在着一些作用很强的结晶抑制物-焦磷酸盐、粘多糖、柠檬酸、镁离子、尿素、丙氨酸、锰离子在及锌离子等。尿石症病人尿中由于缺乏某些结晶抑制物,导致其尿液的结晶倾向增高。例如草酸钙结石病人尿镁浓度的减低,尿石病人尿液柠檬酸排出量减低等均是这方面的例证。
⒎结石基质在尿路结石形成中的作用尿石形成的基质学说认为是基质前身物先转化为基质,再以基质为模板吸附或粘着晶体物质而形成结石。在结石形成者的尿中,粘多糖的硫酸化程度比正常尿中的为高。硫酸粘多糖上的硫酸根可能起着在肾内形成难溶性钙盐的作用。因此,高度硫酸化的粘多糖看来是含钙结石形成的重要因素,基质物质A为基质主要成分。
⒏其他因素关于水质硬度与尿石发生的关系报告尚不一致,广东东莞、花县以及贵州六盘水地区尿石的发生与水质硬度有关。维生素A缺乏易导致过饱和的碱性尿液,还可使上皮脱落,刺激基质蛋白的分泌。精神紧张与交感神经反射的刺激易引起肾石发生。尿石病人尿中尿激酶活性比正常人低,而尿激酶能促进纤维蛋白溶解,保持尿路的通畅。
关于各种尿石的生化成因及特点列于表6-10
(三)尿中防治的临床生化(表6-11)
表6-10 各种尿石的生化成因及特点
| 结石种类 | 生化成因 | 尿液及结石的特点 |
| 草酸钙结石 | 原发性高草酸尿症(先天性酶缺陷) | 尿钙高 |
| 甲状旁腺功能亢进 | 尿草酸增高 | |
| 尿镁及尿磷降低 | ||
| 继发性高草酸尿(膳食性) | 尿pH5.0-6.5 | |
| 维生素B6、B1缺乏,Mg2+缺乏,回肠 | 尿中有草酸钙结晶 | |
| 切除,胆盐重吸收减弱,胆盐分解放出 | 成堆现象 | |
| 甘氨酸,转化成草酸,形成草酸盐结 | 尿沉淀加盐酸溶解 | |
| 石,多数特发性草酸钙石原因未明 | 尿石同心层明显,二水草酸 | |
| 钙白而脆,一水草酸钙褐色坚硬 | ||
| 磷酸钙结石 | 特发性高钙尿症 | 尿钙增高 |
| 甲状旁腺功能亢进症 | 尿pH>7.0 | |
| 肾小管性酸中毒,长期卧床 | 尿沉淀和醋酸溶解 | |
| 骨肿瘤引起高钙尿症尿液 | 结石白色松脆 | |
| 偏碱,尿中粘蛋白改变等 | 碳酸磷灰石加HCl有气泡生成 | |
| 磷酸镁铵结石 | 尿路感染,细菌尿素酶将尿素分解为氨 | 尿细菌培养阳性 |
| 与CO2形成碳酸铵,再与镁及磷酸盐形 | 尿可见磷酸镁铵结晶尿pH>7.0 | |
| 成磷酸镁铵沉淀 | 尿沉淀加醋酸溶解 | |
| 尿酸结石 | 高尿酸尿症 | 尿中尿酸可增高 |
| 尿pH偏酸,尿酸沉积 | 尿液pH<5 | |
| “特发性尿酸结石”机制未明 | 尿沉淀加碱溶解 | |
| 尿中可见尿酸结晶 | ||
| 尿石呈金黄色,同心层明显 | ||
| 胱氨酸结石 | 胱氨酸、鸟氨酸、精氨酸、赖氨酸在肾 | 尿中胱氨酸增加 |
| 及小肠转运障碍,肾重吸收障碍,尿中 | 尿pH偏低 | |
| 大量排泄胱氨酸,沉积而成为胱氨酸结 | 尿中可有胱氨酸结晶 | |
| 石 |
表6-11 防治尿石的生化原理
| 结石种类 | 防治药物 | 防治尿石的生化原理 |
| 氧化镁 | 纠正镁失调,Mg2+与Ca2+竞争草酸根,阻止尿石生长,增 | |
| 加草酸钙溶解度 | ||
| 草酸钙结石 | 维生素B6 | 纠正氨基酸代谢紊乱,降低内生性草酸 |
| 无机磷酸盐 | 增加尿中焦磷酸盐,抑制结石生长 | |
| 亚甲蓝 | 阻止基质与晶体结合,妨碍基质的产生, | |
| 磷酸纤维素 | 聚合与钙形成不吸收的复合物,降低尿钙 | |
| 酪氨酸 | 降低内生性草酸,阻断羟脯氨酸生成草酸 | |
| 别嘌呤醇 | 抑制尿酸生成,防止草酸钙结合长大 | |
| 四环素 | 与金属离子螯合,与亚甲蓝作用类似 |
续表
| 结石种类 | 防治药物 | 防治尿石的生化原理 |
| 磷酸钙结石 | 氢氧化铝胶 | 在肠道与磷形成不溶性磷酸铝减少吸收 |
| 柠檬酸 | 在尿中与钙形成可溶性物质 | |
| 葡萄糖醛酸 | 形成葡萄糖醛酸甙,利于钙结石溶解 | |
| 萜烯类 | 与葡萄糖醛酸结合成葡萄糖醛酸甙 | |
| 酸性磷酪盐与NH4+Cl- | 酸化尿液,促进磷酸钙溶解,降尿钙 | |
| 磷酸纤维素 | 与钙形成不吸收的复合物,降低尿钙 | |
| 磷酸镁铵结石 | 抗生素 | 控制尿路感染,减少尿中尿素酶及尿铵 |
| 乙酰羟肟酸 | 尿素酶抑制剂,减少尿素分解 | |
| 丙氨酸 | 防止尿粘蛋白的聚合 | |
| 尿酸结石 | 别嘌呤醇 | 抑制体内黄嘌呤氧化酶,从而抑制尿酸生成 |
| 柠檬酸盐 | 碱化尿液至pH6.5,使尿酸结石溶解 | |
| 胱氨酸结石 | 青霉胺 | 青霉胺与半胱氨酸结合,形成溶解度很大的复合 |
| 物,减少胱氨酸的生成 | ||
| 柠檬酸盐 | 碱化尿液至pH7.5,促进胱氨酸结石的溶解 |
第二节 镁代谢及其异常
镁在人体内的总量约为21-28g,居构成机体元素的第11位。Mg2+是体内含量较多的阳离子(居第4位)。体内57%的镁存在于骨中,约40%存在于软组织中,其余存在于体液中(表6-12)。
表6-12 新鲜组织和体液中镁的浓度
| 组织 | 浓度(mg/kg) | 体液 | 浓度(μg/L) |
| 骨 | 18.00 | 唾液 | 10.8-65.0 |
| 脑 | 4.83 | 胃液 | 25-590 |
| 骨骼肌 | 1.92 | 胆汁 | 50-183 |
| 心 | 1.92 | 胰液 | 25-33 |
| 胰腺 | 1.83 | 肠液 | 483 |
| 肝 | 1.67 | 脑脊液 | 200 |
| 小肠 | 1.67 | 乳汁 | 192-208 |
| 肾 | 1.50 | 汗液 | 0.25-19.2 |
一、镁在体内的动态及其生理功用
(一)镁的吸收与排泄
镁存在于除脂肪以外的所有动物组织及植物性食品中,日摄入量约为250mg,其中2/3来自谷物和蔬菜。小肠对镁的吸收是主动转运过程,吸收部位主要在回肠。每日镁的吸收量约为2-7.5mg/kg体重,未吸收部位随粪便排出。摄入量与排出量存在一定正比关系。消化液中也含有多量的镁,成人每日可从消化液的吸收过程中回收约35mg的镁。长期或短期大量丢失消化液是造成缺镁的主要原因。消化道手术或造瘘术后未及时补充镁,便会出现缺镁综合征。
体内镁的主要排泄途径是肾,每日经肾小球滤过镁1.8g,再由肾小管(特别是髓袢)将大部分滤过的镁重吸收,仅有2%-5%由尿排出。男性每日由尿排镁100mg,女性为90mg,分别相当于每排出1mg肌酐排出0.068mg及0.076mg的镁。镁的排泄量因摄入量不同及地区差异而不同。红细胞中的镁约为血清镁的3倍,故测血清镁时应防止溶血。
(二)血镁
血清镁:正常人血清镁约为0.81mmol/L(0.75-1.00mmol/L),男略高于女。血清镁若低于0.75mmol/L即为低镁血症,高于1.0mmol/L即为高镁血症。血清镁有三种存在形式:Mg2+约占血清总镁量的55%;与重碳酸、磷酸、柠檬酸等形成的镁盐约占15%;蛋白结合镁约占30%。前两类属于可滤过镁,离子镁具有生理活性,红细胞镁可作为细胞内镁的指标进行测定,其结果可用于了解镁在体内的动态,正常人每升红细胞中含镁56mg。
(三)肌肉镁
在核细胞中存在的镁约有80%存在于肌肉中。肌肉是维持镁平衡的重要组织。急性镁缺乏实验所引起的低镁血症不波及肌肉镁。慢性镁缺乏病人虽然血镁在正常范围,但肌肉镁却显著降低。急、慢性高镁血症时肌肉镁均不增加。
(四)镁的生理功能
镁一半以上沉积于骨中。Mg2+对神经、肌肉的兴奋性有镇静作用,血清Mg2+与血清Ca2+在生理作用上有相互拮抗的关系。Mg2+是近300种酶的辅助因子,Mg2+与ATP分子的β-和γ-磷酸基构成螯合物,降低ATP分子的电负性,参与一切需要ATP的生化反应。Mg2+还通过与磷酸基的络合作用维持DNA双螺旋的稳定性。Mg2+还参与维持tR-Na和核蛋白体的构象。核蛋白体和mRNA及氨基酰tRNA之间的相互作用需要Mg2+,Mg2+还参与氨基酸的活化、核蛋白体循环中转肽及核蛋白体移位等重要步骤。因此Mg2+与体内重要的生物高分子蛋白质、核酸、酶的结构、代谢与功能都有密切关系,在维持机体内环境的相对稳定和维持机体的正常生命活动中起着重要的作用。
二、镁代谢异常
镁代谢异常包括低镁血症(包括镁缺乏)和高镁血症两方面。低镁血症时处于镁缺乏状态。其原因可能是镁摄取不足,吸收不良,由肾脏丧失,镁向细胞内移动,经消化道等途径丧失等。高镁血症的原因可能是外因或内因性镁负荷的增加或肾对镁排泄的障碍,较为多见的则是肾功能不全时投予镁制剂。
镁代谢异常的病因学分类见表6-13
(一)低镁血症与镁缺乏症
低镁血症的发病原因较多,且常伴有其他电解质的紊乱。
⒈肾疾病 肾疾病可因肾小管对镁的重吸收能力降低而引起低镁血症。钠利尿同时也伴有镁排出增多。急性肾功能不全多尿期可出现低镁血症,髓袢利尿剂、噻嗪类利尿剂、渗透压利尿剂引起利尿时均可造成尿镁排出增多,因此,长期服用利尿剂的慢性心功能不全病人和高血压病人可同时发生镁与钾的缺乏。
⒉内分泌紊乱 原发性醛固酮症时尿中排镁增加。原发性甲状旁腺功能亢进时可出现症候性镁缺乏症,这是由于高钙血症导致肾保留镁的能力降低所致,此时尿镁的丢失增加。肠管对钙和镁的吸收具有相互竞争作用,肠管对钙的吸收增加,势必造成镁吸收的降低。糖尿病酸中毒用胰岛素治疗,由于镁向细胞内转移,可造成低镁血症。
表6-13 引起镁代谢异常的疾病
| 低镁血症(镁缺乏) | 高镁血症 | |
| 肾脏 | 肾盂肾炎,肾盂积水,肾硬化症,肾小管性酸中毒,肾病综合征,高度利尿,急性肾功能不全多尿期,肾移植后利尿期 | 急性肾功能不全少尿期,慢性肾功能不全末期(尿毒症)、血液透析 |
| 内分泌紊乱 | 原发性醛固酮症,甲状腺功能亢进症,甲状旁腺功能亢进症,糖尿病酮症酸中毒(胰岛素治疗) | 艾迪生病,肾上腺摘除,甲状腺功能低下症糖尿病酸中毒 |
| 消化系 | 慢性腹泻,脂肪性腹泻,长期呕吐,小肠切除,消化液长期引流,消化道造瘘,急性胰腺炎,酒精中毒性肝硬化,蛋白-能量营养不良综合征,镁摄入不足等 | 先天性巨结肠症 病毒性肝炎 投予含镁的制酸药 缓泻剂等 |
| 恶性肿瘤 | 溶骨性恶性肿瘤,肾上腺皮质癌,胸腺淋巴肉瘤,骨癌,慢性骨髓性白血病 | 慢性淋巴细胞白血病多发性骨髓瘤,淋巴瘤 |
| 神经系 | 癫痫、帕金森病、肌营养不良症、慢性酒精中毒,Huntington舞蹈病 | |
| 循环系 | 动脉粥样硬化症、心肺旁路 | |
| 药物 | 利尿剂、泻剂长期使用,抗痉挛剂、大量的低镁输液,毛地黄中毒,大量氯化铵,细胞毒性药物,庆大霉素、维生素B1过量 | 含镁泻剂过量,锂治疗维生素D过剩,含镁制酸剂,抗醛固酮剂使用,生长激素等 |
| 其他 | 妊娠、长期哺乳、特发性低镁血症,饥饿,寻常性鱼鳞癣,急性间歇性卟啉症 | 乳-碱综合征,低温麻醉,慢性感染,血液浓缩,组织破坏等 |
⒊消化系统疾病 镁在小肠及一部分结肠被吸收。脂肪泻时由于消化管内的镁与脂肪结合成不能被吸收的碱性复合物而引起低镁血症。镁在肠管中吸收较慢,其吸收速度与镁在肠管中通过的时间及肠管内镁的浓度成比例关系。慢性腹泻、小肠切除均可因镁在肠管内通过时间过短而导致吸收减少。每升消化液中含镁12mg,引流、瘘管等造成的消化液的丢失也是缺镁的重要原因。下消化道肠液中镁的浓度较高,溃疡性结肠炎、细菌性肠炎、蛋白-能量营养不良综合征以及长期服用泻剂都可引起镁的丢失过多。
此外,一些抗生素如庆大霉素可因肾的损害造成大量钾和镁的丢失。慢性酒精中毒可造成大量镁的丢失。各种原因引起的饥饿状态下,尿镁的排出并不因进食不足而减少,肾不具备防止体内镁丢失的防御机制。不含镁的高能量输液有症状性缺镁的可能,至少每日补充96mg的镁才有可能抵消肾镁的丢失。
缺镁的主要表现是神经-肌肉障碍和精神与行动的异常,表现为乏力、衰弱感、体温调节不良。严重时可有神经过敏、震颤、搐搦、肌肉痉挛、眼颤以及吞咽困难等。
尿镁含量测定发现,尿镁低于36mg/L(0.88mmol/L),用镁补充治疗,其尿镁也不增加时,可以协助诊断镁缺乏症。简化的镁平衡试验是:对病人做初步的肾功能检查后,静脉输入40mmol硫酸镁(约4.8g),于60-90分钟内输完,由注射开始收集48小时尿液。肾功能正常的病人,48小时内至少排出35mmol的镁(约4.2g硫酸镁)。病人体内缺镁时,排出量可低于25mmol(相当于3g硫酸镁),这种方法可协助镁缺乏的诊断。
(二)高镁血症
肾功能不全(尿素症)及急性肾功能不全少尿期,由于肾清除作用降低,血浆及红细胞内镁含量均增高,可出现高镁血症。当肌酐清除率在30ml/min以下时,血镁含量显著增高。高镁血症的临床表现有心动过速、各种心传导阻滞、血压降低、腱反射消失、肌肉瘫软、啫唾,重者转入昏迷。治疗高镁血症不外乎改善肾功能、加速镁的排泄,可用钠利尿剂以增加尿镁的排泄,还可给以葡萄糖钙等钙剂以与镁相拮抗。钙、镁两种离子由于化学上甚为接近,因此可与同一神经细胞的生化系统相互竞争,增加Ca2+浓度就能排斥Mg2+与该生化系统的结合,从而起到治疗的作用。
第三节 微量元素的作用及其与疾病的关系
微量元素在当代以其广泛的生物学作用、生理功能及临床诊断治疗价值吸引了地球化学、医学地理、农业、环境保护、分子生物学、生物化学、营养学、免疫学、遗传学、药理学、地方病学、老年医学、肿瘤学等许多基础医学及临床医学领域的学者,并取得许多令人瞩目的进展。微量元素在许多疾病的病因学、发病学、诊断学、防治学方面具有重要的意义。
人体内必需的微量元素有铁、锌、铜、锰、铬、钼、钴、硒、镍、钒、锡、氟、碘、硅;非必需的微量元素中属于可能必需的有铷、砷、锶、硼、锗;属于无害的则有钡、钛、铌、锆等;有害的微量元素有铋、锑、铍、镉、汞、铅、铝等。上述微量元素(占人体总重量的1/10000以下者)与宏量元素(占人体总重量1/10000以上者,包括碳、氢、氧、氮、钙、磷、镁、钠、钾、氯、硫)相比,只占人体总重量的0.05%左右。
一、微量元素的分布及其生理功用
(一)人体内微量元素的含量(表6-14)
表6-14 人体内微量元素的含量
| 微量元素 | 体内存在量 | 微量元素 | 体内存在量 |
| 铝(Al) | 60mg | 锰(Mn) | 12-20mg |
| 砷(As) | 1-2mg | 汞(Hg) | 13mg |
| 钡(Ba) | 20mg | 钼(Mo) | 10mg |
| 镉(Cd) | 50mg | 镍(Ni) | 10mg |
| 铬(Cr) | 6mg | 铷(Rb) | 320mg |
| 钴(Co) | 1.5mg | 硒(Se) | 6-12mg |
| 铜(Cu) | 80-100mg | 硅(Si) | 2-3g |
| 氟(F) | 3g | 锶(Sr) | 320mg |
| 碘(I) | 11mg | 锡(Sn) | 17mg |
| 铁(Fe) | 3-5g | 钒(V) | 1.5mg |
| 铅(Pb) | 120mg | 锌(Zn) | 2-3g |
(二)微量无素的生理功用
⒈微量元素与酶的关系 酶是一种生命现象及生物化学反应的基础。人体内发现的近1000种酶中有50%-70%的酶含有微量元素或以微量元素的离子作为激活剂。已知锌与上百种酶有关,铁与数十种酶有关,锰和铜亦与数十种酶有关。钼与黄嘌呤氧化酶等有关,硒与谷胱甘肽过氧化物酶等有关。因此微量元素常作为酶的组成成分或激活剂。
⒉微量元素构成体内重要的载体及电子传递系统 铁参与组成血红蛋白、肌红蛋白,运输和储存氧;铁构成的细胞色素系统(细胞色素b、C1、C、aa3,b5、P450等)是重要的电子传递物质;铁硫蛋白作为呼吸链中的电子传递体。
⒊参与激素和维生素的合成 钴组成维生素B12,碘构成甲状腺激素T3、T4,因而微量元素与代谢的调控有密切关系。
⒋微量元素影响免疫系统的功能,影响生长及发育 锌影响生长发育,能增强免疫功能,硒能刺激抗体的生成,增强机体的抵抗力。
二、微量元素与疾病的关系
不论必需微量元素缺乏或过多,有害微量元素接触、吸收、贮积过多或干扰了必需微量元素的生理功能和营养作用,都会引起一定的生理及生物化学过程的紊乱而发生疾病。反之,在各种疾病情况下,会对微量元素的吸收、运输、利用、储存和排泄产生一定的影响。
微量元素缺乏或过多,可导致某些地方病的发生。例如缺碘与地方性甲状腺肿及呆小病有关;低硒与克山病和大骨节病有关;缺锌与伊朗乡村病和肠原性肢端皮炎有关。接触或吸收过量的有害微量元素还可引起种种职业病,即使是必需微量元素,像铁、铜、钴、锰等进入机体过多也会引起急性或慢性中毒。如接触六价铬可引起特征鹰眼状铬溃疡及鼻中隔穿孔;砷过多引起砷性皮肤癌及中毒;还有锰中毒、铁中毒、锌中毒等。
微量元素的检测还可用作某些疾病的诊断指标,对于某些微量元素缺乏症还可用补充微量元素的方法进行治疗。
(一)微量元素的缺乏症与过多症
⒈微量元素的缺乏症 微量元素的缺乏可导致缺乏症的出现,现将重要的微量元素缺乏症列于表6-15
表6-15 微量元素的功能与缺乏症
| 微量元素 | 功能 | 缺乏症 |
| 锌(Zn) | 细胞分裂,核酸代谢,生长,辅助因子 | 伊朗乡村病,肠原性肢端皮炎 |
| 铁(Fe) | 造血原料,构成细胞色素类 | 缺铁性贫血 |
| 铜(Cu) | Hb合成,结缔组织代谢 | 贫血,毛发异常,脑功能障碍 |
| 铬(Cr) | 构成葡萄糖耐量因子 | 耐糖能力↓,糖尿病,动脉硬化 |
| 碘(I) | 构成T3、T4,促进生长、发育 | 地方性甲状腺肿,呆小病 |
| 钴(Co) | 构成维生素B12,造血 | 恶性贫血,甲基丙二酸尿症 |
| 硒(Se) | 抗氧化 | 克山病,大骨节病 |
| 锰(Mn) | 多种酶的激活剂 | 生长发育迟缓,中枢神经系异常 |
| 氟(F) | 骨、牙齿形成 | 龋齿 |
⒉微量元素的过多症 例如铁过剩的血色沉着病(hemochromatosis)时,铁吸收过多,在心、胰腺、睾丸、肝内沉积,导致纤维化,造成心肌损害、糖尿病、性腺功能不全及肝硬化。先天性铜代谢异常的Wilson病时由于转运铜的铜蓝蛋白生成减少,使过剩的铜在脑的基底核和肝中沉积,出现神经症状及肝硬化。锌过多可引起发热;锰过多可导致中枢神经障碍、运动失调;钴过多可造成心脏病、甲状腺功能异常、听觉障碍等;锡过多可造成呕吐、腹泻、腹痛及肝脏损伤;汞中毒时发生“水俣病”;镉中毒造成疼痛、肾损伤及骨折的“疼痛病”。
(二)微量元素测定在疾病诊断中的应用
由于原子吸收光度法的应用,使得医院检验部门逐步开展体液、毛发中微量元素的检测工作,微量元素的检测在疾病的诊断中的价值开始受到应有的重视。铁、铜、锌、硒等的资料已日渐增多。现将几种重要微量元素的检测对疾病的诊断价值及这些微量元素测定值增高或降低的临床意义列于表6-16。
表6-16 微量元素检测在诊断上的应用(血清)
| 微量元素 | 检测方法 | 正常参考值 | 低值 | 高值 |
| 铁(Fe) | 比色法 | 男80-200μg/dl | 缺铁性贫血 | 再生障碍性贫血 |
| (OPT法) | 女70-180μg/dl | 出血性贫血 | 铁粒幼细胞贫血 | |
| Banti综合征 | 血色沉积病 | |||
| 慢性感染 | 急性肝炎早期 | |||
| 肿瘤等 | ||||
| 铜(Cu) | 比色法 | 男80-130μg/dl | Wilson病 | 毛细胆管性肝炎 |
| 原子吸收光度法 | 女100-150μg/dl | MenkesⅡ型综合征门脉性肝硬化 | 胆汁性肝硬化白血病、贫血 | |
| 尿10-60μg/日 | 肾病综合征 | 感染性疾病 | ||
| 铜吸收障碍 | 恶性肿瘤(淋巴 | |||
| 瘤、骨肉瘤等) | ||||
| 锌(Zn) | 原子吸收光度法 | 男63-147μg/dl | 锌缺乏(肠原肢端皮 | 溶血性贫血 |
| 女63-122μg/dl | 炎、高卡量输液)肝胆疾患(肝硬化、肝炎、肝肿瘤等)血液病(白血病、恶性贫血,再障,缺铁性贫血、多发性骨髓瘤等)、癌、恶性淋巴瘤、肉瘤、心肌梗死、肾病综合征,肾炎,多发性神经炎、脑血管疾病,Addison病,糖尿病肾症,肺炎等 | 红细胞增多症 | ||
| 嗜酸性细胞增多症 | ||||
| 甲状腺功能亢进症 | ||||
| 原发性高血压 | ||||
| X线照射后 | ||||
| 硒(Se) | 原子吸收光度法 | 80-180μg/L(血清) | 溶血性贫血 | 硒接触过量 |
| 克山病、心肌缺血、癌、肌营养不良症、多发性硬化症、糖尿病性视网膜病变、白内障等 | ||||
| 锰(Mn) | 原子吸收光度法 | 5-15μg/L | 慢性淋巴性白血病 | 锰中毒、心梗、急性白血病 |
续表
| 微量元素 | 检测方法 | 正常参考值 | 低值 | 高值 |
| 铬(Cr) | 原子吸收光度法 | 0.16-150μg/L | 糖尿病 | 急、慢性铬中毒 |
| 0.05-4μg/L | 冠心病 | |||
| 镍(Ni) | 原子吸收光度法 | 0.1-1.5μg/L | 急性心肌梗死镍中毒 |
三、重要微量元素的生物学作用及代谢
(一)锌(Zn)
⒈锌的生物学作用 锌是当代微量元素研究中非常活跃的课题之一。
⑴锌可作为多种酶的功能成分或激活剂:锌是碳酸酐酶、DNA聚合酶、RNA聚合酶,胸嘧啶核苷激酶、碱性磷酸酶、亮氨酸氨肽酶等含锌酶的组成成分。
⑵促进机体生长发育,促进核酸及蛋白质的生物合成:缺锌后创伤溃疡难愈合,生长发育不良,性器官发育不全或减退,成为缺锌性侏儒或肠原性肢端皮炎。
⑶增强免疫及吞噬细胞的功能:缺锌后免疫功能减退。
⑷抗氧化、抗衰老及抗癌作用:锌亦是超氧化物歧化酶的组成成分,能防止自由基对细胞膜造成的损伤,减少过氧化脂质的生成,某些肿瘤病人及衰老过程中有缺锌的倾向,因此锌可能具有抗氧化、抗衰老及抗癌作用。
⒉锌在体内的代谢 正常成人体内含锌2-2.5g,平均2.3g。男性比女性稍高,锌以视网膜、前列腺及胰腺中的浓度最高,在肌肉及骨骼中贮存。肌肉内储锌全身锌的62.2%,骨中的锌占全身体内锌的28.5%。正常人从普通膳食中每日摄取10-15mg锌,吸收率为20%-30%,主要在小肠内和胰腺分泌的小分子量配体-前列腺素E2结合后,经小肠上皮细胞吸收。锌进入毛细血管后由血浆运输至肝及全身。锌主要由粪便、尿、汗、头发及乳汁排泄,每天由尿排泄的锌不超过1mg。
含锌丰富的食物有谷类、粗粮、蛋黄、瘦肉、鱼、牡蛎和坚果等。
(二)铜(Cu)
⒈铜的生物学作用
⑴参与造血及铁的代谢:铜主要影响铁的吸收,促进储存铁进入骨髓,加速血红蛋白及铁卟啉的合成。铜还促进幼稚红细胞的成熟,使成熟红细胞从骨髓释放进入血液循环。
⑵构成体内许多含铜的酶(如丁酰辅酶A脱氢酶、酪氨酸氧化酶、尿酸酶、超氧化物歧化酶等)及含铜的生物活性蛋白质(如血浆铜蓝蛋白、血铜蛋白、肝铜蛋白、乳铜蛋白等)。
⑶与DNA结合,在DNA两条链中形成架桥,形成金属络合物,与维持核酸结构的稳定性有关。
⑷铜参与赖氨酸氧化酶的组成,促进弹性蛋白及胶原纤维中共价交联的形成,维持组织的弹性和结缔组织的正常功能。
⑸含铜酶大部属氧化酶类,如细胞色素C氧化酶、酪氨酸酶、多巴胺-β-羟化酶、胺氧化酶等,这些酶类参与儿茶酚胺类激素的代谢、黑色素的生成以及神经递质的代谢,因而对中枢神经系统的功能、智力及精神状态、防御功能及内分泌功能等均有重要影响。
临床上可测定血浆铜蓝蛋白的氧化酶活性,亦可测定血浆铜蓝蛋白,还可测血清铜。
血浆铜蓝蛋白:正常成人在25-43mg/dl之间,在肝豆状核变性(Wilson病,为铜在肝、脑等组织中沉积的慢性内源性铜中毒,为常染色体性隐性遗传性疾病)和Menke综合征(以中枢神经病变为主的、头发卷曲色浅为特征的婴幼儿缺铜性遗传疾病)时明显减低。
血浆铜蓝蛋白氧化酶活性:正常人在66-140单位/L之间。心肌梗死、传染病、肝癌及转移性肿瘤患者明显升高,Menke综合征及Wilson病时显著降低。
血清铜:成人在105-114mg/dl之间,女性稍高于男性。
尿铜:24小时在70μg以下,肝豆状核变性、肾病综合征、急性铜中毒、急性病毒性肝炎、肝硬化时尿铜增加。
毛发铜含量较为恒定,能反映机体营养状态,正常成人平均值9.5-23μg/g。
⒉铜的代谢 铜主要参与造血及酶的合成。正常人体内含铜100mg-200mg,平均150mg左右,约50%-70%的铜存在于肌肉及骨骼内,20%存在于肝。肝是重要的储铜库,5%-10%的铜分布于血液中,微量的铜以酶的形式存在于组织中。一般成年人每日从食物摄取2mg铜已能满足生理需要,富含铜的食品是牡蛎、蛤类、小虾及动物肝肾等。正常人每天从各种渠道排泄铜2mg左右,肠管可能通过含铜复合物的上皮细胞的脱落而排泄铜。
(三)硒(Se)
⒈硒的生物学作用
⑴硒是谷胱甘肽过氧化物酶(GSH-Px)的必需组成成分:每摩尔的酶含有4毫摩尔的硒,该酶催化的反应为:
2GSH+H2O2→GSSG+2H2O
2GSH+ROOH→ROH+GSSG+H2O
通过GSH-Px的作用,在清除自由基、分解过多的H2O2,减少过氧化物、保护细胞膜、保护细胞敏感分子(DNA、RNA)中占有重要地位。
⑵硒参与辅酶A和辅酶Q的合成,促进α-酮酸脱氢酶系的活性,在三羧酸循环及呼吸链电子传递过程中发挥重要作用。
⑶硒与视力和神经传导有密切关系,虹膜及晶状体含硒丰富,视网膜的视力与含硒量有关。硒是光电管基础物质之一,硒在视网膜、运动终板中可能起着整流器及蓄电器的作用。
⑷硒能拮抗某些有毒元素及物质的毒性:硒可在体内外减低汞、镉、铊、砷等的毒性作用。
⑸硒能刺激免疫球蛋白及抗体的产生,增强机体对疾病的抵抗力。
⑹硒与心血管结构和功能的关系:硒可防止镉引起的实验性高血压,并可防止冠心病及心肌梗死。硒参与保护细胞膜的稳定性及正常通透性,抑制脂质的过氧化反应,消除自由基的毒害作用,从而保护心肌的正常结构、代谢和功能。投予亚硒酸(每周0.5-1mg)可有效地防治克山病的发生。
⑺硒能调节维生素A、C、E、K的代谢。
⑻硒的抗肿瘤作用:许多报道表明结肠癌、乳腺癌、前列腺癌、直肠癌及白血病等的死亡率与其居住地区土壤的硒含量、日摄取量以及血硒水平呈逆相关关系。动物致癌试验中观察到硒对动物的实验性皮肤癌、肝癌、结肠癌、乳癌、肺癌等均有显著的抑制作用,且此种抑癌效果还受食物中维生素A、C、E等含量的影响。关于硒化合物的抗癌作用机制尚未阐明,可能与硒能抑制致癌物质的致突变性、或改变致癌物的代谢、或通过抗氧化作用而抗癌。有报道硒化合物能促进肝癌细胞及白血病细胞的再分化,通过促分化而抗癌。
⒉硒的代谢 1957年Schwarz证明硒是动物体内必需的微量元素。人体内硒的含量约14mg-21mg,以肝、胰腺、肾中的含量较多,人类对硒的摄入量受环境及食物含硒量的影响。富含硒的食品有蒜、芝麻、啤酒、酵母、蘑菇、小虾、鱼类、肝、肾等,富硒中药有黄芪、地龙等。人对硒的摄入量不应低于40μg/d,一般认为成人每日从食物中约摄取60μg-350μg的硒,每日由尿约排泄约50μg,由粪排泄80μg,由汗排泄20μg,在血浆内的硒主要与α及β-球蛋白结合而运输。
我国不同地区报道的成人全血硒为0.028-0.033μg/ml,还有报道为0.099-0.160μg/ml。克山病区成人及儿童全血硒显著低下,分别为0.018-0.025μg/ml及0.017-0.024μg/ml。癌瘤时血硒明显减少,特别是消化道癌肿病人血硒降低更为显著,往往表明癌的转移、多发或低分化程度。
尿硒成人平均为11.9±2.6μg/24h,克山病区成人尿硒明显降低,平均为7.9±2.6μg/24h。
(四)铬(Cr)
⒈铬的生物学作用
⑴铬形成葡萄糖耐量因子(GTF),使胰岛素与膜受体上的-SH基形成-S-S-键,协助胰岛素发挥作用。
⑵降低血浆胆固醇及调节血糖。
⑶促进血红蛋白的合成及造血过程。
⒉铬的代谢 成人体内含铬6mg,日摄入量5-115μg,进入血浆的铬与运铁蛋白结合运至肝脏及全身。富含铬的食品有红糖、麦麸、鱼、葡萄汁、啤酒酵母等,铬主要随尿排泄,3-60μg/d。
(五)钴(Co)
⒈钴的生物学作用 人类不能利用钴合成维生素B12,主要从能合成维生素B12的动物及细菌摄取,维生素B12在人体内参与造血。维生素B12参与体内一碳单位的代谢,参与脱氧胸腺嘧啶核苷酸的合成,维生素B12缺乏可导致叶酸的利用率下降,造成巨幼红细胞性贫血。维生素B12促进铁的吸收及储存铁的动员,它还促进锌的吸收,提高锌的活性。
⒉钴的代谢 正常成人体内含钴1.1-1.5mg,一般从普通膳食中摄入钴150-450μg/d,吸收率63%-97%,每日吸收钴190-290μg,钴通过小肠进入血浆后由三种运钴蛋白(transcobalbmin Ⅰ、Ⅱ、Ⅲ)结合后运至肝脏及全身,主要由尿排泄,每日排泄量约等于吸收量。当内因子缺乏、运钴蛋白缺乏、摄入量不足或因消化系统疾病而干扰吸收时,可造成钴及维生素B12缺乏。全血及血清钴10.8±6.0ng/ml,在恶性贫血、急性白血病时降低,慢性粒细胞白血病、淋巴肉芽肿时血钴升高。
富含钴的食品有小虾、扇贝、肉类、粗麦粉及动物肝脏。
检测血清不饱和维生素B12结合力(UBBC)及血清维生素B12正常人血清UBBC为0.7-1.6μg/L,血清维生素B12为0.16-0.75μg/L,这两项指标有利于肝癌的诊断。有些AFP不增高、HBsAg阴性的肝癌病例,UBBC及维生素B12亦可显著增高。
(六)锰(Mn)
⒈锰的生物学作用
⑴锰的多种酶的组成成分及激活剂:锰是精氨酸酶、脯氨酸酶、丙酮酸羧化酶、RNA聚合酶、超氧化物歧化酶的组成成分,又是磷酸化酶、醛缩酶、半乳糖基转移酶等的激活剂,与蛋白质生物合成、生长发育有密切关系。
⑵参与造血、卟啉合成、改善机体对铜的利用。
⑶构成Mn-SOD,有抗衰老作用:衰老时Mn-SOD活性降低,肿瘤细胞内Mn-SOD活性亦降低。故Mn具有抗衰老抗肿瘤作用。
⒉锰的代谢正常成人体内含锰12-20mg,分布于一切组织,每日从一般食物摄入锰0.7-22mg,吸收率为3%-4%。吸收的锰经小肠壁进入血流,与β1-球蛋白或“运锰蛋白”结合后,迅速运至富含线粒体的细胞中,血锰5-15μg/L,人体内的锰主要由肠道、胆汁、尿液排泄。坚果、茶叶、叶菜、谷类富含锰。
(七)钼(Mo)
⒈钼的生物学作用
⑴钼是构成黄嘌呤氧化酶、醛氧化酶、亚硫酸氧化酶等氧化酶的组成成分,可解除有害醛类的毒性。
⑵参与电子的传递及铁从铁蛋白的释放及铁的运输。
⑶钼有抗癌作用,缺钼地区食管癌发病率高,钼构成亚硝酸还原酶(植物),降低环境中亚硝酸含量,减少致癌物亚硝胺的生成。
⑷钼与心血管疾病有关,洋地黄类施用钼肥可提高产量及强心甙疗效。
⒉钼的代谢 成人体内含钼9mg左右,分布于全部组织及体液中。一般成人每日由普通膳食中摄入钼300μg,吸收率为40%-60%,随食物及饮水进入消化道的钼化物可迅速(10分钟)被吸收,80%与蛋白质结合,血清钼590ng/dl,而在食管癌高发区血清钼明显减低为220ng/dl-290ng/dl,癌症病人、心律不齐病人均可有血钼降低,白血病及缺铁性贫血时血清钼增高。
富含钼的食品有豆荚、肝、肾、酵母、牛乳及粗麦粉等。
高钼地区痛风发病率高,可能与黄嘌呤氧化酶活性增高、尿酸生成增多有关。
(八)氟(F)
⒈氟的生物作用 氟为骨骼牙齿的必需组分,与牙齿及骨的形成有关,可增加骨硬度及牙齿的耐酸蚀能力。缺氟易生龋齿,氟过多可引起骨密度增加和斑釉齿,氟在细胞内可抑制多种酶的活性,干扰胶原的合成。
⒉氟的代谢 成人体内含氟量2.6g左右,主要在骨骼、牙、指甲、毛发中。每日约摄入2.4mg的氟,大部分由尿排泄,每日排出约2.4mg。
富含氟的食品有海味(牡蛎)、葱、豆类、茶叶等。
(九)碘(I)
⒈碘的生物学作用 碘是1850年即已发现的必需微量元素,主要构成甲状腺激素T3、T4。甲状腺激素维持正常生长发育及智力发育,调节能量代谢。缺碘可发生地方性甲状腺肿和呆小病,碘过多可造成碘中毒,刺激甲状腺功能亢进。
⒉碘的代谢 人体含碘量约为11mg,每日成人需摄入50-100μg的碘,为防治地方性甲状腺肿,应普遍食用加碘食盐。
此外,近年来对锗这一微量元素的研究日益增多,据报道有些有机锗(如Ge-132)化合物可以提高人体免疫力,诱导干扰素生成,有抗衰老抗肿瘤的作用。有人主张锗是可能必需微量元素。
总之,微量元素在生命科学中的作用、微量元素与地方病、微量元素与职业病、微量元素与衰老、微量元素与肿瘤等领域的研究近十余年来极为活跃,是临床生物化学中令人瞩目的篇章。
第七章 诊断酶学
本世纪初临床就开始测定体液中的酶来诊断疾病,如Wohlgemuth早在1908年就测定尿液中淀粉酶(AMY)以诊断急性胰腺炎;30年代临床测定碱性磷酸酶(ALP)用于诊断骨骼疾病,随后发现不少肝胆疾病特别在出现梗阻性黄疸时此酶常明显升高。这些酶成为当时临床实验室的常规测定项目,直到60年代ALP仍是世界上测定次数最多的酶。但在50年代以前,酶测定在检验科常规工作中只占很少一部分。诊断酶学的真正发展还是从50年代用分光亮度法建立了连续监测酶活性浓度方法开始,它可以测定不少用旧的“固定时间法”不能测定的酶,并用于诊断疾病。结果发现乳酸脱氢酶(LD)、天冬酸氨基转移酶(AST)和α-羟丁酸脱氢酶(HBDH)在诊断急性心肌梗死(AMI)上的灵敏度远远超过其他诊断方法。在60年代初又肯定了肌酸激酶(CK)在诊断AMI比上面几个酶更早出现增高,特异性也高,目前此酶已取代ALP成为世界范围内测定次数最多的酶。同时发现丙氨酸氨基转移酶(ALT)、AST对肝炎诊断不仅敏感度高,而且早在肝炎黄疸前期就明显升高。这些成就引起了当时临床和实验室工作者广泛的兴趣和注意,先后进行了大量临床和实验工作,尝试和评价过成百种酶测定的临床意义,其中十种左右酶已成为目前检验科常用的重要测定项目。酶测定约占目前临床化学总工作量的1/4到1/2。
随着广泛地应用和研究,也发现总酶活性浓度测定对疾病诊断的特异性远不如人们所开始预期的那样高。从70年代开始,学者逐渐将注意力集中到同工酶测定上来,发现CK-MB和LD1诊断AMI比上述总酶特异性更高,CK-MB已成为公认的诊断AMI的“金指标”,此二项同工酶测定也成为各大医院检验科必测项目。
80年代以来,发现组织中同工酶进入体液后,有可能出现变化。如Ck-MM可进一步分为Ck-MM1、MM2和MM3,Ck-MB可分为MB1和MB2。在诊断AMI上优于CK总酶和同工酶,成为目前临床酶学上的一个研究热点。
从70年代起,随着免疫学和技术方法的发展,用抗原抗体反应有可能直接测定微量的酶蛋白,为酶学在临床医学上的发展开拓了一个新的领域。
本章将以血液中酶变化为重点,首先研究其变化的总规律,其次将从临床角度来探讨这些酶测定在临床诊断疾病、判断疗效和疾病预后中的价值。
第一节 概述
长期以来临床将血清酶变化的机制理解得很简单:即病变细胞将其细胞中高浓度的酶释放到血液中,二者间酶浓度梯度越大,则血清中酶升高程度越大。这种理解远不能解释各种各样的临床现象,例如肝中AST绝对量约是ALT的4倍,但在急性肝炎时ALT增高程度远大于AST,而在慢性肝病特别是肝硬化时血中AST又比ALT高,单从上述浓度梯度理论显然很难说清。必须全面了解各种影响血清酶变化的因素。首先要了解血清酶的分类,因为不同类型酶变化模式将是不一样的。
一、血清酶的分类
虽然绝大多数血清酶含量极低,在血液中没有任何功能,但也确有一小部分酶在细胞内合成后分泌到血液中,并行使一定功能。其典型例子就是一些与凝血过程有关的酶,如凝血酶原、Ⅹ因子、Ⅻ因子等,还有与纤溶有关的酶如纤溶酶原、纤溶酶原活化因子等。它们一般以失活或酶原状态分泌入血,在一定情况下被活化,引起一系列病理或生理变化。它们在血中浓度往往很高,甚至超过大多数器官细胞内浓度,因此在血中的变化常不是升高而是下降。它们大都在肝脏合成,并以恒定速度释放入血,肝实质病变时,血中浓度明显下降,常作为肝功能试验的一部分。这类对临床有价值的酶还有胆碱酯酶(CHE)、铜氧化酶、脂蛋白脂酶等,在血中含量都以mg%计,人们将此类酶命名为血浆特异酶。
表7-1 临床常用的酶
| *EC编号 | 推荐名 | 简写 |
| 1.1.1.27 | 乳酸脱氢酶 | LD、LDH |
| 1.1.1.37 | 苹果酸脱氢酶 | MD、MDH |
| 1.1.1.41 | 异柠檬酸脱氢酶 | ICD、ICDH |
| 1.4.1.3 | 谷氨酸脱氢酶 | GDH、GLDH |
| 2.3.2.2 | γ-谷氨酰基转移酶 | GGT、γ-GT(GGTP) |
| 2.6.1.1 | 天门冬酸氨基转移酶 | AST、GOT |
| 2.7.3.2 | 肌酸激酶 | CK(CPK) |
| 3.1.1.3 | 脂肪酶 | LPS |
| 3.1.1.8 | 胆碱酯酶 | CHE |
| 3.1.3.1 | 碱性磷酸酶 | ALP(AKP) |
| 3.1.3.2 | 酸性磷酸酶 | ACP |
| 3.2.1.1 | α-淀粉酶 | AMY(AMS) |
| 3.4.11.2 | 氨基酸芳香酰胺酶 | LAP |
| 4.1.2.13 | 果糖二磷酸醛缩酶 | ALD |
| 2.1.3.3 | 鸟氨酸氨甲酰基转移酶 | OCT |
| 3.1.3.5 | 5′-核苷酸酶 | 5′-NA(5′-NT) |
*EC:国际酶学委员会
其它酶可归为非血浆特异酶,它们在血中浓度很低,常以微克计算,并且无何功能。可进一步分为分泌酶和代谢酶,一些外分泌器官分泌的酶可有小部分入血,如α-淀粉酶(AMY)、脂肪酶(LPS)、胃蛋白酶原等等,它们在血中一般也以失活状态存在,疾病时可以升高,但是如分泌细胞破坏,血中浓度也可下降,往往将ALP、酸性磷酸酶(ACP)也归到此类,认为ALP由骨细胞分泌,ACP由前列腺分泌。
其余绝大多数酶都参与细胞内代谢,随正常细胞的新陈代谢,极少数进入血液,细胞内外浓度差异悬殊,病理情况下极易升高,一般很少考虑其浓度下降的临床意义。
有些书还讲一步将代谢酶分为一般酶和组织专一酶,看来无此必要,从临床观点看,单测总酶变化就要得出病变存在组织或器官的结论,显然是很困难的。
二、血清酶变化的病理生理机制
前面已提到不同类型的酶,其变化机制会有所不同,如把所有可能影响因素都考虑进去,可以得到一个总的酶由细胞内进入血液以及在血中变化的总的模式图(图7-1):
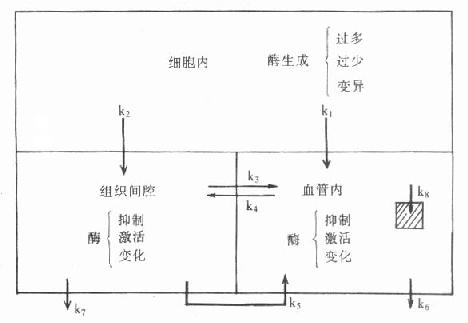
图7-1 血清酶变化机制
式中k1,k2分别代表细胞内酶进入细胞间隙或(和)直接进入血液的速率,如某些种类细胞直接与血液接触,不需经过组织间隙就直接进入血液,则血中酶变化不仅出现早而且明显。K3和k4分别代表酶从两个不同方向通过毛细血管壁的速率,某些组织或器官中毛细血管壁很致密,这些值较低,则可能有相当一部分酶经由淋巴管才进入血液,此速率常数为k5。而k6、k7则代表了酶在细胞间隙和血液的清除速率。K8代表酶被血中细胞或网状内皮系统细胞摄入的速率常数。少数酶属于血浆特异酶和分泌酶,如细胞出现增生性病变,则酶可以产生增多,并进入血液,反之也可能产生减少,从而引起血中酶浓度下降。
不同组织或器管中酶进入血中途径不一,清除方法也有差异,这就构成不同疾病酶变化的多样性,也只有在总的规律基础上,掌握各种器官疾病的特殊性,才能解释和掌握酶变化规律,正确用于临床。从临床角度,可以将上述各种因素归纳为以下四个方面加以叙述。
(一)细胞酶的释放
细胞靠细胞膜来维持其完整性,细胞膜代谢十分活跃,依靠膜上一系列ATP依赖的离子泵来维持细胞内外Na+、K+和Ca2+浓度的差异,这过程需要耗费大量能源,当缺氧或能量代谢障碍、ATP供应减少、离子泵功能障碍时,无法维持正常离子的梯度差,改变了细胞的内渗透压,从而引起细胞肿胀,特别是Ca2+进入细胞内,引起细胞膜的泡状突出,膜孔隙增大,酶开始从细胞内向外溢出,其速度和数量受多种因素影响。主要的有:
⒈细胞内外酶浓度的差异 对于非血浆特异酶,细胞内外浓度差可在千倍以上,因此只要有少量细胞坏死或者细胞有轻度病变,血中酶浓度就可能明显升高。有人计算过只要有1/1000肝细胞坏死,所释放的酶可使血中酶增加一倍。鸟氨酸氨甲酰基转移酶(OCT)在细胞内外浓度差异可达到105:1,此酶在肝脏病变时变化极为明显,可惜的是,由于测定方法不方便,临床应用不多。但对于血浆特异酶而言,由于细胞内外浓度差异小,细胞病变很少引起血中酶浓度明显升高。
⒉酶在细胞内定位与存在形式 从上述酶释放的机制不难理解最容易释放入血的是胞质中游离的酶,如ALT,LD等。而在细胞亚显微结构中的酶则较难溢出,除非细胞病变进一步加重,不局限于细胞膜。特别是线粒体酶,由于有两层致密的线粒体膜,往往当细胞出现坏死病变时,才开始释放入血。在一个典型的AMI病程中,线粒体AST是最后一个出现升高的酶,而且到达峰值时间也最迟。临床通过线体酶的测定,有助于判断疾病的不良预后。又如肝细胞中AST大部分存在于线粒体,虽然其绝对量超过ALT,但在急性肝炎时,由于细胞病变较轻,胞质中含有大量ALT,故血中ALT往往超过AST。而在肝硬化时,主要病变为肝细胞坏死,线粒体中AST大量溢出,血中往往AST大于ALT。
细胞膜上也含有多种酶,如γ-谷氨酰基转移酶(GGT)大量存在于肝中毛细胆管上皮膜上,当胆道梗阻、胆汁潴留在肝中时,胆汁酸盐有表面活性剂作用,可将GGT从细胞膜上洗脱下来,而此时不一定伴有细胞膜病变。正因为血中不同酶变化机制有差异,这样GGT和ALT在各种肝胆疾病时的变化常不一致。
⒊酶蛋白分子量的大小 不少实验都证实酶的释放速度大致与酶的分子量成反比。由于临床上测定的十余种酶之间分子量差异不太大,此因素对血中酶浓度高低影响恐不如上述因素,但对酶在血中出现升高时间先后有相当大影响。例如在AMI时,血中最先升高的CK分子量为85000,而分子量为125000的LD出现升高明显推迟。
(二)酶在细胞外间隙的分布和运送
细胞中的酶经过三种途径进入血液:一种如血细胞和血管内皮细胞中的酶,不经过稀释就直接进入血液。第二种途径,细胞酶既和组织间隙也和血液直接相接触。如肝脾,它们释放的酶很快直接入血,另有小部分进入组织间隙。第三种途径,大多数组织或器官中,由于存在着结构致密的毛细血管,所释放的酶大部分进入组织液。除一小部分通过毛细血管壁进入血液外,主要经由淋巴系统进入血液。由于血液只占细胞外液的20%,淋巴液和血液一天交换量可达50%-100%,其结果通过此途径进入血液的酶量不仅增高程度较低,在血中出现增高时间也较迟。临床医师不能忽视淋巴系统对血清酶浓度的影响。在一组动物实验中,单纯肌肉损伤加以固定,血中酶浓度变化不大;如加上被动运动,则AST明显升高;如移去实验动物胸导管,即使有被动运动,AST也不升高。
有作者认为在坏死病变时血中线粒体酶很少达到像胞质酶这样高的程度,也是因为坏死病灶无淋巴液或很少淋巴液流动,这样大量线粒体酶堆积在坏死区,只有少部分坏死边缘区酶通过淋巴进入血液。剧烈运动后血中酶升高与其说是由于细胞损伤引起,还不如说与酶在不同体液中重新分布有关。首先运动引起血压升高,血浆容量减少,血液浓缩;更重要的是运动加速了淋巴液回流入血,大量组织液中酶进入血液。因此即使细胞中酶无明显释放入血,血液酶浓度也可增高。
不同入血途径还可引起别一种差异,通过第三途径入血的往往是一些分子量小的单纯酶蛋白,通过前两种途径入血的不仅有单纯酶蛋白、大分子量的酶多聚体和抗体的复合物,乃至一些和细胞碎片结合的酶都易进入血液,临床上所观察的巨酶血症常与肝病有关,可能原因在此。
(三)血中酶的清除
为了很好解释临床上复杂情况,还必须了解不同酶从血中清除率的差异以及清除机制。弄清酶的清除率有助于理解,为什么同一疾病不同酶升高持续时间有差异。一般以血中酶的半寿期来代表酶从血中清除快慢,表7-2是一些常用酶的半寿期数据。
表7-2 血浆中酶的半寿期
| 酶 | 半寿期 |
| AST | 17±5h |
| ALT | 47±10h |
| GLD | 18±1h |
| LD1 | 113±60h |
| LD5 | 10±2h |
| CK | 约15h |
| CK-MM | 17±4h |
| Ck-MB | 12±4h |
| CK-BB | 约5h |
| ALP | 3-7天 |
| GGT | 3-4天 |
| CHE | 约10天 |
| AMY | 3-6h |
| LPS | 3-6h |
从表7-2中不难理解为什么在急性肝炎恢复期时AST先于ALT恢复正常,也很好解释在AMI时CK-MB持续时间最短,因其半寿期只有6小时,而LD1因其半寿期长达100余小时,持续时间最长。
有关酶从血中清除的机制,可说是目前最不清楚的问题,酶虽然也是蛋白质,但它们的半寿期明显比一般血清蛋白质为短,说明酶蛋白除遵守一般蛋白质代谢规律外,还有其特殊的清除机制。目前发现对于一小部分分子量小于60000的酶,如AMY可以从肾小球滤过一部分,从尿中排出,肾脏严重疾病时AMY升高也证实此一推论。但对于大多数酶而言,这种清除机制显然是不存在的。
过去曾强调,通过胆汁可能排泄一部分酶,并认为这是梗阻性黄疸时,血中ALP、亮氨酸氨基肽酶(LAP)的升高机制。而目前更多倾向认为此时升高机制是肝细胞受刺激合成更多的酶,因为如给以抑制蛋白合成的药物,虽有黄疸,这些酶也不升高。
目前有人强调单核吞噬细胞系统在清除酶中的作用,它们可将酶迅速吞噬入细胞中,并进一步分解破坏。有学者用Riley病毒选择性地感染小鼠单核吞噬细胞系统,引起血中LD、AST、AMY和CK等酶升高,用其它化合物封闭单核吞噬细胞系统也得类似结果。使用乙芪酚刺激单核吞噬细胞系统可加速这些酶的清除,但是也不能过高评价这种结果,就在同一试验中,一些酶如LD1和ALT就不受影响。此外,从临床角度来看,目前,尚无任何证据显示任何人的病毒感染疾病引起酶清除机制的障碍。
(四)酶合成异常
对于血浆特异酶,细胞内酶合成下降是引起血中酶变化的重要因素,这些酶大多在肝脏合成,因此当肝功能障碍时,胆碱酯酶常与白蛋白同时下降。酶合成减少和变异还见于不少遗传疾病,由于酶基因变异,可引起特定的酶合成减少乃至消失,如肝-豆状核综合征患者,血中铜氧化酶活性可明显下降乃至于零。在增生性疾病如骨骼疾病时,可因为骨细胞增生,合成分泌更多的ALP,引起血中此酶升高。恶性肿瘤患者血中酶升高有一部分可能与肿瘤细胞中酶合成增加有关,如前列腺癌细胞可产生大量酸性磷酸酶。
酶的诱导作用也可引起血中一些酶浓度增高,最明显例子是服用苯巴比妥后常可引起肝中GGT合成增加,血中浓度升高并不意味着肝细胞有什么病理变化,停药后GGT就会下降至正常。乙醇、巴比妥类、杜冷丁类以及双苯内酰脲类药物都有此种诱导作用,诱导的酶除GGT外还可以是ALP。
(五)其它
实验室所测到的酶活性浓度值主要和酶量多少相关,但还受到其它物质特别是抑制剂和活化剂的影响。在病理情况下,应考虑有无抑制作用,因为某些药物和毒物有抑制酶的作用。当使用某些药物治疗肝炎后,还应考虑此药物有无可能抑制ALT活性,此时血中ALT量不一定下降,只是活性受到抑制,使所测活性下降而已。有机磷中毒时所测的血清胆碱脂酶和红细胞中真性胆碱酯酶活性浓度可以很低,此时并不是酶含量降低,只是和有机磷结合,而有机磷是这些酶的不可逆抑制剂,使酶活性无法发挥。
三、血清酶的生理变异
临床医师常通过两个途径来判断血清酶的测定结果是否异常,有无临床价值。一是将所测值与同一被测对象在正常条件下所测值进行比较,二是将所测值与实验室提供的参考值进行比较。但不论用什么方法都需知道,不仅是病理因素,还有不少生理因素也能引起所测值变化,这些变异并无临床或病理意义。
(一)性别
大多数酶在男女之间无大差异,但少数酶如CK、GGT在男女之间有明显差异,因此不能以一个参考值作为判断标准。建议以130U/L为男性正常参考上限,100U/L为女性参考值上限。这是因为CK在肌肉收缩中起重要作用,大量存在于肌肉组织中,从总体说男性肌肉比女性发达,所以血清CK在男女之间差异较大,在衡量个体CK变化时,恐怕肌肉发达程度是一个很重要的因素。GGT是另一个男女差别明显的酶,常以GGT30U/L(30℃)为女性上界值,50U/L(30℃)为男性上界值,这可能与雌激素抑制GGT合成有关。嗜酒者可诱导GGT合成,而嗜酒者男性多于女性。
(二)年龄
不少酶在儿童时期与成人有所不同,例如新生儿的CK、LD、苹果酸脱氢酶(MD)、ACP和谷氨酸脱氢酶(GLD)等常为成人2-3倍,尤其前两项CK、LD是临床常用的酶。CK出生24小时内可为成年人的3倍,到婴儿时降为两倍,到青春期降到成年人值。LD在出生时也为成人两倍,逐渐下降到14岁和成年人值一致。同时LD1值儿童期也比成年人高,正常儿童也可出现LD1>LD2,如不注意此点,简单以正常成年人参考值为诊断标准,易导致儿童心肌炎的误诊。
年龄引起酶变化最明显的酶是ALP,新生儿值略高于成年人。至周岁增至成年人的2-3倍。然后逐渐下降,到10岁左右,发育长高期ALP又明显升高,可达到成人的3-5倍。这可能与该时期软骨细胞、成骨细胞代谢活跃密切相关,切勿认为有肝脏病变,必要时应测ALP同工酶以资鉴别。
也有少数酶如AMY,新生儿比成年人低。当进入老年期,有些酶也可出现变化,如ALP、GGT等都有轻度升高。
(三)进食
大多数血清酶不受饮食影响,故测酶活性不一定空腹采血。但应注意酗酒者常引起GGT明显升高,日本学者认为可根据血中GGT升高判断酗酒程度,如未累及肝脏,戒酒后一周GGT可降至正常。此外如禁食数天可导致血清AMY下降。
(四)运动
剧烈运动可引起血清中多种酶升高,升高程度和运动量及持续时间有关,也与运动者是否经常锻炼相关。训练有素的运动员,其血清酶升高幅度小。升高的酶多为肌肉中含量丰富的CK、LD、AST、醛缩酶(ALD)和ALT等,这些酶变化机制和意义是运动医学的一个重要研究项目,从临床角度,抽血化验前不宜过度运动,以免引起解释困难。
(五)妊娠与分娩
妊娠期出现一系列生理变化,也可引起一些酶升高,如不注意有可能引起误诊。例如由于胎盘产生耐热的ALP可引起血中ALP值超过正常,LAP也会明显升高,但都无临床诊断价值。铜氧化酶在妊娠期升高,若胎儿死亡,此酶很快降至正常,有学者在妊娠期监测此酶以观察胎儿情况。分娩时,由于子宫收缩,肌肉剧烈活动,可导致CK、CK-BB、AST、LD等升高,这些变化与心肌损伤无关。
四、测定方法、标本处理等对测定结果的影响
除了生理条件可引起酶测定值的变化外,还有其它一些非病理因素也会引起实验室测定结果的变化。
(一)酶测定方法
酶测定方法大致经过三个历史发展阶段,50年代以前大都使用“固定时间法”,即让酶与底物作用一段固定时间,一般为半小时左右,然后停止酶反应,用光电比色测产物生成量或底物消耗量,从而计算出酶催化反应的平均速度。再据此按各作者自己定义的“单位”向临床报告测定结果。这些所报的单位往往在前面冠以作者的姓名命名,例如ALP根据不同测定方法就有金氏单位、鲍氏单位、国际单位等等。同一标本不同单位结果从表面数值看可以差异很大。如ALP上列三种单位参考值分别为3-13金氏单位,0-3鲍氏单位,20-80国际单位。
50年代中期,国际临床实验室开始采用“连续监测法”,就是习惯上所称的“动态法”。需要使用生化分析仪,可以测酶反应的初速度,其结果远比“固定时间法”所测平均速度准确,在高浓度标本时尤为明显。这样同一疾病患者用两种方法所测酶的结果并不一定平行一致。在疾病急性期,用新法测定结果增加倍数常明显超过老法。例如在肝炎用赖氏法测ALT很少超过参考值上限的十倍。改用新法,在肝炎急性期可得到超过参考值上限数十倍乃至百倍以上的结果,由于后者结果和临床病理变化相一致,结果准确。目前在发达国家中,新法已几乎取代了老法,我国也已开始广泛应用新法。本书各论在讨论各种疾病酶变化时,将主要根据新法加以叙述。用新法也就是“连续监测法”所得结果,目前已基本统一使用1964年国际生化协会所规定的国际单位(IU):即以每分钟能催化1微摩尔底物的酶量为1个国际单位。如表示血清中酶浓度多以U/L表示之,即IL标本(血清)中含的酶量,我国医学界对此表示方法已经熟悉习惯。我国现在规定SI制为计量的法定单位,SI制的酶单位为Katal,即每秒能催化1总分子底物的酶量为1个Katal,此单位不仅我国医学家不熟悉,国际上应用不多,如有可能可同时用两种单位报告结果,或者注明转换系数,即1U=16.67μKatal。
但即使同使用连续监测法,同用IU/L报告结果,不同实验室测定结果仍可出现较大差异。这是因为上述二法是通过测酶催化速度来间接表示酶量高低,而酶催化的速度还受很多其它因素影响。首先是温度,在国际单位定义中并未规定测定温度,而反应速度随温度升高而加速,一般说每增加10℃,反应速度将增加一倍。因此同一标本、同一方法,在37℃所得结果将显著高于30℃。虽然国际临床化学协会(IFCC)推荐30℃为酶测定温度,但临床实验室由于实验工作方便等因素,愈来愈多使用37℃。因此本书各酶测定参考值将以37℃所测结果为准,某些数据如在30℃测定将在结果后面用(30℃)表示。
除温度外,测定时条件的不同也可能引起结果明显差异。最突出例子是ALP测定,用二乙醇胺为缓冲液测出ALP结果比用氨基甲基丙烷为缓冲液的高2-3倍。又如在ALT测定时,如在底物中加入磷酸吡哆醛比不加者结果可增高20%-100%。正因为如此,为防止临床医生产生错觉,认为IU/L结果在国际上应一致。目前实验室常以U/L,而不以IU/L向临床报告。
总之,临床医师在分析酶测定结果(U/L)时应以给报告实验室的参考值为准,盲目套用文献结果或与其它实验室结果相比较,有可能得到不恰当的结论。
前面已提到从70年代以来,随免疫技术发展,出现利用酶的抗原性,通过抗原抗体反应直接测定酶的质量。测定结果不是以U/L报告而是直接用ng/ml,μg/L等报告酶蛋白含量高低。如临床医师看到这样的报告,就可知道此时使用了最新的免疫学方法测酶,其临床意义有可能与前两种方法结果有差异。例如国外公认,同样测CK-MB,用免疫学方法所测结果诊断价值最高,此问题将在后专门叙述。
(二)标本的采集、处理与贮存
在实验室测定酶之前,标本还要经过采集、分离血清和贮存等一系列处理过程。而酶在血中是处于一个动态变化过程,血液离开体内后,还会有一定变化。因此在其中任何一个阶段处理不当,都有可能引起测定值变化。
采取血液标本时最容易引起实验室误差的是抽血不当,或者实验室急于分离血清,因此体外溶血。由于大部分酶在细胞内外浓度差异明显,少量血细胞的破坏就有可能引起血清中酶明显升高。最明显例子是LD,红细胞中LD约为血清的100倍,这样,只要有轻度溶血,如1/100红细胞破坏,就足以使血清中LD升高一倍,常使原为正常的血清LD值超过参考值上限。其它如CK,虽然红细胞中无CK,但含有丰富腺苷酸激酶(AK),进入血清能使某些用“连续监测法”测定CK的值假性升高,红细胞中酶的干扰,不仅表现在溶血影响上,采血后如不及时将血清和血凝块分离,血细胞中酶同样可以透过细胞膜进入血清,所以采血后1-2小时实验室必须及时离心,分离血清。
除非测定与凝血或纤溶有关的酶,一般都不采用血浆而采用血清作为测定标本。大多数抗凝剂都在一定程度上影响酶活性,例如EDTA为金属离子螯合剂,在去除Ca2+同时也去除其中Mg2+、Mn2+等离子,Mg2+是ALP、CK和5′-核苷酸酶(5′-NA)作用的激活剂,其它如草酸盐、枸橼酸盐乃至肝素都对一些酶有一定程度抑制。有必要时,可考虑使用肝素为抗凝剂,在常用抗凝剂中它对酶影响最小的。
酶蛋白不稳定,易失活,ACP是最明显的一个例子。此酶在中性或碱性环境中极不稳定,血液在37℃放置1小时,ACP活性可下降50%。大部分酶在低温中比较稳定,分离血清如不能及时测定,应放冰箱保存,表(7-3)是常用酶在不同温度情况下的稳定性。
从表7-3可看出-25℃将血清冻结并没有太大优点,相反有些酶如ALD、ALT在融冻时被破坏。有文献报道用液氧在-195℃贮存血清,常用酶如ALT、AST、ALP、CK、LD、GGT和AMY,在10个月活性变化不大。个别酶如LD在低温反而不如室温稳定,即所谓的“冷变性”。
表7-3 酶在不同温度储存的稳定性(活性变化小于10%)
| 酶 | 室温(25℃) | 冰箱(0-4℃) | 冰冻(-25℃) |
| ALD | 2天 | 2天 | 不稳定※ |
| ALT | 2天 | 5天 | 不稳定※ |
| AST | 3天 | 1周 | 1月 |
| ALP | 2-3天 | 2-3天 | 1月 |
| GGT | 2天 | 1周 | 1月 |
| CHE | 1周 | 1周 | 1周 |
| LAP | 1周 | 1周 | 1周 |
| CK | 1周 | 1周 | 1周 |
| LD | 1周 | 1-3天 | 1-3天 |
| ICD | 1天 | 2天 | 1天 |
| AMY | 1月 | 7月 | 2月 |
| LPS | 1周 | 3周 | 3周 |
| ACP | 4小时¥ | 3天§ | 3天§ |
| 亚铁氧化酶I | 1天 | 2周 | 2周 |
| (铜氧化酶) |
※酶不耐融化;
¥ 标本未酸化;
§标本加枸橼酸到pH5
五、免疫方法测酶质量的临床应用
用免疫学方法测酶质量和经典的测酶活性方法相比,不仅灵敏度高,并可能测定一些以前不易测定的酶。但更为有意义的是:有可能为临床上的应用提供新的资料和信息。从疾病论断的角度来看,至少提供下列三个新的可能性:
(一)酶活性和酶质量变化的不平行
这二者的变化在不少情况下是互相平行的,但在一些情况下可以出现不一致。早在1987年Robert就提出可以用免疫学方法直接测量CK的质量,并声称:“由于RIA法是如此的敏感,并可测定血循环中无活性的CK-MB,毫不奇怪此法可比其它方法更早查出心肌梗死,一般在疼痛3小时就可升高。”此后有些学者在急性心肌梗死时比较测CK-MB活性和RIA法CK-B亚单位的结果,发现二者之间存在着不平行关系。CK-MB活性升高持续较短,3天左右;而CK-B亚单位酶蛋白升高却在7天左右。近几年来发展了用免疫学方法测CK-MB酶质量(mass)方法与测酶活性(activity)方法的结果相比较,在检测心肌坏死上,测质量方法明显优于测活性方法。
国内有人报告了用单扩法测酶活性的方法,测定了在各种患者血中超氧歧化酶(SOD)含量,证实有些患者血中存在着无活性的SOD。
在一些疾病情况下,甚至出现二者变化完全相反的情况。例如在狗实验性胰腺炎,用免疫学方法测弹性蛋白酶浓度,出现迅速而急剧地上升,但同时酶活性却出现平行地下降。作者解释在释放酶蛋白入血同时,有大量酶抑制剂也释放入血,以致出现相互矛盾结果。前面提到的用两种方法测前列腺酸性磷酸酶(PACP)来诊断前列腺癌的阳性率不一致,有作者也认为可能是肿瘤组织合成一些无活性或活性较低的酸性磷酸酶。
如果说在这方面尚有争论,那么在遗传学方面的意义则是比较明确的,将此两大类方法结合起来可以研究酶缺陷遗传疾病的发生机制,用免疫学方法可以测出灭活的酶蛋白量。由于基因缺陷引起的酶缺陷如果仅仅影响酶蛋白合成下降,酶活性与酶蛋白量的比率即所谓的“免疫比活性”无变化;如果由于基因缺陷合成了变异的酶蛋白,则不仅总酶活性下降,免疫比活性也将变化;如果合成了一种活性正常但结构不稳定的酶,则幼稚细胞中酶活性及质量都无变化,而在衰老细胞中将出现酶缺陷。酶稳定性的变异常见于红细胞内酶缺陷症。这是由于红细胞寿命较长,成熟红细胞无细胞核不能制造新蛋白质,所以酶稳定性的缺陷容易在衰老红细胞中表现出来。
在最严重的情况,变异的基因甚至能合成无活性的酶蛋白,如胆碱酯酶遗传变异中,有一类型所谓“不活动等位基因”(“silent”allele)就是典型例子。又如在Lesch-Nyhau综合征中存在这免疫活性但无催化活性的次黄嘌呤转磷酸核糖基酶。
(二)以免疫方法取代测酶活性方法
免疫学方法和经典测酶活性方法相比最突出的优点是:免疫学方法能测定一些不表现酶活性的酶蛋白质,这在以前是不能想象的。首先是各种酶原,还有去辅基酶蛋白(apoenzyme)以及一些灭活的酶蛋白裂解产物等等。例如已有用RIA法测胰蛋白酶原。也有用免疫学方法测定线粒体和胞质谷草转氨酶,这其中包括无酶活性的去辅基谷草转氨酶。
免疫学方法另一特点是特异性高,且不受体液中其它物质,特别是抑制剂、激活剂的干扰。不少蛋白水解酶或者对基质特异性不高,或者易受血中一些蛋白质如α1-抗胰蛋白酶、α2-巨球蛋白的抑制作用。用测酶活性方法不易准确定量甚至无法测定。在近年来用免疫学方法陆续建立了RIA测胰蛋白酶、弹性蛋白酶的方法。
一般说来和测酶活性相比,免疫学方法灵敏度更高一些。一些既往由于方法学灵敏度差,不易测定的酶可以尝试改用免疫学方法,脂肪酶是其中例子之一。经典测酶活性方法由于灵敏度差,酶温育时间长达24小时,几经修改仍需4小时,现已有RIA法脂肪酶试剂盒。近年来随着优生学发展,羊水中各种成分包括酶的测定日益受到重视。例如在Tay-Sachs综合征时,胚胎组织细胞中氨基已糖苷同工酶A或(和)B合成障碍,免疫学方法由于灵敏度高,可以比测酶活性方法早几周在胎儿的羊水中查出此酶的缺陷。
(三)同工酶测定
免疫学方法用于测定同工酶比经典的理化方法简单方便,所以愈来愈多同工酶测定改用免疫学方法,国外已逐步取代电泳或层析法。除上文已提到的CK-MB、LD1、LD5测定外,还可用免疫学方法测半乳糖酰基转移酶同工酶A和B,碱性磷酸酶的Regan同工酶。同工酶有可能作为肿瘤的标记物(tumor marker),已证实γγ-烯醇化酶或神经元特异烯醇化酶(NSE)可作为APUD系统肿瘤的一个很好标记物。用RIA测血中NSE在肺小细胞性肿瘤患者中阳性率超过80%,γγ-烯醇化酶在大细胞肺癌阳性率也超过70%。可以预期随科学技术发展,免疫学方法将成为临床上测定同工酶的主要方法。
六、检测同工酶及其亚型的临床意义
从50年代中期开始,临床酶学有了迅速的发展,几乎把当时能测的酶都在临床上试用过,企图通过测定某一种或某几种酶来诊断一些特定疾病或特定组织器官的病变。通过10余年广泛的研究,发现酶测定诊断疾病的特异性并不高,因为机体中各种细胞的代谢有很大的相似性,从临床角度很难说存在着所谓器官特异酶。当时曾有学者想找出一些肿瘤特异酶,但很快发现找不到一种只存在于肿瘤组织并能进入血液而正常组织又不存在的酶。
在此同时,Market等通过对LD在电泳时可分为5个区带的深入研究,在1959年首先提出“isozyme”的术语来描述一类酶,它们催化同一生化反应,但在结构和理化性质上有所不同,目前广泛使用国际生化协会推荐术语“isoenzyme”。我国目前多翻译为同工酶,临床很快将此新概念、新发现用于临床,发现同工酶测定可以弥补上述测酶总活性的不足之处,因为不同同工酶在组织分布、细胞内定位、发生发育方面都可能有所差异,临床可利用这些差异来增加诊断的特异性。目前不论在诊断肿瘤,还是诊断心脏、肝等疾病上都找到一些较总酶活性测定特异性、灵敏度高的同工酶测定项目,同工酶正逐步成为酶学中一个重要分支。
80年代发现某些同工酶从组织进入体液后,进一步变化分为数个不同类型即所谓同工酶的亚型(isoform)。CK-MB的亚型MB1、MB2,CK-MM亚型MM1、MM2和MM3在AMI的诊断和溶栓疗效判断上都优于总酶和同工酶。
同工酶是一个复杂的生物现象,至今在分类、概念上尚有一些问题有待研究澄清。从临床诊断角度可将同工酶理解为一个包括有多种能催化相同生化反应的酶族,在这族中虽然不同酶都催化同一反应,但在理化性质上有差异,如它们对底物的亲和性、特异性不同,对抑制剂出现不同的反应,所带电荷、对热耐受性有差异等等。生化学者根据这些差异用各种物理、化学方法将同工酶分离测定,从本质上说这些差异与酶蛋白结构有关,或者是构成酶蛋白的氨基酸在组成或排列次序上出现差异,或者由于二级、三级乃至四级结构不同而造成上述不同的理化性质,同时这些结构差异又可引起酶蛋白抗原性的差异。目前正在大力发展这种用免疫方法测同工酶的技术,并取得明显进展。
同工酶之所以有较高的诊断价值与下面一些因素有关:
首先是某些同工酶有明显的组织分布差异,以目前临床应用最多的CK和LD同工酶而言,CK大量存在于三种肌肉组织中,单独总CK升高很难判断。CK升高的组织来源为:骨骼肌中主要为CK-MM,平滑肌中为CK-BB,心肌中虽然大多数仍是CK-MM,但却含有14%-40%的其它两种肌组织中没有或仅含少量的CK-MB,这样只要能测CK同工酶,根据同工酶变化,不难判断出释放CK的器官或组织。
LD虽然几乎存在于全身各种细胞中,但其五种同工酶在体内分布情况并不相同。如LD1主要存在于心肌和红细胞中,LD5则主要存在于肝脏和骨骼肌中,正常血清中同工酶分布为LD2>LD1>LD3>LD4>LD5,这样虽然心脏和肝的多种疾病都能引起总LD升高,但对血清LD同工酶影响却大不相同。如AMI时,LD1明显增高以致LD1>LD2,肝病时将出现LD5>LD4,AMI患者在LD1>LD2基础上,又同时出现LD5>LD4,可怀疑是否有右心衰竭,引起肝淤血。有些酶的同工酶与上述情况不同,只是细胞内定位不同,临床上有诊断意义的主要是线粒体同工酶,线粒体中有一些酶在结构和性质上往往与细胞质中同工酶有明显差异,临床应用较多的是线粒体AST。轻度病变时,由于线粒体有两层致密的膜,此同工酶很难进入血清,但在细胞坏死病变时,血清中线粒体同工酶常明显升高。近年来发现ALT也存在着两种同工酶,肝脏疾病时ALT经常升高,ALT同工酶测定应该对肝病预后判断有一定价值。
在胚胎期,和器官的发生发育一样,这一时期酶带有早期种族进化的痕迹,与出生后人体的酶往往有差异。当人得肿瘤后,肿瘤细胞可以出现返祖现象,有可能出现胚胎期同工酶,测定这类同工酶有助于诊断肿瘤,例如胚胎期CK主要为CK-BB,约有10%恶性肿瘤患者血清可查出CK-BB,又如一些肿瘤患者血中可查到胎盘性ALP等等。
总之,随着科学技术的发展,测定项目的增加,测定方法的简化,同工酶测定将在诊断疾病、判断疗效和预后方面起着愈来愈大的作用。
第二节 临床诊断中常用的血清酶类及其同工酶
上一节介绍了血清中酶浓度变化的机制以及在判断血清中酶测定结果的意义时必须注意的一些事项,但在具体应用各种酶测定来诊断疾病时,还各有其特点。
临床医师多从疾病出发,将酶测定结果和其它各种检查结果综合起来,分清主次,辨证地加以应用;对一个检验医师而言,单从这方面考虑显然是不够的,还应对具体每个酶有一个较全面的了解,这样才能为临床提供咨询,解释好各项测定的临床应用价值。
本节以常用的具体酶为中心,系统介绍该酶在临床论断中的应用,包括该酶的作用、性质特点、组织分布与其临床应用价值。
一、肌酸激酶(CK)及其同工酶
肌酸激酶(CK)催化下列反应:
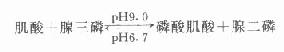
以前临床常习惯将此酶称为肌酸磷酸激酶(CPK),此名称不确切,目前国外已摒弃不用。此反应逆向反应速度约为正向反应速度6倍,与大多数激酶一样,Mg2+为它的辅基,需双硫键维持酶的分子结构。测定酶活性时必须加入巯基化合物,N-乙酰半胱氨酸(NAC)是目前最常用的激活剂。
CK作用生成的磷酸肌酸含高能磷酸键,是肌肉收缩时能量的直接来源,在3种肌组织和脑组织中含量最高。
CK是由两种不同亚基(M和B)组成的二聚体,这样正常人体组织常含3种同工酶,按电泳速率快慢顺序分别为:CK-BB(CK1),CK-MB(CK2)和CK-MM(CK3)。
除细胞质外,在心肌、骨骼肌和脑等组织细胞的线粒体内还存在另一种结构不同的CK,它也是二聚体,常简写为CK-MiMi,电泳时速度最慢,故命名为CK4。
【组织分布】
CK主要存在于骨骼肌、心肌、脑组织中,此外还存在于一些含平滑肌的器官如胃肠道,子宫内。而在肝、红细胞中含量极微或者没有。
骨骼肌无论在每克组织含量(328U/G湿重)还是绝对含量上都大大超过其它组织和器官。其中主要为CK-MM,不含CK-BB,仅有少量CK-MB(<3%)。心肌中CK虽只有骨骼肌的1/10(313U/G湿重),但CK-MB占CK总量的14%-42%,这在人体中是绝无仅有的。脑组织以及其中含平滑肌器官中的CK则几乎全部是CK-BB,它同时也是人胚胎中CK的主要存在形式。
【生理变异】
年龄、性别和种族对CK含量都有一定影响。
新生儿CK常为正常成年人的2-3倍。可能与分娩时骨骼肌损伤和缺氧有关,过6-10周可逐步下降接近成年人值。
CK含量和肌肉运动密切相关,其量和人体肌肉总量有关,男性参考值高于女性可能与这点有联系。国外调查,白种男性CK均值只为黑种人的66%,可能与种族有关,但也不排除两个人种之间体力劳动的差别。
【标本的采集、处理和贮存】
虽然红细胞中不含CK,但含有大量腺苷酸激酶(AK),目前常用的测CK的酶偶联法的反应体系中含有ADP。AK催化下列反应:
![]()
生成的ATP在指示酶6-磷酸葡萄糖脱氢酶作用下,可使NAD+生成NADH,340nm处吸光度升高,引起CK人为地升高。为去除AK的干扰,不少试剂盒中加入AK的抑制剂二腺苷-5-磷酸(DAPP)和AMP。但也有些试剂中未加入DAPP,此时溶血标本将产生明显干扰。
除肝素外,其它常用抗凝剂都能抑制CK活性。如不能及时测定,可将血清放冰箱贮存,-20℃中可长期保存。
【参考值范围】
成年男性参考上限为180U/L(37℃),女性为130U/L(37℃)。
正常血清中用电泳法查不到CK-BB,CK-MB不超过5%,如用免疫法测CK同工酶,依方法不同结果有异,可参考有关文献。
【临床应用】
CK及其同工酶是目前世界上临床测定次数最多的酶,80年代初期统计分别达一亿次和一千万次。这是因为CK在骨骼肌、心肌和脑疾患时常明显升高,如同时测定同工酶还有助于疾病的鉴别诊断。表7-4是各种疾病时CK的变化。
此酶测定主要用于早期诊断AMI和判断溶栓治疗的疗效以及判断疾病预后,特别在无Q波型AMI,此时心电图无Q波变化,常需其它检查来协助临床医师诊断和鉴别诊断AMI。目前有些国外学者认为在诊断AMI不需要同时作许多酶,只要单测CK一项即可。这是因为:
⑴心肌细胞含有大量CK:按每克组织含酶量计算,CK超过天冬氨酸转氨酶(AST,即谷草转氨酶)和乳酸脱氢酶(LD)等。所以当发生AMI、心肌细胞坏死时,释放出来的CK量超过其它酶。
⑵CK由于分子量不大,而且大量存在于胞质中,所以在各种酶中最早进入血液:一般在AMI后4-8小时就可能超过参考值上限。LD由于分子量大(约为125000),故增高明显迟于其它酶。同样线粒体中的酶,如线粒体AST在血中浓度升高明显推迟。
表7-4 总CK和CK同工酶在疾病时的变化
| 疾病 | 总CK | CK同工酶 | |
| 急性心肌梗死(AMI) | 常用酶中CK增高最早(4-8h),24h达峰值,2-3d恢复正常,中度升高,常在3000U/L以下 | 诊断AMI“金标准”(>6%),增高稍早于总CK(?),2-3天恢复正常 | |
| 心肌炎(病毒性,风湿性) | 急性期轻度升高,可达5倍正常上限 | CK-MB可增高 | |
| 增 | 心导管,冠状动脉造影 | 可有中度升高 | 一般无变化 |
| 运动试验) | 可轻度升高 | CK-MB正常 | |
| 肌肉损伤(挫伤、手术,肌注药物、癫痫、酒精和毒素中毒,过度运动) | 升高程度和损伤程度有关,严重病例可达10000U/L以上,肌注药物引起CK升高,一般不超过正常上限2倍,持续不超过一天 | 主要为CK-MM升高,CK-MB绝对值虽可升高,但百分比<6% | |
| 肌萎缩(Duchenne和Becker型) | 有症状患者CK可达50倍正常上限,女性携带者CK可为3-6倍正常上限,可查CK以筛选携带者 | 有些患者也可出现CK-MB升高 | |
| 多发性肌炎与肌炎 | CK明显增高 | 主要为CK-MM | |
| 高 | 神经性肌肉疾患 | CK正常 | 无变化 |
| 脑血管意外(脑出血,脑血栓) | 部分患者血中和CSF中CK升高 | CK-BB可增高 | |
| 恶性肿瘤 | 约10%病例CK-BB升高 | ||
| 甲状腺功能低下 | 可高达50倍正常上限 | 主要累及CK-MM | |
| Reye综合征 | 明显升高可在达70倍正常上限 | 主要为CK-MM | |
| 降 | 卧床患者 | 程度不等地下降 | 主要为CK-MM |
| 甲状腺功能亢进 | 程度不等地下降 | 主要为CK-MM | |
| 低 | 激素治疗 | 个别患者可下降 | 主要为CK-MM |
| 变 | Ⅰ型巨CK血症 | 电泳位于CK-MB和CK-MM之间可能是CK和IgA,或IgG复合物 | |
| 异 | Ⅱ型巨CK血症 | 电泳位于CK-MM后面,为线粒体CK和其聚合体 |
(3)由于CK主要存在于肌肉组织中,其它脏器如肝、肾、血细胞中含量极微:在诊断AMI时,其特异性远比一般代谢酶如LD、AST等为高,特别是溶血标本对CK测定结果影响较小。
近年来国内外使用动态法测定酶活性,大大提高了CK测定的准确性和重复性,并且可以在很短时间(约5分钟)测定完毕,特别适合于急诊患者。但CK测定的局限性也很明显,由于CK大量存在于3种肌肉组织中,尤其是骨骼肌中CK含量不论按浓度含量或按全身总量计算都大大超过心肌。因此骨骼肌局部或全身疾病都很容易引起血中总CK活性升高。血CK的极度增高主要见于全身疾病。曾有一例横纹肌溶解症患者,血中CK含量高达100000U/L,而在AMI时,就是大面积梗死,血中CK含量也很少超过3000U/L。肌注某些药物也可能引起CK一过性升高,但一般不超过正常上限值的2倍,持续时间不超过1天。因此国外有些医院将CK参考值上限的2倍作为诊断AMI的判断值,以减少假阳性。尽管CK测定诊断AMI的灵敏度很高,但特异性却明显偏低,其假阳性率达15%-30%。CK在体内的半寿期明显比其它酶短,心肌组织一次梗死后,CK急剧升高并很快恢复正常(48-72小时),这样在亚急性心肌梗死时CK可能正常,但其它酶如AST,特别是LD很可能居高不下。
以往对CK诊断AMI常强调下列几点:①AMI发病后8小时内查血CK不高,不应轻易除外AMI诊断,并以此结果作为基础值,和以后测定值作比较,任何怀疑AMI者应尽早抽血送检;②发病后24小时CK测定结果临床意义最大,因为此时正当CK的峰值时间,如不超过正常值上限可排除AMI诊断;③发病后48小时内应多次测CK活性,如不出现一个典型升高和下降的过程,应怀疑AMI的诊断。
近年文献指出,某些AMI患者血中CK可以不超过正常值,特别是不超过参考值上限的2倍,这些病例又多见于无Q波心肌梗死。长期以来认为CK峰值高低对AMI预后判断有一定价值。
CK-MB由于大量存在于心肌组织中,其它组织和器官中含量很少,所以CK-MB是目前诊断AMI的一个极其可靠的生化指标,特异性可达95%乃至更高。一般认为其灵敏度比CK总活性稍差,但也有文献报道,约有12.5%AMI患者总CK不高,但CK-MB超过正常。
CK-MB测定方法和试剂盒很多,同一标本不同实验室往往结果不同。从测定原理来分,可分为测酶活性与酶质量两大类方法。第一类方法先用物理、化学和免疫等各种手段将CK-MB和其它同工酶分开,然后根据酶的催化活性测CK-MB浓度,用活性单位/升(U/L)或百分比报告结果。测酶质量方法则是利用CK-MB的抗原性,根据抗原抗体反应测CK-MB酶蛋白浓度,单位为μg/L。两者结果有时不一致,因为如酶变性失活但仍保留抗原性,此时用测酶活性的方法结果不高,但用测质量方法可以增高。80年代早期德国学者根据此结果认为AMI患者血中存在着灭活的CK。但最近用改进方法测CK-MB,发现两法差异不大。国内普遍使用测酶活性方法,目前较多使用电泳方法分离CK-MB。电泳法特点是准确可靠,当出现一些异常同工酶如巨CKⅠ、巨CKⅡ等,从电泳图谱上很容易发现,并可通过光密度计进行定量测定,不会将CK-BB和各种异常同工酶误认为是CK-MB而误诊。其主要缺点是灵敏度较差。
近年来国内一些单位用免疫抑制法测CK-MB,其原理为抗M亚单位的抗血清可以和CK-MM及CK-MB中的M亚单位形成抗原抗体复合物,从而完全抑制M亚单位的酶活性,因为正常血清中几乎无CK-BB,故将此值乘以2可以认为大致代表CK-MB的活性。此法简单迅速,缺点是特异性差,如患者血清中还存在着CK-BB或者异常CK时,都将出现假阳性。故可作为过筛试验,如有疑问时用电泳法核实。另外此法正常值明显高于其它方法。如Sclavo公司试剂盒的正常值为18U/L,而电泳等其它方法都是<5U/L。有人认为这是因为CK-MM还可以进一步分为若干亚型,一般抗MM血清对MM3抑制较好,但对MM1抑制较差,而正常情况下血中MM亚型的存在以MM1为主,故导致正常值明显偏高。AMI第一天血中以MM3为主,但第二天以后则以MM1为主,因此使用此类方法诊断AMI和解释结果时应谨慎。
国外从80年代初建立了多种测CK-MB酶质量的方法。应用最多的是美国Hybriteeh公司的ELISA方法,正常人在5μg/L以下,此法结果和电泳法相关良好。缺点是测定时间长达半天,只适合成批标本。近两年来应用单克隆技术制备出特异性极高的只和CK-MB作用的抗血清,并已有多种试剂盒供应。最近Deerson以CK-MB≥10μg/L以及CK-MB指数≥3.0(μg/UCK×100)为AMI诊断标准。CK-MB单克隆抗体方法简单,特异性高,是目前重点发展的方法,近来有人认为此法有助于不稳定型心绞痛的诊断,可惜国内应用不多。
CK-MB的局限性在于骨骼肌损伤时也可能释放出一定量的CK-MB。虽一般认为骨骼肌含有的CK-MB在总CK中不超过5%,但也有例外。Evans报道1例横纹肌肉瘤的CK-MB高达274U/L,占总CK的28%。因此当CK-MB增高时,临床也应考虑有无非心肌来源的可能性。
骨骼肌中有大量CK,所以CK明显升高(3000U/L)大都见于肌肉疾病。此时CK测定用于鉴别肌萎缩病因,如Duchenne型和Becker型肌萎缩患者,常在临床前期血中CK就可中度和高度增加。该病是伴性遗传,在75%女性Duchenne型肌萎缩携带者和50%女性Becker型肌萎缩携带者血中也可查到CK升高。此外各种原因(病毒,细菌和寄生虫)引起的肌肉感染性疾病都能引起CK升高。但由于神经疾病引起的肌萎缩症,CK常不增高。
脑中虽含有较大量CK,脑血管意义时有大量CK释放入组织间隙,但由于血脑屏障存在,只能在部分病例中查到血中和脑脊液中CK升高,以及出现CK-BB,但阳性率不高,这妨碍了CK在这方面的临床应用。
人胚胎期主要为CK-BB,有人根据“癌胚”理论,提出CK-BB可作为肿瘤标记物,可惜的是阳性率不高,只占恶性肿瘤的10%左右。
二、乳酸脱氢酶(LD)及其同工酶
乳酸脱氢酶(LD)酶催化下列反应:
![]()
不同方法测出LD结果差异较大,这是因为LD有5种不同的同工酶,每种同工酶的最适反应条件不同。例如同为催化乳酸变为丙酮酸,LD1的最适pH为9.8,而LD5的最适pH为8.8。很难找出一个测定血清总LD的最适条件,检验医师在解释测定结果时应加以注意。
LD催化反应是无氧糖酵解的最终反应,广泛存在于人体各组织中,故测总酶临床意义不大,但LD是由两种不同亚基(M,H)组成的四聚体,形成5种结构不同的同工酶,其中H型亚基中酸性氨基酸较多,电泳时负电荷多,因此电泳速率较快。按电泳向阳极泳动快慢,分别命名为LD1(H4),LD2(H3M),LD3(H2M2),LD4(HM3)和LD5(M4)。
【组织分布】
LD虽广泛存在于人体各组织中,但是不同组织中同工酶组成有差异,大致可分为三类:一类以LD1为主,心肌为此类代表,LD可占总酶活性50%以上,此外有红细胞等。另一类以LD5为主,以横纹肌为代表,此外有肝脏等。第三类以LD3为主,脾、肺为此类代表。LD主要存在于细胞质中,线粒体中未查到。
【生理变异】
血清LD高低和性别关系不大,婴儿酶活性可达成年人两倍,儿童和少年活性比成年人高10%-15%,血清LD同工酶目前常用电泳法测定,由于具体方法差异,各学者报告的结果出入较大,但在成年人存在着如下规律:LD2>LD1>LD3>LD4>LD5,值得注意的是有学者报告,部分正常儿童血中LD1可大于LD2。
【标本的采集、处理和贮存】
由于血小板中也含大量LD,血清中和血浆所测LD有一定差异,一般都选用血清为测定标本,采血后应迅速分离血清,因红细胞中LD含量比血清高100倍以上,不宜用溶血血清为测定标本。
LD尤其是LD4和LD5与其它酶不同,不是热变性而是冷变性,在4℃贮存活性下降快于室温25℃,一般说25℃放2-3天LD活性变化不大,有条件者最好还是在采血后24小时内测定,标本应贮存于室温。
【参考值范围】
根据催化反应方向的不同,有两大类测LD方法,一大类为国际临床化学协会(IFFC)推荐方法,以丙酮酸为底物,其参考值范围为80-500U/L(37℃);另一大类以乳酸为底物,在我国应用较普遍,其参考值范围为50-150U/L。
【临床应用】
LD及其同工酶是临床上常用酶之一,尤其是LD同工酶和CK-MB是临床常规实验室最常测的两种同工酶。临床常用于诊断和鉴别诊断心、肝和骨骼肌的疾病。表7-5是LD及其同工酶在这些疾病时的变化情况。
表7-5 总LD和LD同工酶在疾病的变化
| 疾病 | 总LD | LD同工酶 | |
| AMI | 常测酶中,升高最慢(8-10h),但升高时间最长(5-10d),峰值可达10倍正常上限 | LD1>LD2,并可持续到两周(部分病例总LD正常) | |
| 充血性心衰,心肌炎 | 可升高达5倍正常上限 | LD1>LD2 | |
| 增 | 巨细胞性贫血 | 可升高达5倍正常上限,治疗正确迅速下降 | LD1>LD2 |
| 病毒性肝炎 | 部分病例可升高达5倍正常上限 | LD5>LD4(部分病例总LD正常 | |
| 肝硬化 | 常轻度升高 | LD5明显升高,LD5>LD4 | |
| 高 | 原发性肝癌 | 部分病例升高 | LD5明显升高,LD5>LD4 |
| 梗阻性黄疸 | 不定 | 常LD4>LD5 | |
| 肌肉损伤 | 视损伤程度不同而异 | 以LD5升高为主 | |
| 肌萎缩 | 可高达10倍正常上限 | 以LD5为主 | |
| 某些恶性肿瘤(白血病,结肠癌……) | 可升高,个别病例高达5倍正常上限 | 以LD3为主,LD3>LD1 | |
| 变异 | 某些病倒,电泳出现LD6,常显示预后不佳 |
实用中此酶用于诊断AMI,特别是亚急性AMI。在AMI时,LD由于分子量较大(125000),在常用酶中升高最迟,但其半寿期较长,增高持续时间可达5-10天,此时其它酶活性已恢复正常,在亚急性AMI诊断上有一定价值。可惜的是,LD总酶诊断AMI的特异性较差,很多其它疾病,此酶都升高。LD同工酶则不同,诊断特异性可达95%以上,仅次于CK-MB,甚至有学者认为,只要测此二同工酶,不需作其它酶学检查就可诊断AMI。LD同工酶的高特异性是因为人体各组织器官中LD同工酶的分布情况并不相同,除心肌外,只有少数器官如红细胞中的LD是以LD1为主,绝大多数的AMI患者血中的LD同工酶都可出现LD1>LD2变化,即所谓的“反转”(flip)类型变化,其持续时间甚至可超过总LD升高时间,达两周之久。临床上可见部分患者在恢复期,所有酶活性都降至正常,但仍有“反转”类型的LD同工酶变化,这些患者常有复发倾向,除AMI和心肌损伤,此种“反转”类型变化还见于溶血性贫血和巨细胞性贫血,但此类疾病在临床上不难与AMI相区分。
60年代临床上曾经用过所谓“α-羟丁酸脱氢酶”的测定来诊断AMI。实际上并不存在这种酶,所测的主要是LD1的活性,目前国外已较少测定此酶而改用更精确的测LD同工酶的方法。
其次,LD及其同工酶常用于肝脏疾病的诊断和鉴别诊断,由于肝细胞中主要为LD1,在肝实质病变时常出现LD5>LD4,但由于正常血清中LD5含量很少,不少病例就是出现同工酶异常LD5>LD4时,总LD活性仍为正常,故LD同工酶检查阳性率远高于总LD,一般在胆道疾患或梗性黄疸早期未累及肝实质时,不出现LD5>LD4,仍为LD4>LD5。值得注意的是,在骨骼肌疾病和损伤时,也会出现类似变化,临床上诊断各型肌萎缩也常测定LD及LD同工酶,早期常为LD5升高,但在晚期LD1和LD2也升高。
恶性肿瘤可由于肿瘤细胞恶性增长和坏死而引起血清LD升高,如转移到肝脏往往伴有LD4和LD5升高,此外一部分肿瘤,如白血病等常有LD3和LD4的增高。
三、氨基转移酶(ALT,AST)及其同工酶
氨基转移是氨基酸代谢中基本生化反应之一,在机体内存在着多达60种氨基转移酶,丙氨酸基转移酶(ALT)和天冬氨酸氨基转移酶(AST)是其中最重要的两种。
ALT催化下列反应:
![]()
AST催化下列反应:
![]()
它们都需要磷酸吡哆醛(维生素B4)为辅基,不含磷酸吡哆醛的酶蛋白称为脱辅基酶蛋白,没有催化活性。血清除含有有活性的全酶外,还有部分不含磷酸吡哆醛的酶蛋白,如在测定前,先加入足量磷酸吡哆醛,所测血清转氨酶活性常有明显升高。
AST有两种受不同基因控制的同工酶分别存在于细胞质(c-AST)和线粒体(m-AST)中,而一般认为ALT不存在同工酶,我国学者证实在人组织和血清中也存在类似AST的两种同工酶,即细胞质ALT(c-ALT)和线粒体ALT(m-ALT)。
【组织分布】
AST广泛存在于多种器官中,按含量多少顺序为心脏、肝、骨骼肌和肾,还有少量存在于胰腺、脾、肺及红细胞中,肝中AST大部分(70%)存在于肝细胞线粒体中。
ALT也广泛存在于多种器官中,含量最多的不是心脏,而是肝,顺序为肝、肾、心、骨骼肌等,与AST相比,在各器官中含量都比AST少,肝中ALT绝大多数存在于细胞质中,只有少量在线粒体中。
【生理变异】
此二酶生理变异较小,性别、年龄、进食、适度运动对酶活性无明显影响,每天虽有生理性波动,但无统计学意义。
表7-6 ALT和AST在疾病时的变化
| 疾病 | ALT | AST | AST/ALT |
| 病毒性肝炎 | 随不同病期和严重程度而异,常明显升高,可达10-100倍正常上限 | 同ALT,但程度没有ALT明显,恢复到正常早于ALT | <1.0 |
| 重症肝炎 | 不超过20倍正常上限,出现“肝疸分离” | 增高程度常超过ALT | >1.0 |
| 肝硬化 | 变化不定,常轻度增高 | 同ALT,但增高程度常超过ALT | >1.0 |
| 右心衰竭合并肝溢血 | 正常或轻度升高,个别可高达10倍正常上限 | 增高程度常超过ALT | >1.0 |
| 梗阻性黄疸 | 变化不足,常不超过5倍正常上限 | 同ALT | 不定,常<1.0 |
| Gilbert综合征 | 无变化 | 无变化 | |
| 溶血性黄疸 | 无变化 | 无变化 | |
| AMI | 正常或轻度升高 | 明显升高,与CK和LD相比,无何优点 | >1.0 |
| 心肌炎 | 正常或轻度升高 | 急性期可轻度升高 | >1.0 |
| 肌肉损伤 | 正常或轻度升高 | 可高达2-5倍正常上限 | >1.0 |
| 肌萎缩 | 明显上升,可达8倍正常上限 | 同ALT | >1.0 |
【标本的采集、处理和贮存】
红细胞中AST和ALT分别为血清含量的15倍与7倍,所以明显溶血标本不宜测此二酶。
宜采用血清为测定标本,此二酶在4℃冰箱中贮存一周,活性无明显变化,最好不要冰冻,因为在融冻时很容易破坏酶的活性。
【参考值范围】
一般临床使用方法中不加入磷酸吡哆醛,其参考值范围较加入磷酸吡哆醛的为低,ALT为5-40U/L(37℃),AST为8-40U/L(37℃)。
【临床应用】
根据此二酶在人体器官中分布情况,临床医师习惯将ALT用在诊断肝脏疾病,测定AST诊断AMI。在AMI时,不论在出现升高时间,还是升高持续时间,其变化介于CK和LD之间,无特殊价值,随诊断AMI的新试验日益增多,国外已建议不用。ALT在AMI时一般不增高或轻度增高,明显升高常说明有右心衰竭合并肝淤血,m-AST变化比总酶更慢,但其增高程度与坏死病变程度密切相关,是判断AMI预后的一个很好指标。
目前转氨酶测定主要用于肝胆疾病的诊断和鉴别诊断。ALT是我国目前测定次数最多的酶,假如能同时测定AST,并计算出文献上常提到的Deritis比值,即AST/ALT之比,在诊断和鉴别诊断上将是很有用的。
(一)丙氨酸氨基转移酶(ALT)
以前常简称GPT,近年来国外多以ALT为此酶缩写。ALT是我国测定次数最多的酶,这是因为我国肝炎较多,而肝是含ALT最丰富的器官,且大部分存在于肝细胞的胞质中。肝炎时,细胞膜通透性增加,由于肝细胞中ALT浓度约比血清高7000倍,只要有1/1000的肝细胞中的ALT进入血液就足以使血中ALT升高1倍,故此酶是肝损伤的一个很灵敏指标。肝炎时早在黄疸前期就升高,峰值可达数千单位,为正常上限的百余倍。一般而言在急性肝炎时,血清ALT活性高低与临床病情轻重相平行,又由于ALT半寿期较长,往往是肝炎恢复期最后降至正常的酶,是判断急性肝炎是否恢复的一个很好指标。此外在慢性肝炎特别是慢性活动性肝炎ALT也经常升高。因此临床医师对急慢性肝炎患者经常检查ALT并据此诊断和判断病情。
应注意两种情况,重症肝炎时由于大量肝细胞坏死,此时血中ALT可仅轻度增高,临终时常明显下降,但胆红素却进行性升高,即所谓的“胆酶分离”,常是肝坏死征兆。另一种情况是少数人血清中ALT长期持续升高,肝穿无明显病理改变,预后良好,对此现象目前仍在研究之中。
肝炎时ALT常有明显变化,但不能反过来认为凡ALT升高者就有肝炎,其它肝胆疾病如胆石症、胆囊炎、肝癌和肝淤血时也可升高。由于肝脏以外不少器官也含有ALT,因此在AMI、多发性肌炎、急性肾盂肾炎等患者血中ALT也可升高,但一般而言,这些疾病ALT升高很少超过正常上限10倍,常以400U/L为界,超过此值绝大多数可诊断为肝炎。但也有例外,有人报告个别肝淤血、胆石症等患者可超过此界,但在这种情况时,ALT变化往往很快,过几天复查,ALT就可降至400U/L以下。
(二)天冬氨酸氨基转移酶(AST)
以前常简称GOT,现多以AST为缩称,既往认为AST主要存在于心肌中,主要用于诊断AMI。但目前由于AST在AMI时升高迟于CK,恢复早于LD,故诊断AMI价值越来越小,国外不少学者认为诊断AMI,完全可以不测AST。
另一方面肝中AST含量虽低于心肌,但绝对含量(U/mg)仍高于ALT。肝中AST和ALT含量比值约为2.5:1.0。只是由于ALT主要存在于细胞质中,AST主要存在于线粒体中,病变较轻的肝脏疾病如急性肝炎时血中ALT升高程度高于AST,但在慢性肝炎,特别是肝硬化时,病变累及线粒体,此时AST升高程度超过ALT。故在国外对疑是肝炎患者常同时测AST和ALT,并计算AST/ALT比值,正常约为1.15,急性肝炎第1、2、3和4周分别为0.7、0.5、0.3和0.2。如比值有升高倾向,应注意有无发展为慢性肝炎可能,慢性肝炎时可升到1.0以上,肝硬化时可达2.0,此比值对判断肝炎的转归特别有价值。
四、碱性磷酸酶(ALP)
碱性磷酸酶(ALP)是指一组底物特异性很低,在碱性环境中能水解很多磷酸单酯化合物的酶,需要镁和锰离子为激活剂。近年为认为ALP真正作用是将底物中磷酸基团转移到另一个含羟基基团化合物上,磷酸乙醇胺有可能是ALP在体内作用的底物。
ALP确切的生理作用目前仍不十分清楚,一般认为骨中ALP和骨的钙化作用密切相关,成骨细胞中的ALP作用产生磷酸,与钙生成磷酸钙沉积于骨中。ALP还广泛存在于肝细胞血窦侧和胆小管膜上、肾近侧曲管刷状缘和小肠粘膜的微绒毛,据此推测它可能还与物质的吸收和运转过程有关。
各器官的ALP在理化性质都有些差异,临床上长期认为存在着肝、骨、小肠和胎盘等ALP,在病理时还可能出现“高分子”ALP,以及一些和肿瘤有关的变异ALP,如Regan、NagaoALP等。深一步研究发现这些ALP主要受3个基因位点控制,相应生成3大类ALP,即小肠、胎盘和组织非特异ALP,后者包括临床上所谓的骨、肝、肾等组织器官,它们的氨基酸结构类似,抗原性无差别,这些酶电泳速率和对不同抑制剂反应差异来自ALP中所含糖基的不同。
【组织分布】
ALP广泛存在于机体各组织器官中,按每克湿组织ALP含量多少排列,顺序为肝、肾、胎盘、小肠、骨等。
【生理变异】
总论中已经提到ALP变化与年龄密切相关,简言之,新生儿ALP略高于成年人,以后逐渐增高,在1-5岁有一次高峰,可达成年人上限2.5-5倍,以后下降,到儿童身长增高期又再次上升,第二高峰在10-15岁之间,可达成人上限4-5倍。20岁后降至成年人值,到老年期又轻度升高,可能与生理性的激素变化有关。
孕妇血清ALP在妊娠3个月即开始升高,9个月可达峰值,约为正常值的2倍,ALP升高可维持到分娩后1个月,升高的ALP来自胎盘,和胚泡壁的细胞滋养层的发育程度直接相关。
高脂餐后,血清ALP活性升高,尤以血型为O型或B型的分泌型(唾液中可分泌血型抗原者)人群更为突出,增高的ALP多为小肠ALP。
【标本的采集、处理和贮存】
为避免脂肪餐的影响,宜空腹采血,除肝素外,其它常用抗凝剂可与Mg2+作用,引起ALP活性下降。最好用血清为测定标本,红细胞膜上有ALP,明显溶血标本会干扰测定结果。
ALP在室温和冰箱放置后,活性逐渐升高,可增加5%-10%。冷冻血清融冻后,其中活性开始偏低,以后慢慢升高。所以应在采血当日就测定ALP活性。
目前常用的冻干质控血清,复溶后ALP活性常有一个活性升高过程,有些制品复溶24小时后,活性可升高50%-100%,使用时应加以注意。
【参考值范围】
ALP测定结果与所用方法种类和测定温度密切相关,就是同一底物,因缓冲液不同,其结果可有2-3倍差异,目前国内应用较多的方法中以磷酸对硝基酚为底物,2-氨基-2-甲基丙醇为缓冲液,在37℃测定时,成年人参考值范围为20-110U/L。
ALP在不同年龄人群中有较大差异,在以上有关章节中已加以论述,解释结果时必须加以注意。国外文献指出ALP虽然有较大的人群差异,但是个体间差异很小。有条件应将测定结果与被测者的“正常值”进行比较。
【临床应用】
ALP早在本世纪30年代就用于临床,是临床医师非常熟悉的酶之一,在70年代前ALP是世界上临床测定次数最多的酶,主要用于骨骼和肝胆系统疾病的诊断和临床鉴别诊断,可参见下表:
表7-7 碱性磷酸酶在疾病时的变化
(缺)
ALP和骨化过程密切相关,很多骨骼疾病如变形性骨炎(Paget病)、副甲状旁腺功能亢进、佝偻病、软骨症、原发性和继发性骨肿瘤、骨折和肢端肥大症的患者血中ALP都可升高,尤其是变形性骨炎,增高非常明显,可达正常上限值的50倍。此病在我国少见,我国常用于早期诊断佝偻病和软骨病,ALP升高早于血钙、磷变化以及X线检查,是一个很灵敏的诊断指标,但应注意骨质疏松患者血中ALP一般都在正常范围内。
目前ALP测定主要用于黄疸的鉴别诊断,阻塞性黄疸时血清ALP常早期明显升高,甚至可达正常上限值的10-15倍,在完全阻塞性黄疸病例中,80%病例血中ALP超过正常上限5倍。一般而言血清ALP持续低于正常上限值2倍时,阻塞性黄疸可能性较小,肝性黄疸时ALP轻度升高,一般不超过正常上限的2-3倍,约有半数原发性肝癌血中ALP升高,升高程度也常较明显,甚至可达正常上限值的15-20倍。如在无黄疸肝脏疾病人血中发现有ALP升高应警惕有无肝癌可能。
ALP升高还可见于其它疾病,如急慢性胰腺炎、慢性肾衰、肠梗塞、肺梗塞等疾病,但无太大临床价值,某些药物如孕激素、雌激素、胎盘白蛋白等也可引起血中ALP升高。
血ALP下降见于甲状腺功能低下、恶性贫血和遗传性碱性磷酸酶减少症等。
五、γ-谷氨酰基转移酶及其同工酶
γ-谷氨酰基转移酶(GGT)是一种底物特异性不高的酶,可作用于一系列含谷氨酰基的化合物。在体内可能催化下列反应:
![]()
由于有多个多肽参与反应,历史上曾命名为γ-谷氨酰基转肽酶,简写为GGTP。
本酶为含SH基的糖蛋白,其生理功能尚不十分清楚。Meister认为细胞外游离氨基酸需通过γ-谷氨酰基循环的一系列生化反应方能进入细胞,位于细胞膜上的本酶起关键作用,与组织中氨基酸和肽的分泌吸收,合成过程有关。
不同器官GGT的理化性质有差异,用琼脂糖电泳可将GGT分为四个区带。但目前认为控制GGT的基因位点只有一个,上述差异是由于酶蛋白中糖基组成不同而引起。
【组织分布】
人体各器官中GGT含量按下列顺序排列:肾、前列腺、胰、肝、脾、肠、脑等。大部分酶与细胞的膜结构结合,例如在肝细胞胆管侧的细胞膜上。虽然此酶在肾脏含量最高,学者相信血清中GGT主要来自肝脏。
【生理变异】
年龄与妊娠与否对GGT影响不大,不似ALP常明显升高,如同时测此二酶,有助于临床医师判断肝脏是否有病变。
男性血中GGT含量明显高于女性,可能与前列腺有丰富的GGT有关。酗酒会引起GGT明显升高,升高程度与饮酒量有关,诊断疾病时必须排除这一因素。
【标本的采集、处理和贮存】
GGT是一个比较稳定的酶,室温中2天、4℃放1周活性无变化,-20℃可储存1年,红细胞中GGT含量很低,溶血对本酶测定干扰不大。
【参考值范围】
以γ-谷氨酰-3-羧基-4-对硝基苯胺和双甘肽为底物的动态法,在37℃测定,成年男性的参考值为7-56U/L,成年女性为0-30U/L。
【临床应用】
GGT在疾病时的变化可参阅表7-8:
表7-8 γ-谷氨酰基转移酶在疾病时的变化
| 增高 | 无变化 | |
| 胆道疾病 | 明显增高,可达5-30倍正常上限 | 骨疾患 |
| (胆石症、炎症) | 阳性率高,为85%-93% | 妊娠 |
| 肝实质疾病(肝炎,肝硬化) | 轻度增高,可达2-5倍正常上限 | |
| 肝癌(原发,继发) | 明显增高且阳性率高 | |
| 诱导作用 | 轻度升高,停药后降至正常 | |
| (乙醇,苯巴比妥,抗抑郁药,抗癫痫药,含雌激素避孕药 |
GGT测定在我国临床应用很广,可能是因为我国肝脏疾病发病率高,而GGT又是肝胆疾病中阳性率最高的酶。德国内科学会通过对13000例肝胆患者调查,发现95%病例GGT升高,其次才是ALT,阳性率为83%,第三位是胆碱酯酶,如同时测此三种酶,阳性率可达99%。
胆道疾病如胆石症、胆道炎症、肝外梗阻时,GGT不仅阳性率高,而且升高明显,可高达正常上限的5-30倍,肝脏实质疾病如肝炎、脂肪肝、肝硬化时GGT一般只是中度升高(2-5倍),这点有助于肝胆疾病的鉴别诊断。
GGT又是诊断恶性肿瘤有无肝转移的一种很有用的试验,肿瘤患者如GGT升高常说明有肝转移可能。
当ALP升高时,同时测GGT有助于鉴别ALP的来源,如GGT正常说明此时ALP升高常来自肝外的器官,如骨骼疾病等。
临床在使用GGT时一定要注意它是一种诱导酶,不少药物能使血中GGT活性升高,如巴比妥类药物、抗癫痫的苯妥英钠、抗抑郁症的三环化合物、抗热解痛类的对乙酰氨基酚可引起GGT升高;抗凝药香豆素、含雌激素的避孕药以及治疗血脂升高的氯贝丁酯(法脂乙酯)都可引起GGT轻度升高。还有酗酒可使其升高,这些因素限制了此酶的应用价值。医师在应用此酶诊断疾病时,必须先除外这些非病理因素引起GGT增高的可能,国外有些国家由于酗酒人多,以致不测或少测此酶。
六、淀粉酶(AMY)及其同工酶
AMY对食物中多糖化合物的消化起着重要作用。作用于多种糖化合物,如淀粉、糖原等。它不同于植物中的β-淀粉酶,仅作用于多糖化合物的末端,α-淀粉酶可以随机作用于多糖化合物内部的α-1,4葡萄糖苷键,产生一系列分子不等的产物:糊精、麦芽四糖、麦芽三糖、麦芽糖和葡萄糖。
AMY是一种需钙的金属酶,其最适pH在6.5-7.5之间,卤素和其它阴离子有激活作用(Cl->Br->NO3->I-)。其分子量较小,约55ku,易由肾脏排出,尿中AMY活性浓度常高于血。
两个不同基因位点分别控制,并生成两种结构和抗原性有明显差异的AMY同工酶:一种由胰腺分泌,习惯上命名为胰腺淀粉酶(P-AMY);另一种主要来自唾液腺,常命名为唾液腺淀粉酶(S-AMY),但并不意味着只有唾液腺产生此种同工酶,其它器官如肺、骨、卵巢和甲状腺也能生成S-AMY。
【组织分布】
机体中胰腺含AMY最多,由腺泡细胞合成后通过胰管分泌入小肠,唾液腺也分泌大量AMY入口腔开始消化多糖化合物,此外AMY还见于卵巢、肺、睾丸、横纹肌和脂肪组织中,在某些肺癌和卵巢癌患者中可查到AMY升高,且主要为S-AMY,肝中AMY很少乃至没有。
【生理差异】
成年人血中AMY与性别、年龄、进食关系不大,新生儿AMY缺乏,满月后才出现此酶,逐步升高,约在5岁时达到成人水平,老年人AMY开始下降,约低25%。
尿AMY浓度由于受尿液浓缩或稀释的影响,随意留尿测定AMY的诊断价值受到一定限制,国外学者多建议留6小时或24小时尿液测其AMY总含量更为可靠。
【标本采集、处理和贮存】
有些实验室常用血浆作为标本测AMY,这样可以更快地报告结果。但由于AMY为需钙酶,大多数常用抗凝剂都将抑制AMY活性,还是以血清为标本较好。收集标本时必需注意避免唾液的污染,否则将引起假阳性。
AMY是一种很稳定的酶,室温中一周,4℃几个月酶活性都无变化。
【参考值范围】
国内大多数实验室仍使用苏木杰(Somogyi)的碘-淀粉呈色法,由于各实验室所用水解淀粉来源不同,不同实验室所测结果有所差异,但大多数血AMY都在200苏氏单位以下。
愈来愈多临床实验室使用人工合成基质,在自动生化分析仪上测定AMY,但目前尚无成熟的AMY推荐法,不同厂家出的试剂盒由于所用基质不同,结果很难一致,其参考值范围可参考各厂家说明书。
【临床应用】
临床上测定血清AMY主要用于诊断急性胰腺炎,此酶常在腹痛后2-12小时升高,20-30小时达到高峰,一般为正常上限值的4~6倍,最高可达40倍。此值的高低和疾病预后关系不大,但此值愈高,急性胰腺炎的可能性就愈大,约在3-4天内AMY活性恢复正常。急性胰腺炎时尿AMY也增高,且持续时间更长,但由于尿浓度还受尿液浓缩稀释影响,很难定出正常和病理界限,定时收尿测排泄AMY总量,结果虽较可靠但收集麻烦。国外学者认为测尿AMY诊断价值很小。临床医师有时要求测胸水或腹水中AMY活性,如显著升高说明此时急性胰腺炎病变累及胸、腹腔。
诊断急性胰腺炎时必需与其它急腹症相鉴别,可惜的是此时AMY也有不同程度升高,一般说来不超过正常上限的4倍或者500苏氏单位。此值常用作诊断急性胰腺炎的判断值,但也不是绝对的,个别急腹症患者AMY可超过此值,甚至达到2000苏氏单位。
唾液腺疾病如腮腺炎患者血AMY也可升高,但临床上不难鉴别,且主要为S-AMY升高,有别于胰腺炎以P-AMY升高为主。
国外报道在1%-2%人群中可出现巨淀粉酶血症,血中AMY和免疫球蛋白(IgG或IgA)形成大分子的复合物,临床化验特点为血中AMY持续升高,尿中AMY正常或下降。进一步检查可发现血中AMY分子量增高,此现象不和具体疾病有关,增高者也多无临床症状,注意应与病理性AMY升高相区分。
七、酸性磷酸酶(ACP)及其同工酶
酸性磷酸酶(ACP)是一组作用类似碱性磷酸酶的酶,不同点是最适pH偏酸,从pH4.5-7.0。
本酶生理作用还不十分清楚,主要存在于细胞的溶酶体中,是溶酶体的标记酶之一,推测此酶参加细胞中的分解代谢。
目前发现有4个不同的基因位点生成4类不同的同工酶,第一个基因位点生成前列腺ACP,其它组织仅有微量存在,有特异抗原性,用免疫学方法很易与其它ACP分开;第二个基因位点生成为溶酶体ACP,存在于细胞的溶酶体中;第三个基因位点生成为红细胞ACP,实际上不仅存在于红细胞中,还广泛存在于细胞质中;第四个基因位点生成ACP存在于破骨细胞和一些吞噬细胞,如肺泡的巨噬细胞,病理时还存在于高雪细胞和白血病的“毛细胞”中。
【组织分布】
它几乎存在于体内所有细胞中,虽然在细胞质中可找到ACP,但主要在于溶酶体,ACP主要组织来源是前列腺、红细胞和血小板。正常男性血清中1/3和1/2ACP来自前列腺,其余部分和女性血中ACP可能来自血小板或破骨细胞。
【生理变异】
男女性血中ACP含量无差异,新生儿ACP活性与成人相似,出生后1个月中血清酶活性甚高,然后随年龄的增长而逐渐下降,青春期又可出现一活性峰值,至20岁降至成人水平。
【标本的采集、处理和贮存】
常用抗凝剂抑制ACP活性,以血清为测定标本较好。
ACP是常用酶中最不稳定的一个,尤其在37℃和偏碱性时灭活更快,此时放置1小时,酶活性可丧失50%,如将pH降至6.5以下,酶比较稳定。最简单方法可按1ml血清加入10%醋酸20μl酸化,此时酶在常温可稳定1天,4℃3天,-20℃稳定几个月。
【参考值范围】
不同方法参考值也不同,国外多推荐使用磷酸麝香草酚为底物的方法,因为前列腺ACP对此底物亲合度高,测定结果基本能反映ACP含量高低,此法参考值范围为0.5-1.9U/L,男女无差异。
【临床应用】
主要用于论断前列腺癌,由于酶不稳定,测定困难,目前正被其它前列腺癌标记物如前列腺特异抗原(PSA)所取代。
第三节 缺血性冠状动脉疾病的酶学诊断
缺血性冠状动脉疾病包括了心绞痛和急性心肌梗死(AMI)等疾病,其中AMI是西方发达国家主要死因之一。近年来随经济发展人们生活水平提高,此病在我国发病率明显增加,早期诊断、适当治疗对于减低此病死亡率和合并症是极其重要的。
WHO在1997年提出诊断AMI三个方面的依据:即第一,临床表现和症状主要为胸部剧烈疼痛;第二,心电图上特异改变,如出现Q波;第三,血清中有关酶浓度出现升高下降的动态过程。一般认为患者具有三大表现中任何两项都可诊断为AMI。目前一些AMI患者可以缺乏典型胸痛症状,临床上称为寂静型AMI(silentAMI),国外文献认为此种类型约占1/4AMI患者。此类患者多见于老年人,尤其是糖尿病患者。另外一些AMI患者心电图检查查不到AMI特有的Q波,病理检查时这些患者的坏死病变不累及或少累及心脏外层,临床上常称为内膜下或非穿壁性心梗。目前更多使用“无Q波心梗”术语取代上述名词,其发病率约占总AMI的1/3,这样血清中有关酶浓度的动态变化对AMI诊断具有了较特殊价值,是诊断和鉴别诊断AMI的重要依据。
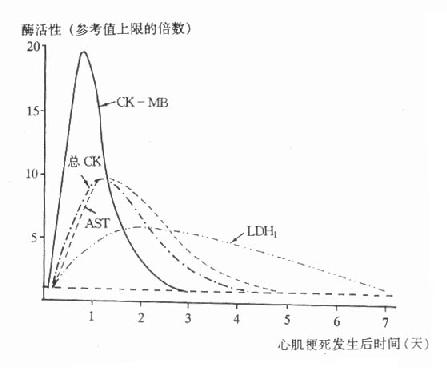
图7-2 血清酶在单纯性AMI后的变化
图7-2 是AMI患者血清酶浓度的典型变化图:
从图7-2可以看到AMI时,任何酶变化都有一个延迟期,也就是说出现心肌梗死时虽然酶已释放到细胞外,但由于病变局部没有血液通过,释放的酶需经由系统缓慢进入血液,大约在4-8小时后,血清酶活性才开始超过参考范围的上限值。在常测酶中首先升高的是CK-MB,升高程度可高达参考值上限的10-25倍,由于骨骼肌中也含有少量CK-MB,但不超过5%,所以临床上更多地愿意用CK-KB占总CK的百分比(<6%)来诊断AMI。CK-MM或者说总CK变化类似CK-MB,但升高和恢复正常都略迟,常在AMI发病8-12小时后才开始升高,24-48小时达到峰值,总酶升高持续7天,LD1持续时间更长达10天,升高程度较低,一般为参考上限的3-4倍。由于心肌中的LD主要为LD1,因此在理论上LD1诊断特异性更高,但这二者的测定都受到溶血干扰,轻度溶血就有可能引起血清LD和LD1非病理性升高,临床上应用此酶必须注意此点。
在我国医院实验室,往往还将天冬氨酸氨基转移酶(AST)和羟丁酸脱氢酶(HBDH)和上述两种酶作为一组测定项目,用于论断AMI。但从图可以看出总AST测定对论断AMI毫无优点,当AST升高时几乎无一例外都伴有CK/或LD升高,如果考虑到AST还大量存在于其它器官,如肝、肌肉内,其测定特异性明显低于CK。
至于HBDH实际上并不存在此种酶,所测的仍是LD的活性,只在当以酮丁酸取代丙酮酸为底物测LD活性时,LD1比其它同工酶对酮丁酸有更大活性,所测结果更能反映出LD1的活性。随着有更好方法,如电泳法、免疫法和化学抑制法测LD同工酶,已无必要再测定此酶以诊断AMI。
随科学技术发展,人们对缺血性冠状动脉疾病的发病机制、分类和治疗有了进一步认识:冠状动脉粥样硬化开始于血管壁上出现富含胆固醇及其酯类的脂肪条纹,病变进一步增大成为粥样硬化斑(atheroma),斑块可以大部分阻塞血管内腔,限制血流,劳累后由于心肌供氧不足,引起心区疼痛,而在休息后或者用血管扩张药后得到缓解,这就是典型的稳定型心绞痛症状。斑块的变化是多种多样的,斑块可钙化变得僵硬和易碎,部分斑块可能进入血流引起下面小血管栓塞,在动脉壁上由于斑块脱落形成溃疡,血小板聚集在溃疡面,释放出强有力的血管收缩因子如TBX-2,有可能在血小板聚集基础上形成血栓,阻塞血流,引起供血障碍,心肌组织病变乃至坏死,出现典型的AMI症状。
栓塞形成后,可能激活体内的溶栓系统活性,促使血栓溶解,血液又重新灌注(reperfusion),但此过程往往已在梗死发生后较长时间后才发生,此时心肌组织已经坏死,如果溶栓出现在心梗发生后6小时内,病变心肌组织有可能不坏死,病变转逆。目前治疗AMI广泛在早期应用溶栓药,如尿激酶、链激酶、组织型纤溶酶原激活剂等,防止心肌坏死形成瘢痕。这些新疗法的出现对实验室提出一些新的要求,如要求早期诊断AMI,用常规测酶方法常不易达到此要求,因为发病后4-6小时正相当这些酶的延迟期,CK同工酶亚型的发现为早期诊断AMI提供新的可能性。
80年代以来发现CK同工酶MM和MB虽然都各由基因位点控制形成组织型的CK-MM3和CK-MB2,但它们释放入体液后在体液中羧基肽酶作用下,分解M亚基羧基端的赖氨酸,改变了MM3和MB2的等电点,可以进一步分为不同亚型(isoform),即CK-MM3可进一步变为电泳速度较快的MM2和MM1,CK-MB2变为MB1。正常血清中亚型含量分别为MM1>MM2>MM3,MB1>MB2,但当AMI时由于组织型MM3和MB2大量释放入血,MM3/MM1和MB2/MB1,比值超过1.0,此变化明显早于CK和CK-MB升高。MB2/MB1在AMI发病后1.5小时达到峰值,MM3/MM1则在3小时达到峰值,Wu在最近提出早期诊断首选的是CK亚型和肌红蛋白,确诊Ami则仍以CK-MB以及最近提出的肌钙蛋白(troponin)为佳,晚期则选用LD和肌钙蛋白。
另一较大进展是使用免疫学方法测CK-Mb的质量(μg/L),而不是用经典方法测CK-MB的活性浓度(U/L)。特别是使用抗CK-MB单克隆血清合并化学发光技术,不仅灵敏度很高,检出值为0.8μg/L,操作简单,时间短,特别适合于急症标本。在发达国家已逐步取代其方法成为测定MB的主要方法,临床上也常以CK-MB指数>6%来诊断AMI,指数按下式计算CK-MB(μg/L)/CK(U/L)。最近有人报告用酶联法测糖原磷酸化酶BB同工酶的质量和CK、CK-MB、肌红蛋白以及肌钙蛋白,比其早期诊断AMI灵敏度高。
使用溶栓药后,需了解用药是否有效,即是否出现再灌注。冠状动脉血管造影是判断是否出现再灌注可靠的方法,但需一定设备,并且是一种损伤性检查,临床医师往往可以根据酶峰值出现时间提早来诊断,如栓塞溶解,出现再灌注,血液通过梗塞部分,可将释放到病变组织中的酶冲洗到血中,酶峰值出现变早。
从冠状动脉粥样硬化的病变和血栓形成、溶栓过程的动态变化,不难理解在典型的心绞痛和心肌梗死之间还存在着各种过渡的病变。例如在按WHOAMI诊断标准未能诊断AMI患者死亡时,可发现有小灶性坏死病变。
最近提出一个新的分类,将此类伴有坏死病变的缺血性心脏疾病命名为IMD(ischemicmyocardialdamage)缺血性心肌损伤,包括两类疾病:一类是符合WHO诊断标准的AMI,另一类缺乏典型心电图和酶学变化,但灵敏的心肌损伤指标如肌损伤MMD(mi-normyocardialdamage)。
另一方面心绞痛发作可不仅限于劳累时,一部分患者在安静时也可以出现心绞痛,其中一部分还可以进一步发展为AMI和出现猝死。临床上将其分类为不稳定型心绞痛,目前认为TnT和CK-MB质量测定有助于临床判断这些患者的预后,一些临床医师主张对这些患者开展一些较积极的抗凝或溶栓治疗。
最近发现一些心肌蛋白可作为心肌损伤的较好指标,尤其是TnT和肌钙蛋白I(TnI)。肌钙蛋白是由三个亚单位组成(TnT,TnI和TnC),它们为心肌的调节单位,在控制心肌收缩中起着重要作用,心肌TnT和TnI的抗原性与骨骼肌有明显差异,可以得到特异性很高的单克隆血清。由于血清中浓度很低,TnT一般不超过0.1μg/L,心肌病变时可成百倍升高,由于分子量较小,升高出现时间都长。目前用TnT诊断AMI,判断不稳定型心绞痛患者预后的价值已为临床所公认,但也发现在肾透析患者血中可有不明原因TnT浓度升高。TnI临床价值仍在探索中,有望成为另一灵敏的心肌损伤指标。
第四节 肝脏疾病的酶学诊断
从1995年卡门(Karmen)等发现急性肝炎患者在黄疸前期,血清中转氨酶(ALT,AST)活性就已明显增高以来,用于诊断肝脏疾病的酶不下几十种。在我国ALT是应用最广泛的酶,其次是碱性磷酸酶(ALP)、γ-谷氨酰基转移酶(GGTP),其它有精氨酸代琥珀酸裂解酶(ASAL)、酰苷脱氨酶(ADA)、5’-核苷酸酶、甘露醇脱氢酶等。总的说来这些酶都能比较确切反映肝胆系统的炎症或坏死性病变,在我国对反映肝脏合成功能方面的酶试验重视较差。公认假性胆碱脂酶(CHE)能反映肝脏蛋白合成能力。还有文献报告卵磷胆固醇酰基转移酶(LACT)也能反肝细胞酶和蛋白的合成能力,但目前此酶所用方法不实用。
1975年德国内科协会年会提出ALT、GGTP和CHE三种酶作为肝脏疾病时的酶过筛试验。三种酶分别反映肝脏的不同病理过程:ALT增高往往说明肝实质细胞的损伤;GGTP增高可以说明胆汁淤积和对肝的“诱导作用”;测定CHE可以检查肝脏的蛋白合成功能。当时德国选用这三种酶是建立在大量的临床观察的基础上,他们统计了10年内13000名肝脏疾病患者的检查结果,发现GGTP增高占病例的95%,GPT为83%,CHE有变化的为74%,其中63%活性下降,11%上升。其余酶阳性率的顺序为AST71%,ALP60%,GLDH47%和LDH34%。如同时测定GGTP、GPT和CHE三种酶可查出99%的肝胆疾病患者。
急性肝炎恢复期患者约有半数ALT是最后一个恢复正常的生化指标,另半数则是GGTP。另有文献报道在肝炎时ADA恢复正常迟于ALT。
大多数中毒病例如果累及肝脏,GGTP灵敏度往往高于转氨酶,尤其是酒精性脂肪肝。血中ALT活性正常而GGTP增高还见于胆石症、胆囊炎、药物的诱导作用以及肝脏慢性淤血等。
很早就知道CHE下降是判断肝脏疾病预后一个很好的指标。而在脂肪肝时随ALT和GGTP的增高,CHE也可能增高。
根据一个酶试验的结果就进行鉴别诊断和判断预后决非易事。所以后来一些学者企图通过比较不同酶变化的结果来提高酶试验应用的价值。DeRitis首先提出AST/ALT比值的诊断意义,ALT主要存在于细胞质中,AST则有一半位于线粒体中,另外ALT在血中半寿期明显高于AST3倍,所以在典型急性肝炎患者血ALT活性不仅升得高并且持续时间长,在急性肝炎时随时间延长DeRitis比值逐步下降。在病程的第1、2、3和4周,其比值分别为0.7、0.5、0.3和0.2。在慢性活动性肝炎和肝硬化时,由于细胞进一步坏死,AST增高程度往往超过ALT,DeRitis比值大于1。肝硬化尤其酒精性肝硬化,此比值可达3.0。文献上还曾介绍LD/AST、GGTP/AST等,但目前已少用。
国内外都有文献报告GGTP和ALP同工酶测定有辅助诊断肝癌作用。LD同工酶试验对肝病诊断也有一定价值。肝中LD以LD5为主,但在正常血中一般不超过10%,所以肝病时测总LD活力价值不大,其阳性率很低,但如测定LD同工酶,则LD5阳性率明显升高,急性肝炎LD5增高患者中有半数LD总活力正常。慢性肝炎中此现象更明显,72%病例LD不增高但LD5异常。一般都认为LD5增高是肝细胞坏死的一个很好指标,可用于判断肝炎治疗效果。重症肝炎时,如LD5持续增高常是需要换血疗法的一个重要指标。
急性肝炎时测定线粒体谷草转氨酶(m-AST)有助于了解肝病严重程度。Ideo等报告28例急性肝炎,血清m-AST/总AST为32.10(9.28U/L)。如果总AST升高明显,而m-AST/总AST比值较低,说明病情较轻,反之,如果比值较高,常有重度线粒体破坏,见于肝坏死。国内有人认为测定m-AST可以在一定程度上取代肝活检。
以往在解释为何肝病时血清酶变化明显时,往往简单地认为是因为肝脏是人体的代谢库,含有多种高浓度的酶,肝细胞病变时细胞膜通透性增加或者细胞坏死,细胞内酶释放入血以致血清中酶活性升高。这显然不能很好解释复杂的临床现象,例如不同的病因(病毒、毒品、淤血等)可以引起类似的肝细胞病变,但酶的变化规律并不相同。这与下列三个因素有关。
(一)肝组织结构和血清酶活性变化
肝有两大类型细胞(肝细胞和非肝细胞),通过近年来定量组织学的研究发现,每立方厘米肝细胞中非肝细胞(枯否细胞、内皮细胞和贮脂细胞)约为0.92亿,肝细胞含量在1.5亿到1.7亿之间,非肝细胞的数数为肝细胞的1/2。非肝细胞体积小于肝细胞,其总体积为肝细胞体积的10%。一般认为量为肝细肝细胞代谢比其它非肝细胞更为活跃,酶含量高于非肝细胞,但已有文献报道按每克蛋白质计算,非肝细胞所含的已糖激酶和葡萄糖-6-磷酸脱氢酶活力高于肝细胞。所以在研究肝脏疾病酶变化规律时,不能只看到肝细胞病变而忽视其它细胞特别是枯否细胞的病变。目前对这方面的研究是很不够的,特别缺乏非肝细胞病变和血清酶变化间关系的文章。值得一提的是腺苷脱氢酶(ADA),急性肝炎时血中此酶一般是中度增高,但是传染性单核细胞增多症时知中ADA增高非常明显,此现象还见于肠伤寒。这些都提示血中ADA活性变化可能和单核吞噬细胞系统病变有关。Neef将肝功能试验分类时,也提出ADA可以反映肝脏间质细胞的反应。还有文献报道肝硬化时N-乙酰-氨基葡萄糖苷酶(ANG)活性增高,此酶主要存在于溶酶体,所以不能排除肝硬化时非肝细胞病变引起ANG增高。
肝细胞与非肝细胞在细胞的亚细胞器方面差异较大。例如肝脏中的线粒体都集中在肝细胞中,非肝细胞中的线粒体只占总数的1.2%。另一方面非肝细胞中含大量的溶酶体,可占总数的43%。膜结构主要存在于肝细胞中,非肝细胞结构面积仅占总面积的8%,但其中高尔基复合体膜面积为总高尔基复合体面积的15%,质膜占27%,溶酶体的膜更多为30%。这从另一方面说明肝细胞中膜结构主要集中于线粒体和内质网。
由于线粒体几乎全存在于肝细胞中,所以检测代表线粒体酶的变化往往能较准确地反映出肝细胞病变程度,尤其是坏死性变化。因为线粒体有两层膜,一般病变并不能促使线粒体中的酶释放入血。谷氨酸脱氢酶(GLDH)和异柠檬酸脱氢酶(以NADP为辅酶)是最能反映肝细胞线粒体苹果酸脱氢酶(m-MDH)的方法,在判断肝细胞病变估计肝病患者预后方面具有一定的价值。
还有人探讨了过氧化物体中的酶,如过氧化氢酶,与内质多有关的酶,如阿司匹林酯酶变化对肝病的诊断价值,但尚看不出有任何明显的临床意义。
(二)肝细胞的不均一性
近年来通过组织化学定性研究,显微分光光度计定量测定、电子显微镜观察等各种手段和技术已经知道,肝脏中不仅存在着肝细胞和非肝细胞的不同,不同的肝细胞也存在着差异,不仅表现在代谢功能上,还表现在细胞结构上。
现常以汇管区肝小叶动静脉终末分枝为中轴进行划分肝小叶,由内向外分为Ⅰ,Ⅱ和Ⅲ带。
不同区带的肝细胞血液供应有差别。氧含量很高的肝动脉血和富含营养物质的肝门静脉血由汇管区的小叶间动脉和静脉进入肝小叶的血窦,依次同过Ⅰ、Ⅱ和Ⅲ带,最后到达中央静脉,所以Ⅰ带的肝细胞氧和营养物质供应最充分。但另一方面通过血液进入肝脏的有毒物质和致病因子也先到达Ⅰ带。这些特点不可能不对各区带的肝细胞产生影响。在电子显微镜下Ⅰ带存在着大而均一的线粒体,Ⅲ带的线粒体少而大小不一。测定Ⅰ带线粒体剖面积,其平均值为0.43μm,嵴突长度的平均值微7.5μm约为Ⅲ带的2倍,其值分别为0.2μm2和0.3μm2。
这促使科学家们去研究不同区带细胞中酶的分布情况。表7-9是综合各学者报告的结果。
表7-9 肝小叶不同区带酶分布情况
| 主要存在于Ⅰ带的酶 | 均匀分布的酶 | 主要存在于Ⅲ带的酶 |
| 细胞色素氧化酶 | 磷酸甘油脱氢酶 | 谷氨酸脱氢酶 |
| 琥珀酸脱氢酶 | 苹果酸酶 | 丙酮酸激酶 |
| 乳酸脱氢酶 | 单胺氧化酶 | 异柠檬酸脱氢酶(NADP) |
| 苹果酸脱氢酶 | 葡萄糖激酶 | 乙醇脱氢酶 |
| 天冬氨酸氨基转移酶 | 已糖激酶 | 3-羟丁酸辅酶A脱氢酶 |
| 丙氨酸氨基转移酶 | 已糖二磷酸激酶 | NADH-四唑还原酶 |
| 糖原合成酶 | 磷酸甘油醛脱氢酶 | NADP-四唑还原酶 |
| 糖原磷酸化酶 | ||
| 磷酸葡萄糖异构酶 | ||
| 葡萄糖-6-磷酸酶 | ||
| 磷酸烯醇丙酮酸激酶 |
Pett和Wmmer还进一步报告了各个酶在Ⅰ带和Ⅲ带浓度间的比值。琥珀酸脱氢酶比值为0.91,苹果酸脱氢酶为1.68,乳酸脱氢酶为1.32,异柠檬脱氢酶为0.52,谷氨酸脱氢酶为0.55,NADH四唑还原酶为0.58,磷酸甘油氧化酶为1.0。
不同病因引起肝的病理变化不尽相同,如右心衰竭时,Ⅲ带供血情况最差,此处肝细胞往往大量坏死,ICD和GLDH大量释放入血。肝炎则往往是肝脏弥漫性病变或者Ⅰ带肝细胞病变更严重些,所以ICD和GLDH变化较小。临床医师注意到右心衰竭时血中氨基转移酶也往往明显增高,如能同时测一下ICD和GLDH活性,往往有助于急性肝炎的鉴别诊断。有学者提出如ICD/AST比值≤1.5则主要是肝细胞坏死,反之如比值≥1.5,则是Ⅲ带肝细胞坏死。
(三)肝血窦的解剖特点
肝血窦缺乏完整的内皮细胞层,肝细胞通过狄氏腔直接和血液相接触,已知狄氏腔有广大的表面积高达600m2,为人肺表面积的7倍,因此即使轻微的肝细胞病变,细胞中的酶也很容易释放到肝血液中,并很快分布到全身,以致末梢血中酶浓度迅速并明显增高。人体其它器官例如肌肉、肾脏中虽也含有大量酶,如肾脏中谷氨酰基转移酶(GGTP)含量甚至是肝组织的25倍,但这些器官病变时,细胞中的酶不可能直接释放入血液,而是先释放入组织液或肾小管中,以后通过毛细管内皮(已知骨骼肌中毛细管通透性很低)或者淋巴系统才能进入血液中。所以这些器官病变时血中酶活力增高较慢也不太明显。升高的酶分子量往往较小,因为大分子的酶通不过毛细血管壁。这些限制因素对肝脏是不存在的,所以肝病时酶活性不仅升得高而且升得快。近年来学者们还注意到肝病时血中往往能查到一些大分子的酶或者酶复合体,研究最多的是高分子碱性磷酸酶。国内外都有文献报道肝病时碱性磷酸酶阳性率很高,梗阻性黄疸时几乎可达100%,这些高分子酶复合体在其它疾病时往往少见。
第八章 肿瘤标志物的临床实验室检查
近二十多年来,为了提高对肿瘤的早期检测、鉴别诊断、疗效观察以及预后判断,人们从肿瘤细胞的化学特性、细胞病理、免疫反应和基因表达产物等诸多方面寻找各种特异性强、灵敏度高的肿瘤标志物,并取得了较快的进展。肿瘤标志物的研究对肿瘤的诊断有极其重要的意义,推动了临床实验室的检测水平的提高和发展。
第一节 概述
一、肿瘤标志物的发展概况
在世界上首先报告肿瘤标志物的是1846年由Bence-Jones发现在尿中有一种随温度变化而改变成凝溶状态的蛋白质,后经证实这是患有多发性骨髓瘤病人的浆细胞所产生,由尿液排泄的蛋白质,并被命名为B-J蛋白。100多年后,人们对这种蛋白质又有了新的认识,其本质是免疫球蛋白的轻链部分,除了在尿液,也可在血清中利用电泳技术将其检测。现已分别检测出多发性骨髓瘤患者浆细胞所分泌的全部的“单克隆系”免疫球蛋白。B-J蛋白的发现,开创了肿瘤标志物的新时期,故常将这一年代称为肿瘤标志物的开创期,或称肿瘤标志物的第一阶段,见表8-1。
表8-1 主要肿瘤标志物的发现阶段
| 年代 | 标志物 | 发现者 |
| 1846 | Bence-Jones | H. Bence-Jones |
| 1928 | 异位激素综合征 | W. H Brown |
| 1930 | 人绒毛膜促性激素 | B. Zondek |
| 1932 | 促肾上腺皮质激素 | H. Cushing |
| 1949 | 血型抗原 | K. Oh-Uti |
| 1959 | 同工酶 | C. Markert |
| 1963 | α-甲胎蛋白 | G. I. Abelev |
| 1965 | 癌胚抗原 | P. Gold and S. Freeman |
| 1969 | 肿瘤基因 | R. Heubner and G. Todaro |
| 1975 | 单克隆抗体 | H. Kohler and G. Milstein |
| 1980 | 肿瘤基因探针和转录 | G. Cooper,R. Weinbeerg and M. Bishop |
| 1985 | 抑癌基因 | H. Harris,R. Sager and A. Knudson |
第二阶段从1928年到1963年,在这段期间发现了与肿瘤相关的标志物,包括激素、同工酶、蛋白质。但是这些标志物的应用,特别是肿瘤所表达的这些物质的理化特性,经过相当的一段时间后,才被人们逐渐认识。尤其是在1963年至1969年期间,即第三阶段中发现并证实,在肿瘤的所产生的蛋白质物质中,某些胎儿期蛋白在肿瘤状态时重新出现,从而认为对这种胎儿蛋白的检测,十分有利于对肿瘤的诊断。第四阶段是1975年起,发现了单克隆抗体,并在肿瘤细胞系中获得了肿瘤抗原和成功地使用癌胚胎抗原臂如CA125、CA153、CA549等。近年来,随着分子遗传学的理论和技术的发展,分子探针的使用,单克隆抗体的筛选成功,基因的定位,包括肿瘤基因和抑癌基因的测定,使肿瘤标志物的检测的内容更广,技术更先进。
二、肿瘤标志物的定义
所谓肿瘤标志物,指在肿瘤发生和增殖过程中,由肿瘤细胞生物合成、释放或者是宿主对癌类反应性的一类物质。这类物质可能是循环物质,可在细胞、组织或体液中出现,人们能利用化学、免疫和分子生物学等技术对血液或分泌物进行定性或定量地检测。通过对这类物质的分析,能帮助人们从正常组织中区别肿瘤或测定肿瘤细胞核、细胞质以及对细胞膜上的特性进行分析,以此作为辨认肿瘤细胞的标志。近年来,分子生物学技术的发展,通过对细胞基因的遗传和表达物质的检测,为研究肿瘤的发生机制以及肿瘤的筛选及早期诊断提供了可靠的标志性的依据。
根据肿瘤标志物的来源以及它的特异性,大致可分为两大类。只是一种肿瘤所产生的特异性物质,称之为该肿瘤的特异性标志物。例如前列腺特异性抗原(PSA)就是前列腺肿瘤所产生的特异性标志物质,只有患前列腺癌时,PSA才会显著性升高。但更多的肿瘤标志物,则是在一类组织类型的相似而性质不同的肿瘤发生时,其含量会有较大的变化,一般称这类物质为肿瘤辅助标志物。这类标志物往往易同良性肿瘤或正常组织发生混淆。但在肿瘤发生时,这类标志物的含量要明显高出良性肿瘤或正常组织。
三、肿瘤标志物的分类
近年来,由于医学检测水平的提高,与肿瘤相关的标志性物质不断地被发现。但是肿瘤标志物的来源和性质非常复杂,所以至今还未有一个统一的肿瘤标志物的分类方法,综合各种专业书籍以及学术报告,肿瘤标导物分类大致有两种:其一按肿瘤标志物的来源,第二是按肿瘤标志物本身的化学特性。本章是按后者的分类进行介绍,主要包括:①肿瘤胚胎性抗原标志物;②糖类标志物;③酶类标志物;④激素类标志物;⑤蛋白质类标志物;⑥基因类标志物。
四、肿瘤标志物的临床应用
近年来,肿瘤标志物在临床上已得到较广泛的应用(表8-2),作为一种良好的肿瘤标志物应该具有下列条件:①标志物的含量变化应与肿瘤的生长、消退、转移有直接的定性或定量的比例关系;②标志物应具有较高的特异性,能比较明显地区别于正常人群和良性肿瘤,但一般的情况下,酶的活性在普通疾病及良性肿瘤状态也会变化,易对肿瘤诊断造成混淆;③检测这类标志物的方法简便,易推广,而且成本较低。尽管目前的标志物或多或少的存在一些遗憾,但是随着检测技术的发展,方法学的改进,特别是标志物项目的增加,结合各种项目检测的微量的变化,经综合评价,为人群的筛选、临床诊断、预后观察以及肿瘤复发、转移评价带来较为有力的证据。这些年来,随着肿瘤标志物与临床诊断的符合率的增加,不少标志物已逐渐成为肿瘤检测中可依赖的指标。
表8-2 肿瘤标志物的临床应用
| 主要存在于Ⅰ带的酶 | 均匀分布的酶 | 主要存在于Ⅲ带的酶 |
| 细胞色素氧化酶 | 磷酸甘油脱氢酶 | 谷氨酸脱氢酶 |
| 琥珀酸脱氢酶 | 苹果酸酶 | 丙酮酸激酶 |
| 乳酸脱氢酶 | 单胺氧化酶 | 异柠檬酸脱氢酶(NADP) |
| 苹果酸脱氢酶 | 葡萄糖激酶 | 乙醇脱氢酶 |
| 天冬氨酸氨基转移酶 | 已糖激酶 | 3-羟丁酸辅酶A脱氢酶 |
| 丙氨酸氨基转移酶 | 已糖二磷酸激酶 | NADH-四唑还原酶 |
| 糖原合成酶 | 磷酸甘油醛脱氢酶 | NADP-四唑还原酶 |
| 糖原磷酸化酶 | ||
| 磷酸葡萄糖异构酶 | ||
| 葡萄糖-6-磷酸酶 | ||
| 磷酸烯醇丙酮酸激酶 |
第二节 常见的肿瘤标志物及其应用评价
肿瘤标志物经过一百多年的发展历史,尽管至今为止,具有明确诊断作用的标志物不是很多,但有不少标志物经过临床实践已被大家熟悉和应用。
一、肿瘤胚胎性抗原标志物
在人类发育过程中,许多原本只在胎盘期才具有蛋白类物质,应随胎儿的出生而逐渐停止合成和分泌,但因某种因素的影响,特别是肿瘤状态时,会使得机体一些“关闭”的基因激活,出现了返祖现象,而重新开启并重新生成和分泌这些胚胎、胎儿期的蛋白(表8-3)。
表8-3 胚胎类肿瘤标志物
| 名称 | 性质 | 相关肿瘤 |
| 甲胎蛋白 | 糖蛋白70ku | 肝细胞、胚细胞(非精原细胞瘤) |
| β-癌胚抗原 | 80ku | 结肠 |
| 癌胚铁蛋白 | 糖蛋白600ku | 肝 |
| 癌胚抗原 | 糖蛋白22ku | 结肠、直肠、胰腺、肺、乳腺 |
| 胰癌胚抗原 | 糖蛋白40ku | 胰腺 |
| 鳞状细胞抗原 | 糖蛋白44-48ku | 肺、皮肤、头和颈部 |
| 组织多肽抗原 | 细胞角蛋白8.18.19 | 乳腺、结肠 |
这类蛋白虽然与肿瘤组织不一定都具有特定的相关性,但与肿瘤的发生存在着内在的联系,故被作为一种较为常见的肿瘤标志物。
(一)癌胚抗原(carcinoembryonicantigen,CEA)
癌胚抗原是1965年Gold和Freedman首先从胎儿及结肠癌组织中发现的,CEA是一种分子量为22ku的多糖蛋白复合物,45%为蛋白质。CEA的编码基因位于19号染色体。一般情况下,CEA是由胎儿胃肠道上皮组织、胰和肝的细胞所合成,通常在妊娠前6个月内CEA含量增高,出生后血清中含量已很低下,健康成年人血清中CEA浓度小于2.5μg/L。
CEA属于非器官特异性肿瘤相关抗原,分泌CEA的肿瘤大多位于空腔脏器,如胃肠道、呼吸道、泌尿道等。正常情况下,CEA经胃肠道代谢,而肿瘤状态时的CEA则进入血和淋巴循环,引起血清CEA异常增高,使上述各种肿瘤患者的血清CEA均有增高。在临床上,当CEA大于60μg/L时,可见于结肠癌、直肠癌、胃癌和肺癌。CEA值升高,表明有病变残存或进展。如肺癌、乳腺癌、膀胱癌和卵巢癌患者血清CEA量会明显升高,大多显示为肿瘤浸润,其中约70%为转移性癌。一般来说,手术切除后6周,CEA水平恢复正常,否则提示有残存肿瘤,若CEA浓度持续不断升高,或其数值超过正常5-6倍者均提示预后不良。连续随访定量检测血清CEA含量,对肿瘤病情判断更具有意义。
有报道在胃肠道恶性肿瘤患者体内存在着CEA的异质体,经等电聚焦电泳检测可显示8-12个CEA峰,已知其中三个峰为癌特异峰,称CEA-S,其余可能属于正常的结肠交叉反应抗原簇或致癌过程中的其它过量产物。
除血液之外,其它生物液体,如胰液和胆汁内CEA定量可用于诊断胰腺或胆道癌;浆液性渗出液的CEA定量可作为细胞学检查的辅助手段;尿液CEA定量可作为判断膀胱癌预后的参考。血清CEA定量结合甲状腺降钙素测定,有助于甲状腺髓样癌的诊断和复发的估计。
(二)甲胎蛋白(alpha-fetoprotein,AFP)
甲胎蛋白是1956年Bergstrandh和Czar在人胎儿血清中发现的一种专一性的甲种球蛋白。1963年G.I.Abelev首先发现AFP主要是由胎盘层其次是卵黄囊合成,胃肠道粘膜和肾脏合成较少。1964年Tatarinov发现肝细胞癌患者血清中检测到AFP。AFP是一种在电场中泳动于α-球蛋白区的单一多聚体肽链的糖蛋白,其分子量平均为70ku,含糖4%,AFP的编码基因位于4号染色体4q11-12,与血清白蛋白、维生素D结合蛋白同属一大家族。近年来已发现了AFP的异质体。妊娠的妇女的血和尿中的AFP含量会持续增高,从妊娠6周开始合成,至12-15周达高峰。胎儿血浆中的AFP值可达到3mg/ml,随后即逐渐降低,出生后,AFP合成很快受抑制,其含量降至50μg/L,周岁末婴儿的浓度接近成人水平,一般健康成人血浆AFP浓度低于25μg/L。
AFP是原发性肝癌的最灵敏、最特异的肿瘤标志,血清AFP测定结果大于500μg/L以上,或含量有不断增高者,更应高度警惕。肝癌患者血清AFP含量变化的速率和程度与肿瘤组织分化程度高低有一定相关性,分化程度较高的肿瘤AFP含量常大于200μg/L。
检测AFP的免疫学方法主要有免疫扩散电泳(火箭电泳)、γ-射线计数125I标记检测法和定性、定量酶免疫方法。用不同的植物凝集素可以检测和鉴别不同组织来源的AFP的异质体。如用小扁豆凝集素(LCA)亲和交叉免疫电泳自显影法,可以检测LCA结合型AFP异质体。
血清AFP含量的检测对其它肿瘤的监测亦有重要临床价值。如睾丸癌、畸胎瘤、胃癌、胰腺癌等患者血清AFP含量可以升高。某些非恶性肝脏病变,如病毒性肝炎、肝硬化,AFP水平亦可升高,故必须通过动态观察AFP含量和ALT酶活性的变化予以鉴别诊断。
(三)胰胚胎抗原(pancreaticoncofetalantigen,POA)
胰胚胎抗原是1974年Banwo等人自胎儿胰腺抽提出的抗原,1979年被国际癌症生物学和医学会正式命名。POA是一种糖蛋白,分子量为40ku,在血清中以分子量900ku复合形式存在,但可降解为40ku。正常人群血清中RIA法测定小于7ku/L。
胰腺癌的POA的阳性率为95%,其血清含量大于20ku/L,当肝癌、大肠癌、胃癌等恶性肿瘤时也会使POA升高,但阳性率较低。
二、糖类抗原标志物
肿瘤标志物相关物质是指由肿瘤细胞表面的抗原物质或者是肿瘤细胞所分泌的物质,这类物质又是单克隆抗体,故又称为糖类抗原(carbohydrateantigen,CA)。这类标志物出现为临床肿瘤的诊断带来方便,糖类抗原标志物产生又可分为两大类,为高分子粘蛋白类(表8-4)和血型类抗原(表8-5)。
表8-4 糖类高分子粘蛋白抗原肿瘤标志物
| 名称 | 性质 | 癌肿 | 常用单克隆抗体 |
| CA125 | 糖蛋白>200ku | 卵巢、子宫内膜 | OC125 |
| CA15-3 | 糖蛋白400ku | 乳腺、卵巢 | DF3和115D3 |
| CA549 | 高分子量糖蛋白 | 乳腺、卵巢 | BC4E549 BC4n154 |
| CA27.29 | 高分子量糖蛋白 | 乳腺 | B27.29 |
| 类粘蛋白 | 糖蛋白350ku | b-12 | |
| 肿瘤相关抗原 | |||
| DU-PAN-2 | 粘蛋白100-500ku | 胰腺、卵巢、胃 | DU-PAN-2 |
表8-5 血型类抗原肿瘤标志物
| 名称 | 性质 | 癌肿 | 常用单克隆抗体 |
| CA19-9 | 唾液酸化Lexa | 胰腺、胃肠、肝 | 116NS19-9 |
| CA19-5 | 唾液酸化Lea和Leag | 胃肠、卵巢 | 116NS19-5 |
| CA50 | 唾液酸化Lea | 胰腺、胃肠、结肠 | Colo-50 |
| CA72-4 | 唾液酸化Tn | 卵巢、乳腺、胃肠、结肠 | B27.3.cc49 |
| CA242 | 唾液酸化CHO | 结肠、直肠、胰腺 | C242 |
| 鳞状细胞抗原 | 糖蛋白 | 子宫颈、肺、皮肤、头颈部 | SCC |
这类抗原标志物的命名是没有规律的,有些是肿瘤细胞株的编号,有些是抗体的物质编号,常用检测方法是单克隆抗体法,有的还同时用两种不同位点的单抗做成双位点固相酶免疫法,这些比一般化学法测定的特异性有很大的提高。而对一些糖类抗原的异质体,则通常用不同的植物凝集素来进行分离检测。
(一)CA125
1983年由Bast等从上皮性卵巢癌抗原检测出可被单克隆抗体OC125结合的一种糖蛋白。分子量为200ku,加热至100℃时CA125的活性破坏,正常人血清CA125中的(RIA)阳性临界值为35ku/L。
CA125是上皮性卵巢癌和子宫内膜癌的标志物,浆液性子宫内膜样癌、透明细胞癌、输卵管癌及未分化卵巢癌患者的CA125含量可明显升高。当卵巢癌复发时,在临床确诊前几个月便可呈现CA125增高,尤其卵巢癌转移患者的血清CA125更明显高于正常参考值。CA125测定和盆腔检查的结合可提高试验的特异性。动态观察血清CA125浓度有助于卵巢癌的预后评价和治疗控制,经治疗后,CA125含量可明显下降,若不能恢复至正常范围,应考虑有残存肿瘤的可能。95%的残存肿瘤患者的血清CA125浓度大于35kU/L。然而,CA125血清浓度轻微上升还见于1%健康妇女,3%-6%良性卵巢疾患或非肿瘤患者,包括孕期起始3个月、行经期、子宫内膜异位、子宫纤维变性、急性输卵管炎、肝病、胸腹膜和心包感染等。
(二)CA15-3
CA15-3是1984年Hilkens等从人乳脂肪球膜上糖蛋白MAM-6制成的小鼠单克隆抗体(115-DB);1984年Kufu等自肝转移乳腺癌细胞膜制成单克隆抗体(DF-3),故被命名为CA15-3。CA15-3分子量为400ku,分子结构尚未清楚。正常健康者血清CA15-3含量(RIA法)小于28kU/L。
30%-50%是乳腺癌患者的CA15-3明显升高,它也是监测乳腺癌患者术后复发的最佳指标,当CA15-3大于100kU/L时,可认为有转移性病变,其含量的变化与治疗结果密切相关。肺癌、胃肠癌、卵巢癌及宫颈癌患者的血清CA15-3也可升高,应予以鉴别,特别要排除部分妊娠引起的含量升高。
(三)CA19-9
CA19-9是1979年Koprowski等用结肠癌细胞免疫小鼠,并与骨髓瘤杂交所得116NS19-9单克隆抗体,它是一种分子量为5000ku的低聚糖类肿瘤相关糖类抗原,其结构为Lea血型抗原物质与唾液酸Lexa的结合物。正常人群的CA19-9血清含量为(RIA法)2-16kU/L。
CA19-9是胰腺癌和结、直肠癌的标志物。血清CA19-9阳性的临界值为37kU/L。胰腺癌患者85%-95%为阳性。当CA19-9小于1000kU/L时,有一定的手术意义,肿瘤切除后CA19-9浓度会下降,如再上升,则可表示复发。结直肠癌、胆囊癌、胆管癌、肝癌和胃癌的阳性率也会很高,若同时检测CEA和AFP可进一步提高阳性检测率。良性疾患时如胰腺炎和黄疸,CA19-9浓度也可增高,但往往呈“一过性”,而且其浓度多低于120kU/L,必须加以鉴别。
(四)CA50
CA50是1983年Lindholm等从抗人结、直肠癌Colo-205细胞株的一系列单克隆抗体中筛选出的一株对结、直肠癌有强烈反应,但不与骨髓瘤细胞及血淋巴细胞反应的单克隆抗体,所能识别的抗原称CA50。Ca50存在于细胞膜内,其抗原决定簇为唾液酸Lea血型物质与唾液酸-N-四氧神经酰胺。在正常人群,CA50血清浓度(RIA法)小于20kU/L。
一般认为,CA50是胰腺和结、直肠癌的标志物,因CA50广泛存在胰腺、胆囊、肝、胃、结直肠、膀胱、子宫,当细胞恶变时,由于糖基转化酶的失活或胚胎期才能活跃的某些转化酶被激活,造成细胞表面糖类结构性质改变而形成CA50,因此,它又是一种普遍的肿瘤标志相关抗原,而不是特指某个器官的肿瘤标志物。所以在多种恶性肿瘤中可检出不同的阳性率。1983年,建立了放射免疫分析法,1987年应用CA50单抗,在国内建立了IRMA技术用于肿瘤的早期诊断,胰腺癌、胆囊癌的阳性检测率达90%,对肝癌、胃癌、结直肠癌及卵巢肿瘤诊断亦有较高价值,在胰腺炎、结肠炎和肺炎发病时,CA50也会升高,但随炎症消除而下降。
三、酶类标志物
酶及同工酶是最早出现和使用的肿瘤标志物之一(表8-6)。肿瘤状态时,机体的酶活力就会发生较大变化,这是因为:①肿瘤细胞或组织本身诱导其它细胞和组织产生异常含量的酶;②肿瘤细胞的代谢旺盛,细胞通透性增加,使得肿瘤细胞内的酶进入血液,或因肿瘤使得某些器官功能不良,导致各种酶的灭活和排泄障碍;③肿瘤组织压迫某些空腔而使某些通过这些空腔排出的酶返流回血液。
在肿瘤标志酶中根据来源可将其分为两类:①组织特异性酶,因组织损伤或变化而使储存在细胞中的酶释放,如前列腺特异性抗原等;②非组织特异性酶,主要是肿瘤细胞代谢加强,
表8-6 酶类肿瘤标志物
| 名称 | 分子量及同工酶 | 相关肿瘤 | |
| 醛缩酶 | 160ku | 3个 | 肝 |
| 碱性磷酸酶 | 95ku | 7个 | 骨、肝、白血病、肉瘤、卵巢等 |
| 淀粉酶 | 45ku | 胰腺等 | |
| 谷胱甘肽转移酶 | 80ku | 多种 | 肝、胃、结肠 |
| 肌酸激酶 | 83ku | 4个 | 前列腺、肺、结肠、卵巢等 |
| γ-谷氨酰转移酶 | 90ku | 12个 | 肝 |
| 乳酸脱氢酶 | 135ku | 5个 | 肝、淋巴瘤、白血病 |
| 神经原特异性烯醇化酶 | 73ku | 2个 | 肺(小细胞)、神经母细胞瘤、类癌、黑色素瘤、嗜铬细胞瘤 |
| 5-核苷酸酶 | 6区带 | 肝 | |
| α-L岩藻糖苷酶 | 230ku | 8个 | 肝 |
| 核糖核酸酶 | 2个 | 卵巢、肺、大肠等 | |
| 前列腺特异性抗原 | 34ku | 前列腺 | |
特别是无氧酵解增强,大量酶释放到血液中,如已糖激酶等。
在酶标志物分析中,同工酶的分辨和检出是提高标志物临床应用的重要的环节,从目前所知的肿瘤标志同工酶可分为三大类型:①异位型同工酶:指某种瘤组织改变了自己的分泌特性,而去分泌表达了其它成年组织的同工酶的类型;②胚胎型同工酶,某些组织在肿瘤状态时,使酶的同工酶谱退化到胚胎时未分化状态,而分泌出大量的胚胎期的同工酶,这种变化往往与肿瘤的恶性程度成正比;③胎盘型同工酶,有些肿瘤组织会分泌出某些原属胎盘阶段的同工酶谱。从目前的资料分析,这类胎盘型同工酶已达20余种。酶的活性变化常常与组织器官的损伤有密切关系。在机体中,能造成酶活性变化的因素太复杂,从而使在诊断肿瘤时特异性受到很大影响。
(一)前列腺特异性抗原(prostatespecificantigen,PSA)
1971年,Hara等首先发现PSA是由前列腺上皮细胞合成分泌至精液中,是精浆的主要成分之一;1979由Wang等从前列腺肥大症患者的前列腺组织中分离出来的丝氨酸蛋白酶,分子量34ku,编码基因定位于19q13,PSA仅存在于前列腺上皮细胞的胞质、导管上皮和粘液内,具有糜蛋白酶样和胰蛋白酶的活性,PSA在正常男性(RIA法、ElA法)含量小于2.5μg/L。
PSA是前列腺癌的特异性标志物,也是目前少数器官特异性肿瘤标志物之一。前列腺癌是男性泌尿系统的主要囊性肿瘤,血清PSA定量的阳性临界值为大于10μg/L,前列腺癌的诊断特异性达90%-97%。
血清PSA除了作为检测和早期发现前列腺癌,还可用于治疗后的监控,90%术后患者的血清PSA值可降至不能检出的痕量水平,若术后血清PSA值升高,提示有残存肿瘤。放疗后疗效显著者,50%以上患者在2个月内血清PSA降至正常。
(二)α-L岩藻糖苷酶(α-L-fucosidase,AFU)
1980年由Deugnier等首先在3例原发性肝癌患者血清中发现AFU活性升高。AFU是存在于血清中的一种溶酶体酸性水解酶,分子量230ku,单个亚基分子量50ku。AFU正常参考值(化学法)为324±90μmol/L。
AFU是原发性肝癌的一种新的诊断标志物,广泛分布于人体组织细胞、血液和体液中,参与体内糖蛋白、糖脂和寡糖的代谢。原发性肝癌患者血清AFU活力显著高于其它各类疾患(包括良、恶性肿瘤)。虽然AFU升高的机制不甚明了,但可能有以下几种:①肝细胞和肿瘤细胞的坏死使溶酶体大量释放入血;②正常肝细胞的变性坏死可使摄取和清除糖苷酶的功能下降;③肿瘤细胞合成糖苷酶的功能亢进;④肿瘤细胞可能分泌某种抑制因子,抑制肝细胞对糖苷酶的清除能力或释放某些刺激因子,促进肝细胞或肿瘤细胞本身合成糖苷酶。总之,血清AFU活性升高可能是由多种因素综合作用的结果,是对原发性肝细胞性肝癌检测的又一敏感、特异的新标志物。
血清AFU活性动态曲线对判断肝癌治疗效果、估计预后和预报复发有着极其重要的意义,甚至优于AFP。但是,值得提出的是,血清AFU活力测定在某些转移性肝癌、肺癌、乳腺癌、卵巢或子宫癌之间有一些重叠,甚至在某些非肿瘤性疾患如肝硬化、慢性肝炎和消化道出血等也有轻度升高,在使用AFU时应与AFP同时测定,可提高原发性肝癌的诊断率,有较好的互补作用。
现发现AFU在用琼脂糖凝胶等电聚焦电泳分析时存在8种不同等电点的同工酶,其范围在3.5-6.5之间,正常人的AFU同工酶有两种类型:低峰值型和Ⅳ主峰型。乙型肝炎患者出现3种AFU同工酶谱:Ⅷ主峰型,Ⅳ、Ⅷ双峰型和Ⅳ主峰、Ⅴ次峰型。原发性肝癌患者血清AFU同工酶变化复杂,有5种类型:低峰值型,Ⅳ、Ⅴ双峰型,Ⅲ、Ⅳ双峰型,Ⅴ型和Ⅵ型。根据各自的峰型特点,AFU同工酶对正常人、肝炎和原发性肝癌患者的鉴别诊断具有一定的临床应用价值。
(三)神经原特异性烯醇化酶(neuron-specificenolase,NSE)
烯醇化酶是催化糖原酵途径中甘油分解的最后的酶。由3个独立的基因片段编码3种免疫学性质不同的亚基α、β、γ,组成5种形式的同工酶αα、ββ、γγ、αγ、βγ。二聚体是该酶分子的活性形式,γ亚基同工酶存在于神经原和神经内分泌组织,称为NSE。α亚基同工酶定位于胶质细胞,称为非神经原特异性烯醇化酶(NNE)。NSE和NNE的分子量分别为78ku和87ku,正常参考范围为0.6-5.4μg/L。
NSE是神经母细胞瘤和小细胞肺癌的标记物。神经母细胞瘤是常见的儿童肿瘤,占1-14岁儿童肿瘤的8%-10%。NSE作为神经母细胞瘤的标志物,对该病的早期诊断具有较高的临床应用价值。神经母细胞瘤患者的尿中NSE水平也有一定升高,治疗后血清NSE水平降至正常。血清NSF水平的测定对于监测疗效和预报复发均具有重要参考价值,比测定尿液中儿茶酚胺的代谢物更有意义,小细胞肺癌(SCLC)是一种恶性程度高的神经内分泌系统肿瘤,约占肺癌的25%-30%,它可表现神经内分泌细胞的特性,有过量的NSE表达,比其它肺癌和正常对照高5-10倍以上。SCLC患者血清NSE检出的阳性率可高达65%-100%,目前已公认为NSE可作为SCLC高特异性、高灵敏性的肿瘤标志物,有报道,NSE水平与SCLC转移程度相关,但与转移的部位无关,NSE水平与其对治疗的反应性之间也有一个良好的相关性。
四、激素类标志物
激素是一类由特异的内分泌腺体或散在体内的分泌细胞所产生的生物活性物质,当这类具有分泌激素功能的细胞癌变时,就会使所分泌的激素量发生异常。常称这类激素为正位激素异常。而异位激素则是指在正常情况下不能生成激素的那些细胞,转化为肿瘤细胞后所产生的激素,或者是那些能产出激素的细胞癌变后,分泌出的是其它激素细胞所产生的激素(表8-7)。衡量异位激素的条件是:①有非内分泌腺细胞合成的激素;②某种内分泌细胞却分泌其他分泌腺细胞的激素;③肿瘤患者同时伴有分泌异常综合征;④这类肿瘤细胞在体外培养时也能产生激素;⑤肿瘤切除或经治疗肿瘤消退时,此种激素含量下降,内分泌综合征的症状改善。
表8-7 激素类肿瘤标志物
| 激素 | 性质 | 相关肿瘤 |
| 儿茶酚胺类 | 嗜铬细胞 | |
| 促肾上腺皮质激素 | 4.5ku | 库欣综合征、肺(小细胞) |
| 抗利尿激素 | 肺(小细胞)、原发性类癌等 | |
| 降钙素 | 3.5ku | 甲状腺髓质 |
| 生长激素 | 21ku | 垂体腺瘤、肾、肺 |
| hGG | 45ku | 胚胎绒毛膜、睾丸(非精原细胞癌) |
| 人胎盘催乳素 | 22ku | 滋养层、性腺、肺、乳腺 |
| 甲状旁腺素 | 肝、肾、乳腺、肺等 | |
| 催乳素 | 22ku | 垂体腺瘤、肾、肺 |
| 胰高糖素 | 3ku | 胰高糖素瘤、嗜铬细胞瘤 |
| 转化生长因子 | 鳞瘤、肾、乳腺 |
一般来讲,异位激素的化学本质与正位激素相似,不同类型的恶性肿瘤可分泌不同种类的异生性激素或分泌出同一种的激素,而同一种肿瘤细胞可分泌一种或多种不同的异生性激素。这给检查带来了难度,常见的可分泌异生性激素的恶性肿瘤是肺未分化小细胞癌、神经外胚层肿瘤及类癌等。根据肿瘤状态、机体内的激素含量的变化,观察这些激素动态变化,无疑会给临床诊断带来标志性的依据。
(一)降钙素(calcitonin,CT)
CT是由甲状腺滤泡细胞C细胞合成、分泌的一种单链多肽激素,故又称甲状腺降钙素,是由32个氨基酸组成,分子量3.5ku。CT的前体物是一个由136个氨基酸残基组成大分子无活性激素原,分子量为15ku,可迅速水解成有活性的CT,人类CT的半寿期只有4-12min,正常情况下它的靶器官是骨、肾和小肠,主要作用是抑制破骨细胞的生长,促进骨盐沉积,增加尿磷,降低血钙和血磷。放射免疫测定为常用方法,正常参考值为小于100ng/L。
目前,甲状腺髓样癌患者的CT一定会升高,因为降钙素的半寿期较短,所以降钙素可作为观察临床疗效的标志物。
肺癌、乳腺癌、胃肠道癌以及嗜铬细胞瘤患者可因高血钙或异位分泌而使血清CT增加,另外,肝癌和肝硬化患者也偶可出现血清CT增高。
(二)人绒毛膜促性腺激素(humanchorionic gonagotropin,hCG)
hCG是由胎盘滋养层细胞所分泌的一类糖蛋白类激素,在正常妊娠妇女血中可以测出hCG。hCG有α、β两个亚基,α-亚基的分子量约为13000,α-亚基的生物特性与卵泡刺激素(FSH)和黄体生成激素(LH)的α-亚基相同。β-亚基的分子量约15000,β-亚基为特异性链,可被单克隆抗体检测,也是一个较好的标志物。在每个亚基上有两条N-糖链,其中3/4是复杂型双天线,1/4是以单天线的形式出现。由此决定了各类hCG激素的生物特性。通常情况下,尿中的hCG的总量(ELISA法)小于30μg/L,血清hCG小于10μg/L,β-hCG小于3.0μg/L。当胎盘绒毛膜细胞恶变为恶性葡萄胎后,hCG会明显增高,这时hCG糖链结构有部分转为三天线和四天线的结构。当发生绒毛膜上皮癌后,除有三、四天线外,还出现更为异常的偏二天线的糖链结构,而且这些异常糖链结构具有与曼陀罗凝集素(DSA)特异的亲和力。正常情况下,结合率为42.3%-72.4%,绒毛膜上皮癌的结合率为53.5%-87.1%。hCG还会在乳腺癌、睾丸癌、卵巢癌增高。当子宫内膜异位症、卵巢囊肿等非肿瘤状态时,hCG也会增高。
(三)儿茶酚胺类及其衍生物
儿茶酚胺类激素是以其结构中均含儿茶酚又属于胺类而得名。正常情况下,它是由肾上腺髓质中的一些交感神经节纤维末梢终止髓质细胞(又称嗜铬细胞)产生和分泌,包括肾上腺素(E)、去甲肾上腺素(NE)和多巴胺(DA)等,它们既是激素又是神经递质。
⒈变肾上腺素(metanephrine)变肾上腺素是儿茶酚胺的甲氧化代谢产物,由于甲基化是在肝脏内微粒体中进行,而儿茶酚胺的形成都是在肾上腺髓质的嗜铬细胞及交感神经末梢处形成,所以,从检测尿中的变肾上腺素浓度可间接地了解儿茶酚胺的分泌。
目前使用高效液相的紫外检测仍是最为有效的方法之一,正常值为0.30-1.50μmol/24h尿。变肾上腺素浓度增高是分泌型嗜铬细胞瘤的主要标志物,它比儿茶酚胺和香草扁桃酸更稳定。
⒉垂草扁桃酸(VMA)香草扁桃酸(3-甲氧-4羟苦杏仁酸,VMA)是肾上腺素和去甲肾上腺素经单胺氧化酶(MAO)和儿茶酚胺-0-甲基转移酶(COMT)的作用下,甲基化和脱氨基而产生的降解产物。VMA主要是从尿中排出。
高效液相电化学检测是常用的方法,正常参考值随年龄增长而增加,成人为5.0-35.0μmol/24h尿。
能合成儿茶酚胺类的肾上腺髓质的嗜铬细胞及交感神经细胞末梢,均源于胚胎期神经嵴,这两种组织含有相同的酶。一旦这类组织增殖,则尿中VMA就会增高,所以它常被认为是神经母细胞瘤、神经节瘤和嗜铬细胞瘤的标志物。
约有70%神经母细胞瘤的患者均有VMA增高,在IV期神经瘤患者VMA/HVA的比值可作为预后评价指标,在儿童的神经母细胞瘤患者中,VMA也是一项重要指标。
VMA又可作为嗜铬细胞瘤的诊断首选标志物,但有时增高程度不稳定,宜同时测定尿中儿茶酚胺和变肾上腺素。
⒊高香草酸(HVA)高香草酸(3-甲氧-4-羟苯乙酸,HVA)是多巴胺的主要代谢产物,儿茶酚在肝脏内经羧化和氨基氧化而成。
常采用高效液相电化学检测方法,正常参考值与VMA相似,也随年龄增长而增加,成人为15-40μmol/24h尿。尿中HVA增加与多巴合成量有关。在神经母细胞瘤、儿童交感神经肿瘤时,常选用HVA作为诊断和随访的一种主要的标志物。
五、其它蛋白质类标志物
蛋白质肿瘤标志是最早发现的标志物(表8-8)。在现有的标志物中,如β2微球蛋白、免疫球蛋白。一般来讲这类标志物特异性稍差,但检测方法相对比较容易,常作常规检测项目。
表8-8 蛋白质类肿瘤标志物
| 名称 | 性质 | 相关肿瘤 |
| β2-微球蛋白 | 12ku | 多发性骨髓瘤、B细胞淋巴瘤、慢性淋巴细胞白血病、Waldenstrom’s巨球蛋白血症 |
| C多肽 | 3.6ku | 胰岛素瘤 |
| 铁蛋白 | 450ku | 肝、肺、乳腺、白血病 |
| 本周蛋白 | 22.5-45ku | 游离轻链病、多发性骨髓病 |
| 免疫球蛋白 | 160-900ku | 多发性骨髓瘤、淋巴瘤 |
| 铜蓝蛋白 | 126-160ku | 肝、胃肠、胰腺 |
| 甲状腺球蛋白 | 670ku | 甲状腺癌 |
(一)β2-微球蛋(β2-microglobulin。β2m)
β2m由Berggard和Bearn于1996年从肾脏患者尿中分离出的一种蛋白质,由于它的分子量仅为1.2ku,电泳时显于β2m区带,故被命名为β2-微球蛋白。β2m是人体有核细胞产生的一种由100个氨基酸残基组成的单链多肽低分子蛋白。β2m血中含量(RIA、EIA法)正常参考范围为3.1±0.96mg/L,尿β2m为0.31±0.34mg/L;脑脊液β2m为1.27±0.11mg/L。
β2m是恶性肿瘤的辅助标志物,也是一些肿瘤细胞上的肿瘤相关抗原。β2m是人类白细胞抗原(HLA)的轻链部分,链内含有一对二硫键,β2m与HLA-A、B、C抗原的重链非共价地相结合而存在于细胞膜上。一般认为除成熟红细胞和胎盘滋养层细胞外,其它细胞均含有β2m。因此,起源于人体间质细胞上皮和造血系统的正常细胞和恶性细胞均能合成β2m。它可从有核细胞中脱落进入血循环,使血液中的β2m升高。血清β2m不但可以在肾功能衰竭、多种血液系统疾病及炎症时升高,而且在多种疾病中均可增高,故应排除由于某些炎症性疾病或肾小球滤过功能减低所致的血清β2m增高。肿瘤患者血清β2m含量异常增高,在淋巴系统肿瘤如慢性淋巴细胞白血病、淋巴细胞肉瘤、多发性骨髓瘤等中尤为明显,在肺癌、乳腺癌、胃肠道癌及子宫颈癌等中也可见增高。由于在肿瘤早期,血清β2m可明显高于正常值,故有助于鉴别良、恶性口腔肿瘤。脑脊液中β2m的检测对脑膜白血病的诊断有特别的意义。
(二)铁蛋白(ferritin,Fer)
铁蛋白是1884年Schmiedeber所发现的水溶性铁贮存蛋白,1937年被Laufberger命名为铁蛋白,1965年Richter等从恶性肿瘤细胞株中分离出铁蛋白,并发现铁蛋白存在于各种组织和体液中。铁蛋白是一种脱铁蛋白组成的具有大分子(450ku)结构的糖蛋白,由24个亚单位聚集而成,每个铁蛋白分子可贮存4500个铁原子。正常血清中含量(RIA法、EIA法)男性为20-250μg/L,女性为10-120μg/L。
铁蛋白具有两个亚基,为肝脏型(L型)和心脏型(H型),不同比例的亚基聚合而成纯聚体和杂合体,可得到不同的同工铁蛋白图谱。在肿瘤状态时,酸性同分异构体铁蛋白增高,一般情况下与白血病、肺癌、乳腺癌有关,当肝癌时,AFP测定值较低的情况下,可用铁蛋白测定值补充,以提高诊断率。在色素沉着、炎症、肝炎时铁蛋白也会升高。
(三)本周蛋白(Bence-Jonesprotein,BJP)
早在1845年由一位内科医生兼化学病理学家HenryBenceJones首次描述了这种蛋白,它可被氨基水杨酸、三氯醋酸、硝酸和盐酸沉淀,加热到45-60℃时,沉淀又再现,故又名为凝溶蛋白。1963年,Schwary和Edelman对骨髓瘤球蛋白轻链的胰蛋白酶水解产物和同一患者的本周蛋白进行比较,结果表明本周蛋白由完整的轻链组成,在大多数病例中,本周蛋白的沉淀系数为3.6s,分子量为45000u,属于游离轻链的双体,当沉淀系数为1.8s时,分子量为22500u,多属于单体。
本周蛋白是多发性骨髓瘤的典型标志物,或称其为“免于球蛋白轻链”标志物。免疫球蛋白的轻链可分为Kappa(κ-Ig)和Lambda(λ-Ig)两类,然而,一个克隆的浆细胞中能产生两种轻链混存于单一抗体分子中。慢性淋巴瘤、骨肉瘤等均会引起本周蛋白阳性,肾病时也会阳性。目前用于检测本周蛋白的方法很多,如:①热沉淀:此种反应易受pH及多种理化因素影响,因此宜用pH4.9醋酸缓冲液调到恒定环境;②醋酸纤维薄膜电泳:可用清晨第一次尿,浓缩尿液50倍左右后,进行CAM电泳,经丽春红染色,BJP区带在α2-γ区间可被显现;③聚丙烯酰胺溶胶电泳:是以聚丙烯酰胺凝胶作支持物的电泳技术,它是一种不连续的凝胶电泳,故能使蛋白质各组份被清楚地分开,BJP呈现的位置与CAME相同;④非浓缩尿与银染技术:由Shate建立的一种不需浓缩尿的银染技术,提高了尿中BJP检测敏感性;⑤免疫电泳;⑥固定免疫电泳:它作为一种更为灵敏的筛选BJP方法,比一般免疫电泳灵敏度提高近10倍。
六、肿瘤标志物的联合应用
用肿瘤标志物测定肿瘤在临床上已应用了许多年,为临床的诊断和疗效观察起了很多的作用,但在应用过程中,确实也存在着特异性不强、阳性率不高等不足。为了提高诊断的阳性差,临床上常将几项相关的标志物组成联合标志物组,同时对某一肿瘤进行检测,应用多变量分析的方法,提高临床的诊断的准确性。
(一)肺癌的诊断的标志物
CEA是最早用于肺癌的诊断,特别对非小细胞肺癌的诊断有一定的意义。目前临床上常将CEA和总唾液酸蛋白(TSA)联合检测,可提高诊断的灵敏度和特异性。或CEA与降钙素以及ACTH联合检测能对治疗的效果提供依据。
在肺癌的基因检测中,往往以检测P53基因和RB基因的表达为主。
肺癌的肿瘤标志物的临床应用如能结合细胞学的检查,其价值就更大。
(二)乳腺癌的诊断的标志物
乳腺肿瘤的标志物有不少,最早使用的是CEA、hCG、铁蛋白等。近年来,癌抗原物质的出现,特别是CA153、CA549标志物的检查为乳腺肿瘤的诊断带来一种较为可靠的依据。在基因检测方面,主要有P53、C-erb-2等。现有学者认为,乳腺肿瘤的患者的家族中存在着一种易感性的基因,这就是BRCA1和BRCA2,这对早期诊断和发现乳腺肿瘤有一定的意义。
(三)肝癌的诊断标志物
到目前为止,AFP仍然是肝癌诊断的最佳标志物,除此之外,还有γ-GT、AFU、GGT-Ⅱ、RNAase同工酶、AKP同工酶、醛缩酶同工酶、β2-微球蛋白相关抗原等。在肝癌的检测中,以几项标志物协同使用,能提高诊断阳性率。
(四)胰腺癌的诊断标志物
胰腺癌的早期诊断比较困难,手术切除率低,从目前的胰腺癌的诊断标志物来看,CA19-9是比较好的诊断标志物,其阳性值与肿瘤大小有一定的相关性。CA19-9又可与CA50或与胰腺癌组织抗原一起,作为胰腺癌诊断的联合指标。
(五)卵巢癌的诊断标志物
从目前的卵巢癌的诊断的单个标志物来看,特异性不高。如能将几个标志物联合检测可提高诊断的阳性率。
现可组合的标志物有:CEA、hCG、SIEX、CA125、CA19-9等单克隆抗体,在基因检测方面有K-ras癌基因等。
第三节 基因类肿瘤标志物的进展及其临床应用
随着分子生物学的理论和技术的发展,癌基因和抑癌基因的检测已成为肿瘤临床诊断新一代的标志物。
正常细胞的生长与增殖是由两大类基因调控的,一类是正向调控信号,主要是起促进细胞生长和增殖,并且阻止其发生终末分化倾向,癌的基因起着这一方面的作用,另一类为负向调控信号,主要是使细胞成熟,促进终末分化,最后是细胞凋亡,抑癌基因则在这方面起作用。正常情况下这两类信号保持着动态平衡,十分精确地调控细胞增殖和成熟。一旦这两类信号中有一类信号过强或过弱均会使细胞生长失控而恶变。
一、癌基因
癌基因或肿瘤基因是指在自然或实验条件下,具有潜在的诱导细胞恶性转化的基因。在研究逆转录病毒时发现,将某些逆转录病毒的基因片段嵌入细胞基因中,并使这些基因迅速地表达,结果是被嵌入的细胞呈恶性转变,特别是如果将这些逆转录病毒进入正常细胞染色体DNA的特定部位,就能很快地改变这些连接部位的基因表达,而使细胞癌变。从目前的资料分析(表8-9),引起细胞恶变功能的基因已达30余种。
表8-9 常见癌基因类肿瘤标志物
| 癌基因 | 细胞株或原发肿瘤 | 相关肿瘤 |
| N-myc | 细胞株 | 神经母细胞瘤、视网膜母细胞瘤、肺癌(小细胞) |
| 原发肿瘤 | 神经母细胞瘤、视网膜母细胞瘤、横纹肌肉瘤 | |
| C-erb-2 | 原发肿瘤 | 胃腺癌、肾腺癌、乳腺癌 |
| N-ras | 细胞株 | 胃腺癌 |
| C-myc | 细胞株 | 乳腺癌、胃腺癌、肺癌(巨细胞) |
| 原发肿瘤 | 急性粒细胞白血病、结肠腺癌 | |
| H-ras | 细胞株 | 黑色瘤 |
| 原发肿瘤 | 膀胱癌、皮肤鳞癌 | |
| K-ras | 细胞株 | 结肠癌、骨肉瘤 |
| 原发肿瘤 | 膀胱癌、胰腺癌、卵巢癌 |
(一)ras基因家族及其表达产物
1980年Langbcheim等通过基因转染实验发现了与Harvery及Kristein小鼠肉瘤病毒相似的细胞癌基因,即c-Ha-ras(1)基因,定位于第11号染色体的11p15区;c-Ha-ras(2)基因为伪基因(pseudogene),定位于X染色体上;c-Ki-ras(1)基因为伪基因,定位于第6号染色体6p11-p12区。Ras基因编码产物为p21ras蛋白,其本质为膜相关的G蛋白,具有GTP酶的活化性,参与信号传导。
当机体发生肿瘤时,编码p21ras蛋白的第12、13及61位氨基酸的核苷酸可以发生点突变,突变型的p21ras蛋白不具有GTP酶活化,无法使GTP水解为GOP。另外尚可在肿瘤中发现p21ras蛋白表达过度。
⒈可用于ras基因检测的方法
⑴PCR-SSCP(单链构象多态性,singlestrandcomformatinpolymorphism)、DGGE(变性梯度凝胶电泳,denaturedgradientgelelectrophoresis)、PCR-ASO(等位基因特异性寡核苷酸杂交,allelespecificoligonucleotide)和测序技术(sequencing):探测点突变。
⑵免疫组织化学:用RAP-5单抗。
⑶Southern印迹法及Northern印迹法。
⑷ELISA法及Western印迹法。
⒉ras基因家族与肿瘤的关系(表8-10)
表8-10 ras基因家族与肿瘤的关系
| 肿瘤类型 | 临床意义 |
| 乳腺癌 | c-Ha-ras基因mRNA水平升高与恶性肿瘤进展期中p21ras水平相关 |
| 结直肠癌 | 50%的肿瘤出现c-Ki-ras 基因点突变 |
| 肺癌 | 20%-30%肿瘤出现ras基因家族成员点突变,其中c-Ki-rad基因点突变与预后不良相关 |
| 胰腺癌 | 90%左右的肿瘤出现c-Ki-ras基因点突变 |
| 胃癌 | 在恶性肿瘤中p21表达水平明显升高,c-Ha-ras基因编码第12位氨基酸突变与肿瘤转移及预后不良相关 |
| 髓性白血病 | 10%-50%的肿瘤中出现c-N-ras基因突变 |
| 膀胱癌 | 部分病例可出现c-Ha-ras基因点突变及p21ras表达过度 |
(二)myc基因家族及其表达产物
1997年Duesberg等发现myc癌基因与禽类MC29病毒具有相似性。Myc基因家族共有6成员:c-myc、N-myc、L-myc、P-myc、R-myc及B-myc。其中c-myc、N-myc及L-myc与一些人类肿瘤相关。c-myc定位于第8号染色体的8q24区,其编码产物为439个氨基酸残基的蛋白质。N-myc定位于第2号染色体的2p23-p24区,其产物为456个氨基酸残基蛋白质。L-myc定位于第1号染色体的1p32区,编码产物为364个氨基酸残基的蛋白质。以上蛋白产物定位于核内,为核转录调节因子,能够与特殊的DNA顺序结合,当机体发生肿瘤时,myc基因家族成员可以发生染色体基因易位、基因扩增以及表达过度。
⒈可用于myc基因检测的方法
⑴标准细胞核型分析:基因易位。
⑵原位杂交:ELISA法。
⑶Southern印迹法及Northern印迹法。
⑷RT-PCR方法。
⒉myc基因家族成员与肿瘤的关系(表8-11)
表8-11 myc基因家族成员与肿瘤的关系
| 肿瘤种类 | 临床意义 |
| 神经母细胞瘤 | 在20%的肿瘤中有N-myc基因扩增 |
| N-myc基因扩增是预后的预测因子 | |
| Burkitt's淋巴瘤 | 几乎100%的Burkitt淋巴瘤病人均有c-myc基因易位,主要有三种表现形式:①与免疫球蛋白重链位点易位:t(8;14)(q24;q23);②与免疫球蛋白κ轻链位点易位:t(8;14)(q24;q23);③与免疫球蛋白γ轻链位点易位:t(8;22)(q24;q11): |
| 急性T细胞性白血病 | 部分病例可见c-myc基因易位,表现为:t(8;14)(q24;q11) |
| 乳腺癌 | 6%-57%的肿瘤中可见c-myc基因扩增。c-myc基因mRNA水平升高与预后不良相关 |
| 结直肠癌 | 10%-20%的肿瘤中可见c-myc基因扩增 |
| 鳞状细胞癌 | c-myc基因扩增与进展期肿瘤相关 |
| 小细胞肺癌 | 30%肿瘤可见L-myc基因过度表达 |
| 视网膜母细胞瘤 | 均见N-myc基因扩增,却与肿瘤预后无关 |
| 胶质母细胞瘤 | 均见N-myc基因扩增,却与肿瘤预后无关 |
| 宫颈癌 | c-myc过度表达与预后不良相关 |
(三)表皮生长因子受体
1984年Downward研究发现表皮生长因子受体与C-erb-B具有相似顺序,首先提出具有致癌潜能。
EGFR基因定位于第7号染色体上,编码产物为P170的糖蛋白,属于受体型酪氨酸蛋白激酶,能够与表皮生长因子及其他配基结合。当机体发生肿瘤时,往往发现EGFR的过度表达。
⒈可用于EGFR的检测方法
⑴竞争配基结合分析(competitiveligand-bindingassay)。
⑵体内显象:用111烟标记的针对EGFR的单克隆抗体。
⑶Northern印迹法及Western印迹法。
⒉表皮生长因子受体与肿瘤的关系(表8-12)EGFR的过度表达与许多临床肿瘤
表8-12 表皮生长因子受体与肿瘤关系
| 肿瘤类型 | 临床意义 |
| 乳腺癌 | EGFR表达过度见于21-33%的肿瘤中,过度表达与预后不良及短期复发相关 |
| 神经胶质瘤 | EGFR表达过度与基因扩增相关,在一些情况下EGFR的EGF结合区截断 |
| 膀胱癌 | 87%的侵袭性肿瘤中有EGFR过度表达,EGFR过度表达与肿瘤分期相关 |
| 肺癌 | 52%-80%非小细胞性肺癌中有EGFR过度表达 |
| 过度表达与预后不良相关 | |
| 卵巢癌 | 49%-64%的肿瘤出现过度表达,并与预后不良相关 |
| 食管癌 | 38%-47%的肿瘤出现过度表达,并与预后不良相关 |
密切相关,尚有研究表明与一些肿瘤的预后也有一定相关。
二、抑癌基因(suppressergene)
机体中有一类对正常细胞增殖起负调节作用的基因称为抑癌基因(表8-13)。当这类基因丢失、失活或变异时,往往会促使细胞失控而呈恶性生长。
表8-13 常见抑癌基因类肿瘤标志物
| 抑癌基因 | 染色体 | 相关肿瘤 |
| RB | 13q14 | 视网膜母细胞瘤、骨肉瘤、乳腺癌、肺癌 |
| WT1 | 11p13 | Wilms肿瘤 |
| NF-1 | 17q11.1q | 神经纤维瘤 |
| DCC | 13q21.3 | 结肠癌 |
| P53 | 17q21-1q | 肺癌、结肠癌、胃癌 |
| Erb-B-2 | 7p | 乳腺癌 |
(一)RB基因及其表达产物
1986年Friend等成功地克隆了RB基因。RB基因定位于第13号染色体的13q14区,共有27个外显子,26个内含子,DNA长度约200kb。其编码的蛋白质产物为p110。
RB蛋白磷酸化为其调节细胞生长分化的主要形式,细胞G1/S其RB蛋白磷酸化受周期依赖性激酶cdk2调节。在肿瘤细胞中突变的RB蛋白失去了同核配体结合的功能。当机体发生肿瘤时,RB基因的主要变化形式有:缺失、突变、甲基化、表达失活及与病毒和细胞癌蛋白结合引起功能性失活。
⒈可用于RB基因检测的方法
⑴PCR-SSCP、DGGE、PCR-ASO及测序技术:检测点突变。
⑵PR-PCR、PCR。
⑶PCR-RFLP限制片段长度多态性(restrictionfragmentlengthpolymorphism)。
⒉RB基因与肿瘤的关系
⑴RB基因突变约见于40%的视网膜母细胞瘤。
⑵RB基因还与成骨细胞肉瘤、软组织肉瘤、小细胞肺癌、乳腺癌、前列腺癌、食管癌及膀胱癌有关。
⑶近期研究表明RB基因还与卵巢癌有关。
(二)p53基因及其产物
1981年Crawford等发现了p53基因,并认为其为癌基因。以后Hinds、Finlay等通过研究发现传染了myc或ras癌基因的细胞中,若存在野生型p53基因,则出现生长抑制。因此提出p53基因属于抑癌基因。
P53基因定位于第17号染色体17p13区,由11个外显子和10个内含子组成,编码393个氨基酸残基的蛋白质即p53蛋白。p53的功能为转录因子,生物学功能为G1期DNA损坏的检查点。人类肿瘤中P53基因突变主要在高度保守区内,以175、248、249、273、282位点突变率最高,不同种类肿瘤其突变类型不同。另外p53变化形式还有缺失、基因重排与肿瘤病毒癌蛋白结合而失活。
⒈可用于p53基因检测的方法:同RB基因检测方法。
⒉p53基因与肿瘤的关系(表8-14)
表8-14 p53基因与肿瘤的关系
| 肿瘤类型 | 临床意义 |
| 乳腺癌 | 40%有p53突变,9%病人血清有p53蛋白 |
| 结肠癌 | 50%-86%表现p53突变 |
| 肺癌 | 45%-70%p53水平升高,57%的小细胞肺癌过度表达p53 |
| 食管癌 | 35%-44%有p53突变 |
| 肝癌 | 50%有p53点突变 |
| 膀胱癌 | 61%有p53基因突变 |
| 慢性髓性白血病 | p53表达的抑制调节造血细胞增殖 |
| 胃癌 | 37%的肿瘤有p53基因突变 |
| 非霍奇金淋巴瘤 | 61%病例有p53蛋白增加 |
| 宫颈癌 | <10%的病例有p53基因突变 |
| 甲状腺癌 | 约24%的肿瘤中有p53基因突变 |
| 神经纤维肉瘤 | 30%的肿瘤有p53基因突变 |
| 脑肿瘤 | <10%的肿瘤有p53基因突变 |
| 卵巢癌 | 50%的肿瘤有p53基因突变 |
| 骨肉瘤 | 33%-76%的肿瘤有p53基因突变 |
第九章 治疗药物监测
第一节 概论
治疗药物监测(therapeuticdrugmonitoring,TDM)是自本世纪60年代起,在临床药理学、药代动力学和临床化学基础上,结合现代分析检测技术,形成和发展的一门应用性边缘学科。其主要任务是通过灵敏可靠的方法,检测病人血液或其它体液中的药物浓度,获取有关药动学参数,应用药代动力学理论,指导临床合理用药方案的制定和调整,以及药物中毒的诊断和治疗,以保证药物治疗的有效性和安全性。
药物是治疗疾病的主要手段之一。任何药物都不会在体内创造一新的生理、生化过程,而是通过调整疾病过程中失调的内源性活性物质量或生理生化过程,杀灭抑制病原体等,达到治疗作用。显然,药物作用靶位浓度不足或过量,势必导致药物治疗的无效或产生新的不良作用,甚可导致药源性疾病的产生,乃至危及生命。因此,如何根据每个病人的具体情况,制定有效而安全的个体化药物治疗方案,长期以来一直是困扰临床医生的一个难题。虽然试图通过按体重、体表面积、不同年龄等方法,计算调整用药剂量,但由于影响药物体内过程的因素众多,具体病人情况千差万别,因此仍未能很好地解决这一问题。本世纪60年代末药代动力学的发展成熟,使人们得以用简练的数学公式表达药物在体内随时间的量变规律。而60年代末和70年代初,相继报告了普鲁卡因胺和地高辛药物效应与血药浓度的关系,形成了以血药浓度为客观依据,调整剂量指导临床用药的设想。随着科学技术的发展,各种高灵敏度、特异性的检测方法的引入,使仅微量存在的药物检测得以进行。另一方面,越来越多的药物的有效血药浓度范围及中毒浓度也相继确定。以血药浓度为客观依据,运用药代动力学理论指导制定合理用药方案的优越性,日益为广大临床医生接受和采用,从而促进了TDM的发展。目前,TDM在欧美等发达国家,已成为临床化学实验室的主要常规工作之一。国内一些有条件的医院也从80年代起,逐步开展了这一工作。近年来,世界卫生组织(WHO)及我国卫生部药物不良反应监测中心的统计资料均显示,因用药不当而致死者远远高于同期死于各种传染病的人数。而用药不当死亡者中,大多是剂量不当所致。可以说随着医疗技术整体水平的提高,在TDM的指导下制定和调整个体化的合理用药方案,是药物治疗学发展的必然趋势。另一方面,也应看到TDM工作的开展,使历来主要为诊断服务的临床化学实验室工作,开辟了积极参与临床药物治疗的广阔新领域。
一、药物在体内的基本过程
(一)生物膜对药物的转运
药物的体内过程包括吸收(血管内给药除外)、分布、生物转化和排泄四过程。在这些过程中都涉及细胞膜、细胞内器膜等生物膜对药物的转运。从基本结构上讲,生物膜均是由镶嵌有蛋白质的双层流动态类脂质分子构成,其间有直径约0.6nm的小孔。生物膜对药物的转运方式根据是否耗能,分做主动转运和被动转运两类。
⒈主动转运生物膜可通过其间镶嵌的某些特异性载体蛋白,消耗能量转运某些药物。主动转运的最大特点是可逆浓度差进行,并在经同一载体转运的药物间存在竞争性抑制。在药物转运上,主动转运仅限于极少数本身即为内源性活性物质,或与内源性物质有极相近结构的药物。
⒉被动转运包括所有不消耗能量,仅能顺浓度差进行的跨膜转运。被动转运包括扩散、滤过和易化扩散三种。由于不能耗能,被动转运均不能逆浓度差进行。除易化扩散外,亦不存在竞争性抑制。
⑴扩散:指穿过生物膜的双层类脂质分子进行的药物跨膜被动转运。影响药物扩散速度的因素除膜两侧的浓度差外,主要为药物脂溶性高低。虽然药物本身的化学结构已决定了其脂溶性,但由于多数药物均为弱酸或弱碱性物质,在一定pH溶液中会发生不同程度的解离。根据Handerson-Hasselbalch公式可推得:

式中pKs为弱酸药或弱碱性药共轭酸的解离平衡常数。
由于对同一物质而言,其解离态脂溶性总是低于分子态。因此,生理情况下膜两侧存在pH差异时(如细胞内、外液间),必然在膜两侧产生以10的指数方次变化的解离程度差异。从理论上讲,只有分子态的药物脂溶性高,才能以扩散方式被动扩散,因此膜两侧有无浓度差仅是指分子态药物而言。当膜两侧存在pH差异时,分子态被动扩散平衡,膜两侧包括解离态的总药物浓度却可有较大不同。
⑵滤过:指通过小孔进行的被动转运。由于生物膜上的小孔直径过小,只有少数分子量小于100的药物如尿素、乙醇等,可以此方式进行。但毛细血管内皮细胞间呈疏松连结,存在8nm左右的间隙,除少数大分子蛋白药物外,允许绝大多数药物自由通过。因此,药物通过毛细血管的吸收、分布,以及通过肾小球排泄时,滤过为主要的转运方式。
⑶易化扩散:借助膜上特异的载体但不耗能的被动转运方式,此种方式在药物转运中极少见。
(二)吸收
吸收(absorption)是指药物从给药部位进入体循环的过程。血管内给药不存在吸收。血管外注射给药时,药物主要通过毛细血管内皮细胞间隙,以滤过方式迅速进入血液。其吸收速度主要受注射部位血管丰富程度和药物分子大小影响。口服药物的吸收大多通过胃、肠道粘膜以被动扩散方式进行。虽然弱酸性药物在酸性胃液中解离少,可有部分被吸收,但由于吸收面积、血液供应及停留时间等的巨大差异,包括弱酸性药物在内,口服药物的主要吸收部位在小肠。影响口服药物吸收的因素众多,主要为药物本身的脂溶性、分子大小等理化性质、药物制剂的崩解速度及溶解度、胃排空速度、肠蠕动等胃肠道功能状态以及胃肠血流动力学状况等。
某些药物口服后吸收过程中,在通过胃肠道粘膜及第一次随肝门静脉血流经肝脏时,可有部分被胃肠粘膜,更主要是被肝细胞中酶代谢失活,从而使进入体循环的量减少。这一现象称“首过消除”(firstpasselimination)或“第一关长效应”。首过消除强的药物,由于不同个体对同一药物代谢能力存在较大差异,可对口服药物吸收度(生物利用度)产生明显影响。
(三)分布
分布(distribution)是药物随血液循环输送至各器官、组织,并通过转运进入细胞间液、细胞及细胞器内的过程。必须指出,药物在体内的分布可达到动态平衡,但往往并不是均匀(浓度相等)的。只有分布到靶器官、组织或细胞的药物,才能产生药理效应。而以被动转运方式分布的药物,其靶位浓度与血药浓度往往是成比例的。药物在体内的分布主要受下列因素影响:
⒈药物的分子大小、pKa、脂溶性等理化性质。
⒉药物与血浆蛋白的结合绝大多数药物都可程度不等地和血浆蛋白以弱的VanderWaals引力、氢键、离子键等迅速形成可逆的结合,并按质量作用定律处于动态平衡。通常弱酸性药主要和白蛋白结合,弱碱性药和α1-酸性糖蛋白或脂蛋白结合。由于蛋白质的大分子性及两性电解质性,与血浆蛋白结合的药物既不能以滤过方式,也不能以被动扩散的方式进行跨血管转运。只有游离的药物才能进行被动转运分布,发挥作用。药物和血浆蛋白的可逆性结合,可视做药物在体内的一种重要的暂时贮存形式及调节方式。药物与血浆蛋白结合可达饱和,此时再加大剂量将会导致游离药物浓度不成比例的升高,甚至中毒。与血浆蛋白同一位点结合的药物间存在竞争性抑制,使游离药物浓度发生改变,这点在高血浆蛋白结合率药物尤应引起重视。如抗凝血药双香豆素的血浆蛋白结合率高达99%,若同时服用竞争同一蛋白结合位点的消炎药保泰松,即使仅使双香豆泰血浆蛋白结合率降为98%,但可发挥作用的游离药物浓度却增加了一倍,势必造成自发性出血等毒性反应。此外血浆蛋白浓度的变化,亦将影响药物的血浆蛋白结合率。基于上述种种原因,理想的TDM应直接测定血中游离部分的药物浓度。
⒊特殊的膜屏障血脑屏障和血眼屏障都是由该处毛细血管内皮细胞间联接紧密、孔隙小,并在其外包裹有一层神经胶质细胞膜形成的脂质膜屏障。只有高度脂溶性的药物才能以被动扩散的方式进入脑脊液、脑组织和房水。而通常所说的胎盘屏障和一般生物膜没有明显的区别,因此,在药物分布上几乎不存在。这也是孕妇用药必须考虑对胎儿影响的原因。
⒋生理性体液pH差异生理情况下细胞外液pH约为7.4,细胞内液为7.0,乳汁更低,约为6.7。由于前述pH对药物解离的影响,弱酸性药将主要分布在血液等细胞外液中,而弱碱性药则在细胞内液和乳汁中分布高。
⒌主动转运或特殊亲和力少数药物可被某些组织细胞主动摄取而形成浓集,如甲状腺滤泡上皮细胞对碘的主动摄取,使甲状腺中I-浓度比血浆高数十倍。另有少数药物对某些组织、细胞成分具特殊亲和力或形成难解难离的共价结合,亦可产生药物在这些部位的高分布。
(四)生物转化
机体对药物进行的化学转化、代谢称生物转化(biotransformation)。不能简单地将生物转化视为药理活性的灭活。事实上,有些药物必须经生物转化才生成具药理活性的代谢物。如可待因需在肝脏脱甲基代谢为吗啡,才能发挥镇咳止痛作用。但生物转化总的结果是使药物极性升高,有利排泄。药物的生物转化主要在肝细胞微粒体混合功能氧化酶(肝药酶)的催化下进行,主要反应类型、该酶系的组成及催化过程,都与肝细胞对内源性物质的生物转化相同,请参阅本书第十章。
现已明确,至少有200余种常用药为肝微粒体混合功能氧化酶的诱导剂或抑制剂。这些药物较长期使用时,对自身及与其同时使用的其他药物生物转化能力的影响,是TDM工作中必须注意的。如使用双香豆素抗凝治疗的病人,服用诱导剂镇静催眠抗癫痫药苯巴比妥30天,可使降血糖的稳态血药浓度由28μg/ml下降至14μg/ml左右;而抑制剂氯霉素使用2天,可使降血糖药甲磺丁脲稳态血药浓度上升近1倍。肝微粒体混合功能氧化酶存在饱和性,当体内药量(血药浓度)超过其最大代谢能力后,将会出现药物消除动力学方式的转化(参见本章第二节)。
(五)排泄
排泄(excretion)是药物及其代谢物排出体外的过程。药物的生物转化和排泄统称为消除(elimination)。药物排泄的主要途径为经肾脏随尿排出。游离的原型药物和代谢物均可通过肾小球毛细血管壁小孔隙滤入原尿中,也有少数弱酸、弱碱药可在近曲小管上皮细胞,以主动转运方式分泌入原尿中。原尿液中的原型药物仍可以被动扩散等方式被肾小管重吸收,此时尿液pH通过对药物解离度的影响,明显改变原尿液中药物被重吸收的量。此亦是弱酸或弱碱性药物中毒时,可通过碱化或酸化尿液,促进药物排泄的原因。而代谢物因极性高,一般不会被重吸收。随原尿逐渐浓缩,其中的药物及代谢浓度均上升,最终可远远超出血中浓度。这种浓集现象是许多药物产生肾毒性的原因,另一方面对用以治疗泌尿道疾患的药物,则有其利于发挥治疗作用的意义。
除经肾脏排泄外,部分药物及其经肝细胞生物转化而成的代谢物,可随胆汁经胆道系统排入十二指肠。进入肠腔的药物及其代谢物可随粪便排出体外,亦有一些药物及其葡糖醛酸或硫酸酯代谢物经肠道细菌水解后,可重新被肠道吸收,形成肠肝循环。某些药物肠肝循环较显著,如强心药洋地黄毒甙在体内可有约20%处于肠肝循环中。此外,挥发性气体药可由肺排泄,而汗液中也可排出少量药物。某些药物特别是弱碱性药,可有相当部分自偏酸性的乳汁中排泄,这点在给哺乳期妇女用药时必须考虑到。
二、药物体内过程与药代动力学
事实上,药物从进入人体内起,即同时在吸收、分布、生物转化和排泄的综合影响下,随着时间而动态变化着的(图9-1)。
显然,孤立地研究上述体内过程中的某环节的变化,笼统地描述某一过程的快慢、强弱,均不能客观全面地反映体内药物随时间的量变及其规律。同样,当取样测定某一体液中的药物浓度,其结果除代表取样瞬间该体液中的药物浓度外,既不能了解在此之前,亦不能预测在此之后的变化情况,实无多大价值。药代动力学则是以必要的数学模型、参数和公式,定量表达某种体液中药物或代谢物在前述体内过程的综合作用下,随着时间的量变规律。此外,应用药代动力学理论,还可了解药物的吸收、分布、消除的规律。如图9-1所示,由于血液中的药物在药物体内过程中起着中心枢纽作用,可将其视作药物体内过程的一面镜子,因此,血液中的药代动力学是最常采用的。在药代动力学理论的指导下,就能够根据血药浓度测定的结果,客观地推测药物的体内过程,判断剂量是否得当,并制定出调整方案。因此,可以说药代动力学是TDM工作的必备重要基础理论。
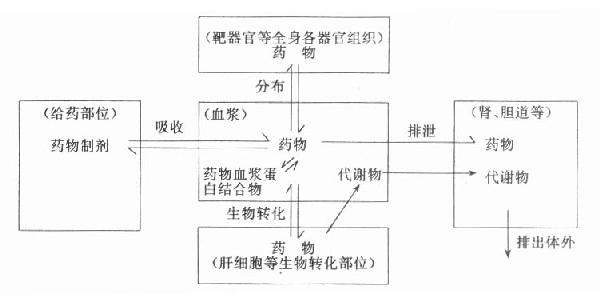
图9-1 药物体内过程及其与血浆中药物的关系示意图
三、血药浓度与药物效应
无论是药物的治疗作用还是不良反应,从本质上说,都是通过药物和靶位上的受体等大分子物质间的相互作用而产生的。这种相互作用符合质量作用定律,因此,药物效应是否出现及其强弱,取决于靶位的药物浓度。从这点上讲,理想的TDM应直接检测靶器官或组织的药物浓度。但大多数药物都是作用于心、肝、肾、胃肠道、中枢及周围神经系统等,从这些部位以损伤性手段取样,在现阶段是困难且不能为病人所接受的。
前已述及,血液中的药物在药物体内过程中起着中心枢纽作用,除直接在靶位局部用药外,到达上述脏器的药物均是从血液分布而至。药物在体内达分布平衡时,虽然血液和靶位的药物浓度往往并不相等,但对绝大多数药物,特别是以被动转运方式分布的药物,其血药浓度与靶位药物浓度的比值则是恒定的。换言之,即药物效应与血药浓度间存在着相关性。这一设想自60年代以来,已为众多研究报告所肯定。根据血药浓度与治疗作用和毒性反应间的关系,不少药物治疗血药浓度范围及中毒水平都已确定。这些工作为TDM的开展,尤其是血液浓度测定结果的解释判断,提供了参考依据。当然,若其他易于获取的体液药物与血液或靶位药物浓度间,也同样存在恒定比值关系,亦可通过检测这些体液中的药物浓度进行TDM。必须指出,上面提到的治疗血药浓度范围和中毒水平,仅是得自群体资料的参考值,由于个体间靶器官、组织或细胞对药物反应性存在差异等原因,因此在解释判断TDM结果时,不能仅拘泥于上述标准,必须结合病人的具体临床表现及治疗效果,作出结论。
第二节 药代动力学基础及有关参数的应用
药代动力学(pharmacokinetics)简称药动学,从广义上讲,泛指研究药物的体内过程即机体对药物的吸收、分布、生物转化和排泄过程及其量变规律。狭义的药动学则是指以数学模型和公式,研究体内药物随时间的量变规律。本节主要介绍后者中与TDM有关的内容。在TDM工作中,药动学主要用于:①建立监测个体的体内药量或药物浓度随时间变化的数学表达式,并求算出有关药动学参数;②应用上述动力学模型、表达式和药动学参数,制定和调整个体化的用药方案,保证药物治疗的有效性和安全性。
一、药动学模型
药动学模型是为了定量研究药物体内过程的速度规律而建立的模拟数学模型。常用的有房室模型和消除动力学模型。
(一)房室模型
房室(compartment)是由具有相近的药物转运速率的器官、组织组合而成。同一房室内各部分的药物处于动态平衡。房室仅是按药物转运动力学特征划分的抽象模型,并不代表解剖或生理上的固定结构或成分。同一房室可由不同的器官、组织组成,而同一器官的不同结构或组织,可能分属不同的房室。此外,不同的药物,其房室模型及组成均可不同。运用房室模型,可将机体视做由一或多个房室组成的系统,从而将复杂的分布过程模型化。
若某药在体内各部位间均有较高及相近的转运速率,可在体内迅速达到分布平衡,则该药属单房室模型。属于单房室模型的药物,在体内达分布平衡后,其血药浓度将只受吸收和消除的影响。而某药在体内不同部位间转运速率存在较大差异的话,则将血液及其他血液供应丰富、并具有较高转运速率的部分,称做中央室,而把其余部分划归周边室,并可依次再分做第一周边室、第二周边室等,此即多室模型。根据划分的房室数,相应称为二室模型、三室模型等。属于多室模型的药物,其首先在中央室范围内达分布平衡,然后再和周边室间达到分布平衡,因此其血药浓度除受吸收和消除的影响外,在室间未达分布平衡前,还受分布的影响。
(二)消除动力学模型
消除动力学(eliminationkinetics)研究体内药物浓度变化速率的规律,可用下列微分方程表示:
dC/dt=-kCn
式中C为药物浓度,t为时间,k为消除速率常数,n代表消除动力学级数。当n=1时即为一级消除动力学,n=0时则为零级消除动力学。药物消除动力学模型即指这两种。
⒈一级消除动力学一级消除动力学(firstordereliminationkinetics)的表达式为:
dc/dt=-kC积分得Ct=Ce-kt
由上指数方程可知,一级消除动力学的最主要特点是药物浓度按恒定的比值减少,即恒比消除。有关一级消除动力学的其他性质及特点,将在本节二、三中详细讨论。
⒉零级消除动力学零级消除动力学(zeroordereliminationkinetics)时,由于n=0,因此其微分表达式为:
dc/dt=-k积分得Ct=C-kt
由此可知,零级消除动力学的最基本特点为药物浓度按恒量衰减,即恒量消除。有关零级消除动力学的其它特点和性质,将在本节四中讨论。
必须指出,并不是某药固定按一级或零级动力学消除。任何药物当其在体内量较少,未达到机体最大消除能力时(主要是未超出催化生物转化的酶的饱和限时),都将按一级动力学方式消除;而当其量超过机体最大消除能力时,将只能按最大消除能力这一恒量进行消除,变为零级消除动力学方式,即出现消除动力学模型转换。苯妥英钠、阿司匹林、氨茶碱等常用药,在治疗血药浓度范围内就存在这种消除动力学模型转移,在TDM工作中尤应注意。
二、单室模型一级消除动力学
(一)单剂静脉注射
⒈模式图及药-时关系单室模型的药物可迅速在体内达到分布平衡,故可不考虑分布的影响。静脉注射用药时,药物直接迅速进入血液,因此也不受吸收的影响。此时体内药量将仅受包括生物转化和排泄在内的消除影响,可建立如下模式图。
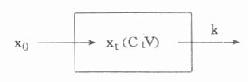
图9-2 单室模型单剂静脉注射模式图
图9-2中x为剂量,xt为t时体内药量,Ct表示t时的血药浓度,V为表观分布容积,k为消除速率常数。当按一级动力学方式消除时,体内药量随时间变化的微分方程为:
dx/dt=-kX式⑴
积分得X=Xe-kt式⑵
因体内药量不可能直接测定,故引入比例常数:表观分布容积V,以便用血药浓度表示,即V=X/C,所以X=VC。代入式⑵可得
CCe-kt式⑶
式⑶取对数得IgC=IgC-kt/2.303式⑷
⑶和⑷式即为单室模型单剂静脉注射给药时的药-时关系表达式。
⒉药动学参数及计算
⑴药-时关系表达式:从式⑷可看出,当血药浓度以对数表示时,与时间t的关系为简单的直线关系。因此,在静脉注射药物后不同时间取血,测定血药浓度。根据血药浓度对数值及相应时间,以图解法或线性回归法(最小二乘方法),即可求得如式⑷的直线方程(图9-3)。
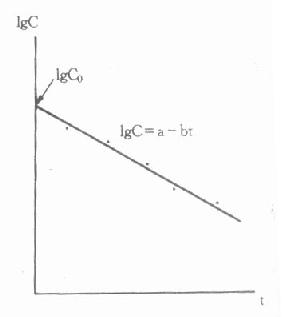
图9-3 单剂静脉注射血药浓度-时间关系示意图
IgC=a-bt
此直线方程与纵轴的截距a=IgC,故C=Ig-1a;而斜率b=k/2.303,可计算出消除速率常数:
k=2.303b。
⑵消除速率常数:消除速率常数(eliminationrateconstant,k)表示单位时间内机体能消除药物的固定分数或百分比,单位为时间的倒数。如某药的k=0.2h-1,表示机体每小时可消除该小时起点时体内药量的20%,此即一级消除动力学的恒比消除特点。此时虽然单位时间消除的百分比不变,但随着时间的推移,体内药量逐渐减少,单位时间内消除的药量也逐渐减少,而不是恒定不变的,消除速率常数是反映体内药物消除快慢的一个重要参数。必须指出,一个药物的消除速率常数在不同的个体间存在差异,但对同一个体来说,若无明显的影响药物体内过程的生理化、病理性变化,则是恒定的,并与该药的剂型、给药途径、剂量(只要在一级动力学范围内)无关。
⑶半寿期:药动学中的半寿期(halflife,t1/2)通常是指血浆消除半寿期,即血浆中药物浓度下降一半所需要的时间。根据这一定义,当t=t1/2时,C=2C,代入式⑷并整理可得
t1/2=0.693/k式⑸
从式⑸可看出,由于一级消除动力学时,k为一常数,半寿期亦为一常数。半寿期恒定不变,是一级消除动力学的又一特征。和消除速率常数一样,半寿期也是衡量药物消除快慢的又一临床常用参数,二者的关系如式⑸所表达。在药物的临床药动学参数资料中,常告知半寿期,只要知道半寿期,根据式⑸即可求得消除速率常数k值。半寿期在指导用药方案的制定中,有较大意义,将在后面讨论。
⑷表现分布容积:如前所述,表观分布容积(apparentvolumeofdistribution,V)是为了用血药浓度计算体内药量而引入的比例常数,表示假设体内药物按血药浓度均匀分布所需要的容积。前已谈到药物在体内分布可达动态平衡,但并非均匀一致,因此表观分布容积仅是一理论容积,并不代表真实的解剖或生理空间。但只要知道某药的表观分布容积V,应用测定的血药浓度,即可根据Xt=Ct·V,计算得实际工作中无法测定的任一时刻体内的药量,并可按上式计算出欲达某一血药浓度C所需使用的剂量X=CV。此外,表现分布容积还可用于评估药物在体内的分布特点。人的总体液量约0.6L/kg体重,若某药的V远远大于0.6L/kg体重,提示该药主要分布于细胞内,被某组织、脏器主动摄取或对某些组织成分有特殊亲和力,致使包括血浆在内的细胞外液中浓度低。大多数弱碱性药由于细胞内液比细胞外液偏酸而存在这一情况,如奎尼丁的表观分布容积可超出2L/kg体重。反之,若某药表观分布容积远远低于0.6L/kg体重,则其主要分布于血浆等细胞外液中。多数弱酸性药便是如此,如水杨酸的表观分布容积仅0.2L/kg体重。
单室模型静脉注射用药时V的求算可采用外推法,即根据前面介绍的药-时关系表达式求得t=0时的C值及注射剂量X,按V=X/C而计算出。其单位最常采用容积单位/kg体重。同前述药动学参数一样,V也是仅取决于药物本身的理化性质、体内分布特点,而与该药剂型、用药方式、并在一级消除动力学范围内与剂量都无关。在所有药动学参数中,V和k是两个最基本的参数。
⑸清除率:药物清除率(clearance,Cl)是指单位时间内机体从血浆中消除某种药物的总能力,其数值即等于该时间内机体能将多少体积血浆中的该药完全消除。与k和t1/2相同,Cl也是衡量体内药物消除快慢的一个药动学参数,但与k和t1/2不同,Cl以具体的解剖生理学概念来表示,可更直观形象地反映机体对药物的消除能力。由于药物在体内按血浆浓度分布的总体积为V,而k表示单位时间内药物被消除的分数,故代表单位时间内机体能消除多少体积血浆药物的清除率可按Cl=Vk计算,单位为体积单位/时间单位。
⑹曲线下面积:血药浓度-时间曲线与纵轴和横轴间围成的范围面积即曲线下面积(areaundertheC-tcurve,AUC),单位为浓度单位×时间单位。由于任何药物不论以何种剂型或途径用药,进入体内后,只要是同一种药物分子,其消除均相同。因此AUC是评估进入体内药量多少的一个客观指标。在后面介绍的生物利用度的计算,以及近年建立的非模式消除动力学分析矩量法(statisticalmomenttheory)中,均有重要意义。
AUC的计算方法有称重法、梯形法和积分法3种。其中称重法为剪下曲线下纸片称取重量,除以单位面积纸片的重量,即为该曲线的AUC。该法较不准确,现已少用。下面介绍后两种计算方法。
1)梯形法:如图9-4所示,可将曲线下范围分做若干个等高梯形,分别计算各个梯形面积累加而成。即:
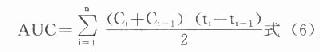
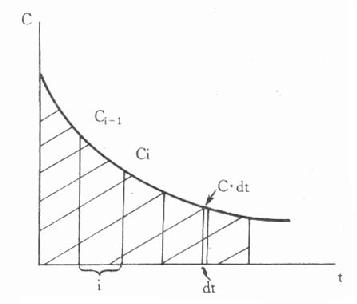
图9-4 单剂静脉注射时的药-时曲线下面积
此法不论何种房室模型及何种途径给药均适用。但本法只能求算测定血药浓度的时间范围内的AUC。
2)积分法:当药-时曲线按足够小的时间间隔dt划分时,可视做若干个矩形,每个矩形的面积分别为C·dt,将其积分得:
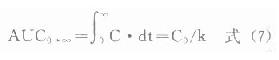
药动学中积分法求算的AUC,均表示曲线随时间无限外延,直至体内药量完全消除时的面积。此外式⑺仅适用于单室模型、一级消除动力学单剂静脉注射给药的情况。
(二)恒速静脉滴注
恒速静脉滴注用药,是临床特别是危重症抢救中常用的方法。此时通过TDM工作,制定和调整滴注药物速度,对确保抢救效果有重要意义。
⒈模式图和药-时关系表达式恒速静脉滴注与单剂静脉注射不同,此时药物一方面以恒速的零级动力学方式进入体内,另一方面又以恒比的一级动力学方式从体内消除(图9-5)。
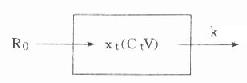
图9-5 单室模型恒速静脉滴注模式图
图中R为滴注速度,R=X/t,X为t时间内滴注入体内的总药量。余参数意义同图9-2。此时体内药量随时间变化的微分表达式为:
dx/dt=R-kX积分得X=R/k(l-e-kt)式⑻
或C=R/Vk(l-e-kt)式⑼
式⑻、⑼即为恒速静脉滴注、单室模型一级消除动力学的体内药量或血药浓度随时间变化的基本表达式。
⒉药动学参数及计算
⑴稳态血药浓度:稳态血药浓度(steadystateplasmaconcentration,Css)指单位时间内自体内消除的药量与进入体内的药量相等时的血药浓度。此时,血药浓度将维持在坪值或波动在一定范围内(多剂分次给药时)。恒速静脉滴注时,只要滴注速度R能使体内药量保持在一级动力学消除范围内,则当t→∞时,式⑼中e-kt→0,式⑼可写作
Css=R/(k·V)式⑽
从式⑽可看出,由于k、V都是常数,恒速滴注时,R也不变,故此时血药浓度亦为一常量,即达到稳态浓度。并且从式⑽还可看出,Css高低仅与R成正比。这也是只要滴注速度得当,长期静脉恒速滴注,血药浓度不会无限上升产生毒性反应的原因。此外,知道某药的k、V值及达到治疗作用所需的Css后,则可根据式⑽计算出所需的滴注速度R=Css·k·V,需指出的是,当恒速静脉滴注药物用于抢救心衰或休克病人时,随着血流动力学的改善,病人的k及V均可改变,必须通过TDM及时调整滴注速度,以保持在所需的Css。
若将时间用半寿期数n表示,即t=nt1/2=0.693n/k,应用前面学过的公式,可得到达稳态前血药浓度C与Css的关系:
C=Css[1-(1/2)n]式⑾
从式⑾可计算出恒速静脉滴注经过5个半寿期,血药浓度可达Css的96.8%,6个半寿期达98.4%。因此,临床上通常视恒速静脉滴注经过5-6个半寿期后,达到了稳态血药浓度。
⑵静脉滴注的负荷剂量:从上可知,为达Css,至少需恒速静脉滴注5-6个半寿期以上。而临床抢救中常需迅速达到有效血药浓度,此时可考虑使用负荷剂量法。负荷剂量(loadingdose,D)是为了迅速或立即达到稳态浓度而首先使用的增大剂量。静脉滴注用药时,有下面两种负荷剂量法。
1)先静脉注射一负荷剂量,立即达Css,继之以恒速滴注维持。根据前面所学知识可得D=Css·V=R/k。故根据治疗浓度确定的所需Css和该药的V,或为达所需Css计算出的恒速滴注速度R和该药的k,即可按上式求得所需D,静脉推注后,立即改为R速度恒速滴注,便可立即达到Css并维持之。
2)先快速滴注t时间,迅速达所需Css水平,再改为恒定的慢速滴注维持。此法较上法安全,尤适用于毒性大、治疗浓度与中毒浓度接近的药物。此时可根据下式(推导从略)计算出所需的负荷速度R*:
R*=R/l-e-kt式⑿
式中R为达所需Css计算出的恒定慢速滴注速度,t为计划的负荷滴注时间。按R*滴注t时间后,血药浓度即可迅速升至Css水平,调整滴注速度为R,即可维持在Css水平。
⑶其它药动学参数计算:若已知某药其它方式用药时的有关药动学参数,前已述及也可用于恒速静脉滴注。当需通过恒速静脉滴注计算药动学参数,可使用终止滴定法。即在恒速静脉滴注t时间后,停止滴注,以t时间为零时,测定随后几个不同的时点(t’)的血药浓度,同前静脉注射法求得直线方程:
IgCt=![]()
应注意此式的t为开始滴注到停止滴注的时间。然后根据下列各式:
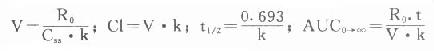
分别计算出各有关药动学参数。
(三)血管外单剂用药
⒈模式图和药-时关系表达式口服、肌肉或皮下注射用药时,和前面讨论的血管内给药不同。此时即存在药物从用药部位吸收进入血液的过程,也存在药物自体内(血液)消除的影响。由于绝大多数药物均是以被动转运的方式吸收,故上述两个过程都按一级动力学方式进行。其模式图如下(图9-6):
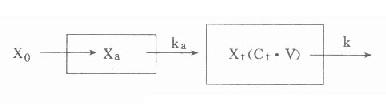
Xa:t时吸收部位药量
Ka:吸收速率常数其它参数同图9-3
图9-6 血管外用药模式图
根据上述关系及模式图,可建立如下微分方程组:
dxa/dt=-Ka·Xa(吸收部位药物衰减速率)
dxa/dt=-Ka·Xa--K·X(血液等药物分布室内药物衰减速率)
解此微分方程组得
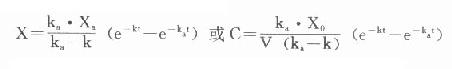
若考虑口服时吸收不完全而引入吸收分数F,则:

式⒀即为单剂血管外用药时,血管浓度随时间变化的基本表达式。
⒉药动学参数及计算通过血管外用药计算药动学参数多用残数法(methodofresidual)。该法基本指导思想是,以血管外用药能获治作用的药物,必然Ka>>k,才有可能在体内达到治疗血药浓度,因此,当t足够大时,首先e-kat→0此时式⒀可写作:
C=A·e-kt取对数得lgC=lgA-kt/2.303
也就是说单剂血管外用药时,经过一段时间后,其血药浓度的变化可视做只受消除的影响,即进入消除相(图9-7)。
此时按前面介绍的单剂静脉注射药动学参数计算法,可求得A、k和消除t1/2。而在进入消除相以前的时间内,血药浓度为吸收和消除两因素共同作用的结果。若将式⒀展开移项则得
A·e-kt-C=A·e-kat,令Cr=A·e-kt-C,
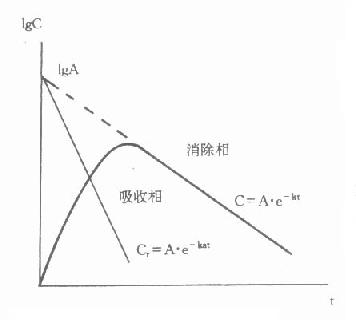
图9-7 单剂血管外用药血药
浓度-时间关系及药动力学求算示意图
Cr为消除相外推段某时点血药浓度减去该时点实测浓度的残数或差值(注意不是对数值相减)。则:
Cr=A·e-kat,取对数得TgCr=IgA-kat/2.303
此即分布相药-时关系的表达式(图9-6),同理可求算得ka和吸收t1/2。
在计算其它药动学参数时,反映药物被机体吸收利用程度的吸收分数F,即生物利用度(bioavailability)是必须首先先知道的。血管外注射用药时,一般均视为F=1。而现在多数口服药在说明书中已告知F值,否则需根据某药口服时AUC与该药同剂量静脉注射时的AUC相比计算出。口服时的AUC可用前述梯形法,或按下列积分法公式求得:
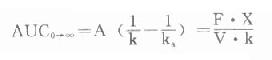
其他药动学参数计算见下。
⑴表观分布容积: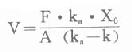
⑵清除率:Cl=k·V
⑶达峰时间(timeofthepeakconcentration,tp):即血管外给药时,达到最高浓度所需时间。由于在此时,血药浓度变化速率dC/dt=0,故可推导出:
![]()
⑷峰浓度(maximumconcentration,Cmax):将tp代入式⒀可得:
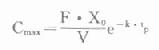
(四)多次用药
为保持或巩固疗效,临床常需反复多次较长期用药。此时体内药量或血药浓度将出现如图9-8所示的波动式上升,每次用药间隔中出现从峰值向谷值的变化。若体内药量不超过一级消除动力学范围,随着用药次数增多,血药浓度逐渐升高,但最终将稳定在一定范围内波动,即进入稳态浓度(推导见后)。指导合理的多次用药方案的制定和调整,使稳态血药浓度波动在治疗浓度范围内,是TDM在临床治疗学中最主要的任务。下面我们将介绍按恒定剂量、固定间隔时间多次用药时与TDM有关的药动学知识。需要指出的是,单剂用药时的有关药动学参数仍适用于多剂给药,并且是多剂用药药动学的基础。
⒈ 剂量函数当按恒量固定间隔时间τ多次用药,无论是静脉注射,还是肌肉注射、口服等血管外用药,均可推导得多剂量函数r(推导从略)。
![]()
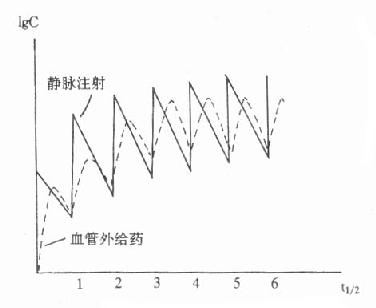
图9-8 多剂用药的血药浓度-时间关系示意图
n:用药次数
Ki:有关速率常数
多剂量函数为多剂用药时,用药间隔时间τ和用药次数n对体内药量或血药浓度的影响的通用函数表达式。具体应用时,只需将单剂用药有关公式中含有速率常数的指数或对数项乘以多剂量函数r即可。但要注意:①此时多剂量函数r中的ki均应换成该项之k或ka;②对数项时,多剂量函数r应放在对数内与有关速率常数相乘;③相应各公式中t应为第n次用药后的时间。如此根据⑶式可得多剂静脉注射用药时,药-时关系表达式为:
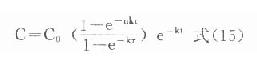
同理根据式⒀可得血管外多剂用药的药-时关系表达式为:
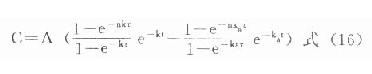
恒速静脉滴注时=0,仍用原式
![]()
⒉稳态浓度和平均稳态浓度当连续多次给药后,n足够大时,多剂量函数式中,则
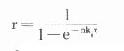
显然此时的多剂量函数式为一常量,此即稳态时的多剂量函数式。分别代入式⒂或⒃中可得:
静脉注射时: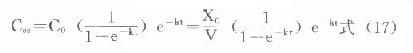
血管外给药时:![]()
以上两式中,由于仅时间t在每次用药间隔中从0→τ的范围变化,血药浓度都将进入在每次用药间隔中,恒定在一定范围内波动的稳态状态。τ越大,波动范围越大。式⒄、⒅则分别为静脉注射、血管外用药时,稳态浓度在每次用药间隔中随时间变化的表达式。实际工作中,当nτ=6t1/2时,血药浓度可达稳态浓度的98.4%。故在连续多剂用药时,一般认为经过6个半寿期以上,即可视做已达稳态状态。此外,无论达稳态否,如果变换剂量,必须再经过6个半寿期以上始能进入新的稳态。
静脉注射时,每次间隔中波动的峰值总是在每次注射完的瞬间(t=0),而谷值则在下次注射前(t=τ)出现,分别代入⒄式可得静脉注射多剂用药时
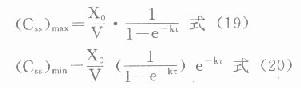
血管外给药时,每次间隔中,谷浓度也将在下次给药前。但由于存在吸收,峰浓度将在达峰时间(t’p)出现。将稳态时多剂量函数代入前述单剂用药tp,求算公式得:
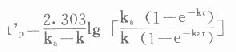
分别以上述t’p或t=τ代入⒅式,并且因ka较大,令e-kaτ→0,可推得血管外给药的
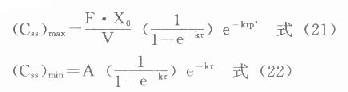
在TDM工作中,运用式⒆-(22)式,选定间隔时间(τ),计算剂量,或选定剂量计算nt-family:TimesNewRoman;mso-fareast-font-family:宋体;mso-font-kerning:1.0pt;mso-ansi-language:EN-US;mso-fareast-language:ZH-CN;mso-bidi-language:AR-SA"lang="EN-US">τ,使(Css)max<最小中毒浓度,而(Css)min>最小有效浓度,是十分有用且经常性的工作。
平均稳态血药浓度(![]() )为稳态时,两剂用药间药-时曲线下面积(AUC)除以间隔时间τ的商值。必须注意,(
)为稳态时,两剂用药间药-时曲线下面积(AUC)除以间隔时间τ的商值。必须注意,(![]() )不是(Css)max和(Css)min的算术或几何平均值。根据稳态浓度的定义可知,此时两剂间AUC就为该剂量单剂给药时的AUC0→∞。根据前面学过的公式,可得
)不是(Css)max和(Css)min的算术或几何平均值。根据稳态浓度的定义可知,此时两剂间AUC就为该剂量单剂给药时的AUC0→∞。根据前面学过的公式,可得
静脉注射: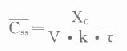
血管外用药:![]()
在TDM工作中,对给药间隔τ不是远远长于半寿期,即稳态时血药浓度波动范围不是太大,且有效血药浓度范围上限与最小中毒浓度有一定差距的药物,以有效血药浓度范围中值或略低定为![]() ,按上式公式制定或调整用药方案,是一简便并且为临床所能接受的方法。
,按上式公式制定或调整用药方案,是一简便并且为临床所能接受的方法。
⒊负荷剂量上面讨论中谈到,多剂用药时,无论间隔时间长短,都需经过6个以上半寿期才可认为已达稳态。对t1/2较长或急需迅速发挥疗效的药物,往往需要使用负荷剂量(X*)。多剂用药时欲使第一次用药后即达到稳态浓度,负荷剂量可按下面公式计算(推导从略)。
静脉注射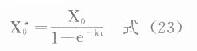
血管外用药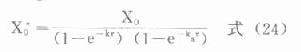
式中X为拟使用的固定剂量。若吸收较快,即ka大,e-kaτ→0,(24)式也可写做
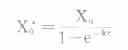
上述各式中1/(1-e-kτ)即前述稳态时的多剂函数式,亦称蓄积指数(accumulationindex),代表稳态时血药浓度峰值或谷值与首剂用药时峰值或谷值之比。蓄积指数实际上反映了达稳态后,每次给药间隔中任一时点血药浓度为首剂用药后同一时点血药浓度的倍数。如τ=t1/2时,蓄积指数为2,X*=2X。实际工作中,根据所需稳态血药浓度水平确定的X及τ,按上述公式计算出负荷剂量X*首剂使用后,再按X及τ,恒量固定间隔用药,可在负荷剂量使用后即达稳态浓度并维持之,获得迅速而稳定的疗效。
三、二室模型一级消除动力学
多室模型和前面讨论的单室模型不同,此时存在着由包括血液在内的中央室向周边室分布达到平衡的过程,影响血药浓度的因素更为复杂,下面以单剂静脉注射为例,简介有关药动学知识。
(一)模式图和药-时关系
静脉注射时,不存在吸收过程,中央室中的药量或血药浓度受中央室与周边室间双向分布,以及自中央室消除的影响。其模式如图9-9所示。
xc中央室药量
xp周边室药量
vc中央室分布容积
vp周边室分布容积
k10中央室向周边室转运速率常数
k21周边室向中央室转运速率常数
图9-9 二室分布静脉注射模式图
中央室药量变化的速率微分方程为:
dkc/dt=k21·xp-k21·xc-k10·xc
对上式积分并引入中央室分布容积Vc,可得中央室(血)药物浓度随时间变化的基本表达式:
C=A·e+B·e-βt式(25)
式中α为分布速率常数,β为消除速率常数,A、B为经验常数。四者都是由模式参数k10、k12、k21组成的混杂参数(hybridparameters)。存在:
α·β=k21·k10α+β=k10+k12+k21
(二)药动学参数计算
二室模型静脉注射药动学参数的求算,类似于单室模型血管外用药,仍采用残数法。即因为α>β,当t充分大时,A·e-αt→0,则式(25)变为
C=Be-βt
此即消除相药-时关系表达式。按前述方法可求得B、β和消除半寿期t1/2β。将式(25)移项可得:
C-B·e-αt=A·e-αt
令Cr=C-B·e-βt,Cr即为消除相以前某时点实测血药浓度减去消除相该点外推浓度的残数。代入上式可得分布相药-时关系表达式
Cr=A·e-αt
同样可求得A、α和分布半寿期t1/2α(图9-10)。
图9-10 二室模型静脉注射血药浓度-时间关系示意图
再根据下列公式,可求得各有关药动学参数。
中央室表观分布容积:Vc=X/(A+B)
周边室向中央室的转运速率常数:
![]()
自中央室消除的速率常数:
k10=α·β/k21
中央室向周边室转运的速率常数:
k12=(α+β)-(k10+k21)
曲线下面积:AUC0→∞=A/α+B/β(积分外推法)
周边室表观分布容积:Vp=Vc·k12/k21
总表观分布容积: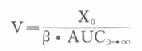 表示体内药量按消除相血药浓度分布的容积)
表示体内药量按消除相血药浓度分布的容积)
稳态表观分布容积:Vss=Vc+Vp
消除率:Cl=k10·Vc=β·V
血管外用药时,由于存在吸收因素,求算药动学参数的基本方法仍为残数法,但此时需进行两次残数处理。有关二室模型血管外用药及二室以上多室模型的药动学,在TDM的实际工作中很少应用,可参阅有关药动学专著。
四、非线性动力学
在消除动力学模型中已介绍,当体内药量(血药浓度)超过机体最大消除能力时,将为恒量消除的零级动力学,而药量(血药浓度)降至最大消除能力以下,将转化为恒比消除的一级动力学(图9-11)。这种存在动力学转换的情况下,药物的消除不能用一种统一简单的线性过程描述,故称非线性动力学(nonlinearpharmacokinetics)消除。若某药使用的剂量能使其在体内的消除由一级动力学转为零级,继续使用该剂量,血药浓度将会出现持续上升,而不能达到稳态浓度。对于安全范围狭窄的药物出现这种情况,是十分危险的。在需进行TDM的药物中,苯妥英钠、氨茶碱等在常用治疗剂量下就存在这种情况。
造成这种药动学方式转化的原因,主要是体内药量(血药浓度)超过了机体生物转化酶系的最大催化能力,即出现了饱和代谢,故亦常用描述酶促反应动力学的米氏方程表达非线性动力学消除的速率,即
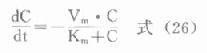
式中Vm为最大消除速率,Km为米氏常数,相当于恰可产生Vm/2时的药物浓度。当C<<Km时,式(26)可变为
dc/dt=VmKm,令0k=Vmdc/kmdt,则dc/dt=-kC
此即前面已介绍的典型一级消除动力学微分表达式。而当C>>Km时,式(26)可写作
dC/dt=-Vm
上式为典型的零级消除动力学微分表达式。
由式(26)可推导得非线性动力学消除时的有关药动学参数计算公式:
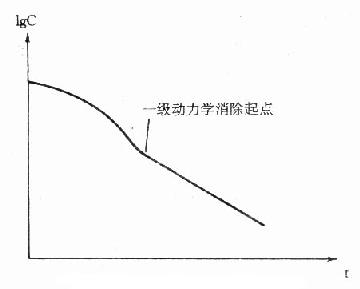
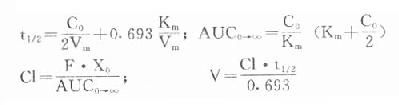
图9-11 非线性动力学消除血药浓度-时间关系示意图
从上述各式中可看出,非线性消除动力学药物的多数参数均为随药物浓度而变化的变量,并非常数。在非线性动力学消除的药物TDM工作中,Vm和Km是两个十分有用的基本常数。必须注意的是,在药物体内过程中,生物转化能力影响因素多,个体差异尤为显著,因此Vm和Km个体差异大。在应用这两个参数的群体均值制定的剂量方案,往往不能达到预期效果。对于需长期用药或完全范围窄的药物,按下面方法确定具体个体的Vm和Km值实属必要。
不同时点取血测定血药浓度,依次求得相邻时点血药浓度及时间差值△C和△t。将式(26)中的dC/dt视做△C/△t,即消除速率V,而以相应两时点血药浓度均值C作为产生相应速率改变的浓度,分别代入式(26),并取倒数整理得:
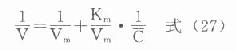
式(27)为1/V随1/C变化的直线方程(图9-12)。该直线与纵轴交点为1/Vm,斜率为Vm/Km,故可分别求出Vm和Km。
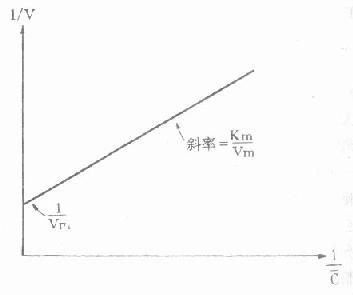
图9-12 双倒数法求算非线性动力学消除的Vm和Km示意图
根据非线性动力学消除的特点,多剂用药时,只有当药物进入体内的速率R(量/d)恰与药物自体内的消除速率相等时,才有可能达稳态浓度Css。借用式(26)可得
R=Vm·Cm/Km+Cm式(28)
由此可得Css=Km·R/Vm-R式(29)
由上两式可计算出非线性动力学消除的药物,欲达某稳态浓度所需的用药速率(每日用药量),或按某速率用药时所能达到的稳态浓度,在这类药物的TDM工作中极为有用。从式(28)、(29)中也可看出,Vm和Km是必须首先求算出的基础。
第三节 合理使用治疗药物监测应考虑的基本因素
从理论上讲,通过TDM工作测定血药浓度,并应用前述药动学理论制定、调整剂量,对任何治疗药物都适用。但在实际工作中,某些药物如青霉素G、多数维生素等,由于本身安全范围大,不易产生严重毒性反应;另一些药物的治疗作用本身所致的生理、生化实验室指标的改变,就是可靠的用药剂量判断指征,如用抗凝血药肝素、双香豆素类时,对出、凝血功能的检测;此外,也出于经济上的考虑,并非所有治疗药物均要进行TDM。一般认为,对存在下列药效学、药动学或其他原因,并且其治疗作用、毒性反应呈血药浓度依赖性,而治疗血药浓度范围和中毒水平已确定的药物,应考虑进行TDM。
一、药效学原因
⒈安全范围狭窄,治疗指数低一些药物的治疗浓度范围和最小中毒浓度十分接近,甚可重叠,极易中毒。只有根据TDM检测的血药浓度调整剂量,才能获得安全而有效的治疗效果。强心甙、大多数抗心律失常药、抗躁狂症药锂盐等,都存在这种情况。如普鲁卡因胺的有效血药浓度范围为4-10μg/ml,一般超过10μg/ml即可出现低血压及多种严重心律紊乱等毒性反应。
⒉以控制疾病发作或复发为目的的用药此类用药的目的不是治疗已存在的疾病,而是巩固疗效或控制发作、复发,因此大多需长达数月或数年的长期用药。如苯妥英钠控制癫痫大发作,环孢素用于器官移植术后抑制排斥反应的发生。此时,除非病症重新出现或毒性反应发生,很难以临床疗效判断剂量是否得当。只有通过TDM将血药浓度控制在有效浓度范围内,以保证长期用药的有效性和安全性。
⒊不同治疗目的需不同血药浓度某些药物随治疗目的不同,所需有效血药浓度将发生改变。如用地高辛治疗心房扑动或心房纤维性颤动时,大多数病人需血药浓度达2ng/ml左右或更高,而不会出现毒性反应;但同样的血药浓度在治疗慢性充血性心力衰竭时,不少患者将发生严重的心律紊乱等毒性反应。显然在这种情况下,借助TDM准确控制药物在治疗目的所需血药浓度范围,是十分必要的。
⒋药物过量中毒多数药物过量中毒,依其特殊的毒性反应表现作出诊断不难。但对少数药物毒性反应表现和该药用以治疗的病症难以区分时,必须依赖于血药浓度检测帮助确诊。如苯妥英钠治疗癫痫,本身过量中毒时亦可致微搐;强心甙可用于治疗心衰和某些心律失常,但其中毒也可表现为心衰加重、出现多种心律紊乱,若仅凭临床表现判断为剂量不足而加大剂量,将会产生严重后果。另一方面,任何药物过量中毒,通过TDM将有助于监控抢救效果,评估预后。
二、药动学原因
⒈治疗血药浓度范围内存在消除动力学方式转换根据非线性动力学消除的特点,若某药按恒定剂量用药,可导致由一级消除动力学转化为零级,仍按此剂量继续用药,将不会达到稳态浓度,血药浓度将持续上升,直至中毒死亡。应根据TDM确定具体病人该药的Vm和Km,计算或调整所需的给药速度R,才能确保血药浓度维持在安全有效的稳态浓度。
⒉首过消除强及生物利用度差异大的药物前面已谈到,在药物体内过程中,生物转化能力由于受遗传、环境及病理因素影响,个体差异巨大。这种差异对首过消除强的药物生物利用度的影响显而易见。如服用相同剂型及剂量的普萘洛尔后,不同个体间的血药浓度差异可达20倍。此外制剂的剂型、质量、胃肠功能状况、空腹或餐后用药等,均可影响药物的生物利用度,改变血药浓度。医学史上就曾有因中途换用同一厂家不同批号地高辛,发生大量中毒性反应的报告。
⒊存在影响药物体内过程的病理情况很多病理情况都可影响药物的吸收、分布、代谢和排泄。腹泻、呕吐将减少药物的吸收;肝脏疾病除降低对药物生物转化的能力外,还可因改变血浆蛋白浓度及比例,改变药物与血浆蛋白的结合率而影响药物的分布及滤过排泄;心衰、休克时血流动力学的改变,将对体内过程各环节都产生影响;肾功能减退,将对药物的排泄,特别是主要以原型药由肾排泄的药物产生明显影响;如肾功能衰竭时,链霉素的半寿期可从正常的2-3小时增加为50-110小时;烧伤时血浆外渗期和利尿期,对药物的分布和排泄以及肌注药物的吸收的不同影响,也是显而易见的。还必须指出,在治疗过程中当上述病理过程发生改变,势必产生相应的药动学参数改变,也应通过TDM调整计算,保证药物治疗的有效性和安全性。
⒋需长期用药及可能产生药动学相互作用的联合用药现已知常用药物中,至少有200余种是肝细胞混合功能氧化酶系的诱导剂或抑制剂,如苯巴比妥、苯妥英钠、利福平、异烟肼等。长期使用这些药,对自身或合并使用的药物的生物转化将产生促进或抑制作用。而不少药物间在与血浆蛋白结合及肾小管分泌排泄上,存在竞争性抑制,同时使用可能产生相互影响。长期用药时,定期进行TDM工作,既可避免因剂量不足而延误病情,或过量产生慢性毒性,也可及时发现因任何影响体内过程的因素产生的血药浓度改变。
除上述药效学或药动学原因外,当药物治疗无效或未达预期疗效时,通过TDM可排除病人是否未按医嘱用药,或服用了假冒伪劣药品,或对该药产生耐受性所致。此外,当涉及某些医学法律问题时,TDM可提供客观依据。如使用氨基甙类抗生素治疗泌尿系统感染时出现肾衰,借助TDM结果,可明确肾衰是由于本身疾病发展还是用药过量所致的毒性作用。
表9-1列出了目前认为需要进行TDM的主要药物,将在本章第七节中进一步讨论
表9-1 需进行TDM的主要药物
| 分类 | 药品 |
| 强心甙 | 地高辛、洋地黄毒苷 |
| 抗心律失常药 | 利多卡因、普鲁卡因胺、奎尼丁、乙胺碘呋酮、因卡胺、异丙吡胺等 |
| 抗癫痫药 | 苯妥因铵、苯巴比妥、乙琥胺、卡马西平、丙戊酸钠 |
| β受体阻断剂 | 普萘洛尔、美托洛尔、阿替洛尔 |
| 平喘药 | 氨茶碱 |
| 抗抑郁药 | 丙咪嗪、阿米替林、去甲替林等 |
| 抗躁狂症药 | 碳酸锂 |
| 解热镇痛药 | 阿司匹林、对乙酰氨基酚 |
| 抗生素 | 庆大霉素、链霉素、卡那霉素、丁胺卡那霉素、氯霉素等 |
| 抗恶性肿瘤药 | 甲氯蝶呤等 |
| 免疫抑制剂 | 环孢素 |
| 利尿药 | 呋塞米(速尿) |
第四节 治疗药物监测的临床应用
TDM对临床合理用药的指导,主要通过以下几方面来实现。
一、获取个体药动学参数
通过前面的介绍,我们可看出药动学模型及参数是反映药物体内过程随时间变化规律的较客观的指标,也是制定用药方案的基础。虽然现在新药上市前均要求进行临床药动学研究,但由于历史原因,目前临床上广泛应用的药物中,不少仍缺乏药动学资料,即便有的,也多得自国外其他人种。近年来遗传药理学研究表明,不同人种间在生物转化及排泄等体内过程上存在着差异。如在对美托洛尔、普萘洛尔等许多心血管药物的氧化代谢,以及异烟肼等药物的乙酰化上,白种人较多的体内存在遗传性缺陷,而在黄种人中则较少见。即便在同一人种间,由于先天因素及后天环境因素和病理情况的影响,也存在巨大的个体差异。因此通过TDM工作,求得具体监测对象的药动学模型及各有关参数,是一重要的基础工作。并且,还可藉以积累我国人群的群体药动学资料。只要确定药物在具体监测对象的房室模型、消除动力学方式及有关药动学参数后,参照本章第二节中分别介绍的有关公式,即可制定出较合理的个体化用药方案。
二、制定用药方案
表9-1所列需进行TDM的药物,其药物效应(包括治疗作用及多数毒性作用)与血药浓度间存在着密切的相关性,并且各药的群体治疗浓度范围及中毒水平均已确定,故在制定用药方案时,可参照有关资料,确定欲达到的稳态浓度水平(静脉滴注)或范围(多剂间隔用药)。应用测定计算得的该个体有关药动学模型及参数,可按公式⑽计算出静脉滴注时的用药速度;而静脉注射或血管外用药等间隔给药时,还需在给药间隔时间>τ和每次用药量X两个参数间,预设定一个,多数情况都是设定>τ,再根据公式⒆-(22),则可计算出另一参数。对于非线性动力学消除的药物,在确定个体的Vm和Km值后,按公式(28)可计算出每日用药量R。
如果不能获得监测病人的具体药动学模型及参数时,可采用有关药物的群体模型及参数均值,作为制定用药方案的依据,但最好能选用同一人种及同一病种的群体资料,以求尽量与接受用药方案的个体接近。此外,对二室及多室模型药物,在制定静脉滴注或多剂用药方案时,一般均按一室模型处理。须强调指出,无论用什么方法制定的用药方案,在实施过程中,仍需通过TDM监测效果,并作出必要的调整。
三、指导调整剂量
通过上述方法制定的用药方案,仅是一理论上的理想方案,实际工作中由于病人具体情况千差万别,在用药过程中任一影响药物体内过程的因素发生改变,均可使血药浓度不是恰在预期水平。即便正好达到预期水平者,也可能在继续用药过程中因上述因素改变,或病情的好转、恶化,使血药浓度改变。因此,通过TDM测定血药浓度,监测用药方案实施效果,指导进行必要的剂量调整,是剂量个体化的必需环节,也是TDM的常规工作。常用的方法有以下两种。
⒈比例法凡属一级消除动力学的药物,假设其剂量调整期间接受治疗的个体体内过程无较大变动,则药动学参数可视做不变,在其达稳态浓度时,血药浓度与剂量间存在正比例关系。因此,根据使用X1剂量或滴注速度达稳态后(5-6个半寿期以上),某次用药后取样测定的稳态血药浓度Css1及在该时刻所需的Css,可计算出调整剂量X=Css·X1/Css1。按调整剂量X用药后,经过5-6个半寿期以上又可达到新的稳态浓度。可如此多次重复定期监测、调整,以达到维持在有效而安全的血药浓度范围水平的目的。
⒉Bayes法该法使用预先按群体药动学资料编制的电脑程序,根据群体药动学参数,结合病人的体质及病理情况,先估算出该个体的药动学参数及用药方案。在按该方案实施过程中,分别在不论是否达稳态的不同时间取血2-4次测定血药浓度,将相应血药浓度和时间输入电脑,用渐近法原理修正出该个体所需的调整方案,经几次反复即可逼近最适方案。该法优点是将前述确定个体药动学参数、制定用药方案及调整剂量多步合在一起完成,并且可同时考虑心、肝、肾功能的影响。但使用本法时,不同药物需不同程序软件,目前仅有地高辛、苯妥英钠、利多卡因等少数药物采用。
四、肝肾功能损伤时剂量的调整
肝脏生物转化和经肾及肝胆系统的排泄,是绝大多数药物消除的主要方式。肝、肾功能的改变将显著影响药物的消除动力学药物的消除动力学,这是TDM工作中必须考虑的。对于肝、肾功能不良的病人,能测定其个体药动学参数或用Bayes法制定用药方案,最为理想。若仅能借用群体资料时,则应通过TDM进行必要的调整。下面介绍应用范围较广的“重复一点法”。
此时,可视做该类个体药动学参数中,仅有消除速率常数k因肝、肾功能损伤而发生改变,而V、F、ka等参数均不受影响。若在按群体资料制定的用药方案实施中,第一次和第二次给药后相同的t时间(选在消除相中)分别取血,测定得血药浓度C1和C2,则此二点间的时间恰等于给药间隔
根据上面计算所得病人k值及群体资料的其它药动学参数,可按下式计算出按此试验剂量和间隔时间用药,所能达的最小稳态浓度。
(Css)min=C1·e-kt/e-kt(l-e-kt)式中t为C1的取样时间
若此最小稳态浓度与欲达到的值不相符,则可按本节三中介绍的比例法,求出达到期望的最小稳态浓度所需的剂量。
必须强调指出,通过TDM指导临床用药时依据的有效治疗血药浓度范围及中毒水平,仅是根据群体资料获得的,并未考虑靶器官、组织或靶细胞对药物反应性的个体差异,以及同时使用的其他药物在药效学上的相互作用(协同或拮抗)。因此,判断病人药物治疗是否有效或发生毒性反应,绝不能仅拘泥于TDM结果,而应结合病人临床表面及其他有关检查,综合分析才能作出正确结论。
第五节 治疗药物监测常用标本及预处理
根据需测定药物的体内过程特点,选用合适的生物体液,在药动学理论及参数指导下确定适宜的时间取样,进行必要的预处理,以具高度特异性及灵敏度的方法,测定药物和(或)代谢物浓度,是TDM工作中最常规的工作。
一、常用标本及收集
⒈血清(浆)第一节中已谈到,在药物的体内过程中,血浆中的药物(血药)起了中央枢纽的作用,可视做药物体内变化的一面镜子,因此有关药动学的资料几乎均是通过对血药的研究获取的。另一方面,绝大多数药物在达到分布平衡后,虽然不是均匀分布,但血药浓度和靶位药物浓度成比例,故也和效应间存在量效依存关系。现已建立了不少药物的治疗血药浓度范围及中毒水平的群体资料,并且血液也易于采集。由于以上原因,血液是TDM工作中最常使用的标本。以血液为TDM标本时,测定血浆或血清中的药物均可,因为药物不和血浆纤维蛋白结合,许多药物的对比研究也证实了血浆和血清中的浓度相等。为避免抗凝剂与药物间可能发生的化学反应及对测定过程的干扰,TDM工作中通常以血清为检测标本。
大多数药物在血液中都程度不等地和血浆蛋白形成可逆结合,显然游离药物浓度和效应间的关系更密切。笼统地测定包括与蛋白结合部分和游离部分的总血药浓度,并不能反映各种原因所致药物血浆蛋白结合率改变而产生的游离药物浓度变化。虽然可通过详细的用药史询问,检测血浆蛋白浓度等方法,协助了解有无影响药物血浆蛋白结合的因素存在,从而在评价总血药浓度时加以考虑,但仍不可靠。尽管直接测定游离血药浓度操作繁杂,在TDM工作中仍日益主张直接测定游离血药浓度,特别是血浆蛋白结合率高的药物。因为此类药物血浆蛋白结合率的轻微改变,将导致游离药物浓度的明显变化而显著影响药效。但本书及文献中报告的有关药物的治疗浓度范围、中毒水平,除特别说明的外,仍是指血清(浆)药物总浓度。
血液标本通常在外周静脉采集。为了能正确反映整个体循环中的药物浓度,静脉注射或滴注用药时,不宜在同一静脉取血,特别是正滴入药物期间、注入药物后短期内或有外漏时。此外肌肉注射或皮下用药后,也应尽量避免在注射部位回流静脉取血。
⒉唾液唾液可无损伤地采集,为病人乐意接受。唾液中的药物除极少数种类可以主动转运方式进入外,大多是由血浆中未与蛋白质结合的游离药物,尤其是高脂溶性的分子态游离药物,以被动扩散的方式进入。另一方面,与血浆相比,唾液中蛋白量甚少,并且为粘蛋白、淀粉酶、免疫球蛋白等不与药物结合的蛋白质,因此唾液中的药物几乎均以游离态存在,并和血浆中游离药物浓度关系密切,用以反映靶位药物浓度较总血药浓度更适合。但是,唾液pH波动在6.2-7.6范围内,平均约6.5。唾液pH的波动将导致与稳定的血浆pH间的差值变动,从而改变药物在两种体液间产生不稳定的解离度和分配比,即唾液药物浓度与血浆游离药物浓度比值出现波动。此外,唾液分泌量及成分受机体功能状态影响,若处于高分泌状态,将产生大量稀薄唾液,一些扩散慢的药物将难以和血药达分布平衡。由于前述pH差异,一般中性或弱酸性药能较快进入唾液,达到分布平衡;而碱性较强的药物则相反,往往出现唾液中的药物浓度较血药浓度滞后的现象。鉴于以上原因,用唾液作TDM标本主要适用于下列情况:①已知唾液药物浓度与血浆药物浓度(总浓度或游离药物浓度)比值较恒定的药物;②在唾液与血浆间能较快达到分布平衡的药物,多数弱碱性、中性及在体内分布属单室模型的药物都属此;③本身或同时使用的药物应无抑制唾液分泌的M胆碱受体阻断作用。丙咪嗪等三环类抗抑郁药、氯丙嗪等吩噻嗪类抗精神分裂症药、苯海拉明等抗组胺药及阿托品等胃肠解痉药,都可抑制唾液分泌,改变唾液中药物浓度,并且收集唾液困难,所以不能用唾液作TDM标本。
有关唾液药物浓度与药物效应间关系的资料极少,因此以唾液为标本进行TDM时,结果的解释评价多通过建立唾液与血药浓度间的关系,再借助后者资料进行。
可用唾液作TDM的药物有对乙酰氨基酚、水杨酸类、苯妥英钠、苯巴比妥、氨茶碱、甲磺丁脲、锂盐等。特别是锂盐,虽是以主动转运方式进入唾液,其唾液浓度可为血浆的2~3倍,但对同一个体,达稳态浓度后,其二者间比值相当恒定,尤宜采用。
唾液标本的收集宜在自然分泌状态下进行。可采用自然吐出,或用特制的负压吸盘采集。咀嚼石蜡块等机械刺激可促进唾液排出,若以维生素C、柠檬酸等置于舌尖,虽可刺激唾液大量分泌,但因可降低唾液药物浓度,改变唾液pH及可能干扰测定,不宜使用。唾液采集后,最好立即测定其pH,以便供解释结果时参考。若为口服用药,应在口服后充分漱口,并不宜在服药后短期内取样,以免残留药物污染干扰。口腔有炎症时,炎性渗出物可能干扰测定,不宜用唾液作为TDM标本。
⒊尿尿液也可无损伤收集。尿中的药物除少数有机酸、碱类药可以在近曲小管被主动分泌入尿外,大多数药物(游离部分)都是从肾小球以滤过方式,由血浆进入原尿中。随着尿液生成过程中的浓缩,尿药浓度逐渐升高,大多远远高出血药浓度,因此易于测定,这也是其优点。
从理论上讲,以滤过方式从肾小球排泄的药物,任一时刻的排泄速率等于该药的肾清除率(Clτ)和该时血浆药物浓度(严格说应为游离药物浓度)(C)之乘积。一般(>τ)恒定,因此药物的尿排泄率与血浆药物浓度成正比。若分时段连续收集尿液,假设每时段内尿药平均排泄速率恰等于该时段中点时刻的排泄速率,则根据该时段平均尿药浓度(C)及尿量(Vu)和时间(t),可计算出该时段中点瞬时尿药排泄速率=C·VEN-US;mso-fareast-language:ZH-CN;msou/t。如果知道该药的Cl,便可求得该时段中点时刻的血药浓度。根据上述理论和方法,还可求得该药的消除速率常数k等有关药动学参数(参见有关药动学专著)。但上述假设与实际有差异,尿液生成不可能是均匀的,并且未考虑影响肾小管对药物重吸收的因素,特别是尿液pH改变对其中的药物解离度的影响所致被动扩散重吸收的变化。事实上,尿液pH随饮食成分、水电解质和酸碱平衡状态的改变而变化,可有较唾液pH更大的波动。因此,在TDM的实际工作中以尿为标本甚少。但对用作治疗泌尿道感染的药物,及可产生肾小管损害的药物,检测尿药浓度则有其特殊意义。
⒋其他体液直接测定脑脊液中的药物浓度,可排除血-脑屏障对药物分布的影响,并且脑脊液中蛋白质少,对作用于中枢神经系统的药物,更接近于靶位浓度。但因取样特别是多次取样难以实现,而有关脑脊液中药物的药动学资料少而不完全,故在TDM工作中不易推广。因同样原因,直接测定其它靶位组织或体液药物浓度,在TDM中也极少应用。
二、取样时间
TDM工作中取样时间对其测定结果的临床价值有较大影响,是开展TDM工作必须考虑的基本问题。应根据TDM的目的及病人具体情况,应用本章第二节中介绍的有关药动学理论,按以下原则确定取样时间。
(一)供计算个体药动学模型及参考
由于两点只能确定一条唯一的直线,此时血药浓度测定中的任何误差,取样时间是否得当,都将对药动学参数产生明显的影响。而血管外用药及多室模型残数法求药动学参数时,消除相方程的计算,都是假设时间t足够大,可不考虑吸收相和(或)分布相的影响而进行的。因此,取样时间的选定应遵循以下两个原则:①在有关血药浓度随时间变化的指数方程中,每一指数项取样不得少于3点,即每一相直线方程的确定至少得有3个或更多的点,此外,在药-时曲线中有关相转折点附近至少有两个点,以便较准确地判明转折点;②消除相取样时间尽量长,时间跨度至少超过两个半寿期。
实际工作中,可根据上述原则,参考有关药物的群体药动学模型及参数资料,具体确定取样时间。理想的是能在一次用药后即连续完成全部取样。若条件不许可,可考虑多剂给药后分次取样累积法,即在多剂用药时,参照群体资料,估计达稳态后,再分别在每剂用药后不同时间取样。假设在取样期间内各药动学参数不发生变化,将每次给药前血药浓度即(Css)min视做零,则可将这些点累积起来,视做一次用药间隔时间内的不同时点处理。此时每次取样时间的选定仍按上述原则进行。但本法较不可靠,特别是消除半寿期接近甚至大于给药间隔者,误差更大。
(二)监测、调整用药方案
应在达稳态浓度后再取样。恒速静脉滴注时,稳态血药浓缩维持在一几乎恒定的水平,因此,只要达稳态后,任何时间取样测定均可。而多剂间隔用药时,稳态血药浓度将波动在一定范围,测定峰值浓度还是谷浓度,应根据临床需要决定。
⒈了解是否达有效血药浓度水平用于控制症状发作、巩固疗效的长期用药,如苯妥英钠控制癫痫大发作等,不能在短时期内靠临床表现作出判断,需依靠TDM回答。这种情况下,若已知病人个体药动学参数,则可在一个给药间隔内的达峰时间(tp)及下次用药前,分别取血测定(Css)max和(Css)min,观察二者是否均在有效血药浓度范围内。二室模型药物(Css)max应为消除相起始段。如果病人个体药动学参数未知,仅参考群体资料制定的用药方案,则群众资料的平均达峰时间tp不一定代表具体病人的tp。但无论如何,任何药物、任一个体每次用药前总是(Css)min,所以,此时最好仅在给药前取样测定(Css)min,根据(Css)min与有效血药浓度范围,特别是下限的关系,调整剂量。
⒉确定是否会致慢性中毒对已达疗效但需了解是否可能产生潜在的慢性毒性作用时,应在tp取样。若不知个体的tp,可在群体tp均值及相邻前后时间分别取样测定,以较可靠地了解血药浓度是否接近或超过最小中毒浓度。
(三)急性药物中毒的诊断及处理
对于前后应立即取样测定,后者则可根据临床需要,在必要时取样,以监测抢救效果。
三、样品预处理
TDM工作中,除少数方法可直接应用收集的标本供测定外,大多需进行必要的预处理。预处理的目的是在不破坏欲测定药物的化学结构的前提下,用适当的方法尽量减少干扰组分,浓缩纯化待测物,以提高检测的灵敏度及特异性,并减少对仪器的损害。预处理包括去蛋白、提取和化学衍生化。
(一)去蛋白
TDM常用的血清(浆)、唾液或尿液等都或多或少地含有蛋白质,并对多种测定方法构成干扰,还可造成仪器污染、损害。去蛋白的方法有沉淀离心法、层析法、超滤法和超速离心法等。其中以沉淀离心法最为简便快捷,并且结合提取的要求,选用合适的酸、碱和有机溶剂,与提取同步进行,故最常选用。由于药物和血浆蛋白的结合,大多是通过离子键、氢键、VanderWaals引力等较弱的作用力形成。当使蛋白质变性沉淀时,这种结合也同时被破坏,释放出药物,因此用沉淀离心法去蛋白处理的体液标本,最后测得的药物浓度应是包括游离药物和与蛋白结合的药物两部分的总浓度。显然,若需要单独测定游离药物浓度时,不能采用此法,而应选用温和但较繁杂、耗时的层析法、超滤法或超速离心法。这样既可去除蛋白,又不至使蛋白结合的药物释出。
(二)提取
为了尽可能选择性地浓缩待测组分,以提高检测的灵敏度,并改善检测方法的特异性,减少干扰,除免疫化学法外,TDM使用的多数检测方法均需进行提取。提取方法有液-液提取和液-固提取两种。
⒈液-液提取由于大多数药物都是有机化合物,并有不少为弱酸、弱碱。它们在pH不同的溶液中,将发生程度不等的解离。因此,应选用对待测物溶解度高、与所用标本不相混溶也不发生乳化的有机溶剂,并根据待测物的酸碱性和pKa,酸化或碱化样本,使待测物尽可能多地以脂溶性高的分子态存在,从而主要分配到有机溶剂中。这样处理,可使在此条件下极性高的干扰成分被排除。离心分离有机相和水相(样本),即可达到提取的目的。若必要,可按上述原理将待测成分再转提到pH适当的水相中,进一步排除高脂溶性的干扰物质。这类方法由于样本和提取介质均为液相,故称液-液提取。
⒉液-固提取又称固相柱提取,是近年发展的一种提取方法。可根据待测物的理化性质选用一合适的常压短色谱柱,TDM中常用疏水性填料柱。待标本(多经去蛋白处理)通过该柱后,以适当强度的溶剂洗脱,选择性收集含待测组分的洗脱液部分,即可达到较理想的提取目的。也可用强度不同的溶剂分次洗脱,仅收集洗脱待测组分。此类提取柱已有数种商品化生产,可供选用。本法虽比液-液提取繁琐,但回收率及提取特异性均高是其优点。
(三)化学衍生化反应
用光谱法和色谱法检测药物时,可根据待测物的化学结构和检测方法的要求,通过化学衍生化反应,特异性地引入显色(可见光分光法)、发光(紫外、荧光、磷光)基团,提高检测的灵敏度和特异性。气相层析时,常需使待测物硅烷化、烷化、卤化和酰化等,以增加待测物的热稳定性和挥发性,改善分离效果和适用于特殊的检测器。用高效液相色谱柱前衍生化分离测定手性异构体药物,则需在待测物中引入手性拆分基团。
第六节 药物浓度测定常用技术
从本质上说,药物都是化学物质,其检测方法均为临床化学的常用技术。但体液中的药物大多以μg/ml或ng/ml水平存在,并且其检测不仅受同时存在的多种结构相似的内源性物质干扰,还受和原型药仅有微小差别的代谢物干扰。而某些药物的TDM除原型药外,还需同时检测具药理活性的代谢物。另一方面,药物的有效浓度范围和中毒水平,是结合大量临床观察确定的,要求任何实验室建立的测定方法应具有高度可比性。下面将仅结合药物测定的特殊要求,对常用临床化学技术作一评估性介绍。
(一)光谱法
多数药物或代谢物本身在紫外光区即存在吸收峰,一些药物或代谢物受激发后,本身即可发射荧光;而另外一些药物还可通过特异的显色反应用分光法检测。但无论可见光分光光度法、紫外光度法还是荧光光度法,用于体液中药物检测时,都存在灵敏度低、特异性差的缺点,特别是易受代谢物干扰。虽然采用提取、双波长、示差、导数分光光度法等试图提高其灵敏度和特异性,仍未能从根本上克服上述缺点。但光谱法操作简便,所需仪器一般临床实验室都具备,检测成本低,便于推广。对治疗血药浓度水平较高,经适当方法改良光谱法能满足临床需要的药物,现阶段仍不失为一可采用的方法,如阿司匹林、对乙酰氨基酚、氨茶碱、苯妥英钠、苯巴比妥钠等。
火焰发射光谱法和原子吸收光谱法特异性及灵敏度均高,操作也较简便,但仅能用于某些含体内仅微量存在的金属离子药物。在TDM中可供锂盐、铂盐检测用。
(二)色谱法
又称层析法。系通过层析作用,使样品中理化性质不同的组分得以分离,若再配以适当的检测器,则可同时完成定性、定量工作。特异性高,可同时检测同一样本中的不同组分,是色谱法的共同优点,这在TDM中尤有意义。薄层色谱法(TLC)虽然不断改进定量点样技术并使用扫描定量,但其灵敏度及重复性仍低于其他色谱法,除用于毒物的检测外,在TDM工作中较少应用。
自60年代末期相继发展成熟的气相色谱法(GC)和高效液相色谱法(HPLC),由于实现了高效层析分离和检测联机,可用微电脑控制层析条件、程序和数据处理,其特异性、灵敏度和重复性均好,并可一次同时完成同一样本中多种药物及其代谢物检测。若采用内标本法定量,还可排除部分操作误差,提高检测结果的可靠性。HPLC和GC相比,由于不需对样品高温气化,一般都可不进行衍生化处理;另一方面,HPLC的固定相种类较多,流动相通过改变其组成成分及比例更是千变万化。二者适当组合,可对绝大多数有机化合物药进行分离测定。事实上,正是GC和HPLC这类先进可靠的检测方法的应用,促进了TDM的形成和发展。特别是HPLC已成为TDM的重要检测技术,并常用作评估其他方法的参考方法。当然,GC和HPLC一般均要求对样品进行预处理,两法所用仪器都昂贵尚难普及。但从发展的观点看,HPLC仍将成为国内TDM的主要分析方法。近年来发展的GC-质谱、HPLC-质谱联机检测,更使这类分析手段的性能极大提高。
(三)免疫化学法
不少药物都是半抗原或抗原,所以可引起过敏反应。若能制备这些药物相应的特异性抗体,即可根据抗原-抗体反应的特异性,应用免疫化学法检测这些药物。TDM应用的免疫化学法一般都采用竞争性免疫分析,即通过定量加入的少量特异性抗体,与标本中相应的抗原或半抗原性药物及定量加入的标记药物间,产生竞争性结合,检测标记药物与抗体结合的抑制程度,和同样处理的标准管比较,便可对样本中的药物定量。免疫化学法灵敏度极高,大多可达ng甚至pg检测水平,可满足所有药物TDM的要求;该法所需标本量少,一般均不需预处理,操作简便,并可制成商品化试剂盒;还可利用一般生化、荧光自动分析仪进行自动化操作。这些都是免疫化学法的优点。
但在TDM中免疫法存在的主要问题是,特异性易受干扰。干扰因素除来自内源性物质外,特殊的是抗原性决定基团未发生变化的待测药物的代谢,或具相同相似抗原性的其他药物及代谢物。如强心药地高辛、洋地黄毒甙、地高辛代谢物二氢地高辛,均可与地高辛抗体结合。显然,在体内几乎不发生代谢转化而原型排泄的药物,则较少代谢物干扰,尤其适用于免疫法检测,如庆大霉素等氨基甙类抗生素。当然,只有具备完全或半抗原性的药物,并要能制备特异性抗体,才能用免疫化学法进行TDM。但总的来说,由于免疫化学法的前述优点,仍是TDM检测技术发展的主要方向。
免疫化学法根据标记物性质不同,可分做放射免疫法、荧光免疫法和酶免疫法三类。根据测定中是否要分离与抗体结合和未结合的标记物,又可分为多相(需分离)和均相(不分离)免疫分析法两种。放射免疫法虽存在放射性污染,又是多相体系,分离结合和未结合标记物操作繁杂,影响因素多,而分离效果又直接影响测定结果,但其灵敏度高,标记技术相对成熟,国内生产的TDM试剂盒多为此类。荧光免疫法按反应体系及定量方法不同,还可进一步分做若干种。与放射免疫法相比,荧光免疫法无放射性污染,并且大多操作简便,便于推广。国外生产的TDM用试剂盒,有相当一部分即属于此类,并且还有专供TDM荧光偏振免疫分析用的自动分析仪生产。酶免疫分析法是目前国外TDM免疫分析中最常采用的方法,特别是均相酶免疫分析法。本法除具有荧光免疫法的优点外,通过选用合适的标记酶,在目前已较广泛普及的多种自动化分析仪即可实现自动化操作,更利于推广。特别是近年来,在TDM的酶免疫分析中,应用酶偶联反应原理,以辅酶黄素腺嘌呤二核苷酸(FAD)标记药物,将葡萄糖氧化酶蛋白、偶联辅助酶-过氧化物酶及底物和显色剂4-氯-1-萘酚固化在薄膜上,制成类似pH试纸样的试条。测定时,只要滴上微量样品,根据成色深浅,即可作出药物浓度的粗略判别。这一方法使病人自我或临床医师病床旁方便快速进行TDM工作成为可能。目前国外已有供茶碱和苯妥英钠测定用的这种试条问市。无疑这一方法的推广,将有助于促进TDM的普及,具有良好的发展前景。
(四)其他检测方法
抑菌试验曾用于测定体液中的抗菌药物浓度。该方法简便易行,可利用临床细菌室即可开展。但其特异性、灵敏度、重复性均差,定量粗糙,并易受同时使用的其他抗菌药物的干扰,在TDM中已较少使用。一些本身即为内源性物质的药物,如钾、钠、钙、激素药等,在临床检验中已有成熟的检测方法,则可借用。
必须指出,多数需进行TDM的药物,都有不只一种方法可供选用。应根据测定药物的有效血药浓度水平所决定的灵敏度要求,是否需同时检测多种药物或活性代谢物,可供选用的仪器设备及检测经济成本等,综合考虑,确定一能满足临床要求的可行方法。
第七节 需测定药物浓度进行监测的主要药物
本章第三节已对需进行TDM的原因和药物作了概括介绍,下面我们将主要讨论目前认为迫切需要进行TDM之药物的药效学、药动学及检测方法的有关问题。
一、强心甙类
强心甙是一类由植物中提取的甙类强心物质。目前供临床使用的主要有毒毛花苷K、去乙酰毛花苷(西地兰)、地高辛和洋地黄毒苷。其中毒毛花苷K和西地兰起效快、消除也较快,药效维持时间短,仅有注射剂型供急症短期用药,一般不需进行TDM。洋地黄毒苷起效慢,消除也慢,临床少用。而地高辛起效及消除均居中,在需长期使用强心甙时,多选用地高辛。故下面只介绍地高辛的有关内容。
(一)药效学及血药浓度参考范围
治疗剂量的强心甙可选择性轻度抑制心肌细胞膜上Na+,K+-ATP酶,使心肌内的Na+更多地依靠Na2+/←→Ca2+交换排出,细胞内Ca2+浓度升高,Ca2+触发的心肌细胞兴奋-收缩耦联增强,产生心肌收缩性增强、心输出量增加、窦性节律降低、房室传导减慢等药理作用。临床上可用于慢性充血性心力衰竭、心房纤颤及心房扑动等的治疗。其主要毒性反应为多种心律失常,并可因此致死,还有中枢神经系统及消化道症状等,均与血药浓度密切相关。
地高辛的治疗血清浓度参考范围为0.8-2.0ng/ml(1.0-2.6nmol/L),安全范围极狭窄,当血清浓度超过2.0ng/ml后,80%以上病人都出现心律紊乱等毒性反应。但治疗心房纤颤和心房扑动时,多数病人可耐受2.0ng/ml甚至更高的血清浓度,因此时即是利用地高辛轻度中毒时产生的房-室传导阻滞等作用,减慢心室率,发挥治疗作用。
(二)药动学
不同强心甙类药物体内过程互异,用药途径也不同,这是造成起效有快有慢、维持时间长短不一的原因。地高辛以片剂和酊剂供口服,多用前者。口服后地高辛在胃肠道以被动扩散方式吸收。片剂的生物利用度约60%-80%,酊剂较高,可达80%-100%。影响片剂生物利用度的主要原因是制剂的崩解、溶出药物速度。因此,在长期使用地高辛时,最好能坚持用同一厂家同批号产品。血液中的地高辛约20%-25%与血浆蛋白结合,其分布属二室模型,8-12h转入消除相。只有在消除相,心肌与血药浓度的比值才较恒定。因此TDM取样时间应选在消除相内(至少服药后12h)。地高辛的表观分布容积约5-10L/kg体重。
地高辛在体内消除主要是以原型药经肾小球滤过,或肾小管分泌排泄,仅约10%左右在肝通过水解、还原及结合反应代谢,另有7%左右处于肠肝循环。但在肾功能减退时,经代谢转化及处于肠-肝循环的比率可明显升高。治疗剂量下,地高辛在体内的消除属一级动力学。消除半寿期成人约36h(30-51h),儿童约30h(11-50h)。
(三)其他影响血药浓度的因素
除肝、肾、心脏及消化系统功能可影响地高辛体内过程外,同时使用奎尼丁、螺内酯(安体舒通)、呋塞米(速尿)、多种钙通道阻滞剂及口服广谱抗生素,都可使地高辛血药浓度增加,特别是奎尼丁,可通过抑制地高辛的肾小管分泌排泄,使其清除率下降。有报道治疗量奎尼丁可使地高辛血药浓度升高达2.5倍,这是极其危险的。此外,甲状腺功能减退症患者血清地高辛浓度升高,心肌敏感性上升,也易出现中毒;低钾、镁、高钙血症均可使心肌对强心甙敏感性提高,有效血药浓度范围内即可出现心脏毒性。
(四)检测技术
地高辛的TDM一般均用血清作标本。虽然已证实唾液和血清地高辛浓度间有高度相关性,但如本章第一节所述,影响唾液药物浓度因素太多,而地高辛治疗浓度水平低,安全范围又太狭窄,故目前仍主张使用血清。取血时间如前所述,一般应在达稳态后(10天以上),并在服药后16h左右采集。但如果病人达稳态前即出现中毒表现,则应立即取血测定。
由于地高辛的血清浓度过低,目前TDM常用的分析方法中,只有免疫化学法的灵敏度能满足其要求。当前商品化试剂盒有放射免疫和酶免疫两种,国内仅有前者生产。
放射免疫法测定地高辛所用的标记物有3H和125I两种。前者半寿期长,试剂保存期长,但测定需昂贵的液闪计数仪,且易受样本中内源性物质化学发光或色淬灭干扰。125I标记试剂盒虽半寿期短,但测定只需用γ计数仪,并且不受上述内源性物质产生的干扰影响。放射免疫法均是多相体系,都存在分离结合和未结合标记药物的步骤。若用右旋糖包裹的活性碳吸附未与抗体结合的标记地高辛,则离心后上清液中的放射性代表与抗体结合的标记药物。而以双抗体法沉淀分离时,离心后上清液中的放射性代表未与抗体结合的标记地高辛。
酶免疫试剂盒系以葡萄糖-6-磷酸脱氢酶为标记物的均相体系,无放射污染,并可省去分离步骤,操作简便,还可利用自动生化分析仪实现自动检测。
放射免疫法的灵敏度可达0.3ng/ml,酶免疫法为0.5ng/ml。两种方法间存在极好的相关性(r>0.9)。变异系数各实验室报告不一,在治疗浓度范围内大多可控制在10%以下。但无论用何种免疫法面临的主要问题都是特异性易受干扰。现已知地高辛的某些尚有部分活性的不全水解代谢物以及无活性的代谢物二氢地高辛、洋地黄毒苷、去乙酰毛花苷(西地兰)等其他强心甙药,螺内酯的某些极性代谢物,内源性皮质激素等,与地高辛抗体有程度不一的交叉免疫性,特别在特异性差的抗体更明显。
二、抗癫痫药
抗癫痫药是一类可通过不同作用机制,控制癫痫发作的药物。现在临床应用的主要有苯妥英、苯巴比妥、酰胺咪嗪、乙琥胺、氯硝基安定、丙戊酸钠等,大多需进行TDM。下面以本类药中最常使用,也是最迫切需要进行TDM的苯妥英为例,介绍有关TDM的知识。
(一)药效学及血药浓度参考范围
苯妥英可通过对大脑神经元胞膜的稳定作用,及增强中枢抑制性递质γ-氨基丁酸作用,阻止大脑异常放电的扩散,用作治疗癫痫大发作的首选药物。对局限性或精神运动性癫痫亦有效,还用于治疗室性心律失常,特别是强心甙中毒所致,也用于多种外周神经痛的治疗。治疗癫痫时,需长达数年用药,临床只能根据癫痫是否发作判断疗效。现已确定,苯妥英钠的治疗作用及不良反应中常见的小脑-迷路症状、精神异常、多种抽搐等毒性反应,以及牙龈增生等,都与血药浓度相关。
苯妥英治疗血清浓度参考范围为10-20μg/ml,最小中毒浓度约25μg/ml。
(二)药动学
苯妥英以其钠盐供临床使用。口服后,苯妥英以被动扩散方式经小肠吸收,吸收缓慢,平均约8h(6-12h)达峰浓度。其生物利用度受制剂质量影响大,但一般均可达90%左右。血液中的苯妥英约90%与白蛋白结合。苯妥英可迅速分布至全身,属一室分布模型,其表观分布容积为0.5-0.7L/kg体重。
苯妥英在体内的消除仅2%以原型从肾排泄,绝大部分经肝细胞生物转化为无活性的代谢物后再排出。苯妥英为肝药酶诱导剂,长期使用可因此加速自身的代谢转化。在治疗浓度范围内,苯妥英存在消除动力学方式的转换,当血药浓度在10μg/ml以下时,一般按一级动力学方式消除;但超过此浓度时,大多数个体转换为零级消除动力学,故其消除半寿期不恒定,随血药浓度而变。成人大多波动在15-30h,儿童为12-22h。文献报告我国癫痫患者Vm均值约为400mg/d,Km均值约5.6mg/L左右。
(三)其他影响血药浓度因素
苯妥英与血浆白蛋白结合率高。老年人、妊娠晚期、肝硬化、尿毒症等时,血浆白蛋白减少,同时服用可与苯妥英竞争白蛋白结合位点的药物丙戊酸钠、保泰松、水杨酸类、磺胺类等以及较高浓度的尿素、胆红素等内源性物质,均可使苯妥蛋白结合率下降,游离药物浓度升高而总浓度无变化。若对测定苯妥英总浓度的结果进行分析解释时,必须考虑上述影响。此外,服用苯妥英期间若同时使用了苯巴比妥、酰胺咪嗪、利福平等肝药酶诱导剂,异烟肼、氯霉素等肝药酶抑制剂,可使苯妥血药浓度降低或升高。肝功能损害者,因对苯妥英生物转化受损,亦可致血药浓度升高,半寿期延长。
(四)检测技术
苯妥英TDM通常以血清为标本。由于唾液中苯妥英浓度依据唾液与血浆pH差值对苯妥英解离的影响进行校正后,与血清游离血药浓度接近,也可考虑采用。由于苯妥英在治疗血药浓度范围内存在消除动力学方式转换,除用药速度(剂量/日)恰等于按第二节中(28)式计算的结果外,无稳态可言。但一般取血仍参照一级消除动力学原则,用药或改变剂量后10天以上服药前取样。
测定苯妥英可用光谱法、HPLC及免疫化学法,分别介绍于后。
⒈分光光度法有多种方法报告,其中较成熟的是衍生化后紫外检测法。其原理是将标本调节至pH6.8后,以二氯甲烷提取及沉淀蛋白,再转溶于NaOH溶液,加KmnO4再加热,使苯妥英氧化为吸光值大的二苯酮衍生物,再以环已烷提取,247nm紫外光比色定量。本法灵敏度、线性范围、重复性均可满足TDM要求,但虽然反复多次提取,仍无法完全排除代谢物干扰。
⒉HPLC法用HPLC检测苯妥英,除具有灵敏度、特异性、重复性均佳的优点外,由于抗癫痫药常合并用药,本法则可对多种抗癫痫药同时检测,是其特有的长处。文献报告方法很多,国内也有实验室建立了可同时检测苯妥英、苯巴比妥、酰胺咪嗪、乙琥胺和去氧苯比妥5种抗癫痫药的HPLC-UV内标法。该法以5-乙基-5甲基-苯巴比妥酸为内标物,ODS柱为固定相,流动相由乙腈:甲醇:水(9:37:54)组成,254nm紫外光检测。当流速在2ml/min时,可在8min内完成对上述5种抗癫痫药及内标物的色谱分离、检测。5种药物的线性范围均可覆盖治疗及中毒血清浓度水平,灵敏度都在2μg/ml以下,变异系数在5%左右,可满足TDM的要求。
⒊免疫化学法供检测苯妥英及其他常用抗癫痫药的放射免疫、酶免疫、荧光免疫检测试剂盒均有市售,以后两种特别是酶免疫为多。无论何种免疫法均与HPLC法有极好的相关性,结果可比性也高。此外,前面已介绍,应用酶辅基标记免疫分析技术,已制成供苯妥英检测用试条。试条虽然使用方便,但定量较粗糙,必要时仍应考虑较精确的方法。
由于苯妥英在治疗浓度范围内存在消除动力学方式转换,在制定或调整用药方案时,应按非线性动力学的有关参数(Vm、Km)及公式处理(参见本章第二节)。此外,对苯妥英等非线性动力学消除的药物,还可用以下两种方法处理。
⑴给病人分别试用两种不同给药速度R1和R2(剂量/日):各自在连续用药2周以后的某次用药前,或用药后相同间隔时间取血,测得的血药浓度C1和C2可视做各自给药速度所达到的稳态浓度,用下列公式求得较准确的Vm、Km,再用第二节中的式(28)计算出欲达所需稳态浓度应该使用的给药速度。
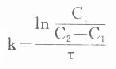
⑵先试用一给药速度R1,2周后某次用药前取血,测得浓度C1,利用群体Km均值及上面介绍的公式求得Vm,再按式(28)计算出欲达所需稳态浓度的较合理用药速度R2。达稳态后再进行监测,如果仍不满意,则根据已获得的R1、R2及相应的C1、C2,按前面⑴中介绍的方法处理。
需要强调的是,无论用何种可靠的方法测定苯妥英浓度,也不论用什么方法调整剂量已达到理想的稳态浓度,由于影响苯妥英血药浓度因素多,又需长达数年连续用药,因此应该坚持定期监测血药浓度,及时发现变化,作出调整。
三、治疗情感性精神障碍药
(一)三环类抗抑郁药
三环类抗抑郁药是目前治疗抑郁症的主要药物,包括丙咪嗪、去甲丙咪嗪、去甲替林、阿米替林、多虑平等。
⒈药效学目前倾向于认为,该类药物可通过抑制脑内突触前神经细胞膜对去甲肾上腺素和5-羟色胺的再摄取,增加突触间隙内上述单胺递质的浓度而发挥抗抑郁症作用。常见不良反应包括M胆碱受体阻断作用所致的阿托品样副作用,以及体位性低血压、严重的心律紊乱、心力衰竭、抽搐、昏迷等毒性反应。其治疗作用和毒性反应均与血药浓度密切相关。
⒉药动学及血药浓度参考范围该类药物脂溶性高,口服后吸收快而完全,但因首过消除强且差异大,故生物利用度不一。血液中的三环类抗抑郁药90%左右与血浆白蛋白、脂蛋白、α1-酸性糖蛋白结合,游离药物能迅速分布至各组织。该类药物绝大部分需在肝脏经过去甲基化、羟化及结合反应代谢后,由肾脏排泄。其中丙咪嗪、阿米替林、多虑平的去甲基化代谢物,都有和原药同样的药理活性,并且去甲丙咪嗪、去甲替林本身也为三环类抗抑郁药。在常用剂量下,该类药物消除均属一级动力学。但对代谢物仍有药理活性者,判断药效持续时间不能仅凭原型药的消除半寿期。同样评价生物利用度时,也应考虑这点。常用三环类抗抑郁药的药动学参数及血药浓度参考范围,参见表9-2。特别要指出,本类药中多数血药浓度存在特殊的“治疗窗”(thera-peuticwindow)现象,即低于“治疗窗”范围无效,而高出此范围不但毒副作用增强,并且治疗作用反下降。
表9-2 常用三环类抗抑郁药药动学参数及参考血药浓度范围
| 丙咪嗪 | 去甲丙咪嗪 | 阿米替林 | 去甲替林 | 多虑平 | |
| 生物利用度(%) | 26-68 | 33-68 | 56-70 | 46-56 | 17-37 |
| 血浆蛋白结合率(%) | 89-94 | 90-93 | 82-96 | 93-96 | >90 |
| 表观分布容积(L/kg) | 9-21 | 26-42 | 6-10 | 14-22 | 12-28 |
| 原型药半寿期(h) | 10-16 | 13-23 | 10-20 | 18-44 | 11-23 |
| 治疗血清浓度(μg/L) | 150-300* | 150-300 | 150-250* | 50-200 | 30-150* |
| 中毒血清浓度(μg/L) | >500* | >500 | >500* | >500 | >500* |
*原型药和有活性的去甲基代谢物总浓度
⒊检测技术一般均以血清为检测标本。取样时间应在达稳态后任一次用药前,以测定其稳态谷浓度(Css)min。抗凝剂、某些塑料试管及橡胶塞中的增塑剂,可改变该类药物在红细胞和血浆中的分配比,应避免使用。玻璃器皿可对该类药物产生吸附,采用同一批号玻璃器皿并置于已烷;正丙烷(99:1)的溶液或超声处理,可减少吸附及吸附差异产生的干扰误差。聚丙烯制作的器皿吸附最小,可考虑选用。
由于三环类抗抑郁药中不少需同时测定其活性代谢物浓度,故以HPLC及GC最为适合,以前者多用。共同测定步骤为碱化血清后,以含有一定浓度内标物的适宜有机溶剂提取,并沉淀蛋白质。移取有机相挥发干,以流动相重溶残留物,取样上柱进行色谱分析。色谱系统大多采用反相,但也有使用正相离子对吸附色谱的。一般都用紫外检测器。由于丙咪嗪及去甲丙嘛嗪本身即有较强的荧光活性,单独测定这两种药可使用荧光检测器,提高灵敏度及特异性。三环类抗抑郁药化学结构相似,建立的检测方法大多可通用于该类药物的各种药品分别或同时分析。
现也有供本类药物免疫化学法检测的试剂盒,操作简便、灵敏度高,并与HPLC法有较好的相关性。主要不足之处是同时服用的三环类抗抑郁药间,以及N-去甲基化等代谢物与原型药间,存在交叉免疫反应,干扰测定。对亦要求同时测定有活性的去甲基化代谢物的阿米替林、丙咪嗪及多虑平单独使用时,使用免疫化学法测定较适合。
(二)碳酸锂
⒈药效学及血药浓度参考范围Li+可抑制脑内去甲肾上腺素的释放,促进其被再摄取,从而降低突触间隙的去甲肾上腺素浓度;Li+还可抑制α1-肾上腺受体激动后胞内信使物质的产生,发挥抗躁狂症作用。碳酸锂过量中毒时可出现意识障碍、肌颤、共济失调、抽搐及低血钾所致的多种心律失常,严重者可致死亡。Li+的治疗作用及毒性反应与血清浓度关系密切,安全范围狭窄。为便于比较评价血清Li+浓度,在TDM中规定在距前晚服药后12h的次晨取血,测得的血清Li+浓度称12小时标准血清Li+浓度(12h-stSLi+)。
12h-stSLi+治疗参考范围为0.8-1.3mmol/L,最小中毒12h-stSLi+参考值1.5mmol/L。
⒉药动学锂盐口服后在胃肠道吸收迅速、完全,不存在首过清除,其生物利用度几乎接近100%。血Li+与血浆蛋白无结合,呈二室分布模型,分布半寿期约1h。中央室表观分布容积约0.2-0.25L/kg,总表观分布容积均值为0.79L/kg。Li+不被代谢,其消除几乎全部通过肾脏排泄,消除半寿期约20h左右,受肾功能影响大。
⒊其他影响血药浓度因素Na+摄入不足可降低Li+排泄,升高血Li+浓度。高Na+摄入则产生相反影响。肾功能减退及肾血流量减少的病症,如充血性心衰、高血压亦可减少Li+的肾排泄,升高血Li+浓度。同时使用茶碱、咖啡因、螺内酯、乙酰唑胺、碳酸氢钠等药物,可促进Li+浓度;而噻嗪类、呋塞米(速尿)等利尿药则有相反作用。
⒋检测技术锂盐的TDM多以血清为标本。如前所述,通常在达稳态后距前晚末次用药12h的次晨取血,测定12h-stSLi+。Li+以主动转运方式进入唾液,唾液中Li+浓度为血清的2-3倍,对同一个体则该比值相对恒定。因此,在确定具体病人二者比值后,可考虑用唾液为标本进行TDM。但唾液Li+浓度影响因素多,测定唾液Li+浓度供TDM是否可靠,尚有分歧。
Li+为简单的金属离子,使用的检测方法有火焰发射光谱和原子吸收光谱法。后法需昂贵的原子吸收分光仪,且在Li+测定上较前法无明显优点。火焰光度计一般临床检验室均具备,在Li+测定上存在灵敏度高(0.3pg/ml)、线性范围、重复性好(C·V<5%)、操作简便的优点,故多用火焰发射光谱法。该法原理为标本适当稀释后(通常1:100),引入雾化器喷射到火焰中原子化。Li+吸收热能后跃迁到激发态,当回复到基态时发射出特有的670.8nm光波,检测该光波强弱定量。本法的干扰主要来自:①标本中的钙、锶和钠离子,前两种元素均可在670.8nm发射带状光谱,为此要求火焰光度计的单色系统可靠;②标本与标准液的粘度、表面张力等物理性质差异,可采用稀释标本,并使标准液其他成分尽量与标本一致来克服;③火焰波动造成的干扰,通过采用双光束内标型仪器,以钾做内标物,可在一定程度上排除这种干扰。
四、茶碱
茶碱通常制成氨茶碱等水溶性较高的盐类供药用,但在体内均解离出茶碱发挥作用,故不论何种制剂,TDM检测对象均为茶碱。下面以氨茶碱为代表讨论。
(一)药效学及血药浓度参考范围
茶碱可抑制细胞内磷酸二酯酶,使β肾上腺素受体激动产生的胞内信使物质cAMP分解代谢受阻而堆积,出现类似β肾上腺素受体激动样作用。临床上主要用于预防和治疗支气管哮喘,治疗早产儿呼吸暂停等。此时,其他β肾上腺素受体激动样作用便成为不良反应,严重者可出现躁动、抽搐等中枢神经兴奋症状,以及多种心律失常及严重呕吐等毒性反应。茶碱的治疗作用、毒性反应与血药浓度关系密切。
茶碱的治疗血清浓度参考范围为儿童及成人10-20μg/ml,新生儿5-10μg/ml;最小中毒浓度参考值为成人20μg/ml,新生儿15μg/ml。其安全范围甚狭窄。
(二)药动学
氨茶碱口服后,茶碱可迅速而完全经胃肠道吸收,约2.5h达峰浓度。成人生物利用度接近100%。茶碱血浆蛋白结合率约56%,可迅速在体内达分布平衡,但部分个体呈二室分布模型。成人表观分布容积约0.5L/kg,新生儿及早产儿增大。茶碱约90%由肝脏代谢,仅8%左右以原型从肾脏排泄。消除半寿期成人均值约6h(3-13h),儿童较短,约3.5-6h,新生儿却长达15~37h。但在15%左右病人,茶碱在治疗血药浓度范围上限可发生转化为零级消除动力学。
(三)其他影响血药浓度因素
吸烟、高蛋白低糖饮食、同时使用苯巴比妥、利福平等肝药酶诱导剂,可促进茶碱消除。充血性心衰、肺心病、肝硬化,以及合并使用异烟肼、红霉素、西米替丁等肝药酶抑制剂,可使茶碱消除减慢,血药浓度升高。
(四)检测技术
茶碱TDM通常用血清为标本。唾液与血清茶碱浓度有极佳的相关性(r=0.99),唾液浓度约为血清浓度的50%,接近于游离血药浓度,需要时也可选用。取样多在达稳态后(通常5天以上)给药前进行,测定稳态谷浓度。
茶碱目前常用免疫化学法、HPLC及紫外光度法检测。其中免疫法多为均相酶放大免疫法(EMIT),荧光偏振免疫法(FPIA),亦有放射免疫法试剂盒,均可实现自动化检测。与苯妥英一样,也有以同样原理制成的试条供茶碱TDM。
茶碱的HPLC检测各种实验室具体方法不尽相同,但大多采用反相HPLC,以β-羟乙基茶碱或8-氯茶碱为内标,紫外检测器λ=275nm(茶碱最大吸收波长)检测。为排除标本中可能存在的与茶碱有相近光吸收峰的某些半合成青霉素、头孢菌素的干扰,样本应以适当方法提取。此外,为保证能分离可能存在的因食入或茶碱代谢生成的其他甲基黄嘌呤衍生物,如咖啡因、可可碱、二甲基黄嘌呤等,在建立方法时即应适当调整流动相,并进行必要的干扰实验证实。
紫外光度法较成熟的是双波长法。其原理为酸化标本并以有机溶剂提取,再反提至0.1mol/LNaOH溶液中,以氯化铵调节至pH10左右。分别在275nm及300nm测A275和A300,以A275-A300差值作为茶碱的吸光度。该法分别酸化标本提取及反提回碱液中,可排除不少药物及其他杂质的干扰。将比色液pH调至10,茶碱吸收峰不变,但可使有相近吸收峰的巴比妥类峰转移而得以排除,并以A300作为样本底吸光度值。经过上述改进,本法灵敏度、特异性都有较大提高,在2-80μg/ml范围内存在极好的线性关系,回收率95%-102%,重复性良好(CV在4%-7%),并与免疫法、HPLC有很好相关性。本法的主要不足之处是不能排除HPLC法中提及的其它甲基黄嘌呤衍生物干扰。但由于本法不需特殊仪器,且能基本满足临床TDM的要求,仍不失为可供选用的一种方法。
此外,在HPLC和紫外光度法中,若以氨茶碱配制标准液时,应注意是每2分子茶碱与1分子乙二胺成盐,故需将氨茶碱重量乘以0.79,正确换算成茶碱重量。
在茶碱TDM中,当发现血药浓度明显高于测算值时,应警惕转换为零级消除动力学,即呈非线性动力学消除的可能。
五、抗心律失常药
抗心律失常药可通过不同作用机制,改变心肌细胞的自律性、传导性、动作电位时程、有效不应期等电生理特性,用于治疗各种心律失常。显然,药物所致上述心肌电生理特性过度改变,将导致新的心律紊乱,因此这类药大多安全范围狭窄。由于心肌血液供应丰富,该类药血清浓度较能反映靶位浓度,并大多与治疗作用和毒性反应,特别是心脏毒性相关。用本类药物治疗的疾病,往往存在循环、肝、肾功能改变,将对该类药的体内过程产生影响。而本类药中普鲁卡因胺、利多卡因等的代谢物仍有药理活性,此外本类中某些药物代谢上存在强、弱代谢型的遗传多态性(polymorphism)。基于上述原因,本类药物大多需进行TDM。由于篇幅限制,我们仅将该类药中目前公认应进行TDM的药物药动学参数、检测方法总结于表9-3,供参考。还应指出,某些情况下心电图对这类药物也为一有效监测手段,但不能替代TDM;反之,TDM的结果也应结合心电图改变及其他临床表现,作出解释和判断。
表9-3 需进行TDM的抗心律失常药药动学参数、血清浓度及检测方法
| 普鲁卡因胺 | 利多卡因 | 异丙吡胺 | 奎尼丁 | |
| 口服生物利用度(%) | 75-95 | / | 72-94 | 70-90 |
| 血浆蛋白结合率(%) | 11-21 | 43-59 | 20-65 | 60-82 |
| 总表观分布容积(L/kg) | 1.6-2.4 | 0.7-1.5 | 0.5-1.7 | 1.5-3.0 |
| 消除半寿期(h) | 2.7(强乙酰化型) | 1.8 | 7.8 | 6.2 |
| 5.2(弱乙酰化型) | ||||
| 治疗血清浓度(μg/ml) | 10-30 | 2-5 | 2-5 | 2-5 |
| 最小中毒浓度(μg/ml) | 30* | 9 | 7 | 6 |
| 常用检测方法 | HPLC | HPLC | HPLC | HPLC、免疫法 |
| 免疫法** | 免疫法 | 免疫法 | 荧光光度法 |
*普鲁卡因胺+活性代谢物N-乙酰普鲁卡因胺总浓度
**有分别供检测普鲁卡因胺和N-乙酰普卡因胺试剂盒
六、氨基甙类抗生素
氯基甙类抗生素包括链霉素、庆大霉素、卡那霉素、丁胺卡那霉素、妥布霉素、乙基西梭霉素等。该类药物化学结构相似,体内过程相近,作用原理及抗菌谱相同,检测方法可通用。除用于抗结核治疗外,目前以庆大霉素最常用,下面将以庆大霉素为例,介绍该类药TDM的有关问题。
(一)药效学及血药浓度参考范围
庆大霉素等氨基甙类抗生素对敏感病原体蛋白质合成的各个阶段均产生干扰,有较强的杀菌作用,广泛用于多种需氧革兰氏阴性杆菌、某些革兰氏阳性球菌感染的治疗,亦是主要的抗结核药。但可产生第八对脑神经损害、肾损害、神经-肌肉接点阻断等毒性反应和过敏反应。其治疗作用及毒性反应与血清浓度关系密切,并且安全范围狭窄。
庆大霉素血清治疗浓度参考范围为0.5-10μg/ml,最小中毒浓度参考值为12μg/ml。
(二)药动学
氨基甙类抗生素为强极性碱性药,主要为肌肉或静脉注射用药。肌肉注射后吸收迅速完全,可在1h内达峰浓度。由于极性大,该类药与血浆蛋白结合率低(除链霉素外,大多<10%),并主要分布在细胞外液中。庆大霉素的表观分布容积成人约0.25L/kg,儿童可增至0.5L/kg或更高。该类药物几乎全部以原型药从肾小球滤过排泄,故其消除属一级动力学,并受肾功能影响大。肾功能正常者庆大霉素消除半寿期约为1.5-2.7h。
(三)其他影响血药浓度因素
心衰、肾疾患影响该类药自肾小球滤过排泄,是影响血药浓度的主要因素,肾功能较正常下降10%即导致该类药半寿期延长。由于该类药主要分布于细胞外液,因此儿童特别是新生儿、烧伤后利尿期前,均可使表观分布容积明显增大,半寿期延长,而失水则产生相反影响。接受血液透析者,可加速该类药消除。
(四)检测方法
氨基甙类抗生素TDM一般均用血清。若用血浆,由于该类药可和肝素形成复合物,故不能用肝素抗凝。
该类药物检测方法包括微生物法、HPLC及免疫化学法。微生物法如本章第二节所述,已较少应用。HPLC测定氨基甙类抗生素,大多需复杂的衍生化反应引入荧光基团,以荧光检测始能达到所需灵敏度,也少用。由于本类药物内几无代谢,故不存在代谢物干扰,尤其适用于免疫法检测。目前已有多种放射免疫、荧光免疫、酶免疫试剂盒供庆大霉素等氨基甙类药TDM用。免疫法特别是荧光免疫和酶免疫法,操作简便,并可实现自动检测。但某些免疫试剂盒抗体特异性较差,在氨基甙类药物间存在交叉免疫性,虽然临床中两种氨基甙类药合并使用可能性不大,但在解释结果时仍应考虑排除这种可能。
七、环孢素A
(一)药效学及血药浓度参考范围
环孢素A为高脂溶性肽类大分子药物,可通过对免疫应答过程多环节的作用,选择性抑制辅助性T淋巴细胞(TH)的增殖及功能,产生免疫调节作用,用于器官移植后的抗排斥反应,及多种自身免疫性疾病的治疗。该药虽较其他免疫抑制剂毒性作用少,但仍存在肝、肾损害、震颤、高血压等毒性反应。环孢素A的治疗作用、毒性反应与血药浓度关系密切,安全范围窄。本药又大多供长期预防性用药,而肾、肝毒性在肾、肝移植时,难以和排斥反应区别。鉴于上述原因,环孢素A需进行TDM。
免疫法测得环孢素A的全血治疗浓度参考范围为0.1-0.4μg/ml,最小中毒浓度参考值0.6μg/ml。
(二)药动学
环孢素A的体内过程随移植器官种类而变,肌肉注射吸收不规则,口服吸收慢而不完全,约4h达峰浓度。生物利用度随移植物不同而有差异,大多为30%左右。该药在血液中几乎全部与蛋白结合,与血细胞(主要为红细胞)结合部分约为与血浆蛋白结合的2倍。环孢素A的分布呈多室模型,并易分布至细胞内。表观分布容积个体差异大,平均约4L/kg。其几乎全部经肝脏代谢为10余种代谢物,再由肾或胆道排泄。消除半寿期随病理状态而变,肝功能正常者约4h左右,亦有长达数十小时者。
(三)检测技术
由于上述血细胞结合特点,本药的TDM多主张用肝素抗凝,作全血浓度测定。取样时间通常在达稳态后用药前,以测定稳态谷浓度。
供该药测定的方法为HPLC和免疫法。两法灵敏度、线性范围、重复性均可满足要求。免疫法早年多用放射免疫法,新近已有荧光免疫试剂盒问市。但免疫法主要问题仍是代谢物可与抗体发生交叉结合反应造成干扰,故其结果高于HPLC法,甚至可高出数倍。用乙醚提取预处理可减少这类干扰。HPLC法特异性高,重复性好,但色谱条件要求高,耗时,较难普及推广。
第十章 肝胆疾病的生物化学与实验诊断
第一节 概述
一、肝细胞的正常代谢功能
肝是人体内体积最大的实质性腺体,是具有重要而复杂的代谢功能的器官。它具有肝动脉和肝静脉双重的血液供应,且有肝静脉及胆道系统出肝,加上丰富的血窦及精巧的肝小叶结构,以及肝细胞中富含线粒体、内质网、核蛋白体和大量酶类,因而能完成复杂多样的代谢功能。
每个肝细胞平均约含400个线粒体,呈圆形、椭圆形或棒形。线粒体与三羧酸循环、呼吸链及氧化磷酸化、脂肪酸的β-氧化及酮体生成、氨基酸的脱氨基、转氨基及尿素合成等有密切关系。线粒体对缺氧特别敏感,易于受损伤。肝细胞的粗面内质网是合成各种蛋白质和酶类的场所,而滑面内质网则与糖原的合成和分解、胆红素、激素、药物、染料及毒物等的生物转化有关。
溶酶体中含10余种水解酶类,它与肝细胞的溶解和坏死、胆红素的分泌以及胆褐素和铁颗粒的代谢有关,具有吞饮、储存、消化和运输细胞内代谢产物的作用。
高尔基复合体与分泌和排泄代谢产物及合成糖蛋白等有关。有人认为高尔基复合体、溶酶体和毛细胆管构成肝细胞的胆汁分泌微小器官,在肝内胆汁淤积时其功能受到损害。
肝细胞的胞质中含有糖酵解、磷酸戊糖通路、氨基酸激活、脂肪酸和胆固醇合成的多种酶类。
肝细胞核染色体DNA及调控蛋白对肝细胞内代谢起调控作用,肝细胞再生时,DNA大量合成和复制。
肝细胞膜由蛋白质和磷脂等构成,具有三种形态:一是两个相邻肝细胞间的细胞膜,依靠指状突起使相邻肝细胞相互连接;面向肝窦的细胞膜则具有微绒毛,能增大与肝窦血液的接触面积,有利于物质交换;在2或3个肝细胞之间,细胞膜皱折形成毛细胆管,毛细胆管与胆红素等胆汁成分的排泌有关。当肝内或肝外胆汁淤积时,毛细胆管发生改变。
肝细胞能合成多种血浆蛋白质(白蛋白、纤维蛋白原、凝血酶原及多种血浆蛋白质)。在血浆蛋白质的处理上肝起着重要作用。白蛋白以外的血浆蛋白质都是含糖基的蛋白质,它们在肝细胞膜上的唾液酸酶的作用下,失去糖基末端的唾液酸,就可被肝细胞上的特异受体-肝糖结合蛋白所识别,并经胞饮作用进入肝细胞而被溶酶体清除。
肝内含有丰富的与氨基酸分解代谢有关的酶类,由食物消化吸收而来的和组织蛋白分解而来的氨基酸大部分被肝细胞摄取,经过转氨基、脱氨基、转甲基、硫和脱羧等反应转变成酮酸及其他化合物。除亮氨酸、异亮氨酸和缬氨酸这三种支链氨基酸主要在肌肉组织降解外,其余氨基酸特别是苯丙氨酸、酪氨酸及色氨酸等芳香氨基酸都主要在肝内进行分解代谢。肝是合成尿素的重要器官,肝细胞功能严重障碍会引起血中多种氨基酸的含量增高,血氨浓度增高、血中尿素浓度降低。
肝脏是维持血糖浓度相对稳定的重要器官,肝有较强的糖原合成与分解的能力,通过糖原的合成与分解而调节血糖。肝是进行糖异生的重要器官,可将甘油、乳酸、氨基酸等转化为葡萄糖或糖原。肝还可将半乳糖、果糖等转化为葡萄糖。
肝在脂类的消化、吸收、运输、合成及分解等过程中起重要作用。肝合成甘油三酯、磷脂及胆固醇的能力很强,并进一步合成VLDL及HDL。某些载脂蛋白(如ApoAⅠ、ApoB100、ApoCⅠ、ApoC等)以及卵磷脂胆固醇酰基转移酶(LCAT)在肝细胞中合成,它们在脂蛋白的代谢及脂类运输中起着重要的作用。肝对甘油三酯及脂肪酸的分解能力很强,是生成酮体的重要器官。
肝在维生素的吸收、储存和转化方面都起着重要作用。维生素A、D、E、K及B在肝内储存,胡萝卜转变成维生素A,维生素D3在C25位上的羟化,由维生素PP合成NAD+和NADP+,由维生素B1合成TPP等过程均在肝内进行。
肝与许多激素的灭活和排泄有密切关系,血中的类固醇激素在肝内灭活,生成种种代谢产物(如17-羟类固醇、17-氧类固醇),有的代谢产物在肝脏进一步与葡萄糖醛酸或硫酸结合,再从尿液或胆汗排出。胰岛素及其他多种蛋白质或多肽激素以及肾上腺素、甲状激素等也都在肝内灭活。因此,当发生严重肝功能损伤时,体内多种激素因灭活而堆积,会导致相应的激素调节功能紊乱。肝的生物转化功能,胆汁酸及胆色素代谢功能在第二、三、四节中介绍。
二、枯否细胞的功能
肝内存在的枯否细胞(Kupffercells,KC)实际是位于肝窦内的巨噬细胞,此类细胞的特点是其内质网及核膜上的内源性过氧化物酶活性。KC除具有吞噬、消灭病原微生物、清除机体内的内毒素、调节免疫和炎症反应等功能外,还能调控组织和基质修复、调控肝细胞、肝储脂细胞的增殖和合成细胞外基质等。这些功能是通过枯否细胞所分泌的多种生物活性因子(如转化生长因子TGF、肝细胞生长因子HGF、胰岛素样生长因子ⅡIGFⅡ、转化生长因子βTGFβ、白介素6IL-6、白介素-1IL-1、肿瘤坏死因子αTNFα、干扰素IFN等)而起作用的。肝内的枯否细胞、储脂细胞等与肝纤维化的形成有密切关系。
三、肝细胞损伤时的代谢障碍
(一)肝细胞损伤时蛋白质代谢的变化
肝细胞合成白蛋白的能力很强,正常人每天能合成10g。当肝功能严重受损时,血浆胶体渗透压可因白蛋白的合成不足而降低,同时球蛋白浓度(尤其γ-球蛋白)反而增高,导致血浆白蛋白与球蛋白的比值(A/G)降低。在重症肝炎及急性黄色肝萎缩时,可见α、β及γ球蛋白降低。肝细胞损伤时血清游离氨基酸增加,甚至从尿中丢失,这可能是由于肝细胞处理氨基酸的能力下降所致。肝细胞损伤时合成尿素的能力降低,可引起血氨增高。
肝细胞损伤时肝细胞(胞质,线粒体等)内多种酶可逸入血中,使血中多种酶活性增高。
(二)肝细胞损伤时的脂类代谢变化
肝细胞损伤时可导致脂肪肝的形成。肝实质细胞内有甘油三酯的蓄积,这是由于甘油三酯在肝细胞内的合成与其向体循环中释放间的平衡失调所致。在脂肪肝的肝细胞线粒体内,ATP、NAD及细胞色素C的含量常显著减少,由于糖代谢障碍而引起脂肪动员的增加。脂肪肝时磷脂酰胆碱显著减少,可能是由于缺氧或氧化磷酸化障碍,致使ATP和CDP-胆碱的形成不足,造成磷脂及VLDL的合成障碍,导致肝内脂肪向体循环的释放不足,促使肝细胞中甘油三酯的堆积。
在重度肝细胞损伤时,肝细胞合成胆固醇的能力降低,这种情况见于严重的肝细胞炎症及变性坏死。肝细胞严重损伤时,胆固醇酯的合成也降低。某些慢性肝损伤时,由于糖代谢障碍,糖利用减少,脂肪分解增加,可导致酮症。
(三)肝细胞损伤时糖代谢的变化
当肝细胞损伤(尤其肝炎)时,肝内糖代谢有关酶类的特征性变化是:活性增高的酶有G6P脱氢酶、6-磷酸葡萄糖酸脱氢酶、磷酸甘油醛脱氢酶、丙酮酸磷酸激酶、乳酸脱氢酶等。
活性降低的酶是葡萄糖-6-磷酸酶、醛缩酶、磷酸甘油脱氢酶、异柠檬酸脱氢酶、琥珀酸脱氢酶、苹果酸脱氢酶等。
由上述酶谱变化可以看出:肝细胞损伤时糖代谢变化的特点是磷酸戊糖途径及糖酵解途径相对增强,严重肝损伤时糖有氧氧化及三羧酸循环运转不佳,血中丙酮酸量可显著上升。慢性肝病时血中α-酮戊二酸量与症状平行地增加。不同肝病时耐糖曲线可呈低平型、高峰型、高坡型等异常曲线。
第二节 肝的生物转化功能
在人整个生命过程中经常有某些外来异物(如毒物、药物、致癌物)进入体内,代谢过程中体内也不断产生一些生物活性物质及代谢物(激素、胺类等)。这些外来及内生物质大部分在肝内进行代谢转化。在肝内有关酶的催化下,一方面使上述物质的极性或水溶性增加,有利于从尿或胆汁排出,同时也改变了它们的毒性或药理作用。通常将这些物质在体内(主要是肝细胞微粒体内)的代谢转变过程称为生物转化作用(biotransformation)。
现已证明:肝、肾、胃肠道、肺、神经、皮肤及胎盘等组织都存在一些使毒物、药物及激素等进行生物转化的酶系,但以肝为最重要,其生物转化功能最强。
随着工业化和科学技术的发展,人们与各种化学物质接触的机会越来越多,常用的化学物质有63000种以上。对于人体来说,这些化学物质有的可能是药物,有的可能是对人体有害的毒物,有的可能是能诱发细胞癌变的致癌物。阐明这些物质在肝内生物转化的规律,对于保障人类的健康是很有意义的。
一、生物转化的反应类型
通常将生物转化反应分为两相反应。第一相反应包括氧化、还原、水解反应,第二相反应即结合反应。每一相反应又各自包括多种不同的反应,分别在不同的部位中进行(表10-1)。
表10-1 生物转化反应的一般类型
| 反应类型 | 反应性质 | 细胞内酶的主要定位 | |
| 羟化反应 | 微粒体 | ||
| 第 | 脱烷基反应 | 微粒体 | |
| 环氧化反应 | 微粒体 | ||
| 一 | 氧化 | 脱硫反应 | 微粒体 |
| 脱卤反应 | 微粒体 | ||
| 醇氧化反应 | 胞液为主、微粒体少量 | ||
| 相 | 醛氧化反应 | 胞液、线粒体 | |
| 脱氨反应 | 微粒体、线粒体 | ||
| 反 | 还原 | 醛还原反应 | 胞液 |
| 偶氮还原反应 | 微粒体 | ||
| 应 | 硝基还原反应 | 微粒体、胞液 | |
| 水解 | 酯水解反应 | 微粒体、胞液 | |
| 酰胺水解反应 | 微粒体、胞液 | ||
| 第 | 葡萄糖醛酸结合 | 微粒体 | |
| 二 | 甘氨酸结合 | 线粒体 | |
| 相 | 结合 | 乙酰化反应 | 胞液 |
| 反 | 甲基化反应 | 胞液 | |
| 应 | 谷胱甘肽结合 | 胞液 | |
| 硫酸结合 | 胞液 | ||
(一)第一相反应
大多数毒物、药物等进入肝细胞后,常先进行氧化反应,有些可被水解,少数物质被还原。经过氧化、还原和水解作用,一般能使非极性的化合物产生带氧的极性基团,从而使其水溶性增加以便于排泄,同时也改变了药物或毒物分子原有的某些功能基团,或产生新的功能基团使毒物解毒或活化,使某些药物的药理活化发生变化,使某些致癌物质活化或灭活。
⒈氧化作用在肝细胞的微粒体、线粒体及胞液中含有参与生物转化的不同的氧化酶系,包括加单氧酶系、胺氧化酶系脱氧酶系(表10-2)。
表10-2 与生物转化有关的几种氧化酶类
| 酶系 | 细胞内定位 | 反应式及举例 |
| 加单氧酶系 | 微粒体 | RH+O2+NADPH+H+→ROH+NADP+ |
| (混合功能氧化酶) | (滑面内质网) | +H2O(如烃类的氧化等) |
| 胺氧化酶系 | 线粒体 | RCH2NH2+O2+H2O→RCHO+NH3+H2O2 |
| (如单胺氧化酶催化组胺、酪胺等的氧化) | ||
| 脱氢酶系 | 胞液 | RCH2OH+NAD+→RCHO+NADH+H+ |
| 线粒体 | RCHO→RCOOH | |
| (如醇脱氢酶及醛脱氢酶催化的反应) |
存在于微粒体中的以细胞色素P450为重要成分的加单氧酶系具有十分重要的生理意义。在该系统所催化的反应中,由于氧分子中的一个氧原子掺入到底物中,而另一个氧原子使NADPH氧化生成水,即一种氧分子发挥了两种功能,故又称混合功能氧化酶,从底物的角度来看,只掺入一个原子的氧,故称加单氧酶。
应当指出:依赖细胞色素P450的电子传递系统存在于各种生物膜系统中。在高等动物组织中有微粒体型及线粒体内膜型两大类。微粒体型又包括单一电子传递系统和复合电子传递系统,后者中既有NADPH参与,又有NADP参与。线粒体内膜型则有铁氧还原蛋白、NADPH及细胞色素P450参加。多样的细胞色素P450系统催化外来异物的羟化和脱烷基反应,还参与类固醇激素的生物合成、灭活胆汁酸的生物合成及维生素D3的羟化反应等。许多毒物、药物或致癌物经过混合功能氧化酶的催化而产生各种羟化反应产物。现将微粒体混合功能氧化酶催化的氧化反应类型列于表10-3。
表10-3 微粒体混合功能氧化酶催化的氧化反应类型

⒉还原作用肝细胞中生物转化的还原反应主要有偶氮还原酶和硝基还原酶所催化的两类反应。硝基还原酶存在于肝、肾、肺等细胞微粒体中,是FAD型还原酶,可使对-硝基苯甲酸、硝基苯、氯霉素等的-NO2还原成-NH2,反应在厌氧条件下进行,由NADH供氢。偶氮还原酶存在于肝细胞微粒体中,由NADPH供氢,中间经氢偶氮复合物最后生成胺,反应可在有氧条件下进行,此酶属P450酶类。
⒊水解作用如某些酯类(普鲁卡因)、酰胺类(异丙异菸肼)及糖苷类化合物(洋地黄毒苷)可分别在酯酶、酰胺酶、糖苷酶等水解酶的作用下被水解。这类酶在体内分布广泛,种类繁多,肝外组织也含有这些酶类。
(二)第二相反应
有机毒物或药物,特别是具有极性基团的物质,不论是否经过氧化、还原及水解反应,大多要与体内其他化合物或基团相结合,从而遮盖了药物或毒物分子中的某些功能基团,使它们的生物活性、分子大小以及溶解度等发生改变,这就是生物转化中的结合反应。结合反应往往属于耗能反应,它在保护有机体不受外来异物毒害、维持内环境稳定方面具有重要意义。结合反应可在肝细胞的微粒体、胞液和线粒体内进行。不同形式的结合反应由肝内特异的酶系所催化。常见的结合反应有葡萄糖醛酸结合、硫酸结合、乙酰基结合、甘氨酰基结合、甲基结合、谷胱甘肽结合及水化等。但其中以葡萄糖醛酸结合最为重要。(表10-4)。
表10-4 结合反应的主要类型
| 结合反应 | 结合基团的直接供体 | 酶类 | 酶定位 | 底物类型 |
| 葡萄糖醛酸结合 | 尿苷二磷酸葡萄糖醛酸(UDPGA) | 葡萄糖醛酸基转移酶 | 微粒体 | 酚、醇、羧酸、胺、羟胺、磺胺、巯基化合物等 |
| 硫酸结合 | 3′-磷酸腺苷-5′-磷酸硫酸(PAPS) | 硫酸转移酶 | 胞液 | 醇、酚、芳香胺类 |
| 乙酰基结合 | 乙酰辅酶A | 乙酰基转移酶 | 胞液 | 芳香胺、胺类、氯基酸 |
| 甘氨酰基结合 | 甘氨酸(Gly) | 酰基转移酶 | 线粒体 | 酰基CoA(如苯甲酰CoA) |
| 甲基结合 | S-腺苷蛋氨酸(SAM) | 甲基转移酶 | 胞液 | 生物胺、吡啶喹啉、异吡唑等 |
| 谷胱甘肽结合 | 谷胱甘肽(GSH) | 谷胱甘肽-S-转移酶 | 胞液 | 卤化有机物、环氧化物、溴酚酜、胰岛素等 |
| 水化 | H2O | 环氧水化酶 | 微粒体 | 不稳定的环氧化物(如环氧萘) |
二、致癌物质的生物转化
(一)致癌物的分类及致癌作用
癌常由各种化学物质所致,已确定至少有约1700种化学物质具有致癌作用,这些致癌物大体有下述几类:
⒈人工合成的化学致癌物
⑴芳香烃类:如苯并芘。
⑵芳香胺类:如2-乙酰氨基芴。
⑶芳香族偶氮化合物:如4-二甲基氨基偶氮苯(奶油黄,DAB)。
⑷杂环化合物:如1-氧-4-硝基喹啉(4-NQO)。
⑸脂肪族化合物:如芥子气。
⑹有机卤化物及无机化合物:如666及羰基镍、砷化物等。
⑺N-亚硝基化合物:如二乙基亚硝胺等。
⒉由微生物产生的化学致癌物一些真菌毒素具有致癌作用,已确定结构的17种黄曲霉毒素中致癌作用最强的是黄曲霉毒素B1。
⒊来自植物的致癌物如苏铁苷、香樟素等。
⒋食物加热过程中产生的致癌物质如谷氨酸加热环缩而成为Glu-p-2。
⒌由肠菌作用所产生的致癌物质例如在肠菌作用下由胆汁酸生成甲基胆蒽,由色氨酸生成酚类,肠菌作用产生的胺类与亚硝酸盐生成亚硝胺类等。
上述致癌物进入人体后常需经过转化而转变为活化型的致癌物,再以DNA和蛋白质为靶分子,使基因产生突变、错位、倒转、插入、重排、断裂等一系列结构损伤。基因是决定细胞增殖,生长,分化的关键因素,其突变无论是致癌剂引起的体细胞基因突变或(和)遗传因素导致生殖细胞突变,或正常基因丢失以及正常细胞分化过程中基因调控异常,均可使基因表达发生紊乱,出现异常表型,影响细胞形态和生物活性,导致细胞癌变。
表10-5 某些致癌物质的生物转化
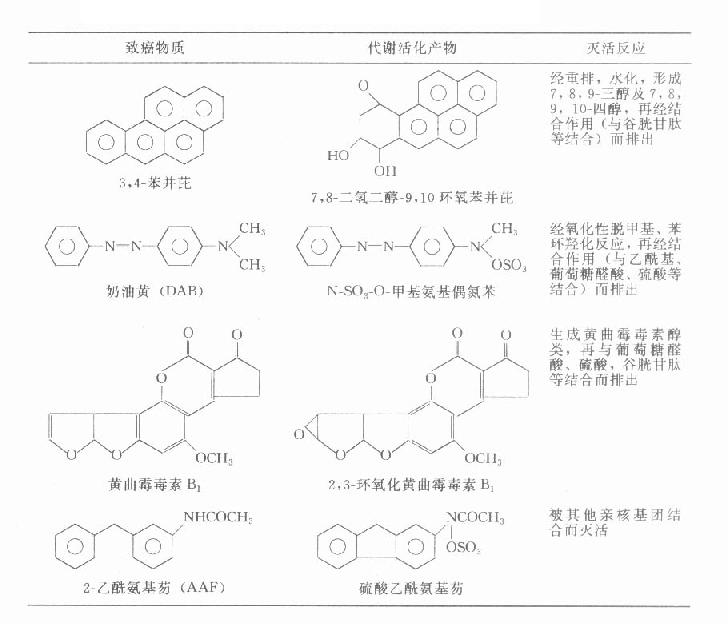
致癌物的代谢活化及灭活反应是转化的两个方面,许多致癌物质是经过肝细胞内微粒体混合功能氧化酶的作用,被代谢活化而变为终致癌物的。致癌物质亦可经生物转化而灭活或被排泄,因此,生物转化具有两重性(表10-5)。
三、药物的生物转化
大多数药物经不同途径被摄入人体后都要发生分子结构的改变,药物的生物转化主要是在肝细胞滑面内质网的混合功能氧化酶系的催化下完成的,反应也包括氧化、还原、水解、结合等反应,通过生物转化,常引起药物药理活性的变化(表10-6)。
表10-6 药物经生物转化引起药理活性的变化

四、毒物的生物转化
毒物在生物转化过程中往往形成活性中间产物,这叫代谢活化,常为第一相反应,与加单氧酶系有关;第二相反应则常与毒物的解毒及促进排泄有关。毒物的生物转化举例如表10-7。
五、有关生物转化与排泄功能的肝功能试验
(一)溴酚(磺溴酞钠,BSP)排泄试验
以溴酚1ml缓慢静注(1min),45min后95%以上可以排出,滞留2%以下。当注入BSP后,与血液中的白蛋白相结合,被肝细胞摄取再在酶的催化下,与谷胱甘肽结合,随胆汁排出。当肝细胞摄取、结合及排泄功能障碍时,BSP出现滞留。急、慢性肝炎及肝硬化、肝昏迷时滞留,可用以判断肝损伤程度及预后,本试验应注意过敏反应及休克。
表10-7毒物的生物转化举例
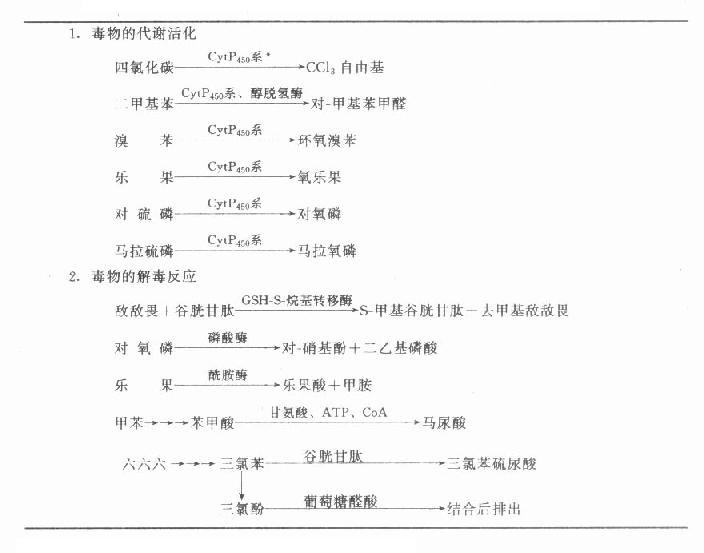
*CytP450系即CytP450依赖性混合功能氧化酶系。
(二)靛青绿(ICG)排泄试验
靛青绿为暗绿色色素,静脉注射后迅速与白蛋白结合,90%以上被肝细胞摄取,以原形从胆汁排出,此试验主要反映肝细胞摄取色素的功能。ICG试验与肝小叶间质系统的病变(假小叶、纤维化)有关,慢性肝炎活动期肝硬化时显著滞留,肝炎时亦出现滞留,恢复期此试验较早正常化。正常人15min滞留10%以下。本试验亦应注意过敏反应及休克。
第三节 肝与胆汁酸代谢
胆汁酸是胆汁中存在的一类胆烷酸的总称。人类胆汁中存在的胆汁酸主要有胆酸(CA)、鹅脱氧胆酸(CDCA)、脱氧胆酸(DCA),还有少量石胆酸(LCA)及微量熊脱氧胆酸(UDCA)。前4种胆酸在胆汁中的比例通常为10:10:5:1。
胆汁酸按其在体内来源的不同可分为初级胆汁酸和次级胆汁酸。在肝细胞内以胆固醇为原料合成的叫初级胆汁酸(包括胆酸及鹅脱氧胆酸),而后在肠道内经肠菌中酶的作用形成次级胆汁酸(包括脱氧胆酸、石胆酸及熊脱氧胆酸等)。胆汁酸主要以结合型形式从肝分泌入肝汁。结合型即指前述胆汁酸与甘氨酸或牛磺酸结合而成的结合胆汁酸。较大量存在的结合胆汁酸有甘氨胆酸、甘氨鹅脱氧胆酸、甘氨脱氧胆酸、牛磺胆酸、牛磺鹅脱氧胆酸及牛磺脱氧胆酸等。无论游离的或结合型的胆汁酸,其分子内部都是既含亲水基团(羟基、羧基、磺酰基),又含疏水基团(甲基及烃核),故胆汁酸的立体构型具有亲水和疏水两个侧面,因而使胆汁酸表现出很强的界面活性。它能降低脂、水两相之间的表面张力,促进脂类形成混合微团,这对脂类物质的消化吸收以及维持胆汁中胆固醇的溶解都起重要作用。
一、胆汁酸代谢异常与疾病的关系
肝在胆汁酸代谢中占重要地位,肝细胞与胆汁酸的生物合成、分泌、摄取、加工转化都有密切关系。因此,当肝细胞损伤或胆道阻塞时都会引起胆汁酸代谢的障碍。在肝胆疾病时首先表现出的是病人血清胆汁酸浓度的增高。在肝实质细胞病变时,胆汁酸的合成功能受损,还会引起初级胆汁酸比值(CA/CDCA)变小甚至出现倒置。
实验性肝内胆汁淤滞可因给动物投给牛磺石胆酸而引起,实验研究结果表明:石胆酸能抑制毛细胆管膜上的Na+、K+-ATP酶的活性,从而导致胆汁分泌障碍。另外,由石胆酸所形成的微团粗大,使胆汁粘度显著增加,这也是石胆酸造成胆汁淤滞的原因。
胆汁酸代谢与胆固醇结石的形成有密切关系(见本章第五节)。
胆汁酸代谢与高脂蛋白血症有密切关系。各型高脂蛋白血症,其血浆胆固醇浓度可有不同程度的升高,胆汁酸代谢可从下述几方面影响体内胆固醇的平衡。
⒈胆汁酸的生物合成是内源性胆固醇的主要代谢去路胆汁酸的生物合成又主要是通过其本身的反馈作用进行调控的。
⒉肝细胞通过胆汁排泄胆固醇,主要依靠胆汁酸的乳化及形成混合微团的作用,因而胆汁酸合成、分泌的质和量都对胆固醇的排泄有影响。
⒊胆汁酸在食物胆固醇的吸收过程中起协助作用,吸收的胆固醇直接调控肠壁细胞及肝细胞内胆固醇的合成。因此,高脂蛋白血症时的代谢紊乱必然涉及胆汁酸的代谢异常。例如IIa型高脂血症时,CA的量明显降低,而CDCA的合成则代偿地增加。
在维持胆汁酸肠肝循环的过程中,小肠起着重要的作用。回肠切除术后,如果胆汁酸的重吸收严重减少,胆汁酸丢失过多,可导致脂肪消化吸收不良(脂肪泻)。
二、血清胆汁酸测定的临床意义
血清胆汁酸浓度很低(总胆汁酸2μg/ml),可用气相色谱、放射免疫、高效液相层析法及酶学分析法(以3α-羟类胆固醇脱氢酶为工具酶)测定NADH生成量。可以测定血清总胆汁酸(TBA),用气相色谱、放射免疫和高效液相层析尚可对胆酸、脱氧胆酸及鹅脱氧胆酸进行分别测定,还可进行胆汁酸负荷试验及尿中硫酸结合型胆汁酸的测定。
测定血清胆汁酸可提供肝胆系统是否正常的重要信息。肝量疾病时周围血液循环中的胆汁酸水平明显增高。急性胆炎早期和肝外阻塞性黄疸时,有些病例可增至正常值的100倍以上。在胆道阻塞时,病人血清中CA及CDCA浓度增加,但CA所占比例较高(CA/CDCA>1)。肝实质细胞损伤时,CA/CDCA<1。故CA/CDCA比值可作为肝胆阻塞性疾病与肝实质细胞性疾病的鉴别指标。
肝胆疾病时血清胆汁酸浓度的升高与其他肝功能试验及肝组织学的变化极为吻合,在肝细胞仅有轻微坏死时,血清胆汁酸的升高常比其他检查更为灵敏。有报告统计,急性肝炎、肝硬化、原发性肝癌、急性肝丙胆汁淤滞、原发性胆汁性肝硬化以及肝外阻塞性黄疸,其血清总胆汁酸均100%出现异常,上述疾病时均有血清胆汁酸含量的增高。关于血中胆汁酸异常的程度与肝胆疾病种类的关系如表10-8所示。
表10-8 血中胆汁酸异常的程度与肝胆疾病种类的关系
| 血中胆汁酸轻度增加(10-20μmol/L) | 血中胆汁酸中度增加(20-40μmol/L) | 血中胆汁酸重度增加(40μmol/L以上) |
| 急性肝炎(恢复期) | 急性肝炎(急性期) | 急性肝炎(急性期) |
| 慢性肝炎(非活动期,活动期) | 慢性肝炎(活动期) | |
| 肝硬化(代偿期) | 肝硬化(代偿期) | 肝硬 |
| 肝癌 | 肝癌 | 肝化(代偿性,非代偿性) 癌 |
| 体质性黄疸(Gilbert病Dubin-Johnson综合征) | 胆汁淤滞性黄疸(肝内、肝外性)重症肝炎 |
血清中胆汁酸含量受肠道吸收的胆汁酸量与肝门静脉中的胆汁酸被肝脏摄取的摄取率所决定。由于肝损伤的存在,则经肝门静脉回肝的胆汁酸因肝细胞功能低下或侧枝循环的形成,导致肝不能充分摄取胆汁酸,则胆汁酸在血中浓度增高。
近年来有用熊去氧胆酸进行的胆汁酸负荷试验,检测肝脏对胆汁酸处理的能力。经口服熊去氧胆酸300mg后,用3α-羟类固醇脱氢酶法测定血中的胆汁酸,结果发现慢性肝炎(活动型)、肝硬化及脂肪病人在负荷后血清总胆汁酸显著增高,表明这些病人清除胆汁酸的能力显著降低。有报告胆汁酸负荷试验结果异常的检出率为:肝硬化100%,慢性肝炎活动型为92%-100%,慢性肝炎(非活动型)为70%-82%。
还有检测尿中硫酸结合型胆汁酸的报告,胆汁酸与硫酸结合反应亦属机体防御机制之一,当胆汁淤滞使血中胆汁酸升高时,一部分胆汁酸即与硫酸相结合,从而使其极性增加,随尿排泄增多。过去测定尿中硫酸结合型胆汁酸用层析法,操作复杂,现在有硫酸酯酶与3α-羟基类固醇脱氢酶(3α-HSD)组成的试剂盒,操作简便。结果表明:正常组为2.0±1.8μmol/g肌酐,在各种肝疾患时上升,急性肝炎122.0±28.6μmol/g肌酐;肝外性胆道阻塞124.5±67.7μmol/g肌酐;非代偿性肝硬化77.8±21.3μmol/g肌酐;肝内胆汁淤滞则为67.2±23.5μmol/g肌酐。这是诊断急性肝炎及肝外胆道阻塞、非代偿性肝硬化等的重要指标。
第四节 胆红素代谢与黄疸
一、胆红素的来源、生成与运输
(一)胆红素的来源与生成
用14C标记的甘氨酸的示踪试验及其他实验研究的结果表明,胆红素的来源不外以下几种:①大部分胆红素是由衰老红细胞破坏、降解而来,由衰老红细胞中血红蛋白的辅基血红素降解而产生的胆红素的量约占人体胆红素总量的75%;②小部分胆红素来自组织(特别是肝细胞)中非血红蛋白的血红素蛋白质(如细胞色素P450、细胞色素b5、过氧化氢酶等)的血红素辅基的分解;③极小部分胆红素是由造血过程中,骨髓内作为造血原料的血红蛋白或血红素,在未成为成熟细胞成分之前有少量分解,即无效造血所产生的胆红素。
胆红素的生成过程包括:①衰老的红细胞在单核吞噬细胞系统被破坏,首先除去珠蛋白而分离出血红素;②血红素在单核吞噬细胞内微粒体的血红素加氧酶的作用下,将血红素卟啉环氧化断裂,释放出CO和铁,并形成胆绿素,血红素加氧酶存在于肝、脾、骨髓或巨噬细胞等单核吞噬细胞系统细胞中,在微粒体内属混合功能氧化酶,反应需要分子氧参加,并需要NADPH、NADPH-细胞色素P450还原酶共同存在;③胆绿素在胆绿素还原酶催化下生成胆红素Ⅸa,胆绿素还原酶存在于单核吞噬细胞系统细胞内的可溶性部分,以NADPH为辅酶。
在体内从血红素形成胆绿素,继而还原为胆红素,并进一步被结合、排泄,这样复杂的过程总共只需1-2min。胆红素生成过程如图10-1所示。
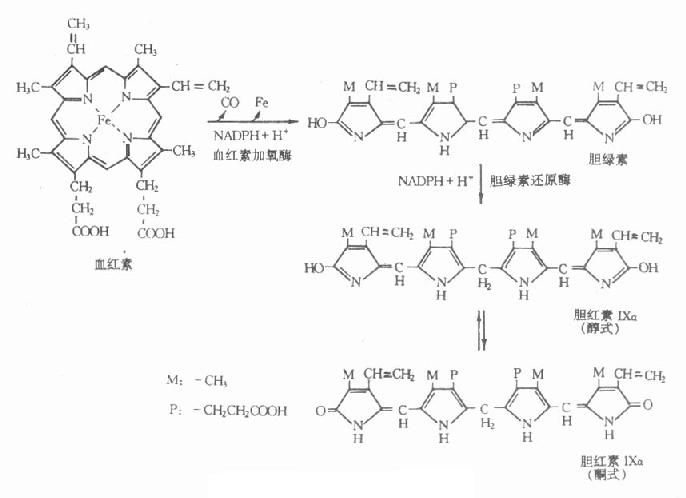
图10-1 胆红素生成过程
(二)胆红素在血液中的运输
在单核吞噬细胞中生成的胆红素可进入血液循环,在血浆内主要以胆红素-白蛋白复合体的形式存在和运输。除白蛋白外,α1-球蛋白也可与胆红素结合。一般说白蛋白与胆红素的结合是可逆的。当血浆胆红素浓度正常时,1分子白蛋白通常结合1分子胆红素,而当血浆胆红素增多时则可结合2分子胆红素。正常成人每100ml血浆中的白蛋白结合胆红素的能力约为20-25mg,所以正常情况下白蛋白结合胆红素的潜力很大。由于胆红素与白蛋白较紧密地结合成复合体,一方面改变了胆红素的脂溶性,另一方面又限制了它自由通过各种生物膜的能力,不致有大量游离胆红素进入组织细胞而产生毒性作用。据报道有一部分胆红素与白蛋白共价结合,可能是白蛋白分子中赖氨酸残基的ε-氨基与胆红素一个丙酸基的羧基形成酰胺键,在血中停滞时间长,称为δ-胆红素。δ-胆红素在肝细胞损伤及胆汁郁滞等高结合胆红素性黄疸时出现,与重氮试剂呈直接反应,可用离子交换柱层析法检测。目前认为δ-胆红素检测的临床意义是:①δ-胆红素与急性黄疸性肝炎的恢复期密切相关,恢复期总胆红素下降,尤其结合胆红素明显降低,而δ-胆红素的相对百分比却显著增高,最后达总胆红素的80%-90%以上,这种表现可作为对急性黄疸性肝炎恢复期观察的可靠指征;②严重肝功不全病人血清中δ-胆红素常小于总胆红素的35%,死前可降至20%以下,患者恢复后δ-胆红素占总胆红素的40%-90%。据此δ-胆红素可作为判断严重肝病预后的指征。
二、肝对胆红素的摄取、转化及排泄
当胆红素随血液运输到肝后,由于肝细胞具有极强的摄取胆红素的能力,故可迅速被肝细胞摄取。肝迅速地选择性地从血浆摄取胆红素的能力与下述机制有关。
⒈位于血窦表面的肝细胞膜上可能有特异的载体蛋白系统,胆红素等有机阴离子与膜上载体结合后,即从膜的外表面转移至内表面,然后进入胞质。当白蛋白-胆红素复合物通过肝窦壁时,胆红素与白蛋白解离,只有胆红素被肝细胞所摄取。
⒉肝细胞内有两种色素受体蛋白即Y蛋白和Z蛋白Y蛋白与胆红素亲和力较高,在肝细胞中含量较大,约占肝细胞浆蛋白的5%,是肝细胞内主要的胆红素载体蛋白;Z蛋白是另一种胆红素载体蛋白,对长链脂肪酸具有很强的亲和力,可能是肝细胞内脂肪酸载体蛋白。Y蛋白与Z蛋白利用其对胆红素的高亲和力,从细胞膜上接受进入胞质的胆红素,并将它运至内质网。
肝细胞对胆红素的转化在滑面内质网上进行。在葡萄糖醛酸基转移酶的催化下,胆红素被转化为葡萄糖醛酸胆红素,胆红素在肝细胞内经结合转化后,其理化性质发生了变化,从极性很低的脂溶性的未结合胆红素变为极性较强的水溶性结合物-葡萄糖醛酸胆红素,从而不易透过生物膜。这样既起到解毒作用,又有利于胆红素从胆道排泄。在肝细胞内,胆红素通过其丙酸基与葡萄糖醛酸结合,主要生成双葡萄糖醛酸胆红素。
结合胆红素被排泄至毛细胆管的过程,有内质网、高尔基复合体、溶酶体等参与,毛细胆管膜上也存在一种以载体为中介的转运过程,这一过程必须对抗浓度梯度。当肝细胞损伤时,可由于结合型胆红素的排泄障碍而造成肝细胞淤滞性黄疸。由于肝细胞内有亲和力强的胆红素载体蛋白及葡萄糖醛酸基转移酶,因而不断地将胆红素摄取、结合、转化及排泄,保证了血浆中的胆红素不断地经肝细胞而被清除(图10-2)。
三、胆红素在肠管中的变化及其肠肝循环
结合胆红素(葡萄糖醛酸胆红素)随胆汁排泄至肠管后,在回肠末端至结肠部位,在肠管菌丛的作用下大部分被水解而脱下葡萄糖醛酸,然后逐步被还原成二氢胆红素、中胆红素、二氢中胆红素、中胆素原、粪胆原及少量d-尿胆原,后三者与重氮试剂的呈色反应相同,统称为胆素原。正常人每天从粪便排出的胆素原为40-280mg,胆素原在肠管下段接触空气后分别被氧化成为尿胆素、粪胆素和d-尿胆素(三者统称为胆素),随粪便排出,成为粪便的主要色素。在小肠下段生成的胆素原约有10%-20%可被肠粘膜重吸收,再经肝门静脉入肝,重吸收入肝的胆素原大部分以原形再排入胆道,构成肠肝循环,小部(每日0.4-4.0mg)经体循环随尿排出。
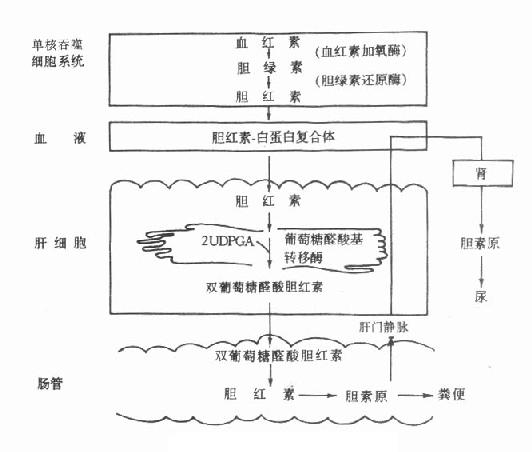
图10-2 胆色素代谢概况
四、黄疸的发生机制
黄疸是指高胆红素血症引起皮肤、巩膜和粘膜等组织黄染的现象。正常人血清胆红素小于1mg/dl(10mg/L),其中未结合胆红素占80%。当胆红素超过正常范围,但又在2mg/dl以内,肉眼难于察觉,称为隐性黄疸。如胆红素超过2mg/dl(可高达7-8mg/dl)即为显性黄疸。
黄疸按原因可分为溶血性、肝细胞性和梗阻性黄疸;按发病机制可分为胆红素产生过多性、滞留性及反流性黄疸;按病变部位可分为肝前性、肝性和肝后性黄疸;按治疗观点又可分为内科性和外科性黄疸。较为合理的是按血中升高的胆红素的类型分为高未结合胆红素性黄疸及高结合胆红素性黄疸两大类,然后再按病因、发病机制等细分。
黄疸发生的机制如下:
(一)胆红素形成过多
胆红素在体内形成过多,超过肝脏处理胆红素的能力时,大量未结合胆红素即在血中积聚而发生黄疸。未结合型胆红素形成过多的原因包括溶血性与非溶血性两大类。临床上任何原因引起大量溶血时,红细胞破坏释放的大量血红蛋白即成为胆红素的来源。非溶血性的胆红素形成过多则多见于无效造血而产生过多胆红素。
(二)肝细胞处理胆红素的能力下降
这包括:①肝细胞对胆红素的摄取障碍;②肝细胞对胆红素的结合障碍(肝细胞中葡萄糖醛酸基转移酶活性降低);③肝细胞对胆红素的排泄障碍(肝内胆汁淤滞、乙醇性肝炎等)。
(三)胆红素在肝外的排泄障碍,逆流入血而引起黄疸
新生儿生理性黄疸的成因有下述几方面:①新生儿肝细胞内葡萄糖醛酸基转移酶活性不高;②胆红素在新生儿体内产生较多;③新生儿肝细胞内缺乏Y蛋白,故摄取胆红素的能力
也比成人差,这些都可能促使新生儿生理性黄疸的发生。
高未结合胆红素血症及高结合胆红素血症的常见疾病见表10-10,10-11。
表10-10 高未结合胆红素血症的疾病
| ㈠胆红素生成增加 |
| ⒈溶血性黄疸 |
| ⑴新生儿溶血性黄疸(血型不合妊娠) |
| ⑵遗传性异常血红蛋白病:镰状细胞贫血、珠蛋白生成障碍性贫血 |
| ⑶红细胞膜异常症:遗传性球形红细胞症、遗传性椭圆形红细胞症 |
| ⑷先天性红细胞酶异常症:丙酮酸激酶缺损症、葡萄糖-6-磷酸脱氢酶缺损症 |
| ⒉早期胆红素的增加 |
| ⑴原发性早期高胆红素血症 |
| ⑵先天性骨髓性卟啉症 |
| ㈡肝内胆红素处理功能异常 |
| ⒈结合酶活性低 |
| ⑴新生儿高胆红素血症 |
| ⑵肝未成熟迁延性新生儿黄疸 |
| ⑶Grigler-Najjar综合征(肝葡萄糖醛酸基转移酶缺陷) |
| ⒉结合障碍 |
| ⑴哺乳性黄疸:母乳中孕烷3α,20β-二醇对葡萄糖醛酸基转移酶的抑制所引起 |
| ⑵Lucey-Driscoll综合征:孕烷二醇等对葡萄糖醛酸基转移酶的抑制所引起 |
| ⒊肝的摄取机制及向肝细胞内转运障碍 |
| ⑴Gilbert综合征:先天性或家族性葡萄糖醛酸基转移酶活性低下,肝细胞膜的异常或肝细胞内色素结合蛋白的异常,引起胆红素在肝细胞内转运的障碍 |
| ⑵先天性甲状腺功能低下症:肝排泄胆红素功能降低,新生儿黄疸症状长期持续 |
| ⒋胃肠道狭窄或闭锁 |
表10-11 高结合胆红素血症的疾病
| ㈠肝细胞损伤 |
| ⒈乳儿肝炎 |
| ⒉急性肝炎 |
| ⑴病毒性肝炎:血清性肝炎、传染性肝炎、水疱疹性肝炎、先天性风疹 |
| 症候群、卵黄囊-B病毒肝炎、腺病毒感染、巨细胞病毒肝炎等 |
| ⑵细菌感染并发的肝炎、败血症、肺炎、肾盂肾炎 |
| ⑶先天性梅毒 |
| ⑷弓形体病 |
| ⒊慢性肝炎 |
| ⒋肝硬化 |
| ㈡由肝细胞向毛细胆管排泄的异常 |
| ⒈Dubin-Johnson综合征 |
| ⒉Rotor综合征 |
| ㈢肝内胆汁淤滞 |
| ⒈先天性肝内胆管闭锁症 |
| ⒉总胆管性肝炎 |
| ⒊原发性胆汁性肝硬化 |
| ⒋Byler病引起的先天性肝内胆管闭锁 |
| ㈣肝外胆汁淤滞 |
| ⒈肝外胆道闭锁症 |
| ⒉总胆管扩张症 |
| ㈤由先天性代谢异常所引起的非溶血性黄疸 |
| ⒈半乳糖血症 |
| ⒉酪氨酸血症 |
| ⒊α1-抗胰蛋白酶缺乏症 |
| ⒋果糖血症 |
五、黄疸的鉴别试验
⒈高结合胆红素性黄疸与高未结合胆红素性黄疸的鉴别依靠结合胆红素与未结合胆红素的分别定量。高结合胆红素性黄疸血中结合胆红素增高,高未结合胆红素性黄疸时则血中未结合胆红素增高;高结合胆红素性黄疸时尿中胆红素阳性,胆素原视病因而异,可以增加(肝炎)、减少或消失(梗阻);高未结合胆红素性黄疸时,尿中胆红素阴性,尿中胆素原增加。
⒉梗阻与非梗阻性(肝细胞性)高结合胆红素性黄疸的鉴别可联合应用反映胆道梗阻(ALP、LAP、γ-GT、血清总胆固醇、总胆汁酸)及肝细胞损伤(ALT、AST、LDH活性、血清总蛋白及白蛋白定量等)的检验指标来加以鉴别。
⒊肝内淤滞与肝外梗阻性黄疸的鉴别可应用病史、肝功能试验、泼尼松治疗试验、苯巴比妥治疗试验等进行鉴别。对于原发性胆汁性肝硬化,其发病可能与免疫机制有关,胆管的抗原与来自肝门静脉的抗体反应,并与补体结合在胆道被吸附,在胆道壁形成免疫复合物。原发性胆汁性肝硬化血中ALP活性增高,γ-GT及5-核苷酸酶活性增高,IgM增高,抗线粒体抗体阳性。关于肝内性胆汁淤滞与肝外梗阻性黄疸的鉴别见表10-12。
⒋溶血性、肝细胞性及梗阻性黄量的鉴别(见表10-13)。
表10-12 肝内胆汁淤滞与肝外梗阻性黄疸的鉴别
| 肝内胆汁淤滞 | 肝外梗阻性黄疸 | ||
| 症状 | 肝炎样发病 | 常有发热,疼痛 | |
| 血沉 | 正常 | 加快 | |
| 白细胞数 | 正常 | 正常或增加 | |
| 血中胆红素 | 不定 | 一般较高 | |
| 血清ALP(KA单位) | 多在30单位以下 | 30单位以上高值 | |
| 血清胆固醇 | 不定 | 一般较高 | |
| 尿中(靛甙)尿蓝母 | 阴性 | 阳性 | |
| 药物引起 | 常有 | 无 | |
| 对肾上皮质激素的反应 | 常有 | 无 | |
| 胆道造影 | 肝外胆管阻塞 | (-) | (+) |
| 肝外胆管扩大 | (-) | (+) | |
| 肝内胆管多球状 | (-) | (+) | |
| 肝活检 | 小叶中心胆汁沉积 | 早期出现 | 早期出现 |
| 毛细胆管胆汁沉积 | (-)晚期出现 | 晚期出现 | |
| 胆汁栓,″胆汁湖″ | (-) | 晚期出现 | |
| 肝门静脉区浮肿 | 不显著 | 显著 | |
| 肝门静脉区粒细胞浸润 | 无 | 显著 | |
| 嗜酸细胞浸润 | 常有 | 不明显 | |
| 胆管扩大 | (-) | 早期出现 | |
| 肝细胞灶状坏死 | 早期可有发现 | 晚期出现 | |
表10-13 溶血性黄疸、肝细胞性黄疸及梗阻性黄疸的鉴别
| 溶血性黄疸 | 肝细胞性黄疸 | 梗阻性黄疸 | ||
| 胆 | 血浆总胆红素 | 多在5mg/dl | 1-70mg/dl | 不全梗阻10-15mg/dl |
| 红 | 浓度 | 以内 | 完全梗阻20-30mg/dl | |
| 素 | 未结合胆红素 | 高度增加 | 增加 | 增加 |
| 代 | 结合胆红素 | 正常 | 增加 | 高度增加 |
| 谢 | 尿胆红素定性 | 阴性 | 阳性 | 强阳性 |
| 试 | 尿中胆素原 | 增多 | 不定,或升高 | 减少或消失 |
| 验 | 粪中胆素原 | 增多 | 减少 | 减少或消失 |
| 血 | 电泳谱 | 正常 | Alb减少,γ-球蛋 | 球蛋白明显升高 |
| 清 | 白升高 | |||
| 蛋 | ||||
| 白 | 脂蛋白X | 阴性 | 一般阴性 | 明显增高 |
| 谷丙转氨酶 | 正常,稍高 | 肝炎急性期增高 | 正常或增高 | |
| 碱性磷酸酶 | 正常 | 正常或轻度增高 | 明显增高 | |
| 亮氨酸氨肽酶 | 正常 | 可增高 | 明显增高 | |
| γ-谷氨酰转肽酶 | 正常 | 可增高 | 明显增高 | |
| 其 | 凝血酶原时间 | 正常 | 延长,不易被维生素K纠正 | 延长,能被维生素K纠正 |
| 胆固醇 | 正常 | 降低,胆固醇酯明显降低 | 增高 | |
| RBC脆性 | 降低 | 正常 | 正常 | |
| 他 | RBC寿命 | 降低 | 正常 | 正常 |
| 网织RBC | 增多 | 正常 | 正常 |
第五节 某些肝病的生化机制
一、乙醇在肝内的代谢及乙醇性肝损伤
乙醇对于人体来说是一种异物,在肠道内由于细菌发酵所产生的乙醇仅以微量存在,因而对机体影响不大。我们所讲的乙醇代谢主要是指通过饮酒而摄入体内的外源性乙醇在体内的代谢。乙醇在胃及小肠上部迅速被吸收(胃30%,小肠上部70%),被摄取的乙醇90%-98%在肝内被代谢,剩下的2%-10%随尿及呼出气而被排泄。人的乙醇代谢率从其在血中浓度的消失率来看为100-200mg(kg·h)。因而在健康成人为每小时10g左右,一日代谢量约为240g 。
乙醇的饮用量与肝硬化的发病率之间有密切关系,饮酒量越高的国家,其肝硬化死亡率也越高。例如饮酒量每人在10升/年以下的瑞典、英国、丹麦等国家,肝硬化的死亡率在10/10万以下;而饮酒量最高的法国人均饮酒量27升/年左右,其肝硬化死亡率则为30/10万以上。由于长期饮酒容易形成乙醇性脂肪肝、乙醇性肝炎、肝硬化,甚至在孕妇还可造成胎儿性乙醇综合征,影响胎儿的发育成长。因此,了解乙醇在人体的正常代谢及乙醇对有机体的影响,在含酒精饮品日益泛滥的今天无疑具有重要的生理意义、临床意义和社会意义。
㈠乙醇在体内的代谢
乙醇在体内的代谢具有下述特征:①乙醇在作为药物(异物)的同时,每克能释放7Kcal(1cal=4.2J)的热能;②被摄取的乙醇的大部分(90%-98%)被代谢,由肾和肺排泄的仅占一小部位;③乙醇的大部分在肝脏内被氧化;④乙醇及其代谢产物不能在体内储存;⑤并不存在调节乙醇氧化速度的特殊的反馈机制。
乙醇的代谢途径包括乙醇脱氢酶(alcohol dehydrogenase,ADH)催化的乙醇氧化体系(即ADH乙醇氧化体系)与微粒体乙醇氧化体系(microsomal ethanol oxidizing system,MEOS),另外还有NADPH氧化酶-过氧化氢酶体系以及黄嘌呤氧化酶-过氧化氢酶体系(表10-14)。这些体系中以ADH乙醇氧化体系与微粒体乙醇氧化体系最为重要。
表10-14 肝脏中的乙醇代谢体系

⒈ADH乙醇氧化体系被摄取至肝内的乙醇大部分被肝细胞液中的乙醇脱氢酶催化脱氢而生成乙醛,乙醛进一步在乙醛脱氢酶催化下脱氢而生成乙酸,后者又形成乙酰辅酶A而进入三羧酶循环,最后生成二氧化碳和水,并释放能量生成ATP。
乙醛脱氢酶可分为两型,Ⅰ型为NAD依赖性的低Km酶,全分布在线粒体内;Ⅱ型为高Km酶,分布在线粒体及微粒体中。乙醇氧化产生的乙醛大部分在线粒体内被NAD依赖性的低Km酶的代谢系统所氧化。
⒉微粒体乙醇氧化体系(MEOS)乙醇的代谢与肝细胞微粒体的功能有很大关系,从形态学上观察到:长期饮酒的人及实验动物的肝细胞滑面内质网显著增加,表明乙醇也可能在微粒体被代谢。ADH乙醇氧化体系与微粒体乙醇氧化体系的组成和性质不同(表10-15)。
表10-15 乙醇脱氢酶(ADH)体系与微粒体乙醇氧化体系(MEOS)的比较
| ADH体系 | MEOS | |
| 细胞内的区域性分布 | 肝细胞胞液、线粒体内 | 肝细胞微粒体 |
| 最适pH | 10.8 | 7.2-7.4 |
| 辅酶 | NAD+ | NADPH |
| 米氏常数(Km) | 2mmol/L | 8.6mmol/L |
| 吡唑的抑制程度 | 抑制99% | 抑制3% |
| (4.4mM/kg体重) | ||
| 投与乙醇后引起的活性变化 | 不变 | 增加 |
| 与乙醇氧化相伴的能量变化 | 与氧化磷酸化相偶连产生氢,释能 | 需NADPH和O2,耗能 |
| 在乙醇代谢中所占的比率 | 75%-80% | 20%-25% |
1970年Lieber与De Carli用大鼠及人肝微粒体对MEOS的特征进行探讨,发现MEOS的反应有以下特征:①最适pH是在生理范围(pH6.8-7.4)内;②对底物(乙醇)的Km为8.2mmol/L;③需要NADPH和O2作为辅助因子,如用其它吡啶核苷酸作为电子供体时则不能反应;④对CO气体较敏感;⑤对过氧化氢酶、ADH两种酶的抑制剂较有耐受性;⑥长期饮酒对MEOS系统可明显产生诱导。
㈡乙醇代谢对机体的影响
乙醇代谢亢进所并发的各种代谢异常可能是由于NADH/NAD+比值的上升,因此先加以叙述。
⒈NADH/NAD+比值的上升乙醇在体内的代谢第一阶段是由乙醇生成乙醛,第二阶段是由乙醛氧化成乙酸。催化这两步反应的乙醇脱氢酶、乙醛脱氢酶的辅酶都是NAD+。因此,在乙醇氧化的过程中NAD+被还原为NADH。通过乙醇代谢产生的过剩的NADH制成NADH/NAD+比值的上升或NAD+/NADH比值的降低。例如给予2.4g/kg体重乙醇处理时,在给乙醇两小时后,NAD+/NADH比值从4.1降至1.5。NADH/NAD+比值的上升(或NAD+/NADH比值的下降)使得肝脏中乳酸的利用降低。另一方面,丙酮酸被增多的NADH还原成乳酸,容易导致乳酸性酸中毒。又可由于酸中毒引起肾排泄尿酸的障碍,导致发生高尿酸血症。
⒉乙醛对机体的影响在乙醇代谢对机体造成影响的因素中,不能忽视乙醇在肝内代谢的中间产物-乙醛的作用。乙醛的化学性质活泼,具有强烈的药理作用。动物实验表明:摄取乙醇四周的大鼠对乙醛的氧化率明显低于对照组,这可能是由于乙醇引起线粒体的功能障碍所致。长期饮酒所引起的肝线粒体的功能不全可能是因为乙醛的毒性所致。乙醛可引起线粒体的功能障碍,使线粒体的呼吸功能、脂肪酸氧化能力受到损伤。在慢性饮酒者可能形成一种恶性循环:持续饮酒可引起肝内产生较多的乙醇,高浓度的乙醛引起肝细胞线粒体的损伤,于是线粒体的乙醛代谢率降低,导致肝内乙醛浓度进一步上升,线粒体功能进一步低下。
乙醛除了对脑内胺代谢、线粒体呼吸功能、心肌蛋白质合成能力等有抑制作用外,还具有下述药理作用或毒性作用:①乙醛具有使内源性儿茶酚胺释放的刺激交感神经样作用,这可能是引起乙醇性心肌病的一个原因;②乙醛可能是四乙秋兰姆化二硫(戒酒硫,disulfiram)引起的中毒作用的原因;③乙醛与儿茶酚胺缩合成了与吗啡生物碱的前身物质结构非常相似的四氢异喹啉,这一物质是酒瘾发病的原因;④乙醛使5-羟色胺代谢发生障碍,结果产生具有幻觉作用的四氢-β-咔啉,可引起酒后的种种精神障碍;⑤乙醛是乙醇引起的戒断症状发病的一个原因;⑥乙醛对肝和脑的辅酶A活性具有相同抑制作用;⑦乙醛是造成慢性饮酒者维生素B6缺乏症的重要成因;⑧乙醛能抑制脑内Na+、K+-ATP酶。L-抗坏血酸、L-半胱氨酸等可防止乙醛对ATP酶的抑制作用。
⒊乙醇对血糖、氨基酸代谢、水电解质平衡、维生素D代谢及药物代谢的影响饮酒后血糖有降低倾向,健康人在较长时间(48-72小时)饥饿状态下给予乙醇能发生显著的低血糖。这可能是由于NADH的增高导致丙酮酸氧化性-脱羧、脂肪酸氧化、三羧酸循环等过程发生障碍,饥饿状态下摄取乙醇时糖异生过程亦发生障碍,加上摄取乙醇时进食不足,造成肝糖原储备降低,这皆是导致乙醇性低血糖的原因。
喂乙醇动物有负氮平衡倾向,乙醇性肝损伤时有蛋白质代谢障碍,慢性投予乙醇,动物血中γ-谷氨酰转肽酶活性增高。嗜酒者血清谷氨酸脱氢酶(线粒体酶)活性显著上升,表明乙醇性肝损伤时,肝小叶中心部肝细胞线粒体发生损伤,因为谷氨酸脱氢酶主要分布于肝小叶中心部位。
饮酒后利尿作用增强,伴脱水症状,引起口渴。乙醇利尿现象是垂体抗利尿激素分泌的抑制所致。血清中各种电解质因脱水而浓缩,饮酒后还引起血液pH降低、重碳酸盐减少,有酸中毒倾向,这主要是由于乙醇代谢时引起的乳酸、乙酸、酮体等增加所致。
乙醇性肝损伤(如乙醇性脂肪肝、乙醇性肝炎、乙醇性肝硬化)时均可观察到血清中25-OH-维生素D的减低,其降低的原因主要是维生素摄取不足、乙醇作用于肠道引起的吸收障碍、肝中25-羟化作用的降低及分泌的降低等。
当给急性乙醇中毒患者镇静剂和安眠药时可见到敏感的反应,有时会产生意想不到的重度中毒症状。另一方面慢性饮酒者可对镇静剂呈抗药性,麻醉剂的效果亦较难出现。在肝内代谢、对肝有毒性的CCl4,如果给予慢性摄取乙醇的动物,则会显示更强的毒性。解热镇痛剂乙酰氨基酚在对照组不发生肝损坏的剂量,对慢性乙醇投入组则可导致肝细胞坏死。乙醇与药物同在微粒体中被代谢,当乙醇与药物同时进入人体时,即可能发生对于其共同氧化系统细胞色素P450的底物的竞争现象。另外大量乙醇可引起药物代谢酶活性的抑制,这样,乙醇与药物两者的代谢是相互抑制的。于是会引起用药时发生意外的严重的中毒症状。因此,大量应用镇静剂、安眠药时必须注意患者有否饮酒嗜好。
(三)乙醇性肝损伤与胎儿性乙醇综合征
本段着重叙述几种重要的乙醇性肝损伤:乙醇性脂肪肝、乙醇性肝炎、乙醇性肝硬化以及妊娠时由于孕妇饮酒而造成胎儿异常的胎儿性乙醇综合征(fetal alcohol syndrome,FAS)。
⒈乙醇性脂肪肝过量摄入乙醇往往会引起脂肪肝的变化。乙醇中毒者脂肪肝的发生率可达70%-80%。此种脂肪肝主要是肝内中性脂肪的增加造成的。中性脂肪的来源包括经口摄取的脂肪、外周组织中的脂肪和肝内所合成的脂肪。
乙醇性脂肪肝形成机制包括:①急性乙醇中毒时脂肪肝动员的增加,一次大量摄入乙醇,通过儿茶酚胺的作用引起储存脂肪的动员,同时伴有高脂血症的发生;②慢性摄取乙醇时乙醇代谢亢进,NADH/NAD+比值上升,使磷酸二羟丙酮向α-磷酸甘油的转化增加,有利于肝内甘油三酯的大量合成;③由于NADH的增加,NAD+的减少,导致三羧酸循环及脂肪酸的氧化被抑制,肝中脂肪酸的氧化降低;④大量摄取乙醇时脂蛋白的合成及分泌减少。
乙醇性脂肪肝是良性的可逆性的病态,它可因戒酒而消退,可以认为乙醇性脂肪肝是由乙醇代谢亢进引起的代谢紊乱造成的后果。
⒉乙醇性肝炎乙醇性肝炎在病理组织学上以肝细胞坏死为主要变化,伴有透明小体形成。关于此种肝细胞坏死的机制可能如下述:①乙醇性肝损伤时的蛋白贮留和蛋白的分泌障碍:乙醇性肝损伤时不仅肝内脂肪量增加,且肝细胞液中有蛋白(白蛋白、运铁蛋白等)贮留;②线粒体与内质网的损伤:乙醇及乙醛均作用于线粒体,造成线粒体损伤,后者与肝细胞坏死有关,长期摄入乙醇后MEOS这一代谢体系由平时占总代谢量的1/4升至1/2,所以内质网内乙醛的产生增加,也造成内质网的损伤;③乙醇在微粒体氧化时,氧自由基使脂质过氧化,导致肝中还原型谷胱甘肽的减少和过氧化脂质的增加;④乙醇引起的代谢亢进状态造成耗氧量的增加。⑤乙醇性肝损伤时,会出现IgA增多、白细胞粘着能力降低等免疫功能异常。
⒊胎儿性乙醇综合征胎儿性乙醇综合征是由于孕妇饮酒造成的胎儿异常,这是一种包含智能障碍的中枢神经系统的功能障碍,由出生前开始的发育障碍以及特有容貌和畸形为特征的综合征。FAS的动物模型表明其胎儿体重、大脑重量均较对照组为低,C-亮氨酸向脑核蛋白的掺入减少,脑内总RNA及tRNA减少。这表明FAS动物脑内蛋白质合成的抑制可解释人类FAS中智能发育延迟的现象。长期大量饮酒不仅危害人类的健康,还影响下一代的发育成长。
二、肝硬化的生化
肝硬化与病毒性肝炎、慢性酒精中毒、中毒性肝损伤有密切关系,在肝硬化的基础上有发生肝癌可能性。
肝硬化时可发生糖代谢异常,如肝糖原的减少,线粒体代谢障碍,使得乳酸、丙酮酸及α-酮戊二酸增多。脂类代谢中胆固醇酯的合成障碍,肝硬化晚期胆固醇的合成亦发生障碍。在蛋白质代谢方面,白蛋白、纤维蛋白原等的合成减少,氨基酸代谢及尿素合成异常,导致尿中出现氨基酸和血氨升高。
此外,肝硬化时肝的生物转化及对激素灭活的功能亦发生障碍。在肝硬化形成过程中,纤维组织形成是重要的形态变化。在缺氧和炎症刺激下,胶原纤维的合成增强。当发生肝硬化时,肝内的酸性粘多糖增加,特别是含有半乳糖胺的硫酸软膏素B显著增加,并与胶原纤维化有关,肝纤维化时增加的胶原以Ⅰ型及Ⅱ型胶原为主。
近十年来关于肝纤维化的研究有较大进展,发现许多与肝纤维化有关的因子,就细胞成分而言,枯否细胞(Kupffer cell,KC)、储脂细胞(fat-storing cell,FSC)在肝纤维化形成过程中都起着非常重要的作用,成纤维细胞与肝门静脉区的结缔组织形成有关。此外,胶原(特别是Ⅰ型、Ⅲ型及Ⅳ型)、弹性蛋白以及细胞外基质(其中所含的结合蛋白质及粘多糖)和某些相关的酶(如金属蛋白酶、胶原酶、透明质酸酶),还有一些肝纤维化的活化因子(如白细胞介素Ⅰ、肿瘤坏死因子、转化生长因子-βTGF-β、干扰素、前列腺素2等)等都与肝纤维化有关。
肝纤维化时枯否细胞可分泌多种细胞因子及胶原酶等生物活性物质,并由此对肝细胞外基质的合成与降解起调控作用,从而在肝纤维化的发生发展中起着重要作用。曾认为储脂细胞的活化是肝纤维化的关键,储脂细胞产生胶原,细胞外基质中的糖蛋白-板层素(LN)及纤维连接蛋白(FN)等的能力使得它在肝损伤后组织修复及纤维化的发生中起重要作用。探讨这几种细胞对细胞外基质形成的影响,阐明胶原合成的调节机制,有助于阐明肝纤维化、肝硬化的生化机制。
三、肝昏迷的生化机制
肝昏迷又称肝性脑病,是由严重的肝病所致的中枢神经系统功能紊乱,出现一系列精神症状直至进入昏迷。肝昏迷是肝功能不全的重危合并症。引起肝昏迷的代谢因素极为复杂。表10-16列举肝昏迷时血液成分的变化,可以看出肝昏迷时的代谢异常是多方面的。
目前认为氨基酸的代谢障碍可能是肝昏迷发生的生物化学基础。下面从氨中毒、假神经递质及氨基酸不平衡三方面来叙述肝昏迷的生物化学机制。
表10-16 肝昏迷时血液成分变化所反映的代谢异常
| 代谢指标 | 变化 |
| ⒈ 含氮代谢物 | |
| ⑴血氨 | 增高 |
| ⑵血液酚,吲哚 | 增高 |
| ⑶血浆氨基酸 | |
| 芳香氨基酸 | 增加 |
| 支链氨基酸 | 减少 |
| ⑷血清胺类 | 增加 |
| ⑸核苷类 | 减少 |
| ⑹未结合胆红素 | 增加 |
| ⑺粪卟啉 | 增加 |
| ⒉糖代谢 | |
| ⑴血糖 | 减少 |
| ⑵丙酮酸,乳酸 | 增加 |
| ⑶α-酮戊二酸 | 增加 |
| ⒊脂代谢 | |
| ⑴血清游离脂肪酸 | 增加 |
| ⑵血清短链脂肪酸 | 增加 |
| ⒋电解质及酸碱平衡 | |
| ⑴血清Na+ | 减低 |
| ⑵血清K+ | 减低 |
| ⑶血清pH | 升高或减低 |
㈠氨中毒与肝昏迷
40多年前即发现肝昏迷与氨代谢有密切关系,其依据是:①多数肝昏迷病人血氨增高,病情好转时血氨降低;②慢性肝病病人摄入高蛋白膳食或含铵药物,常诱发肝昏迷;③肝昏迷病人脑电图变化与血氨平行;④对门腔静脉吻合犬给以高蛋白可引起血氨升高及昏迷症状;⑤给猴注射醋酸铵,其血氨达200-400μg/dl时,显示与人的肝昏迷相似的症状;⑥上述病人及实验动物经过降血氨疗法病情常可好转。上述事实都表明氨中毒与肝昏迷有着密切关系。
关于氨中毒的机制,据认为是由于肝功能不全情况下,血氨的来源增多或去路减少,引起血氨升高,脑组织对氨毒性极为敏感,因而出现脑功能障碍而导致昏迷。正常血氨来自肠菌产氨(每日约4g)、肾泌氨、肌肉组织产氨等,而解除氨毒性的机制主要靠肝内的尿素合成。由于肝严重病变导致肝功能不全,清除氨的能力大为降低,加之门腔静脉短路,使由肠管回血液的氨不经肝解毒而直接进入体循环,造成高氨血症与肝昏迷。饮食蛋白过多、消化道出血、摄入铵盐、放腹水以及应用利尿剂等均可引起血氨的升高或氨毒性增加,从而能诱发肝昏迷。
氨对脑组织的毒性作用在于氨主要是干扰了脑的能量代谢,使高能磷酸化合物(ATP等)浓度降低。氨对脑细胞代谢的干扰有下述几方面:①氨能抑制丙酮酸脱氢酶的活性,影响乙酰辅酶A的生成,既干扰了三羧酸循环的起始步骤,又影响了神经递质乙酰胆碱的生成;②氨中毒时,脑内以形成谷氨酰胺的方式解毒,从而消耗了较多的NADH(α-酮戊二酸经还原性氨基化而生成谷氨酸),影响线粒体氧化磷酸化的正常进行,妨碍ATP生成;③大量氨与α-酮戊二酸结合生成谷氨酸,可使三羧酸循环中的α-酮戊二酸耗竭,妨碍了供能物质在脑细胞中能量的释放与转换。由于α-酮戊二酸及草酰乙酸难于通过血脑屏障,脑内转氨酶活性低,难于使α-酮戊二酸等得到补充,因此氨中毒使脑细胞三羧酸循环发生障碍,ATP的生成减少;④氨和谷氨酸合成谷氨酰胺时增加ATP消耗;⑤氨能激活神经细胞膜上的Na+、K+-ATP酶,并和K+有竞争作用,影响离子分布和神经传导的正常进行。
应当指出,氨中毒现象并不能解释所有肝昏迷的发生,有些病例血氨并不高,降血氨疗法亦不一定有效,尚须探讨其他机制。
㈡假神经递质学说
在肠管内,一部分氨基酸经肠菌的氨基酸脱羧酶作用而形成胺类,如苯丙氨酸及酪氨酸脱羧形成苯乙胺及酪胺,正常情况下可被肝内单胺氧化酶分解而清除。肝功能不全时,由于肝内单胺氧化酶活性降低或门体侧支循环的形成,于是芳香胺类直接经体循环入脑,经脑内非特异羟化酶作用,于是苯乙胺羟化而生成苯乙醇胺,酪胺经羟化而生成鱆胺(β-羟酪胺)由于苯乙醇胺及鱆胺与儿茶酚胺递质(多巴胺、去甲肾上腺素)结构相似,又不能正常地传递冲动,故称假神经递质。
假神经递质被释放后引起神经系统某些部位(如脑干网状结构上行激动系统)功能发生障碍,使大脑发生深度抑制而昏迷。黑质、纹状体通路中的多巴胺被假递质取代后,使乙酰胆碱的作用占优势,因而出现扑翼样振颤,当然,假递质学说也不能解释全部肝昏迷的发生机制(图10-3)。
㈢氨基酸不平衡与肝昏迷
近20年来关于肝昏迷时氨基酸代谢异常的资料表明:在严重肝功能损伤和有门腔静脉短路的条件下,由于种种原因引起体内氨基酸代谢异常。最突出的表现是血中支链氨基酸(缬氨酸、亮氨酸、异亮氨酸)浓度明显降低,芳香族氨基酸(苯丙氨酸、酪氨酸、色氨酸)明显增高,从而引起脑功能障碍。
由于芳香族氨基酸主要在肝内分解,当肝功能不全时,芳香族氨基酸在肝内代谢发生障碍,因而在血中的浓度增高;支链氨基酸主要在肌肉组织中代谢,由于肝功能不全时胰岛素的灭活发生障碍,在高水平的胰岛素的作用下,支链氨基酸大量进入肌肉组织被分解,因此血浆中的支链氨基酸的浓度降低。
芳香族氨基酸进入脑组织,可引起假神经递质的产生增多,色氨酸可使5-羟色胺的生成增
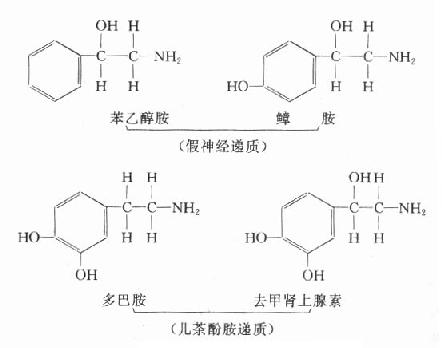
图10-3 假递质学说
多,后者为抑制性神经递质,使中枢进入抑制状态。而支链氨基酸的降低使得血脑屏障上芳香族氨基酸占了优势,缺少了竞争载体的对象,于是大量进入脑内,上述氨基酸代谢的不平衡引起严重的后果。上述三种学说可能解释不同情况下肝昏迷发病机制的主要方面。
肝昏迷(肝功能不全)的检验所见可有:①血清胆红素可呈显著的高值(能达40mg/dl),说明有胆汁排泄障碍;②血清白蛋白减低,表明其蛋白合成能力低;③低胆固醇血症,亦是合成能力降低所致;④AST及ALP由高值转为低值,见于大量肝细胞坏死的情况;⑤BUN呈低值,表示肝合成尿素功能低下;⑥血糖降低,由于肝糖原储备减少;⑦凝血酶原时间延长是由于肝中凝血酶原、Ⅶ因子、Ⅸ因子、Ⅹ因子的合成减低;⑧血浆纤维蛋白原呈低值,在肝内合成减低;⑨血氨增高是尿素合成降低所致;⑩血液pH增高,PCO2降低(呼吸性碱中毒)是因脑水肿引起的换气过度所致。
四、胆石症的生物化学
胆石症的主要组成成分有胆固醇、胆红素、钙及其他无机元素、胆汁酸、结石基质(硫酸化糖蛋白及糖蛋白)等。按胆石的主要成分及形成机制的不同,可将胆石分为胆固醇系结石、胆红素结石及其他胆石三大类,仅将前两大类胆石的形成机制略作介绍。
㈠胆固醇系结石的形成机制
胆固醇系胆石病人的胆汁可有下述异常:
⒈胆汁中胆固醇含量增高含有过饱和胆固醇的胆汁是胆固醇沉淀的先决条件。胆汁中胆固醇的需要量和胆汁酸盐及卵磷脂的含量保持一定的比例。如果胆固醇含量过高或胆汁酸盐及卵磷脂的含量降低,破坏了它们之间的正常比例,就形成了“致石性胆汁”。有报告指出:胆固醇系胆石病人肝内的HMGCoA还原酶(胆固醇合成的限速酶)活性增加,而7α-羟化酶(胆汁酸合成的限速酶)活性降低,即胆固醇的合成增加,胆固醇向胆汁酸的转化减少,这是形成胆固醇过饱和胆汁的代谢原因。
⒉胆汁中胆汁酸盐的减少胆固醇系胆石病人的总胆汁酸代谢池甚小,仅为正常人的一半。这一方面是由于其胆固醇代谢的失调,另一方面可能是由于胆囊对胆汁酸的重吸收增加及肠肝循环障碍所致。此外,必需脂肪酸的缺乏或代谢障碍可导致胆汁酸生成障碍。
⒊胆汁中胆汁酸组成的改变正常人胆汁中胆酸、鹅脱氧胆酸及脱氧胆酸三者的比例为1.3:1.0:0.6,而胆固醇结石病人的胆汁中鹅脱氧胆酸的比例则明显减低。
⒋胆汁中的磷脂降低胆固醇结石病人胆汁中的磷脂只为正常胆汁中的1/3。磷脂/胆固醇比值在正常胆汁为6.6,而在胆固醇结石病人的胆汁则为2.3。胆汁中的磷脂(90%是磷脂酰胆碱)是与胆汁酸盐、胆固醇共同形成混合微团的重要成分,它也与增加胆固醇在胆汁中的溶解有关。
⒌δ电位的降低 δ电位是混合微团吸附层与扩散层正、负离子分布均匀处两点间的电位差,电位差越大,则微团带电荷越多,稳定性越大。正常胆汁中甘氨胆汁酸与牛磺胆汁酸的比例约为3:1,胆固醇结石病人胆汁中牛磺胆汁酸减少,这个比例可达15:1,因而对胆汁酸的δ电位有影响。增加牛磺胆汁酸则可因其负电荷较强的-CH2SO3根而增加胆汁中的δ电位。
在肝生成异常胆汁后,在胆囊中形成肉眼可见的胆石,并逐步长大。在结石形成过程中,作为结石基质的糖蛋白类可能起着把胆固醇结晶及颗粒粘连在一起的网架作用。
㈡胆红素系胆石的形成机制
肝分泌的胆汁中的胆红素是结合型的葡萄糖醛酸胆红素。胆汁中由组织产生的β-葡萄糖苷酸酶活性降低(该酶最适pH5左右,胆汁pH为6.1-8.6),而且受该酶的抑制物葡萄糖二酸-1,4-内酯的抑制,所以正常胆汁中的葡萄糖醛酸胆红素不易被水解而处于良好的溶解状态。胆红素钙结石病人的胆汁中可出现下述异常:
⒈由于蛔虫钻进胆道,带入大肠杆菌,造成胆道感染,胆汁中出现细菌性β-葡萄糖苷酸酶(最适pH6.8-7.2,与胆汁pH一致)的活性高,超过了胆汁中该酶的抑制物β-葡萄糖二酸-1,4-内酯所能抑制的能力,因而可使葡萄糖醛酸胆红素大量被水解,生成游离胆红素而易于沉淀。
⒉胆红素系结石病人胆汁中葡萄糖二酸-1,4-内酯的含量降低在正常对照组胆汁中该物质的含量为200μg/ml,而胆红素钙结合病人胆汁中则仅含40μg/ml,这也是其胆红素易于沉淀的原因,因为β-葡萄糖苷酸酶抑制物的降低更有利于细菌性β-葡萄糖苷酸酶发挥作用。
⒊δ电位降低这是胆红素钙融合集结的条件。钙、钠、钾、镁等无机离子及胆道感染时出现的高分子有机物质,均可使胆盐微团的δ电位降低。
在胆红素结石形成过程中蛔虫残体、蛔虫卵及其他异物均构成结石的核心,细菌性β-葡萄糖苷酸酶催化葡萄糖醛酸胆红素的水解,游离胆红素与Ca+形成胆红素钙,胆红素钙又在前述无机离子的作用下,再加上结石基质(主要是硫酸化糖蛋白)的网架作用,而集结成胆红素钙结石。
五、肝癌的生化机制
㈠某些致癌物质与肝癌发生的关系
⒈乙酰氨基芴(AAF)AAF在肝内经生物转化后被活化成硫酸AAF,与蛋白质分子中的蛋氨酸残基及核酸分子中的鸟嘌呤碱基结合,引起生物高分子的结构与功能的异常。它与DNA结合,使细胞内调控蛋白的合成不足,细菌失去控制,但DNA复制不受影响,从而引起癌变。
⒉黄曲霉毒素 黄曲霉毒素B1被摄入体内后,在肝细菌微粒体混合功能氧化酶催化下转化成环氧化黄曲霉毒素B1,它可与DNA、RNA结合而致癌。
⒊二甲基氨基偶氨苯(DAB,奶油黄)可引起大鼠实验性肝癌。DAB在肝细胞混合功能氧化酶作用下N-脱羟化,再在硫酸转移酶作用下形成N-SO3-O-甲基氨基偶氮苯。后者再与核酸的鸟嘌呤碱基结合而引起癌变。
㈡肝癌时的代谢变化
⒈蛋白质及氨基酸代谢的变化癌组织中蛋白质合成旺盛,宿主其他组织蛋白质的分解增强。肝癌时,肝癌组织中与氨基酸分解代谢有关的酶(如色氨酸吡咯酶、酪氨酸转氨酶、苏氨酸脱水酶、苯丙氨酸转氨酸及组氨酸酶等)活性显著降低,表明癌组织中氨基酸分解代谢减弱,可使氨基酸重新用于蛋白质的合成,此现象可能与癌组织的生长有关。另外,与氨基酸转运有关的γ-谷氨酰转肽酶活性在肝癌组织中显著增高。
在肝癌组织中与尿素合成有关的肝组织特异酶-鸟氨酸氨甲酰基转移酶(OCT)、氨甲酰磷酸合成酶I(CPSI)、精氨酸酶等的活性降低;与核酸合成有关的天冬氨酸氨甲酰基转移酶(ATC),与细胞增殖及多巴胺合成有关的鸟氨酸脱羧酶(ODC)的活性则增高。在肝癌组织中支链氨基酸转氨酶的活性亦增高,这可能与供能有关。肝癌组织中白蛋白的合成降低。上述事实表明;在肝癌细胞中与增殖有关的酶活性增高,与肝细胞特异性功能有关的酶活性降低。肝癌组织还大量合成甲胎蛋白,呈现肿瘤组织的反分化特征。
⒉肝癌时糖代谢的变化肝癌组织中与糖代谢有关的酶活性呈下述变化:①糖异生关键酶(磷酸烯醇式丙酮酸羧激酶、果糖-1,6-二磷酸酶、葡萄糖-6-磷酸酶等)活性降低,癌瘤恶性程度越高,这些酶活性越低;②糖酵解酶系(已糖激酶、磷酸果糖激酶、丙酮酸激酶等)活性增高,癌瘤恶性程度越高,这些酶的活性越高;③同工酶谱的变化呈现胚胎化,能被调节的高Km型同工酶(已糖激酶Ⅰ-Ⅲ型等)的活性上升。肝癌组织中同工酶谱的变化可使ATP失去对糖酵解的调节作用,这可能是癌细胞失去巴斯德效应的原因之一。肝癌时糖代谢变化的主要特点是:糖的有氧氧化降低(正常肝有氧氧化占99%,酵解占1%,肝癌时酵解可占50%),糖酵解增加,糖异生减少,磷酸戊糖途径的代谢增强(表10-17)。
⒊肝癌时的脂类代谢在肝癌细胞中可发现磷脂的减少和甘油三酯的增加。在脂质组成中非脂肪酸增加,构成脂质的脂肪酸中出现C20:4的减少和C18:1的增加。在人的肝癌组织中能检查出非生理性的不饱和脂肪酸,其中最多见的是C20:39。此种不饱和脂肪酸只在血中AFP阳性的肝癌者中出现。
⒋其他据报道:大鼠原发性肝癌、移植性肝癌、癌前期等的肝细胞和正常肝细胞的Na+、K+-ATP酶活性的测定结果是:原发性肝癌和移植性肝癌细胞中该酶的活性都显著高于正常肝细胞,癌前期肝细胞的Na+、K+-ATP酶活性则高于正常肝细胞而低于癌细胞,提示了肝细胞癌变过程中钠泵活性的变化规律。肝癌细胞cAMP合成能力大为降低,肝癌组织中腺苷酸环化酶活性明显低于正常肝组织。
表10-17 正常肝与肝癌组织糖代谢有关酶活性的比较
| 比活性 | |||
| 代谢途径 | 酶 | ||
| 正 常 肝 | 迅速生长的肝癌组织 | ||
| 已糖激酶 | 100 | 500 | |
| 糖酵解 | 磷酸果糖激酶 | 100 | 229 |
| 丙酮酸激酶 | 100 | 449 | |
| 葡萄糖-6-磷酸酶 | 100 | <1 | |
| 糖异生 | 果糖-1,6-二磷酸酶 | 100 | <1 |
| 磷酸烯醇式丙酮酸羧激酶 | 100 | <1 | |
| 丙酮酸羧化酶 | 100 | <1 | |
| 磷酸戊糖通路 | 6-磷酸葡萄糖脱氢酶 | 100 | 751 |
| 糖酵解/糖异生 | 已糖激酶/葡萄糖-6-磷酸酶 | 100 | 8800 |
| 磷酸果糖激酶/果糖-1,6-二磷酸酶 | 100 | 6463 | |
㈢肝细胞癌变原理
关于细胞癌变的原理除了前述化学致癌物对于细胞的DNA的损伤之外,近10余年来随着癌基因与抑癌基因(抗癌基因)研究的进展,人们更加注重肝癌的分子生物学的研究,细胞内本来存在的原癌基因在物理、化学、生物性的致癌因素作用下被激活,通过点突变、基因易位、基因扩增等机制激活后而呈现过度表达,导致更多的癌基因产物(癌蛋白)的产生,造成细胞内基因表达调控的失常,最终导致细胞癌变。在这一癌变过程中不仅有原癌基因的变化,同时伴有抑癌基因的缺失或失活,并且癌变还是多阶段一步步地发生,这即多阶段癌变学说。所谓多阶段癌变学说即是说机体的癌变过程是长期多阶段地发生的,正常细胞在癌基因,抑癌基因等的异常的积累过程中,逐渐地离开了正常的细胞增殖控制机制的轨道,分阶段地向癌细胞转化,且恶性程度逐步增加。
近年来,关于肝炎(乙型及丙型)与肝癌的关系日益受到重视。乙型肝炎病毒(HBV)与肝癌的发生关系已基本阐明,HBV基因组中的X基因与肝癌有密切关系,用转基因小鼠实验证明此X基因可诱发肝癌的发生。X基因编码的X蛋白使肝细胞的DNA合成提高到发癌的准备状态,在多阶段癌变过程中起到促进作用。X蛋白具有促进DNA合成的作用,在肝细胞癌变过程中,X基因的高水平的持续性的表达是必要的,由于癌变过程中往往几种癌基因同时表达,因此人们都强调肝细胞癌的发生可能是多种癌基因协同作用的结果。如N-ras表达异常可导致跨膜信号传递的改变,进而启动了ets-2、C-myc、P53等核内表达产物异常,使细胞分裂增加,细胞处于活跃增殖状态。IGF-Ⅱ和fms表达增强可通过“自分泌”作用使细胞处于不断生长状态,多种因素综合作用最终导致细胞增殖失控而形成肝细胞癌,关于肝细胞癌的多阶段癌变学说如图10-4所示。
丙型肝炎病毒与肝癌的发生亦有密切的关系。丙型肝炎感染后20年左右导致肝硬化, 30年左右发展成肝癌,在日本肝癌有15%与乙型肝炎有关,有80%与丙型肝炎有关。本室用PCR技术检测肝细胞癌中乙型、丙型肝炎病毒核酸的结果,表明HBV-DNA阳性率为69.5%,HCV-RNA阳性率为30.4%,在我国丙型肝炎与肝细胞癌的关系亦正在日益受到重视。
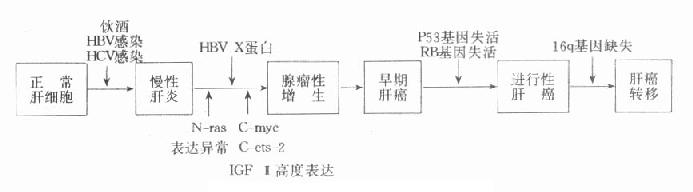
图10-4 肝细胞癌的多阶段癌变学说
第六节 肝细胞损伤时的肝功能试验
一、肝细胞损伤时的肝功能检验指标(表10-18)
表10-18肝细胞损伤时蛋白质、糖、脂类代谢变化的检测指标
| 类别 | 检 测 指 标 | 临 床 意 义 |
| 蛋白质代谢 | 血清总蛋白测定 | 严重肝炎及肝硬化时减少 |
| A/G比值测定 | 肝硬化时降低 | |
| 血清蛋白电泳测定 | 急性肝炎:白蛋白、α2-球蛋白降低 | |
| 肝硬化:白蛋白及α2-球蛋白降低,γ-球蛋白增加 | ||
| 免疫球蛋白测定 | 肝硬化时IgG、IgA、IgM增加 | |
| 脑磷脂胆固醇絮状试验(CFT) | 灵敏地反映急性肝损伤 | |
| 麝香草酚浊度试验(TTT) | 肝炎、肝硬化、肝癌时增高 | |
| 甲胎蛋白(AFP)测定 | 原发性肝细胞癌时显著增高 | |
| 癌胚抗原(CEA)测定 | 转移性肝癌时阳性率高 | |
| 血浆纤维蛋白原测定 | 反映功能性肝细胞的多少 | |
| 血及尿中氨基酸测定 | 严重肝细胞坏死时增高 | |
| 血中尿素测定 | 严重肝功能不全时降低 | |
| 血氨测定 | 严重肝损伤、肝硬化、肝昏迷时显著增高 | |
| 尿米伦(Millon)反应 | 重症肝炎、肝硬化时阳性(酪氨酸增加) | |
| 糖代谢 | 血糖测定 | 肝功能不全时血糖降低 |
| 静脉葡萄糖耐量试验 | 肝病时多呈异常耐糖曲线 | |
| 半乳糖耐量试验 | 肝细胞损伤时耐量降低 | |
| 血中丙酮酸的测定 | 肝昏迷时增加 | |
| 血中乳酸的测定 | 反映肝清除乳酸的能力 | |
| 脂类代谢 | 血清总胆固醇测定 | 阻塞性黄疸时增高,严重肝损伤时降低 |
| 血清胆固醇酯测定 | 肝细胞损伤时降低 | |
| 胆固醇酯/总胆固醇比测定 | 肝细胞损伤时降低 | |
| 血清磷脂测定 | 胆道阻塞时增高,肝实质损害时降低 | |
| 血清甘油三酯测定 | 脂肪肝及胆道阻塞时增高 | |
| α-脂蛋白测定 | 重症肝炎、阻塞性黄疸时减少 | |
| β-脂蛋白测定 | 慢性脂肪肝及肝炎时增高 | |
| 前β-脂蛋白测定 | 急性脂肪肝及肝炎时增高 |
续表
| 类别 | 检 测 指 标 | 临 床 意 义 |
| 高密度脂蛋白2(HDL2) | 胆管炎、Banti综合征时增高,实质性肝细胞损害时降低 | |
| 低密度脂蛋白(LDL) | 肝硬化时呈低值 | |
| 载脂蛋白AⅠ | 急性肝炎↓↓,阻塞性黄疸↓↓ | |
| 载脂蛋白AⅡ | 急性肝炎↓↓,阻塞性黄疸↓↓ | |
| 载脂蛋白B | 阻塞性黄疸↑ | |
| 载脂蛋白CⅡ | 原发性胆汁性肝硬化↑↑ | |
| 载脂蛋白CⅢ | 肝细胞癌↓↓,阻塞性黄疸↑ | |
| 载脂蛋白E | 原发性胆汁性肝硬化↑↑↑,肝炎↑↑ | |
| 载脂蛋白X | 肝外胆道阻塞、原发性胆汁性肝硬化↑ | |
| 血中胆汁酸(胆酸与鹅去氧胆酸)测定 | 肝炎、肝硬化、肝细胞癌时↑ | |
| 卵磷脂胆固醇脂酰基转移酶(LCAT) | 反映肝合成酶蛋白能力 |
二、急性肝细胞损伤的检验指标
能引起急性肝细胞损伤的因素有感染(如病毒性肝炎)、中毒(药物及化学毒物中毒)、乙醇等。反映肝细胞损伤的检验指标有:血清酶类的测定,血清铁的测定,F抗原等。
(一)血清酶活性的测定
常用来反映肝细胞损伤以及判断损伤程度的酶有丙氨酸转氨酶(ALT)、天冬氨酸转氨酶(AST)、乳酸脱氢酶(LDH)、谷氨酸脱氢酶(GLDH)、腺苷脱氨酶(ADA)、鸟嘌呤酶(GU)等。这些酶中常用的重要的酶仍为ALT和AST,它们能敏感地提示肝细胞的损伤及损伤的程度。反映急性肝细胞损伤时,以ALT为最敏感,而反映损伤程度时AST较为敏感。一般认为AST大部分存在于肝细胞线粒体中(ASTm),当肝细胞损伤较严重时ASTm释放入血液循环中,AST/ALT>1。有人主张当AST、ALT活性增高(达正常20倍)情况下,AST/ALT的比值不同类型的肝细胞损伤其比值不同。急性病毒性肝炎、药物性肝损伤时AST/ALT<1;重症肝炎、乙醇性肝炎时AST/ALT>1;正常人血清AST/ALT比值平均为1.15;急性病毒性肝炎早期或轻型肝炎时比值降至0.56,至恢复期比值逐渐上升;肝硬化代偿期AST/ALT比值为1.5-0.7,失代偿期为1.8-0.7;肝癌时比值明显上升,半数病例>3.0。
AST同工酶的测定有助于判断肝损伤病变程度。AST同工酶包括细胞质AST(ASTc)和线粒体AST(ASTm),当肝细胞通透性增加时表现为ASTc的增高;当肝细胞坏死或线粒体崩解时,血清中ASTm明显增高;慢性肝病时若发现有ASTm升高则提示有活动性肝细胞损伤。
乳酸脱氢酶及其同工酶的测定亦可作为肝细胞损伤的诊断指标,急性肝炎时LDH同工酶谱的特点是LDH5增高,LDH1、LDH2降低。
酒精性肝炎有γ-GT增高,认为酒精可造成肝细胞微粒体损伤,导致血清中γ-GT的活性增高。
(二)血清铁测定
急性肝炎时血清铁增高,一般认为是由于肝细胞坏死而导致铁从肝细胞中逸出,引起血清铁增高。
(三)F抗原
F抗原又称F蛋白,是一种主要存在于肝细胞质中的蛋白质。正常人血清中F抗原水平很低(10ng/ml),肝细胞损伤时血清F抗原浓度升高,是一项较灵敏的细胞损伤的指标。有报道F抗原在药物引起的肝细胞损伤时反映灵敏,药物损伤肝小叶中心的肝细胞,而这一区域F抗原浓度高。肝癌时F抗原亦升高。
三、慢性肝细胞损伤的检验指标
(一)慢性肝细胞损伤的酶学指标
γ-GT在反映慢性肝细胞损伤及其病变活动时较转氨酶敏感。γ-GT活性75%存在于肝细胞微粒体,当慢性肝病有活动性病变时,诱导微粒体酶合成增加。在急性肝炎恢复期ALT活性已正常,如发现γ-GT活性持续升高,即提示肝炎慢性化;慢性肝炎即使ALT正常,如γ-GT持续不降,在排除胆道疾病情况下,提示病变仍在活动;慢性持续性肝炎(CPH)γ-GT轻度增高;慢性活动性肝炎(CAH)γ-GT明显增高;肝细胞严重损伤,微粒体破坏时,γ-GT合成减少,故重症肝炎,晚期肝硬化时γ-GT反而降低。
此外,鸟氨酸氨甲酰基转移酶(OCT)、腺苷脱氨酶等的活性在慢性肝细胞损伤时可以增高,而卵磷酯胆固醇酰基转移酶(LCAT)、胆碱酯酶(ChE)活性则在慢性肝病时因酶合成减少而降低。
(二)慢性肝细胞损伤时血浆蛋白的变化
血浆白蛋白可反映肝脏合成功能,代表肝的储备功能。此外前白蛋白(PA)及抗凝血酶Ⅲ(AT-Ⅲ)亦能很好地反映肝脏的储备能力,藉以判断慢性肝细胞损伤的病变程度。γ-球蛋白增高的程度可评价慢性肝病的演变及预后,慢性持续性肝炎的γ-球蛋白正常或基本正常,慢性活动性肝炎及早期肝硬化时γ-球蛋白呈轻、中度升高,若γ-球蛋白增高达40%时提示预后不佳。γ-球蛋白增高的机制是枯否细胞功能减退,不能清除血循环中内源性或肠源性抗原物质,后者刺激B细胞产生大量抗体IgG,以致γ-球蛋白增高。ICG排泄试验测15分钟滞留率可用于筛选慢性职业中毒肝损伤、追踪慢性肝病的恢复和急性肝病的慢性化过程。
四、肝纤维化的生化诊断
肝纤维化是肝硬化前期的必经阶段,目前常规的肝功能试验无法诊断肝纤维化或早期肝硬化。因此肝纤维化的血清标志物在临床上有重要诊断价值。肝纤维化的实质是细胞外间质的结缔组织增生,其成分主要是胶原蛋白,还有各种糖蛋白和蛋白多糖等。
肝组织中肝实质细胞约占78%,肝窦壁细胞约占6%-7%,由上述细胞合成和分泌的细胞外基质(extracellular matrix)占肝的5%左右。在肝硬化时细胞外基质成分较正常肝增加2-20倍。细胞外基质成分中的胶原蛋白、有关的代谢酶、代谢产物等有的作为肝纤维化的标志物,用于肝纤维化的诊断,现将有临床应用价值的肝纤维化标志物列于表10-19。
表10-19 肝纤维化的诊断标志
| 诊断标志物 | 来源或性质 | 临床意义 |
| ㈠Ⅲ型胶原 | ||
| ⒈Ⅲ型前胶原氨基端肽(PⅢP) | 纤维增生之前即出现增高,炎症坏死时胶原降解亦增加,分子量45000,RIA法检测 | 与肝纤维化及肝脏炎症坏死有关 |
| ⒉Ⅲ型前胶原(PCⅢ) | 纤维型胶原,可由人胎皮中提取,用RIA法检测 | 与肝纤维化明显相关,慢活肝,肝硬化明显增高 |
| ㈡Ⅳ型胶原可测其7S片段或羧基端片段 | 为构成基底膜主要成分与板层素有高度亲和性,RIA法检测 | 肝纤维化肝硬化时显著增高 |
| ㈢Ⅰ型胶原(CⅠ)及Ⅰ型前胶原肽(PIP) | 可由健康人皮肤提取CⅠ,用RIA法检测,Ⅰ型为纤维型胶原属膜型胶原 | 反映肝纤维化程度 |
| ㈣Ⅳ型胶原(CⅥ) | 属膜型胶原 | 慢活肝及肝硬化时显著升高 |
| ㈤脯氨酰羟化酶(PH) | 为胶原合成关键酶,由亚单位构成四聚体,可测其亚单位(酶联法) | 主要反映肝纤维化的活动性 |
| ㈥脯氨酸肽酶(PLD) | 为二肽酶,水解含脯氨酸,羟脯氨酸的二肽,胶原富含脯氨酸及羟脯氨酸 | 与肝纤维化有关 |
| ㈦板层素(LN) | LN又称基膜粘连蛋白或层粘联蛋白,为基膜特有成分,肝纤维化时LN与Ⅳ型胶原结合形成内皮基底膜,形成肝硬化 | 慢性活动性肝炎和肝硬化时血中LN显著升高 |
| ㈧纤维连结素(FN) | 为一种糖蛋白,包括血浆FN及细胞FN,还可测FN受体(FNR) | FNR与肝纤维化程度相关 |
五、肝癌的生化诊断
原发性肝癌的诊断标志见表10-20。
表10-20 原发性肝癌的诊断标志
| 诊断标志 | 来源和性质 | 临床意义 |
| 甲胎蛋白(AFP) | 胎肝、卵黄囊、肝癌细胞,为癌胎儿性蛋白 | 肝细胞癌、卵黄囊肿瘤、肝硬化、慢肝时增高 |
| AFP异质体 | 因AFP糖链不同,对凝集素亲和力不同,而形成异质体,原发性肝癌AFP异质体上有较多岩藻糖与扁豆素(LCA)有较大亲和力 | 诊断原发性肝癌及鉴别原发性肝癌与胚胎性肿瘤,还可检测小肝癌及AFP阴性的肝细胞癌 |
| 异常凝血酶原(DCP、γ-脱羧凝血酶原前体) | 肝脏合成的凝血酶原前体,因肝癌时丧失羧化酶基因表达而使上述前体不能羧化而形成异常凝血酶原 | 诊断原发性肝癌、转移性肝癌,与AFP联合检测可提高肝癌检出率 |
| γ-谷氨酰转肽酶同工酶(γ-GT) | 在γ-GT中有三种同工酶(γ-GTⅠ,Ⅱ,Ⅲ)与肿瘤有关,Ⅰ即γ-GTⅡ,肝癌时该酶基因脱抑制,导致富含唾液酸的γ-GT产生,电泳时即γ-GTⅡ增加 | 诊断肝癌 |
| 岩藻糖苷酶(AFU) | AFU属溶酶体酸性水解酶类,参与含岩藻糖苷的糖蛋白、糖脂的降解、代谢,肿瘤组织中岩藻糖转换率增加,AFU活性增高 | 原发性肝癌诊断标志,与AFP联合检测可提高肝癌检出率 |
续表
| 诊断标志 | 来源和性质 | 临床意义 |
| α-抗胰蛋白酶(AAT)及其异质体 | AAT存在于肝细胞中,其异质体中LCA亲合性AAT在肝癌时比例明显升高 | AAT-LCA结合型与AFP联合检测有助于原发性肝癌诊断 |
| 5-核苷磷酸二酯酶同工酶Ⅴ(5-NPD-Ⅴ) | 5-NPD-Ⅴ在肝癌时出现,为肝癌特异区带 | 可能有助于肝癌诊断 |
应当指出:上述肝癌诊断标志虽具有一定的特异性、敏感性,但有的还难以鉴别原发性肝癌与转移性肝癌。因此,进行几种肝癌诊断标志物的联合检测,可以互补而提高原发性肝癌诊断的阳性率,减少假阳性及假阴性的发生,提高诊断的准确性。
第十一章 肾功能不全的实验室生物化学诊断
机体新陈代谢过程中产生的代谢终产物,多余的水、无机物以及药物、异物,经血液循环,主要通过肾脏,以尿的形式排出体外。由于从肾脏排出的物质种类较多,并可随着机体的不同情况而改变尿量和尿中的物质排出量,从而维持机体内环境的相对稳定,因而可以说肾脏是机体内最重要的排泄器官。
第一节 概述
肾为实质器官。外层为皮质,内层为髓质。髓质由肾锥体组成,开口于肾小盏,肾小盏合成肾盂,肾盂向下逐渐缩小续于输尿管。
肾单位(nephron)是肾脏结构和功能的基本单位,它与集合管共同完成泌尿功能。人的两侧约有170万-240万个肾单位,每个肾单位由肾小体和肾小管组成。肾小管中的远曲小管和集合管相连,集合管不包括在肾单位内,但在尿液浓缩与稀释的过程中起着重要作用,可把集合管视为肾小管的终末部分。
肾单位可分为两类:一类为皮质肾单位,特点是肾小球体积小,入球小动脉口径比出球小动脉的粗,毛细血管内压较高,与尿液生成有关。另一类为近髓肾单位,体积较大,髓袢较长,入球小动脉与出球小动脉口径无明显差异。出球小动脉离开肾小球后分成两种小血管:一种形成网状小血管,包绕在邻近的近曲小管和远曲小管周围;另一种形成细长的直小血管,与近髓肾单位的长髓袢相伴行。直小血管之间有吻合支,其中血流可以互相流通,近髓肾单位在尿液的浓缩与稀释的功能中起着重要的作用。
肾小球旁器(juxtaglomerularapparatus)是指位于肾小球入球小动脉与出球小动脉间的一群细胞,包括肾小球旁细胞、间质细胞和远曲小管致密斑。
肾脏的血液供应十分丰富。正常人安静时每分钟约有1200ml血液流过两侧肾脏,相当于心输出量的20%-25%。其中94%左右的血液分布在肾皮质,5%-6%分布在外髓,其余不到1%供应内髓。通常所说的肾血流量(renal blood flow)主要指的是肾皮质血流量。肾血流量既能适应肾脏泌尿功能的需要,又能与全身的血循环相配合,前者主要靠自身调节,后者主要靠神经调节和体液调节,尤其在应激状态时,参与全身血流量的重新分配的调节,以适应整体生理活动的需要。
肾脏基本生理功能包括排泄废物、调节体液以及酸碱平衡、分泌激素。其结果是维持机体的内环境稳定,保证新陈代谢正常进行。
⒈排泄体内代谢产物,如尿素、肌酐、尿酸等。
⒉排泄外来物质,如药物、毒物等。
⒊保留体内所需的物质,如蛋白质、氨基酸、葡萄糖等。
⒋排泄过多的体液,以保持水的平衡。
⒌调节体液的电解质、渗透压和酸碱平衡。
⒍内分泌功能,如分泌肾素、促红细胞生成素、前列腺素、羟化的维生素D3及其它一些活性物质(内皮素、内皮细胞源性舒张因子等)。
一、肾小球的滤过功能
肾小球(毛细血管球)和肾小囊组成肾小体,构成肾单位的一部分。肾小球的滤过是形成尿液的第一个环节。滤过是指当血液流过肾小球毛细血管网时,血浆中的水和小分子溶质,包括少量的分子量较小的血浆蛋白,通过滤膜滤到肾小囊的囊腔内,形成滤液(原尿)的过程。24小时内约有1700L血液通过肾脏,其中约有180L血浆溶液在肾小球滤过生成原尿,除了不含血细胞和血浆蛋白质外,其余成分和血浆中相同。
决定肾小球滤过作用的因素主要有三方面:滤过膜通透性是滤过的结构基础;有效滤过压是滤过的动力;肾血浆流量(renal plasma flow)是滤过的物质基础。
滤过膜的超微结构由三层组成:①内层为毛细血管的内皮细胞层,可见大量的圆形窗孔,孔径40-100nm,对血浆蛋白几乎不起屏障作用;②中间为非细胞性的基膜层,是由微纤维织成的网状结构,可让水及部分溶质通过,但限制大分子的血浆蛋白质滤出,是滤过膜中主要的屏障;③外层是肾小囊皮细胞,由突起的足细胞构成,足突附着在基膜,足突间有裂隙,称为裂孔,裂孔上有一薄层裂孔膜,是滤过膜的最后一道屏障。滤过膜的屏障作用由两部分组成:①机械性屏障:分子直径小于2nm的物质可自由通过肾小球滤过膜,随着分子直径的增大,通过滤过膜的能力减小,机械屏障的大小与滤过膜上的孔径大以及构型有关;②电荷屏障:正常情况下,血浆白蛋白不能通过肾小球滤过膜,而与白蛋白分子直径相同的中性右旋糖酐则易通过。这是因为白蛋白在正常血浆pH时带负电荷,而且肾小球滤过膜含有涎酸、硫酸肝素等多糖,使滤过膜也带有负电荷,该负电荷形成了电荷屏障,阻止带负电荷的白蛋白滤出。在某些病理状态下,滤过膜上的负电荷消失,使大量白蛋白经滤过膜滤出,形成蛋白尿。
有效滤过压(effectivefiltration pressure)由三种力组成,根据三种力作用方向的不同,可列出下式:
肾小球有效滤过压=肾小球毛细血管血压-(血浆胶体渗透压+囊内压)
肾小球毛细血管血压平均为6.0Pa,囊内压为1.3Pa,血浆胶体渗透压约为2.7Pa,有效滤过压正常时为2.0Pa,依靠此滤过压可使血浆透过肾小球滤膜,进入肾小囊,形成原尿。
肾小球的滤过量大小可用肾小球滤过率(glomerular filtration rate,GFR)表示,单位时间内两肾生成的滤液量称为肾小球滤过率。据测定,体表面积为1.73m2的人肾小球滤过率为125ml/min左右,一昼夜从肾小球滤出的血浆量可达180L,约为体重的3倍,由此可见,肾小球滤过功能在肾的排泄功能中占有重要位置,肾小球滤过率可作为衡量肾功能的重要指标。
二、肾小管的重吸收功能
肾小管长而弯曲,分为三段:①近端小管,包括近曲小管和髓袢降支粗段;②髓袢细段,包括降支细段和升支细段;③远端小管,包括髓袢升支粗段和远曲小管。近曲小管和远曲小管位于皮质,管道弯曲。髓袢(Henle’s loop)呈U字形,从皮质直行进入髓质不同深度后再折返皮质。
重吸收是小管上皮细胞将小管液中的水分和某些溶质,部分地或全部地转运到血液的过程。比较原尿和终尿的量和质可发现,成人每天生成的原尿量约有180L,但终尿量每天只有1.5L左右,表明肾小管的重吸收量达99%。肾小管和集合管和重吸收是有“选择性”的,原尿中葡萄糖、氨基酸和少量蛋白质全部被肾小管重吸收,水和电解质大部分被吸收,尿素只有小部分被重吸收,肌酐则完全不被重吸收。
近曲小管是重吸收最重要的部位,原尿中的葡萄糖、氨基酸、维生素及微量蛋白质等,几乎全部在近曲小管重吸收,Na+、K+Cl-、HCO3-等也绝大部分在此段重吸收。近曲小管对葡萄糖的重吸收是有一定限度的,当血糖浓度在10mmol/L以下,近曲小管对葡萄糖的重吸收率可随血浆浓度的升高而增加,但当血糖浓度超过10mmol/L时,血糖浓度再增加,重吸收不再增加,尿中出现葡萄糖,这个浓度界限称为肾糖阈。
髓袢主要吸收一部分和氯化钠,在降支内水的重吸收大于溶质吸收,使管内的渗透压逐渐升高,形成渗透梯度,最高可达1200mOsm/kg以上。升支不透水,而溶质却不断被重吸收,形成逆向的渗透梯度,即从1200mOsm/kg又逐渐下降到等渗,甚至低渗。此段渗透压变化的过程称为“逆流倍增”,在尿液的浓缩稀释等功能中起重要作用。
远曲小管和集合管可继续重吸收部分水和Na+等,此段的重吸收量受抗利尿激素和醛固酮的调节,在决定尿量和终尿质方面起着十分重要的作用。其主要功能为参与机体对体液及酸碱等的调节,在维持机体内环境稳定中等起主要作用。
肾小管重吸收的基本方式可分为被动重吸收和主动重吸收两类。被动重吸收是一种顺电化学梯度进行的扩散过程,不需要消耗细胞的能量。例如尿素、水和Cl-。主动重吸收是逆电化学梯度进行的转运,需要由细胞本身提供能量,是肾小管上皮细胞主动活动的结果。如葡萄糖、氨基酸和大部分Na+的重吸收都是主动的。
三、肾小管、集合管的排泄作用
肾小管和集合管的上皮细胞具有将细胞产生的或血液中已存在的某些物质转运到管腔的能力,这一过程称为分泌或排泄。能够从肾小管和集合管上皮细胞分泌的主要物质有H+、K+和NH3等。
肾小管在调节机体酸碱平衡方面起着重要作用,通过分泌H+、重吸收NaHCO3。通过增加或减少体液中的NaHCO3的浓度以调节H+的浓度。在近端小管、远端小管和集合管都能分泌H+,发生H+-Na+交换,达到分泌H+而重吸收NaHCO3的目的。
远端小管和集合管分泌NH3,NH3主要由谷氨酰胺在谷氨酰胺酶的催化下脱氨而来,与H+结合成铵离子(NH4+)排出。铵盐的生成不仅促进了排H+,也促进了NaHCO3被重吸收。酸中毒时,近端小管也能分泌NH3,同时碳酸酐酶活性增强,谷氨酰胺活性也增强,氨的分泌增多。这样,起到促进酸的排出和NaHCO3重吸收的作用,从而调节机体酸碱平衡。若肾小管上皮细胞分泌NH3功能障碍,可导致酸中毒。
尿中的K+主要是由远曲小管和集合管分泌的。一般当有Na+的主动吸收时,才会有K+的分泌,两者转运方向相反,称为K+-Na+交换。K+-Na+交换和H+-Na+交换有相互抑制现象,即H+-Na+交换增多时,K+-Na+交换将减少,这就是酸中毒时血钾升高的原因之一。在远曲小管和集合管即存在钾的重吸收,又有钾的分泌,而且,一般是钾的分泌量大于其重吸收量,故尿中排出的K+主要来源于钾的分泌过程。
正常机体产生的肌酐和对氨基马尿酸等,既能从肾小球滤过,又能由肾小管分泌。此外,进入体内的某些物质,如青霉素、酚红等,则主要通过近端小管的分泌排出体外,临床上常用酚红排泄试验来检查肾小管的分泌功能。
四、肾功能的调节
肾的功能在于肾小球的滤过和肾小管、集合管的重吸收及分泌功能。
肾小球的滤过功能主要取决于肾血流量及肾小球有效滤过压。除了自身调节和肾神经调节外,还有球管反馈和血管活性物质调节。
自身调节是指当肾脏的灌注压在一定范围内变化时(10.7-24kPa),肾血流量及肾小球滤过率基本保持不变。
肾神经末梢主要分布在入球小动脉、出球小动脉及肾小管,刺激肾神经可引起入球、出球小动脉收缩,但对入球小动脉作用更为明显,导致肾小球滤过率的下降。
球管反馈(tubuloglomerularfeedback,TGF)是指到达远端肾小管起始段NaCl发生改变,可引起该肾单位血管阻力发生变化,从而引起肾小球滤过率的改变。该反馈的感受部位为致密斑,效应器官主要为入球小动脉和出球小动脉。高速灌流髓袢时,主要表现为入球小动脉收缩;低流量灌注时,入球、出球小动脉同时收缩。TGF的生理意义为在肾单位水平上,通过调节肾小球滤过率,使远端肾小管流量维持在一个狭小的变化范围内,以便对更远端的肾小管作更为精细的调节。
血管活性物质有血管紧张素、腺苷、前列腺素、激肽、心钠素、内皮素。血管紧张素可引起入球、出球小动脉收缩,GFR下降;腺苷为调节肾血管阻力的重要因素;前列腺素是在有血管紧张素和肾神经兴奋等缩血管作用因素存在时,产生增加,对抗缩血管引起的GFR下降;激肽(如缓激肽)可使肾血管扩张;心钠素主要使出球小动脉收缩,入球小动脉舒张,引起GFR的升高;内皮素具有很强的缩血管作用,主要产生于内皮细胞的多肽。内皮素可引起GFR的显著下降,还可与各种血管活性物质相互作用,促使前列腺素、内皮细胞源性舒张因子(扩血管作用)和心钠素的分泌。
对于肾小管和集合管功能的调节,主要是神经和体液因素对肾小管上皮细胞的重吸收水分和无机离子的调节功能,这在保证体内水和电解质和动态平衡、血浆渗透压及细胞外容量等的相对恒定均有重要意义。其中最重要的是抗利尿激素和醛固酮的调节作用。抗利尿激素(ADH)是一种多肽,它的主要生理作用是提高远曲小管和集合管上皮细胞对水的通透性,从而促进水的重吸收。醛固酮是肾上腺皮质所分泌的,其作用是促进远曲小管和集合管重吸收钠而分泌钾,有间接保水的作用,维持细胞内外的钾、钠浓度相对稳态和保存细胞外液的量。此外,还有血管紧张素、心钠素、甲状旁腺激素的调节作用。
第二节 常见肾脏疾病的病理生物化学
一、急性肾小球肾炎
急性肾小球肾炎(简称急性肾炎)临床表现为急性起病,以血尿、蛋白质、高血压、水肿、肾小球滤过率降低为特点的肾小球疾病。大多数为急性球菌感染1-3周后,因变态反应而引起双侧肾弥漫性的肾小球损害。
典型的临床病例表现为:70%病例有水肿,是因肾小球滤过率减低,水钠潴留引起;约半数病人有网眼血尿,同时伴有程度不同的蛋白质,但多数每日<3.0g;大多数病人有高血压,除水肿外,它是最主要的临床表现,为水钠潴留、血容量扩大所致。
实验室检查:血尿为急性肾炎重要表现,为肉眼血尿或镜下血尿;尿渗透压大于350mOsm/kgH2O;尿蛋白定量通常为1-3g/d,尿蛋白多属非选择性。
血液和尿液中出现FDP,意味着体内有纤维蛋白形成,纤维蛋白原及纤维蛋白分解代谢增强,尿FDP的测定能正确地反映肾血管内凝血。尿FDP浓度增高也与肾小球滤过膜通透性有关,可随血中FDP的增高而增高。
急性期肾小球滤过率(GFR)下降,肌酐清除率降低,肾血流量多数正常。
肾小管功能相对良好,肾浓缩功能仍多保持。尿钠减少,一般可有轻度高血钾。血浆白蛋白可因水、钠滞留及血容量增加致血液稀释而轻度下降,血清蛋白电泳多见白蛋白降低,γ球蛋白增高,少数病例有α或β球蛋白增高且往往并存高脂血症。急性肾炎病程早期有血总补体及C3的明显下降,可降至正常50%以下,其后逐渐恢复,6-8周时恢复正常,此种动态变化在急性链球菌感染后肾炎表现典型,可视为急性肾炎病情活动的指标。
二、肾病综合征
肾病综合征(nephroticsyndrome)的典型表现为大量蛋白尿(每日>3.5g/1.73m2体表面积)、低白蛋白血症(血浆白蛋白<30g/L)、高脂血症及水肿。大量蛋白尿及其导致的低蛋白血症是诊断肾病综合征的必备条件,肾病综合征是持续性大量蛋白尿的后果,其它表现都是在持续大量蛋白尿的基础上发生的。
凡能引起肾小球毛细血管滤过膜损伤的各种疾病,均可发生肾病综合征。肾病综合征不是一组独立的疾病,而是许多疾病过程中损伤了肾小球毛细血管滤过膜的通透性而发生的一个综合症状。可分为原发性肾病综合征和继发性肾病综合征。
原发性肾病综合征是原始病变发生在肾小球的疾病,急性肾小球肾炎、急进性肾小球肾炎、慢性肾小球肾炎及肾小球肾病都可在疾病过程中出现肾病综合征。
继发性肾病综合征在我国以系统性红斑狼疮、糖尿病、过敏性紫癜最为常见。
⒈临床表现大量蛋白尿是肾病综合征的标志主要由于肾小球毛细血管壁对蛋白质的通透性增加,肾小球滤过屏障发生异常所致。肾小球滤过屏障异常可分为电荷的异常及通透性的异常。电荷屏障的缺陷在光镜下肾小球结构无异常,白蛋白清除率增加,能显示电荷屏障缺陷的程度;通透性屏障缺陷在光镜下常可见到肾小球结构异常,选择性蛋白尿的测定,对判断通透性屏障缺陷的损害程度有一定的帮助,因为尿蛋白的成分可反映肾小球毛细血管壁病变的程度。肾小球基底膜的通透性与蛋白质分子大小成反比,在肾病综合征尿中发现的蛋白质,相对分子量都低于200000,其中包括白蛋白、α1-球蛋白、转铁蛋白和γ-球蛋白;相对分子量在1000000以上的,不能在尿中发现;相对分子量为300000-1000000的蛋白质,仅有少量;而尿中出现最多的是相对分子量在70000以下的蛋白分子。在光镜下不表现肾小球异常的病例,尿中含白蛋白和相对分子量小的球蛋白,特别是转铁蛋白。γ-球蛋白等;尿中可出现较大相对分子量的球蛋白,如α2-球蛋白和一些β-球蛋白,这表明肾小球有更严重的渗漏。这就是肾小球疾病测定蛋白尿选择性指数的理论依据。目前常用IgG(分子量160000)清除率与转铁蛋白(Tf,分子量88000)清除率之比来表示蛋白尿的选择性,即CIgG/CTf,此值≤0.1提示高选择性,如≥0.2提示非选择性。由于肾小球对蛋白质的渗漏,使血浆中相对分子量较小的白蛋白和α1-球蛋白大为降低,而α2-球蛋白、β-球蛋白和纤维蛋白原相对增加,醋纤薄膜电泳呈特征性图谱:白蛋白下降至50%以下,γ-球蛋白也相对减少,α2-球蛋白和β-球蛋白比例明显升高,出现两端下陷、中间增高的图谱。
⒉低蛋白血症是指肾病综合征患者血清总蛋白浓度降低其中主要的是白蛋白浓度降低(<30g/L)。主要原因是尿中蛋白大量丢失,从血浆中丢失的白蛋白的量常超过尿白蛋白的量,这主要是肾小球滤过的白蛋白被肾小管上皮细胞重吸收后,在肾组织内部分降解成肽和氨基酸;机体的其它部位白蛋白降解也增加,例如胃肠道丢失白蛋白显著高于正常人,其机制可能是胃肠道基膜炎症性损害及淋巴管扩张所致。
⒊肾病综合征时免疫球蛋白特别是相对分子量较小的IgG可从肾小球中大量滤出如同白蛋白一样,IgG除了尿中大量丢失以外,肾小管的吸收和分解亦增加。IgG的合成能力的下降可能也是肾病综合征患者血浆IgG下降的重要原因。IgM相对分子量较大,肾小球不能滤过,几乎所有肾病综合征患者都有IgM的相对增高,提示患者体内免疫刺激增强。血浆中其它大分子物质如纤维蛋白原,α2、β球蛋白等,滤过虽未增多,但合成却增加,血浆浓度增高,此为肾病综合征患者血沉增快的原因。转铁蛋白的相对分子量与白蛋白相近,尿中丢失和分解都增多,血浆浓度降低,这是肾病综合征贫血的原因之一。补体激活旁路B因子的缺乏可损害机体对细菌的调理作用,为肾病综合征患者易感染的原因之一。此外严重低蛋白血症可导致持续性的代谢性碱中毒。
⒋高脂血症是指肾病综合征的主要临床表现之一脂代谢异常的特点为血浆中几乎各种脂蛋白成分均增加,血浆胆固醇(Ch)和LDL升高,甘油三酯(TG)和VLDL升高,HDL浓度可以升高、正常或降低;HDL亚型分布异常,即HDL3增加而HDL2减少,表明HDL3的成熟障碍。在疾病过程中各脂质成分的增加出现在不同的时间,一般以Ch升高出现最早,其次才为磷脂及TG。除数量改变外,脂质的质量也发生改变,各种脂蛋白中胆固醇/磷脂及胆固醇/甘油三酯的比例均升高。肾病综合征时也常有载脂蛋白(Apo)的异常,特征性改变为:ApoB明显升高。ApoC和ApoE轻度升高,ApoCⅡ虽在尿中丢失但血中浓度升高,ApoCⅢ/ApoCⅡ升高。HDL的主要结构蛋白-ApoAⅠ和ApoAⅡ降低或正常,ApoAⅠ/ApoCⅢ降低,ApoAⅠ/ApoB降低,另外还有ApoCⅡ和ApoE从HDL向LDL再分布。脂质异常通常与蛋白尿和低蛋白血症的程度有关,因为:①肝脏合成Ch、TG及脂蛋白增加:低蛋白血症是其合成增加的原因,血浆胶体渗透压下降是触发肝合成蛋白增加的启动因素;代谢延迟可能在高甘油三酯血症的发生中起重要的作用。②脂质调节酶活性改变及LDL受体活性数目改变导致脂质的清除障碍:其中脂蛋白酶活性降低30%-60%,肝脏甘油三酯脂酶活性降低可使VLDL向LDL转变减少,VLDL清除障碍;另外卵磷脂胆固醇酰基转移酶(LCAT)活性降低,可致HDL向HDL3转变减少;LDL受体活性降低并缺乏受体的相互作用,从而使脂质代谢减少,血中浓度升高。③尿中丢失HDL增加,构成HDL的载脂蛋白ApoAⅠ丢失50%-100%,而且患者血浆HDL3增加而HDL减少,说明HDL在转变为较大的HDL3颗粒之前已在尿中丢失。
⒌高凝状态是肾病综合征又一症状由于血浆中的一些凝血因子和纤维蛋白原、因子Ⅴ、Ⅶ、Ⅷ和Ⅹ等的相对分子量都较大,不能从肾小球滤过,而体内合成又相对增加,故血浆中浓度常明显增高。抗凝血酶Ⅲ为血浆中主要的抗凝因子,相对分子量和白蛋白相近,可从肾病综合征患者尿中大量丢失而严重减少,这是高凝状态的重要原因。血小板集聚力亦增高,可能由于高脂血症改变了血小板膜,血小板集聚力与白蛋白浓度呈负相关。集聚的血小板释放β-血栓球蛋白,抑制血管内皮前列腺素分解而加重高凝状态。尿纤维蛋白降解产物(FDP)的增加反映了肾小球滤过的改变(主要)和肾小球内的凝血(次要)。由于高胆固醇血症及高纤维蛋白原血症的联合影响,血浆粘稠度多增加。总的来说:血凝中凝聚及凝集的各种因子增强,而抗凝集及纤溶作用的机制受损。当血管内皮受损或血液淤积时,加上上述因素,肾病综合征患者易于产生自发性血栓形成。
⒍水肿的出现及其严重程度一般来说与低蛋白血症的程度呈正相关由于血浆白蛋白的减低,使血浆胶体渗透压大大下降,因而引起水分和小分子的可扩散溶质由血浆转入组织间隙,则血浆容量减少,促使交感神经兴奋,儿茶酚胺释放,及肾素-血管紧张素-醛固酮系统活性增加,又激发抗利尿激素分泌增多,排钠因子受抑,致使水和钠潴留,水肿进一步加剧。肾病综合征的水肿虽与血浆白蛋白过低有比例关系,但决定因素还是肾脏对水和盐的排泄率。肾病综合征的钠滞留主要是由于心钠素对肾小管作用的障碍造成肾脏调节钠平衡的障碍。(图11-1)。

图11-1 肾病综合征水肿发生机理
三、急性肾功能衰竭
任何原因引起的急性肾功能损害,使肾单位丧失调节功能,不能维持体内电解质平衡和排泄代谢产物,导致高血钾、代谢性酸中毒及急性尿毒症(指血尿毒氮、肌酐及其它代谢产物迅速增高,并出现一系列症状和体征)的患者,统称为急性肾功能衰竭(acute renal failure,ARF)。与日俱增的进行性血尿素氮和血肌酐的升高(通常每日血肌酐可增加88.4-176.8μmol/L,尿素氮升高3.6-10.7mmol/L)是诊断急性肾功能衰竭的可靠依据,其肾衰为可逆性。
典型的急性肾功能衰竭是急性肾小管坏死(acute tubular necrosis,ATN)、急性缺血或急性肾中毒所引起的急性肾病变。急性肾缺血引起者占绝大多数,其中以创伤、手术及严重感染引起者居多。肾毒性物质通常分为两大类:外源性毒物和内源性毒物。外源性肾毒性物质主要有三类:肾毒性药物(如氨基糖甙类药物、四环素族和二性霉素B等)、有机溶剂(如四氯化碳等)和重金属(主要有汞、砷、金、银、锑和铜等);内源性肾毒性物质包括肌红蛋白、血红蛋白、尿酸和钙,肌红蛋白是由于出血、休克、感染和创伤组织释放的,血红蛋白由烧伤引起大量细胞外液缺失和发生血管内溶血而造成的。
ATN在临床上常与急性肾衰这一术语交替使用,ATN的典型临床表现为在创伤、手术或误输异型血管情况后(数小时至48小时),突发尿量减少。血肌酐、尿素氮等代谢物逐渐增高,二氧化碳结合力降低,病人常有尿毒症症状。临床过程常分为少尿期、多尿期和恢复期。
㈠少尿期
⒈尿液的改变 病人可表现为少尿(<400ml/24h)或无尿(<100ml/24h),尿比重常为1.010-1.015,尿渗透压为280-00Osm/L,尿蛋白+~++。
⒉氮质血症由于肾清除氮质代谢废物的功能障碍,后期血尿素氮可高达71.4mmol/L。一般的ATN病人,其血肌酐和尿素氮分别为每日44.2-88.4μmol/L和3.57-7.14mmol/L。
⒊代谢性酸中毒 代谢性酸中毒在ATN患者中很常见。由于非挥发性酸性代谢产物如无机磷酸盐等的排出障碍,以及肾小管分泌H+及氨功能的丧失,导致体内酸性代谢物的积聚和血内碳酸氢根离子浓度的下降,而产生高阴离子隙的代谢性酸中毒。
⒋水中毒和钠潴留 ATN病人如给予过多的液体,可发生水中毒。临床主要表现为中枢神经系统症状,重者可发生昏厥或昏迷而死亡。水中毒时,可发生稀释性低钠血症,血钠浓度常低于125mmol/L。除了少尿导致水潴留和补液过多可致低钠血症外,如不严格控制钠盐的摄入,则可发生钠潴留,导致体重增加,周围水肿,高血压和心力衰竭,后者是ATN的主要死亡原因。
⒌高钾血症由于组织创伤、感染性休克、溶血和高分解代谢状态等导致细胞释放钾过多,而代谢性酸中毒又促使细胞内钾向细胞外转移,血钾浓度增高。ATN时,由于肾排钾功能障碍,故少尿数日常后出现高钾血症,这是本病的突出问题,可严重威胁病人生命。通常血钾每日递增约0.3mmol/L,高血钾是急性肾衰最严重的并发症,是起病第1周最常见的死亡原因,应尽早诊治高钾血症。
⒍高磷血症和低钙血症由于肾排磷功能受损,常有高磷血症,尤其是广泛组织创伤等造成的高分解代谢病人,血磷可高达1.9-2.6mmol/L。由于高磷血症,肾生成1,25-(OH)2D3及骨骼对PTH的钙动员作用减弱,因而低钙血症也较常见。中度的高镁血症在ATN病人中也较常见。
⒎尿毒症症状 ATN病人,由于体内氮代谢产物迅速积累,少尿期数日后即可出现尿毒症症状。尿毒症症状的严重程度与血中尿素氮及肌酐增高的浓度相一致,故临床上以血清尿素氮及肌酐增高的程度与速度作为尿毒症严重性的主要指标,主要症状有:
⑴胃肠道症状:胃肠道症状是ATN最常见的临床表现,病人可有食欲减退、恶心、呕吐、腹泻等症状。
⑵心血管系统症状:ATN时,心血管系统的主要并发症是高血压和心力衰竭,其主要原因是水、钠负荷过重。
⑶神经精神症状:神经系统功能障碍的原因包括中枢神经系统抑制药物的使用、电解质和酸碱平衡紊乱以及氮质代谢产物的积聚等多种因素联合作用的结果。
⑷血液系统表现:ATN常有正细胞正色素性贫血,随着肾功能的恶化、氮质代谢产物的积累,贫血有加重的趋势。贫血的主要原因是细胞生成减少和不同程度的血管外溶血。由于骨髓产生血小板的减少,在ATN的早期,常有血小板减少。血小板的减少和功能障碍以及凝血功能异常,与ATN的出血倾向有关。
⑸感染:感染是ATN最常见的并发症,且为ATN的主要死亡原因之一。感染的好发部位包括呼吸道、泌尿道和手术伤口。感染的高发生率与ATN时免疫功能损害致机体抵抗力下降、正常解剖屏障的破坏以及不恰当地使用抗生素有关。
㈡多尿期
当ATN病人每日尿量增致400ml以上时,提示进入多尿期。此时,肾功能已有恢复,能排出尿液。在多尿期的早期,肾单位功能仍未完全恢复,不能充分地排出血中的氮质代谢废物、钾和磷。血中上述物质仍可继续上升,所谓多尿早期是以血清肌酐及尿素氮持续增高为标志。
㈢恢复期
多尿期过后,肾功能已显著改善,尿量逐渐恢复正常。血肌酐、尿素氮此时基本恢复正常水平,但肾小管尚有轻度障碍。
尿生化分析:尿指标的测定可估计ATN的肾小管功能。尿指标包括尿渗透压和比重、尿钠测定、尿肌酐/血浆肌酐、滤过钠排泄分数(FeNa)等。
⒈尿浓缩能力测定 尿比重ATN<1.010,尿渗透压<350mOsm,尿渗透压/血浆渗透压<1.1,自由水清除值>-1ml/min,被认为是肾小管功能的敏感指标。ATN时肾浓度功能丧失,自由水清除值升高而接近0,是一个较肌酐清除率和滤过钠排泄分数异常更早出现的ATN指标,不过单独测定自由水清除值无多大意义,自由水清除值增高接近于0,而肌酐清除率急剧降低才提示ATN。
⒉GFR和总的肾小管重吸收能力肌酐清除率在血清肌酐和尿素氮尚在正常范围时,已显著降低,说明是ATN早期诊断的灵敏指标。尿肌酐/血肌酐的比值反映了肾小管重吸收从肾小球滤过水分的能力。因为肌酐不会被肾小管重吸收,故尿肌酐浓度愈低,则肾小管吸收水分的能力愈差。
⒊肾小管处理溶质的能力 尿钠和FeNa。在ATN时,肾小管受损,不能很好地重吸收钠,故尿钠浓度高,常大于40mmol/L。ATN中,FeNa>1,对于诊断ATN一般认为较有价值。
在诊断ATN时一般以尿浓度功能测定比较常用,因为肾小管受损时,首先损害的是肾浓度能力,当损害进一步发展时,钠重吸收才减少。
⒋肾小管损害的尿标记 β2-微球蛋白、LDH、NAG、LYS、THP等均有增高,但这些指标都是非特异性的。
四、慢性肾功能衰竭
慢性肾功能衰竭(chronicrenal failure,CRF简称慢性肾衰)是在发生各种慢性肾脏疾病基础上,由于肾单位逐渐受损,缓慢出现的肾功能减退以至不可逆转的肾衰。其临床主要表现为肾功能减退,代谢废物潴留,水、电解质和酸碱平衡失调,以致于不能维持机体内环境的稳定。慢性肾衰较常见,情况预后严重,它不是一种独立疾病,而是一个临床综合征。
肾功能减退可分为以下四个阶段:
⒈肾贮备能力丧失期 正常肾小球滤过率(GFR)约为120ml/min,此期的GFR减少至30-60ml/min。此时,肾贮备能力虽已丧失,但肾排泄代谢废物、调节水电解质和酸碱平衡的能力仍能维持机体内环境的稳定,因而临床上并无症状。血生化检查正常,血肌酐和尿素氮通常比正常范围轻微升高。
⒉氮质血症期 是慢性肾衰的序幕。此期GFR减少至25ml/min左右,肾维持机体内环境稳定的能力有一定程度障碍。常有氮质血症(血肌酐和尿素氮常升高,血肌酐>177mmol/,血尿素氮>7.0Lmmol/L),肾浓缩功能有轻度损害(夜尿和多尿),轻度贫血。
⒊肾功能衰竭期 当GFR>10ml/min左右时,肾功能已严重受损,不能维持机体内环境的稳定,出现明显的氮质血症(血肌酐、尿素氮明显升高,血肌酐>442μmol/L,尿素氮>17.9-21.4mmol/L),肾浓缩和稀释功能显著障碍。水、电解质和酸碱平衡失调,表现为轻或中度代谢性酸中毒,水、钠潴留,低钙血症和高磷血症等。由于肾排钾的能力可勉强维持平衡,故此期可不出现高钾血症,有较明显的贫血。
⒋尿毒症期 GFR<10ml/min,就进入慢性肾衰晚期,即尿毒症期。血肌酐、尿素氮显著升高,水、电解质失调严重,常有明显的代谢性酸中毒、低钠血症和高钾血症,血钙明显降低,血磷升高。体内多个系统均受累而出现相应的症状,尤其是胃肠道、心血管和中枢神经系统症状更明显,甚至昏迷。
慢性肾衰的病因很广,各种肾脏疾病的晚期都可以出现慢性肾衰,慢性肾小球肾炎是最常见的一种。
第三节 肾功能不全的生化诊断及评价
一、蛋白尿
肾脏疾病时,尿液的常规检查是不可忽视的,不少肾脏病变早期就可出现蛋白尿,或者尿沉渣中出现有形成分。
蛋白尿是尿液中出现超过正常量的蛋白质,当肾小球通透性增加,或血浆中低分子蛋白质过多,这些蛋白质大量进入原尿,超过了肾小管的重吸收能力时,产生蛋白尿。前者称为肾小球性蛋白尿,后者称为血浆性(或溢出性)蛋白尿。此外,当近曲小管上皮细胞受损,重吸收能力降低或丧失时,则产生肾小管性蛋白尿。可以对尿蛋白进行定性和定量检查,是肾脏疾病诊断的一个粗筛试验。
二、肾功能试验
㈠肾小球滤过功能检查
肾“清除”率是1928年Van Slyke制定的,表示肾脏在单位时间内(每分钟)将多少亳升血浆中的某物质清除出去。清除率对于了解肾脏各部位的功能很有帮助。以公式表示如下:
C=UV/P
C-清除率(ml/min)
V-每分钟尿量(ml/min)
U-尿中测定物质的浓度(mmol/L)
P-血中测定物质的浓度(mmol/L)
由于此公式计算得到的清除值是被测者个体的结果,但个体大小、高矮、胖瘦、年龄等差异很大,必须标准化,以标准的体表面积1.78m2校正。A代表个体的体表面积。
校正后的清除值 C=UV/P×1.73/A
体表面积的计算:
logA(m2)=0.425log[体重kg]+0.725log[身高cm]-2.144
利用清除率分别测定肾小球滤过率、肾血流量、肾小管对各物质的重吸收和分泌作用(表11-1)。
血浆中所含某一物质小部分经肾小球滤过,不被肾小管重吸收,而且血中剩余部分又可全部由肾小管分泌,使这一物质通过肾后几乎全部排出,那么它的清除率既代表肾血浆流量(RPF),又可反映肾小管的分泌功能,如对氨基马尿酸、碘锐特、酚红和青霉素等。
若另一物质能全部经肾小球滤过,肾小管对其不吸收、不排泄,则其清除率可反映肾小球的滤过率(glomerular filtration rate,GFR),如菊粉、肌酐等。
某物质经肾小球滤过后,完全被肾小管重吸收,其清除值等于0,例如葡萄糖。在血浆浓度接近肾糖阈时,利用清除值公式,可计算出滤液中被重吸收的葡萄糖量即肾小管葡萄糖最大重吸收量(TMG),用以反映近端肾小管的重吸收功能。
表11-1 肾清除试验及其临床意义
| 肾脏对物质的清除方式 | 物 质 | 清除值临床意义 |
| 从肾小球滤过,肾小管对它即不重吸收又不分泌 | 肌酐、菊粉 甘露醇、硫代硫酸钠 |
反映肾小球滤过功能 |
| 小部分从肾小球滤过大部分从肾小管分泌 | 对氨基马尿酸酚红、青霉素 | 反映肾小管分泌功能代表肾血流量 |
| 经肾小球滤过后,相当一部分被肾小管重吸收 | 尿酸 | 尿量多少影响重吸收,尿量减少,尿素重吸收增加,消除值变异较大,不是理想的肾功能试验 |
| 经肾小球滤过后,在近曲小管几乎完全被重吸收,远曲小管可以被分泌 | 钾离子 | 清除值无意义 |
| 经肾小球滤过后大部分被肾小管重吸收 | 各种电解质 氨基酸 |
清除值很低,C<10ml/min |
| 经肾小球滤过后完全被肾小管重吸收 | 葡萄糖 | 清除值为0 |
⒈肾小球滤过率
⑴菊粉清除率测定:菊粉(inulin)是一种植物多糖,相对分子量为5200左右,完全由肾小球滤过,不被肾小管重吸收或分泌,是理想的测定GFR的物质,其清除率(125ml/min)可准确反映肾小球滤过率。其它类似的物质有肌酐、甘露醇、硫代硫酸钠和51Cr-EDTA等,在这些物质中菊粉最为适合,但菊粉是外源性物质,测定方法麻烦。临床上多测定内生肌酐清除率,它具有测定方法简单的优点。
⑵内生肌酐清除率(Ccr):人体肌肉以每分钟1mg速度将肌酐排入血中,血浆肌酐浓度比较稳定,受外界因素如蛋白质的摄入等影响较小。从肾小球滤过后,不被肾小管重吸收和分泌,只要同时测定血和尿中肌酐浓度,并记录每分钟尿量就可计算出内生肌酐清除率。
每分钟肌酐清除率:

参考值:根据体表面积校正后,范围为80-120ml/min/1.73m2。
在严格控制条件下,尿中肌酐排泄量相当稳定,故可将24小时法改用4小时法测定Ccr。
测定内生肌酐清除率来估计肾小球滤过率从理论上说不如菊粉,因为当血中肌酐明显增高时,可有一小部分肌酐由肾小管分泌到尿中。此时测出的肌酐清除率高于实际的肾小球滤过率。又由于血浆中肌酐浓度较低,常用的碱性苦味酸试剂显色法(Jaffe反应)有其它干扰因素存在,常使血浆测定值偏高,而使清除值低于菊粉清除值。
肾小球病变时,一部分肾小球破坏,滤过面积减少,肾小球滤过率可明显下降,但由于肾脏有强大的贮备能力,余下的肾单位仍能排出日常机体所产生的尿素和肌酐等代谢产物,血浆中这些物质浓度变化不大。如图11-2说明肾小球滤过率与血中尿素、肌酐浓度变化间的关系。
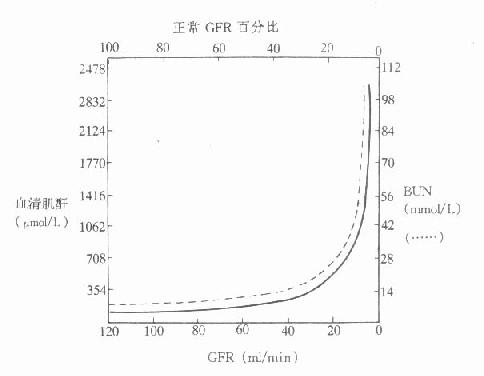
图11-2 肾小球滤过率与血肌酐、尿素浓度的关系
从图11-2可见,只有当肾小球滤过率下降到正常的50%以下时,血浆中尿素及肌酐浓度才出现增高,当肌酐高达618.8-707.2μmol/L时,肾小球滤过率已明显下降到仅及正常的10%。说明测定肾小球滤过率比测定血浆尿素和肌酐含量更为灵敏可靠。
⒉血肌酐和尿素浓度测定 血肌酐(serumcreatinine,Scr)和血尿素(bloodureanitrogen,BUN)的浓度取决于机体氮的分解代谢与肾脏的排泄能力。在摄入食物及体内分解代谢比较稳定的情况下,其血浓度取决于肾排泄能力,因此,Scr和BUN浓度在一定程度上可反映肾小球滤过率功能的损害程度,是常用的肾功能指标。
⑴尿素的测定方法可分为两大类:一类直接法,尿素直接和某试剂作用,测定其产物,最常见的为二乙酰一肟法;另一类是尿素酶法,用尿素酶将尿素变成氨,然后用不同的方法测定氨。
1)尿素酶法(直接法):尿素酶法利用尿素酶催化尿素水解生成铵盐,铵盐可用纳氏试剂直接显色、酚-次氯酸盐显色或酶偶联反应显色。
尿素测定目前多采用尿素酶偶联法:用尿素酶分解尿素产生氨,氨在谷氨酸脱氢酶的作用下使NADH氧化为NAD+时,通过340nm吸光度的降低值可计算出尿素含量。

此反应是目前自动生化分析仪上常用的测定原理。此外,尿素酶水解尿素产生氨的速率,也可用电导的方法进行测定,其电导的增加与氨离子浓度有关,反应只需要很短的时间,适用于自动分析仪。
2)酚-次氯酸盐显色法:尿素酶水解尿素生成氨和酚及次氯酸盐,在碱性环境中作用形成对-醌氯亚胺,亚硝基铁氰化钠催化此反应:
![]()
对-醌氯亚胺同另一分子的酚作用,形成吲哚酚,它在碱性溶液中产生蓝色的解离型吲哚酚:
![]()
此反应敏感,血清用量少(10μl),无需蛋白沉淀,一般用于手工操作测定中。
3)纳氏试剂显色法:尿素经尿素酶作用后生成氨,氨可与纳氏试剂(HgI2.2KI的强碱溶液)作用,生成棕黄色的碘化双汞铵。
尿素酶法的优点是反应专一,特异性强,不受尿素类似物的影响,缺点是操作费时,且受体液中氨的影响。
⑵二乙酰一肟法(直接法):尿素可与二乙酰作用,在强酸加热的条件下,生成粉红色的二嗪化合物(Fearom反应),在540nm比色,其颜色强度与尿素含量成正比。二乙酰不稳定,用二乙酰一肟代替,后者遇酸水解成二乙酰。
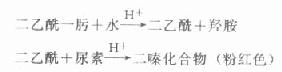
试剂中加入Fe3+或Cd2+及硫氨脲,可提高灵敏度,增加显色稳定性,其中Fe3+和Cd2+有氧化作用,还能消除羟胺的干扰作用。提高酸的浓度可增加灵敏度。二乙酰一肟与尿素的反应不是专一的,与瓜氨酸也有显色。本法灵敏、简单,产生的颜色稳定,缺点是加热时有异味释放,一般临床已很少使用此方法。
尿素测定用血清或血浆,体液中尿素的浓度常用尿素中含有的氮来表示,称为尿素氮。如欲换算成尿素,可根据60g 尿素含有28g氮计算,即1g尿素相当于0.467g尿素氮,或是1g尿素氮相当于2.14g尿素。
正常参考值:血清尿素氮为2.8-7.1mmol/L,相当于尿素1.8-6.8mmol/L。
⑶肌酐与碱性苦味酸试剂反应产生红色(Jaffe反应),仍是目前常用的测定方法。为提高其准确性现已改为动力学测定法。
1)Jaffe反应法:测定肌酐最常用的方法是尿、血清中的肌酐与苦味酸盐作用,生成黄红色的苦味酸肌酐复合物。此法的缺点是特异性不高,维生素C、丙酮酸、丙酮、葡萄糖、乙酰醋酸、果糖、氨基马尿酸、蛋白质、胍基醋酸酰胺及许多化合物也能与碱性苦味酸盐生成红色。这些不是肌酐而能起反应的物质称为假肌酐,相当数量的假肌酐存在于红细胞中,故在测定血液肌酐时用血清和血浆较好。
为去除假肌酐的影响,现在常用速率法来测定血肌酐。速率法亦称动力学方法,它是根据肌酐与碱性苦味酸形成复合物的速度与假肌酐不同,且肌酐的反应速度与浓度成正比的原理。如乙酰乙酸在20s内已与碱性苦味酸反应完成,其他多数干扰物则在80s后才与苦味酸有较快的反应,而20-80s之间主要是肌酐的反应。因此,血清与苦味酸混合后,分别读取510nm在20s及80s时的吸光度Ao和At,At-Ao除以间隔时间的值与肌酐浓度成正比,借标准液与样品同样测定即可求取样品中的肌酐量。现在速率法逐渐成为常规分析法,因为它不需去蛋白,方法简单、快速,可自动扣除血清及试剂空白吸光度。
2)酶联法:肌酐经肌酐水合酶催化生成肌酸,肌酸与肌酸激酶、丙酮酸激酶、乳酸脱氢酶偶联反应,使NADH变成NAD+,测量在340nm处吸光度的降低,其降低程度与肌酐含量呈正比例,反应式如下:

此法特异性高,标本不需去蛋白,特别适用于自动分析。但工具酶过多、价格昂贵。
正常参考值:血浆肌酐44-133μmol/L。
血尿素浓度除受肾功能影响外,还受到蛋白质分解代谢引起的变化,如高蛋白饮食、胃肠道出血、口服类固醇激素等都可使血尿素浓度增高。而肌酐摄入、生成量恒定,故血肌酐测定较血尿素测定更能准确地反映肾小球功能,但反应较迟钝。
肾功能不全的代偿期可见BUN轻度增高(>7.0mmol/L),肌酐可不增高或轻度增高;肾功能衰竭失代偿期,BUN中度增高(17.9-21.4mmol/L),肌酐也中度增高(442.0μmol/L);尿毒症时BUN>21.4mmol/L,为尿毒症诊断指标之一,肌酐可达1.8mmol/L。
⒊血清尿酸(serum uricacid,SUA)尿酸是嘌呤类的终末产物,血尿酸主要从肾脏排出,肾功能减退时SUA增高。尿酸从肾小球滤过后在肾小管中重吸收和分泌,最后排出滤过量的8%,在严重衰竭时肾小管分泌大增,可达滤过量的85%被排出,慢性尿毒症时SUA的增高程度不明显。
尿酸测定方法可分为两大类:磷钨酸还原法和尿酸酶法,目前以尿酸酶法为主。
⑴磷钨酸法:是利用在碱性环境中尿酸具有还原性,无蛋白血滤液中的尿酸可使磷钨酸还原生成蓝色的钨蓝,可进行比色测定,反应如下:
尿酸+磷钨酸→尿囊素+CO2+钨蓝
此法的不足之处是特异性不高,显色褪色速率变化不定,灵敏度低。
⑵尿酸酶法:尿酸酶测定方法可分为三种类型:
1)紫外分光法:尿酸在282-292nm处有特异吸收峰,当其经尿酸酶作用后,产物在此波长范围无吸收峰,测量酶作用前后吸光度之差,经标准品、测定样品同时处理,可计算尿酸含量。该法灵敏度高,特异性强,其它物质无干扰,可用血清直接测定,不需沉淀蛋白,易于自动化,具有简单、快速的优点。
2)酶联比色法:尿酸经尿酸酶作用生成的H2O2,可用偶联的过氧化物酶(POD)使H2O2氧化还原色素原,成为氧化型而显色,用酚和4-氨基安替比林作生色原。反应如下:

反应生成的红色醌亚类化合物,在500nm处有最大吸光度,吸光度的增加与样品中酸含量呈正比,可进行比色测定。此法敏感,反应所产生的颜色比用酚作色素原时产生的颜色强度大4倍,可用血清直接测定。
3)酶联-紫外分光法:尿酸酶将尿酸氧化时生成的H2O2的存在下同乙醇作用生成乙醛,后者被偶联的醛脱氢酶(ALDH)进一步氧化生成乙酸,伴随着NAD+变成NADH,在340nm测定由NAD+还原产生的光吸收增加与样品中尿酸含量成正比。反应式如下:

此反应第一步是特异的,但后面的氧化还原反应易受干扰,因体内有许多脱氢酶反应,亦可氧化内源性底物而伴有NAD+还原生成NADH,导致结果偏高。除以上方法外,固相酶技术的发展使测定更简单,更易自动化。尿酸酶的固相化有以下几种类型:①固相尿酸酶与氧电极相联,测定尿酸氧化时的耗氧量;②固相尿酸酶及过氧化物酶与显色法结合;③夹心固相酶-荧光法,使产生的H2O2与对羟基苯乙酸反应产生荧光,此法具有酶反应的特异性及荧光法的灵敏度。
正常参考值:血尿酸:男性148.7-416.4μmol/L,女性89.2-356.9mol/L.
㈡肾血流量测定
测定对氨基马尿酸(PAH)清除率或碘锐特清除率均可反映肾血流量.对氨基马尿酸(PAH)主要由近端小管分泌排出.当血浆中PAH浓度很低时流经肾脏,90%从肾脏清除而排入尿中,即流经肾脏的PAH大部分被清除.PAH清除率相当于流经肾脏的血浆量,称为有效肾血浆流量(ERPF)。PAH为外源性物质,操作复杂,临床上多不采用。放射性核素(同位素)肾图能比较敏感地反映肾的血浆流量,目前临床上将其列为肾功能的常规检查。
㈢肾小管功能检查
肾小管的功能比较多,除了具有强大的再吸收水分与一些物质的能力外,还具有选择分泌与排泄一些物质的能力。到目前还没有理想的肾功能试验适用于临床。生理研究中的肾小管葡萄糖最高再吸收率(TmG)和对氨基马尿酸最大排泄率(TmPAH),可较好地代表肾小管再吸收与排泄的功能,但操作麻烦,不适合临床应用,多以尿液浓缩-稀释试验和酚红排泄试验作为肾小管的功能试验。近年来尿中酶的测定和自由水清除值等的测定逐步使肾小管功能试验简单、灵敏、快速,更适用于临床。
⒈近端小管功能检查 酚红排泄率可作为判断近端小管排泄功能的粗略指标。酚红注入体内后,94%由近端小管上皮细胞主动排泄,从尿液排出。由于酚红排泄试验受肾血流量及其它肾外因素影响较大,对肾小管功能敏感不高,故目前基本不用。以测定尿β2-微球蛋白及溶菌酶等估计近端小管功能。
⒉肾浓缩稀释试验 肾浓缩和稀释尿液功能主要在远端小管和集合管进行。在日常或特定饮食条件下观察病人尿量和尿比重的变化,称为浓缩稀释试验,作为判断远端小管功能的指标。(以上二试验在临床检验中已详尽叙述,本章略。)
⒊尿渗透压测定 液体的渗透压是由溶液中溶质的毫摩尔浓度决定的,尿液渗透压反映尿液中溶质的摩尔数。尿比重和尿渗透压都能反映尿中溶质的含量,但尿比重易受溶质微粒大小和性质的影响,如蛋白质、葡萄糖等均可使尿比重增高;而尿渗透压则反映尿中各种溶质微粒的总数目,而与溶质分子相对重量、微粒体积大小无关,因而测定尿渗透压较测定尿比重更好,更能反映肾浓缩和稀释能力。
目前多采用尿液冰点下降法测定渗透压,也可用蒸气压渗透压计算法测定。溶液的渗透压增加,使其冰点降低。以纯水的冰点0℃为标准,任何溶液的冰点都比纯水为低,均为负值。冰点愈低,负值愈大,表明溶液中的渗透浓度越大。1渗量的溶质可使1kg水的冰点下降1.858℃,因此溶质渗透压等于冰点下降数除以1.858。
渗透压(Osm/kgH2O)=测得溶液冰点下降度(0C)/1.858
测定尿渗透压(Uosm)的方法有三种:①直接测定尿渗透压;②测定尿、血浆渗透压(Posm)的比值;③自由水清除率。
正常尿渗透压为600-1000 Osm/kg H2O。
尿渗透压和血浆渗透压相比,当尿渗透压高于血浆渗透压时,表示尿已浓缩,称为高渗尿;低于血浆渗透压表示已稀释,称为低渗尿;若与血浆渗透压相等为等渗尿,反映肾脏浓缩功能严重损害。
⒋自由水清除率 自由水清除值指单位时间内使尿液达到等渗,而应从尿中减去或加入的纯水量。在尿浓缩时,排出的尿量等于渗透尿量减去被总吸收的纯水量;在尿稀释时,排出的尿量等于等渗透尿量加上血浆中清除的纯水量。等渗尿量实际上就是渗透性溶质清除率,又称渗量清除率(Cosm)。它表示单位时间内肾脏能够将多少血浆中的渗透性溶质清除出去。
Cosm=Uosm/Posm×V(单位时间尿量)
由于V=Cosm+CH2O,因此CH2O的计算公式是:
CH2O=(l/Uosm/Posm)×V
自由水清除率正值代表肾稀释能力,负值代表肾脏浓缩能力;若CH2O等于或接近于0,则表示肾不能浓缩和稀释尿液,是肾功能严重损害的表现。因此,进行尿液浓缩试验时,自由水清除率的正值变小而接近于0,表示肾稀释功能受损,正常人常为-25-100ml/h。因在急性肾功能衰竭早期CH2O趋于0,CH2O呈现负值大小可反映肾功能恢复的程度,所以此测定对急性肾功能衰竭的早期诊断及病情变化有一定价值。
⒌肾小管对尿液的酸碱调节功能——H+总排泄量测定 正常饮食情况下,每天代谢产生50-100ml非挥发性酸,并从尿液中排出,主要经过肾小管分泌。若这么多酸都以游离的H+排除,则尿液的pH应小于2,但实际上正常尿液的pH在5-6之间,因此大部分H+是被缓冲后排出的。
肾小管以三种方式排酸:
⑴直接排H+,这部分可用pH计进行测定。
⑵和磷酸根、硫酸根及其它有机化合物结合。如HPO43-+H+→H2PO42-,pH4.5时,尿中磷酸盐几乎全部以H2PO42-存在,这部分酸加上游离H+可通过酸碱滴定来测定,称为可滴定酸度(UTA)。
⑶以NH4+形式排出,NH3在肾小管腔中与H+结合为NH4+排出体外。假定尿中无HCO3-排出,则H+总排泄量(UH)为:UH=UTA+UNH4+;当尿中有HCO3-排泄时,则机体每排泄1mmol HCO3-,必回收1mmol H+。所以肾的H+总排泄量应为:UH=UTA+UNH4--HCO3-在肾脏某些疾病时,肾小管排酸能力可能出现障碍,血液中有磷酸盐、硫酸盐、有机酸滞留,导致代谢性肾性酸中毒,主要见于各型原发性肾小管酸中毒症。
⒍尿钠的测定 尿钠浓度可作为估计肾小管坏死程度的指标。尿钠排泄量多少取决于胞外液量及肾小管重吸收的变化,在鉴别急性肾功能衰竭和肾前性氮质血症时有意义。肾前性氮质血症是由于肾血流量灌注不足引起的肾功能损害,若缺血严重或持续时间延长(超过2小时),则可引起急性肾小管坏死,是急性肾功能衰竭的前奏曲。在急性肾衰时,肾小管功能受损,不能很好地重吸收钠,故尿钠浓度>40mmol/L;而肾前性氮质血症的肾功能没有损坏,由于血容量不足,肾小管最大限度地重吸收钠,以维持血容量,故尿钠浓度<20mmol/L;若尿钠在20-40mmol/L之间,则表明病人正在由肾前性氮质血症向急性肾衰发展。
滤过钠排泄分数(FeNa)对鉴别肾前性氮质血症和急性肾衰有特别意义。以尿钠浓度表示肾小管功能状况不正确的理由是易理解的。尿钠浓度与自由水清除值呈反比,而醛固酮和抗利尿激素可使尿钠浓度向相反方向转变。FeNa则不受上述因素影响,故能正确地反映肾小管的功能。FeNa的定义为肾小球滤过的钠经肾小管重吸收后,由肾排出的百分率。

尿钠和血钠的单位为mmol/L,尿肌酐和血肌酐单位为μmol/L
尿标本、血标本应为同一时间采集的。在急性肾衰时,FeNa>1(主要是根据急性肾衰时,尿钠浓度增高和尿肌酐/血肌酐比值下降的原理)。肾前性氮质血症FeNa<1。
三、早期肾损伤的生化诊断
蛋白尿是肾脏疾病的一个重要指标,在某些肾脏病早期,尿常规测定常为阴性,尿中蛋白质含量实际已有微量的增加。为早期发现肾脏疾病,必须做一些尿中微量蛋白的检测,以监测肾脏以及某些其它器官的功能状态,提供可靠的生化指标。随着免疫化学技术的发展,已能检测纳克/ml(ng/ml)水平的微量蛋白尿,对肾脏及肾脏有关疾病的早期诊断具有重要的意义。
尿微量蛋白是指常规定性或定量难以检出的一些尿蛋白,其理化性质、合成部位、生理功能都各不相同。正常情况下,尿中这些蛋白质总含量仅为微克至毫微克水平,某些肾脏疾病或尿路其它部位分泌的蛋白可在病理乃至正常情况下出现于尿中。目前最常检查的有:白蛋白(albumin,Alb)、转铁蛋白(transferrin,Tf)、α1-微球蛋白(α1-microglobulin,α1m)、β2-微球蛋白(β2-microglobulin,β2m)、溶菌酶(lysozyme,LYS)、TH糖蛋白(Tamm-Horsfallprotein,THP)、纤维蛋白降解产物(fibrindegradation products,FDP)、C3、IgG、IgA、IgM、本周蛋白(Bence-Jones protein,B-JP)、视黄醇结合蛋白(retinal binding protein,RBP)、N-乙酰-β-氨基葡萄糖苷酶(N-acetyl-β-D-glu-cosaminidase,NAG)、碱性磷酸酶(alkaline phosphatase,ALP)、α2巨球蛋白(α2-macroglobulin,α2-MG)等。
㈠肾小球性微量蛋白
尿微量蛋白检测目的在于早期发现肾脏疾病。肾小球滤过膜通透性增加和滤膜的静电屏障受损,使肾小球滤液中的蛋白质增加,若超过肾小管重吸收的阈值,尿中出现高分子蛋白,构成肾小球性蛋白。肾小管性蛋白尿包括Alb、IgG、IgA、IgM、C3、Tf、α2-MG等出现或增多,对各类肾小球病变具有特异性鉴别诊断价值。相对分子量大小反映肾小球滤膜通透性的改变程度,尿Ig检测有助于肾脏疾病分期及预后判断。
⒈选择性蛋白尿 肾小球通透性改变可用选择性蛋白尿表示。选择性蛋白尿是指肾小球滤膜对血浆蛋白能否通过具有一定的选择性。相对分子量较大的蛋白质不易滤过,相对分子量较小的蛋白质较易滤过,即选择性滤过。尿中仅有少量大分子蛋白质排出,这些蛋白尿称为选择性蛋白尿。非选择性蛋白尿是指不论蛋白质相对分子量大小,以同样的速率滤过,此时尿中有大量大分子蛋白质排出。
蛋白尿选择性估计,在临床上一般有两种方法:
⑴选择性指数(selectiveproteinuria index,SPI):即测定IgG清除率与转铁蛋白清除率的比值。
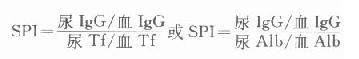
SPI<0.1者为选择性蛋白尿,>0.2者属非选择性蛋白尿。
⑵测定尿中两种大小悬殊的蛋白质(如Tf与IgM或α2-MG):以其相对清除率对数与相对分子量对数绘制的直线斜率θ角来表示。通常先代入直线方程,求出直线斜率K,查三角函数表或计算θ角。θ>64°为选择性好,53°-64°为一般选择性,<53°为非选择性。
以上无论IgG、Tf、Alb的测定,由于高灵敏度、简单、稳定的酶联免疫法(ELISA)替代了放射免疫法(RIA)在临床的应用,为快速、准确的判断提供了可行性。
SPI可推测病理类型:蛋白尿选择性可反映肾小球滤过膜的通透性,在某种程度上与肾小球疾病的病理组织学改变有一定关系。可预测治疗反应及估计预后:选择性高者预后好,反之预后差。
⒉白蛋白、免疫球蛋白测定应用ELISA法测定免疫球蛋白(IgG、IgA、IgM)及白蛋白(Alb),检出量可达到ng/ml水平,在肾脏病早期,尿常规阴性时,即肾小球滤膜通透性改变而肾小管未受累,尿中β2m和THP含量正常时,检测尿微量蛋白为肾小球病变的早期诊断提供线索。
尿微量蛋白的测定可推测肾小球病变的严重性。肾小球轻度病变时尿中Alb增高;当肾小球进一步受损时,尿IgG及IgA增高;肾小球严重病变时尿中IgM增高。尿中Alb及IgG出现提示病变向慢性过渡,尿中IgM出现对预测肾衰有重要价值。全身性疾病累及肾脏,尿中出现Alb及IgG作为肾小球受损的一个过筛试验,尿IgA增高可见于炎症性小管及间质性病变尿常规阴性者。
⒊纤维蛋白降解产物(FDP)测定 任何原因所引起的体内凝血功能亢进,均可同时促发纤维蛋白溶解(纤溶)系统,以保持动态平衡。肾小球肾炎是一种免疫性损伤,在发病过程中存在着肾小球毛细血管局部凝血以及纤维蛋白沉着,继而导致纤溶亢进,尿纤维蛋白尿降解产物(FDP)生成增多。FDP属肽类碎片,正常人尿液中无FDP,尿FDP的出现意味着肾内有凝血和纤溶现象,也提示存在炎症,可用来鉴别肾炎和肾病。肾病患者尿中FDP阴性约占80%,肾炎患者尿FDP的含量往往与尿蛋白成正比,而肾病患者尿FDP和尿蛋白无明显关系。动态地观察尿FDP的变化,对肾移植后排斥反应的诊断也有一定意义。一般说来,肾移植后尿中FDP持续升高的,往往提示即将出现排斥反应。FDP的测定方法目前多采用免疫学方法。
㈡尿低分子量蛋白
尿低分子量蛋白是一组能自由通过肾小球滤过膜而在肾近曲小管全部吸收的蛋白。此组蛋白尿排量增加是肾近曲小管受损的标志。此组蛋白主要有视黄醇结合蛋白(RBP)、β2-微球蛋白(β2m)、α1-微球蛋白(α1m)、和Tamm-Horsfall蛋白(THP)。
⒈β2-微球蛋白(β2m)测定 β2-微球蛋白(β2m)是一种相对分子量小的蛋白质,分子量为11800,主要由淋巴细胞产生,肿瘤细胞合成β2m的能力非常强。由于β2m相对分子量小,进入血循环的β2m可从肾小球自由滤过,约99.9%被近端小管重吸收,仅0.1%由终尿排出体外。β2m几乎全部在肾进行分解代谢而不会以原形重吸入血而影响浓度。肾病患者β2m合成速度比正常高4-7倍。
测定血清和尿液β2m目前主要用酶联免疫抑制试验。
肾小球滤过率(GFR)及肾血流量降低,则血清β2m升高,β2m与GFR呈直线负相关。当肾小球滤过功能减退,β2m即开始上升,故测定血清β2m能较好地了解肾小球滤过功能,并且较血肌酐浓度增高更早、更显著;肾移植成功后血清β2m很快下降,甚至比血肌酐浓度下降更早,当发生排异反应时,由于肾功能下降及排异引起的淋巴细胞增多而使β2m合成增加,血清β2m常升高,且往往较血肌酐升高早更明显。
尿液β2m升高是反应近端小管受损的非常灵敏和特异的指标:近端小管是β2m在体内处理的唯一场所,故近端小管受损时尿β2m浓度明显增加,说明肾小管重吸收障碍,称为肾小管性蛋白尿,以区别于以白蛋白为主的肾小球蛋白尿,可用来鉴别上、下尿路感染。上尿路感染时,尿β2m浓度明显增加,而下尿路感染时则正常。肾移植时无排异反应者,尿β2m不高,当出现急性排异反应,在排异期前数天即见尿β2m明显升高,在排异高危期定期测定有一定价值。在判断尿β2m升高的临床意义时,必须考虑血β2m浓度。在肾小球损伤、恶性肿瘤及自身免疫性疾病等致血清β2m明显升高,超过肾小管重吸收极限时,尿中β2m均增加。
⒉α1-微球蛋白(α1m)α1-微球蛋白(α1m)是相对分子量为26000-33000的糖蛋白,由于该蛋白的产生较恒定,较容易透过肾小球滤过膜,滤过的绝大部分又被肾小管重吸收,且其测定不受尿pH等因素的影响,因此在肾脏病诊断方面被认为具有重要价值。
血α1m、β2m与血肌酐呈明显正相关;尿α1m增高与肾小球滤过膜的通透性改变或肾小管重吸收改变有关。尿β2m是主要反映肾小管功能受损的指标,而肾小管对α1m重吸收障碍先于β2m,因此,尿α1m比β2m更能反映肾脏早期病变。
⒊视黄醇结合蛋白(RBP) 视黄醇结合蛋白(RBP)是存在于血液中的一种低分子蛋白,相对分子量约为21000。在正常人当血液中的RBP一旦经肾小球滤过后,则在肾近曲小管重吸收,因此,正常人尿中RBP排量极少(约100μg/24h)。尿RBP排量升高能敏感的反映肾近曲小管的损害程度。
用双抗体夹心酶免疫法(ELISA)检测尿RBP含量,正常值为0.11±0.07mg/L,尿RBP排量与小管间质损害程度有明显相关,可作为监测病程,指导治疗和判断预后的一项灵敏的生化指标。
⒋Tamm-Horsfall蛋白(THP)正常时THP是肾小管髓袢厚壁升支及远曲小管细胞合成和分泌的一种糖蛋白,它作为一隐秘抗原只存在于上述细胞膜上,而不暴露于免疫系统。当小管间质病变,THP漏入间质引起免疫反应产生抗THP抗体。尿中THP检测用于诊断、监测肾小管损伤(如毒物、肾移植排异反应)。肾实质病变、肾单位大量减少时尿中排出减少,单纯下尿路感染时排量正常。血中THP抗体高低可区别上下尿路感染,在急性肾盂炎时,血清THP抗体的IgG组份显著增高,而膀胱炎则无此现象。血清THP抗体IgG组份的测定有助于泌尿道感染的定位。
㈢尿酶的检测
正常尿含酶量极少,肾脏疾患时血液中以及肾组织中的某些酶可在尿中出现,从而使尿酶活性发生改变,这些改变和肾脏病变有关。目前已知尿酶有40多种,认为对肾脏疾病较有诊断价值的尿酶约有10多种,主要有:乳酸脱氢酶(lactate dehydrogenase,LDH)、碱性磷酸酶(ALP)、亮氨酸氨基肽酶(leucine aminopeptidase,LAP)等属于反映代谢的酶;溶菌酶(LYS)、β-葡萄糖苷酸酶(β-glucuronidase,β-GLU)、N-乙酰-β-氨基葡萄糖苷酶(NAG)等为溶酶体的酶;γ-谷氨酰转肽酶(γ-glutamyl transpeptidase,γ-GT)和丙氨酸氨基肽酶(alanine aminopeptidase,AAP)是反映近端肾小管刷状缘功能的酶。
⒈乳酸脱氢酶(LDH)是一种糖原醇解酶,相对分子量120000,广泛存在于各种器官、组织细胞及体液中。70%肾疾患者尿LDH均可升高,故缺乏特异性,主要用于随访肾实质病变的进展。
⒉溶菌酶(LYS)相对分子量为15000,可从肾小球自由滤过,几乎全部被肾小管重吸收,故尿中含量极微(<2μg/ml)。在肾盂肾炎和肾小管-间质性疾病,由于肾小管重吸收功能障碍,尿LYS明显升高而血清浓度正常。肾移植后发生排异反应时,尿LYS亦升高。
⒊N-乙酰-β-氨基葡萄糖苷酶(NAG) NAG是一种溶酶体酶,相对分子量约130000-140000,广泛分布于各组织中,血液中的NAG因相对分子量大,不能经肾小球滤过,肾小球功能正常时,尿中NAG不是来自血浆。肾组织特别是肾小管上皮细胞含有丰富的NAG,其浓度远高于输尿管及下尿道,一般认为尿中NAG活性增高可作为肾损伤的标志。测定尿NAG常能发现早期的肾毒性损害。肾移植急性排异反应时,尿NAG常明显升高,甚至早于肾功能的改变。
⒋丙氨酸氨基肽酶(AAP) AAP是一种刷状缘酶,相对分子量为240000,不能经肾小球滤过。尿中AAP主要来自近端小管上皮细胞,任何原因致近端小管损伤均可致尿AAP增高,其增高常缺乏特异性,目前多用于监测药物等引起的肾毒性反应。
⒌β-葡萄糖苷酸酶(β-GLU)尿中β-GLU主要来自肾小管和膀胱上皮细胞的溶酶体。相对分子量为230000。尿中β-GLU不受血清β-GLU影响,尿β-GLU在活动性肾盂肾炎和活动性肾小球肾炎时中度升高,急性肾小管坏死、肾移植急性排异时显著升高。亦可区别良恶性肿瘤,90%恶性肿瘤患者尿β-GLU升高。
⒍γ-谷氨酰转肽酶(γ-GT)γ-GT存在于许多组织中,以肾脏中含量最高,主要在近曲小管刷状缘。正常人尿γ-GT较血清高2-6倍,是肾功能指标之一。多数肾小球肾炎γ-GT增高,慢性肾盂肾炎γ-GT正常,肾肿瘤γ-GT含量小于正常肾脏,肾移植排异时γ-GT升高。
⒎亮氨酸氨基肽酶(LAP)LAP在人体各组织中广泛存在,在毒性物质或疾病影响到富含LAP的近端小管时,尿LAP活性最高。肾小球基底膜通透性增高、肾小管上皮细胞损害、药物致中毒性肾损害和肾肿瘤时LAP增高。肿瘤治疗后尿LAP增高提示肿瘤复发。对210例各种肾脏病例进行分析,发现LAP阳性率最高。
⒏碱性磷酸酶(ALP)正常人尿液中ALP主要来自肾小管上皮细胞,当肾小球滤过功能障碍、肾缺血、肾小管上皮细胞坏死或过度脱落时,尿中ALP即可显著增高。ALP可作为药物性肾损害的早期诊断指标。
在这些尿酶中间以尿NAG在尿中比较稳定,检测方法相对简易,检测值可靠,并较其它尿酶更敏感地反应肾脏病变。LYS、NAG、β-GLU等测定均可反映近端肾小管的损伤,LYS主要反映重吸收功能的缺陷或损伤;NAG、β-GLU主要反映其急性损伤,是病变活动的灵敏指标。尿酶的检测应注意:各种体内外因素均会影响结果,如标本贮存或冰冻、尿液本身pH、稀释浓缩状态、尿中成分以及患者用药的影响。
四、肾小球-肾小管损伤标志物的组合分析
肾小球滤过功能一般以肌酐清除率作为常规评价指标,选择性尿白蛋白的测定是肌酐清除率的协同指标,这两个指标的应用对肾小球滤过功能的早期损伤的评价已比较完善。
血尿素氮、血肌酐和血尿酸的测定,仍为临床常用的反应肾小球功能的标志。由于肾脏可通过肾小管排泄肌酐,故肾脏疾病早期血清肌酐通常是不高的,直至肾功能实质性损伤时,血清肌酐值才增高,所以血肌酐测定对晚期肾脏病临床意义较大。同时测定尿素氮和肌酐对临床诊断有帮助。正常情况下尿素氮与肌酐之比为(15-24):1。在肾脏疾病,血清尿素氮增高比肌酐更明显,由于肾前原因(特别是严重肠道出血)引起尿素氮值明显增高。由于尿道阻塞而使非蛋白含氮化合物滞留,则将出现尿素氮及肌酐值同时成比例增高。在严重肾小管损害时,尿素氮与肌酐之比可降低至10:1。
肾小管重吸收功能一般以α1m、β2m和RBP等作为评价指标,这类低相对分子量蛋白质容易通过肾小球滤膜,绝大部分又被肾小管重吸收。一旦尿中出现,即反应了肾小管重吸收功能障碍。
临床可选用白蛋白作为肾小球滤过功能标志物,α1m作为肾小管功能标志物,以弥补常规尿酶联试纸和镜检漏检的肾小球性和肾小管性蛋白尿。
对于近端小管的损伤可用NAG、ALP作为标志,NAG较灵敏,非特异性的ALP可作为近端小管的补充标志物。髓袢损伤标志物以THP为主。
这些标志物的应用(表11-2)使肾脏疾病在可逆转的阶段就可得到诊治,对肾脏疾病的早期诊断治疗提供了可能性。
表11-2 肾小球-肾小管损伤的标志物
| 损伤部位 | 可检出的标志物 |
| 肾小球选择通透性 | 白蛋白(Alb)、运铁蛋白(Tf)、IgG、α2-巨球蛋白(α2-MG) |
| 肾小管重吸收 | α1-微球蛋白(α1m)、β2-微球蛋白(β2m)、视黄醇结合蛋白(RBP)、溶菌酶(LYS) |
| 近端小管刷状缘 | γ-谷氨酰氨基转移酶(γ-GT)、碱性磷酸酶(ALP)、丙氨酸氨基肽酶(AAP) |
| 近端小管溶酶体 | N-乙酰-β-氨基葡萄糖苷酶(NAG)、β-葡萄糖苷酸酶(β-Glu) |
| 肾小管胞质 | 乳酸脱氢酶(LDH) |
| 肾小管髓袢厚壁升支 | Tamm-Horsfall蛋白(THP) |
第十二章 内分泌疾病的生物化学诊断
第一节 概述
内分泌是指机体某些腺体或散在的特化细胞,能合成并释放具有生物活性的物质,随血液循环输送到其他部位的靶器官、靶细胞、传递细胞间信息,调节这些器官或细胞的代谢和功能的过程。这类生物活性物质称激素。内分泌系统和神经系统是调节机体的各种正常代谢和功能的两个既互相影响又协调的主要系统。体内的各种激素是在神经系统的参与下,通过复杂而精细的调节机制,保持在与机体发育阶段及功能状态相适应的水平。其中以反馈调节方式,通过下丘脑-垂体-内分泌腺或细胞-激素系统进行的调控,是普遍而主要的调节机制(图12-1)。该调节系统任一环节异常,都将导致激素水平紊乱。产生相应的内分泌病。
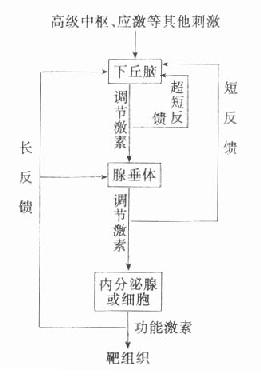
图12-1 激素分泌的下丘脑-垂体-内分泌腺(细胞)调节轴示意图
因此,有关内分泌紊乱性疾病的临床生化检查主要用作①确定病人是否存在某一内分泌功能紊乱;②若存在紊乱,则进一步确定病变部位和性质。
有关内分泌病的临床生化诊断方法主要有以下三类:
⒈某内分泌腺特有的或其分泌的激素调节的生理、生化过程的检测如甲状腺功能紊乱时131I摄取试验或测定基础代谢率,甲状旁腺功能紊乱时血钙测定。这类检测可协助诊断有无某一内分泌功能异常。
⒉直接检测体液中某一激素或其代谢物的水平或转运蛋白浓度可对判断有无某一种类内分泌病提供有诊断价值的客观指标,在内分泌病诊断中普遍应用。
⒊动态功能试验(dynamic function test)即应用对激素分泌的前述反馈调节系统中某一环节具特异性刺激或抑制作用的药物、激素,分别测定使用前后相应靶激素水平的动态变化。动态功能试验结合其他检查手段,对导致内分泌紊乱的病变部位(环节)及病变性质的诊断很有价值。
近年来,发现某些非内分泌组织的肿瘤细胞可分泌异源性激素(ectopic hormone),产生异源性内分泌病。异源性激素分泌,均有不受上述下丘脑-垂体-内分泌细胞调节轴影响、而呈“自主性”分泌的特点。有人提出,可分泌异源性激素的肿瘤细胞组织,在胚胎发育上与正常内分泌组织均起源于神经嵴外胚层,这类可分泌激素的组织细胞称为胺原摄取及脱羧细胞(amine precursor uptake and decarboxylation cell,APUD细胞)。肿瘤组织中的APUD细胞分化不完全,故具有产生异源性激素的内分泌功能。此外,一些调节内分泌腺功能的激素存在交叉效应,如促甲状腺激素释放激素除促进垂体释放促甲状腺激素外,还可增加垂体催乳素和生长激素分泌。这些在有关内分泌紊乱的诊断中,必须考虑到。
激素按化学结构分作氨基酸及肽、蛋白类,类固醇类和儿茶酚胺类三类。其中氨基酸及肽或蛋白类主要有甲状腺激素、甲状旁腺激素、下丘脑激素、垂体激素、降钙素、某些胎盘激素、心肌激素和胃肠道激素等;类固醇类激素包括性激素和肾上腺皮质激素;儿茶酚胺类激素则主要指肾上腺素和去甲肾上腺素。其中参与Ca2+、水和Na+、K+及糖代谢调节的激素紊乱,已分别在第三、五、六章中介绍,胎盘激素将在第十四章中讨论。本章主要介绍甲状腺激素、肾上腺皮质及髓质激素、下丘脑-垂体激素、性激素等紊乱的临床生化诊断的有关内容。
第二节 甲状腺功能紊乱的临床生化
一、甲状腺激素的生理、生化及分泌调节
㈠甲状腺激素的化学及生物合成
甲状腺激素为甲状腺素(thyroxine,T4)和三碘甲腺原氨酸(3,5,3′-triiodothyronine,T3)的统称。从化学结构看均是酪氨酸的含碘衍生物(图12-2)。
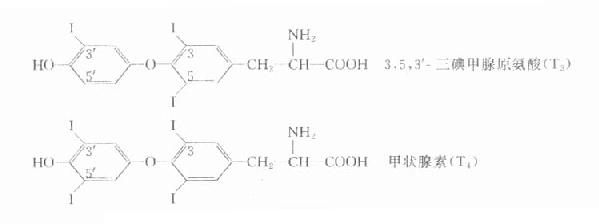
图12-2 甲状腺激素化学结构示意图
T3、T4均是由甲状腺滤泡上皮细胞中甲状腺球蛋白上的酪氨酸残基碘化而成。其生物合成包括:①碘的摄取和活化:甲状腺上皮细胞可通过胞膜上的“碘泵”主动摄取血浆中的I-,造成I-在甲状腺浓集,正常情况下甲状腺中的I-为血浆浓度的数十倍。甲状腺上皮细胞中的I-在过氧化酶催化下,氧化成形式尚不清的活性碘。②酪氨酸的碘化及缩合:活性碘使甲状腺上皮细胞核糖体上的甲状腺球蛋白中的酪氨酸残基碘化,生成一碘酪氨酸(MIT)或二碘酪氨酸(DIT)残基。然后再在过氧化酶催化下,一分子DIT与一分子MIT缩合成一分子T3,两分子DIT缩合成一分子T4。含T3、T4的甲状腺球蛋白随分泌泡进入滤泡腔中贮存。
㈡甲状腺激素的分泌、运输、代谢及调节
在垂体促甲状腺激素刺激下,含T3、T4的甲状腺球蛋白被甲状腺上皮细胞吞饮,并与溶酶体融合,在溶酶体蛋白水解酶催化下水解出T3、T4,释放至血液中。血液中99%以上的T3和T4均与血浆蛋白可逆结合,主要与血浆中肝合成的一种α球蛋白-甲状腺素结合球蛋白(thyroxine binding globulin,TBG)结合,此外尚有少量T3及10%-15%的T4可与前白蛋白结合,约5%T4及近30%的T3可与白蛋白结合。只有约占血浆中总量0.4%的T3和0.04%的T4为游离的。但只有游离的T3、T4才能进入靶细胞发挥作用,这是T3较T4作用迅速而强大的原因之一。与血浆蛋白结合的部分,则对游离T3、T4的相对稳定起着调节作用。
甲状腺激素的代谢包括脱碘、脱氨基或羧基、结合反应。其中以脱碘反应为主,该反应受肝、肾及其他组织中特异的脱碘酶催化。此酶对T3作用弱,主要催化T4分别在5’或5倍脱碘,生成T3和几无生理活性的3,3’,5’-三碘甲腺原氨酸,即反T3(reverse triiodothyronine,r T3)。血液中的T3近80%来自T4脱碘。T3及r T3可进一步脱碘生成二碘甲腺原氨酸,T3和T4尚可脱氨基、羧基,生成相应的低活性代谢物。少量T3、T4及上述各种代谢物均可在肝、肾通过其酚羟基与葡萄醛酸或硫酸结合,由尿及胆汁排泄。
甲状腺激素的合成和分泌主要受前述下丘脑-垂体-甲状腺轴的调节。血液中游离T3、T4水平的波动,负反馈地引起下丘脑释放促甲状腺激素释放激素(thyrotropin-releasinghormone,TRH)及垂体释放促甲状腺激素(thyroiodstimulated hormone,TSH)的增多或减少。TRH为下丘脑产生的一种三肽激素,主要作用为促进腺垂体合成和释放TSH,亦有弱的促生长激素和催乳素释放作用。TSH为一种含α和β两亚基的糖蛋白,可通过β亚基特异地和甲状腺细胞膜上的TSH受体结合,活化腺苷酸环化酶,通过腺苷酸环化酶-cAMP-蛋白激酶系统,刺激甲状腺细胞增生和甲状腺球蛋白合成,并对甲状腺激素合成中从碘摄取到T4、T3释放的各过程均有促进作用。在上述调节过程中,血液游离T3、T4水平对腺垂体TSH释放的负反馈调控最重要。此外肾上腺皮质激素可抑制TRH释放,并和生长激素均能降低腺垂体对TRH的反应性,减少TSH分泌;而雌激素可敏化腺垂体对TRH的反应,促进TSH释放;甲状腺激素本身和I-浓度对甲状腺功能也有自身负反馈调节作用,应激状态等亦可通过不同途径影响甲状腺激素的分泌;人绒毛膜促性腺素(hCG)也具一定TSh 样活性。近年发现,多种滋养层源组织肿瘤如绒毛膜上皮癌、睾丸胚胎瘤等亦可产生TSH和hCG。
㈢甲状腺激素的生理生化功能
大多数组织细胞核染色体的某些转录启动区上,存在甲状腺激素受体,该受体对T3亲和力远比T4高,这也是T3作用强的一个原因。T3、T4与该受体结合后,可促进某些mRNA的转录,增加Na+、K+-ATP酶等相应蛋白质合成,产生下列作用。
⒈三大营养物质代谢提高大多数组织的耗氧量,促进能量代谢,增加产热和提高基础代谢率。该作用与甲状腺激素增加Na+、K+-ATP酶活性、促进ATP分解供能产热有关。但甲状腺激素对三大营养物质代谢的具体影响较复杂,对糖代谢既可促进糖的吸收和肝糖原分解,又可促进组织细胞对糖的有氧代谢。甲状腺激素可促进体脂动员;对胆固醇代谢有重要的调节作用,能促进肝脏合成胆固醇,而促进胆固醇代谢为胆汁酸的作用更显著。生理浓度的甲状腺激素可通过诱导mRNA合成,增强蛋白质的同化作用,呈正氮平衡。但过高的甲状腺激素反致负氮平衡,特别是肌蛋白分解尤为显著。
⒉骨骼、神经系统发育及正常功能维持甲状腺激素可与生长激素产生协同作用,增强未成年者的长骨骨骺增殖造骨,以及蛋白质同化作用,促进机体生长发育。另一方面,甲状腺激素可刺激神经元树突、轴突发育,神经胶质细胞增殖,髓鞘的形成,影响神经系统的发育。甲状腺激素对长骨和神经系统生长发育的影响,在胎儿期和新生儿期最为重要。对成人则可维持中枢神经系统的正常兴奋性。
⒊其他作用甲状腺激素可产生类似肾上腺素β受体激动样心血管作用,加快心率,提高心肌收缩力,增加心肌氧耗,扩张外周血管。
二、甲状腺功能紊乱
㈠甲状腺功能亢进症
甲状腺功能亢进症(hyperthyroidism)简称甲亢,系指各种原因所致甲状腺激素功能异常升高产生的内分泌病。病因复杂多样,约75%为弥漫性甲状腺肿伴甲亢,即Graves病,该病现倾向于为一种自身免疫病;另有约15%为腺瘤样甲状腺肿伴甲亢;近10%为急性或亚急性甲状腺炎;垂体肿瘤、滤泡性甲状腺癌性甲亢及异源性甲亢等均属少见。
甲亢的病理生理生化改变多为前述甲状腺激素功能病理性增强的结果。主要表现为:①高代谢综合征:由于三大营养物质及能量代谢亢进,食多但消瘦,怕热多汗,基础代谢率明显升高。对胆固醇分解代谢的促进使血清胆固醇降低。甲状腺激素水平过高时,蛋白质特别是肌蛋白的分解代谢增强,出现肌肉萎缩、乏力、血及尿中肌酸明显升高、超出正常上限数倍等负氮平衡表现。②神经系统兴奋性升高,烦躁易激动,肌颤等。③心率加快,心输出量增多,收缩压升高而脉压差增大,可出现心律失常。④突眼症及甲状腺肿大等。
㈡甲状腺功能减退症
甲状腺功能减退症(hypothyroidism)简称甲减,指由各种原因所致甲状腺激素功能异常低下的多种内分泌病统称。其中有各种原因,如甲亢或癌肿放射治疗、手术切除过多、慢性甲状腺炎症、低碘或高碘、抗甲亢药过量等,直接影响甲状腺T3、T4合成分泌减少所致的甲状腺性甲减占绝大多数,其次为肿瘤、放疗、手术等损伤下丘脑或垂体,使TRH和(或)TSH释放不足所致甲减,但此时多为复合性内分泌紊乱。遗传性甲状腺激素受体缺陷性甲减极为罕见。
由于甲状腺激素对骨骼和神经系统生长发育的作用,使甲减的临床表现按起病年龄不同而有特殊的症状,并因此分作:甲减始于胎儿及新生儿的呆小症,始于性发育前少儿的幼年型甲减,及始于成人的成年型甲减三类。成年型甲减主要表现为甲状腺激素对三大营养物质和能量代谢调节、维持神经系统及心血管系统正常功能等作用减弱的各种表现。如精神迟钝、情况低下、畏寒少汗(皮肤干燥)、基础代谢率低、乏力、心脏功能抑制,以及性腺和肾上腺皮质内分泌功能减退等。其特殊表现为:此时虽然绝大多数蛋白质同化作用减弱,但细胞间粘蛋白合成却增多。粘蛋白富含阴离子,可大量吸引正离子和水分,形成非凹陷性粘液性水肿,亦可因此导致心肌、脑、肝、肾、骨骼肌等组织和器官发生间质性水肿,出现相应症状。此外50%以上甲减病人可有轻中度贫血,脑脊液蛋白量常见升高,半数病人血清谷丙转氨酶轻中度升高,因甲状腺激素促胆固醇分解代谢的作用减弱,出现血清胆固醇、甘油三酯及低密度脂蛋白均升高等一般实验室检查改变。另有约80%甲减者血清肌酐激酶(CK)主要是CK-3同工酶活性明显升高,平均可达正常上限5倍,原因不明。
起病于肿儿及新生儿期的甲减,除可表现上述成人型表现外,由于对此期骨骼和神经系统生长发育的影响,出现体格及智力发育障碍的特征性改变,故称呆小病或克汀病(cretinism)。幼年型甲减则视发病年龄而表现上述两型程度不一的混合表现。
必须指出,甲状腺功能紊乱实际上是一种常见而至今仍易被漏诊的疾病。据病因临床内分泌医师学会1996年1月报告,在对美国11个城市4.6万人的普查中发现,有11%的人存在未被诊断出的甲状腺功能紊乱,应引起足够重视。
三、甲状腺功能紊乱的生化诊断
㈠血清甲状腺激素测定
前已提及,T3、T4在血中均99%以上和TBG等血浆蛋白结合,游离部分更能可靠反映甲状腺激素的生物活性,因此,血清甲状腺激素测定包括总T3(TT3)、总T4(TT4)、游离T3(FT3)和游离T4(FT4)测定。此外,为排除血浆蛋白结合率改变的影响,尚有测定甲状腺激素结合比值(131I-T3树脂摄取率)、游离T4、T3指数等方法。
⒈血清TT4、TT3及FT4、FT3测定 由于即便TT4、TT3在血液中亦仅微量存在,T4、T3本身又具有一定抗原性,因此对四者的检测目前均用免疫化学法。TT3、TT4早期多用125I标记T3或T4的放射免疫法测定,现在用多相酶联免疫法(ELISA)、均相酶放大免疫法(EMIT)及数种荧光免疫法分别测定血清TT4或TT3的技术已广泛应用,并能实现自动化检测。测定FT4、FT3以平衡透析前后免疫化学测定法为标准参考方法,但本法耗时且成本高。在临床实验室中采用的是非透析免疫化学测定。在测定TT4、TT3时,血清需用8-苯胺-1-萘磺酸(ANS)及巴比妥缓冲液等预处理,使与血浆蛋白结合的T4、T3解离出再测定。FT4或FT3抗体结合的标记T4或T3类似物法;亦有采用抗体标记法;还有将相应抗体固定于测定池壁上,待血清中FT4或FT3与其结合后,除去血清再加入标记FT4或FT3,根据竞争性结合原理,测定血清中FT4或FT3的“两步法”。
血清TT4、TT3浓度受血中TBG水平影响。TBG正常者,不同年龄段TT4及TT3水平亦有较大差异,其正常值参考范围见表12-1。
FT4及FT3由于血清浓度甚低,受检测方法、试剂盒质量、实验室条件等的影响显著,文献报告正常值差异大。FT4平均为19-30pmol/L(1.5-3ng/dl),FT3为6.0-11.4pmol/L(0.39-0.74ng/dl)。
血清TT4、TT3及FT4、FT3在甲亢和甲减的诊断、病情严重程度评估、疗效监测上均有价值。但影响上述指标的因素较多,除测定中的各种因素外,还受血浆TBG浓度、多种非甲状腺功能紊乱疾病、抗甲亢药或甲状腺激素治疗等的影响。正常成人血清TBG浓度为15-34mg/L,当TBG升高或降低时,TT4、TT3亦相应升高或降低。雌激素及含雌激素避孕药、病毒性肝炎、妊娠、遗传性高TBG症等时,血清TBG增多;而雄激素、糖皮质激素、肾病综合征、各种原因致蛋白营养不良及应激状态等,均可使血清TBG减少。而严重心、肝、肾疾患,使用抗心律失常药乙胺碘呋酮等脱碘酶抑制剂,可因影响T4在外周组织脱碘代谢成T3及rT3,使TT4、FT4升高,TT3、FT3减少,T3/T4比值下降。此外,各种影响血浆白蛋白浓度的因素,除可改变T4、T3与白蛋白结合率外,尚可因TT4及TT3免疫法测定的抗体,系用T3或T4与白蛋白结合后免疫动物制取的,故亦可影响测定。因此,在评价甲状腺激素测定结果时,应考虑上述因素的影响。从理论上讲,FT4及FT3,特别是后者,较少受影响TBG浓度及T4脱碘为T3因素干扰,且为甲状腺激素能发挥生物效应的主要形式,更有意义。但由于前述影响FT4、FT3测定可靠性原因及未普遍开展,故临床仍多用TT4及TT3测定。为减少TBG改变时TT4、TT3测定结果的影响,可考虑采用下列方法校正。
表12-1 不同年龄血清TT4、TT3正常参考值
| 脐血 | 新生儿 | 婴儿 | 1-5岁 | 6-10岁 | 11-60岁 | >60岁男 | >60岁女 | |
| TT4(nmol/L) | 101-169 | 130-273 | 91-195 | 95-195 | 83-173 | 65-156 | 65-130 | 72-136 |
| TT3(nmol/L) | 0.5-1.1 | 1.4-2.6 | 1.4-2.7 | 1.5-4.0 | 1.4-3.7 | 1.8-2.9 | 1.6-2.7 | 1.7-3.2 |
注:换算为μg/dl的惯用单位换算因子TT4为0.077、TT3为0.065
⒉甲状腺激素结合比值(thyroidhormone-binding ratio,THBR)试验旧称T3树脂摄取率(T3resin uptake ratio,T3RUR)试验。其原理为血清中的T4与TBG的结合较T3牢固,因此在血清中加入过量125I-T3,125I-T3将只能与未被T4及T3结合的游离TBG结合,以树脂或红细胞摄取游离的125I-T3后计量,与正常标准血清比较,可计算出待测血清的THBR。THBR可间接反映TT4及TBG浓度,当血清TBG浓度正常时,TT4及TT3与THBR平行。正常参考值为0.98-1.00。
⒊游离T4指数(free T4index,FT4I)和游离T3指数(free T3index,FT3I)根据TT4、TT3和THBR结果,分别按FT4I=TT4×THBR及FT3I=TT3×THBR计算得。TT4以μg/dl表示时,FT4I正常值参考范围为2.2-14.0;TT3以ng/dl表示时,FT3I正常值参考范围为130-165。非甲状腺功能紊乱者,TBG增多或减少,TT4、TT3虽亦相应升高或降低,但同时THBR却会向相反方向变动,因而FT4I及FT3I不会有明显改变。而甲亢、甲减者的TT4、TT3异常不是TBG改变的结果,THBR亦将有同样的变化,故FT4I及FT3I将改变更显著。FT4I及FT3I可在一定程度上校正TBG改变所致TT4、TT3变化,近似反映FT4、FT3水平。其中FT3I对甲亢诊断符合率高,FT4I对甲减的诊断较有价值。
㈡甲状腺分泌调节功能测定及意义
⒈促甲状腺激素(TSH)检测TSH均用免疫化学法测定。早年由于试剂盒灵敏度不能达到临床要求,限制了其推广应用。90年代以来,许多第三代商品化试剂盒的“功能性灵敏度”(相当于20%批间C.V的TSH浓度)已可达0.01-0.02mU/L,足以满足临床检测要求,故血清TSH测定已成为甲状腺功能紊乱的常规临床生化手段。其正常值参考范围为:脐血3-12mU/L,儿童0.9-8.1mU/L,60岁以下成人2-10mU/L,60岁以上者男女性分别为2-7.3mU/L和2-16.8mU/L。TSH水平不受TBG浓度影响,亦较少受影响TT4、TT3的多种非甲状腺疾病的干扰。单独或配合甲状腺激素测定及动态功能试验,对甲状腺功能紊乱及病变部位诊断上很有价值。美国临床内分泌学会及许多国家学者均推荐将TSH测定作为甲状腺功能紊乱实验室检查的首选项目。
甲状腺性甲减者,TSH水平升高,但甲状腺激素水平低于同龄正常值下限;甲状腺激素受体缺陷及存在T4、T3自身抗体时,TSH和甲状腺激素均升高;而TSH及甲状腺激素均低下者,多为下丘脑性TRH分泌不足或压迫TSH分泌细胞的垂体催乳素瘤等所致的继发性甲减。甲状腺性甲亢者,甲状腺激素升高而TSH低下;若二者水平均高,提示为垂体TSH分泌细胞腺瘤或异源性TSH分泌综合征所致甲亢。但TSH分泌存在昼夜节律,血液峰值出现在夜间10-11点,谷值见于上午10点左右,二者差别可达2-3倍。因此在决定取样时间及结果解释时,必须考虑这点。此外下列非甲状腺功能紊乱情况可影响TSH分泌:乙胺碘呋酮、大剂量无机碘等含I-药剂及锂盐的长期应用、缺碘地区居住、阿狄森病等,可致TSH分泌增多;而活动性甲状腺炎、急性创伤、皮质醇增多症及应用大剂量糖皮质激素、全身性危重疾病、慢性抑郁症等精神失常,可使TSH水平降低。
⒉甲状腺功能动态试验
⑴放射性碘摄取试验:利用甲状腺的聚碘功能,给受试者一定剂量131I后,测定甲状腺区的放射性强度变化,以甲状腺摄取碘的速度和量(摄取率)间接反映甲状腺合成分泌T4、T3能力。甲亢者将出现对131I摄取速度加快(峰前移)及量增多(摄取率提高),甲减者则峰平坦且摄取率下降。但本法易受富碘食物、含碘药物、缺碘、单纯性甲状腺肿等影响并且较粗糙,现已少用。但若同时扫描发现边缘模糊的“冷结节”可有助甲状腺癌的诊断。
⑵甲状腺激素抑制试验:正常者,甲状腺激素对下丘脑-垂体-甲状腺轴有敏感的负反馈调节作用,甲亢者因长期处于高甲状腺激素水平作用下,对外源性甲状腺激素的反应弱。在连续给予T4或T3一周前后,分别测定131I摄取率。正常人和伴131I摄取率高的缺碘者和单纯性甲状腺肿者,甲状腺131I摄取率将抑制达50%以上,甲亢者则变化不大,抑制率<50%。
⑶TRH兴奋试验:TRH可迅速刺激腺垂体释放贮存的TSH,因此分别测定静脉注射200-500μg TRH前及注射后0.5h血清TSH,可反映垂体TSH贮存能力。正常人基础值参考范围见前,注射TRH后,儿童TSH可升至11-35mU/L,男性成人达15-30mU/L,女性成人达20-40mU/L;或正常男性可较基础值升高约8mU/L,女性升高约12mU/L。甲状腺性甲亢病人不但TSH基础值低,并且垂体TSH贮存少,注射TRH后血清TSH无明显升高(<2mU/L);异源性TSH分泌综合征性甲亢,TSH基础值高,并且因其呈自主性分泌,所以对TRH无反应;垂体腺瘤性甲亢虽然TSH基础值高,TRH兴奋试验可呈阳性,但藉临床表现及TT4、TT3测定等,不难与甲减鉴别。甲状腺性甲减时,TSH基础值升高,TRH兴奋后升高幅度多比正常人大;下丘脑性及垂体性甲减者,虽然二者TSH基础值均低,但后者对TRH兴奋试验几无反应,而前者可有延迟性反应,即若注射TRH后除0.5h外,还分别在1及1.5h取血测定TSH,其峰值约在1或1.5h时出现。TRH兴奋试验较其他动态功能试验省时、安全、影响因素少,又可同时完成TSH基础水平测定,在病变部位的诊断上有较大意义。现认为是甲状腺功能紊乱的临床生化检测项目中最有价值和可靠的。
㈢其他方法
⒈基础代谢率(basicmetabolic rate,BMR)测定BMR早年曾广泛用于甲状腺激素功能紊乱的诊断,但影响因素众多,不可靠,现已少用。
⒉自身抗体检测现已肯定某些甲状腺功能紊乱与自身免疫反应有关。多数弥漫性甲状腺肿伴甲亢病人血清中存在多种抗甲状腺细胞成分的自身抗体,包括长效甲状腺刺激物(long-acting thyroid stimulator,LATS)、甲状腺刺激免疫球蛋白(thyroid-stimulating immunoglobulin,TSI)等。其中TSI除可保护LATS免受血清中相应抗体中和外,本身亦可和TSH竞争与胞膜上受体结合,产生持久TSH样作用,约在95%的Graves病人中可检出。检测TSI可用于其他方法无法确诊的Graves病的辅助诊断,也用作评估疗效和预后的指标。
⒊血清rT3测定前面甲状腺激素代谢中已介绍,rT3为T4在外周组织脱碘的产物,虽然生物活性很低,但其代谢慢,血清浓度男女间无差异,有人主张检测血清rT3用于甲状腺功能紊乱的诊断及疗效评估。正常人血清rT3浓度参考范围为0.6-0.77nmol/L(40-50ng/dl)。甲亢者rT3明显升高,平均可达103ng/dl;甲减者大多伴有rT3下降,特别是胎儿及新生儿,T4主要代谢为rT3,下降更明显。但影响rT3水平的因素较多,很多非甲状腺功能紊乱疾病、酸碱失调、多种药物均可改变血清rT3水平。因此其诊断价值尚有待进一步证实。
表12-2小结了常见甲状腺功能紊乱时,主要临床生化检测的改变,供参考。
表12-2 常见甲状腺功能紊乱主要临床生化检测所见
| 项目 | 甲状腺功能亢进症 | 甲状腺功能减退症 | |||||
| Graves病 | 甲状腺腺样瘤 | 垂体腺瘤 | 异源性 | 甲状腺性 | 垂体性 | 下丘脑性 | |
| 血清甲状腺激素 | 升高 | 升高 | 升高 | 升高 | 降低 | 降低 | 降低 |
| 血清TSH | 降低 | 降低 | 升高 | 升高 | 升高 | 降低 | 降低 |
| TRH兴奋试验* | 阴性 | 阴性 | 阳性 | 阴性 | 强阳性 | 阴性 | 延迟反应 |
* 以TSH为观察指标
第三节 肾上腺功能紊乱的临床生化
肾上腺是由中心部的髓质和周边部的皮质两个独立的内分泌器官组成。下面将分别讨论肾上腺髓质和皮质的内分泌功能紊乱的临床生化有关内容。
一、肾上腺髓质激素及功能紊乱
㈠肾上腺髓质激素
肾上腺髓质从组织发育学上可看做是节后神经元特化为内分泌细胞(嗜铬细胞)的交感神经节,不同的嗜铬细胞可分别合成释放肾上腺素(epinephrine,E)、去甲肾上腺素(norepinephrine,NE)、多巴胺(dopamine,DA),三者在化学结构上均为儿茶酚胺类。后两者亦为神经递质,但作为递质释放的NE和DA绝大部分又重新被神经末梢及其中的囊泡主动摄取、贮存。肾上腺髓质释放的E约为NE的4倍,仅分泌微量DA,因此血液及尿液中的E几乎全部来自肾上腺髓质分泌,NE及DA则还可来自其他组织中的嗜铬细胞及未被摄取的神经递质。
儿茶酚胺类激素以酪氨为原料,经下列酶促反应生成。由于各种组织中存在的酶有不同,故分别合成E、NE或DA。
肾上腺髓质合成的E和NE贮存于嗜铬细胞的囊泡中,其释放受交感神经兴奋控制。作为激素释放的E和NE,亦具有交感神经兴奋样心血管作用及促进能量代谢、升高血糖等作用。进入血液的E和NE均迅速被单胺氧化酶及儿茶酚胺氧位甲基转移酶等代谢灭活,与临床生化检测有关的主要代谢产物如下:

㈡肾上腺髓质嗜铬细胞瘤及其临床生化诊断
肾上腺髓质嗜铬细胞瘤(pheochromocytoma)最好发部位。由于过量的E及NE释放入血液中,作用于肾上腺素受体,产生持续性或阵发性高血压,并伴有血糖、血脂肪酸、基础代谢率升高等代谢紊乱。本病的临床生化检查主要有两类。
⒈儿茶酚胺类激素及其代谢物测定 由于检测技术限制,早年主要用比色法或荧光光度法测定24h尿中总游离儿茶酚胺类物质或VMA量。正常成人24h尿游离儿茶酚胺类物质总量<1.65μmol/d(280μg/d),VMA量为15.7-88.5μmol/d(3.1-17.6mg/d),二者明显超过正常值上限有助于嗜铬细胞瘤诊断。但血和尿中儿茶酚胺类主要为递质性NE及DA,而VMA为NE及E共有的代谢物,因此这两项指标对嗜铬细胞瘤,特别是发生于肾上腺髓质者特异性不高。并且该类测定影响因素多,香蕉、茶、咖啡等含香草的食品糖果、四环素、红霉素、多种拟肾上腺素药、抗抑郁症药及含多巴结构的药等可致假阳性;芬氟拉明、甲基葡胺造影剂可产生假阴性。随后采用检测仅为E代谢物的3-氧-甲基肾上腺素的24h尿排量,虽仍受上述因素影响,但特异性有所提高。3-氧-甲基肾上腺素的成人正常参考值范围为<5.07μmol/d。
近年来,由于采用HPLC-电化学检测法的灵敏度已可满足需要,故应用本法直接分离测定血浆中E及NE,对本病的诊断价值更高。但包括静脉穿刺取血在内的紧张及各种应急状态,甚至体位改变,均可刺激肾上腺髓质释放E及NE;而取血后室温下5min内不除去红细胞,将使E和NE浓度迅速下降。因此应在清晨平卧时,插入保留式静脉取血管(indwelling catheter),至少30min后待病人安静时再取血,转入盛有冰冻过的抗凝剂和抗氧化剂的试管中,迅速低温离心分离血浆进行测定。按上述条件测得的正常成人血浆参考值范围为E:109-437pmol/L(20-80pg/ml);NE:0.615-3.240nmol/L(104-548pg/ml)。嗜铬细胞瘤时,二者明显升高,若E升高较NE显著则提示可能为肾上腺髓质嗜铬细胞瘤。
⒉动态功能试验诊断肾上腺髓质嗜铬细胞瘤的动态功能试验方法较多。目前兴奋试验常用胰高血糖素激发试验,即在疑为本病者非发作期,按上述步骤及方法取血及测量血压后,静脉注射胰高血糖素1mg,注毕每15s量血压,1-3min内再取血,分别测定给药前后血浆E及NE。由于胰高血糖素可迅速刺激肾上腺髓质释放E及NE,因此肾上腺髓质嗜铬细胞瘤者,血浆E和NE可较基础对照值升高明显,达3倍以上,血压也急剧上升,可达26.2/21.3kPa(200/160mmHg)。本法禁用于糖尿病人。抑制试验则多用可乐定(clonidine)抑制试验。降压药可乐定可抑制递质性儿茶酚胺释放,但不影响嗜铬细胞释放E及NE。对有高血压而怀疑本病者,给予可乐定0.3mg一剂口服前及服药后3h,分别取血测定血浆NE。非嗜铬细胞瘤性高血压者,血浆NE将降低50%以上,嗜铬细胞瘤性高血压者,NE仅轻度减少。本法尤适用于有持续性高血压,其他检测结果又在边缘范围者。由于多种降压药及三环类抗抑郁药可干扰本试验,故需停用上述药至少12h后才能进行。
二、肾上腺皮质的内分泌功能
㈠肾上腺皮质激素及类固醇激素的生物合成
肾上腺皮质可分泌多种激素,按生理生化功能及分泌组织,可分做三类:①球状带分泌的盐皮质激素(mineralocorticoide),主要是醛固酮(aldosterone)和脱氧皮质酮(deoxycorticosterone);②束状带分泌的糖皮质激素(glucocorticoide),主要有皮质醇 (cortisol)及少量的皮质酮(corticosterone);③网状带分泌的性激素,如脱氢异雄酮(dehydroepiandrosterone)、雄烯二酮(androstenodione)及少量雌激素。从化学结构上看,这三类激素及性腺合成的其他性激素,均是胆固醇的衍生物,故统称类固醇激素(steroid hormones),而上述三类肾上腺皮质激素又合称皮质类固醇(corticosteroids)。
类固醇激素在人体内均是以胆固醇为原料,经过一系列酶促反应而合成的,只是由于某些酶活性在某些内分泌腺或同一腺体不同的组织中特别高,从而生成不同的激素。类固醇激素的主要合成途径及产物见图12-3。图中标示出了酶活性存在腺体差异性的反应步骤,以及临床常见的先天性酶缺陷所影响的步骤,有助于有关酶缺陷的临床生化诊断参考。
有关盐皮质激素的临床生化在第五章已作介绍,性激素的临床生化将在本章第五节讨论,故本节将只介绍有关糖皮质激素的临床生化。
㈡糖皮质激素的运输及代谢
释放入血液中的糖皮质激素主要为皮质醇及10%左右的皮质酮。二者均约75%左右与肝脏合成的一种α1-球蛋白,即皮质素转运蛋白(transcortin),亦称皮质类固醇结合球蛋白(corticosteroid-bindingglobulin,CBG)可逆结合,15%与白蛋白可逆结合,仅10%左右以游离形式存在。CBG对糖皮质激素的亲和力高,但每分子CBG仅有一个结合部位,且血浆浓度低,故其结合容量有限。白蛋白虽然与糖皮质激素亲和力低,但可有多个结合位点,血浆浓度又高,因此结合容量大。当血中皮质激素浓度明显升高时,与CBG结合易达饱和,将出现与白蛋白结合部分及游离部分比率不成比例的升高。只有游离糖皮质激素才能进入靶细胞发挥生理生化作用及反馈调节自身分泌。
糖皮质激素的代谢主要在肝细胞中进行。主要反应方式为C-3酮基及环节中双键被加氢还原,生成多种加氢代谢物,以四氢皮质醇最多,此外尚有少量二氢、六氢代谢物。90%氢化皮质醇等代谢物及少量原型糖皮质激素,与葡糖醛酸或硫酸结合成相应的酯化物,从尿中排出,亦有少量可随胆汁排入肠道随着粪便排泄。以游离原型从尿中排出的皮质醇仅为血液总量的1%左右。
图12-3 类固醇激素生物合成的途径
㈢糖皮质激素的生理生化功能
游离皮质醇等糖皮质激素可经靶细胞扩散入胞质内,与其受体结合。糖皮质激素-受体复合物转运入细胞核内,可启动某些DNA片段转录,生成的mRNA进入胞质,指导合成特异的酶和脂皮素(lipocortin)等蛋白质或肽类介质,产生广泛的生理生化作用。生理性浓度下,糖皮质激素的主要功能为:
⒈调节糖、脂肪、蛋白质三大营养物质的代谢 对糖代谢,糖皮质激素可促进糖原异生,增加肝糖原和肌糖原含量,另一方面又抑制除脑和心脏外其他组织对糖的利用,使血糖升高。对蛋白质代谢,可促进除肝脏外多种器官、组织的蛋白质分解,抑制蛋白质的合成,升高血中氨基酸,出现尿酸、尿素氮排泄增多等负氮平衡表现。糖皮质激素能激活四肢皮下的脂酶,促进这些部位的脂肪分解,血脂肪酸升高,并使脂肪呈向心性重新分布。
⒉影响水电解质代谢 糖皮质激素有弱的盐皮质激素样潴钠排钾作用,亦有弱的促尿排钙排泄及抗利尿激素作用。
⒊允许作用(permissibleaction) 机体内其他一些激素、神经递质等生物活性物质的作用,需有适当浓度的糖皮质激素存在,才能正常表达,此即糖皮质激素的“允许作用”。主要为对肾上腺素及胰高血糖素的作用。
但高浓度的糖皮质激素如药用或肾上腺皮质功能亢进等,则除上述作用增强外,还可表现出抑制炎症、免疫反应,影响血细胞等作用,将在肾上腺皮质功能亢进症中介绍。
㈣糖皮质激素分泌的调节
和甲状腺激素分泌调节相似,肾上腺糖皮质激素的合成和分泌亦主要受图12-1所示的下丘脑-垂体-内分泌腺调节轴的控制。血液中游离糖皮质激素水平的变化,负反馈地引起下丘脑及垂体分别释放促肾上腺皮质激素释放激素(corticotropin releasing hormone,CRH)和促肾上腺皮质激素(corticotropin 或adrenocorticotropic hormone,ACTH)的增多或减少。CRH为下丘脑产生的一种含41个氨基酸残基的多肽,可选择性地促进腺垂体释放ACTH。ACTH是腺垂体促肾上腺皮质素细胞释放的39肽激素,可通过作用于肾上腺皮质束状带或网状带细胞膜上的ACTH受体,激活腺苷酸-cAMP-蛋白激酶系统,促进细胞增殖,合成和分泌糖皮质激素、性激素增多。持续的高ACTH状态仅早期一过性地引起盐皮质激素分泌增加,无持久影响。ACTH和CRH亦可负反馈地调节下丘脑CRH的释放。和甲状腺激素分泌调节不同,在肾上腺皮质激素的分泌调节中,最主要的是血液中游离糖皮质激素对下丘脑CRH释放的负反馈调节。ACTH和糖皮质激素的分泌存在明显的昼夜节律,分泌高峰见于晨6-8小时,低谷在午夜22-24时。此外,糖皮质激素是机体应激反应时释放的主要激素,因此,各种伤害性刺激均可通过高级神经中枢-下丘脑-垂体-肾上腺皮质轴,促进糖皮质激素的分泌。
除垂体外,一些垂体外的肿瘤主要是肺燕麦细胞癌,其次为胸腺癌、胰岛细胞癌、类癌、甲状腺髓样癌、嗜铬细胞瘤等,亦可分泌异源性ACTH。但这些异位肿瘤ACTH的分泌既不受血液糖皮质激素水平的负反馈调控,也不受CRH促进。此外,近年还发现有少数肿瘤可不受糖皮质激素反馈调节地释放异源性CRH。
现已明确,无论是腺垂体还是异位肿瘤分泌的ACTH,均和γ-黑色细胞刺激素(γ-melanocyte stimulating hormone,γ-MSH)、β-促脂解素(β-lipotropichormone,β-LPH)、β-内啡肽(β-endophin)等,来自同一由265个氨基酸残基组成的大分子前体蛋白,称阿片皮质素原(proopiomelanocortin,POMC)。ACTH等有关多肽激素与POMC的关系见图12-4。
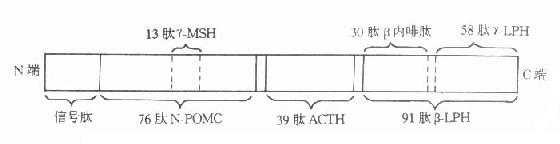
图12-4 阿片皮质素原及其多肽激素裂解产物的关系
三、肾上腺皮质功能紊乱
㈠皮质醇增多症
皮质醇增多症(hypercortisolism)又称库欣综合征(Cushing’s syndrome),是各种原因致慢性糖皮质激素分泌过多而产生的症候群统称。按病因可分做:①垂体腺瘤及下丘脑-垂体功能紊乱,ACTH过量释放产生的继发性皮质醇增多症,又称库欣病,约占70%。其中主要为不伴蝶鞍扩大的微腺瘤,其病因定位诊断主要依赖临床生化检测。②肾上腺皮质肿瘤或结节性增生所致的原发性者,其中以皮质腺瘤多见,约占总病例的近20%,皮质腺瘤约占5%,结节性增生少见。此类病人糖皮质激素分泌一般呈自主性,不受ACTH调控。③异源性ACTH或CRH综合征,由垂体、下丘脑以外的癌瘤细胞分泌释放异源性ACTH或CRH而致。前者以肺燕麦细胞癌最多见,其次为胸腺癌、胰岛细胞癌等;后者可见于肺癌及类癌。早年统计,异源性ACTH、CRH综合征约占皮质醇增多症的5%,但随着对此症的警惕及诊断手段提高,近年发现发病率上升,甚至有报告高达占皮质醇增多症20%的。此外,药源性皮质醇增多症虽临床常见,但因有明确的大剂量糖皮质激素应用史可查,不在此讨论。
皮质醇增多症时,因糖皮质激素病理性持续高水平,导致上述生理作用扩大、增强,产生一些共同的临床表现,如向心性肥胖,皮肤、肌蛋白大量分解而致萎缩,并因此使皮下微血管显露呈对称紫纹,骨质疏松,高血压等。因同时伴有性激素(主要是雄激素)分泌增多,女性可见多毛、月经失调、甚至男性化改变。常规临床生化检查可见血糖升高,葡萄糖耐量降低,血Na+升高,血K+、Ca2+降低,并可出现低钾性代谢性碱中毒的改变,血、尿肌酸、尿素氮明显升高等负氮平衡表现等。高浓度的糖皮质激素可通过诱导磷脂酶A2抑制性多肽脂皮素合成,减少细胞因子白三烯类、血小板活化因子及前列腺素生成等作用,抑制炎症反应及免疫反应。患者对感染的抵抗力降低,并出现各种体液和细胞免疫功能检查指标低下。还可刺激骨髓造血功能,红细胞、血红蛋白、血小板及嗜中性粒细胞均增多,但淋巴细胞和嗜酸性粒细胞明显减少等血液系统改变。库欣病及异源性ACTH综合征者,特别是后者,由于大量阿片皮质素原产生,伴有黑色细胞刺激素释放增多,出现皮肤色素沉着。
㈡肾上腺皮质功能减退症
肾上腺皮质功能减退症(adrenal corticalinsufficiency)是指慢性肾上腺皮质分泌糖皮质激素不足产生的综合征。本病较少见,包括原发性及继发性两种。原发性者又称阿狄森病(Addison’s disease),多因肾上腺结核、自身免疫性肾上腺皮质萎缩、转移性肾上腺癌肿、手术切除等破坏肾上腺皮质,造成糖皮质激素和(或)盐皮质激素分泌不足致病。临床所见除心血管系统、消化系统、神经系统、生殖系统等功能低下,以及低血糖、低血Na+、高血K+、Ca2+等一般实验室检查改变外,由于低糖皮质激素水平负反馈引起ACTH释放增多,而前已介绍,ACTH前体物阿片皮质素原(POMC)中同时含有黑色细胞刺激素多肽片段,故原发性肾上腺皮质功能减退症者可出现特征性皮肤粘膜色素沉着,并可藉此与继发性者鉴别。继发性肾上腺皮质功能减退症指因各种原因,如颅内肿瘤压迫、浸润,垂体前味缺血坏死、手术切除、放疗等,造成下丘脑及垂体不能正常释放CRH、ACTH而致。此时多为多内分泌腺功能减退,极少仅单独表现为肾上腺皮质功能不足,并且无上述皮肤粘膜色素沉着出现。
㈢先天性肾上腺皮质增生症
先天性肾上腺皮质增生症(congenital adrenalhyperplasia,CAH)为常染色体隐性遗传性疾病,系因肾上腺皮质激素合成中某一酶先天性缺陷,肾上腺皮质激素合成受阻,反馈性引发CRH及ACTH分泌增多,致肾上腺皮质弥漫性增生。此时多伴有肾上腺皮质性激素分泌亢进,故CAH常表现为肾上腺性性征异常症。由于任何酶缺陷都将使其所催化的底物堆积并大量释放入血液,被代谢后从尿中排出,因此血和尿中的此类物质可作为该酶缺陷的生化标志物。有关酶缺陷影响的生物合成步骤参见图12-3。表12-3列举了常见的引起CAH的先天性酶缺陷类型、主要临床表现、血液及尿中有诊断意义的生化标志物。测定观察血或尿中这些标志物的变化,有助于诊断CAH及其类型。
表12-3 CAH的酶缺陷类型、主要临床表现及血和尿中的生化标志物
| 酶缺陷种类 | 主要临床表现 | 血生化标志物 | 尿生化标志物 |
| 21-羟化酶 | 轻型:女性假两性畸形,男性假性早熟 重型:同上,并出现阿狄森病 |
17-羟孕酮 | 17-羟孕酮硫酸或葡萄糖醛酸酯、孕三醇 |
| 胆固醇裂解酶 | 肾上腺皮质功能衰竭,早夭 | 无皮质激素 | 无皮质激素及代谢物 |
| 3-β羟类固醇脱氢酶 | 男女均呈假两性畸形 | 脱氢异雄酮 | 16-羟脱氢异雄酮,孕烯三醇 |
| 17-α羟化酶 | 高血钠,低血钾,低血糖,高血压,性幼稚症 | 孕酮 | 孕二醇 |
| 11-β羟化酶 | 高血压,女性假两性畸形,男性假性早熟 | 11-脱氧皮质醇 | 四氢脱氧皮质醇 |
四、肾上腺皮质功能紊乱的临床生化诊断
前述肾上腺皮质功能紊乱时出现的血液电解质、血糖等一般生化指标改变,对肾上腺皮质功能亢进或减退的诊断有一定的价值。下面我们将介绍诊断肾上腺皮质功能紊乱的一些特殊的临床生化检测项目。
㈠血、尿中糖皮质激素及其代谢物测定
⒈尿17-羟皮质类固醇、17-酮类固醇测定 尿中17-羟皮质类固醇(17-hydroxycorti-costeroids,17-OHCS)测定是指对尿中C-17上有羟基的所有类固醇类物质的测定。该类内源性物质在人类主要为肾上腺皮质所分泌的糖皮质激素皮质醇,及其活性更强的代谢产物去氧皮质醇,以及二者的二氢、四氢、六氢代谢产物。上述物质大多以葡糖醛酸酯或硫酸酯的结合形式排出。24h尿中以17-OHCS排出的糖皮质激素及其各种代谢物约占每日分泌量的25%-40%。尿17-OHCS测定一般均收集24h尿,量取体积后取样加酸水解,释放出游离17-OHCS,这样该类皮质类固醇中的二羟丙酮侧链可与硫酸溶液中的盐酸苯肼反应显色,而以分光光度法测定。成人24h尿17-OHCS正常值参考范围为:男性21.3-34.5μmol/d(7.7-12.5mg/d),女性19.3-28.2μmol/d(7.0-10.2mg/d)。儿童低于成人,约在青春期达成人水平。皮质醇增多症或肾上腺皮质功能减退症时,尿17-OHCS将分别明显增多或减少。但影响本测定的因素较多,如应激状态、营养不良、慢性消耗性疾病、肝硬化、肾功能不良、多种可干扰测定的药物及食物等,故其灵敏度及特异性均差。约有15%的皮质醇增多症者不能观察到尿17-OHCS明显升高,而其诊断皮质醇增多症的假阳性率也近15%。
尿17-酮类固醇(17-ketosteroids,17-KS)指尿中出现的所有C-17为酮基的类固醇类物质。人类尿中排出的内源性17-KS包括雄酮、异雄酮、脱氢异雄酮等及其代谢物,此外有少量皮质醇可在肝脏发生C-17羟基脱氢氧化成17-KS,由尿中排出。和17-OHCS一样,尿中上述17-KS大多也以葡糖醛酸酯或硫酸酯的结合形式存在。尿内源性17-KS中男性约2/3来自肾上腺皮质,1/3来自睾丸;女性则几乎全部来自肾上腺皮质,卵巢仅产生少量。因此,尿17-KS在女性青春期前可较粗略地代表肾上腺皮质的内分泌功能,男性则反映了肾上腺皮质和睾丸二者的内分泌功能状态。尿17-KS测定也需先酸解以释放出游离17-KS,提取后,在碱性环境中,通过其结构中的酮-亚甲基(-CO-CH2-)与间二硝基苯反应显色,再以分光法测定。24h尿17-KS正常成人参考范围为:男性28.5-61.8μmol/d(8.2-17.8mg/d),女性20.8-52.0μmol/d(6-15mg/d),青春期前儿童低于成人。尿中存在的氯丙嗪、甲丙氨酯(眠尔通)以及多种有色药物,有色食品饮料,严重肝、肾疾患,睾丸或卵巢内分泌功能紊乱,均可影响本测定结果。故在诊断肾上腺皮质功能紊乱上,尿17-KS比尿17-OHCS特异性更低。据统计,在诊断皮质醇增多症上,尿17-KS可出现约45%假阴性和10%的假阳性。
由于以上原因,在诊断肾上腺皮质功能紊乱的临床生化检测中,单独测定尿17-OHCS及17-KS,特别是后者已较少应用。但若出现尿17-KS显著升高,且不被地塞米松抑制(见后),则有助于肾上腺皮质腺瘤或癌的诊断。因为这两种情况,特别是后者可产生大量17-KS类雄激素。此外21-羟化酶、3-β羟化酶及11-β羟化酶缺陷所致先天性肾上腺皮质增生症者,尿17-KS也明显升高。
⒉血皮质醇及24小时尿游离皮质醇测定 血液中皮质醇浓度直接反映肾上腺糖皮质激素分泌情况,而尿中游离皮质醇(urinefree cortisol,UFC)由血液中游离皮质醇经肾上球滤过而来,因此其量与血浆中真正具生物活性(包括调节自身分泌)的游离皮质醇浓度成正比。血浆(清)皮质醇或24h UFC测定现被推荐为是否存在肾上腺皮质功能紊乱的临床生化检查首选项目。目前皮质醇测定方法有荧光光度法、免疫化学法、HPLC、GC、GC-MS等。其中荧光法因受多种内源性皮质激素及其前体物如皮质酮、11-去氧皮质醇(甲吡酮抑制试验时可能大量出现)等干扰,特异性较差,测定结果较其他方法偏高。HPLC、GC、GC-MS法虽然特异性、灵敏度均高,但难以在临床常规工作中使用,仅用作参考方法。免疫化学法除特异性及灵敏度均满足要求外,其操作简便、快速,且有商品试剂盒供选用,为目前最常使用的方法。
血浆(清)皮质醇测定,是检测包括蛋白结合和游离两部分的总皮质醇浓度,并不能排除CBG、白蛋白浓度改变等各种影响皮质醇蛋白结合率因素对游离皮质醇浓度的影响,因此其浓度不一定和游离皮质醇浓度平行。正常人皮质醇的分泌存在昼夜节律,可由于导致单次取样测定因取样时间在分泌峰或谷浓度,产生假阳性或假阴性结果。皮质醇增多症者,该昼夜节律多消失,并为诊断依据之一。故现在均主张分别在早晨8点及午夜12点分别取血测定,代表峰浓度、谷浓度。并且为避免因住院、静脉穿刺等产生应激性皮质醇分泌因素的影响,采血宜在住院至少3天后,并以保留式静脉取血套管进行。免疫法测得血浆(清)皮质醇正常值参考范围为晨8点165.6-441.6nmol/L(6-16μg/dl),午夜12点55.2-165.6nmol/L(2-6μg/dl),并且早晚的昼夜节律波动范围>2。血皮质醇浓度无性别差异,儿童较成人高。皮质醇增多症者,早晚血皮质醇浓度,尤其是午夜值显著高于正常,且二者比值<2。肾上腺皮质功能减退者亦多出现昼夜节律性波动消失(早/晚比值<2),且皮质醇浓度均显著低于正常值下限,特别是晨8点结果意义更大。但若同时患有任何严重的非肾上腺疾病,各种原因产生的应激状态,以及妊娠、肥胖、甲状腺功能紊乱、慢性肝病、抑郁症、吩噻嗪类抗精神失常药、苯妥英钠、利血平和长期使用糖皮质激素类药,均可影响皮质醇水平或昼夜节律。在解释结果时应予以注意。
24h UFC测定不受昼夜节律影响,并可靠地反映游离皮质醇水平。特别是当皮质醇增多症时,血皮质醇浓度超过CBG结合容量,游离皮质醇浓度将不成比例地升高,UFC亦将出现较血浆总皮质醇、尿17-OHCS和17-KS更明显的改变。故在诊断皮质醇增多症上,24h UFC测定更为敏感可靠。但本测定除同样受上述影响血浆皮质醇测定的因素影响外,还受肾功能影响。若同时测定尿肌酐排泄量,以24h UFC/24h尿肌酐表示,可在一定程度上排除肾功能对24h UFC测定的影响。免疫法检测24h UFC成人正常值以参考范围为27.6-276nmol/d(10-100μg/d)或30-99μg/g肌酐。24h UFC亦无性别差异,但儿童年龄越小越低。若成人24h UFC>552nmol/d,大多提示存在皮质醇增多症。但24h UFC存在较大的天间变动,不如血浆皮质醇浓度稳定,最好分别测定3日UFC,综合判断。
㈡下丘脑-垂体-肾上腺皮质轴功能检测及动态功能试验
上述血、尿中皮质醇及皮质激素代谢产生检测,在肾上腺皮质功能紊乱的生化诊断中,仅起着较粗糙的筛选作用,确诊及对治疗方案有指导意义的病变部位、性质的判定,往往需进行下丘脑-垂体-肾上腺皮质调节轴功能检测及必要的动态试验。
⒈血浆ACTH及N-POMC测定 ACTH为腺垂体分泌的微量多肽激素,现大多以免疫化学法检测。正常ACTH分泌存在与皮质醇相同的昼夜节律,并且在肾上腺皮质功能紊乱时,ACTH分泌的昼夜节律也大多消失。异源性ACTH分泌也无昼夜节律,故ACTH测定也和血浆皮质醇测定一样,应在早上8点及午夜12点各取血测定,既观察昼夜节律有无,也避免取样时间造成的影响。有关影响本测定的因素、注意事项及取血方法,参见血浆皮质醇测定。血浆ACTH正常成人参考范围为:早上8点2.2-22.0pmol/L(10-100pg/ml),午夜12点1.1-4.4pmol/L(5-20pg.ml),二者比率>2。在阿狄森病、先天性肾上腺皮质增生症、下丘脑及垂体性皮质醇增多症、特别是异源性ACTH综合征时,午夜ACTH明显高于正常范围,并且昼夜节律消失。而继发性肾上腺皮质功能减退症、肾上腺皮质腺瘤或癌所致的原发性皮质醇增多症者,则晨8点血浆ACTH明显降低(<1.1pmol/L),昼夜节律也消失。在高水平ACTH时,为鉴别ACTH属垂体性或异源性,可采用静脉插管,分别同时采集岩下窦及外周静脉血,测定二者ACTH。若岩下窦血ACTH为外周血2倍以上,则提示为垂体源性;而岩下窦血ACTH反低于外周血ACTH水平,则可确定为异源性ACTH综合征。这一方法在其他检查无法确定癌肿灶时,尤有价值。
前已介绍,POMC为ACTH的前体物,其N端76肽水解自然N-POMC与ACTH等分子数产生,但其降解速度慢,血中浓度高,更易检测。近年有主张以N-POMC测定代替ACTH检测的趋势。
⒉兴奋试验 肾上腺皮质功能试验紊乱的兴奋性动态功能试验有多种,目前较成熟的为ACTH兴奋试验。该试验是根据ACTH可刺激肾上腺皮质合成并迅速放贮存的皮质醇等皮质激素原理,分别检测使用ACTH前后体液中皮质醇或其代谢物的变化,反映肾上腺皮质的内分泌功能状况。通常用0.25mg合成ACTH肌肉或静脉注射,分别在注射前和注射后0.5、1h采集静脉血,测定和观察血浆皮质醇浓度变化。正常人注射ACTH后,峰浓度在0.5h出现,该时血浆皮质醇浓度较注射前基础值至少增加157.5nmol/L(7μg/dl)以上,或峰浓度值高于550nmol/L(20μg/dl)。未用皮质激素治疗的阿狄森病患者,基础值低,且对ACTH刺激无反应,有时甚至反下降;继发性肾上腺皮质功能低下者,基础值亦低,但对ACTH可有延迟性反应。肾上腺皮质腺瘤或癌性皮质醇增多症者,其皮质醇分泌呈自主性,对ACTH刺激亦多无反应,但其皮质醇基础水平高,且临床表现与阿狄森病迥异,不难鉴别。下丘脑垂体性皮质醇增多症则出现强阳性反应,而异源性ACTH综合征者,肾上腺皮质无病变,对ACTH刺激亦呈阳性反应。亦可以注射前、注射日及次日24h尿17-OHCS或UFC作为观察指标,正常人注射日或次日,前者应较基础增加1-3倍,后者增加2倍以上。近年还有根据CRF可促进腺垂体释放ACTH,但对异源性分泌ACTH的癌瘤细胞无影响的原理,检测注射CRH前后血浆ACTH变化,鉴别皮质醇增多症的病因尚未普遍开展。
⒊抑制试验 最常用地塞米松抑制试验。地塞米松(dexamethasone,DMT)为人工合成的强效糖皮质激素类药,对下丘脑-垂体-肾上腺皮质调节轴可产生皮质醇样但更强的负反馈调节作用,其影响部位主要是抑制腺垂体释放ACTH,进而间接抑制肾上腺皮质激素的合成和释放,故可用于判定肾上腺皮质功能紊乱是否因下丘脑垂体功能异常所致。具体实施方案很多,如单剂、48h及标准DMT抑制试验。其中单剂(1mg)DMT抑制试验易造成假阴性,而对类似皮质醇增多症的单纯性肥胖者却产生假阳性。标准DMT抑制试验过于耗时、繁琐,现多采用48h小剂量DMT抑制试验,即在连续两日收集24h尿作基础对照后,第三日开始DMt 0.5mg/每6h,连续两日(总剂量4mg),并分别收集这两日的24h尿。分别测定每日24h UFC或17-OHCS。正常人包括单纯性肥胖者,用DMT后尿17-OHCS或UFC均应降至基础值的50%以下。皮质醇增多症者抑制程度达不到50%,甚至几无反应。若再连续两日服用DMt 2mg/每6h,并也分别收集这两日24h尿,测定17-OHCS或UFC,即为标准DMT抑制试验。下丘脑垂体性皮质醇增多症者,使用DMT的第3或4日的24h17-OHCS或UFC可较基础值水平降低50%以上;而异源性ACTH综合征、肾上腺皮质腺癌或瘤性皮质醇增多症者则一般不受抑制。若有条件,也可用晨8点取血测定ACTH或皮质醇,作为观察指标,判断标准同上。但较长期服用有肝药酶诱导作用的药物,如苯妥英钠、苯巴比妥钠、利福平等,可加速DMT代谢灭活,产生假阴性。近期曾较长期使用糖皮质激素类药物,显然不宜进行本试验。此外,若机体处于任何原因所致的应激状态时,亦可能干扰本试验,均应避免。
甲吡酮(metyrapone,美替拉酮)抑制试验对皮质醇增多症病因与定位诊断上亦有一定价值。甲吡酮为皮质醇合成中,催化11-脱氧皮质醇转化为皮质醇的11-β羟化酶特异性抑制剂。使用后可使皮质醇合成、释放减少,反馈性诱导ACTH分泌增多,肾上腺皮质中11-脱氧皮质醇(亦属17-OHCS)大量堆积,尿中排泄增多,而皮质醇并不见增加。在试验日前收集二日24h尿,给予甲吡酮500-750mg/每4h口服共6次,再收集服药日及次日24h尿,分别测定各24h 17-OHCS。正常人可较基础值水平升高2-4倍。垂体性特别是下丘脑性皮质醇增多症者,用药后17-OHCS可升高4倍以上,也有部分垂体腺瘤者,虽然ACTH释放增多,但尿17-OHCS升高不能达诊断水平。肾上腺皮质腺瘤或癌及异源性ACTH综合征者,一般均无反应,后者若垂体源性ACTH在皮质醇分泌中仍有一定调节作用,可有一定反应,但很少能达到正常反应上限。若以直接测定血浆ACTH代替尿17-OHCS作为观察指标,则更为可靠。
肾上腺皮质功能紊乱的有关特殊临床生化检查所见总结于表12-4。
表12-4 肾上腺皮质功能紊乱的临床生化检查所见
| 皮质醇增多症 | 肾上腺皮质功能减退症 | |||||
| 下丘脑垂体性 | 肾上腺皮质腺瘤 | 肾上腺皮质腺癌 | 异源性ACTH | 阿狄森病 | 继发性 | |
| 尿17-OHCS | 中度升高 | 中度升高 | 明显升高 | 明显升高 | 减少 | 减少 |
| 尿17-KS | 升高 | 略升高 | 明显升高 | 明显升高 | 减少 | 减少 |
| 血皮质醇或UFC | 升高 | 升高 | 明显升高 | 明显升高 | 减少 | 减少 |
| 血浆ACTH | 升高 | 降低 | 降低 | 明显升高 | 升高 | 减少 |
| ACTH兴奋试验 | 强反应 | 无或弱反应 | 无反应 | 多无反应 | 无反应 | 延迟反应 |
| DMT抑制试验 | 无或有反应* | 无或弱反应 | 无反应 | 无或弱反应* | ||
| 甲吡酮抑制试验 | 强反应 | 无或弱反应 | 无反应 | 无或弱反应 | ||
* 标准地塞米松(DMT)抑制试验时才可能部分患者有一定反应。
第四节 下丘脑-垂体内分泌功能紊乱的临床生化
一、下丘脑-垂体内分泌功能及调节
㈠垂体分泌的激素
垂体即脑垂体,为位于颅底蝶鞍中的重要内分泌器官,由茎状垂体柄与下丘脑相连。从组织学上可将垂体分做腺垂体及神经垂体。腺垂体包括前部、结节部和中间部,神经垂体由下丘脑某些神经元直接延续而成。垂体分泌的激素相应分做腺垂体激素和神经垂体激素两类。有关激素的生理作用已在生理学中介绍。表12-5概括了重要的垂体激素及其主要生理功能。
上述激素均为肽类或糖蛋白,其中TSH、LH和FSH均是由α和β两个多肽亚基组成的糖蛋白。这三种激素的α亚基具高度同源性,氨基酸残基亦较接近,其生理活性主要取决于β亚基。在用免疫化学法检测时,往往存在交叉免疫反应而互相干扰。
表12-5 主要的垂体激素及生理作用
| 激 素 名 称 | 主要生理作用 |
| 腺垂体激素 | |
| 生长激素(growth hormone,GH) | 促进机体生长 |
| 促肾上腺皮质激素(corticotropin,ACTH) | 促进肾上腺皮质激素合成及释放 |
| 促甲状腺素(thyrotropin,TSH) | 促进甲状腺激素合成及释放 |
| 卵泡刺激素(follicle-stimulating hormone,FSH) | 促进卵泡或精子生成 |
| 黄体生成素(luterzilizing hormone,LH) | 促进排卵和黄体生成,刺激孕激素、雄激素分泌 |
| 催乳素(prolactin,PRL) | 刺激乳房发育及泌乳 |
| 黑色细胞刺激素(melanocyte stimulating hormone,MSH) | 促黑色细胞合成黑色素 |
| 神经垂体激素 | |
| 抗利尿激素(antidiuretic hormone,ADH) | 收缩血管,促进集尿管对水重吸收 |
| 催产素(oxytocin,OT) | 促进子宫收缩,乳腺泌乳 |
㈡下丘脑激素
下丘脑的一些特化的神经细胞可分泌不同的调节腺垂体有关激素释放的调节激素(因子)。从组织结构上看,这些分泌性神经细胞的轴突组成结节-漏斗束,终止于垂体柄内垂体门脉系统的初级毛细血管网周围。借助特殊的垂体门脉系统,这些分泌性神经细胞释放的调节激素,可迅速直接地被输送至腺垂体发挥作用。下丘脑调节激素均是多肽,这些激素的名称、缩写及受其调节的腺垂体激素见表12-6。从表中可看出,下丘脑调节激素的作用通过其名称即可知。但也存在某些交叉,如TRH还可促进生长激素和催乳素释放,而GHIH也能抑制腺垂体TSH、ACTH及胰腺胰岛素的释放。近年还发现,下丘脑外的某些神经细胞及一些脏器组织细胞,也可产生某些下丘脑激素。这些下丘脑外活性多肽的功能尚不清。
表12-6 下丘脑分泌的主要调节激素
| 激素名称 | 调节的腺垂体激素 |
| 促甲状腺激素释放激素(thyrotropin-releasing hormone,TRH) | TSH(主要),GH,PRL,FSH |
| 促性腺激素释放激素(gonandotropin-releasing hormone,GnRH) | LH,FSH |
| 促肾上腺皮质激素释放激素(corticotropin-releasing hormone,CRH) | ACTH |
| 生长激素释放激素(growth hormone-releasing hormone,CRH) | GH |
| 生长激素抑制激素(growth hormone-inhibiting hormone,GHIH) | GH(主要),TSH,ACTH,PRL |
| 催乳素释放激素(prolactin-releasing hormone,PRH) | PRL |
| 催乳素抑制激素(prolactin-inhibiting hormone,PIH) | PRL |
| 黑色细胞刺激素释放激素(melanocyte stimulatinghormone-re-leasing hormone,MRH) | MSH |
| 黑色细胞刺激素抑制激素(melanocyte stimulatinghormone-in-hibiting hormone,MIH) | MSH |
㈢下丘脑-腺垂体激素分泌的调节
下丘脑-腺垂体激素分泌的调节,主要受腺垂体各种促激素作用的靶腺分泌的激素之反馈调节(长反馈),其中甲状腺激素的长反馈调节主要作用于腺垂体,而其他外周激素长反馈调节作用部位则主要为下丘脑水平(图12-1)。前已谈到长反馈调节的主要方式为负反馈,但在月经周期中排卵期前,当雌激素水平达最高峰时,可正反馈地调节下丘脑相关激素的释放(短反馈),在GH分泌的调节中,短反馈为主要方式。而下丘脑激素或腺垂体激素,还可负反馈地调节下丘脑或腺垂体对自身的合成和分泌(超短反馈)。此外,应激状态、某些外周感觉神经冲动以及边缘系统的情绪活动等,均可通过下丘脑以外的中枢神经系统,影响下丘脑-垂体的的激素分泌,并进而影响外周内分泌腺功能。这种神经系统对内分泌的控制,还表现为多种内分泌功能的昼夜节律。
有关下丘脑-腺垂体调节激素的紊乱,将分别在本章有关外周内分泌腺功能紊乱中介绍,而ADH及OT的临床生化分别在第五章和第十四章中讨论。本节主要讨论有关GH紊乱的临床生化,并简单介绍催乳素瘤。
二、生长激素及生长调节素
㈠生长激素的化学、分泌调节及作用
生长激素(growth hormone,GH;somatotropin,STH)是腺垂体嗜酸细胞分泌的,由191个氨基酸残基组成的直链肽类激素。其结构与PRL相似,并有一定交叉抗原性。释放入血液中的GH不与血浆蛋白结合,以游离形式输送到各靶组织发挥作用。Gh 的生理作用最主要的是对成年前长骨生长的促进。现已明确,这一作用是通过生长调节素(so-matomedin,SM)的介导,促进硫酸掺入到骨骺软骨中,及尿嘧啶核苷、胸腺嘧啶核苷分别掺入软骨细胞RNA或DNA中,加速RNA、DNA及蛋白粘多糖合成及软骨细胞分裂增殖,使骨骺板增厚,身材得以长高。GH亦参与代谢调节,主要表现为与生长相适应的蛋白质同化作用,产生正氮平衡;促进体脂水解,血游离脂肪酸升高;对糖代谢则可促进肝糖原分解,升高血糖。此外,GH对维持正常的性发育也有重要作用。
GH的分泌主要受下丘脑GHRH和GHIH的控制。除GH和SM可反馈性调节GHRH和GHIH释放外,剧烈运动、精氨酸等氨基酸、多巴胺、中枢α2肾上腺素受体激动剂等,可通过作用于下丘脑,垂体或下丘脑以外的中枢神经系统,促进GH的分泌。正常情况下,随机体生长发育阶段不同而有不同的GH水平。而每日生长激素的分泌存在昼夜节律性波动,分泌主要在熟睡后1h左右(睡眠脑电图时相3或4期)呈脉冲式进行。
㈡生长调节素
生长调节素(SM)即生长激素依赖性胰岛素样生长调节因子(GH-dependent insulin-like growth factor ,IGF),曾称硫化因子(sulfation factor)。SM为一类在GH作用下,主要在肝脏也由多种GH靶细胞合成的多肽,分子量6000~8500。现至少已确定A、B、C三种亚型,均具胰岛素作用。其中SM-C即IGf I,其结构与胰岛素有近一半的氨基酸残基相同。和其他肽类激素不同,血液中的SM几乎全部均和高亲和力的SM结合蛋白形成可逆结合而运输。如前所述,现至少肯定GH的促生长作用必须通过SM介导,也有认为GH的代谢调节作用也依赖于SM。从这一意义上说,SM水平反映GH的生物活性比GH本身更为直接。
三、生长激素功能紊乱的生化诊断
㈠生长激素功能紊乱
⒈生长激素缺乏症 生长激素缺乏症(growthhormone deficiency)又称垂体性侏儒症(pituitary dwarfism),是由于下丘脑-垂体-GH-SM中任一过程受损而产生的儿童及青少年生长发育障碍。按病因可分做:①原因不明,但可能在胚胎发育或围产期下丘脑损伤,致GHRH合成、分泌不足,或垂体损伤产生的持发性GH缺乏症,约占70%,大多伴有其他垂体激素缺乏症;②遗传性GH缺乏症,以不同的遗传方式所致的单一性GH缺乏为多见,极少数病人也表现为包括GH在内的多种垂体激素缺乏症。近年还发现有少数病人表现为遗传性SM生成障碍,其GH反增多;③继发性GH缺乏症,由于下丘脑、垂体及周围组织的后天性病变或损伤,如肿瘤压迫、感染、外伤、手术切除等,致GH分泌不足。
GH缺乏症的突出临床表现为生长发育迟缓,身材矮小,但大多匀称,骨龄至少落后2年以上。若未伴甲状腺功能减退,则智力一般正常,以别于呆小症。此外性发育迟缓,特别是伴有促性腺激素缺乏者尤显。患儿大多血糖偏低,若伴ACTH缺乏者更显著,婴幼儿甚至可出现低血糖抽搐、昏迷。
⒉巨人症及肢端肥大症 巨人症(gigantism)和肢端肥大症(acromegaly)由GH过度分泌而致。若起病于生长发育期表现为前者,而在成人起病则表现为后者,巨人症多可继续发展为肢端肥大症。病因大多为垂体腺瘤、癌或GH分泌细胞增生而致;也有少数系可分泌GHRH或GH的垂体外肿瘤产生的异源性GHRH或GH综合征,包括胰腺瘤、胰岛细胞癌、肠及支气管类癌等。单纯巨人症以身材异常高大、肌肉发达、性早熟为突出表现。同时存在高基础代谢率、血糖升高、糖耐量降低、尿糖等实验室检查改变。但生长至最高峰后,各器官功能逐渐出现衰老样减退。肢端肥大症者由于生长发育已停止,GH的促骨细胞增殖作用表现为骨周增长,产生肢端肥大和特殊的面部表现,及包括外周内分泌腺在内的广泛性内脏肥大。亦有高血糖、尿糖、糖耐量降低、高脂血症、高血清钙等实验室检查改变。粥样动脉硬化及心衰常为本病死因。病情发展至高峰后,亦转入同巨人症一样的衰退期。
㈡GH紊乱的临床生化检查
⒈血清(浆)GH测定均用免疫化学法测定。一般在清晨起床前,空腹平卧安静状态下取血测定作为基础值。正常参考范围为新生儿15-40μg/L,2岁儿童平均约为4μg/L,2-4岁儿童平均约8μg/L,4岁以上儿童及成人为0-5μg/L,女性略高于男性。若测定结果远远超出正常水平,结合临床所见,有助于巨人症或肢端肥大症、以及遗传性SN生成缺陷所致的GH缺乏症诊断。但由于前述GH每日分泌主要在夜间熟睡中,呈脉冲式释放的生理性波动特点,而其半寿期又仅20min。因此若在非脉冲式释放期取样测定,GH水平再低也无多大意义。故在诊断GH缺乏症时,最好在病儿熟睡后1-1.5h取血测定。更为严格的是插入留置式取血导管后,进行24h或晚8点到次晨8点内每0.5h取血测定Gh ,了解全天或夜间GH分泌的总体情况。若测定结果为低,则还需应用下列兴奋试验证实。
⒉动态功能试验 GH释放的兴奋试验,方法较多,常用的有以下几种:
⑴运动刺激试验:可合作年龄儿童空腹取血作基础对照后,剧烈运动20-30min,运动结束后20-30min取血测定。因剧烈运动及可能存在的血糖水平偏低均可刺激腺垂体释放GH,故运动后,正常者血清GH值应较基础对照值明显升高或≥10μg/L;GH缺乏症者,运动后GH水平<5μg/L。
⑵药物刺激试验:可刺激腺垂体释放GH的药物很多,目前常用的药物及方法为:①胰岛素-低血糖试验(insulin-hypoglycemiatest),因低血糖应激状态可刺激腺垂体释放GH、ACTH、PRL等多种激素,故在清晨空腹卧床采血作对照后,按0.1μ/kg体重静脉注射普通胰岛素后30、60、90、120及150min分别取血,测定GH水平,必要时可同时检测ACTH、PRL,以发现复合性垂体前叶功能减退;②其他药物刺激试验均是在同上抽取清晨卧床空腹血后,给予L-多巴(促GHRH释放)500mg(儿童10mg/kg体重)一剂口服,或可乐定(促GHRH释放)4μg/kg体重(或150μg/m2体表面积)一剂口服,或盐酸精氨酸(促进垂体释放GH)0.5g/kg在30min内静脉滴注。分别在上述药物使用后30、60、90和120min取血,测定血清(浆)GH水平。正常人在使用上述刺激剂后,GH分泌峰多在60或90min出现,胰岛素可推迟到120或150min出现,峰值应比对照基础值升高5-7μg/L以上,或峰值浓度≥20μg/L。若两项以上刺激试验峰浓度均<5μg/L,则为GH缺乏症。而峰值浓度≥20μg/L,则可排除GH缺乏症,但GH受体缺陷等所致SM遗传性生成障碍者,GH基础值反可升高,并且对上述兴奋试验可有正常人样反应,此时只有通过SM测定进行鉴别。
⒊GH分泌的抑制试验对于多次测定基础GH值约>10μg/L的疑为巨人症或肢端肥大症者,应考虑进一步作高血糖抑制GH释放试验。即按上述方法抽取空腹基础静脉血,口服含100g(儿童1.75g.kg体重)葡萄糖的浓糖水后,分别在30、60、90和120min取血,测定各血清GH水平。正常人服用葡萄糖后血清GH最低应降至2μg/L以下,或在基础对照水平50%以下。垂体腺瘤性或异源性GH所致巨人症或肢端肥大症者,因呈“自主性”GH分泌,不会被明显抑制,最低浓度>5μg/L,或在基础对照水平50%以上。但本试验可有假阴性出现,特别在治疗可能出现的高血压、高血糖,使用了可乐定、α-甲基多巴等中枢α2肾上腺素受体激动剂或降血糖药者,应注意避免,最好停用上述药物一周以上再行本试验。
⒋SM-C及SM结合蛋白测定前面已介绍GH的促生长作用等,均需通过SM介导。SM为一类多肽物质,现已分离出A、B、C三亚型。其中SM-C(IGf I)为中性肽,血浆中SM-C几乎均为GH作用于肝细胞膜受体后,诱导肝细胞合成释放的。由于SM-C的血浆浓度不随GH分泌的脉冲式波动而变动,前述各种刺激或抑制GH释放的因素,均不能在短时内引起SM-C的浓度改变,因此其水平较稳定。单次取样测定即可了解较长一段时间的GH功能状况。再加之其半寿期长,血浆浓度高,易于检测。现在推荐以免疫化学法检测单次血样中SM-C浓度,作为判断GH功能状况的简便而可靠的筛选方法。血清SM-C正常值参考范围为:青春期前儿童0.1-2.8u/ml,青春期少年0.9-5.9u/ml,成人男性0.3-1.9u/ml,女性0.5-2.2u/ml。任何GH缺乏症,包括高GH水平的遗传性GH受体缺陷性者,SM-C均低于同龄正常水平的下限;巨人症及肢端肥大症者则远远高于正常水平。但恶病质、严重营养不良及严重肝病者,SM-C可降低;青春期少年有时可超出正常值上限。
由于血浆中SM几乎均与高亲和力的特异结合蛋白SMBP(IGFBP)结合,亦有主张检测SMBP间接反映GH功能者,但尚未成熟。
四、催乳素瘤
催乳素瘤(prolactinoma,PRL瘤)为功能性垂体腺瘤中最常见者,好发于女性,多为微小腺瘤,临床表现为泌乳、闭经、多毛、痤疮及不育等。男性则往往为大腺瘤,临床以性功能减退、阳萎、不育及垂体压迫症状为主,偶见泌乳。临床生化检查可见血清PRL极度升高。正常人清晨血清基础值男性<20μg/L,非妊娠及哺乳期女性即便在月经周期的黄体期亦<40μg/L。不论男女,若血清PRL基础值>200μg/L,应高度怀疑本病,>300μg/L则可确诊。对于PRL水平在100-300μg/L的高催乳素状态者,可以TRH、氯丙嗪或灭吐灵兴奋试验协助鉴别。正常人及功能性高催乳素血症者,上述兴奋试验可使血清PRL较基础对照明显升高,而催乳素瘤者因呈“自主性”高分泌,故反应低下或无,即便有弱反应也峰值推迟。
第五节 性激素紊乱的临床生化
一、性激素的生理与生化
(一)性激素的化学、生物合成及代谢
性激素(sex hormones)包括雄性激素(androgens)、雌激素(estrogens)和孕激素孕酮(progesterone)三类,后二者合称雌性激素。雄性激素主要为睾酮(testosterone)及少量的脱氢异雄酮(dehydroepiandrosterone,DHEA)和雄烯三酮(androste-nedione)。雌激素则主要为雌二醇(estrodiol)及少量雌酮(estrone)、雌三醇(estriol)。从化学上看,所有性激素都是类固醇类激素。有关类固醇激素生物合成在本章第三节已一并介绍(图12-3)。由于某些酶活性在不同腺体及组织中存在差异,所以睾酮主要在男性睾丸间质细胞中生成,女性血液中少量睾酮则为DHEA的代谢产物;DHEA及雄烯二酮由肾上腺皮质、睾丸和卵巢分泌,并为女性的主要雄性激素;雌二醇主要由卵巢滤泡,黄体及妊娠时胎盘生成,极少量由睾丸产生或为睾酮代谢产物,并为男性的雌激素主要来源;雌酮大多为雄烯二酮代谢物;雌三醇除孕妇胎盘可直接分泌外,均为雌二醇的代谢产物;孕酮虽为类固醇激素合成的中间代谢产物,但血液中的孕酮几乎都是由黄体或胎盘所分泌。
血浆中的性激素90%以上都和血浆蛋白形成可逆结合。其中雄性激素和雌激素主要与肝合成的一种β球蛋白-性激素结合球蛋白(sexhormone binding globulin,SHBG)结合,旧称睾酮-雌激素结合球蛋白(testosterone-estrogen bindingglobulin,TEBG),孕酮及少量雌二醇则可与皮质类固醇结合球蛋白(CBG)结合。性激素主要在肝脏代谢,除少量可直接和葡糖醛酸或硫酸结合成相应的酯类排泄外,大部分需经历类固醇环上的化学转化,再与上述两种酸结合成酯,由尿或胆汁(少量)排泄。睾酮的主要代谢产物为雄酮及初胆烷醇酮,为尿中17-KS的主要来源。雌二醇和雌酮的主要代谢产物为雌三醇及2-甲氧基雌二醇。孕酮则主要代谢为孕烷二醇,故测定孕烷二醇尿排量可作为黄体功能指标。
但睾酮在睾丸及其他靶组织中存在的5α-还原酶作用下,可生成5α-二氢睾酮,其与受体亲和力比睾酮还强,被认为是睾酮的活性形式,在胚胎期及出生后男性生殖器分化、形成和发育上有重要作用。先天性5α-还原酶缺陷者,可发生男性假两性畸形,5α-二氢睾酮的进一步代谢产物为雄烷二醇。
(二)性激素的生理功能与分泌调节
⒈雄性激素的生理功能与分泌调节 雄性激素主要是睾酮,其生理功能可概括为:①刺激胚胎期及出生后男性内外生殖器的分化、形成和发育,参与男性性功能及第二性征的出现和维持;②促进蛋白质合成的同化作用,使机体呈正氮平衡,对男性青春期的长高起着重要作用;③促进肾脏合成促红细胞生成素(erythropoietin),刺激骨髓的造血功能。
睾丸的内分泌功能主要通过睾酮对下丘脑GnRH释放及腺垂体LH和FSH分泌的负反馈调节来控制。LH可与睾丸间质细胞膜上的受体结合,促进睾酮的合成、分泌。而FSH则在LH诱导下分泌的适量睾酮参与下,促进精子的生成。非青春期睾酮分泌的昼夜节律不甚明显,清晨约比傍晚高20%。但进入青春期的男孩,可能因松果体分泌的降黑素减少,GnRH出现约每2h一次的脉冲式分泌,特别在夜间尤著,促使LH及FSH释放增多。出现青春期特有的性及体格发育完善,第二性征的形成。男性青春期一般始于11-13岁,18-24岁发育成熟。
⒉雌性激素的生理功能及分泌调节 雌二醇等雌激素的生理功能主要有:①促进女性生殖器官的发育及功能形成、第二性征的出现和维持,并与孕激素协同配合,形成月经周期。②对代谢的影响,包括促进肝脏合成多种血浆中的转运蛋白,如运铁蛋白、甲状腺素结合蛋白、皮质类固醇结合蛋白等;降低血浆胆固醇,但促进HDL的合成,并减少动脉壁弹性硬蛋白,故生育期妇女不易发生动脉粥样硬化及冠心病;还可促进钙盐在骨沉积,促进肾上管对钠和水的重吸收等。孕激素的作用则主要为与雌激素协同作用于子宫内膜,形成月经周期;还可松弛子宫及胃肠道平滑肌,促进乳腺腺泡和导管的发育,促进水钠排泄,并在排卵后使基础体温升高约1℃。
雌激素的卵巢分泌在青春期前,主要受雌激素对垂体LH、FSH分泌的负反馈调节而控制,少量孕激素可由肾上腺皮质分泌。但女性进入青春期(13-18岁)后,下丘脑出现约60-90min一次的强脉冲式GnRH分泌,促进腺垂体大量释放LH和FSH。女性内外生殖器发育成熟,第二性征出现,并诱导卵泡细胞膜上的FSH受体及卵泡内膜、颗粒细胞膜上的LH受体增多,周期性地每次出现一个成熟卵泡,而雌激素和孕激素的分泌亦出现与卵泡周期性变化有关的波动,形成月经及周期性排卵,标志着女性性功能发育成熟。月经周期中,排卵前分别由卵泡的内膜细胞及颗粒细胞合成分泌雌激素和少量孕酮,排卵后则由黄体颗粒细胞及黄体卵泡内膜细胞大量合成释放孕酮和雌激素。月经周期中卵巢内分泌活动的周期性变化,也受下丘脑-腺垂体-卵巢内分泌细胞调节轴的控制,但不同于其他内分泌,其反馈调节方式较复杂,简述如下:①当前次月经中的黄体萎缩后,血中雌、孕激素急剧下降,负反馈地促进下丘脑GnRH及垂体LH、FSH释放逐渐增多,刺激卵泡发育和雌激素分泌逐渐增加,子宫内膜出现增生期变化;②随着卵泡发育成熟,高浓度雌激素反而对下丘脑GnRH释放产生脉冲式强正反馈调节,并进而引起腺垂体LH、FSH分泌高峰,诱发排卵;③LH、FSH在排卵后迅速下降,排卵后破裂的卵泡形成的黄体在LH作用下,继续分泌雌激素及大量分泌孕激素,约于排卵后一周出现雌激素的第二次高峰及孕激素高峰,子宫内膜由增生期转变为分泌期;④若未受孕,则高雌激素水平在同时存在的孕激素水平协同下,对下丘脑及垂体产生负反馈调节,GnRH、LH和FSH分泌进一步减少,黄体萎缩,血中雌、孕激素骤降,子宫内膜也随之缺血、坏死脱落形成月经。图12-5总结了月结周期中有关激素浓度的变化。有关妊娠时性激素的分泌及调节,将在第十四章中介绍。
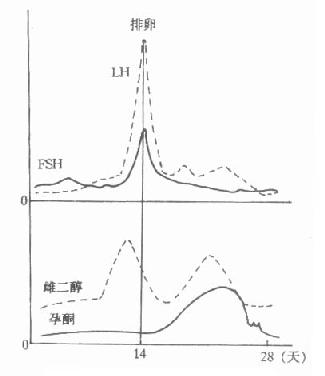
图12-5 月经周期中有关雌性激素变化示意图
由此看出,血中性激素水平,特别是雌性激素水平,在不同的发育阶段及月经周期的不同期,有不同的参考范围。表12-7列举了正常人血清性激素水平的参考范围。
表12-7 正常人血清主要性激素水平参考范围
| 男 | 女 | |||
| 睾酮 | 儿童 | <8.8nmol/L | <0.7 nmol/L | |
| 成人 | 14-25.4 nmol/L | 1.3-2.8 nmol/L | ||
| 妊娠期 | 2.7-5.3 nmol/L | |||
| 雌二醇 | 成人 | 29-132pmol/L | 卵泡期 | 17-330pmol/L |
| 排卵期 | 370-1850 pmol/L | |||
| 黄体期 | 184-881 pmol/L | |||
| - | 绝经期 | 37-110 pmol/L | ||
| 孕 酮 | 成人 | 0.38-0.95nmol/L | 卵泡期 | 0.6-1.9 pmol/L |
| 排卵期 | 1.1-11.2nmol/L | |||
| 黄体期 | 20.8-103.0 nmol/L | |||
| LH | 儿童 | 1.6-2.0 IU/L | 1.5-2.3 IU/L | |
| 成人 | 6-23 IU/L | 卵泡期 | 5-20 IU/L | |
| 排卵期 | 75-150 IU/L | |||
| 黄体期 | 3-30 IU/L | |||
| 绝经期 | 30-130 IU/L | |||
| FSH | 儿童 | 2.0-2.5 IU/L | 2.1-2.9 IU/L | |
| 成人 | 3-30 IU/L | 卵泡期 | 5-20 IU/L | |
| 排卵期 | 10-90 IU/L | |||
| 黄体期 | 5-12 IU/L | |||
| 绝经期 | 40-250 IU/L |
二、性腺功能的临床生化检测
(一)血清(浆)性激素测定
性激素的分泌虽不似肾上腺皮质激素和生长激素的分泌那样存在明显的昼夜节律,但每日中仍有一定波动。通常清晨高于下午,青春期这种差异更大。为便于比较,一般均在晨8点取血。必须指出,上述时间取样测定的结果,虽能代表取样时的血清浓度,但是并不一定能真实地反映性腺的内分泌功能,特别是女性,更存在着月经周期中不同期的每一天的差异,有时这种天间差异可相当巨大,如排卵日与前后日间。因此为真实地了解性激素内分泌功能,除确定病人的发育阶段、生育期女性取样日处于月经周期的什么阶段外,大多需进行必要的动态功能试验,观察有关性激素的变化,才有利于诊断。目前性激素的检测大多采用免疫化学法,测定包括游离和结合两部分的总浓度,主要性激素的血清正常值参考范围见表12-7。此外,在血清性激素测定结果的评价中,应注意可能存在的干扰因素。甲亢、肝硬化等疾患时,肝脏合成SHBG增多,血清中睾酮、雌二醇总浓度升高,但能发挥作用的游离部分可能并无变化。而甲减、极度营养不良及多种严重疾患时,可减少SHBG合成,产生相反的影响。女性使用避孕药除因其所含的性激素可能干扰下丘脑、垂体的反馈调节外,还可因所含雌激素诱导SHBG的合成,产生影响。
(二)性腺内分泌功能的动态试验
在判断性腺内分泌功能紊乱的有无,特别是病变部位的确定上,性腺内分泌动态功能试验有较大的意义。
⒈GnRH兴奋试验 GnRH为下丘脑释放的一种十肽调节激素,可迅速地促进腺垂体释放贮存的LH及FSH,并刺激LH和FSH的合成。本试验主要检测腺垂体促性腺激素的贮备功能。在抽取静脉血做基础对照后,静脉注射GnRH100μg,注射后20和60min再分别取血测定血清LH和FSH,与基础对照值比较。正常人GnRH刺激后,峰值应在20min出现。LH的变化为:正常男、女性青春期的峰值应为基础水平的3倍以上;正常成人男性峰值约为基础值的8-10倍,而女性成人卵泡中期峰浓度约为基础水平的6倍,黄体中期约为基础值的3倍左右;男性成人峰值约为基础对照值的2.5倍;女性成人卵泡中期峰值约为对照值的2倍,黄体中期约为2.5倍,也以排卵前期增加最显著。若有垂体病变所致性激素功能紊乱者,GnRH兴奋试验反应缺乏或低下;下丘脑病变所致者,反应正常或峰值延迟至60min始出现;单纯性青春期延迟者,虽然基础对照值低,但反应正常。
⒉绒毛膜促性腺素兴奋试验 人绒毛膜促性腺激素(human chorionic gonan-dotropin,hCG)为胎盘分泌的一种糖蛋白激素。其化学结构和生物学效应均类似LH(见第十四章)。本试验即利用其可促进睾丸间质细胞合成及释放睾酮的作用,了解睾丸间质细胞合成及贮存睾酮的功能状况。在第一日晨8点取血作对照后,开始每日肌肉注射hCg 2000IU,每日1次连续4日,分别于第4、5日晨8点再采血,测定对照及hCG刺激后血清睾酮浓度。睾丸内分泌功能正常者,第4日血清睾酮浓度为对照基础值的3倍左右,且第5日比第4日还高。因睾丸本身病变或畸形所致的原发性睾丸功能减退者,无反应或仅有弱反应,而继发性者则大多有正常反应。但本试验禁用于前列腺癌或肥大者。
⒊氯米芬间接兴奋试验 氯米芬(clomifene)又称氯底酚胺,为雌激素受体的部分激动剂,其内在活性很低,但和下丘脑GnRH分泌细胞上的雌激素受体结合后,可阻断雌激素(雌二醇等)对GnRH释放的负反馈调节作用,因此可用了解调节性腺功能的下丘脑-腺垂体轴的功能状况。常与GnRH兴奋试验配合,用作性腺功能减退症的定位诊断。具体方法是育龄女性在月经周期的第6日轴血作基础对照后,开始口服氯米芬50-100mg/d,连服5日,分别在开始服药的第3、5、7日取血。男性则可随时开始。测定服药前、后各血样的血清LH和FSH浓度。下丘脑-腺垂体调节轴功能正常者,男性第7日血清LH及FSH水平应较对照基础值分别升高50%和20%以上。女性开始服用氯米芬的第3日血清LH和FSH水平应较对照基础值分别升高85%和50%以上。性腺功能低下者,若对本试验及GnRH兴奋试验均无反应或仅有弱反应。提示病变发生在垂体水平;若本试验无反应或仅有弱反应,而GnRH兴奋试验反应正常或呈延迟反应,则表明病变在下丘脑水平。
⒋雌激素-孕激素试验本试验原理是通过使用雌激素和孕激素类药物,人工造成近似于月经周期中性激素水平的变化,观察有无月经出现,协助诊断育龄期女性闭经原因。闭经者给予已烯雌酚1mg,每晚1次口服,连服20日,并于开始服用已烯雌酚的第16日起,每日肌注黄体酮1mg1次,连续5日,随后同时停用雌、孕激素药、观察1周内有无月经。有月经,提示闭经是子宫以外的病变所致;无月经,则表明闭经原因是子宫内膜病变,如子宫内膜萎缩等。
三、性激素紊乱性疾病的有关临床生化诊断
性腺功能异常性疾病种类多,包括先天性性分化异常及遗传性性基因异常所致的各类畸形,后天性性发育异常及性腺功能紊乱性疾病。前一类疾病的诊断大多依靠临床表现及性染色体鉴定,将不在本书述及。现只主要讨论与临床生化诊断关系密切的性激素紊乱所致的有关疾病。
(一)性发育异常
指各种原因所致的出生后性腺、第二性征及性功能发育异常的统称。包括性早熟、青春期延迟及性幼稚症。
⒈性早熟 性早熟(sexualprecocity)即青春期提前出现。正常男女性青春期约于13岁左右开始,但受社会环境、文化教育等的影响可有较大差异。一般认为,女性在9岁以前出现包括第二性征在内的性发育,10岁以前月经来潮,男性在10岁以前出现性发育,即为性早熟。女性较男性多见。若性早熟系由于各种原因导致下丘脑-腺垂体-性腺轴对性发育的促进提前发动者,称真性性早熟。其中以下丘脑提前发动脉冲式大量释放GnRH而致的特发性性早熟最多见。此外,多种神经系统肿瘤、疾病亦可引发下丘脑或垂体提前产生青春期样GnRH及LH、FSH分泌。而某些原发性甲减及肾上腺皮质功能减退症的少儿,因TSH及ACTH释放增多可伴有LH和FSH释放增多,亦可引起真性性早熟。若性早熟不是依赖于下丘脑-腺垂体-性腺调节释放的促性腺激素或性激素所致者,称假性性早熟,多为睾丸、卵巢或肾上腺肿瘤“自主性”大量分泌性激素,或其他肿瘤组织产生的异源性LH、FSH所致。也有医源性者,而近年来国内因食用含性激素的保健品或饮料而致者,亦不少见。
性早熟的诊断根据临床表现一般不难作出。但真性性早熟和假性性早熟的临床处置及预后明显不同,二者的鉴别则有赖于临床生化检测及CT等检查。
性早熟者,检测血中性激素毫无例外均远远超出同龄同性别正常值,达到青春期或成人水平,甚至更高。若同时测定促性腺激素LH及FSH水平仍在同龄同性别正常范围或更低,则提示为假性性早熟中由于性腺肿瘤或分泌异源性性激素的其他部位的组织器官肿瘤而致。当性激素及促性激素水平均达到或超出青春期或成人水平,则应进一步作动态功能试验。如果GnRH兴奋试验或氯米芬间接兴奋试验出现正常成人样阳性反应或更强,提示为真性性早熟;若上述兴奋试验无反应或仅有弱反应,则应考虑为分泌异源性促性激素的肿瘤所致的假性性早熟,此时必须进一步确定并治疗原发病灶。
⒉青春期延迟及性幼稚症 青春期延迟(delayed puberty)指已进入青春期年龄仍无性发育者。根据我国人群的体质及文化、社会环境,一般规定为男性到18岁,女性到17岁以后才出现性发育者。性幼稚症(infantilism)则指由于下丘脑-垂体-性腺轴任一环节病变,出现男性20岁、女性19岁后,即进入性成熟期后,而性幼稚症若不及时处置,则可能终身不会性成熟。特别在青春期及时鉴别二者,对治疗方案的制定和预后均有重要意义。但此时仅凭临床表现,二者无法区别,而临床生化检查则可对二者作出权威性鉴别诊断。
青春期延迟绝大多数均为特发性(体质性),并往往有家族史,少数可由各种全身慢性消耗性疾病或营养不良引起。青春期延迟者有关性激素及促性腺激素LH、FSH测定,虽和下述的继发性性幼稚症者一样,均显示低下同龄同性别的正常值,但对GnRH和氯米芬兴奋试验,青春期延迟者都有正常反应。据此可与包括继发性性幼稚症在内的各种性幼稚症鉴别。
性幼稚症按病因可分为由性腺的各种先天发育异常、遗传缺陷及后天病损所致的原发性性腺功能低下,以及由各种下丘脑或腺垂体疾患、损伤所致的继发性性腺功能不足。性幼稚症诊断,根据临床所见不难作出,但还应通过检测性激素和促性腺激素血清水平作为筛选试验,必要时配合动态功能试验,进一步确定病变部位(环节),以指导治疗。下面分别介绍:
无论下丘脑病变还是垂体病变所致的继发性性幼稚症者,实验室检查均可见除性激素水平低下外,尚可出现促性腺激素LH、FSH水平亦低下。但应注意有时可仅表现为原因不明的单纯LH降低而FSH正常,或FSH降低而LH正常。若出现后者,性激素水平可无明显降低甚在正常水平。GnRH兴奋试验和氯米芬间接兴奋试验有助于病变部位诊断。二者均无反应或反应低下,提示病变部位为垂体性;若GnRH兴奋试验反应正常,而氯米芬间接兴奋试验无反应或反应弱,则病变往往在下丘脑水平。
(二)性激素合成酶缺陷性性功能紊乱
性激素合成酶缺陷都是遗传性先天缺陷。本章第三节中已谈及,包括性激素、肾上腺皮质激素在内有类固醇激素生物合成中,由胆固醇开始的起始步骤及多数中间反应,均由相同的酶催化。只是催化某些旁路反应和终末步骤的酶活性分布,存在组织、器官的差异,才造成肾上腺皮质、睾丸和卵巢分别主要合成不同的类固醇激素(参见图12-3)。在性腺和肾上腺皮质中活性无差异的酶缺陷所致的性功能紊乱,已在第二节中先天性肾上腺皮质增生症中一并介绍。此处仅简要讨论酶活性主要存在于性腺的性激素合成酶缺陷的临床生化诊断。
⒈C-17,20裂链酶缺陷 如图12-3所示,该酶催化类固醇激素合成中,17-羟孕烯醇酮转变为脱氢异雄酮(DHEA)及17-羟孕酮转化为雄烯二酮的反应。该酶缺陷将导致性腺及肾上腺皮质中各种性激素合成均受阻,出现性发育障碍、性幼稚症。由于睾酮在胚胎期还参与男性生殖系统的分化发育,故男性还可出现假两性畸形。该类病人实验室检查所见符合原发性性幼稚症的改变,即血中性激素水平极低,促性腺激素LH、FSH却很高。特异性的是血及尿中17-羟孕烯醇酮及17-羟孕酮大量出现。当使用ACTH及hCG兴奋试验时,这两种中间代谢物更明显升高,但性激素水平无明显变化,对本病有确诊价值。
⒉17-β羟类固醇脱氢酶缺陷 该酶催化性激素合成的最后一步反应,即由雄烯二酮还原为睾酮及雌酮还原为雌二醇的步骤。本病均见于男性,因此均存在程度不等的假两性畸形,甚至外生殖器完全女性化。临床生化检查除可见前述原发性性幼稚症改变外,若以HPLC法检测时,可观察到血及尿中雄烯二酮及雌酮特别是前者大量出现。hCG兴奋试验时,睾酮多无明显改变,但雄烯二酮升高极为明显。
⒊5α-还原酶缺陷 5α-还原酶未参与类固醇激素合成,主要分布于睾丸及其他睾酮的靶组织中,催化睾酮加氢还原为具高生物活性的二氢睾酮,发挥生理作用。本症只见于男性,因此亦都存在假两性畸形或外生殖器完全女性化。不同于其他任何类型性幼稚症之处在于,血睾酮水平正常甚至反升高,LH及FSH亦正常或升高。若以HPLC或免疫化学法同时检测血睾酮(T)及二氢睾酮(DHT),可见DHT明显减少,比较T/DHT比值,则可发现该比值明显增大。正常人比值为8-16,本症患者可高达35-84。根据上述临床生化的指标改变,可作出本症的诊断。
(三)青春期后性功能减退症及继发性闭经
青春期后性功能减退症(postpubertal hypogonadism)指男性性成熟后,因各种原因致雄性激素分泌不足产生的症群。继发性闭经(secondary amenorrhea)则指生育期女性已有规则月经者,出现月经持续停止6个月以上者。
青春期后性功能减退可因靶组织中不能产生雄激素受体激动效应(雄激素抗药综合征)、睾丸、腺垂体及下丘脑病变而致。均可以阳萎、第二性征减退甚至呈女性化等性功能低下表现为临床所见,大多还可见睾丸变小变软。临床生化检查可帮助确定病因或病变部位。雄激素抗药综合征者,血液性激素、促性腺激素改变与5α-还原酶缺陷症改变相似。若同时出现T/DHT比值明显增大,则可能为5α-还原酶缺陷所致。其他原因产生的青春期后性功能减退症,都会出现血睾酮水平低下,此时应结合LH及FSH测定,并配合必要的动态功能试验,按性幼稚症中确定病变部位的方法和标准,确定病变是在睾丸,还是腺垂体或下丘脑水平。
继发性闭经除外妊娠、哺乳等生理性因素后,则应考虑为子宫内膜、卵巢与腺垂体或下丘脑病变所致。雌激素-孕激素试验仍不能诱发月经,则提示可能为子宫内膜萎缩等子宫内病变所致;若有月经形成,则应考虑病因为下丘脑-腺垂体-卵巢轴中某一环节发生病变或功能失调。可参照性幼稚症中确定病变部位的方法,通过检测血清雌激素、孕激素及LH、FSH水平,配合动态功能试验,协助诊断可能的致病环节,以指导治疗。
(四)其他
性激素紊乱性疾病除上述外,较常见的还有女性多毛症(hirsutism),即女性出现男性样分布的体毛。若多毛症同时伴有男性第二性征出现,则称男性化(virilization)。二者皆因雄激素异常增多所致,并已证实女性多毛症及男性化的表现程度,与血中雄激素主要是睾酮的水平密切相关。女性体内的少量雄激素由卵巢和肾上腺皮质分泌。其中卵巢能合成释放一定量的睾酮和雄烯二酮,肾上腺皮质仅合成释放活性较低的脱氢异雄酮(DHEA)和雄烯二酮。多毛症和男性化均为卵巢和(或)肾上腺皮质合成释放雄激素异常增多的结果。
伴发于皮质醇增多症的多毛症,临床生化诊断见本章第二节。肾上腺男性化肿瘤所致者,系癌瘤细胞大量合成释放DHEA及雄烯二酮的结果。DHEA及其代谢物为女性尿17-KS类物质的主要来源,藉血中DHEA及尿17-KS测定结果异常升高,并且对地塞米松抑制试验无反应,可做出诊断。见于类固醇合成酶缺陷(先天性肾上腺皮质增生症,CAH)的多毛症或男性化,可参见表12-3,测定有关血及尿生化标志物水平明显升高,特别是ACTH兴奋试验后,有关标志物水平较基础值升高2倍以上,应考虑为相应的类固醇合成酶缺陷所致。主要产生雄激素的卵巢肿瘤导致的多毛症及男性化患者,血清睾酮浓度明显升高,并且对GnRH兴奋试验无或仅有弱反应。同时出现血中雌激素水平低下,促性腺激素多正常或升高。而同样可产生大量雄激素的多囊卵巢病(综合征)出现的多毛症或男性化,虽然亦可出现血睾酮及LH轻度升高,但雌激素水平多正常,FSH正常或降低,并且GnRH兴奋试验时,血睾酮及LH呈增强反应,因此不难与主要产生雄激素的卵巢男性化肿瘤鉴别。
第十三章 神经、精神疾病的生物化学
第一节 概述
人类医学科学的迅速发展,神经科学已成为医学和生命科学的前沿,近10多年以来,神经生物学的进展很快,正在整体、离体细胞及分子水平上深入研究,以揭示脑的奥秘。人类神经系统为自然界最复杂的系统,大脑内的神经元总数估计为1010-1012个,胶质细胞数较此多出10-50倍,而mRNA的表达比其它器官高出3-5倍。在神经细胞内及细胞间持续进行的信息传递是脑行使其功能的基础,机制涉及多种具有特殊神经功能的蛋白质(如受体、离子通道、信使蛋白等)以及多种具有调控功能的化学物质(如中枢神经递质)。神经分子生物学的研究使人们对脑在各种疾病状态时的功能改变了更为深入的认识,成亿的神经元以神经化学物质传递的方式相互作用,维持中枢神经系统的功能。任何原因导致中枢神经系统功能和结构改变以及与其它系统相互关系的不平衡,都可表现出精神活动的异常,神经化学物质对维持人类正常精神活动起着极其重要的作用,其功能改变则与精神疾病的发生有关系。因此,神经生化的研究对探讨精神疾病的病因具有极其重要的作用。
一、神经系统的生物化学特点
中枢神经系统主要由神经元和神经胶质细胞构成。神经元是神经组织的结构单位。神经元之间的相互作用以化学物质(神经递质)传递的方式进行,前一个神经元在神经冲动时从末梢向突触间释放神经递质,后者与突触后膜上的受体发生作用,引起一系列生理反应。由于中枢神经系统功能十分独特,其代谢亦具特点。
(一)糖代谢
大脑含葡萄糖量为112±37mg/100g,人脑组织糖原含量仅为0.1%脑组织重。脑组织利用的葡萄糖主要靠血液提供,人脑对血糖浓度的波动极敏感,血糖浓度正常时,血脑屏障对葡萄糖的易化转运能力颇强,脑对葡萄糖的需要不受脑毛细血管转运的限制。据计算,人脑平均葡萄糖的利用率为31μmol/100g脑组织/分钟,其中26μmol用于氧化供能,其余用于合成脑内糖脂、糖蛋白的原料,或转变成其它脑组织有用之物。葡萄糖在中枢神经组织的氧化形式主要为有氧氧化和无氧酵解,占脑中葡萄糖分解率的90%-95%,其次是磷酸戊糖途径,占5%-10%,1克分子葡萄糖在脑细胞内彻底氧化供能可生成38克分子ATP,因葡萄糖氧化过程中形成的NADH+H+主要通过苹果酸-天冬氨酸穿梭进入线粒体内。脑龄不同,葡萄糖氧化方式不尽相同,胎脑组织葡萄糖酵解供能,出生不久逐渐以糖的有氧氧化来提供能量。正常情况下,人脑乳酸生成量很低,为2.7μmol/(100脑组织·min),丙酮酸生成量0.6μmol(100g脑组织·min)。当脑供血及供氧不足时,丙酮酸、乳酸生成明显增加,可危害大脑功能。一旦氧供应恢复,脑内LDH1催化乳酸生成丙酮酸,后者进入线粒体内转变成乙酰CoA参加三羧酸循环而彻底氧化。当血糖降至1.0mmol/L(20mg%)以下时,对大脑耗糖量会产生明显限制性影响,此时耗氧量减少20%,可出现严重低血糖症状。当血糖降至0.5mmol/L时,耗氧量仅为正常的58%,则产生低血糖昏迷,危害大脑功能。大量注射胰岛素,使血糖显著降低,引起脑内糖供应不足而产生低血糖休克,用以治疗躁狂型精神病人,对防止病人自伤或危及他人安全有一定效果,但低血糖可损害脑功能,不宜频繁使用。脑组织除利用葡萄糖外,还可利用甘露糖及半乳糖氧化供能。婴幼儿以乳汁为主要能源,因此婴幼儿脑内半乳糖的分解代谢显得比成人重要。
脑组织糖代谢酶类的定位与分布亦有其特征,酸性磷酸酶主要分布于神经胶质细胞和髓鞘内,碱性磷酸酶活性主要见于神经元内,而醛缩酶主要存在于大脑灰质和小脑内,即使在同一神经元内,胞核与胞浆的糖代谢酶类布局也有不同。胞核中已糖激酶及6-磷酸葡萄糖脱氢酶活性高于胞浆,相反胞核内的磷酸果糖激酶活性低于胞浆。这种酶活性分布的差异,其意义还不清楚。对神经胶质细胞糖代谢酶活性的研究,发现糖代谢酶与年龄有关,则出生动物的6-磷酸葡萄糖脱氢酶及琥珀酸脱氢酶活性均较高,而随年龄增长活性呈下降趋势,这反映了髓鞘形成时期对能量需要大。此外,脑损伤的恢复期,6-磷酸葡萄糖脱氢酶活性旺盛,磷酸戊糖途径代谢活跃,生成的NADPH+H+可用于脑组织的修复。
(二)脂类代谢
脂类在脑内含量较丰富,且相当稳定,更新率缓慢。脑组织可利用葡萄糖分解产物乙酰CoA作原料合成脂酸,但能力不强。脑内生成的23碳、25碳长链奇数碳原子脂肪酸是构建脑组织类脂成分及合成某些脑活性物质的重要原料,α-羧脂酸是脑内脑苷脂和脑硫脂的重要组分。脑组织亦具有进行α-氧化的能力。先天性脂酸α-氧化代谢缺陷病人,不能使植烷酸进行α-氧化,而大量堆积于血浆或组织中,导致在髓鞘中堆积,植烷酸还可抑制其它脂酸正常代谢,如抑制软脂酸转变成软脂酰CoA,严重影响脑组织结构及功能,临床上称为Refsum综合征。
脑组织利用α-磷酸甘油和脂酰CoA合成溶血磷脂酸,再与一分子脂酰CoA作用生成磷脂酸、磷脂酸在磷酸酶作用下磷酸解生成甘油二酯,甘油二酯与CDP-胆碱反应合成卵磷脂,脑内不能直接利用脑磷脂与S-腺苷蛋氨酸反应生成卵磷脂。神经磷脂中脂酸碳链较长(C18-26)为其特点之一。
脑内鞘脂分为鞘磷脂和鞘糖脂两类,均含鞘氨醇,不含甘油醇。脑组织鞘糖脂主要存在于脑灰质中,其脂酸通常为硬脂酸。脑内神经节苷脂的N-脂酰鞘氨醇部分有疏水性,糖基部分有亲水性。神经节苷脂中含数目不等的唾液酸分子,唾液酸可起屏蔽作用,抵御神经节苷脂被糖苷酶酶解,现已从脑组织中分离出30余种神经节苷脂,为神经元尤其是突触膜的重要成分。神经节苷脂的亲水性糖基与唾液酸构成神经元的膜激素受体及神经递质受体,参与神经细胞间的识别与信息交流,发挥其重要的功能。神经节苷脂在溶酶体内被β-半乳糖苷酶或已糖胺酶降解,若先天性缺乏这两种酶则发生GM1-神经节苷脂沉淀病或GN2-神经节苷脂贮积病(黑朦性痴呆)。神经髓鞘含16%半乳糖脑苷脂及4%脑硫脂,神经鞘磷脂占5%髓鞘干重。半乳糖脑苷脂与鞘形成有关,前者可被半乳糖基磷脂酰胺-β-半乳糖苷酶水解生成神经酰胺和半乳糖,缺乏此酶时,半乳糖苷脂沉积于组织中,可导致Krable球样细胞脑白质营养不良症。脑硫脂降解需硫酸脑苷脂酶催化脱去硫酸,缺乏此酶可致脑硫脂贮积,发生异染性脑白质营养不良。神经鞘磷脂可被溶酶体中的神经鞘磷脂酶降解,缺乏此酶引起神经鞘磷脂沉淀症,出现患儿痴呆,肝脾肿大,易夭折。
脑组织能合成胆固醇,也能摄取血液胆固醇,用于构建其膜系统。幼年动物脑髓鞘化活跃期,HMGCoA还原酶活性较强,脑合成胆固醇旺盛;成年期比酶活性锐降,脑内胆固醇合成率明显降低。脑组织缺乏降解胆固醇酶系,因此,其更新十分缓慢。
脑内乙酰乙酸硫激酶(AAT)和琥珀酸单酰CoA转硫酶(SUT)活性高,因此,脑组织可利用肝脏脂酸β-氧化所形成的酮体作为能源,实验证明长期饥饿的动物脑,其25%-50%的能量来自酮体的氧化。
(三)氨基酸代谢
中枢神经系统存在两个氨基酸代谢池:一是神经胶质细胞内代谢池,其更新率较快;另一个是神经元代谢池,其更新率缓慢。脑组织可利用葡萄糖代谢的中间产物碳骨架经转氨基作用合成非必需氨基酸,又可从血液直接摄取氨基酸。已知血液中的氨基酸进入脑内需经血液屏障的转运系统,其转运系统转运氨基酸的能力随脑发育成熟而变化。正常情况下氨基酸净入脑率似乎不受血脑屏障的转运饱和度控制,而受脑中氨基酸代谢率的限制,因脑毛细血管对氨基酸转运到脑内的饱和度大大超过正常血浆中氨基酸的浓度。此外,还存在各种氨基酸入脑率相互竞争机制,例如:苯丙酮尿症病人,苯丙氨酸在脑内蓄积可抑制必需氨基酸(色氨酸)的入脑率,严重影响以色氨酸为前体的5-羟色胺神经递质的合成,使病人脑部神经、精神异常症状加重。脑组织中游离氨基酸约75%-80%是天冬氨酸、谷氨酸、谷氨酰胺,其中以谷氨酸浓度最高,为10mmol/L,此外,还有N-乙酰天冬氨酸、牛磺酸及γ-氨基丁酸。
代谢池内的氨基酸可用于合成脑特殊蛋白质,如合成与降解神经递质的酶蛋白;在轴突末梢可用于合成少量结构蛋白,如微管蛋白、神经微丝及膜蛋白,保证使脑细胞的蛋白成分处于不断的更新状况。幼年动物髓鞘形成期蛋白质合成旺盛,成年期缓慢更新。脑组织存在多种蛋白水解酶(以酸性和中性蛋白酶为主),蛋白酶将脑内衰老变性的蛋白质水解成各种肽类,后者在内肽酶及外肽酶作用下,降解成氨基酸而进入脑氨基酸代谢池。
脑内谷氨酸脱氢酶活性虽仅次于肝和肾上腺皮质,但谷氨酸脱氢酶催化反应的平衡常数实际上有利于谷氨酸的生成。
脑细胞缺乏合成尿素的酶等,脑内生成的氨不能转变成尿素,而只能用于合成氨酰胺,再运送到肝或肾。
(四)核酸代谢
脑组织可利用甘氨酸、天冬氨酸、谷氨酰胺、CO2及一碳单位作原料,从头合成嘌呤核苷酸,又可从补救合成途径合成嘌呤核苷酸。但脑内缺乏合成嘧啶环的氨甲酰磷酸合成酶Ⅱ,不能从头合成嘧啶核苷酸,只能从补救途径合成。脑内RNA含量最为丰富,其代谢速度的快慢与其神经系统所处功能状态有关,急性电休克可加速脑组织核苷酸代谢率,其中以GTP及UTP在脑内浓度增高和更新率加速最明显。DNA主要存在于神经细胞核内,成熟的神经元内DNA含量相当恒定。线粒体DNA含量少,更新缓慢。生长激素及神经生长因子能促进脑内核酸的合成与更新。不同脑区核酸更新率存在差别,大脑半球灰质的核酸更新不及白质的快,小脑、丘脑和脑干比大脑转换的更快。脑组织含有丰富的核酸与其蕴藏大量的遗传信息有关,通过合成大量的神经递质、神经肽类激素及各种激素释放因子和抑制因子协调全身代谢。
(五)能量代谢
正常情况下,人脑呼吸商为1,清醒时流经脑的血流量为52±12ml/(100g脑组织·min),脑的耗氧量为3.5ml/(100g脑组织·min)。明显高于机体其他组织的耗氧量。成人脑组织全脑代谢率按人脑平均1400g计算,相当于每分钟需耗氧46ml,需氧化76mg葡萄糖,流经脑组织的血液每分钟需有750-1000ml。人脑按重量只占体重的2%左右,其需氧量几乎占全身的20%-25%,血流量占心输出量的15%,证明脑组织耗氧量大,是体内能量代谢十分活跃的器官之一。生长发育期脑需氧比例更大,四岁前幼童脑耗氧量占全身的50%以上。脑对缺氧耐受力极差,3-5分钟严重缺氧对大脑产生明显的功能损害,处于完全缺氧状态5分钟后,神经元功能难以恢复,缺氧30分钟后造成永久的不可逆的神经损害,尤其是脑皮层及皮层下视觉通路神经元最不耐受缺氧。脑对缺氧耐受性差与脑内ATP高稳定水平有关,即脑ATP迅速生成及迅速利用。1/2的ATP在3秒钟内即可变成ADP,有的脑区甚至不需3秒钟。在基础状况下,ATP/ADP比值为10-20,低于此比值,脑内腺苷激酶催化2克分子ADP生成1克分子ATP和1克分子AMP,增加可利用的ATP,以应付急需。AMP可促进ATP生成,为正调节剂,并具有放大效应。脑内ATP丰富时,肌酸激酶活跃,可生成磷酸肌酸而贮存能量,脑内肌酸激酶为BB型同工酶。
二、神经递质的生物化学基础
神经递质的研究是始于外周神经,从1869年Schmiedeberg等首先发现毒蕈碱对心脏的抑制作用与刺激迷走神经的效果很相似的研究开始,到目前为止已发现的神经递质有人估计可能达200种。由于它们直接作为递质参与神经调节,或调制传统神经递质的活动,从而使人们对神经调节的传统概念重新加以修改和补充,提出神经递质(neuro-transmitter)和神经调质(neuromodulator)的概念。
神经递质是神经系统进行信息传递过程的媒介物,是化学传递的物质基础。其主要特征为:①在神经细胞内合成,存在(贮存)于突触前神经末梢,在中枢呈不均一分布。②在神经受刺激时释放,作用于突触后膜上的特异性受体。③在效应细胞引起特定的功能改变或电位变化后,一段时间内迅速失活。④直接外加于突触可引起与刺激神经同样效应,并可被特异性拮抗剂所阻断。
神经调质与神经递质不同,在于前者不直接触发所支配细胞的功能效应,只是调制传统递质的作用。其特征:①可为神经细胞、胶质细胞或其它分化细胞所释放,对主递质起调制作用。本身不直接负责跨突触信号传递或不直接引起效应细胞的功能改变。②间接调制主递质在突触前神经末梢的释放及其基础活动水平。③影响突触后效应细胞对递质的反应性,对递质的效应起调制作用。
目前将单胺、乙酰胆碱和氨基酸三类一般认为是神经递质,而神经肽则多认为神经调质。一些材料说明,脑内的一些神经肽与传递递质共存,但由于中枢神经细胞密集,结构复杂,当前还很难用实验方法确定传统神经递质和神经肽在末梢共同释放,更难以确切证明它们所引起的生理效应并用药理学方法加以验证,有的就统称神经递质。
⒈神经递质的分类(表13-1)
表13-1 神经递质的分类
| 中文名称 | 英文名称 | 英文缩写 |
| 乙酰胆碱 | Acetylcholine | Ach |
| 单胺类 | ||
| 肾上腺素 | Epinephrine | A |
| 去甲肾上腺素 | Norepinephrine | NE;NA |
| 多巴胺 | Dopamine | DA |
| 5-羟色胺 | Serotonin | 5-HT |
| 组胺 | Histamine | HA |
| 氨基酸类 | ||
| β-氨基丁酸 | β-Aminobutyric Acid | GABA |
| 甘氨酸 | Glycine | Gly |
| 谷氨酸 | Glutamic Acid | Glu |
| 天冬氨酸 | Aspartic Acid | Asp |
| 神经肽类 | ||
| 下丘脑释放激素 | ||
| 促甲状腺素释放素 | Thyrotropin Releasing Hormone | TRH |
| 促黄体生成素释放素 | Lutinising Hormone Releasing Hormone | LHRH |
| 生长激素抑制素 | Somatostatin | SST |
| 促肾上腺皮质激素释放素 | Corticotropin Releasing Hormone | CRH |
| 生长激素释放素 | Growth Hormone Releasing Hormone | GRH |
| 垂体肽 | ||
| 促肾上腺皮质激素 | Adrenocorticotropic Hormone | ACTH |
| β-内啡肽 | β-Endorphine | β-E |
| α-促黑素细胞激素 | α-Melanocytestimulating Hormone | |
| 血管加压素 | Vasopressin | |
| 催产素 | Oxytocin | OT |
| 脑肠肽 | ||
| 亮氨酸脑啡肽 | Leucine Enkephalin | |
| 蛋氨酸脑啡肽 | Methionine Enkephalin | |
| P物质 | Substance P | SP;P |
| 胆囊收缩素 | Cholecystokinin | CCK |
| 血管活性肠肽 | Vasoactive Intestinal Polypeptide | VIP |
| 神经降压肽 | Neurotensin | |
| 胰岛素 | Insulin | |
| 肠胰高血糖素 | Enteroglucagon | |
| 胰泌素 | Secretin | |
| 蛙皮素 | Bombesin | |
| 胃泌素 | Gastrin | |
| 其他 | ||
| 血管紧张素Ⅱ | Angiotensin Ⅱ | |
| 徐缓激肽 | Bradykinin | |
| 肌肽 | Carnosine | |
| 前列腺素 | Prostaglandin | PG |
| 脑钠肽 | Brain Natriuretic Peptide | BNP |
| 降钙素 | Calcitonine |
⒉某些中枢神经递质的代谢及作用
⑴乙酰胆碱(Ach):胆碱能神经利用乙酰CoA和胆碱合成乙酰胆碱,其合成酶是胆碱乙酰化酶,该酶存在于突触胞浆中,脑内胆碱乙酰化酶浓度与Ach含量平行。合成的Ach约有一半贮存于囊泡,一半游离于胞浆中。神经元兴奋时,释放Ach至突触间隙,与突触后膜受体结合发挥作用后,以极快的速度使突触前膜和后膜上的乙酰胆碱酯酶(AchE)水解失活。Ach在中枢神经系统中分布广泛,其中纹状体、下丘脑、杏仁核及脑干网状结构等含量较高,大脑皮质和小脑皮质则低。胆碱能受体分毒蕈碱受体(M型)和烟碱样受体(N型),Ach与前者关系密切,激活这一受体可引起两种不同的细胞内信号系统的活动,从而将其又分为两种亚型,即M1和M2.M1与细胞内第二信使磷酸肌醇有关,M2通过偶联蛋白Gi与腺苷酸环化酶偶联。Ach对各级中枢神经的作用有:感觉功能,感觉特异投射系统的第二级、第三级神经元很可能是胆碱能神经元;运动功能,锥体外系运动中枢纹状体中含M型胆碱能中间神经元,此中间神经元与人类的僵住症(强直性晕厥)和帕金森症密切相关;学习、记忆与意识功能,海马胆碱能系统的兴奋是学习、记忆和意识的基础,大脑皮层感觉区含M1胆碱受体是清晰-睡眠周期活动的密切相关部位,大剂量M胆碱拮抗剂东莨菪碱致人麻醉,抑制大脑皮层和海马M胆碱功能,人意识消失,近期记忆力缺乏,相反胆碱能激动剂具有增强学习与记忆的功能,Ach对行为、脑电、摄食、饮水、体温及血压调节均有一定的作用。
⑵去甲肾上腺素(NE):NE能神经元可利用酪氨酸为原料,经酪氨酸羟化酶(TH)及多巴β-羟化酶(DβH)的作用生成NE。合成的NE在囊泡内与ATP按4:1的比例结合而贮存,当动作电位到达末梢,囊泡中的NE、ATP、嗜铬颗粒蛋白A及可溶性DβH一起排入突触间隙。NE作用终止的主要方式是75%-95%释放量被突触前膜再摄取,另一小部位进入血液,被非神经组织摄取,经MAO及儿茶酚胺氧位甲基移位酶(COMT)作用而降解,外周组织中,NE代谢产物以3-甲氧-4-羟苯乙醇酸(VMA)为主,在CNS中,则以3-甲氧-4-羟苯乙二醇(MHPG)为主。NE能神经元在中枢神经系统中分布广泛,其胞体主要集中分布于延髓和脑桥。中枢NE能受体分为α-受体和β-受体,α-受体可分为α1和α2两型。NE具有中枢效应,能维持脑电和行为的觉醒,NE能神经元适当兴奋可产生兴奋与欣快情绪,过度兴奋则导致躁狂与攻击行为。NE与精神活动有关,利血平降压可使NE耗竭而出现抑郁症,NE类似物可产生拟精神病的发作,情感性精神病人体液中NE及其代谢物可异常。NE还与体温调节、摄食、记忆和血压等调节有关。
⑶多巴胺(DA):DA与NE合成基本相似,但DA的囊泡中不含DβH,脑内DA的主要产物是高香草酸(HVA)。脑内DA能神经元胞体位于中脑和下丘脑。DA受体根据功能差别可分为两个亚型:DA-1型受体,其活性与腺苷酸环化酶有关;DA-2型受体,DA受体阻断剂对DA-2型受体具有较强的亲和力,凡用于抗精神病的药物及治疗帕金森症的麦角碱均通过DA-2型受体起作用。此外,突触前神经元胞体或树突上还存在DA自身受体(DA-3型受体),该受体能对同一个DA能神经元释放的DA产生反应,反馈性调节DA能神经元的活动。
中枢神经DA的作用表现多方面:锥体外系统的DA与躯体运动功能有关,是一切躯体运动的基本条件,用伪递质6-羟多巴胺注入动物纹状体内可发生类似于人类的帕金森氏症状的震颤和强直症状;脑内DA在影响机体的一般行为和精神情绪活动上起着重要作用,将DA注入动物脑室,可产生与人类精神分裂症相似的行为变化,服用DA前体左旋多巴,可改善抑郁症,下丘脑的DA神经元对垂体的内分泌活动,特别是对促性腺激素的分泌活动具有控制作用。
⑷5-羟色氨酸(5-HT):5-HT能神经元以色氨酸为原料,在色氨酸羟化酶及5-羟色氨酸脱羧酶作用下合成5-HT。色氨酸羟化酶特异性较高,只存在于5-HT能神经元中,且含量少,活性较低,是合成5-HT的限速因子。5-HT贮存于囊泡中,释放后被再摄取及MAO降解而终止作用。5-HT主要经MAO灭活生成5-羟吲哚乙酸(5-HIAA),而在松果体内,5-HT经羟基吲哚氧位甲基移位酶(HIOMT)及芳香烃胺氮位甲基移位酶(AANMT)作用生成黑色紧张素(褪黑素),后者能抑制垂体促性腺激素的分泌。5-HT能神经元胞体位于低位脑干中线附近的中缝核。目前认为脑内有三种5-HT对大脑表现抑制性影响与睡眠有关,当脑内5-HT减少时则出现失眠。中枢5-HT有提高痛阈作用,具有镇痛功能。脑内5-HT与情绪和精神活动有关,当脑内5-HT代谢失调,可导致智力障碍和精神症状,近年发现精神分裂症病人脑脊液中存在5-甲氧色胺(5-MT),当给予大量蛋氨酸作为甲基供体,可加重精神分裂症状。急性青春型和其它急性兴奋型精神病患者,其血液5-HT含量显著低于健康人,5-HT水平低下者有自杀意念。这些表明5-HT有助于维持精神、情绪的稳定,中枢5-HT功能不足是情绪紊乱的体质因素。脑发育不全,智力愚钝患者,血中5-HT含量较低,这可能由于苯丙氨酸过多,抑制色氨酸羟化酶,使5-HT合成减少所致。脑内5-HT有抑制肾上腺激素分泌和黄体生成素的分泌,促进催乳素的分泌,说明5-HT与性激素、性行为有关。
⑸组胺(HA):HA系氨基酸脱羧生成。主要分布于丘脑、乳头体和视上核,脑内合成的HA贮存于神经元和肥大细胞内。在肥大细胞内组胺与肝素、碱性蛋白硫酸多糖形成复合物,此种形式的组胺更新缓慢,神经元内组胺更新较快。组胺主要在组胺-N-甲基转移酶作用下生成甲基组胺,再经MAO作用转化为3-甲基咪唑乙酸。中枢神经系统中HA能神经元集中在下丘脑、中脑、纹状体及黑质。脑内HA受体分为H1和H2两型,H1型激动时产生兴奋效应,H2型激动时则产生抑制效应。此外还有H3受体,很可能是神经末梢的自身受体,抑制HA的释放。直接向脑室注射HA,可出现血压升高、心率加快、体温下降及促进抗利尿激素的分泌,外周H1型受体兴奋使支气管、肺动脉、小动脉的平滑肌松弛,引起扩张血管作用。保持锥体外系神经功能的正常有赖于DA和Ach能神经元及5-HT和HA能神经元这两个系统的动态平衡,DA及5-HT能神经元为抑制性神经元,而Ach能及HA能神经元为兴奋性神经元,两体系失衡可引起锥体外系疾病,如帕金森症。
⑹γ-氨基丁酸(GABA):中枢GABA的合成部位在神经末梢。谷氨酸经谷氨酸脱羧酶(GAD)作用生成GABA。GABA合成后与线粒体膜或突触体膜结合而贮存。释放的GABA经再摄取而终止其作用,在GABA转氨酶作用下脱氨基生成琥珀酸半醛,后者经琥珀酸半醛脱氢酶作用生成琥珀酸,而进入三羧酸循环被彻底氧化。GABA主要分布于脑灰质内,尤以黑质、苍白球含量最高。已知GABA受体有两种亚型:GABA-A型受体,在小脑集中于颗粒细胞层,为突触后膜上的受体;GABA-B型受体,主要集中于小脑胶质细胞,为突触前受体。GABA是中枢皮层的主要抑制性递质,对所有神经元都呈抑制作用,睡眠时皮层释放GABA增加。癫痫发作的强度与大脑皮层内GABA含量降低程度一致,基底神经节中GABA降低与帕金森综合征及亨延顿舞蹈病(Huntington disease,HD)有关。GABA降低,使抑制性神经冲动不足,DA功能亢进,可促发精神分裂症。
⑺谷氨酸(Glu):脑内谷氨酸和天冬氨酸含量很高,对神经元有极强的兴奋作用,是兴奋性递质。谷氨酸在脊髓中特异性分布,即背根高于腹根,而天冬氨酸腹根高于背根,故认为谷氨酸是初级传入纤维的兴奋性递质,天冬氨酸是中间神经元兴奋性递质。脑内存在一个神经末梢与胶质细胞谷氨酸循环轮回系统,神经胶质细胞内含有谷氨酰胺合成酶,它能将从突触间隙摄取的谷氨酸转化成谷氨酰胺,后者可转运到神经末梢中,经谷氨酰胺酶脱氨生成末梢内的谷氨酸,在神经兴奋时释放至突触间隙起递质作用。谷氨酸受体以不同激动剂来分,有海人藻酸受体(KA-受体)、使君子氨酸受体(QA-受体)及N-甲基-D-天冬氨酸受体(NMDA-受体)。目前还有的提出谷氨酸的代谢性受体,为一种跨膜蛋白核成员,与G蛋白有关,这类受体可激活磷脂酶C,产生肌醇三磷酸(IP3),使Ca2+从神经胞内钙池流出,影响神经元的兴奋性。也有人认为QA受体和KA受体的功能与动物的定位、学习能力、嗅觉的学习有关。谷氨酸和天冬氨酸几乎对所有的神经元都有兴奋作用,其特点是作用快、消失快。脑内谷氨酸和谷氨酰胺的转化是对NH3的解毒作用。谷氨酸也能促进脑组织合成乙酰胆碱、GSH及推动脑内糖的有氧氧化。兴奋性氨基酸的代谢紊乱和在神经组织中的积聚可通过兴奋性作用引起脑组织损伤,尤其是谷氨酸的神经毒性可能与人类某些神经系统疾病如:早老性痴呆和亨延顿病中出现的神经退化相关。谷氨酸是味精的主要成分,自60年代末报道有其神经毒性作用以来,对食用味精是否安全的问题一直存在争论,某些外国人食用味精后出现某些异常反应,从而在国外提出“中国餐馆综合征”的概念,争论并未解决,仍待深入研究。
⑻内啡肽和脑啡肽:内啡肽早先称为内原性鸦片样物质(EOLS),现已知EOLS包括内啡肽和脑啡肽,其受体与鸦片受体共存。内啡肽主要存在于脑和垂体中,为大分子的吗啡样肽,有α-、β-、γ-和δ-内啡肽四种,其中以α-和β-内啡肽(前者为16肽,后者为31肽)较重要。β-内啡肽与蛋氨酸脑啡肽共存于β-脂酸释放激素(β-LPH)中。脑啡肽分为蛋氨酸脑啡肽(H-酪-甘-甘-苯丙-蛋-OH)与亮氨酸脑啡肽(H-酪-甘-甘-苯丙-亮-OH),二者均为五肽神经激素。脑内存在单独的内啡肽途径,在下丘脑和支配中脑与边缘系统神经轴索中一组细胞有β-内啡肽系统。β-LPH、ACTH与β-内啡肽存在于同一脑神经元,鸦片黑素皮质素原可能为这三种物质的前体。脑啡肽对酶解不稳定,N-末端极易水解掉一个酪氨酸而成为无活性4肽。β-内啡肽较为稳定。内啡肽及其受体可以影响针刺止痛效应,针刺使人脑脊液中内啡肽含量增高,其增高的幅度与针刺镇痛效果平行,α-内啡肽有镇定和轻度镇痛作用,而β-内啡肽却具有较强的镇痛作用,β-内啡肽通过抑制多巴胺能神经元的活动,引起运动减少,有资料报告,β-内啡肽直接刺激脑内DNA的合成,与脑内蛋白质合成有关。此外,内啡肽还可促进垂体前叶分泌催乳素和生长激素,在中枢神经系统中发挥神经调质作用。β-内啡肽与精神分裂症有关,在脑内β-内啡肽可直接降解为α-内啡肽(α-E)及γ-内啡肽(γ-E),γ-E可再降解成α-E。γ-E和α-E分别在降解酶作用下生成有精神安定特性的N-端脱酪氨γ-内啡肽(DTγE)片段及有精神刺激特性的脱酪氨酸α-内啡肽(DTαE),当脑内DTγE及DTαE生成缺陷,而α-、β-内啡肽过多可致精神分裂。脑啡肽神经元分布广泛,其末梢主要分布于杏仁核、丘脑背内侧核、脑室及导水管周围灰质和脊髓背角等部位,与鸦片受体分布相关性大,脑啡肽在脑内起抑制性递质作用,具有镇痛作用,但不及β-内啡肽强。脑啡肽参与动物自我刺激和奖赏功能,并与学习、记忆行为有关。近年研究发现,β-内啡肽和脑啡肽能作用于从活体到体外的一系列免疫过程,包括有丝分裂原引起的淋巴细胞增殖、自然杀伤细胞活性、化学趋化作用以及T细胞释放淋巴因子的过程。
强啡肽(Dynorphins,DyN)为内源性吗啡的第三族,由前体-前强啡肽转化而来。强啡肽为鸦片样受体族中KaPa亚型受体强有力选择激动剂,存在于中枢神经系统各部位,以垂体神经下丘脑最高。其生理功能主要与神经分泌调节有关,例如:对伤害刺激冲动的调控、触觉、渗透压及化学感受的调控等有关。
⑼P物质(SP):P物质由11个氨基酸残基组成的多肽,分子量1340,耐热、抗酸,可被多种蛋白水解酶裂解而失活。人脑以黑质、丘脑下部和松果体内含P物质最高。主要集中于神经末梢的突触体内。P物质是传入纤维末梢释放兴奋性神经递质,它又具有明显的镇痛作用,其镇痛作用可被鸦片拮抗剂纳洛酮所阻断,因而有人据此把它归入内源性鸦片肽的一种。P物质在中枢对去甲肾上腺素能神经纤维具有兴奋作用,在外周可促进NE的释放。P物质可提高脑内多巴胺的更新率,降低多巴胺水平,它与5-HT共存于一个神经元,降低中枢5-HT的更新率。P物质在某些神经元内与乙酰胆碱共存,在受体水平两者互相拮抗。
⑽前列腺素(PG):PG是脑内一种内源性活性物质,对神经元的体液介导或刺激调控具有独特的作用,现已发现有一系列结构相似的化合物,推测为其一大类神经递质或神经激素。CNS内磷酯在磷酯酶作用下生成花生四烯酸,后者经PG合成酶催化生成RG。其特点是在需要发生生理效应时才合成,在局部释放及局部产生生物学作用,故贮存不多,主要含于突触体部分,脑内以PGF2a最多,其次是PGE2、PGE1、PGE1a和PGD2等,它们主要存在于大脑皮质、小脑和下丘脑等部位。PG类分解主要在PG-△13还原酶作用下分解成活性极低或无活性产物,为6-酮-PGF1a,随尿排出。脑内PGE1有明显的致热效应,细菌性致热原或内毒素产生发热是通过PG生成而实现,PGE(E1、E2及E3)对动物有镇静作用,以7-20μg/kgPG注入脑室,可使动物昏睡与木僵状态,少量皮下注射仅有轻微安定作用。PGF2可促进NE、DA的释放,PGD2抑制黄体生成素释放,诱导垂体5-HT生成与更新。PGE可抑制摄食,此外,PG类对脑血循环也有影响。
⑾一氧化氮(NO):最近研究表明,NO很可能是一种内源性的神经递质,是细胞间信息交流的载体,它具有广泛的生理功能。体内精氨酸在NO合成酶(NOS)作用下生成NO和胍氨酸。生成的NO与含鸟苷酸环化酶的血红素基团结合,形成NO-血红素-鸟苷酸环化酶复合体,此为鸟苷酸环化酶活化形式,从而使细胞内cGMP水平升高,发挥生理功能。神经元中的NO是中枢和外周神经系统的信使物质,可能与脑细胞的发育、学习和记忆过程、垂体后叶分泌加压素和催产素,以及保护脑细胞避免毒物的攻击与脑缺血调整脑血循环等方面有关。故认为脑内NO的生成与分解与神经精神活动有关,其代谢异常会影响精神状态。有关更多的资料还待深入研究。
三、精神病的生物化学基础
(一)神经生化是精神生化的重要基础
本世纪70年代以来,神经生物学研究的重大突破,尤其是对中枢神经递质的研究,如:胺类递质的神经元通路、中枢递质的合成与降解、分子水平上的代谢调控机制、递质受体的结构与功能都渐渐比较清晰。cAMP与cGMP应用于解释中枢兴奋、信息媒介的传播机制方面,对于认识神经冲动的兴奋与抑制过程起到重要作用。80年代,对神经元膜系统上的肌醇磷酸酯的生化转变与信息传递相互联系的研究,把神经元的分子生物学研究推向新的台阶。现已清楚cAMP、cGMP及钙离子等神经递质和某些肽类激素的第二信使和第三信使,在神经递质受体、神经系统膜结构的变化中所起的作用。80年代末,由于基因工程应用于神经生物学及精神病遗传的研究中,从DNA分子水平明确了一些重要的先天性遗传性神经、精神病遗传缺陷的关键。像采用重组DNA技术,发现亨延顿舞蹈病的致病基因位于第4号染色体上;利用限制性片段长度的多态性(RFLP)的DNA片段发现双相(躁狂与抑郁)情感性障碍的致病基因位于第11号染色体短臂末端;近年报告老年性痴呆(阿尔采默病,Alzheimer disease,AD)的病理基因位于21号染色体上臂的q21区;自毁容貌综合征的病因,次黄嘌呤-鸟嘌呤磷酸核糖转移酶(HGPRT)缺陷神经、精神活动的规律从分子水平的研究也取得巨大进步。例如:研究动物学习与记忆的分子学基础,提出记忆过程、短时记忆(几分钟到几小时记忆)是通过对脑内原有与记忆有关的蛋白质共价修饰完成,其中涉及蛋白激酶、递质释放、Ca2+、cAMP等。而长时记忆则有赖于脑内合成新蛋白质,该蛋白质的诱导合成建立在短时记忆基础上,产生修饰调节物,后者激活有关基因,合成新的与记忆及联想有关的蛋白质,人类这种高级神经活动与精神活动密切相关。由于对人的思维活动、记忆与学习、睡眠与觉醒化学本质的研究,人们才能对精神病人发生的遗忘、幻视、幻听,谵妄、偏执、狂暴及抑郁等症状,甚至某些行为心理的变态发生的物质基础有较深入的理解,从而正确区分精神病的类型,对治疗与预防都有积极的意义。
(二)神经内分泌与精神病
内分泌激素,尤其是神经内分泌的研究极大的丰富与充实了精神生物化学的内容。精神病,广义是指那些心理功能异常而本质上又与正常人不同的人,这些人丧失了与现实接触的能力,出现幻觉、幻视及幻听等症状,即在并无外界刺激的情况下,产生犹如真实身临其境的感受,对不符合实际的问题却坚信不移,产生妄想,并作出异常反应,行为异端,这就是精神病的特点。
器质性和功能性精神病之间的区别,在于前者有肯定的脑结构性变化的疾病,或具有足以引起精神功能障碍的代谢性疾病(如肝或肾疾病)。器质性精神病患者的智力、获取、储存新信息的能力均受到严重的损害,疾呆是由于脑结构的改变引起,而精神错乱常继发于脑外其他代谢障碍,多属可逆、功能性精神病常保持有智力、获取及侦破新信息的能力,据此推测,功能性精神病的障碍是特定的神经系统的特有化学抑制障碍,例如:对Cush-ing病和Addison病的研究发现,下丘脑-垂体-肾上腺(HPA)轴功能异常可能与原发性情感障碍的发病有关,并成为该病的特征,50%的Cushing病患者有情感障碍,10%伴有精神病或自杀、认知障碍。常见知觉障碍与皮质醇浓度相关,当皮质醇水平下降后情绪和精神症状戏剧性地恢复正常。Addison病常有情感淡漠、社会性退缩、睡眠障碍、注意障碍和疲劳,用糖皮质醇治疗可改善上述症状。甲状腺功能亢进病人,70%表现为焦虑不安、识别能力衰退,而甲状腺功能低下的出现呆小症,并存在精神与智力障碍。胆囊收缩素(CCK)在大脑皮层、边缘系统和下丘脑浓度很高,参与行为的调节,抑制食物的摄取,引起饱胀感和缓解疼痛,有人报告精神分裂症病人CSF中CCK的含量降低,而抗精神病药物有CCK类似的作用。上述研究说明内分泌疾病与精神病发生率之间有显著相关性,从而检查患者血液、尿液及CSF内各种内分泌激素的改变为诊断提供信息。
从70年代起精神病医师就认识到免疫系统从易感素质和发病诱因两方面对精神病发生起中介作用。神经内分泌因素是通过调节免疫反应,参与心理变化,而对精神病的发生产生影响。如:动物在拥挤、固定制动、噪音及面临更凶恶的天敌动物面前,能促发机体对那些免疫系统影响的疾病易感性。现已知重型精神病有免疫功能的异常,情感障碍、精神分裂症、酒中毒、Alzheimer病、孤独症等。
(三)精神药物与精神病
精神病的生物化学概念始于20世纪50-60年代,主要是萝芙木生物碱、蛇根碱,用于治疗高血压病时产生一些副作用而提出。萝芙木具有降压作用,1954年Wikins发现其降压有效成分为利血平,在治疗高血压病人中,可发生严重的抑郁性疾病。人工合成的利血平类似物-四苯嗪也使人出现严重的抑郁,后经动物实验证明,利血平及四苯嗪可导致动物镇静与退缩状态,据此有人提出利血平和四苯嗪导致的抑郁可能与脑内单胺递质耗竭有关的理论。支持该理论的实验有:单胺氧化酶抑制剂,异烟酰异丙肼能提高动物脑内NE及5-HT浓度,大白鼠预先服用此药后,再给予利血平则不发生通常出现的镇静和退缩状态,而呈现兴奋反应。由于异烟酰异丙肼减慢单胺类物质的降解,脑内单胺类物质水平增高所致,因而异烟酰异丙肼为一种有效抗郁药。
1958年Connell等报告苯异丙胺慢性滥用者,发生与急性偏执型精神分裂症表现极为相似的精神症状,其证据有:苯异丙胺对低级哺乳动物引起嗅、舔、啃的刻板行为综合征,在高等哺乳动物这种综合征更为复杂,可出现各种先天性遗传的和后天获得的行为异常,这与脑内神经递质多巴胺释放有关;一些拟交感神经胺,如:甲基苯异丙胺、苯甲噁嗪、利他灵及麻黄碱等都能引起与苯异丙胺所致的相似精神症状;妄想型症状可迅速发展成特发性精神分裂症出现的特有症状。这些都支持多巴胺学说。
精神药物及精神理学的研究,有力地促进了精神病生物化学的研究,在临床上,检查各种类型精神病人的尿液、血液和脑脊液中神经递质及其代谢物浓度,寻找其异常代谢环节。同时,开发与研制出各种提高或降低脑内NE、DA及5-HT等神经递质或神经调质浓度的药物,改善病人的精神症状,此过程中也为精神生物化学进一步发展提供了丰富的资料。
第二节 某些神经疾病的生物化学
一、神经肌肉性疾病的生物化学
神经-肌肉兴奋传递过程中,任何部位病变均可引起神经肌肉疾病,根据受累部位可分为神经肌肉接头病和肌肉疾病。现就进行性肌营养不良的生化改变进行介绍。
进行性肌营养不良是一组原发于肌肉的遗传性变性疾病,主要临床特点是骨骼肌进行性加重、对称性的无力和肌萎缩,多发生在肢体的近侧端、躯干和肢带的肌群,起病多在幼年期或少年期。
(一)肌肉的生化改变
⒈收缩蛋白及代谢以在室温下是否可溶于碱性溶液可将肌蛋白分为不溶的胶原蛋白和可溶的肌纤维蛋白。正常成人肌肉中后者占90%以上,肌病时可溶性蛋白下降,晚期达50%以下,而结缔组织胶原蛋白增多。假肥大型肌营养不良的肌肉中,肌球蛋白的ATP酶活性下降,肌动球蛋白与Ca2+结合能力比正常人低。从肌病或带基因者的肌肉提取的核蛋白体中,蛋白质合成率比正常人高,自动放射显影法探测病人肌肉的肌浆中亮氨酸掺入蛋白质的速度比正常人快,以肌动蛋白和肌球蛋白的主要成分3-甲基组氨酸为指标,发现假肥大型肌病儿童尿中排出量比同年龄正常儿童高3倍,说明肌病时肌蛋白的合成率增加。
⒉肌酶类 肌病早期时,组织蛋白酶A和B、二肽酶、芳基硫酯酶及核糖核酸酶等酶活性明显升高,而组织蛋白酶D、酸性磷酸酶、β-乙酰葡萄糖胺苷酶、岩藻糖苷酶和甘露糖苷酶等酶活性在肌病晚期才开始升高。一般认为这是一种非特异性和继发性改变,是肌纤维对某种有害因素的反应。在糖酵解和糖代谢酶方面,先发现肌病病人肌肉的糖酵解率比正常人的下降,继而发现α-磷酸化酶、磷酸葡萄糖变位酶(PGM)及醛缩酶活性下降,其醛缩酶活性仅为正常人的一半以下。CPK和ATP酶也有明显下降,尤其是少年型较明显。腺苷酸激酶(AK)、AMMP脱氨酶、LDH、丙酮酸激酶(PK)及磷酸甘油醛脱氢酶(GAP-DH)等酶活性亦较正常肌肉中低,而6-磷酸葡萄糖脱氢酶及6-磷酸葡萄糖醛酸脱氢酶活性升高,提示磷酸戊糖途径活跃,这是由于肌肉中再生纤维增长,需要较多的核酸合成之缘故。LDH5在肌营养不良患者的肌肉中只占LDH的17.7%(正常为31.4%)明显下降,而CPK-MM则比正常肌肉中有所升高。在假肥大型肌病时出现异常的已糖激酶Ⅱ型(HK-Ⅱ)。肌病晚期,线粒体的氧化磷酸化下降。肌病中,ATP和CrP的含量都减少,ATP/ADP及CrP/Cr比例也下降,辅酶Q缺乏,但与NADP+有关的异柠檬酸脱氢酶及谷胱苷肽还原酶在肌病中的活性却增高,可能与磷酸戊糖途径活动增强有关。
有关蛋白裂解酶的研究发现,假肥大型肌病时Ca2+激活中性蛋白酶(CANP)在病肌的匀浆中明显上升,CANP可裂解某些肌原纤维蛋白,如肌钙蛋白(TN-1、TN-C)。因为肌病时,由于肌浆有缺陷或受损,以致Ca2+从细胞外流入肌细胞内使CANP激活,而CANP在正常肌纤维由于Ca2+很低几乎无活性。此外,与溶酶体相伴的碱性蛋白酶、二肽酶Ⅳ在肌萎缩时活性亦增高。酸性或碱性核糖核酸酶活性在假肥大肌中显著上升,脂酶活性也见增高。
有关肌病时肌肉酶活性改变机制还不太清楚,多数认为是肌浆膜对酶的通透性有变化,酶大量漏入血中。有人用同位素掺入法研究酶的转换率,发现肌病时酶蛋白合成率下降,降解率常增高。还有人认为,营养不良的肌肉酶活性与发育期胎儿肌肉酶活性相似,说明由于运动神经对肌肉的冲动减少所造成的后果。
⒊肌膜的生化改变用电子显微镜观察肌营养不良病人的肌纤维膜,发现肌浆膜的内外含蛋白质的颗粒大量消失。生物化学方面,从分离出的肌膜上ATP酶活性有改变,如Na+、K+-ATP酶活性降低,而Ca2+、Mg2+-ATP酶活性升高,腺苷酸环化酶活性降低,5′-核苷酸酶活性增高,表明肌纤维膜的生化缺陷。假肥大型肌病中鞘磷脂较多,而胆碱磷脂和氨基乙醇磷脂较正常儿童减少。用同位素标记注入法观察到病肌肌膜对同位素的保留能力明显低于正常肌肉。
在对假肥大型病肌肌节的肌浆网培养中,发现其对Ca2+再摄入的能力比正常肌肉明显降低,肌浆网对Ca2+的亲和力也下降。PAGE分析中发现病人肌浆网蛋白成分出现两种异常组型,提示假肥大型肌病存在有不同类型。对有关肌膜上蛋白质异常的研究,逐渐阐明了肌营养不良的肌原纤维膜上缺乏一种分子量大而含量极少的蛋白质致营养不良素(dystrophin),这种蛋白质基因定位在X染色体的短臂上(XP21)。严重肌营养不良病例中该蛋白质几乎全部缺乏或含量极低。
(二)血液的生物化学改变
⒈血清酶最早发现醛缩酶在假肥大型肌病时可增高10位以上,此后又发现CPK、转氨酶、LDH、PK、磷酸葡萄糖变位酶(PGM)、磷酸葡萄糖异构酶(PGI)、6-磷酸葡萄糖脱氢酶、磷酸丙糖异构酶(PTI)、磷酸甘油酸激酶(PGK)、磷酸甘油酸脱氢酶(GAPDH)、磷酸已糖异构酶(PHI)、苹果酸脱氢酶(MDH)、肌激酶(AMP激酶,MK)、腺苷酸激酶(AK)等在肌营养不良时都可增高,以假肥大型肌营养不良时最显著。CPK活性改变是肌病时最敏感的酶指标。假肥大型肌病初期CPK活性即可升高,可作为早期假肥大型肌营养不良症的诊断指标。一般病儿在出生后即可发现CPK升高,大约生后14-22个月达最高峰,有的在临床症状未出现以前CPK就已上升。CPK活性还可用于探测假肥大型肌病的带基因者,该型是伴X染色体遗传,病儿绝大多数是男性,女性往往是带基因者,用血清CPK方法检测病儿的母亲或姐妹,大约2/3的带基因者可被检出,20-30岁与40-50岁的女的平均值最高。妊娠早期常伴CPK值暂时性下降,其他类型肌病CPK活性也可升高,但不如假肥大型明显,恶性高热症患者常伴有肌病,血清CPK增高,多发性肌炎亦可明显升高。CPK-MB在假肥大型肌病病人中升高,同时可见LDH1-3的升高,而女性带基因者主要是LDH5升高。PK活性一般在肌病早期轻微升高,随着病程进展逐渐上升,严重期持续于高值。有人认为,用PK和CPK两种酶作交叉检测对肌病诊断更为可靠,CPK可检出2/3的带基因患者,其余1/3左右可用PK活性检测。
⒉血清蛋白类 肌病患者中α2-球蛋白增高,有时伴α1-球蛋白升高。用免疫电泳法在β1-球蛋白范围内出现一条额外的抗肌病带,位置与血液结合素相近。假肥大型肌病病人血中肌红蛋白明显增高,其浓度变化与病程起伏及对治疗的反应有一定相关,且在临床症状出现变化之前即可测出改变。肌红蛋白的测定对假肥大型带基因者的诊断也是比较可靠的指标之一。
⒊其他肌病病人血红细胞膜可发生改变,膜脆性、渗透性增高,膜脂质成分、ATP含量、K+流出速度、磷脂酶活性、膜Ca2+-ATP酶与底物的亲和力及对Ca2+影响的敏感性等与正常人RBC膜有明显异常。假肥大型病人血小板对5-HT摄入速度降低,肌肉组织中Na+增多,K+减少,细胞内水分减少。肌营养不良病人血中Mg2+含量显著低下,血糖一般在正常范围,葡萄糖耐量试验仅个别病人出现迟缓型曲线。
(三)尿的生化改变
⒈肌酸尿 肌酸代谢障碍主要表现为:①尿中出现大量的游离肌酸。②尿中肌酐的排出减少。③肌酸的耐量改变。
肌营养不良病人早期可出现肌酸尿,患者尿中肌酸含量明显增多,但肌酸尿的程度与疾病的严重程度不成比例,如:晚期有明显肌萎缩者肌酸排出反而很少,肌酐的排出大为减少。因此,病人尿中肌酸指数(肌酸/肌酐比值)一般升高,而且在不同类型的肌病及不同时期中指数有所差异,这具有鉴别诊断的意义。在同一病人的病程中,临床症状的好转或恶化,也可见肌酸指数相应地下降或上升。
⒉核糖尿肌营养不良病人尿中有时可出现戊糖,但这是一种非特异性、非恒定的改变。病人排出的戊糖主要是核糖,24小时内比正常人多1倍。假肥大型病病人1/3左右尿中排出NAD+增多,有人认为核糖尿是由于肌物质耗损,核苷酸类降解的结果。
⒊氨基酸尿肌营养不良病人出现氨基酸尿,较多见于甘、亮、缬、苏、精、脯、组氨酸及代谢中间物(牛磺酸、甲基组氨酸)。其中一部分与肌酸的合成代谢有关。但假肥大型肌病尿中羟脯氨酸显著减少,这由于结缔组织增生,使排出量减少。氨基酸尿可能因蛋白质-氨基酸代谢紊乱所引起,而不是肾小管吸收功能的影响。
⒋肌红蛋白尿进行性肌营养不良病人尿中有肌红蛋白排出,这是肌萎缩的非特异性改变。
肌营养不良的生物化学改变虽在肌肉、血液及尿中出现异常,其检测对临床诊断有一定的帮助,但多为非特异性改变,一些特异性的实验诊断指标有今后深入探讨。
二、多神经炎的生物化学
多神经炎(多发性周围神经炎)又称末梢性神经炎,是一类周围神经的多病因性疾病,为肢体远端的多发性神经损害。临床表现主要有肢体远端对称性的感觉、运动障碍,伴有肢体的出汗、血管舒缩和皮肤营养的损害。其病因有各种急或慢性感染、营养缺乏与代谢障碍、毒物、变态反应、胶原疾病、癌症及遗传性疾病。其中营养和代谢障碍最为多见。
(一)营养性多神经炎
丙酮酸是糖代谢的中间产物,丙酮酸氧化脱羧过程中需有丙酮酸脱氢酶复合体(包括:丙酮酸脱氢酶、二氢硫辛酰胺转乙酰酶及二氢硫辛酰胺脱氢酶)和辅酶A、NAD+、FAD、硫辛酸、焦磷酸硫胺素(TPP)及Mg2+等辅因子参加。当毒物(砷化物)进入体内,可抑制二氢硫辛酰胺转乙酰酶活性,若体内缺乏维生素B1,使TPP生成障碍,则抑制丙酮酸脱氢酶活性,其结果是丙酮酸氧化脱羧反应受阻,神经组织中可见大量丙酮酸蓄积。同时,三羧酸循环中的琥珀酸氧化脱羧反应亦受阻,导致糖代谢障碍。人类的维生素B1缺乏性神经炎,在急性期血中丙酮酸常增高。临床症状不明显的病人,使用丙酮酸耐量或葡萄糖负荷试验可测出丙酮酸含量增高或耐量减低。几乎有50%的病人在60分钟或90分钟后血中丙酮酸含量仍然高于正常,其中部分病人血中乳酸含量也增高。红细胞中转酮基酶(TKA)活性比正常人明显下降,这是因为磷酸戊糖途径中TKA需要TPP作为辅因子。维生素B1缺乏,TKA活性降低,影响磷酸戊糖途径的代谢。正常神经组织中TKA活性以脑桥和脊髓白质最高,当脑中TKA活性降低到正常值的58%以下时,神经系统症状和体征即不可恢复,说明转酮基作用的缺陷似乎比丙酮酸代谢异常的逆转更为困难。
长期饮酒可引起多神经炎,这主要是由于慢性胃炎或胃肠功能紊乱,以致呼吸和利用维生素B1的功能失调所致。酒精性神经炎的病人几乎1/2左右常伴有胃酸减低或缺失,部分病人血中硫胺或TPP的水平下降,同时血中丙酮酸含量明显增高。
烟酸及维生素B12缺乏亦可有多神经炎出现,有些还可有周围和中枢神经脱髓鞘病理改变,其发病机制还不太了解。
(二)代谢性多神经炎
糖尿病是全身性的代谢病,并发神经炎很可能是继发于糖和脂代谢的紊乱,其中维生素B1缺乏和血管变性等因素可能有一定作用,与周围神经中枢神经递质的代谢和转运也有密切关系。从实验性糖尿病模型发现:①糖类在周围神经中积累:实验动物的坐骨神经中有大量糖类物质累积,如葡萄糖浓度达正常时的6倍,果糖为12倍,山梨醇达23倍,而血中葡萄糖仅为正常时的4.5倍。周围神经中肌醇类含量减少,若在膳食中加入肌醇则神经传导速度可以好转。②有髓纤维内脂和蛋白质合成下降:用同位素标记示踪研究发现有髓纤维的脑苷脂、甘油磷脂和胆固醇合成速度明显减慢,但降解速度保持不变。标记亮氨酸掺入蛋白质实验发现蛋白质合成速度下降。③神经递质的轴浆转运受阻:链脲佐菌素注入大鼠四周后,其周围神经轴素对神经递质及有关酶的转运作用明显受损,标记亮氨酸掺入蛋白质的速度在脊神经根中也下降40%,甚至逆向性转运也受干扰。
卟啉性多神经炎多见于急性间歇性卟啉症,患者体内卟啉代谢紊乱,尿中排出大量卟啉或卟啉原,同时出现四肢疼痛和对称性的运动感觉型周围神经病损,严重者3-5天内可发展成四肢弛缓性瘫痪。实验室检查尿中胆色素原排出增多,红细胞中尿卟啉原Ⅰ合成酶活性明显降低,仅为正常值的50%左右。患者的神经轴素和髓鞘发生变性,其机制不太清楚,推测可能是卟啉类色素在神经组织中积累过多,影响神经的传导功能和髓鞘的正常代谢。
遗传性共济失调型多神经炎(Refsum病)是由代谢障碍所引起。由于α-氧化酶缺乏,植烷酸在体内不能降解,沉积于神经组织中,临床表现为儿童期开始感觉运动型多神经损害,同时伴视、听力减退、共济失调、皮肤鱼鳞癣及骨骼发育异常等,病人血清植烷酸明显升高,CSF中蛋白质偏高。
脂蛋白代谢异常型多神经炎现已发现有两种类型:①α-脂蛋白减低(Tangier病),是一种常染色体隐性遗传病,特点是血清中HDL含量明显减少,磷脂和胆固醇也减少,而TG含量增高。②β-脂蛋白缺失(Bassen-Kornzweig病,棘状红细胞增多症),也是一种常染色体隐性或显性的遗传代谢病,其特征是儿童或青年期出现周围神经损害,有下肢感觉型共济失调,眼底网膜色素变性。患者血清LDL缺乏或降低,胆固醇和磷脂类低于正常值。
三、帕金森病的生物化学
帕金森病(Parkinson′s disease,PD)又称震颤麻痹,多见于中年人以上的中枢神经系统变性疾病,主要病变在黑质和纹状体,其临床特征是随意运动减少,肢体震颤,肌强直及正常姿势反射丧失。而由脑炎、动脉硬化、梅毒、化学物质中毒或基底节肿瘤所引起的另称帕金森氏综合征。帕金森病的代谢缺陷主要表现在黑质及纹状体通路中DA能神经元的退变和消失。近年的研究发现该病不仅仅限于DA能神经元,可牵涉到十多种神经递质和肽类。
早在半个多世纪以前,本病可见黑质-纹状体中神经元的退变和消失,近20多年的研究进展,发现患者尸体脑中纹状体内的DA和NE含量比正常人减少。在一侧帕金森病患者脑中也表现相应侧DA含量明显下降。证明病人脑组织存在单胺类,特别是DA代谢缺陷。在尾核、壳核等部位同样发现多巴脱羧基酶(DOPA-DC)活性下降,代谢物高香草酸(HVA)含量减少。黑质胞体和纤维的缺失愈严重,酶活性和HVA的改变也愈明显,病人CSF中HVA含量普遍较低。DA及HVA减少的程度与黑质细胞丧失的程度成正比,即与PD的主要病理改变成正比。
在新纹状体中存在抑制性的DA和兴奋性的Ach之间的功能平衡机制,PD患者,由于DA释放减少,而Ach的功能相对占优势,应用有中枢性作用的胆碱能药物时,PD症状加重,用抗胆碱能药物时则改善。有人提出,PD的发生与5-HT组胺系统平衡失调有关,PD患者的5-HT减少而组胺相对增多,应用5-HT前体5-羟色氨酸或抗组胺药物可治疗PD。
抑制性递质GABA大量存在于黑质及新、旧纹状体中,新纹状体的小神经元合成GABA再运输至黑质。PD患者这些部位中的GABA含量减少,可能与小神经元丧失有关。
PD病人大脑皮层及皮层下NE及其代谢产物MHPG(3-甲氧-4-羟基苯乙二醇)均降低,可能由于脑干蓝斑神经元破坏,导致背侧上行NE能神经元变性。大脑皮层特别是蓝斑的NE减少对出现的智能障碍及抑郁症状可能起一定作用。
神经肽的失调可能与PD有关。缩胆囊肽(CCK-8)在黑质中大约减少30%。甲硫氨酸脑啡肽(met-en)在黑质中减少70%,在壳核及苍白球中减少50%。亮氨酸脑啡肽(Len-en)在壳核及苍白球区减少30%-40%。P物质在黑质与苍白球区减少约30%-40%,提示在苍白球及纹状体黑质系中含这种肽的神经受损。生长抑素在额叶及海马皮层减少30%-50%,这与智能衰退有关。
80年代初,有些吸毒者注射不纯的海洛因制剂而产生非常类似PD的症状,经研究发现其不纯物N-甲基-4-苯基-1,2,3,6四氢吡啶(MPTP)进入中枢神经系统后,在神经胶质细胞及5-HT神经元内,经MAO-B催化形成MPP+(1-甲基-4-苯吡啶离子),后者与单胺递质的特异性物质结合进入DA及NE神经元,从而抑制线粒体的NADH:CoQ还原酶(复合体Ⅰ)的活性,干扰ATP合成,导致神经元的慢性死亡。
近年来,发现PD病灶氧自由基清除酶活性降低,黑质的脂质过氧化物增多。MAO-B活性随年龄而增强,使H2O2形成增多,其结果是局部氧自由基过量形成,导致线粒体DNA损伤,复合体Ⅰ活性降低。
目前,已确信PD病人除有中枢DA明显降低外,同时有多种神经递质的失调及不少神经肽的减少。根据神经递质和神经肽的降低,才能了解临床复杂的症状。
四、癫痫的生物化学
癫痫症是最常见的神经系统疾病之一,其患病率在一般人口中每1000人约有5人。癫痫症不是一个独立的疾病,是由各种不同病因所引起的一种反复发作的临床症候群。有关癫痫的生化代谢研究虽然从17世纪就开始进行探索,研究资料很多,但其系统的理解与探讨仍有限。
(一)脑能量代谢变化
癫痫发作活动的开始几分钟内,大脑葡萄糖消耗及糖原酵解率增高,脑乳酸盐聚积。在持续发作整过程中,即使大脑氧供应充分,脑乳酸盐也增加,局部脑皮质及小脑在发作时葡萄糖利用率均增高。由于大脑葡萄糖代谢增快,氧需要量增加,因而发作的头几分钟内大脑葡萄糖及氧含量迅速下降,在发作2小时内脑皮质葡萄糖浓度一直低。癫痫全身发作头几秒钟内,脑磷酸肌酸浓度降低,而肌酸升高,ATP浓度稍降低。实验证明,癫痫发作时,脑血流量(CBF)、脑氧新陈代谢率(CMRo2)及葡萄糖利用率(CMRg1)增加与持续发作时间有关。有关大脑生化改变见表13-2:
表13-2 癫痫发作时生化变化
| 大脑 | 早期 | 晚期 | 系统 | 早期 | 晚期 |
| 乳酸盐 | ↑ | ↑ | 动脉氧分压 | ↓ | → |
| ATP | ↓ | → | 乳酸盐 | ↑ | ↑ |
| GABA | → | ↑ | 血糖 | ↑ | ↓ |
| 氨 | ↑ | ↑ | 〔K+〕 | → | ↑ |
| 谷氨酸盐 | → | ↓ | 非酯化脂肪酸 | ↓ | ↓ |
早期:发作5分钟晚期:发作后30-120分钟
↑增加;↓降低;→无改变
(二)神经递质的改变
⒈5-HT 有人报告利血平能减少5-HT合成,促使癫痫发作,适量使用5-HT可抗癫痫。测定5-HT代谢产物羟吲哚乙酸(5-HIAA),可见癫痫患者CSF内5-HIAA含量降低,其中原发性及头痛型癫痫病人脑脊液内5-HIAA降低更为显著。
⒉多巴胺测定癫痫病人脑脊液中儿茶酚胺发现患者脑脊液内多巴及多巴胺显著降低,而酪胺及对羟苯-β-羟乙胺却增加。因为脑内酪氨酸→多巴胺→肾上腺素代谢途径活性降低,酪氨酸→酪胺→对羟苯-β-羟乙胺侧路途径活性代偿性增加所致。对癫痫大脑皮质病灶切除组织中多巴胺的测定,呈现癫痫病灶的多巴胺比其周围组织显著减少。有人报告癫痫鼠(EI)新纹状体及横隔核内多巴胺含量低,脑室内用氯化钙后含量增加,多巴胺含量增加的脑区就是钙调素及酪氨酸羟化酶高水平分布区。EI鼠脑生物胺含量低可能与其通过中枢性钙-钙调素依赖性生物胺合成机制的钙离子水平受损有关,因而增加了EI鼠癫痫抽搐的易感性。临床测定癫痫病人脑脊液中的多巴胺代谢物HVA可见降低。
⒊乙酰胆碱(Ach)多数人认为脑内Ach变化可能是激发癫痫发作的一个因素,分别给猫及鸡脑内注入少量Ach可导致癫痫发作。将Ach与抗胆碱酯酶药共同作用于大脑皮质,可观察到皮质细胞有规律地产生癫痫样放电。癫痫发作时,患者脑脊液及血液中Ach含量均显著增高,尤在即将发作时Ach上升更显著。有人对癫痫患者血清、脑脊液胆碱酯酶活性与各种临床特征进行比较分析,评价Ach-AchE系统在癫痫发病中的作用,其实验结果是86.8%患者血清胆碱酯酶活性显著升高。有疗效者AchE活性高,无疗效者酶活性较低。9例手术者中7例术前AchE活性升高,8例术后酶活性降低。
⒋γ-氨基丁酸(GABA)钴引起猫实验性癫痫发作时,大脑皮质GABA含量降低,降低程度与癫痫发作程度一致。GABA局部置于大脑皮质或注入颈动脉及脑室内能提高化学性癫痫阈值与听源性发作阈值。脑池内注射GABA能抑制脑内因注射谷氨酸与吡哆醛-5-磷酸所引起的癫痫发作。癫痫患者脑脊液中GABA水平有明显降低,而且脑脊液中GABA水平与癫痫患者发作频率有一定关系,抗癫痫药物可使癫痫患者脑脊液GABA水平提高。
⒌谷氨酸(Glu)谷氨酸是脑内含量最多的一种氨基酸,约为GABA的4倍,对中枢神经系统所有神经元起兴奋作用,现已确定癫痫性脑损害是兴奋性神经递质谷氨酸盐及天冬氨酸盐的兴奋性机制,破坏中枢神经元势能的神经毒素引起。正常状态下脑组织也有积聚谷氨酸的能力,但在癫痫条件下加剧谷氨酸的积聚。有关谷氨酸能受体,近年来发现兴奋性氨基酸(EAA)能作用于N-甲基-D-天冬氨酸盐(NMDA)受体而有抽搐活动的作用,因此将EAA视为癫痫触发因子。有人认为癫痫发作强度与谷氨酸量相关,同时与谷氨酸脱氢酶活力升高相一致。
⒍脑肠肽类目前认为脑肠肽是抗癫痫药物最有希望的一类神经肽,其中缩胆囊素(CCK-8)与癫痫发作有关。CCK-8能明显促使培养中的大鼠脑皮质突触体释放GABA。对遗传易感性听源性癫痫动物脑室内注入微量CCK-8则产生癫痫发作抑制作用,放射免疫法测定癫痫患者颞叶病灶内CCK含量均见减少。
(三)癫痫与环核苷酸
近年研究发现由刺激诱发抽搐及药物诱发抽搐时,脑内cAMP值及cGMP上升,实验性癫痫病灶组织内cAMP或cGMP值上升。环核苷酸在中枢神经系统内参与癫痫发病机制已有较多实验资料证明,动物癫痫抽搐及人类癫痫明显影响脑内环核苷酸水平。目前认为脑组织癫痫样放电,细胞内钙离子浓度增加,激活鸟苷酸环化酶,致癫痫脑内cGMP升高,同时从细胞内释放出生物胺及腺苷作用于突触,引起胞内cAMP含量增加,cAMP又作用于胞膜,改变胞膜离子通透性,减弱神经兴奋性,使发作停止或减弱,因而脑内cAMP水平提高有抗癫痫作用。脑内cGMP在抑制癫痫样放电及扩散的机制中有作用。有人测定癫痫患者脑脊液中cAMP及cGMP含量,其结果是cAMP在发作后第二天明显升高,而cGMP值与癫痫发作准备状态有关。
(四)癫痫与钙离子和钙调素
现已证实钙离子与癫痫的关系,钙离子细胞内流是癫痫发病的基本条件。在细胞水平上,癫痫的共同病理基础是脑神经元异常放电,这种自发性放电是短暂加速的钙离子内流和缓慢钙离子内流引起的细胞膜去极化。在癫痫状流首先产生缓慢去极化,这种去极化达到一定程度时就会触发钠离子内流,从而爆发一系列的迅速的去极化过程。在癫痫发作前,突触前末梢活性区域中高电子密度颗粒实验上是一种钙离子通道,它为钙离子进入细胞并触发癫痫发作提供了条件。钙离子内流也导致兴奋性氨基酸的释放,谷氨酸操纵钙离子通道,具有神经毒性和致痫性,而抑制性氨基酸GABA通过抑制钙离子通道,产生对中枢抑制作用。
钙调素(CaM)是人体重要的钙结合蛋白,为钙离子发挥作用的受体。CaM对神经兴奋性的影响是通过对突触部位的Ca2+-CaM依赖的蛋白激酶Ⅱ(CaMPKⅡ)活性调节进行的。直接将活化的GaMPKⅡ注入动物的神经元中可以直接改变神经细胞膜对离子通透性和神经细胞的兴奋性。由CaMPKⅡ介导的细胞膜通透性改变与癫痫状态下神经细胞膜的自动去极化有关。癫痫病人的神经兴奋性与CaMPKⅡ活性呈反比关系,CaMPKⅡ活性长时间降低可以导致癫痫发作。CaM作为钙离子这个第二信使的受体,其重要作用是调节细胞内cAMP和cGMP浓度,而cAMP和cGMP水平的改变与癫痫发作有关。CaM又能促进神经递质的合成和释放。已证实凡是DA水平上升幅度较大的脑区,CaM和酪氨酸羟化酶活性较高。有人报道,脑室内注入CaM拮抗剂,抑制CaMPKⅡ的活性,可减少GABA在脑脊液中的释放。
第三节 精神性疾病的生物化学
一、精神分裂症的生物化学
精神分裂症是一组以思维、情感、行为之间不协调,精神活动和现实脱离为主要特征的一为常见精神病。起病多在青壮年,病程迁延,缓慢进展,可发展为衰退。本病是精神病中患病率最高的一种,我国1982年资料统计,终身患病率为5.69‰,常见类型有妄想型、幻觉妄想型、单纯型、青春型及紧张型。精神分裂症的发生可能与病人体内代谢异常的假说,在近二三十年来已成为本病理论研究最活跃的领域之一。有关精神分裂症的生化基础研究有以下几方面:
(一)多巴胺功能亢进假说
主要认为精神分裂症病人有中枢DA功能的亢进或异常,其药理学证据有:苯丙胺致精神病作用。苯丙胺具有中枢兴奋作用,主要是抑制DA的再摄取,使受体部位的DA含量增高,功能亢进。长期服用大量苯丙胺的病人,引起慢性苯丙胺中毒而出现偏执性精神病,其表现类似精神分裂症的阳性症状,特别有丰富的幻觉,提示精神分裂症有中枢DA功能亢进。左旋多巴治疗帕金森病时病人出现精神症状,左旋多巴为DA的前体物质,可使DA合成增加。抗精神病药物氟哌啶醇对脑内D2受体有特异性阻断作用,通过阻断DA受体使其过高的DA功能降低,达到治疗效果,临床疗效与阻断D2受体的效价成正比。
据报道未经治疗的精神分裂症病人血浆DA代谢产物HVA升高,其HVA浓度与病人的阳性症状及治疗反应呈正相关,而精神分裂症病人外周DβH及MAO活性降低,可能为精神疾病易感性的一种遗传标志。
关于DA受体的研究,有报告指出:精神分裂症病人中具有阳性症状者D2受体密度增加,而具有阴性症状者D2受体密度减低或不变。Crow等(1981)提出精神分裂症有二型:Ⅰ型以阳性症状幻觉、妄想为主,有DA功能亢进,对阻断DA受体的神经阻断剂治疗反应好,可能以D2受体增多为病理基础;Ⅱ型以阴性症状(情感淡漠、主动性缺乏)为主,DA功能改变不明显,对神经阻断剂反应不良,常伴有脑器质性改变。有关DA功能改变原因,Waytt(1985)认为精神分裂症病人中枢DA系统可能存在易损性,由遗传决定。当受到强烈或长期应激性刺激时,其中枢DA末梢受到损伤,早期表现为DA更新率升高,出现阳性症状,当DA末梢进行性损伤出现有神经组织缺失时,DA更新率下降,表现出阴性症状。
中枢DA系统的功能与NE、5-HT系统存在着复杂的相互作用,与神经肽类物质关系也密切。单纯用DA功能改变解释精神分裂症的机制尚欠不足。因此,对其它神经递质与精神分裂症的相关亦受到重视。例如5-HT假说提出5-HT的类似物麦角酸二乙酰胺(LSD)有明显的致幻作用,它通过阻断5-HT受体而抑制5-HT的功能,其表现与精神分裂症阳性症状类似。5-HT耗竭剂利血平可缓解精神分裂症的孤僻、行为退缩和情感不协调等症状。NE假说指出:NE功能不足使犒赏系统失调,快感和意向活动减低,这可能与精神分裂的阴性症状有关,NE活动亢进则与偏执性症状有关。有人报告,在精神分裂症患者脑的特定区域,尤其是富含NE的前脑边缘系统,NE含量升高。在妄想型精神分裂症病人的伏隔核和乳头体内NE含量较对照组约高3倍。多巴胺能系统和谷氨酸系统功能不平衡假说认为精神分裂症是由于皮层下DA功能系统和谷氨酸功能系统不平衡所致。PCP(Phencyclidine)是一种拟精神病制剂,也是谷氨酸的非竞争性拮抗剂,可产生模拟精神分裂症的症状,主要作用部位是通过离子通路与N-甲基-D-天冬氨酸(NMDA)受体联系,后者是谷氨酸的主要受体。PCP可引起儿茶酚胺(CA)的释放,而皮层纹状体谷氨酸通道则使CA的释放受抑制。谷氨酸能系统的功能缺陷或PCP等谷氨酸拮抗剂促使CA释放所引起类似精神分裂症的症状,可看作是一种多巴胺-谷氨酸反馈调节系统中枢神经递质平衡所致的综合征。
(二)神经肽与精神分裂症
精神分裂症病人脑脊液中内啡肽含量增高,且随病情改善而随之下降。有关神经肽在精神分裂症的机制虽有大量研究,但有二种不同的看法:一类认为精神分裂症病人神经肽(包括内啡肽、脑啡肽和强啡肽)功能过高,用拮抗剂纳洛酮可改善其症状;另一类意见则认为本病有阿片肽功能不足,补充这类肽可能有益,大量临床研究未取得一致。近年较重视CCK在精神分裂症的作用,CCK在脑中特异性地与DA存在于共同的神经元内,有调节DA系统功能的作用。当脑中CCK不足,DA功能呈现过高。Ferrier等(1983)研究6种神经肽的变化,发现精神分裂症Ⅱ型病人在海马、杏仁核和额叶中CCK含量明显降低。有资料报道精神分裂症病人脑脊液中CCK水平下降。这些结果提示CCK的功能障碍以及由它调节的DA功能改变可导致精神分裂症。
(三)精神分裂症的分子遗传学研究
半个多世纪以来系统的家谱调查,证明遗传因素在精神分裂症的发生中具有一定作用,精神分裂症患者亲属中的患病率比一般居民高得多,与病人的血缘关系愈近,患病率愈高。现有遗传学研究资料推测遗传方式主要有单基因和多基因两种。主张单基因遗传者认为精神分裂症的发生必需有一个病理基因,基因可以是显性、隐性或中间性。Slater提出精神分裂症是单基因显性遗传伴有外显率低的假说,认为精神分裂症只少数是纯合子,97%是杂合子,杂合子的外显率低,为26%,只有1/4携带显性基因的人表现有病。多基因遗传是由许多基因积累作用造成,无显性、隐性基因的明显遗传规律,但有一个阈值,超过这个阈值就称异常。Gottesman和Schield认为精神分裂症的发生是遗传的易感性和环境因素共同作用的结果,多基因遗传无特异性基因。遗传因素对产生遗传性状和遗传疾病的影响程度称遗传度,东北大庆地区算出精神分裂症的遗传度为75.7%,辽宁铁岭地区为70%-80%。有关多态位点在家系中是否与精神分裂症的传递有关已有报道,Basset认为第5号染色体5q11.2-13.3片断的三体与精神分裂症的发生有关。Sherrington注意到Basset的报告,用位于第5号染色体的二个基因探针D5s76及D5s39对5个冰岛精神分裂症家系,2个英国家庭进行限制性片断长度多肽性(RFLP)分析,,获得慢性最大优势计数(Lod Score)为2.45,证实连锁存在。Sherrington的此一研究成为精神分裂症分子遗传学领域的首次阳性结果,引起了广泛的反应。Crow(英国)依据:①两性发病的临床表现不同,男性起病早,症状严重。②先证者的亲属中,同性别患者率较异性别者高。③精神分裂者的性染色体异常远较对照组高。④精神分裂症家族中的遗传具有相当程度的常染色体遗传特征等四个方面提出精神分裂症的素质基因位于性染色体假常染色体区域的假说。他用Dxys14区域(位X、Y染色体的假常染色体区域)的基因探针进行患病同胞配对分析,其结果支持精神分裂症与性染色体假常染色体区域的基因探针有连锁存在。目前对精神分裂症的基因定位尚无定论。需进一步研究。
二、老年性痴呆的生物化学
痴呆是非正常性急剧的神经系统老化出现全面的认知障碍的一种临床综合征,是一种精神状况的病态改变,常见的包括人格的变化、智力的消失及记忆和推理的变异。痴呆的原因有:①缺乏维生素。②长期低血糖、低甲状腺素及慢性肝病。③颅脑外伤。④三期梅毒。⑤癫痫。⑥脑内占位性病变。⑦Huntington舞蹈病。⑧Alzheimerdisease(早老性痴呆,AD)。⑨血管病变型(老年性痴呆,又名多发性痴呆症)。
老年期的痴呆患病率国内几个小型调查为0.61%-3.2%,多发于60岁以后,但随着年龄增加,患病率亦增加,80岁以上人群,患病率达10.6%。但亦见于40-60岁的患者,女性多于男性。Alzheimer型痴呆和多发性痴呆是老年期痴呆的两个主要类型,占老年期痴呆总数的70%-80%。有人认为早老性痴呆与老年性痴呆相比,无论临床还是病理生化变化均相同,只是轻重程度有所不同,两者仅是疾病的前后过程,故有人将Alzheimer病又称为Alzheimer型老年性痴呆。为揭示老年性痴呆发病机制,近年来,对早老性痴呆患者体内生化变化进行了研究,发现中枢神经递质、淀粉样蛋白、神经节苷脂的改变等与本病有关。
(一)神经肽
早老性痴呆患者神经肽变化的最大特征之一,是大脑皮层和脑脊液中的生长抑素含量显著减少。生长抑素含量不足与智能和记忆功能障碍直接相关。在不并发Alzheimer病(AD)的“单纯”衰老,大脑皮质生长抑素含量没有改变,提示生长抑素的神经元变性,不是由于年龄增长所引起的特有现象,生长抑素改变在早老性痴呆发病机制中起重要作用。
加压素(VP)或精氨酸加压素(AVP)参与记忆和学习的信息加工、记忆巩固和条件反射形成,特别是与长时记忆有关,并认为该激素具有神经调剂作用,能持续地抑制神经系统的兴奋性。有人报告Alzheimer病患者海马、苍白球和副核加压素含量减少,而海马是一个参与记忆形成的主要部位,又是加压素受体密度最高的脑结构,故加压素含量下降,在Alzheimer病的记忆障碍发生中起决定作用。加压素和催产素同为下丘脑核分泌,加压素具有增强记忆的作用,而催产素是加压素功能的拮抗剂,对记忆巩固过程有抑制作用。Alzheimer病患者海马的催产素含量增高,加压素-催产素系统平衡明显失调,使老年性痴呆病人的学习和记忆过程障碍加重。
Beal等人发现Alzheimer病的大脑皮层和海马P物质水平普遍下降20%-30%,其中额叶下降最明显。以智力障碍为主,运动障碍不明显的Alzheimer病患者P物质变化不大,而有明显运动功能性障碍的痴呆患者则有明显下降,提示P物质可能与运动功能有密切关系。目前似乎可以肯定,大脑皮层和脑脊液中的生长抑素含量显著减少,在Alzheimer病的发病机制上可能有重要意义,是该病神经肽表现中最大特征之一。加压素-催产素系统的改变,在Alzheimer病值得注意,其它脑神经肽的作用尚待进一步研究。
(二)其它神经递质的改变
老年性痴呆患者的前脑内侧Mcynert底核的胞体减少25%-90%,大量尸检和活检发现老年性痴呆患者大脑皮层和海马中胆碱乙酰酶(chAT)活性及乙酰胆碱酯酶(AchE)活性降低,chAT活性可降至正常同龄者的35%-40%。在记忆形成测定中chAT活性降低与智力损害程度有关。AchE是AD患者脑脊液中的主要胆碱酯酶,也是观察胆碱能神经元功能的另一重要指标。AD患者脑脊液AchE活性显著降低提示患者脑中胆碱能神经元减少或胆碱代谢紊乱。胆碱递质的减少与智力丧失程度相一致。许多学者认为老年性痴呆的NE能系统受损,表现为儿茶酚胺浓度下降及含NE和5-HT的神经细胞减少,测定AD患者脑中多巴胺、NE、高香草酸及5-HT含量均见减少,脑脊液中DβH活性降低,而MAO水平可高于同龄正常人。有人报道,在AD患者中这些神经递质改变程度,胆碱能系统所受影响最大,5-HT系统次之,肾上腺素和多巴胺系统受影响最小,所以病人表现为睡眠障碍、抑郁症等。
(三)异常蛋白质
AD患者脑中明显的病理改变是神经炎斑(老年斑),神经原纤维缠结及脑血管壁淀粉样变性。神经炎斑是在脑内神经细胞间形成以淀粉样蛋白为核心的病变,与血管壁发生的淀粉样变性都是β-淀粉样蛋白沉积,神经原纤维缠结的超微结构是双股螺旋细丝,其化学成分主要是异常磷酸化的T形蛋白,这些均为异常蛋白质。
β-淀粉样蛋白很难溶解,从神经炎斑中分离的淀粉样蛋白作氨基酸序列分析,发现与AD患者脑血管斑分离的β-蛋白属同系物,其基本结构中含40-42个氨基酸的多肽,命名为β-淀粉样蛋白或βA4蛋白(β-amyloidprotein)。通过分子杂交技术对人脑cDNA基因文库进行筛选,找到β-淀粉样蛋白前体(β-amyloid precursor protein,APP)基因,这个基因在含有一个能合成695个氨基酸的开放读码区,进一步通过体细胞杂交系,将该基因定位于人类第21号染色体上。β-淀粉样蛋白是淀粉样前体蛋白的一种亚单位,它们同受一个宿主基因编码。
目前对淀粉样前体蛋白的生物学功能推测有:①APP具有调节细胞生长和粘附力。有些学者认为APP可以维持神经细胞的生长,在AD的发病过程中,淀粉样蛋白作为一种“生长刺激因子”的角色而大量形成,以弥补神经元的变性死亡或由于某种缺失使神经细胞失去常态而产生变性,变性的神经细胞将膜上的淀粉样蛋白释放出来,互相融合形成难溶的斑块。②建立或保持神经元之间的连接功能。淀粉样前体蛋白以跨膜方式存在同细胞膜保持联系。③神经营养和神经毒性的双重作用。Yanker(1989年)证实AD患者的淀粉样前体蛋白的碎片具有神经毒性作用,Richards(1991年)等学者进一步指出淀粉样蛋白对一些未成熟的神经细胞具有营养作用,而对那些过于成熟的神经元则起毒性作用。其作用最为关键的部位位于第25-35位氨基酸之间的残基。还有人报道淀粉样蛋白的作用可被P物质的拮抗物所模仿,并被P物质阻滞。
1986年发现AD和正常老年人也有淀粉样蛋白沉积,只不过AD的神经病理学特征在正常老人其分布部位较局限,程度较轻微。二者病理改变的类似性,尤其是沉积的淀粉样蛋白氨基酸顺序的一致,提示这种病态的痴呆与正常高龄以后出现记忆力下降以至于痴呆可以是相似过程,即淀粉样蛋白的沉积本来是一个正常的神经老化过程,只不过在AD变得异常严重和广泛。目前认为,淀粉样蛋白沉积是发生于AD形成的早期,引起轴突异常生长和其它神经病理学特征的病灶。
AD患者脑中的另一重要特征是形成神经原纤维缠结(NFT),NFT发展到一定程度,可使神经元细胞变性、破坏,残留下嗜银的原纤维斑块。AD患者的痴呆程度与NFT的数量成正比。NFT的成分有:①泛素(ubiquitin),是组成NFT的蛋白质成分,由76个氨基酸组成的多肽,具有高度恒定性,能参与蛋白质的二价修饰作用。泛素是一种与其它结构蛋白变性有关的蛋白。②T形蛋白(protein tau),T形蛋白是一种微管状结合蛋白,至少有4种同型物,在AD患者脑中,其同型物被异常磷酸化,在NFT中也可见到被异常磷酸化的高分子量神经元丝状物积聚。③Alz68,通过氨基酸序列分析和免疫学研究证明Alz68和T形蛋白的形成有极密切联系,前者可经磷酸化作用而修饰。Alz68包括T形蛋白的抗原决定部位,它在AD患者脑中增多,但正常衰老的脑中不增多。
(四)其它
神经节苷脂在早发型AD患者的额叶和颞叶皮层、海马及尾核均减少(P<0.001)。死亡较早的病人,其神经节苷脂的下降最为显著,晚发型AD患者,脑内神经节苷脂只在颞叶皮层和海马下降,且下降比较轻(P<0.05)。
对老年痴呆及AD的研究,发现在分裂中的细胞生长素和某些神经递质可加速及早反应蛋白(immediate early response protein,IE)的出现,这对细胞的生长、生存及分裂都有促进作用,因此IE的出现与细胞衰老成反比。生长因子和神经递质能促进IE因子生成,而IE因子又可促进神经递质合成酶的形成。老年性痴呆和AD患者大量的神经递质损失,其IE的合成受到障碍,引起大量的神经细胞退化。有些学者提出AD与神经细胞内钙离子有关,细胞内Ca2+的增高可引起神经轴突退化,导致细胞死亡。也可导致纤维缠结的产生,故认为AD患者的神经病理现象可能与Ca2+增高有关,此外Ca2+可激活神经细胞中的谷氨酰胺转移酶,该酶可与微丝蛋白链以共价键结合,从而降低微丝蛋白的溶解度,造成AD的病理变化。
免疫假说:衰老伴有细胞-递质和体液-递质的免疫机制低下,而AD是此免疫过程低下更为严重的疾病,NFT和老年斑中的淀粉样蛋白沉积,是在机体免疫功能改变的情况下发生。老年斑核心存在免疫球蛋白,有报道老年期痴呆AD型(SDAT)病人血清中出现异常免疫球蛋白,其水平与认知功能显著相关。
遗传因素:有人调查了299名老年性痴呆患者亲属患病危险率,父母为7.0%±1.6%,同胞为5.5%±1.1%,子女为4.3%±1.2%。多数研究发现患者家庭成员患AD的危险率比一般人群高3-4倍,推测可能为常染色体显性遗传传播。AD可能具有遗传的异质性,早发者可能位于21号染色体上为常染色体显性基因,晚发者有相同常染色体显性遗传基因,但可能在19号染色体上。
目前人均寿命延长,老年人口迅速增长,随之老年性痴呆患病率也有所上升,其中AD病人约占所得痴呆例数的一半以上,把AD列为老年疾病的重点研究项目,对老年医学及老年精神病学具有重要意义。
第四节 神经、精神疾病生化诊断
神经性或精神性疾病的诊断,根据其致病原因可采用各种不同的检查方法,其中有的实验室生化检查对某些神经或精神性疾病的诊断可提供非常有价值的依据,但大多只是作为一种辅助检查,提供诊断的信息。测定的标本常采用CSF,因血、尿或其它体液中的物质含量不能确切地反映脑内情况。检测的内容多为蛋白质、酶和神经递质及其代谢产物。近10多年来,由于神经分子学的发展,用分子生物学技术对神经性或精神性疾病进行基因诊断已有资料报道。
一、蛋白质检查
(一)总蛋白质
CSF是存在于脑室及蛛网膜下隙内的一种无色透明液体。正常CSF中的蛋白质80%以上通过超滤作用来自于血浆,但不同部位的CSF标本,其蛋白质有差异。正常不同部位CSF蛋白质总量为:脑室液50-150mg/L;脑池液150-250mg/L;腰池液150-450mg/L。CSF蛋白质总量随年龄增长而增高,儿童为100-200mg/L,20-50岁约为150-450mg/L,老年人可达600mg/L。足月新生儿生后6天为300-2000mg/L,6-30天为300-1500mg/L,1-6个月为300-1000mg/L,早产新生儿4000mg/L。但正常成人超过450mg/L,一般是病理性的增高,可见于感染、出血、占位性病变及蛛网膜粘连等。多次电休克治疗后可见CSF总蛋白质增高。
CSF蛋白电泳呈现前清蛋白含量异常增高时,多见于脑积水、脑外伤、脑萎缩及中枢神经系统退行性变等疾病。含量下降见于脑内炎性疾病。CSF免疫电泳可提示血脑屏障渗透性改变的严重程度,如β-脂蛋白升高,证明渗透性明显增加;α1-巨球蛋白升高,提示渗透性中等增加;IgA升高表示渗透性轻度增加:γ-球蛋白增高,常见于脱髓鞘性疾病(多发性硬化及麻痹性痴呆)。
(二)蛋白指数
⒈白蛋白指数 CSF中的白蛋白完全通过血脑屏障来自于血浆。其白蛋白指数计算公式为:
当指数<9时,认为血脑屏障无损害;若指数为9-14,有轻度损害;指数为15-30,中度损害;指数为31-100,严重损害;指数>100,表明屏障完全崩溃。
⒉IgG和白蛋白比率 在脱髓鞘疾病时,由于鞘内合成免疫球蛋白增加,CSF中免疫球蛋白亦增加,测定CSF中IgG和白蛋白比率对脱髓鞘疾病诊断有一定价值。
比率=CSF中IgGmg/dL/CSF中白蛋白mg/dL
70%多发性硬化病例比率>0.27。
⒊免疫球蛋白指数 CSF免疫球蛋白指数计算公式:
指数参考范围:0.30-0.77,指数>0.77,证明鞘内IgG合成增加,90%以上多发性硬化病例指数>0.77。
(三)特异性蛋白质
⒈β-淀粉样蛋白(β-Amyloid Protein)AD患者脑脊液中存在β-淀粉样蛋白,测定该蛋白质对AD的诊断有重要价值。颅脑外伤亦可出现β-淀粉样蛋白。
⒉APP(β-淀粉样蛋白前体,β-AmyloidPrecursor Protein)AD患者脑脊液、血浆及血小板中均存在APP,为β-淀粉样蛋白的前体物质。
⒊C反应蛋白 正常人脑脊液中无C反应蛋白,当中枢神经系统急性感染时,患者血清和CSF中C反应蛋白升高。
⒋β2-微球蛋白(β2-M)正常成人CSF中β2-M含量为11.5±37mg/L,当颅内感染,癫痫、肿瘤、脑梗塞、格林-巴利综合征等疾病时可见β2-M含量升高。
⒌免疫球蛋白 CSF中IgG、IgA及IgM的检测对某些神经系统疾病的诊断或鉴别诊断有意义,如结核性脑膜炎,IgA增高显著;化脓性脑膜炎,IgM显著增高;病毒性脑膜炎时IgA及IgM无明显改变。
⒍神经组织特异蛋白 测定神经组织特异蛋白首先需分离、纯化及鉴定,再制备成多克隆或单克隆抗体,用双部位放免法进行测定。①神经胶质纤维酸性蛋白(GFAP)增加,可见于Alzheimer型痴呆、脊髓空洞症、神经胶质瘤、脑星形细胞病、血管性疾病及海绵状脑病。②髓鞘碱性蛋白(MBP)增加,见于多发性硬化、髓鞘损伤性疾病。
二、酶活力测定
CSF中酶活力测定可反映中枢神经系统的病变,因CSF中酶浓度一般不受中枢神经系统之外疾病的影响。
⒈LDH及同工酶增高见于癫痫、脑肿瘤、痴呆、脑膜炎、脑积水、肌萎缩侧束硬化、脑血管性疾病及脑外伤等。
⒉GOT增高见于痴呆、癫痫、脑外伤、小脑病变、脑肿瘤、周围神经病及多发性硬化。
⒊CPK(CK)同工酶增高见于早期假肥大型肌营养不良症、脑肿瘤、癫痫、脑膜炎、脑损伤及脑血管性疾病等。
⒋酸性磷酸酶增高可见于脑萎缩、脑肿瘤、脑膜炎及多发性硬化等。
⒌核糖核酸酶(RNase)增高见于癫痫、脑肿瘤、痴呆、脱髓鞘性疾病及脑膜炎等。
⒍多巴胺-β-羟化酶(DβH)精神分裂症患者血中DβH活性增高,而老年性痴呆患者CSF中DβH活性降低。
⒎蛋白酶及鸟嘌呤酶增高见于多发性硬化。
⒏乙酰胆碱酯酶(AchE)及假胆碱酯酶(BchE)Alzheimer病患者脑脊液中AchE活性降低,但血浆及脑脊液中BchE活性增高。癫痫患者血清AchE显著增高。
⒐β-葡萄糖苷酶增高见于脱髓鞘性疾病、糖尿病性神经病、癫痫、脑肿瘤及细菌性脑膜炎等。
三、神经递质的检查
神经递质对中枢神经系统功能及人的精神活动起着重要的作用,某些神经系统的疾病及精神病可表现出神经递质代谢的变化,临床上检测神经递质及其代谢物对其诊断具有一定的意义。
⒈Ach 癫痫发作时患者血液及脑脊液中Ach含量显着增高,急性颅脑损伤,脑脊液中Ach含量升高。
⒉DA及HVA 帕金森病(Parkinsondisease,PD)患者CSF中DA的代谢产物HVA含量降低;癫痫病人CSF中DA及HVA显着降低;精神分裂症病人血浆中HVA升高。
⒊5-HT及5-HIAA 精神分裂症病人血中5-HT含量减少,抑郁性精神病患者CSF中5-HIAA含量降低;PD及癫痫患者CSF中5-HT及5-HIAA降低;精神发育迟滞的病儿血中5-HT含量减少,CSF及尿中5-HIAA含量亦降低;颅脑外伤及脑血管疾病患者CSF中5-HIAA含量上升。
⒋GABA 癫痫病人CSF中GABA含量明显下降。
⒌谷氨酸 PA病人CSF中谷氨酸含量减少。
⒍神经肽
⑴内啡肽:躁狂症患者CSF中β-内啡肽含量升高,症状缓解时下降;精神分裂患者CSF中β-内啡肽含量明显升高,急性精神分裂症时其含量超过正常的10倍。而AD患者CSF中β-内啡肽却减少。
⑵生长抑素(14肽):Alzheimer型老年性痴呆患者CSF中生长抑素明显减少。
⑶脑啡肽:癫痫患者CSF中L-脑啡肽(亮氨酸脑啡肽)显著增高。
⑷P物质:大多数抑郁症病人CSF中P物质明显升高。帕金森病患者CSF中P物质含量一般下降,但病情严重时反而明显增高。
⑸胆囊收缩素(CCK):精神分裂症患者CSF中CCK水平下降。
四、基因诊断
由于分子生物学理论和技术的快速发展,分子生物学方法已成为神经、精神科学研究方法的重要组成部分,被用于神经、精神性疾病的诊断。例如:Goate(1991年)通过PCR方法扩增APP基因,进行核苷酸序列分析,发现第17外显子的第2149碱基对位点发生C→T替换,使蛋白质多肽链的第717位点氨基酸发生Val→Ile,并且突变形成了一个新的限制性内切酶位点,此位点可检测Alzheimer患者APP基因的突变,作为基因诊断。用假肥大型肌营养不良(DMD)基因的cDNA克隆检测病人,证实DMD病人中有65%以上有缺失突变,5%有复制性突变,30%没有基因缺失,后者可利用PCR方法扩增DMD基因内部多态性位点及DMD基因3’端小卫星的DNA(-CACA-)后,再用RFLP分析可检测出携带者以及进行产前诊断。Gusella等人(1983年)从一个噬菌体载体克隆的人类DNA随机片段中发现一个与亨迋顿病的缺陷基因位于第四对染色体上,这是人类第一次仅仅应用DNA标志的连锁分析将一种遗传病的缺陷基因定位于特定染色体,并应用于临床诊断。
目前,用基因诊断的神经性疾病有苯丙酮尿症、亨迋顿病、Alzheimer病、线粒体肌病和脑肌病、遗传性共济失调、神经肌肉病、神经纤维瘤病、癫痫、X连锁智力低下及肝豆状核变性等,其检查方法常用分子杂交、RFLP及PCR等技术
第十四章 妊娠的临床生物化学
妊娠是胎儿在母体内发育成长的一个复杂的生理过程。卵子受精是妊娠的开始,胎儿及其附属物的排出是妊娠的终止。为适应妊娠的需要,母体各器官系统均处于应激状态,发生了相应的功能及代谢改变。这一系列改变主要源于新增加的器官-胎盘所分泌的激素以及甾体激素中的雌激素和孕激素的影响。因此孕妇的营养需要,物质代谢过程及内分泌状态与未妊娠时都有许多区别。利用孕妇血样、尿液及羊水等作为标本,进行有关项目的测定是作出早孕诊断、了解胎儿在宫内发育成熟状态及发现遗传性疾病的依据,并对开展遗传咨询、优生、计划生育及保护母子健康都起到指导性作用。
第一节 妊娠期母体生物化学特征
妊娠期内,孕妇的各器官、系统均发生明显变化,其中最主要的是生殖器官的局部变化和母体各器官相应的功能及代谢改变。就生物化学变化特征而言,突出表现在以下几方面。
一、妊娠期营养需要的特点
胎儿通过胎盘不断地从母体吸取各种营养物质(氨基酸、葡萄糖、少量游离脂酸、酮体、各种维生素及必需的无机盐)与水分。妊娠期母体的血容量增加,血凝与纤溶系统也发生不同程度的改变,母体因妊娠而发生的一系列的重大的生理、生化方面的变化,使妇女在孕期有特殊的营养需要。孕妇营养不良可导致胎儿生长发育迟缓、发育不良,严重的营养不良可导致胎儿畸形或胎死宫内。
(一)热量
孕期营养的热量需求应根据孕妇的年龄、身高、体重、职业性质以及劳动的情况而不同。一般情况下,孕早期3个月与非孕妊期大致相同,每日需热量为8372kJ(2000kcal);孕中期3个月为8790kJ(2100kcal);孕晚期3个月为9418kJ(2250kcal)。此外,整个孕期孕妇热量消耗比未孕时要多,平均每日多消耗1256kJ(300kcal),比未妊娠时提高12%左右,因此对糖类的摄入量也要适当增加才能满足此时的需要。一般情况下,孕妇的食欲增加完全能够保证每日热量调整的需求。
(二)蛋白质
在蛋白质供应方面,妊娠期蛋白质的需要量比非孕期增加较多,平均每日需要60-80g,双胎妊娠需要90g,比未孕时多补充30g左右。对于身材瘦弱的年轻孕妇合理地补充蛋白质尤为重要。孕期补充的蛋白质一方面用于满足胎儿的氨基酸需要,另一方面用于子宫、胎盘、乳房组织的增长。由于胎儿不能合成多种必需氨基酸,故补充蛋白质时还应兼顾到食物所含氨基酸的种类。
动物蛋白质含有丰富的氨基酸,是生命的必需物质,每日150g瘦肉可满足妊娠期的平均需要量。鱼类、乳制品和蛋类也含有丰富的蛋白质,而且还具有易于消化和利用的优点。含钙的乳类蛋白(色氨酸、赖氨酸)容易通过机体转化,在维生素D的协同下有利于骨组织代谢。可见,乳制品可替代肉类食物,而肉类不能代替乳制品。因此建议孕妇在补充蛋白质时可肉类与乳制品交替食用。一些豆类及其加工后食品,如豆浆、豆腐等,含有与肉类相似的植物蛋白质。一些统计资料表明,充足平衡地摄入植物性蛋白质可减少妊娠贫血及其它妊娠并发症的发生。
妊娠期妇女,如果膳食中的蛋白质供应不足,则胎儿的生长发育迟缓,孕妇也较衰弱,产后恢复慢,乳汁较稀少。为了保证孕妇和胎儿的健康,孕妇的膳食一定需要足够的蛋白质。
(三)脂类
妊娠期每日需要脂类大约60-80g,奶类、动物油、植物油及人造奶油等均可提供所需的脂类。恰当地摄入含脂类食品可以避免脂溶性维生素A、E、D、K的缺乏。
脂肪酸的营养价值已被肯定,其中亚油酸列为首位,是高等动物机体内不能合成,但又必不可少的必需脂肪酸之一。亚油酸主要来自一些植物油,在体内可转化为花生四烯酸,后者参与细胞膜系统的脂蛋白的合成,并在神经细胞和神经系统的髓鞘磷酸脂的形成中起着重要的作用。因此,胎儿神经系统等发育需要提供一定量的亚油酸。妊娠期胎儿所需要的亚油酸完全靠母体膳食提供,出生后由母乳或新生儿食品供给。由此可见,妊娠期孕妇增加含脂肪酸的膳食有利于胎儿的发育,尤其是神经系统的发育,也为优质的哺乳做好准备。
(四)碳水化合物
碳水化合物是胎儿生长发育的最基本营养物质,每日所需来源有两种,即长效糖类与单糖类。长效糖类主要指米饭、面食、土豆等谷类食物。此类碳水化合物的特点是可以缓慢地释放热能。如果孕妇每日摄入的副食品如肉、鱼、蛋等充足,其每日对长效糖类的需要量是:早孕期200g,孕中期250g,孕晚期300g。各种食用糖和含糖的各种饮料、甜食等食物中的糖属于单糖类,正常孕妇每日只需摄入50g,该糖类在妊娠期不宜多食,过多地食用常引起孕妇超常肥胖。
(五)维生素
维生素对孕妇和胎儿都是必不可少的重要的营养物质。妊娠期如果维生素摄入量不足或缺乏可导致流产及死胎。不同维生素缺乏造成的影响各异。
由于胎盘屏障作用,孕妇血浆脂溶性维生素(A、D、E、K)运往胎儿受到一定程度的限制,因此胎儿血浆内脂溶性维生素的浓度等于或略低于母体血浆中的浓度。维生素A是维持机体抵抗力、防止夜盲、促进生长的重要物质。妊娠期孕妇对其需要量比非孕期多30%-50%。维生素A缺乏可导致早产或死产,以及产后感染率增加。传统认为妊娠期需要补充大量的维生素D和钙剂。但近年来的研究指出,妊娠期不需要过多地补充维生素D和钙。相反,当给予大剂量维生素D以后,可造成胎儿心脏瓣膜综合征(supravd-syndrome)。胎儿可有肺动脉及主动脉狭窄,出生后其智商也低,应当予以重视。母体中缺乏维生素E可使胎儿死亡或流产,所以国外一些产科医生对每位就诊孕妇的系统地补充维生素E。维生素K能促进肝合成凝血酶原及凝血因子Ⅶ、Ⅸ、Ⅹ。维生素K缺乏可造成新生儿严重的出血性疾病,而且从胎儿期到出生,维生素K缺乏是最常见的。在分娩前一周对孕妇进行维生素K肌肉注射可以预防新生儿出血性疾病的发生。脂溶性维生素多存在于植物油脂内,动物的肝、蛋、鱼肝油、牛奶中含量也较多,只要注意定期食用这些食品,可避免脂溶性维生素的缺乏。
相反,水溶性维生素易于通过胎盘,其在胎儿血浆中的浓度高于母体血浆的浓度,同时这也反映胎儿对其需要量大、摄取较多。维生素C可使孕妇对疾病的抵抗力增加,并可辅助治疗一些过敏性、中毒性及传染性疾病。新鲜蔬菜和水果是维生素C的最好来源。维生素B参与糖、蛋白质、脂类代谢,可预防神经炎和维持组织正常的功能。当B族维生素严重缺乏时子痫的发生率增加,维生素B6对妊娠期肌肉痉挛疗效显著。粗粮和白菜、瘦肉等物质中含有丰富的维生素B。叶酸是参与DNA和RNA合成的重要辅酶,所有生长发育或新陈代谢旺盛的组织均需要有足够的叶酸供应。正常妊娠每日最低需从食物中得到叶酸500-600μg,以供胎儿和母体的需要,双胎妊娠时对叶酸的需求量更大。妊娠期叶酸容易缺乏,从而导致孕妇发生妊娠贫血。虽然母体缺乏叶酸不会造成胎儿、新生儿叶酸缺乏性贫血,但在国外的一些观察中发现,母体极度缺乏叶酸时新生儿出生体重及出生后最初6个月生长发育都低于正常同龄儿。另有报道可致早产率增加,并已证实可引起某些中枢神经系统的畸形(脊柱裂、无脑儿)、面部表情呆傻等。人体不能合成叶酸,主要靠食物供给。绿叶蔬菜中叶酸的含量最多。偏食、蔬菜煮沸过久和营养不良均可造成叶酸缺乏性贫血。正常非孕妇女每日需2μg维生素B12,妊娠妇女则需4-5μg。维生素B12是DNA、蛋白质合成时重要的辅酶,妊娠期很少缺乏。
总之,胎儿所需维生素全部来自母体血液,如果孕妇维生素摄入不足,血浆维生素低下,就会使进入胎儿体内的维生素不能满足胎儿生长与发育的需要。轻者影响胎儿在宫内正常生长与发育,影响孕妇自身体质;重者会导致某些严重的妊娠并发症,如先兆流产、早产低体重儿(或超体重儿)、胎盘滞留等危害母子健康的后果。
(六)微量元素
妊娠期孕妇对必需微量元素(碘、钴、锌、硒、镁及锰等)的需要量增加。
锌是生物体内许多蛋白质和酶的组成部分,参与DNA和RNA合成及蛋白质积累,对胎儿和婴儿出生后的生长发育也颇为重要。妊娠晚期3个月内母体大约每日以3.82μmol/kg的锌通过胎盘向胎儿输送。在这一时期内母体锌摄入量不足或胎盘缺乏充足的血液灌注皆可使胎儿处于低锌状态。严重缺锌可导致胎儿宫内发育迟缓、胎死宫内、流产、先天性畸形(中枢神经系统和骨骼)、胎儿脑组织萎缩、羊水抗菌活性下降、分娩障碍等。孕妇血锌低于8.03μmol/L,毛发中锌含量不到157.5-250μg/L是胎儿宫内缺锌的危险指标,为补锌的标准。推荐孕妇每天饮食中锌含量应为20mg。
镁离子是某些酶的激活剂,而且在糖和蛋白质的代谢中有着极其重要的作用。镁离子对神经系统有抑制作用,低镁可使神经肌肉兴奋性增高。正常人每日镁的需求量为200mg左右,妊娠期镁的需要量增加造成镁缺乏相对常见。缺镁可引起妊娠期间的某些疼痛综合征。国外曾报道,孕妇若镁摄入不足导致镁的负平衡,可引起子宫胎盘循环系统的血管挛缩,成为引起妊高征及胎儿宫内发育迟缓的原因。
碘是甲状腺素的组成部分,而甲状腺素能促进蛋白质的生物合成,促进胎儿的生长发育。妊娠期孕妇甲状腺素功能亢进,对碘的需要量也增加。缺碘可引起孕妇甲状腺肿大,儿童甲状腺肿可表现基础代谢降低及矮小痴呆,故妊娠时应注意食含碘量较高的海产品。
妊娠期对铜、锰及其它元素的需求量目前还没定论。
二、糖代谢特点
葡萄糖不仅是胎儿主要的能量物质,而且也是胎儿合成糖原、脂肪及非必需氨基酸的原料。胎儿的葡萄糖全部依靠母体供给,足月胎儿每日约需摄取26-30g葡萄糖。葡萄糖可以自由地通过胎盘从母血运送到胎儿血循环中。妊娠期母体内分泌的改变使孕妇体内糖代谢发生变化,血糖和糖耐量试验与非孕期不同,以孕中、末期更加显着。
从早孕最初几周开始,胎儿-胎盘单位分泌的雌孕激素增多,胰岛逐渐胎大,β细胞增生使胰岛素释放增加,血浆胰岛素水平进行性升高,组织对胰岛素的敏感性也增强,机体对葡萄糖利用加速,使空腹血糖浓度降低。孕妇空腹血糖为3.0-3.3mmol/L,比非孕期大约低0.82-1.1mmol/L,此时常易发生饥饿性低血糖。在妊娠中期,孕妇的早孕反应减轻、食欲增加、胰岛素释放增多;但同时胎盘分泌的人类胎盘催乳素(hPL)和人类胎盘绒毛膜促性腺激素(hCG)以及其它蛋白类、固醇类激素合成增加,垂体泌乳素、可的松和胰高血糖素分泌也增加,对抗胰岛素的作用加强,使血葡萄糖上升以供应胎儿需要。晚期妊娠时,胎盘抗胰岛素激素的敏感性仅为非孕期的1/5,胰岛素清除葡萄糖的能力却减低,血糖升高,使有充分的葡萄糖供给胎儿。总之,妊娠期机体对胰岛素的需要量增加,胰岛细胞增殖分泌较多胰岛素,同时此期胎盘甾体激素增加,皮质醇等抗胰岛素分泌增多均可引起胰岛素分泌亢进,使孕妇外周血中胰岛素含量升高。孕妇静脉注射葡萄糖后,血中胰岛素含量升高要比非孕妇高,说明孕妇胰岛β细胞功能活跃,分泌旺盛。孕妇注射胰岛素后血糖下降要比非孕妇少,说明孕妇对胰岛素的作用不敏感。这些变化确保了母体能源源不断地供给胎儿足够的葡萄糖。
妊娠期孕妇血糖变化的特点是:①孕妇空腹血糖浓度的基线值下降,而且分娩前下降得更多,如非孕妇女空腹血糖基线值为4.62mmol/L,而妊娠34-36周时则降至4.29mmol/L,分娩前甚至降到3.85mmol/L。②孕妇餐后血糖呈持续性升高,且升高的峰值较非孕妇女高。即孕妇餐后1-2小时血糖值可达7.7mmol/L,而非孕妇女仅高到6.6mmol/L左右,但在下一次餐前,孕妇血糖往往又低于非孕妇女。孕后血糖水平的这种改变为胎儿从母体吸取葡萄糖创造了有利条件,饭后持续高血糖可使母血中的葡萄糖顺浓度差不断地转运给胎儿,以致餐前孕妇血糖呈低值。胎儿消耗葡萄糖的量可大到足以使母体产生低血糖的程度。
孕妇口服50g葡萄糖后的耐量试验也会发生两种变化:一是高峰迟缓现象,即血糖达到最高值的时间推迟。二是血糖最高值高于非孕妇女。这是由于妊娠时孕妇血中升高血糖的激素水平都增高,它们拮抗胰岛素的降血糖功能,而且胎盘产生的胰岛素降解酶使胰岛素降解加快。虽然这些激素促使胰岛β细胞分泌亢进,胰岛分泌量比未孕时增高60%-80%,但机体仍处于胰岛素相对不足的状态,对葡萄糖的不耐受性增加。如果孕妇胰岛素分泌的潜力不足则可发生妊娠期糖尿病。
虽然妊娠时胰岛分泌亢进是维持孕妇糖代谢及胎儿葡萄糖供应的重要生理性反应,但是妊娠期因多种激素及代谢的改变,致使有很强的致糖尿病倾向,甚至认为妊娠本身就是一个致糖尿病因子。当胰岛功能不全如隐性或轻型糖尿病患者,在早孕时胰岛功能尚可代偿,可以满足产生较非孕时多的胰岛素保持母体血糖稳定。但到妊娠后期由于胰岛功能不足,满足不了逐渐增长的需要,即产生相对性胰岛素不足,导致血糖增高,临床出现糖尿病。产后当胰岛的应激状态被解除,血糖又可以恢复正常。糖尿病患者妊娠后可使病情加重或复杂化,妊娠晚期常发生酮症酸中毒和低血糖。酮症酸中毒是孕妇和围产儿死亡的主要原因。
妊娠期血容量增加血液稀释,使血胰岛素浓度相对下降:同时由于肾小球滤过率增加,肾小管回吸收率不增加或受抑制而减少,自妊娠4个月起肾小球对葡萄糖的滤过率多已超过肾小管的回吸收率,使肾糖阈降低,且孕末期可有乳糖排出,因此,约有20%-30%的正常孕妇出现间隙性糖尿,其中75%的糖耐量正常。如果尿中排糖量增加显著,还可导致孕妇低血糖。
约一半妇女口服避孕药后可以出现轻度的糖耐量降低,可通过口服或静脉葡萄糖耐量试验检测。空腹血糖一般尚不受影响,大多数妇女停药后这个现象即消失。这一现象主要由于避孕药中雌激素的组分所致。
三、脂类代谢特点
妊娠期血脂全部升高,其中孕早期与非孕时相差不大,约为6.0g/L(未孕时为5.0g/L),孕中期以后逐渐增高,到孕末期明显增高,分娩前可增到10.0g/L。其中胆固醇从未孕时的5.18mmol/L升高到6.48mmol/L,其它脂类如甘油三酯、游离脂肪酸均呈现较高值。妊娠期促使脂肪动员激素,如皮质醇、胰高血糖素、生长激素等分泌增加,孕妇血中水平增高,脂肪组织中激素敏感性脂肪酶活性增强,加速孕妇血中雌激素水平升高,促使肝合成极低密度脂蛋白(VLDL)增强,故孕妇血中富含甘油三酯的脂蛋白(前β脂蛋白)水平增高,加速孕妇体脂贮存,导致孕末期有高脂血症倾向。孕妇β脂蛋白较α脂蛋白增加的多,使二者的比例由非孕时的2:1增到5:1。
妊娠期母体的高脂血症倾向并非病理现象,而是一种生理适应性措施,它有利于胎儿从母体血中吸取更多的游离脂肪酸及其脂类,作为胎儿发育、胎脑组织及肺表面脂类活性物质合成原料。母体脂解作用增强产生的过多酮体还能经胎盘输送到胎血供胎脑、胎肾组织利用。此外在妊娠30周前,孕妇还有脂肪蓄积,分布于腹壁、背及大腿部,为妊娠晚期、分娩及产褥期供应必要的能量作贮备。妊娠晚期母血中游离脂肪酸增高,母体将其作为能源而将葡萄糖节约下来,保证了胎儿的需要。一般情况下,分娩时血脂比妊娠末期还高,产后逐渐下降,产后6天大约半数产妇接近非孕水平,产后2-6周几乎全部恢复正常。脂类中以甘油三酯下降最早,其它脂类逐渐下降。
口服避孕药因含有雌激素等,长期服用也可影响妇女的脂类代谢,造成血浆脂蛋白组分改变。血浆脂蛋白质组分的变化可有:①空腹血浆VLDL、甘油三酯的水平升高,血浆LDL-胆固醇的轻度增加。②血浆HDL-胆固醇水平的下降。有人认为这些改变如出现早而持久,有增加血栓形成的危险,据英国、瑞典、丹麦三国资料分析,避孕药的复合制剂中,雌激素含量高者血栓发生率较高。
四、蛋白质代谢特点
蛋白质是构成人体组织的基本物质,胎儿、胎盘、母血、子宫等组织主要由蛋白质组成。妊娠期间母体需要大量蛋白质,如足月妊娠时胎儿、胎盘、羊水约含蛋白质500g,母血的血红蛋白、血浆蛋白、子宫及乳房所增加的蛋白质约500g,故母体内蛋白质合成增加而分解也旺盛,但总体上是合成大于分解,呈正氮平衡。母体内氮的储存,不仅仅是供给胎儿发育、子宫及乳腺增大所用,而且也为分娩消耗及产后乳汁分泌作好准备。
氨基酸是蛋白质的基本组成单位,孕妇氨基酸代谢与胎儿蛋白质的合成密切相关。胎早期胎儿肝、肾及脑等重要脏器含水量较大,蛋白质含量少,蛋白质合成速度较慢,因此母体氨基酸代谢变化不显著,随着妊娠月份不断进展,到3-4个月时,胎儿肝合成蛋白质、酶的种类增多,到妊娠末期肾、脑等重要脏器迅速发育,大量合成与蓄积蛋白质,因此胎儿摄取母血氨基酸速度加快。此时母体氨基酸代谢的主要特点反映在血浆氨基酸水平变化上是:除丙氨酸外,空腹血氨基酸基础值下降,蛋白质餐后的高氨基酸血症不能持久,常迅速地下降,说明母血中的氨基酸被胎儿大量摄取,但由于胎儿在不同的发育期合成不同的器官蛋白质,所需氨基酸种类与数量因而有些区别,故母血在不同的妊娠期通过胎盘输送给胎儿的氨基酸无论种类和数量均有不同。例如妊娠3-4个月胎肝合成旺盛期,母体供应较多的谷氨酸、半胱氨酸用于胎肝蛋白的合成,到妊娠7-8个月后胎儿摄取母血中的酪氨酸、脯氨酸、丝氨酸、蛋氨酸等。若妊娠期蛋白质、热量供应不足或供应蛋白质的必需氨基酸种类不全,孕妇就分解肌肉、内脏蛋白质以维持胎儿对氨基酸的需要。若母体分解自身蛋白质仍不能满足胎儿的需要,则会发生宫内胎儿生长迟缓,如胎儿低体重,脑组织核酸及蛋白质含量低下。因此孕妇处于负氮平衡,对孕妇及胎儿健康都仍为不利。
妊娠时孕妇血浆游离氨基酸浓度低于胎儿血浆氨基酸浓度,而胎儿血浆氨基酸浓度又低于胎盘内游离氨基酸浓度,这是因为胎儿体内蛋白质合成十分旺盛,他一方面从母血中大量地摄取氨基酸,同时自身又合成非必需氨基酸,造成胎儿血浆氨基酸浓度反而高于母血。氨基酸从母血进入胎盘的机制类似于小肠粘膜细胞、肾小管上皮细胞转运氨基酸的过程,而位于胎盘滋养叶外层合体细胞膜上的氨基酸载体能将氨基酸逆浓度差从母体携带到胎盘,然后释放到胎儿血液内。另外胎盘内还存在有活性较强的γ-谷氨酰转肽酶(γ-glulamyl-transpeptidase,γ-GT)和谷氨酰胺合成酶。γ-GT在谷胱甘肽协助下能将氨基酸从母血转移到胎盘合体细胞内。谷氨酰胺合成酶能将不易通过胎盘的谷氨酸转变成容易通过胎盘的谷氨酰胺,协助谷氨酸向胎盘转运,便于被胎儿利用。
多数孕妇尿氨基酸排出增加,出现氨基酸尿。妊娠期肾小管对肾小球滤过的氨基酸重吸收率下降,其中以组氨酸、苏氨酸、丝氨酸及丙氨酸排泄最多,到临产前为未孕时的3-5倍。其次以赖氨酸、半胱氨酸、牛磺酸、苯丙氨酸、亮氨酸及酪氨酸排泄量也增加,但它们不持续到孕末期,其它氨基酸尿排泄量变化不显著。
妊娠期随着机体多系统的变化,孕妇血容量增加,血液中多种蛋白质成分均发生了一系列变化,如血浆蛋白降低、多种凝血因子含量明显增加,纤溶系统的成分也改变,血液处于高凝及纤溶系统活性降低的状态,并出现妊娠相关性蛋白质,有关的详细内容见本章第三节。
五、矿物质代谢与酸碱平衡特点
妊娠期孕妇的血容量增加,造血功能活跃,加上胎儿骨骼生长发育等都需要无机盐,孕妇对钠、钾、钙、磷及铁的需要量都有所增加,如果无机盐供应不足,也会产生许多危害孕母与胎儿的并发症,如缺铁性贫血等。
铁是血红蛋白、肌红蛋白、细胞色素酶类以及多种氧化酶的组成成分,它与血液中氧的运输和细胞内生物氧化过程有着密切的关系。妊娠期胎儿发育生长及母体组织的变化,对铁的需要量增加。妊娠期母体红细胞增加250-450ml,需要铁约500mg;新生儿体内铁含量约300mg,所以产前胎儿和母体红细胞生成需铁共约800mg。妊娠期对铁的需要主要在后半期增加。妊娠最后3个月胎儿除了造血以外,胎儿的肝与脾还需要贮存一部分铁,因而向胎儿供给的铁量增加,单靠从食物中吸收以及动员母体贮存的铁常不能满足需要,如不补充外源铁,孕妇血清铁含量会下降,进而发生贫血。
铁缺乏是造成孕妇贫血最常见的原因,缺铁性贫血约占妊娠期贫血的90%。妊娠各期孕妇对铁的需求不一样,妊娠中期以后多数妇女每日需铁量为25-50mg。由于一般饮食可提供的铁约10-15mg,而且肠道仅能吸收其中约4%-11%,故孕妇常有缺铁征象,临床上多表现出乏力、苍白、心慌、气短,感染的危险也随之上升。贫血的孕妇虽然铁和叶酸仍能运至胎盘及胎儿,但早产和死产发生率、新生儿发病率均高于正常孕妇。在孕期及时纠正贫血对胎儿、婴儿的预后均要好得多。维生素C和稀盐酸有利于铁的吸收,应同时补充。
妊娠期胎儿骨骼的形成及胎盘形成都需要更多的钙和磷。妊娠末期胎儿体内约含钙25g、磷14g,且绝大部位都是妊娠最后2个月才贮存下来的,所以早产儿易发生缺钙。孕妇每日对钙、磷的需求量为1-2g,钙和磷需求比为1:1。如果妊娠妇女每日钙和磷摄入不足或吸收不良,胎儿所需的钙和磷就必须从母体骨质中获取。妊娠晚期孕妇血钙比未孕或早孕时减低,故妊娠后半期应补钙。维生素D具有调节钙磷代谢,促进钙吸收的作用,故在补钙的同时每日应补充维生素D400U。牛奶及奶制品中含有较多钙,建议孕妇多食奶制品。
妊娠期体内水分潴留较突出,平均约7.5L,其中胎儿、胎盘、羊水、子宫、乳房和血流量增多的水分潴留约6L,其余为组织间液。研究发现:血压正常而仅有下肢凹陷性水肿属生理现象。这些孕妇所分娩的新生儿的体重往往比无水肿者大,围产儿死亡率也较低,即比无水肿者更有利。钠是细胞外液的主要电介质,在水潴留的同时必然要贮留一定量的钠。孕妇在妊娠期贮钠约1000mmol。整个妊娠期不必忌盐,以保证孕妇有足够的钠供应。正常妊娠期间水、钠潴留与血液动力学及某些激素有关,如雌激素、醛固酮有助于水、钠潴留。
正常孕妇较非孕妇女换气量大,排出CO2多,使血中H2CO3含量减低,可以引起呼吸性碱中毒。但孕妇通过血浆重碳酸盐呈代偿性降低,使H2CO3/NaHCO3比值不变,仍保持血液pH值正常或稍微上升。
综上所述,孕妇在妊娠初期代谢即出现变化,但要到妊娠末期胎儿迅速生长发育时,母体内三大营养物质代谢才发生显著变化,其特点可归纳为以下两方面:①“容易较早发生饥饿”妊娠末期妇女餐后容易较早出现饥饿状态。空腹和餐前的血糖、血氨基酸基础值低于正常非妊娠妇女。这是由于胎儿大量地吸收母血中的葡萄糖、游离氨基酸所致。与此同时贮脂动员,在脂解激素(生长素、胎盘生乳素等)升高而胰岛素作用被拮抗的情况下脂解加速,母血的甘油、游离脂酸、酮体增高。上述母体代谢变化有利于胎儿更好地利用母血中的营养物。②“促进合成代谢”妊娠末期孕妇餐后出现高氨基酸血症、较长时间的餐后高血糖及乳糜微粒不易及时廓清,致使发生高脂血症倾向。此时孕妇血中含较丰富的营养物,经胎盘输送到胎血供胎儿合成之用。由于妊娠期末期母体这种特殊的代谢状态,充分保证了胎儿营养物质的供应,促进胎儿正常的生长发育。
第二节 妊娠期的内分泌特点
妊娠期激素可以来自母体,也可来自胎儿内分泌腺。此外,胎盘可以视为一个重要的不完全的内分泌器官,它可以利用母体及胎儿的代谢前身物质-如来自母体胆固醇,来自母体及胎儿肾上腺的脱氢表雄酮硫酸酯(DHEAS),以及由胎儿合成的16α-羟DHEAS及母体的大量氨基酸合成类固醇和蛋白类激素。因此母体血循环中的激素变化往往可以反映胎儿及胎盘的功能状态。
一、妊娠期黄体功能
受精卵植入子宫内膜后,母体卵巢的黄体仍继续分泌孕酮以维持早期妊娠子宫内膜(蜕膜)的发育。但其分泌量从妊娠2-3周开始逐渐下降,至7-8周胎盘滋养层即能分泌足够的孕酮以维持妊娠,妊娠3个月后黄体分泌孕酮功能基本消失而完全由胎盘供应足够孕酮以维持正常胎儿-胎盘复合体需要。
二、胎盘激素
妊娠期胎盘能合成大量甾体激素如雌激素(雌二醇、雌酮、雌三醇)、孕激素(孕酮、孕二醇)等,此外胎盘还合成大量蛋白激素如绒毛膜促性腺激素、胎盘生乳素等。人类胎盘合成分泌的主要激素有:
(一)人类胎盘绒毛促性腺激素
人类胎盘绒毛膜促性腺激素(human chorionicgonadotropin,hCG)为蛋白,分子量约为39000u,由α-亚基与β-亚基以11-12个二硫键互联而成,它在胎盘合体滋养层细胞内合成,为胎盘独有。α-亚基分子量为14900,含92个氨基酸及40%糖;β亚基则由145氨基酸组成,其分子量为23000u,含糖约30%。黄体生成素(LH)、卵泡刺激素(FSH)及促甲状腺激素(TSH)的α-亚基结构与hCG的α-亚基结构大致相同,这四种激素的区别仅在于β-亚基结构不同。胚胎着床后hCG的生成量呈对数递增,到6-8周达高峰,妊娠60-80天达到最高峰,随后逐渐下降,到妊娠160-180天时下降到最低点,此后稍又回升继续保持到分娩。妊娠早期hCG的主要功能是维持卵巢的黄体分泌孕酮以维持早期胚胎发育的需要;它在胎儿-胎盘复合体中还促进类固醇激素的生物合成。
(二)人类胎盘催乳素
人类胎盘催乳素(human placenta lactogen,hPL)也称人类绒毛膜促生长催乳素(human chorionicsomatamammotropin,hCS),为单链多肽类激素,其分子量为21600u,由190个氨基酸残基合成,含有两条链内二硫键。人胎盘催乳素与人生长激素有160个相同的氨基酸残基序列,因此二者在免疫学与生物学方面密切相关。hPL也由胎盘合体滋养层细胞合成与分泌,分泌后大部分进入绒毛间隙和胎盘血窦,很少出现于胎体内。
妊娠5-6周即可用放射免疫法测出血浆中的hPL,以后分泌量缓慢上升,自15-30周时即迅速增高,到妊娠34周时达高峰,约7.75mg/L-10.6mg/L以后维持此水平直到分娩。hPL的半寿期在21-23分钟之间,分泌量与胎盘体积成正比;足月妊娠时,每日每百克胎盘约分泌0.5g hPL,以此计算整个胎盘每日分泌1-2g 左右的hPL,产后迅速下降,3-6小时即测不出。
hPL的功能是在孕妇体内与胰岛素、皮质激素协同作用促进乳腺发育及促进正氮平衡,有利于妊娠期蛋白质的蓄积。hPL还抑制脂肪沉积作用,促进脂肪分解使血中游离脂酸升高。当血液中游离脂酸较葡萄糖占优势时,肌组织主要以游离脂酸作为能源,减少对葡萄糖的摄取,有利于胎儿从母血大量地摄取葡萄糖。hPL这一功能是胎儿迅速生长发育的重要条件。hPL在母血中含量与胎盘重量及胎儿体重相关,可直接反映胎盘的功能状态。
(三)孕酮
孕激素主要有孕酮及其代谢产物孕烷二醇,属甾体激素。孕酮由卵巢和肾上腺合成。
胎盘能利用孕母血中的胆固醇合成孕酮,从妊娠36天起它即能生产足够孕酮。非孕妇血浆孕酮值在0-15ng/ml,孕妇于妊娠第5周时血清中的浓度为24±7.3ng/ml,到孕32周时增高到125.2±37ng/ml,到37孕周达到最高峰为150ng/ml,一直保持到临产前才稍降。待胎盘娩出后迅速降至10-20ng/ml。这证明妊娠末期孕妇血清高水平的孕酮来自胎盘分泌。妊娠早期孕酮日分泌为30-50mg左右(未孕妇仅1-25mg/d),主要为妊娠黄体分泌,到孕末期分泌量为250-300mg/d。胎盘合成的孕酮大部分进入母体与胎儿并进行代谢,主要代谢产物为孕烷二醇,后者主要在肝转变成葡萄糖酸苷及硫酸酯等结合形式,结合型水溶性增加,易从尿中排出,妊娠期末期孕妇尿液中孕烷二醇日排泄量约为35-40mg。血中孕烷二醇的水平随孕酮水平而变动,尿中排泄量也与孕妇血中孕酮量成正比。胎盘内孕酮的合成过程以图14-1简示。
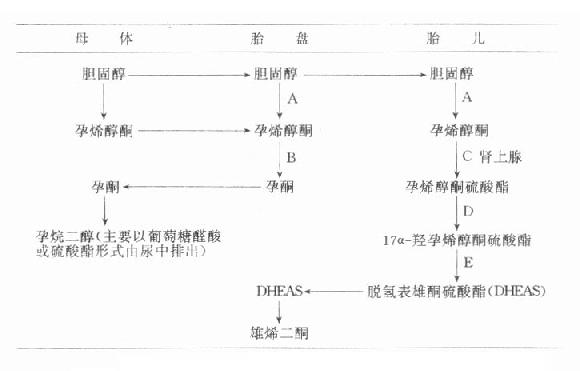
图14-1 胎儿-胎盘复合体孕酮合成途径
有关酶类:A=C20-22裂解酶;B=3β-脱氢酶;C=硫酸酯激酶;
D=17α-羟化酶;E=C17-20裂解酶
从图14-1可见,胎盘合成孕酮的原料是胆固醇,它来源于母血供应。母血还可直接输送孕酮的前身物-孕烯醇酮,用于合成孕酮。
(四)雌激素
雌激素属甾体激素,非孕妇女主要由卵巢、肾上腺皮质合成。雌激素主要有雌酮(E1)、雌二醇(E2)及雌三醇(E3)。由于胎盘组织也能合成雌酮、雌二醇及雌三醇,故正常妊娠妇女的雌激素水平随妊娠月份进展而不断增高,到妊娠第7周时胎盘生成的雌激素已超过50%,因此妊娠期妇女除妊娠头几周外,胎盘才是雌激素的重要合成器官。
正常早期孕妇,雌酮、雌二醇主要来源于卵巢,雌三醇是前两者在外周组织的代谢产物。妊娠期雌激素75%-95%来源于胎儿-胎盘复合体。这是因为胎盘中合成雌激素的某些酶活性极低,而这些酶在胎儿肾上腺、胎肝内却十分丰富。因此要完成整个雌激素的合成与代谢,必须依赖正常的胎儿与胎盘的共同活动,即把胎儿与胎盘视为一个完整的功能单位与统一体,称之为胎儿-胎盘复合体。但此复合体又与母体不可分割,因为合成雌激素的前体来自母血供应。所以母体与胎儿-胎盘复合体在合成雌激素时三位一体,只不过胎盘是雌三醇最终生成部位而已。
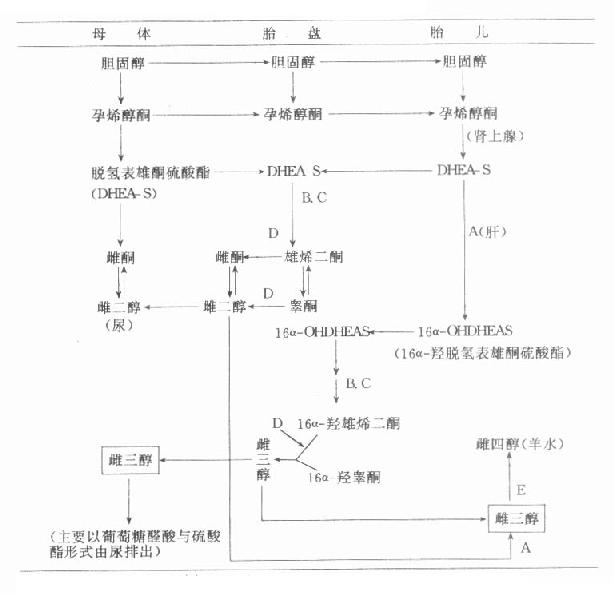
图14-2 胎儿-胎盘复合体雌激素合成途径
有关酶类:A=16α-羟化酶;B=硫酸酯酶;C=3β-羟化酶;D=芳香化酶;E=15α-羟化酶
从图14-1可见,胎儿也能利用从胎盘输送来的胆固醇,或自身合成的胆固醇作为原料,在C20-22裂解酶作用下转变成孕烯醇酮。胎儿肾上腺中存在活性很强的硫酸酯激酶,后者经17α-羟化酶作用下生成17α-羟孕烯醇酮硫酸酯,继而它又在C17-20裂解酶催化下生成脱氢表雄酮硫酯(dehydroepiandrosteronesul-fate,DHEAS)。胎儿生成的DHEAS可返回胎盘在硫酸酯酶及3β-羟化酶作用下生成雄烯二酮,成为合成雌激素的原料。
雄烯二酮可转变成睾酮,睾酮经胎盘内的芳香化酶作用生成雌二醇。雄烯二酮还可直接芳香化而成雌酮,胎盘生成的这两种激素可返回到母体内,成为孕妇血中激素最重要来源。胎儿体内生成的DHEAS还可在胎肝内存在的16α-羟化酶作用下生成16α-羟脱氢表雄酮硫酸酯(16α-OHDHEAS)。16α-OHDHEAS进入胎盘后生成16α-羟雄烯二酮及16α-羟睾酮,这两种雄激素经胎盘内芳香化酶作用转变成雌三醇,雌三醇可返回母体或进入胎儿体内(图14-2)。
胎盘生成的雌二醇在胎儿体内经16α-羟化酶作用转变为雌三醇,进入胎儿体内的雌三醇,在胎肝内经15α-羟化酶作用最科均生成雌四醇(图14-2)。胎儿生成的雌四醇可出现在羊水等体液内,孕妇尿液中的雌四醇的最主要来源也是最终经胎肝生成的。
正常妊娠期妇女血液激素组分的变化可参见表14-1。
表14-1 正常妊娠期血液激素组分的变化
| 激 素 | 非妊娠妇女(范围) | 妊娠后期妇女(范围) |
| 黄体生成素(LH mIU/ml) | 5-25 | <2 |
| 促卵泡成熟激素(FSH mIU/ml) | 2-15 | <2 |
| 垂体生乳素(PRL ng/ml) | 5-25 | 100-300 |
| 生长激素(GH ng/ml) | <5 | <7 |
| 促肾上腺皮质激素(ACTH ng/ml) | 7.8-120 | 12-60 |
| 总T4μg/dl | 5-12 | 7-8 |
| 总T3μg/dl | 50-250 | 120-280 |
| 可的松μg/dl | 5-25 | 10-40 |
| DOC ng/dl | 4-16 | 50-200 |
| 醛固酮ng/dl | 2-10 | 40-150 |
| 雌三醇ng/dl | 0.07-0.3 | 5-25 |
| 孕酮ng/dl | 1-25 | 7.0-250 |
| 1,25-(OH)2D3pg/ml | 10-90 | 20-200 |
三、其它内分泌腺的变化
妊娠期间,孕妇的各器官、系统均发生了明显变化以适应胎儿生长发育的需要。就内分泌系统而言,胎盘与胎儿所合成分泌的激素发挥了相当大的作用,但这些并不能满足母体与胎儿的需要。因此母体内分泌腺在妊娠期亦积极参与适应性变化。
(一)垂体
妊娠期垂体的体积和重量均增加,体积约比妊娠前增加20%-40%,重量几乎增加一倍。垂体前叶分泌的垂体生乳素(PRL)、促甲状腺素(TSH)、促肾上腺皮质激素(ACTH)和黑色细胞刺激素(MSH)增多,生长激素(GH)的分泌无改变,促性腺激素(FSH、LH)的分泌减少,垂体后叶不论催产素-加压素的组织结构或功能在孕期均无特殊改变。PRL于孕7周就开始增多,并随孕周的增加而逐渐升高,血清PRL值可由非孕时的100ng/L上升为足月妊娠时的2μg/L。PRL有促进乳房发育的作用,其分泌增加是产后哺乳的条件。MSH分泌增多,使面颊、乳状、乳晕、腹中线、外阴等处有色素沉着。由于FSH、LH分泌减少,妊娠期卵巢中的卵泡不再发育成熟,也无排卵。
(二)甲状腺
妊娠期垂体前叶分泌的TSH和胎盘分泌的促甲状腺激素释放激素(TRH)和hCG共同引起甲状腺组织增生、肥大,使甲状腺激素合成和分泌增加,致使①基础代谢率(BMR)由非妊娠时的-10%~+15%增加至+20%~+30%,增加20%-30%。②血浆蛋白结合碘(PBI)由非妊娠时的276-630nmol/L增至946-1103nmol/L。③受大量雌激素的影响,肝产生较多的甲状腺素结合球蛋白(TBG),血清TBG浓度由130-250μg/L增至300-500μg/L,为非孕时的25倍。且TBG与甲状腺素(T4)和三碘甲腺原氨酸(T3)的结合力增加,故血浆中结合型的T3、T4量增多,而游离型的T3、T4减少。妊娠期甲状腺功能变化如表14-2。
表14-2 妊娠期甲状腺功能与非妊娠期的比较
| 检查项目 | 非妊娠期 | 妊娠期 |
| 基础代谢(BMR %) | -10%~+15% | +20%~+30% |
| 血清蛋白结合碘(PBI μg/L) | 40~80 | 60~180 |
| 甲状腺结合球蛋白(TBG μg/L) | 130~250 | 300~500 |
| 血清总T3(TT3μg/L) | 152±0.4 | 稍增加 |
| 血清总T4(TT4μg/L) | 60~90 | 100~180 |
| 血清游离T3(FT3ng/L) | 3.9~7.4 | 稍增加 |
| 血清游离T4(FT4ng/L) | 20~40 | 稍增加 |
(三)甲状旁腺
甲状旁腺激素(PTH)的主要功能是调节Ca、P代谢,维持Ca、P的自身稳定和平衡。PTH的主要作用部位是肾和骨质,它可增强肾小管的重吸收能力,对P的重吸收能力减少,尿磷排量增加,Ca的排量减少,最后表现为血磷降低,血钙增高。甲状旁腺在妊娠时呈增生状态,孕24周开始血浆中PTH的浓度逐渐升高以满足妊娠期及胎儿对钙质的需要。
(四)肾上腺
妊娠期间,由于大量雌激素的影响,孕妇肾上腺皮质分泌活动增加,孕妇血皮质醇水平随孕期的进展而逐渐升高。醛固酮每日分泌量由非孕时的139mmol-695mmol(50-250μg)上升为足月时的2780mmol(1mg)左右,是原来的4倍以上。醛固酮与肾素、血管紧张素一起,调节控制血容量、血钠、血钾平衡及血压。妊娠期间肾上腺皮质中层囊状带分泌的糖皮质激素-皮质醇是非孕时的3倍;网状带分泌的性激素睾丸酮也略增加,故孕妇的阴毛、腋毛均可增粗、增多。而孕期肾上腺髓质所产生的肾上腺素和去甲肾上腺素却无改变。
(五)胰腺
妊娠期由于胎盘滋养层细胞产生的胎盘生乳素、雌激素、孕激素、胎盘胰岛素酶及肾上腺皮质激素分泌增多等,均为妊娠期特有的抗胰岛素因素,它们使胰岛素发生代偿性功能亢进,即胰岛增大,β细胞增多、增加胰岛素的分泌来维持体内的糖代谢。胰岛素分泌增多从孕中期开始,至分娩前达高峰。
第三节 妊娠的生物化学诊断
妊娠期胎盘及胎儿-胎盘复合体生成多种激素(孕酮、睾酮、雌激素、hCG、hPL及P-Sβ1G)、多种酶类,它们的含量均随妊娠进展而变动,其中有许多可出现在孕妇体液内。因此测定孕妇血样、尿液及其它体液内这类物质的含量可作为早孕诊断及监护胎盘、胎儿-胎盘复合体功能,了解胎儿宫内发育,成熟及安全的手段。
一、妊娠的内分泌检查
(一)妊娠试验
目前妊娠早期诊断开展最普遍的项目是测定血清及尿液绒毛膜促性腺激素(hCG)。早孕时hCG迅速上升,在受精卵着床后1.7-2天血清中hCG就增加一倍,一周后即从未妊娠时的25ng/ml(或低于此值)上升到100ng/ml。两周后显着增高,到12-13周达高峰,妊娠末期稍下降,hCG在早孕明显升高的特点,已广泛用于早孕的诊断。表14-3显示了孕妇尿hCG测定情况。
表14-3 孕妇尿hCG测定
| 妊娠时间 | 尿hCG含量(IU/L) |
| 排卵后17天 | 625 |
| 妊娠6周 | 5000 |
| 妊娠10-12周 | 8万-32万 |
| 妊娠12-14周后 | 1万-2万 |
血,尿hCG测定的其它临床价值是:①双胎妊娠血清hCG值比单胎增加一倍以上,妊娠10周的单胎妊娠妇女血清值为10000-12000ng/ml,而双胎者为20000-40000ng/ml。②若孕妇血中hCG水平低值或连续测定呈下降趋势,如妊娠55-110天时低于1000ng/ml者预示先兆流产。③放射免疫法测定hCG的β-亚基(hCG-β)能早期发现异常妊娠性hCG-β显著高值可能为滋养叶细胞疾病。葡萄胎、绒毛膜上皮细胞癌及合体滋养叶细胞坏死、崩解情况。④异位妊娠时hCG值低于同期正常妊娠值。
孕妇血尿中的hCG是内分泌测定诊断妊娠的基础。目前临床测定血、尿中hCG常见方法为免疫学方法,可分为乳胶凝集抑制试验、单红细胞凝集抑制法、放射免疫法。测定时可进行定性、定量、稀释、浓缩试验。定性试验主要用于诊断早期妊娠、先兆流产预后的判定。稀释试验常用于诊断滋养层细胞肿瘤。浓缩试验是葡萄胎流产后,恶性葡萄胎绒毛瘤化疗或手术后随访治疗结果,了解有否残留病变或复发的重要检查方法,如浓缩300ml尿试验为阴性,一般认为体内无活跃的滋养层细胞。
(二)孕激素的测定
妊娠8周以后孕激素主要来源于胎盘,妊娠晚期孕酮的产生量为180-280mg/d,且孕酮量大致与产生它的胎盘重量成比例,故孕妇血中孕酮水平能反映胎盘的状况。血清孕酮在妊娠各期的水平见表14-4。
孕酮的主要代谢产物孕烷二醇E2经尿液排泄,因此尿中孕烷二醇的排泄量与血中孕酮量成正比,测定尿孕烷二醇排出量能间接反映胎盘功能。妊娠期尿E2值见表14-5。妊娠后尿中E2排出量随着妊娠周期数增加而增加,直至妊娠36周时达最高峰,以后保持恒定直至分娩,分娩后迅速减少,产后4-5天降至正常水平。
表14-4 妊娠期血孕酮量
| 妊娠周数 | 孕酮(nmol/L) |
| 7 | 78.4 |
| 8 | 91.5 |
| 9-12 | 133 |
| 13-16 | 145.6 |
| 17-20 | 202.6 |
| 21-24 | 354.8 |
| 25-34 | 528.9 |
| 35-40 | 645 |
表14-5 妊娠尿E2值
| 妊娠周数 | 尿E2(μmol/d) |
| 16 | 16-65 |
| 20 | 19-81 |
| 24 | 37-100 |
| 28 | 59-160 |
| 32 | 69-206 |
| 36 | 41-240 |
| 40 | 72-197 |
血中孕酮低值及尿液孕烷二醇低值可预示早产。但鉴于此法个体差异较大,在作胎盘功能的估计时,临床意义不如雌三醇价值大。
(三)胎盘催乳素测定
目前认为胎盘激素(hPL)是与胎儿生长有关的主要激素,并可作为胎盘功能测定的指标。
由于在末次月经后5-6周时,应用放射免疫法即能检测出血中胎盘催乳素(hPL),故可作为早孕诊断的检查项目。hPL为胎盘独有,半寿期较短,能迅速反映胎盘功能状态。它在临床上的实用价值是①诊断滋养叶细胞疾病:葡萄胎患者血中hPL值较正常妊娠值低,但hCG值反而增高。所以hCG/hPL比值比正常妊娠时高100倍,而且在恶变时hPL更加降低,故同时测定这两种激素可诊断葡萄胎与随访此病患者。②早孕时如连续测定hPL可衡量先兆流产的预后,hPL值偏低或呈下降趋势,提示妊娠将要或已经中止,可能会发生流产。③高危妊娠的监护:用放射免疫法测定妊娠35周后孕妇血浆hPL,若其值低下4.0μg/ml提示有先兆子痫、胎盘功能不良或胎儿宫内窒息等情况。必须注意到,hPL值和胎盘大小有一定关系,糖尿病合并妊娠和母儿血型不合病例,由于胎盘较大,hPL数值可以偏高。母儿血型不合时还由于溶血首先累及胎儿,其次才累及胎盘,故hPL改变较晚。
测定hPL的方法有放射免疫法、血细胞凝集抑制反应、补体结合试验等,这些方法均可进行微量测定。
(四)雌激素的测定
雌激素的测定是卵巢与胎盘功能检查的主要项目。雌激素在人体卵巢、肾上腺、胎盘内产生,到肝内灭活,经肾由尿液排出,故可供检查雌激素的标本有血、尿及羊水。雌激素可分为雌酮、雌二醇、雌三醇。雌三醇是前二者的代谢产物,为检查的重点。未孕妇女雌激素检查常用于判断卵巢功能,而妊娠期其测定意义就广泛得多。
孕妇血与尿液雌激素的测定,是判断胎儿、胎盘功能及预测胎儿预后较可信的方法。应用放射免疫法测定孕妇血中雌三醇(E3)值可了解胎儿状态。妊娠中期、末期尿液E3系雌激素排泄量的500-1000倍,E3的前体脱氢表雄酮硫酸酯、16α-羟脱氢表雄酮等均来自胎儿,故随妊娠月份增长尿液E3排泄量也相应增加。妊娠20周时E3排泄量最低值为2mg/24小时尿液,以后每2周至少增加1mg,至妊娠40周时E3排泄量至少为12mg/24小时尿液,一般正常妊娠时应在30mg左右。若低于12mg/24小时尿液,提示胎盘功能不良,出现宫内胎儿生长迟缓、先兆子痫、胎儿畸形、先天性肝、肾上腺功能不全及葡萄胎等情况。
由于血浆E3含量随妊娠进展而增加,到妊娠22-23周时已达8μg/dl,33-39周时为20μg/dl,直到40周时其含量才稍降。因此有人主张测定血清E3值监护胎儿安全。当孕妇血清E3下降而雌二醇(E2)反见增高时预示早产。
对于高危妊娠的监护须连续测定E3才有诊断价值,过期妊娠的E3值下降缓慢,先兆子痫与宫内生长迟缓则E3下降较快。糖尿病孕妇常易发生胎儿-胎盘功能急骤变化,致胎儿突然死亡,故需每日作E3测定,并以测定血浆游离E3为妥。目前提倡高危妊娠时于妊娠26周就需作E3的测定。
除测血、尿中的E3外,尿中雌四醇(E4)对于判断胎肝发育有特殊价值。因为雌四醇前体主要为雌二醇(或雌三醇),雌二醇经胎肝独有的15α、16α-羟化酶作用生成雌四醇。如孕妇血、尿中雌四醇低值,常意味胎肝先天性缺陷,可发生宫内死胎。因此测定妇女尿中雌四醇排泄量可作为宫内诊断(intrautering diagnosis)指征,能了解胎肝发育状况。
临床上常以雌三醇来表示雌激素的含量,这是因为:妊娠期雌三醇主要由胎儿胎盘复合体产生,测定孕妇尿中雌三醇的排泄量,可以反映胎儿胎盘的功能状态;雌三醇在妊娠32周后尿中排量急剧上升,正常足月妊娠时尿中雌三醇的排出量平均值约24.4mg/24h尿(15.35-33.25mg/24h尿),而雌酮与雌二醇极少,尿中雌三醇可以代表雌激素整体水平。
尿中雌三醇含量减少常见于:①胎儿肾上腺皮质功能减退,如先天性肾上腺皮质发育不全、无脑儿、孕妇服用皮质类激素致胎儿肾上腺被抑制。②胎盘缺乏硫酸酯酶,不能产生雌三醇。③孕妇肝肾功能不全。④生活在高原区的孕妇。
尿中雌三醇含量增多的原因有:①多胎妊娠及胎儿体重大,肾上腺皮质功能增加。②胎儿患先天性肾上腺皮质功能亢进症。③糖尿病合并妊娠时胎儿体重过大,雌三醇排出量偏高(可正常)。④母儿血型不合。
由于血浆中雌三醇值逐日变化很大,测定方法不够普及,故有待进一步实践。
(五)硫酸脱氢表雄酮负荷试验
给孕妇静脉注射硫酸脱氢表雄酮(DHEAS)50mg,注射前后分别测定24小时尿雌三醇(E3)及17酮类固醇(17-Ks),观察E3/17-Ks比值的变化。正常妊娠末期注射DHEAS后E3、17-Ks同时显著增高,但E3增高的幅度大,E3/17-Ks比值从注射前的3.3上升到注射后的5.97,说明胎盘功能正常,胎儿安全。若E3/17-Ks比值下降,则说明胎盘转化DHEAS为E3的能力下降,提示胎盘功能不佳,易发生低体重儿、新生儿窒息、死胎等恶果。
二、血清酶类检查
正常妊娠时从孕妇血清中可测到多种胎盘产生的酶,且其含量能反映胎盘的功能状态,临床应用较多的是:
(一)催产素酶
主要为胱氨酸氨肽酶(L-cystine-aminopeptidase,CAP)此酶由胎盘合体滋养层和细胞产生,其作用是可使催产素分子中胱氨酸与酪氨酸之间的共价键断裂,使催产素灭活。CAP由胎盘合成分泌后仅见于孕妇血中,脐血和羊水中测不到,连续测定CAP对估计胎盘功能有一定价值。
CAP活性自妊娠2个月即可在孕妇血中测出,且随妊娠进展而逐渐增加。表14-6显示了血清催产素酶的正常值。胎盘娩出后24小时,血清CAP活性减半,产后7天恢复到非妊娠期水平。
表14-6 血清催产素酶的正常值
| 妊娠周数 | 催产素酶活性(U/L) |
| 非妊娠期 | 15-35 |
| 20 | 15-65 |
| 25 | 25-90 |
| 30 | 35-125 |
| 35 | 45-175 |
| 40 | 65-240 |
孕妇血清催产素酶活性超过正常值常预示双胎妊娠;如持续低值则表明胎盘功能不良,有分娩低体重儿的可能;如突然发生急剧降低则说明胎盘功能有急性障碍,有可能早产或死胎。
(二)耐热性碱性磷酸酶(HSAP)
胎盘产生的碱性磷酸酶与来源于肝、骨及肠的碱性磷酸酶不同,它具有抗热性。当加热到65℃,历时30分钟后,其它来源的碱性磷酸酶均失活,但不影响胎盘碱性磷酸酶活性,故称为耐热性磷酸酶(heat-stablealkaline phosphatase,HSAP)。通常用比色法测定血清HSAP。方法是先测定碱性磷酸酶总活性,然后加热再测定HSAP活性。妊娠16-20周时即可在孕妇血清中测出HSAP活性,并随妊娠期进展而持续增高,直至分娩。HSAP正常值与孕周关系如下:
| 孕周数 | 26 | 30 | 34 | 38 | 40 |
| HSAP活性(μmol/L) | 0.4 | 0.64 | 0.96 | 1.28 | 1.36 |
血清HSAP突然增高常见于胎盘梗死时大量滋养叶细胞崩解、破坏,预示胎儿处境危险。相反,HSAP活性持续下降可能为死胎。畸形与低体重儿。妊娠晚期连续测定HSAP对估计胎儿预后有一定价值。由于单凭此酶活性高低判断胎儿预后准确性受限,故应结合其它测定一起判断。
目前已应用凝胶过滤法、等电点聚集等生化技术成功地分离出血清胎盘型碱性磷酸酶,为研究妊娠时此酶的临床意义提供了新的手段。
(三)组氨酸酶
也叫二胺氧化酶(diamine oxydase,DAO)。由胎盘蜕膜产生,妊娠后9-18天即能在血清中测出,妊娠6周开始逐渐增高,其含量随妊娠进展而增加,妊娠20周时达峰值,较非妊娠时增加1000倍,产后下降。DAO妊娠8周和12周的平均值为1.5U/L和5.2U/L。
DAO测定可作为观察胎盘功能的指标,对妊娠早、中期预后有诊断价值。血清DAO水平低下则提示胎儿代谢不良,常出现宫内发育迟缓、早产等不良预后。
三、妊娠期血浆蛋白质的变化及其临床意义
妊娠期孕妇体内蛋白质代谢处于正氮平衡状态,尤其是孕中、晚期更为显着。妊娠后半期孕妇每天可储存约2-3g氮,其中50%供给胎儿、胎盘的发育生长,50%供给子宫、乳腺及母体其他组织增生肥大用。此外胎儿-胎盘复合体的成熟,妊娠期内分泌的变化及血容量、血凝与纤溶系统的变化等,均可致孕妇血浆蛋白质组成与非孕妇女有所不同。足月妊娠时血液的部分改变见表14-7。
表14-7 足月妊娠时血容量、血清蛋白、电解质的改变
| 正常妇女 | 足月妇女 | 增减百分比 | |
| 血容量(ml) | 3930 | 5332 | +35% |
| 血浆容量(ml) | 2580 | 3655 | +42% |
| 红细胞容量(ml) | 1353 | 1677 | +24% |
| 红细胞 | 4.2×1012/L | 3.6×1012/L | -12% |
| 白细胞 | 7.0×109/L | 10.0×109/L | +43% |
| 血清蛋白(g/L) | 77 | 69 | -10% |
| 血清白蛋白(g/L) | 44 | 35 | -20% |
| 血清钠(mmol/L) | 139.5 | 138 | |
| 血清钾(mmol/L) | 4.3 | 4.2 | |
| 血清氯(mmol/L) | 103.5 | 103 |
近年来随着对血浆蛋白质的深入研究,尤其是应用免疫电泳法及色谱法,发现孕妇血浆蛋白质成分主要改变如下:
(一)血清蛋白质成分的主要变化
⒈血清蛋白 妊娠期血清总蛋白水平逐步降低,至妊娠晚期达最低,较非孕期降低约13%。其中血清白蛋白轻度降低,α、β球蛋白升高,γ球蛋白下降。白蛋白与球蛋白的比值由非孕时的1.32:1降至妊娠3个月时的1.21:1;足月时降至0.84:1。由于血清总蛋白降低,使血浆胶体渗透压下降,孕妇有形成水肿的倾向。造成其降低的原因主要是孕妇血中氨基酸大量地输给胎儿、胎盘、子宫及乳腺等组织供生长发育使用。其次妊娠期血容量增加,血清蛋白质被稀释也加重血清蛋白质的降低。
⒉凝血因子血浆凝血因子为可溶性蛋白质,妊娠期多种凝血因子含量明显增加,妊娠晚期除XIII、Ⅺ因子外,几乎所有的凝血因子都有不同程度的增加,如表14-8所示。
表14-8 妊娠晚期凝血因子的改变
| 凝血因子 | 非妊娠期 | 妊娠期 |
| 纤维蛋白原 | 2~4.5g/l | 4~6.5g/l |
| 因子Ⅱ | 75%~125% | 100%~125% |
| Ⅴ | 75%~125% | 100%~150% |
| Ⅶ | 75%~125% | 150%~250% |
| Ⅷ | 75%~150% | 200%~500% |
| Ⅸ | 75%~125% | 100%~150% |
| Ⅹ | 75%~125% | 150%~250% |
| Ⅺ | 75%~125% | 50%~100% |
| Ⅻ | 75%~125% | 100%~200% |
| XIII | 75%~125% | 37%~75% |
| AT-Ⅲ | 85%~110% | 75%~100% |
| 抗因子Ⅹa | 85%~110% | 75%~100% |
妊娠期纤维蛋白原的变化最为明显,其含量于妊娠4个月后逐渐增高,足月妊娠时达高峰,由非孕时的平均约2~4g/L增至4~6g/L,增加约50%。增高的纤维蛋白原约有10%用于分娩时消耗。参加外源性凝血系统的Ⅶ因子(即血清凝血酶原转变加速素,SPCA)增加,约为非孕妇女的120%~180%,促使组织凝血活酶素(又叫组织凝血活酶)生成增加,从而加速外源性凝血过程。Ⅷ因子(抗甲种血友病球蛋白,AHG)增加达正常非孕妇女的200%,它使血小板磷脂上的Ⅸa-Ca2+-XIII复合物生成增多,加上Ⅱ、Ⅹ因子增加,内源性凝血过程也加速,致使孕妇处于高凝状态。血液于妊娠期处于高凝状态,是预防分娩期失血的有利因素。
抗凝血因子-抗凝血酶Ⅲ(AT-Ⅲ)在妊娠早期就降低,在妊娠晚期仅轻度降低。
⒊纤溶系统 妊娠期随着纤维蛋白原的增加,纤溶系统中的纤溶酶原与此同时也相应的增加,到孕末期纤溶酶原达正常非孕妇的两倍。但孕妇血管内皮释放的纤溶激活物(纤溶激活酶)却减少,致使无活性的纤溶酶原转变成能降解纤维蛋白的有活性的纤溶酶减少,故实际上纤溶性是下降。此外,孕妇血浆的α2-巨球蛋白增加20%,由于它有强大的抗纤溶活性,所以它的升高加重了纤溶活性的降低。再者,随胎盘的成熟,纤溶抑制物释放入血,阻断尿激酶引起的纤维蛋白降解,更使纤维蛋白降解减少。以上因素都促进了孕妇由于凝血因子增加而造成的高凝状态。妊娠期纤溶活性降低至足月分娩时达高峰。
⒋甲状腺素结果球蛋白(thyroxinebinding globulin,TBG)水平增高,故血浆总T4、T3水平相应增高,游离T4水平不变,T3变化不定。
⒌妊娠期α1-抗胰蛋白酶 水平增高两倍,它的功能是防止母体内的蛋白酶类对胎盘滋养层细胞的消化。
⒍血浆转铁蛋白 随妊娠期进展而呈比例增高,保证了孕妇小肠铁吸收后的转运,到妊娠末期转铁蛋白水平比末期妊娠时增高了70%,为分娩失血作好了充分准备。
⒎β-脂蛋白(LDL)水平增高180% 致使孕妇易发生动脉粥样硬化症及血栓栓塞。
⒏免疫球蛋白 在孕妇血中IgG轻度下降,IgD增高,IgA与IgM水平基本不变。孕妇血中IgG浓度下降与其分子量小(160ku,沉降系数7S),能通过胎盘进入胎血成为胎儿免疫球蛋白有关。而IgM分子量大(1000ku,沉降系数19S),难以通过胎盘,故母体内它的浓度不变。
口服避孕药后引起血浆蛋白质组分改变的报道较多,多数由雌激素成分引起。它包括①血浆白蛋白的降低;②载体蛋白中,类固醇性激素结合球蛋白、皮质素结合球蛋白、甲状腺素结合球蛋白、铜蓝蛋白及转铁蛋白等的增加,因此,可以出现相应的血浆中被上述球蛋白结合的类固醇激素及金属含量的变化;③凝血因子,Ⅱ、Ⅶ、Ⅹ成分及纤维蛋白原的增加,血小板凝集倾向的增加,此外,有报道α1-巨球蛋白的增加,载脂蛋白的增加以及血浆结合球蛋白含量的降低,其重要性及机制尚待进一步研究。
(二)妊娠相关性蛋白质及其临床意义
妊娠期孕妇体内及血液中将会出现一些妊娠异蛋白质,它们是:
⒈妊娠特异性β1-糖蛋白(pregnancy-specific-β1-glycoprotein,PSβ1G)也称为妊娠相关性血浆蛋白(pregnacy-associatedplasma protein C,PAPP-C)它与hCG、hPL一样,不会在非孕妇女血浆中找到,含量随妊娠进展而增加。PSβ1G在胎盘滋养层合体细胞生成,其分子量为29ku,含糖量29%,等电点为4.5。妊娠8-9周时血浆PSβ1G值为2.75±2.0μg/ml,20-21周时为53.0±16.9μg/ml,到40周时猛增到212.4±88.9μg/ml。
β1-糖蛋白(PSβ1G)为妊娠所特有。血浆PSβ1G于妊娠开始第一周即可测出,故也可用于早孕诊断,而尿中则最早于妊娠3周后才能测出,故对早孕诊断尚不及尿中hCG测定。但是PSβ1G在血流中代谢缓慢,每日变动范围小,故在妊娠期能较好地反映胎盘功能,PSβ1G低值提示宫内死胎、分娩低体重儿及妊娠中毒症。因为胎儿生长发育与体重均从属于胎盘,故从PSβ1G水平可间接了解胎儿状况。
⒉妊娠相关性血清蛋白A(pregnancy associated plasma protein A,PAPP-A)胎盘滋养层合体细胞产生的α2球蛋白。体外试验发现它有抑制补体及纤溶酶活性的特性,在滋养层局部保持胎儿免遭免疫排斥的危害,故PAPP-A与免疫和纤溶有关。
⒊妊娠相关性血清蛋白B(pregancy-associated plasma protein B,PAPP-B)为由胎盘产生并分泌到孕妇血内的胎盘蛋白,分子量为100万。孕妇血中PAPP-B低值说明有功能的胎盘组织减少,于胎儿安全不利。
⒋胎盘蛋白5(placental protein 5,PP5)为已鉴定出的14种胎盘蛋白中的重要蛋白质,PP5分子量小,仅约39ku,随妊娠期进展而增高,但其分泌量少,仅用放射免疫法才能发现。孕妇血中PP5异常高值可作为胎盘早剥的预测指征,而绒毛膜上皮细胞癌患者PP5消失,孕妇血浆胎盘蛋白5(PP5)若明显增高可能为葡萄胎。
⒌甲胎蛋白(AFP)甲胎蛋白是一种胎儿的特异性α1球蛋白,主要在胎儿的肝和卵黄囊中合成,此外,胎儿的胃粘膜和胸腺也能产生少量。胎儿产生的AFP经胎盘进入母体,因此在胎儿血、羊水、及母体血中的浓度差别很大。AFP早在胎儿6.5周时就已出现,13周时达高峰(28.0mg/L),21周时开始下降,至40周时达最低值。母体血中的AFP则于妊娠16周时才开始上升,32-34周时达高峰,约450-500μg/L,以后逐渐下降,一般孕妇血AFP含量在100-200μg/L以下。AFP是一种抑制母体免疫反应的物质,妊娠时测定的意义见羊水测定。
⒍妊娠带蛋白(pregnancy-zoneprotein,PZP)此蛋白也见于口服避孕药妇女血浆中,它在体内功能尚不十分确切,体外试验发现PZP与免疫抑制有关。表14-9列举人类胎盘合成的一些具有生物活性的蛋白质。
表14-9 人类胎盘合成的某些蛋白质
| 蛋白激素 | ⒈人绒毛膜促性腺激素(hCG) |
| ⒉人胎盘催乳素(hPL) | |
| ⒊人绒毛膜促甲状腺激素(hCT) | |
| 酶类 | ⒈耐热碱性磷酸酶(HSAP),即胎盘型碱性磷酸酶 |
| ⒉催产素酶,即胱氨酸氨肽酶 | |
| ⒊组氨酸酶,即二胺氧化酶 | |
| 妊娠相关性蛋白质 | ⒈妊娠特异性β1-糖蛋白(SPβ1G) |
| ⒉胎盘蛋白5(PP5) |
四、羊水的检测及其临床意义
妊娠期充满羊膜腔的液体称为羊水,它是孕妇子宫内的重要组成部分。妊娠早期的羊水主要是由母体血浆通过胎膜进入羊膜腔的透析液,为澄清液体,成分与血浆相似。妊娠中期以后,胎儿尿可能成为羊水的重要来源,以后更有胎儿代谢物、分泌物进入,成分逐渐复杂,足月妊娠时羊水呈乳白色、混浊、不透明,可见胎脂、上皮细胞及毳毛等有形物质。由于胎儿尿液混入羊水,羊水逐渐变为低渗(钠浓度低),渗透压由妊娠早期的280mmol/L降为255-260mmol/L,但尿酸、肌酐、尿素浓度却比母体血浆逐渐升高。羊水量在妊娠38周前随胎龄而增加,但个体差异大。妊娠8周时羊水量为5-10ml,20周为200ml,36-38周达高峰,约1000-1500ml,以后逐渐减少。羊水pH值8-9,密度1.006-1.020g/cm2。
妊娠前半期羊水的成分基本上与母体血浆相似,只是蛋白质含量低,甲胎蛋白(AFP)含量高,羊水中几乎没有成形的微粒。妊娠16周以后,由于胎儿吞咽、呼吸及排尿功能的建立,使羊水成分发生了很大变化。羊水中葡萄糖含量较母体血中低,约为2.04-2.79mmol/L,而足月妊娠时葡萄糖含量明显下降,可能与胎盘通透性降低有关。羊水中脂肪含量约为4.5-5.9g/L,其中50%为脂肪酸,磷脂为0.39-0.59mmol/L,胆固醇为0.52-2.50mmol/L。羊水中蛋白质占有机物的50%,平均2.6g/L。妊娠22周时羊水蛋白质为10g/L,随妊娠进展其含量逐渐下降,到36周时为5g/L。羊水蛋白以白蛋白为主。羊水中无机物主要为气体和电解质。当胎尿进入羊水后,羊水逐渐由开始的近似等渗液变为低渗,其中钠、氯浓度下降,钾略上升。此外随妊娠的进展,羊水胆红素浓度下降,至妊娠37周时接近于0,这反映胎儿肝脏的酶系统发育已逐渐完善。
羊水存在整个妊娠过程中,包括分娩时,对胎儿和母体都具有重要保护作用。由于羊水与母体血浆进行着物质交换,所以它与母体、胎儿的关系都很密切。近年来通过穿刺采取羊水标本进行各种检查,了解胎儿性别,胎儿成熟度;从羊水成分的变化窥视胎儿的安全状态与有否先天性缺陷或遗传性疾病。因此羊水是产前诊断的良好材料,羊水检查正在越来越多地应用于临床。下面列举几项羊水检查项目。
(一)甲胎蛋白测定
火箭电泳法测定羊水AFP水平,发现妊娠13-14周达高峰,但正常低于30μg/ml。羊水AFP检测的临床价值是:①用于神经管开放(未闭)的产前诊断,这种先天性遗传病胎儿的甲胎蛋白从胎儿脑脊液经脑脊膜毛细血管向羊水渗入增加,故羊水含量增高可达10倍。②胎儿脐膨出与消化道畸形(开放性腹壁缺损),内脏与羊水接触、羊水的ADP含量增高,故可测AFP作此病的产前诊断。③诊断宫内死胎:死亡胎体渗液进入羊水,加之此时胎盘屏障通透性增高,羊水(还有孕妇血浆)的AFP剧增。④无脑儿、共济失调毛细血管扩张症及胰腺纤维囊性变化的胎儿,羊水AFP也呈高值。⑤葡萄胎中缺乏胎儿组织,故羊水AFP低值为诊断此病指征之一。
(二)羊水卵磷脂/鞘磷脂比值测定
胎儿肺泡表面的脂类活性物质主要为卵磷脂与鞘磷脂,系维持肺稳定性的重要物质,两者均可进入羊水内。正常妊娠34周之前羊水中卵磷脂(lecithin,L)和鞘磷脂(sphin-gomyelin,S)含量基本一致,卵磷脂/鞘磷脂比值(L/S比值)低于2。35周后肺泡上皮Ⅱ型细胞产生的卵磷脂合成迅速加快,鞘磷脂稳定,使L/S比值逐渐等于2或大于2,说明此时胎肺已发育成熟。若此时L/S比值仍低于2则说明胎肺功能发育不健全,娩出的新生儿易发生胎儿呼吸窘迫综合征(respiratory distress syndrome,RDS)。故测定妊娠35周后羊水的L/S比值可预测新生儿及宫内胎儿RDS。
(三)羊水肌酐测定
羊水中的肌酐来自胎尿,因而它的含量能反映胎肾发育成熟度。羊水的尿酸、肌酐、尿素随妊娠进展而增加。尿素由早期妊娠的3.84mmol/L增至足月妊娠的5.01mmol/L。肌酐含量由28周的88.4μmol/L上升至足月妊娠的176.8μmol/L,若肌酐浓度达194.48μmol/L,尿酸浓度达595μmol/L,提示肾脏已发育成熟。此外羊水中的肌酐浓度随孕酮的增高而上升。妊娠36周时的正常值为132.6μmol/L,37周后超过176.8μmol/L,也说明胎肾已发育成熟。不过胎肾发育有时虽不够成熟,但由于孕妇血浆肌酐增加,也会使羊水肌酐呈高值,故在判断时还应考虑到这点。
(四)睾酮测定
妊娠17周时羊水中睾酮达高峰。一般在妊娠中期孕男胎者羊水睾酮正常平均值为224±11pg/ml,孕女胎者羊水睾酮平均值仅39±2pg/ml,两者显著差异,据此可预测胎儿性别。
(五)雌激素测定
羊水中含有多种激素,如皮质醇、雌三醇、孕酮、睾酮、催乳素、绒毛膜促性腺激素及前列腺素等,它们来源于胎儿和胎盘,其含量直接反映胎儿-胎盘复合体功能。此外,羊水中还含有ACTH、FSH、LH及促甲状腺激素等。这些激素的作用可能与发动宫缩有关。
羊水中的雌三醇(E3)与孕妇血中与尿液E3相关性良好,妊娠末其羊水E3正常值为80-120μg/dl,雌四醇(E4)为16-65μg/dl。正常的E3/E4比值等于3,说明胎盘功能良好。若E3低值,此比值缩小提示胎盘功能不良,可能为胎儿呼吸窘迫综合征,宫内生长迟缓。如E3突然下降可能为先兆流产。
(六)乙酰胆碱酯酶测定
乙酰胆碱酯酶(AChE)即真性胆碱酯酶,主要来自胎儿的兴奋性细胞,如嗜铬细胞、神经节细胞、运动细胞、中枢神经系统神经细胞和肌细胞,反映神经系统成熟度。胎儿神经管闭合
■[此处缺少一些内容]■
交,再用放射自显影检查是否有α-珠蛋白基因存在即可。
(八)用限制性内切酶法检测羊水细胞的有关基因
此法比DNA杂交法优越,可直接从20ml羊水细胞中提取DNA,不需培养,且可用于妊娠早期作产前诊断。由于基因突变可以造成某种限制性内切酶切点的丧失或新切点的出现,这样使内切酶切出的DNA片断长度与正常基因的有差别。应用此法已对镰状细胞贫血作出确切的产前诊断。其方法是人类正常的珠蛋白DNA用限制性内切酶HpaⅠ来切割时,切点在距β-珠蛋白基因3′-末端5000个核苷酸处,切下的β基因包含在76000个碱基对(7.7kb)的DNA片段中。镰状红细胞血红蛋白S基因是β链第6位密码子密码突变,这一改变使得HpaⅠ切点发生变化,因而切割出的变异β基因存在于13000碱基对(13.0kb)片段中。这样通过用HpaⅠ切点的改变,观察能否在羊水细胞DNA切下13.0kb片段,即能诊断胎儿是否为镰状细胞贫血患者。此法还能预测α-珠蛋白生成障碍性贫血,方法是用限制性内切EcoRI或(BgⅡ)消化羊水细胞后,正常胎儿α肽链基因有23kb DNA片段,β肽链基因有5.5kb的DNA片段。而α珠蛋白生成障碍性贫血胎儿只有β链的基因而无α-肽链基因,故可准确地筛选出患此病的胎儿。
羊水生物化学检测远不止于这些,目前用它作氨基酸分析可检出精氨酰代琥珀酸尿症、胱氨酸尿症及遗传性烟酸缺乏症等遗传性代谢病。检测羊水中的粘多糖还可诊断粘多糖沉积病,测定羊水内的酶类活性能确诊先天性酶缺陷。由此可见羊水生化检查在产前诊断中具有重要的地位。
总之,妊娠时监测各项与胎盘功能、胎儿-胎盘复合体功能有关的生物化学指标,对于维持孕妇及胎儿的健康与安全,开展遗传优生咨询,搞好计划生育都具有非常实用的临床价值。表14-10列举了一些临床上常用的监护孕妇与胎儿安全的生化检查项目,实践中可酌情选择应用。
表14-10 常用的胎儿、胎盘功能及产前诊断的生物化学检查项目
| 胎盘功能 | ⒈激素①血、尿中绒毛膜促性腺激素(hCG) |
| ②血、尿中孕激素(血孕酮及雌二醇) | |
| ③血中胎盘催乳素(hPL) | |
| ⒉血清酶 ①催产素酶 | |
| ②耐热碱性磷酸酶(胎盘型碱性磷酸酶) | |
| ③组氨酸酶(二胺氧化酶) | |
| ⒊胎盘蛋白(妊娠特异β1-糖蛋白即PSβ1G、胎盘蛋白5即PP5) | |
| 胎儿-胎盘 | ⒈血尿及羊水中雌三醇(E3) |
| 复合体功能 | ⒉尿雌激素/17-酮类固醇(E/17-KS) |
| ⒊硫酸去氢表雄酮负荷试验 | |
| ⒋血、尿中15α-羟雌三醇(雌四醇即E4) | |
| ⒌尿雌激素/肌酐比值(E/C) | |
| ⒍羊水甲胎蛋白(AFP) | |
| ⒎羊水卵磷脂/鞘磷脂比值(L/S) | |
| ⒏羊水胆碱酯酶活性 |
第十五章 遗传性疾病的生物化学与分子生物学诊断
第一节 概述
生物是多样的,人类也不例外。除一卵双生外,不可能存在二个遗传完全相同的个体。在分子水平分析这些遗传学上决定性差异的性质和效应是人类生化遗传学的主要内容。
遗传的基本生物学单位是基因。基因的功能有遗传与变异两个方面,复制、转录、修复体现了遗传特性,即保守性;基因突变和重排则体现了变异。变异的积累引起生物的多态性,生物得以发展和进化。但变异也有许多情况给人类带来负面影响,产生疾病。这些变异通过遗传保守性传给下一代,造成遗传性疾病的发生。
遗传性疾病的发生是遗传物质变异所致,但发病是通过遗传物质的表达物质蛋白质(酶)表现所致。
1949年,Pauling等发现镰状细胞贫血患者的血红蛋白与正常血红蛋白的电泳迁移速度不同,他们认为这是由于两种血红蛋白分子间存在化学差异所致,首次提出了“分子病”的概念。分子病是指蛋白质分子合成异常引起的疾病,但是蛋白质分子的合成是由基因控制的,是由DNA分子上的碱基序列决定的。如果DNA分子的碱基序列或调控元件发生改变,那么由它所控制合成的蛋白质分子的结构或合成数量也就发生相应的变化,由此可能引起一系列的病理生理变化,导致疾病。这种疾病是由于核酸分子结构是异常导致蛋白质分子结构异常,核酸调控表达异常导致蛋白质分子合成数量异常,故称分子病。遗传性疾病中由于人体内某种酶的遗传缺陷而产生的疾病,一般称为先天性代谢缺陷。但酶就是蛋白质分子,因此先天性代谢缺陷和分子病严格来说很难区分,只是不同年代用不同研究方法提出的概念而已。目前一般来说酶分子异常导致的疾病称先天性代谢缺陷,结构蛋白缺陷称分子病。但其本质都是核苷酸分子的异常。迄今发现有上千种,如血红蛋白病、血浆蛋白病。由于酶合成异常所致的先天性代谢缺陷病有:酶缺陷而导致的中间产物积累的半乳糖血症;导致底物积累的糖原贮积症I型;导致代谢终产物缺乏的白化症;导致旁路代谢加强副产物积累的苯丙酮尿症和酶活性升高导致产物生成增多的痛风症等。其中苯丙酮尿症和血红蛋白病是研究较多、较深入的疾病。(表15-1)
表15-1 遗传性疾病举例
| 蛋白质变异(分子病) | 病症 |
| 血红蛋白 | 镰状红细胞病 |
| 珠蛋白 | 珠蛋白生成障碍性贫血 |
| 酶变异(先天性代谢缺陷) | |
| 已糖激酶 | 已糠激酶缺乏型溶血性贫血 |
| 半乳糖-1-磷酸尿苷转移酶 | 半乳糖血症 |
| 酪氨酸酶 | 白化病 |
| 磷酸化酶激酶 | 糖原贮积症 |
| β-葡萄苷酸酶 | β-葡糖苷酸酶缺乏症 |
| 苯丙氨酸羟化酶 | 苯丙酮尿症 |
| 黄嘌呤氧化酶 | 黄嘌呤尿症(或痛风) |
第二节 常见遗传性疾病的发生与基因诊断
一、血红蛋白分子病
血红蛋白由四种珠蛋白肽链组成,它们分别是α、β、δ和γ肽链,由它们不同的组合组成各种血红蛋白。除α肽链的基因位于16号染色体外,其余β、δ、γ肽链的基因连锁在11号染色体。血红蛋白异常导致的分子病就是珠蛋白结构基因的DNA分子结构异常所致。
⒈点突变 DNA分子的碱基转换或颠换造成单个碱基替代,导致三联体遗传密码改变,使得相应表达翻译的肽链中氨基酸发生改变而导致蛋白质结构改变,最终引起该蛋白的生理功能发生改变。如β链第6位应是谷氨酸,其对应的碱基密码是GAA,当颠换成GUA时,表达的氨基酸改为缬氨酸,当转换成AAA时,表达的氨基酸改为赖氨酸。在目前发现的血红蛋白异常中,绝大多数属于这种类型。
⒉终止密码子突变 mRNA翻译蛋白质的终止密码有UAA、UAG、UGA。当终止密码子发生点突变,如UAA转换为CAA时,终止密码翻译为谷胺酰胺,肽链无法终止导致肽链延长。相反,肽链中途某一密码突变终止密码时,肽链就提前终止变短。如在中国人中发现有β珠蛋白基因突变,使转录mRNA密码子17由AAG→UAG,使翻译的β珠蛋白过早终止,造成β链过短,而无正常功能,导致珠蛋白生成障碍性贫血发生。
⒊移码突变 由于DNA分子三联体密码子之间是无标点符号的,因此当DNA的碱基序列中缺失或插入一个核苷酸,就相当于多了或少了一个碱基,使得在突变位置以后的三联体密码子均依次发生变化,包括终止密码。所以移码突变可以是肽链的氨基酸发生改变,也可是肽链长短发生改变。而且这种突变位置越靠近翻译起始端(5′端)其后果越严重。如在中国人中发现有β珠蛋白基因转录的密码子41-42产生缺失,造成β链合成提前终止,这种β链不稳定而导致β珠蛋白生成障碍性贫血发生。
突变类型检测可用核酸测序,人工合成寡核苷酸探针作产前诊断。前者多用于研究,后者可应用于临床。现可利用PCR法扩增突变区域,再行测序或杂交分析,大大提高了灵敏度和特异性,可省去目的基因的克隆,方法也得到一定的简化。
⒋密码子缺失和插入它与移码突变不同的是生殖细胞在减数分裂时,染色单体发生错配或不等交换,造成在基因DNA分子中,缺失或插入的核苷酸不是一个,而是一部分,即多了或少了一部分密码子。当然其肽链也相应缺失或插入一个以上的氨基酸。如珠蛋白生成障碍性贫血发现二种类型,一种为右侧缺失型,缺失了涉及α2和α1基因3.7kb的片段,而左侧缺失型则缺失了α2及左侧区域4.2kb的片段。(α2基因排列在左,α1基因排列在右)。该类变异可用Southern blot法,RFLP法加以诊断,参见诊断分子生物学基本技术有关章节。
⒌融合基因由于减数分裂时不同珠蛋白肽链的基因之间发生不等交换,结果造成某一珠蛋白基因同时融合两种不同珠蛋白基因的部分碱基,由此合成的肽链也含有两种不同珠蛋白的氨基酸序列。
血红蛋白病的诊断可根据临床症状,如溶血性贫血等现象。有些没有临床症状,须依赖实验室诊断。在细胞水平可以通过血液学检查,蛋白分子水平也可通过电泳,热稳定试验等方法检查。
血红蛋白病的治疗目前尚无根治办法。该病的起因是基因突变所致,其治疗有待于基因工程技术来矫正基因的缺陷,即用基因治疗的方法加以根治。但要进行基因治疗,必须建立完善基因诊断的技术,这是医学检验工作者面临的新课题。目前基因诊断技术可利用核酸探针进行分子杂交或进行核酸测定来分析基因突变和突变的位置。由于mRNA分子直接转录了DNA分子上基因的信息,又直接指导蛋白质合成,在细胞内又有一定的量,故易于分离纯化和分析,因此对内源性基因的分析,其分析对象多为mRNA分子。mRNA分析最常使用的分子杂交技术有斑点杂交和Northern blot技术以及逆转录PCR(RT-PCR)技术。
二、先天性代谢缺陷病
(一)苯丙酮尿症
苯丙酮尿症(PKU)因患者尿中含大量苯丙酮酸而得名病因是患者肝缺乏苯丙氨酸羟化酶,使由食物摄入体内的苯丙酮酸不能正常代谢为酪氨酸,导致血清中苯丙氨酸浓度升高,可高达50-100mg/dl(正常参考值为1-3mg/dl)。大量的苯丙氨酸使旁路代谢活跃,经苯丙氨酸转氨酶作用生成苯丙酮酸。
苯丙氨酸羟化酶是在肝细胞中合成的,即在肝细胞中专一表达而在胎儿的绒毛细胞或羊水细胞中并不表达,给PKU的产前诊断带来了困难。
由于诊断分子生物学技术的发展,1983年胡流清等人完成了人苯丙氨酸羟化酶cDAN探针的制备。他们利用探针和Southern blot技术进行分子杂交,通过限制性片段长度多态性(restriction fragment-length polymerphism ,RFLP)分析实现了PKU的产前基因诊断。他们先用MSP-I限制性内切酶消化正常人基因组DNA,经核酸电泳后,将DNA片段转移到一张硝酸纤维膜(或尼龙膜)上,再用32p标记的人苯丙氨酸羟化酶cDNA探针杂交后,再经放射自显影技术显示正常人群有23kb和19kb两种限制性片段长度。同时他们调查了一个PKU家系,进行比较发现,同样用Msp-I内切酶消化,父母同时有23kb和19kb DNA,患儿只有19kb片段。说明在该家系中,苯丙氨酸羟化酶突变基因是与19kb片段连锁的。连锁简而言之就是指同一条染色体上的基因联合遗传的现象。因此在该家系中的第2胎,如果只出现19kb DNA片段将是患儿,而同时出现23kb和19kb DNA片段或只出现23kbDNA片段的都是正常个体,而且只出现23kb DNA片段者不携带突变基因,不是突变基因的携带者。正常人出现19kb仅代表蛋白质和酶的多态。PKU家系的患儿为19kb,正常儿为23kb说明该PKU家系的基因突变与19kb连锁。(表15-2)
表15-2 人苯丙氨酸羧化酶基因核酸电泳与cDNA探针杂交自显影
| 正常人多态 | PKU家系 | |||||
| 父 | 母 | 患儿 | 正常胚胎 | |||
| 23kb | - | - | - | - | - | |
| 19kb | - | - | - | - | ||
1986年胡流清等进一步证明苯丙氨酸羧化酶的表达异常是基因点突变所致,并确定了突变点位置,并由此设计了正常和突变的各含21个核苷酸的寡核苷酸探针。
↓
正常探针5′TCCATTAACAGTAAGTAATTT3′
突变探针5′TCCATTAACAATAAGTAATTTT3′
↑
用这两个探针分别与PKU家系成员的DNA杂交,证实与正常探针杂交者为正常个体,与突变探针杂交者为患者,同时可与两个探针杂交者为正常个体,但携带突变基因。此法已成功用于产前诊断。(图15-1)
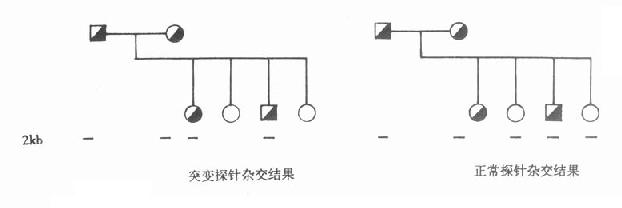
图15-1 正常与突变苯丙氨酸羟化酶探针与PKU家系成员的DNA杂交结果
杂交技术由于费时、操作复杂而难以在基层实验室推广。现在多用聚酶链反应(PCR)扩增技术进行基因诊断,或在PCR基础上再进行探针杂交,在提高灵敏度的同时又提高特异性,而且快速、经济、简便实用。
(二)嘌呤代谢紊乱与痛风
尿酸是嘌呤核苷酸分解代谢的重要产物,血和尿中尿酸浓度的检测是嘌呤代谢紊乱的重要化学指标。可由经典的磷钨酸还原法和高度特异的尿酸酶等化学方法检测。
健康成年男子每天约生成尿酸600-700mg,其中60%-70%经肾排出,剩余约200mg排入肠道由细菌降解。体内尿酸池维持在1200mg尿酸水平。肾排出尿酸的机制是由于尿酸分子量小(168u),可全部经肾小球滤过,但至少有98%被近曲小管重吸收,再经远曲小管主动分泌,因此随尿排出的尿酸主要由肾小管所分泌。肾每天约排出尿酸400-500mg(2.4-3.0mmol/L),相当于肾小球原滤液中含尿酸的4%-5%。
在血浆pH为7.4时,尿酸几乎完全以单钠尿酸盐的形式存在,溶解度有限,约为0.42mmol/L(7.0mg/dl);而尿酸的溶解度更低,在pH5的尿液中,比尿酸难溶20倍。健康成年男子的血清尿酸浓度约0.3mmol/L,女子约低20%。血清尿酸水平随年龄增加而增高,男性比女性更明显,可达0.46mmol/L。血清尿酸超过0.42mmol/L为高尿酸血症(hyperuricemia)。在血清尿酸浓度超过尿酸盐的溶解度时,就有可能尿酸钠的针状结晶沉淀于关节、肌腱、韧带、肾锥体的间质组织等软组织。足够数量的尿酸盐结晶可引起急性炎症反应,如沉积于关节腔,形成急性关节炎。这是由于尿酸盐结晶被白细胞吞噬后,破坏溶酶体膜,使膜内酶释放损伤白细胞和周围组织,引起关节炎症状。表现为关节剧烈疼痛,反复发作,此即痛风(gout)。沉积于软组织的结晶称为痛风石,周围发生炎症反应则构成痛风结节。原发痛风可由于遗传所致的核苷酸代谢中某一种酶表达异常所致,继发性痛风则继发于多种疾病。继发性病因可能有大量服用葡萄糖、果糖与甘露糖,使体内嘌呤合成增加;或多发性骨髓瘤、红细胞增多症、恶性贫血、牛皮癣和广泛转移的恶性肿瘤,使核蛋白转换率增快,而尿酸生成过多;或细胞毒药物或放射治疗时,核酸分解亢进使尿酸盐进一步增高。
先天性遗传原发通风病因是次黄嘌呤-鸟嘌呤磷酸核糖转移酶(HGPRT)缺失所致,部分缺失时,临床表现为尿酸过多的痛风特征;当完全缺失时,表现为高尿酸血症、精神发育迟缓等特征的自毁容貌综合征。
HGPRT蛋白质分子为相同亚基的四聚体,每个亚基由217个氨基酸残基组成。HG-PRT基因定位于X染色体长臂远端,因此该病表现为X连锁。HGPRT基因长约34kb,而成熟的mRNA的长度只有1.6kb,可见DNA分子内含有大量的内含子。HGPRT的突变基因及表达产物(mRNA,蛋白质)已被分离、研究,和血红蛋白一样有许多突变类型,如有的109位丝氨酸被亮氨酸代替,有的103位丝氨酸被精氨酸代替,还有报告基因发生缺失、重排的严重发病患者。1983年Wilson等发现一例DNA上的TagI限制性内切酶的切点发生突变,使在正常情况下可切出2.0kb片段的电泳区带,由于切点突变而失去酶切作用,变成4.0kb的区带出现。在蛋白质水平进一步研究发现50位的精氨酸被甘氨酸取代。
三、诊断分子生物学在生化遗传学实验室的发展前景
在我国医学检验领域,60年代蛋白质研究的兴起,70年代酶学研究的形成,80年代单克隆抗体的开发为临床生化检验建立灵敏、特异的检测方法奠定了基础。许多检验指标在临床诊断中起着非常重要的作用。
遗传性疾病的发病基础是核酸分子结构变异与核酸的表达产物蛋白质、酶分子结构改变所致。前者可通过染色体形态观察加以分析,后者可通过电泳方法或酶活性测定加以分析。这些方法与免疫方法结合,使检测方法特异性更好,灵敏度更高。这些已建立的方法在遗传性疾病的诊断,蛋白质、酶的多态分析中起重要作用,目前仍为实验室所广泛采用。核酸研究的发展与诊断分子生物学的形成,使生化遗传学研究进入到核酸分子水平,给先天性遗传病治疗带来了希望。
90年代随我国对外开放力度不断加大,对外交流日益加强,诊断分子生物学在我国形成,发展异常迅速,不断地向临床各学科各领域渗透。过去认为遗传性疾病无法根治,但基因研究在临床研究与应用的迅速发展,基因治疗的初步形成,已有治疗成功的先例。基因治疗的发展必然给我们基因诊断提出新的课题、新的要求。诊断分子生物学的发展速度,基因诊断的水平必将影响基因治疗在临床开展的进程。
近年来,分子生物学的新技术、研究成果和学科生长点频频出现,不仅仅在解决人类的疾病诊断、治疗方面成绩显著,在解决粮食、环境等棘手问题也带来新的希望。分子生物学研究领域很多,简而言之主要涉及两大方面内容:基因结构与基因调控。基因结构研究就是揭示人基因组的序列分析,该项工作耗资巨大,取得很多成果,但人基因组非常复杂,有待新的快速测定方法开发,加快研究速度。基因调控研究更为复杂,参与基因调控的元件、因子之多,令人眼花缭乱。分子生物学技术及研究手段也有多种多样,但目前在临床应用得较多的主要有杂交技术和扩增技术。
第三节 基因治疗
80年代初美国加州大学Cline首次对β-蛛蛋白生成障碍性贫血患者进行基因治疗尝试时遭到人们的非议,但时隔10年,分子生物学飞速发展,改变了人们的思维。1990年9月14日,在马里兰州贝塞斯达国家保健研究所医疗中心,一位因患遗传性腺苷脱氨酶(adenosine deaminase,ADA)缺乏症导致抵抗力下降的4岁女孩,接受了W、费仁兹、安德烈等医生用经处理的腺病毒为载体,将正常ADA基因导入患儿的白细胞中,并表达了正常的ADA,成为第一个接受基因治疗并获成功的患者。此后两年中,她仅患过一次感冒。
近来基因研究不断深入,基因治疗的概念也有所发展,不仅可导入正常基因以纠正遗传性疾病的基因缺陷,也可导入特定的DNA、RNA片段以封闭或抑制特定基因的表达,即反义技术治疗肿瘤或病毒感染等疾病。
实施基因治疗首先应具备以下条件:了解疾病发生的分子机制,诊断分子生物学技术的正确应用与基因诊断临床应用,在此基础上分离、克隆正常基因或有治疗作用的核酸片段。选择高效的载体系统与目的基因重组。选择适当的受体细胞,接受正常基因并回输到患者体内适度表达,发挥特定的治疗作用。
反义技术基因治疗是利用反义核酸细胞作用,导入细胞内抑制或封闭基因表达的技术。反义核酸是指与异常表达或过度表达的目的基因或目的基因mRNA互补,并以碱基配对的方式与目的基因序列结合的核酸,它可以在复制、转录核酸剪接加工,mRNA转运及翻译水平上抑制目的基因的表达,达到治疗的目的。
反义核酸用于基因治疗发展较快,如反义核酸最佳作用靶点的选择,采用化学修饰方法提高反义核酸在细胞内的稳定性,常用硫(S)修饰,形成硫代反义核酸,为了便于反义核酸导入细胞,反义核酸常只有10-30bp大小。有时还用脂质体或微囊包装反义核酸,或以逆转录病毒作为载体等,提高反义核酸的稳定性和导入效率,从而提高治疗效果。
虽然基因治疗已部分应用于临床并获得成功,但尚属研究阶段,导入基因的稳定性,表达时效等问题尚待解决。特别是外来基因导入引起的伦理讨论尚无定论,在此仅做简述,但我们深信基因治疗在遗传性疾病,肿瘤等疾病的治疗上,将产生深远的意义。基因治疗的迅速发展给基因诊断提出了新的课题、新的要求,我们必须努力开发简便易行的诊断分子生物学技术,以满足发展的需要。
第十六章 常用分析技术在临床生物化学中的应用
第一节 光谱分析技术的应用
一、光谱分析技术的分类
利用各种化学物质(包括原子、基团、分子及高分子化合物)所具有的发射、吸收或散射光谱系的特征,来确定其性质、结构或含量的技术,称为光谱分析技术。根据光谱谱系的特征不同,可把光谱分析技术分为发射光谱分析、吸收光谱分析和散射光谱分析三大类。
在临床生化检验中,光谱分析是一类十分重要的技术,应用极为广泛。应用吸收光谱原理进行分析的主要有可见光及紫外光分光亮度法,以及原子吸收分光亮度法。红外分光亮度法虽然也属于吸收光谱法,但在临床生化上应用不多。应用发射光谱原理进行分析的主要有荧光分析法和火焰亮度法。应用散射光谱原理进行分析的主要是比浊法,包括免疫比浊法等。
二、光谱分析的常用方法
(一)吸收光谱分析法
1、可见光及紫外光分光亮度法
(1)标准曲线法:将一系列浓度不同的标准溶液按照一定操作过程显色后,分别测吸亮度,以吸亮度为纵坐标,浓度为横坐标绘制标准曲线。在相同条件下处理待测物质并测定其吸亮度,即可从标准曲线找出相对应的浓度。
(2)对比法:将标准样品与待测样品在相同条件下显色并测定各自的吸亮度。由于测定体系温度、厚度以及入射光波长是一致的,所以标准与待测样品K值及L相等,可应用下式比较计算待测样品浓度:Cx=Cs×As/Ax。
(3)差示法:有色溶液浓度太浓或太稀(透光度超过90%-10%)时,测定结果会产生较大误差,此时可采用差示法(differentialspectrophotometry)。①高浓度样液差示法:用标准品制备浓度稍低于试样的参比溶液,先将仪器光门关闭,调节T%=0,再将参比溶液置于光路上,打开光门使T%=100,然后测定样品溶液的透光率即可。例如某一样品溶液原来的透光率读数为5%(10%以下),用差示法后读数为50%,这实质是把透光率标尺扩展了10倍,从而减少了测量误差。②低浓度样液差示法:用标准品制备浓度稍高于试样的参比溶液,将它放在光路上,打开光门,调节透光率至0%,然后换以空白溶剂,调节刻度至100%。此后测定样品的读数即可。
(4)多组分混合物的测定:当试样中有两种或两种以上的组分共存,可根据各组分的吸收光谱的重叠程度,选用不同的定量方法。如果混合物各组分的吸收峰互不干扰,这时可按单组分的测定方法,选择测定波长,分别测定各组分的含量;若各组分的吸收峰相互重叠,可采用解联立方程法、等吸收点法、双波长法等解决量中的干扰问题。
(5)利用摩尔吸光系数进行检测:①吸光系数:Beer定律的数学表达式为A=kbc,若溶液的浓度c以g/L为单位,b为光径以cm为单位,则常数K称为吸光系数,以a表示,其单位为升/(克·厘米)[L/(g·cm],A=kbc可写成A=abc。②摩尔吸光系数:公式A=kbc中的以为1mol/L,b为1cm时,则系数k称为摩尔吸光系数,以ε表示,单位为升/(摩尔·厘米)[L/(mol·cm)],A=kbc可写成A=εc。在实际工作中,不能直接用1mol/L这种高浓度的溶液测定吸光度,而是在稀释成适当浓度时测定吸光度进行运算。ε值与入射光波长、溶液的性质等因素有关。如NADH在260nm时ε为15000,写成ε260NADH=15×103;在340nm时ε为6220,写成ε340NADH=6.22×103。③比吸光系数:如公式A=kbc中的c是百分浓度(w/v)b为cm,则常数k可用E%表示,称为比吸光系数或百分吸光系数,A=kbc可写成A=E%bc。当待测物的化学结构是已知者可用ε值分析,若所测物的化学结构是未知的,则ε无法确定,此时用比吸光系数分析就很方便。a、ε和E常用作粗定量分析,主要用于定性分析。
近年来由于新的灵敏度高、选择性好的显色剂和掩蔽剂不断出现,一般不经分离过程即可直接比色测定。几乎所有的无机离子和有机物都可直接或间接用可见光及紫外光分光光度法进行定量测定。
2、原子吸收分光光度法 原子吸收光光度法是基于元素所产生的原子蒸气中,待测元素的基态原子,对所发射的特征谱线的吸收作用进行定量分析的一种技术。具有灵敏度高、选择性好、操作简便、分析速度快等优点,对大部分元素,其灵敏度约为10-8~10-10g/ml,是微量元素检测的一个十分有铲的方法之一。
(1)分析条件的选择
1)分析线:原子吸收法中常选择待测元素的共振作分析线,但并不是任何情况下都应使用共振线,例如As、Se、Hg的共振线在远紫外区,该区域火焰吸收强烈,不宜选用共振线作分析线。在选择分析线时,首先扫描空心阴极灯的发射光谱,然后喷入试样溶液,观察谱线的吸收和干扰情况,一般选用不受干扰且吸收最强的谱线作为分析线。常用元素分析线见表16-1。
表16-1 常用元素分析线
| 元素 | 分析线 | 元素 | 分析线 | 元素 | 分析线 |
| Al | 3093.3082 | Ca | 4227.2309 | Cd | 2288.3261 |
| Cu | 3248.3274 | Fe | 2483.3523 | Hg | 2537 |
| K | 7665.7699 | Mg | 2852.2796 | Na | 5890.3303 |
| Pb | 2167.2833 | Sn | 2246.2796 | Zm | 21393.3076 |
2)狭缝宽度的选择:适宜狭缝宽度可由实验确定:将试样喷入火焰,调节狭缝宽度,测定不同狭缝宽度时的吸光度,达到一定宽度后,吸光度趋于稳定,进一步增加狭缝宽度,当其他谱线或非吸收透过狭缝时,吸光度立即减小。不引起吸光度减小的最大狭缝宽度,就是最适宜的狭缝宽度。
3)原子化条件的选择:对于火焰原子化法,火焰的种类和燃助比的选择是很重要的。当燃气和助燃气选择好后,可通过下述方法选择燃助比:固定助燃气流量,改变燃气流量,测量标准溶液在不同燃助比时的吸光度,绘制吸光度-燃助比关系曲线,以确定最佳燃助比。
对于石墨炉原子化器的使用。应注意干燥是一个低温去溶剂的过程,可在稍低于溶剂沸点的温度下进行。灰化是为了破坏和去除试样基体,故在保证试样无明显损失的前提下,将试样加热到尽可能高的温度。原子化阶段应选择最大吸收信号的最低温度。总之,根据试样的性质确定各阶段所选定的温度与加热时间。
4)试样量的选择:火焰原子化法在一定范围内,喷入的试样量增加,原子吸光度增大,但在超过一定量后,由于试样不能完全有效地原子化及试液的冷却效应会使吸光度不再增大甚至有所下降。因此在保持一定的火焰条件下,测定吸光度随喷入试样量的增加达到最大吸光度时的喷雾量,就是适宜的试样量。石墨炉原子化一般固体取样0.1~10mg,液体取样量为1~50μl,主要依石墨管容器的大小而定。
(2)定量方法:常用的定量方法有标准曲线法、标准加入法和内标法。
1)标准曲线法:同紫外可见标准曲线法。但由于燃气流量和喷雾效率的变化,单色器波长的漂移等因素可导致样品测试条件与标准曲线测定条件不同,所以,在测定未知样品时,应随时对标准曲线进行检查,每次实验都要重新制作标准曲线。
2)标准加入法:在标准曲线法中,一般情况下要求标准溶液和未知溶液的组成保持一致。但在实际工作中不是总能做到的,采用标准加入法可以克服这个缺点。本法把未知试样溶液分成体积相同的若干份,留其中一份,其余分别加入不同量的标准样品,然后测定各溶液的吸光度,以吸光度为纵坐标,标准样品加入量为横坐标绘制标准曲线(图16-1)。由于未知样品中含待测组分,故直线不通过原点而是在吸光度高于原点的A处与纵轴相交,且直线外推法使工作曲线延长交横轴处于B点,则OB所对应的浓度Cx就是未知试样中待测组分浓度。
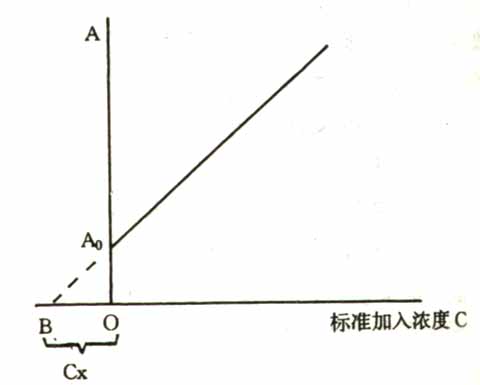
图16-1 标准加入法工作曲线
标准加入法的优点是能够更好地消除样品中其它成分对测定的影响。
3)内标法:在系列标准样品和未知样品中加入一定量试样中不存在的元素(内标元素),然后测得As和Ax,以As/Ax比值对标准样品中待测元素浓度C绘制标准曲线,再根据未加内标元素的试样A与加入内标元素的试样As比值(A/As)即可在标准曲线上求得试样中待测元素浓度。
本法要求内标元素应与待测元素有相近的物理和化学性质。此外,内标法只适用于双通道型原子吸收分光光度计。
(二)发射光谱分析法
1、荧光分析法 许多物质都是光致发光的,即它们可以吸收电磁辐射,然后又重新发射出相同或较长波长的辐射。光致发光最常见的类型是荧光(fluorescence)和磷光(phosphorescence)。利用荧光强度进行分析的方法,称为荧光法(fluorimetry)。在荧光分析中,待测物质分子成为激发态时所吸收的光称为激发光,处于激发态的分子回到基态时所产生的荧光称发射光。荧光法测定的是受光激发后所发射的荧光的强弱,而不是测定激发光的强弱。凡能产生荧光的化合的,均可采用荧光分析法进行定性或定量。
(1)荧光强度的影响因素
1)溶剂:增大溶剂的极性,将使π-π※跃迁的能量降低,荧光增强。在水、乙醇、环已烷等这些常用溶剂中常含有荧光杂质,影响测定,必须在使用前作净化处理。
2)荧光物质的浓度:对于某一荧光物质的稀溶液,在一定频率和一定强度的放射光Ⅰ。照射下,如果光被吸收的百分率不太大,且溶液的浓度很小,当溶液的厚度不变时,则它所发生的荧光强度F和该溶液的浓度C成正比,即F=φⅠ。ECL式中φ为荧效率。当荧光物质浓度高时,会发生分子间碰撞,使荧光效率降低。因为温度增高后使分子间碰撞次数增加,消耗分子的内部能量。
3)温度:大多数情况随温度升高时,荧光效率降低。因为温度增高后使分子间碰撞次数增加,消耗分子的内部能量。
4)溶液的PH值:当荧光物质本身为弱酸或弱碱时,溶液的pH值改变对溶液荧光强度产生影响较大,因为有些物质在离子状态时无荧光,而有些则相反,也有二者均有荧光,但荧光光谱有所不同。
(2)荧光定量分析法:荧光定量分析法通常有标准对比法和标准曲线法。操作和计算可参照比色法,但应注意,空白溶液的荧光强度F。往往不在零点,用上述两种方法时应先测定F,再从标准品Fs和试样Fx中减去F。后进行计算。
多组分混合物在荧光分析也可根据荧光峰相距情况采取不同的方法,如各组分荧光峰相距颇远,可分别在不同波长测定各个组分的荧光强度,然后直接求出各组分浓度。如果各组分荧光光谱相互重叠,利用荧光强度的加和性质,在适宜荧光波长处,测得混合物的荧光强度,再根据被测物质各自在适宜波长处的最大荧光强度,列出联立方程式求算各自的含量。
对较高浓度的荧光物质可用差示荧光法测定。
(3)荧光分析技术的应用:荧光分析法灵敏度高,选择性好、取样量少,因此已广泛应用于各领域,在临床生化检验方面可用于某些无机物与有机物的分析。
无机化合物能直接产生荧光并用于测定的为数不多,但与有机试剂络合后进行荧光分析的元素已达60余种,常见无机物的荧光测定见表16-2。
表16-2 常见无机物的荧光测定
| 元素 | 荧光试剂 | 激发波长(nm) | 荧光波长(nm) | 灵敏度(μg/ml) |
| Ag | 四氯荧光素 | 540 | 580 | 0.1 |
| Al | 桑色素 | 430 | 500 | 0.1 |
| Br | 荧光素 | 440 | 470 | 0.002 |
| Ca | 乙二醛-双-(4-羟苄基腙) | 453 | 523 | 0.0004 |
| Cl | 荧光素+AgNO3 | 254 | 505 | 0.002 |
| CN | 2',7'-双(乙酸基汞)荧光素 | 500 | 650 | 0.1 |
| Fe | 曙红+1,10-二氮杂非 | 540 | 580 | 0.1 |
| Pb | 曙红+1,10-二氮杂非 | 540 | 580 | 0.1 |
| Zn | 8-羟基喹啉 | 365 | 520 | 0.5 |
| F | 石榴茜互R-AI络合物 | 470 | 500 | 0.001 |
某些有机化合物的荧光测定应用较多,如糖类、胺类、甾族化合物、DNA与RNA、酶与辅酶、维生素等。常见有机化合物的荧光测定法见表16-3。
表16-3 常用有机化合物荧光测定法
| 待测物 | 试剂 | 激发波长(nm) | 荧光波长(nm) | 灵敏度(μg/ml) |
| 核酸 | 溴化乙啶 | 360-365 | 580-590 | 0.1 |
| 蛋白质 | 曙红y | 紫外 | 540 | 0.06 |
| 氨基酸 | 氧化酶等 | 315 | 425 | 0.01 |
| 肾上腺素 | 乙二胺 | 420 | 525 | 0.001 |
| NAD(P)H | 自身为荧光物质 | 340 | 450 | 10-6mol/L |
| ATP | 已糖激酶、6-磷酸葡萄糖脱氢酶、6-磷酸葡萄糖 | 340 | 450 | 2×10-6mol/L |
| 维生素A | 无水乙醇 | 345 | 490 | 0.001 |
2、火焰光度法火焰光度法是利用火焰中激发态原子回降至基态时发射的光谱强度进行含量分析的方法。它在仪器结构和分析操作上与火焰原子吸收法相似。
在火焰光度法中,试液和助燃气一起进入雾化室,雾化后喷入火焰,雾粒在火焰中蒸发和激发,激发态原子降落到低能态时发生光辐射,经单色器分光后到达检测器,然后由显示系统显示其发射光强度。
试样中的待测元素激发态原子的发射光强度I与该元素浓度C成正比关系,即I=aC。式中a为常数。a与试样的组成、蒸发和激发过程有关。
火焰光度法同样存在各种因素干扰,如供气压力,试样导入量、有机溶剂和无机酸的影响,以及金属元素间的相互作用等,某些干扰因素的消除方法同原子吸收法。对于金属离子间的相互作用可以下述方法予以消除:
(1)阳离子的干扰:第二阳离子的存在可使待测阳离子的电离作用降低而导致以元素形式存在居多,结果发射强度增大,这种现象称为阳离子增强效应。例如测定钙时有钾存在,钾可抑制钙的电离,干扰钙的测定。消除这种干扰的办法是在标准溶液及试样中加入本身易电离的金属如铯和锂。
(2)阴离子干扰:草酸根、磷酸根和硫酸根可与某些阳离子在火焰温度下形成仅能缓慢蒸发的化合物而抑制原子激发,结果导致待测元素发射强度降低。消除这种干扰的办法是用释放剂。释放剂的作用是同干扰阴离子牢固结合,使待测阳离子的激发行为不受干扰,或与待测阳离子形成更稳定而易挥发的配合物。故尽量避免使用磷酸、硫酸、草酸做试剂。
此外,应避免环境污染测试体系。使用的器皿应为塑料制品以防止玻璃器皿中金属溶出干扰测定。
火焰光度法通常采用的定量方法有标准曲线法、标准加入法和内标法。临床检验工作中前两种应用较多,而且测定血液或血清中钠和钾已成常规。应用火焰光度法测定某些元素的波长及检测限见表16-4。
表16-4 火焰光度法测定某些元素的波长与检测限
| 元素 | 测定波长(A) | 检测限(mg/L) | 元素 | 测定波长(A) | 检测限(mg/L) |
| Li | 6708 | 10-4 | Ca | 4227 | 3×10-3 |
| Na | 5890 | 10-4 | Sr | 4607 | 0.02 |
| K | 7665 | 10-3 | Ba | 4934 | 0.2 |
| Rb | 7800 | 0.05 | Mg | 3852 | 0.1 |
| Cs | 8521 | 1.0 |
(三)散射光谱分析法
散射光谱分析法主要测定光线通过溶液混悬颗粒后的光吸收或光散射程度的一类定量方法。测定过程与比色法类同,常用法为比浊法。但颗粒的大小和形状及悬液的稳定性对比浊结果有较大的影响,因此不能完全按比色法的规律进行测定,否则就会引起误差。
1、仪器和测定方法 由于测定仪器和方法的不同,比浊法又可分为散射测浑法(turbidimetry)和浊度测定法(nephelometry)二类。前者利用一般的光电比色计和分光光度计,其原理是利用光线通过混悬溶液时,由于颗粒的散射使通过的光线减弱,根据光线减弱的程度测定溶液中颗粒的浓度,所以,实际上可将散射测浑法看成是比色分析的一种特殊情况。后者则是直接测定混悬溶液中颗粒散射光的强度,由于一般光电比色计中光源和光电管在一直线上,无法测定散射光线的强度,需要特殊的浊度计,如激光比浊仪。测定方法可采用速率法或终点法进行。
速率散射比浊法是一种动力学测定方法,1977年由Seternbery首先用于免疫测定,在一定条件下,抗原和相应的抗体很快结合成抗原抗体免疫复合物颗粒,速率比浊法就是在一定时间内抗原抗体结合过程中,测定二者结合的最大反应速度,即反应达顶峰峰值。终点散射比浊法用于免疫测定时,在一定时间内,通常是抗原抗体反应达到平衡,复合物的浊度不再受时间的影响,但必须在聚合形成絮状沉淀之前进行浊度测定。
2、影响比浊法测定的因素比浊法的突出问题是颗粒的大小对浊度和浊度曲线有较大的影响。因此,在比浊法中必须力求作到:①混悬液中微粒的分散度也就是颗粒的大小应尽可能相同,并易重复。标准管和测定管中颗粒大小应力求一致。②混悬液在一定时间内,至少10分钟内应维持稳定,也就是颗粒应该不易相互聚集,变粗变大。为此,关键在于制备混悬液必须严格控制条件,一般应注意;①沉淀剂或抗体的浓度,一般情况下浊度随其浓度的增高而增大。②混匀方法和速度,一般而言,缓慢加入试剂,逐滴加入,不断摇匀,易产生粗颗粒沉淀;而迅速加入,迅速摇匀,易产生胶态溶液。③温度,温度升高可加快分子间的碰撞,常使某些在室温细小均匀的颗粒易变为粗大的絮状沉淀。④pH溶液,pH可影响沉淀的形成及颗粒的大小,如蛋白质、酶类,在其等电点pH值时最易形成颗粒并沉淀。⑤混悬液的稳定性和测定时间,大多数混悬液随放置时间的延长颗粒变粗而沉淀,吸光度下降,因此应及时比浊。如出现沉淀太快,可以加入保护性胶体,如聚乙烯吡咯烷酮、表面活性剂等,但应注意加入后可能会引起颗粒及光学性质的变化。⑥其它电解质和非电解质存在的干扰。由此可见,比浊法易受外界各种因素的影响,因此在建立一种比浊测定方法时,必须认真探索其反应规律,力求控制各种影响因素以克服比浊法重复性和准确性较差的缺点。
3、比浊法的应用目前在临床生化检验中使用最多的比浊法是免疫比浊法,如免疫球蛋白、载脂蛋白和补体等项目均已大部分用免疫比浊法进行快速定量。
早在1838年Libby等人把比浊法测定就应用到抗原抗体反应上。所谓免疫比浊法是利用抗原和抗体的特异性结合形成复合物,通过测定复合物形成量的多少,对抗原或抗体进行定量的方法。在介质溶液中形成复合物需要一定的条件,包括:①要有特异性抗体。作为组织或体液中蛋白质种类相当多,若要快速特异测定,就需要有单价特异抗体,也就是说,某一种蛋白有其特异抗体才能与该抗原结合,形成免疫复合物,再定量。若抗体不纯或混有另一种或两种少量抗体,这种免疫复合物就不是单一复合物而是大杂烩,结果偏高。②抗原抗体的比例要适当。因免疫复合物的形成有三个阶段,第一阶段是初步形成抗原抗体二元复合物;第二阶段是复合物交联成大的网络状结构;第三阶段是复合物聚合产生絮状沉淀。只有在抗原与抗体等价时即无过剩抗体,此时,复合物的结合和解离处在平衡状态,其混浊程度达高峰,在抗体过量时,随抗原量的增加而复合物的形成也增加,成正比关系,其测定只能在以应曲线的左侧进行。同时,抗原也不能过量,因为抗原过量,则复合物减少,光散射或光吸收就减少,检测结果偏低。③溶液介质适宜,一般要求溶液中有非离子性亲水多聚体促进免疫复合的形成,如聚乙二醇6000等,溶液pH以6.5~8.0之间为宜,离子强度大比小易形成复合物,对离子的种类也有一定的要求。
在免疫比浊过程中,由于抗原与抗体结合有三个阶段,从而导致吸光度与浓度之间不呈线性关系,一般是三次方程曲线关系。如果要将抗原与抗体两个变量之间的变动特征恰当地反映出来,需要经三次方程拟合成近似直线化的曲线方程,再进行运算。方法可采用终点法和速率法,用五个不同浓度进行定标,经三次曲线方程求出一条能反映真实情况的浓度与吸光度的关系曲线方程。作为定量的工作曲线。
三、影响光谱分析的主要因素
光谱分析的基础是被测物质的浓度与吸亮度成正比,但在实际测定中,往往容易发生偏离直线的现象而引入误差,导致误差的主要原因有化学因素、主观因素和光学因素等三类。
(一)化学因素
化学因素是指被测溶液不遵守光吸收定律所引起的误差。主要有以下几种影响。
1、被测物浓度的影响由于有色物质的电离、水解、缔合等原因,当溶液稀释或增浓时,有色物质颜色的深浅并不按比例降低或增高,因而也就不完全符合光吸收定律。
2、溶液pH的影响 有些溶液的颜色对pH值的改变十分敏感,因而溶液pH不同时常会产生误差,如溴甲酚绿法测定血清白蛋白。
3、杂质的影响如被测物溶液中含有杂质而且这种杂质本身是有色的,或能和显色剂生成有色化合物或沉淀,或能与被测物质发生化学反应等,都会影响吸光度的变化。
4、放置时间的影响某些有色物质能迅速生成,但却不太稳定,放置后色泽消退或起变化,而另一些有色物质须经过相当长的时间后,反应才能完全,因此,若在不恰当的时间内进行比色,将会造成相当大的误差。
此外,还有溶剂、温度、溶液体系的均匀性等都会引起测定的误差。
(二)主观因素
由于操作不当,特别是在分离、稀释和加显色剂等过程中,没有遵守一定的条件,也会引入不同程度的误差。因为有些有色物质的生成和其颜色的深浅,往往随加入试剂的量、加入顺序、试剂浓度、反应温度和反应时间等因素而发生改变。
(三)光学因素
光学因素主要是指分析仪器的性能低劣所引起的误差,分析仪器应用最多的是分光光度计,它们的性能好坏直接影响到测定结果的可靠性和精密性。因此对分析仪器均应有质量保证措施,这里主要介绍分光光度的质量保证方法。
1、分光光度计的性能检查
(1)波长校正:使用光电比色计或分光光度计,在更换光源灯、重新安装、搬运或检修后,以及仪器工作不正常时,都要进行波长校正。就是正常工作的仪器,每隔一个月也要检查一次波长,必要时进行校正,这样才能保证波长读数与通过样品的波长符合,保证仪器的最大灵敏度。方法常用谱钕滤光片校正法,适用于721型仪可见光区的波长校正,常以585nm或529nm处的吸收峰或T%为标准。鉴于721型仪器的出射光波长较宽,不易将573nm和585nm的两峰或两谷分开,校正时易产生误差,故推荐用529nm处的峰或谷为标准来进行波长校正。校正时,将仪器按要求预热。要求电源电压稳定。波长度盘置580nm处,T%调至最大,在比色杯处放一白纸条,观察是否有光强均匀、边缘无光晕或杂光的光斑,如不符合要求,可调节灯泡位置使其符合要求,是为波长校正的粗调。再把灵敏度扭置于“1”(最低档),波长度盘对准529nm,电表机械零点为零,在光路空白时调T%为100%T,并反复查零点和100%T稳定情况。将谱钕滤光片插入光路,慢慢旋转波长度盘,找到透光率最低的一点(向左右微旋波长度盘时,该点透光率值均增加),这一点即为波长529nm。检查波长度盘的指示值是不是529nm,如指示值为534nm,此时波长工误差为5nm,超出规定(±1nm)必须进行调整。
调整方法:将波长度盘对准529nm,从光路取出谱钕滤光片,光路空白时调电表指针到100%T,再将谱钕滤光片插入光路。打开仪器左侧小盖板,找到波长校正螺丝(3个中左侧柄长的一个);反时针方向微微调节(负误差时顺时针方向),使电表指针的指示T%为最低。反复检查波长误差情况,直到符合仪器技术指标为止。盖好左侧小盖板,校正结束。
有些高档仪器如岛津UV-260型双光束分光光度计,可使用氢灯或置谱钕滤光片于测定比色槽内,编入滤工扫描程序后,仪器即可自动地在一定波长范围内进行波长校正。高档分光光度仪的光学系统密封在一个单元组件内,若发生故障,波长不准,常须请制造商派专人修理。其它尚有谱线校正法、干涉滤光片校正法和有色溶液校正法等,可参考有关资料。
(2)线性检查:线性检查包括仪器线性及测定方法线性两个方面的检查。线性误差表现为溶液的浓度与吸光度不成线性关系,出现正偏离或负偏离的现象。这种偏离,按比耳定律的现象来自两个方面:一是溶液本身不符合比耳定律,这种现象叫做化学偏离;二是仪器本身各种因素的影响,使吸光度测定值与浓度之间不成线性关系,这种现象叫仪器偏离。此处研究的是后者。仪器偏离的因素很多,如杂光、有限带宽、检测器噪声、环境条件变化、波长的变动、比色杯的误差、辐射光的非平行性、检测器本身的非线性等。
仪器线性检查常用一种在一定波长及一定浓度范围内确知其服从比耳定律的有色物质,配成不同浓度的溶液,来检查仪器本身是否能如实地反映有色物质的浓度变化。这种检查方法与任何被测物质呈色反应等方法学上的问题无关。用测得的吸光度对浓度作图,在理想情况下应是一条直线。常用的方法是用0.8、1.6、2.4、4.0mg/L伊文蓝浓度系列来检查,波长用610nm。
(3)杂光(散光)检查:在吸光度测定中,凡检测器感受到的不需要的辐射都称为杂光。杂光对吸光度测定法的准确性有严重的影响,但却往往被忽视。杂光的来源有:①仪器本身的原因,如单色器的设计、光源的光谱分布、光学原件的老化程度、波带宽度以及仪器内部的反射及散射等;②室内光线过强而漏入仪器,仪器暗室盖不严;③样品本身的原因,如样品有无荧光、样品的散射能力强弱等。
杂光检测方法:①紫外光区的杂光监测可用10±0.1g/L的碘化钠溶液,在240nm处测定的吸光度应大于2.00。此外,也可用12g/L的氯化钾溶液,在220nm处测其透光度,即为杂光量,一般应小于1%T。以上测定均用石英杯,以蒸馏水校零。②在可见光区可使用谱钕滤光片或硫酸镍法检查。先用谱钕滤片校正波长,然后用黑纸片挡住比色杯光路(进口仪器带有比色杯样黑色标柱)之后调整0%T,再用空气做空白调100%T。插入谱钕滤光片,在585nm处测定,其测定值即为杂光水平。
(4)比色杯的质量检查:比色杯一般由玻璃、石英或荧石制成,光径1.0或0.5cm,光线通过时有一部分光为空气与玻璃接触面的反射而损失(4%),另一部分(很少)为玻璃吸收。比色杯的质量除其原料外,再就是玻璃的厚度均匀,上下一致,各杯彼此相配。厂家多以四个一套供应,且杯口上部外面标有箭头,箭头对光源侧使用。尽管如此,在使用前还是应作质量检查。常用伊文蓝法:将一定量的2.0mg/L的伊文蓝溶液注入比色杯中,比色杯内液面应相等。用一个比色杯作标准,用红色滤光片(波长600~610nm),用水校零后将此杯的读数准确调至50%T,接着读取其余杯的透光度。透光度相差在±0.5%T范围内者为合格。
(5)稳定性检查:检查方法:将分光光度计及附件接于0.5kVA以上的可调变压器,先调电压为220V,波长固定在650nm,在光路比色杯装以空白液,调读数为90%T处,再将电压升至230V及降至190V,观察透光度的漂移值。若介于88.5%~91.5%T之间为合格。在电源电压不变的条件下,在3分钟内其读数漂移不应超过标尺上限值的±0.5%。
(6)重复性检查:在波长、工作状态、电源电压、比色杯配套等合格的前提下,进行重复检查。在用交流电源供电时,仪器对一种溶液重复测定的读数值差应小于或等于标尺上限值的1%。试验方法:将分析纯重络酸钾于120~150摄氏度烘干2小时,干燥器中冷却。精称509.1mg于1000ml容量瓶中,以蒸馏水溶解并加至刻度,混匀(此液180mg/L铬)。就用液为30、60、180mg铬/L。取波长440nm,将应用液各浓度管连续测3~5次,各浓度管中最大误差小于1%T为合格。
(7)灵敏度检查:将重铬酸钾液配制成30和32.5mg铬/L及120和122.5mg/L铬的4种应用液(浓度差两组各为2.5mg/L铬)。波长440nm,以水校零点,将上述应用液连续测3次吸光度,两组2.5mg/L铬浓度差的吸光度值差都不小于0.01为合格。
2、分光光度计日常工作状态监测在可见光区范围内,用一种有色溶液检查仪器是否处于较好的工作状态(包括波长、杂光水平等),常用硫酸镍水溶液。该液在400~700nm之间有吸收峰,峰值在510nm。检查时,在400、460、510、550及570nm处测定硫酸镍溶液的透光度值,记录并保存。待一定时间后用该溶液再检测。比较前后两次的数据,即可知仪器的工作状态。
硫酸镍溶液的制备:将硫酸镍溶于0.1%的硫酸中,用量的多少以在510nm测得的透光度达80%T为准,以0.1%硫酸校零点,配好后密封备用。
监测的意义:在400、700nm处的测定可以监测杂光,这些波长处的测定值升高说明杂光增加,其中任一值增加3%就需要进行检修。510nm处的测定值可以监测带宽,这个波长的测定值减小说明带宽加大。光源强度减弱或波长不准可以产生这种结果,如降低2%T,对于带宽为20nm的仪器就需要修理。460与550nm处的读数主要作为监测波长之用,称二次波长校正。这两个波长的读数相互关联,一个升高另一个就降低。如460nm的测定值(透光度)降低,而550nm的测定值升高,说明透过比色杯样品的波长小于波长度盘指示的波长(图16-2左)。反之,如460nm的测定值升高,而550的测定值降低,说明透过比色杯样品的波长大于波长度盘指示的波长(图16-2右)。
图16-2 硫酸镍溶液监测仪器工作状态
实线代表波长正确时的吸收光谱,虚线代表波长不正确时的吸收光谱。箭头1表示510nm处的透光度,左右图均下降;箭头2表示460nm处的透光度,左图下降,右图升高,箭头3表示550处的透光度,左图升高,右图下降。
仪器工作状态测定后,若波长与杂光符合要求时,记录并保存各项数据,以备以后测定时对比。一般应每月监测一次,当400nm及700nm处杂光增强时,可能的原因包括:①激发光源陈旧,变黑或表层模糊不清。②透镜有灰尘。③色散元件失效等。如510nm处透光度减弱,可能原因为光源灯丝故障,比色池出光缝失灵或光栅损坏。
第二节 电泳技术的应用
在直流电场中,带电粒子向带符号相反的电极移动的现象称为电泳(electropho-resis)。1809年俄国物理学家Peнce首先发现了电泳现象,但直到1937年瑞典的Tiselius建立了分离蛋白质的界面电泳(boundaryelectrophoresis)之后,电泳技术才开始应用。本世纪60-70年代,当滤纸、聚丙烯酰胺凝胶等介质相继引入电泳以来,电泳技术得以迅速发展。丰富多彩的电泳形式使其应用十分广泛。电泳技术除了用于小分子物质的分离分析外,最主要用于蛋白质、核酸、酶,甚至病毒与细胞的研究。由于某些电泳法设备简单,操作方便,具有高分辨率及选择性特点,已成为医学检验中常用的技术。
一、电泳技术的基本原理和分类
在电场中,推动带电质点运动的力(F)等于质点所带净电荷量(Q)与电场强度(E)的乘积。
F=QE
质点的前移同样要受到阻力(F)的影响,对于一个球形质点,服从Stoke定律,即:
F′=6πrην
式中r为质点半径,η为介质粘度,ν为质点移动速度,当质点在电场中作稳定运动时:
F=F′即QE=6πrην
上式交换后可写成
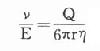
ν/E的含意为单位电场强度下的移动速度,这里可用迁移率μ表示,即
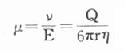
从上式可见,球形质点的迁移率,首先取决于自身状态,即与所带电量成正比,与其半径及介质粘度成反比。除了自身状态的因素外,电泳体系中其它因素也影响质点的电泳迁移率。
电泳法可分为自由电泳(无支持体)及区带电泳(有支持体)两大类。前者包括Tise-leas式微量电泳、显微电泳、等电聚焦电泳、等速电泳及密度梯度电泳。区带电泳则包括滤纸电泳(常压及高压)、薄层电泳(薄膜及薄板)、凝胶电泳(琼脂、琼脂糖、淀粉胶、聚丙烯酰胺凝胶)等。
自由电泳法的发展并不迅速,因为其电泳仪构造复杂、体积庞大,操作要求严格,价格昂贵等。而区带电泳可用各种类型的物质作支持体,其应用比较广泛。本节仅对常用的几种区带电泳分别加以叙述。
二、影响电泳迁移率的因素
⒈电场强度 电场强度是指单位长度(cm)的电位降,也称电势梯度。如以滤纸作支持物,其两端浸入到电极液中,电极液与滤纸交界面的纸长为20cm,测得的电位降为200V,那么电场强度为200V/20cm=10V/cm。当电压在500V以下,电场强度在2-10v/cm时为常压电泳。电压在500V以上,电场强度在20-200V/cm时为高压电泳。电场强度大,带电质点的迁移率加速,因此省时,但因产生大量热量,应配备冷却装置以维持恒温。
⒉溶液的pH值溶液的pH决定被分离物质的解离程度和质点的带电性质及所带净电荷量。例如蛋白质分子,它是既有酸性基团(-COOH),又有碱性基团(-NH2)的两性电解质,在某一溶液中所带正负电荷相等,即分子的净电荷等于零,此时,蛋白质在电场中不再移动,溶液的这一pH值为该蛋白质的等电点(isoelctric point,pI)。若溶液pH处于等电点酸侧,即pH<pl,则蛋白质带正电荷,在电场中向负极移动。若溶液pH处于等电点碱侧,即pH>pl,则蛋白质带负电荷,向正极移动。溶液的pH离pl越远,质点所带净电荷越多,电泳迁移率越大。因此在电泳时,应根据样品性质,选择合适的pH值缓冲液。
⒊溶液的离子强度电泳液中的离子浓度增加时会引起质点迁移率的降低。其原因是带电质点吸引相反符合的离子聚集其周围,形成一个与运动质点符合相反的离子氛(ionic atmosphere),离子氛不仅降低质点的带电量,同时增加质点前移的阻力,甚至使其不能泳动。然而离子浓度过低,会降低缓冲液的总浓度及缓冲容量,不易维持溶液的pH值,影响质点的带电量,改变泳动速度。离子的这种障碍效应与其浓度和价数相关。可用离子强度I表示。
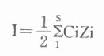
式中S表示溶液中离子种类,Ci和Zi分别表示每种离子的摩尔浓度与化合价。最常用I值在0.02-0.2之间。
⒋电渗在电场作用下液体对于固体支持物的相对移动称为电渗(electro-osmosis)。其产生的原因是固体支持物多孔,且带有可解离的化学基团,因此常吸附溶液中的正离子或负离子,使溶液相对带负电或正电。如以滤纸作支持物时,纸上纤维素吸附OH-带负电荷,与纸接触的水溶液因产生H3O+,带正电荷移向负极,若质点原来在电场中移向负极,结果质点的表现速度比其固有速度要快,若质点原来移向正极,表现速度比其固有速度要慢,可见应尽可能选择低电渗作用的支持物以减少电渗的影响。
三、电泳分析常用方法
(一)醋酸纤维素薄膜电泳
醋酸纤维素是提纤维素的羟基乙酰化形成的纤维素醋酸酯。由该物质制成的薄膜称为醋酸纤维素薄膜。这种薄膜对蛋白质样品吸附性小,几乎能完全消除纸电泳中出现的“拖尾”现象,又因为膜的亲水性比较小,它所容纳的缓冲液也少,电泳时电流的大部分由样品传导,所以分离速度快,电泳时间短,样品用量少,5μg的蛋白质可得到满意的分离效果。因此特别适合于病理情况下微量异常蛋白的检测。
醋酸纤维素膜经过冰醋酸乙醇溶液或其它透明液处理后可使膜透明化有利于对电泳图谱的光吸收扫描测定和膜的长期保存。
⒈材料与试剂醋酸纤维素膜一般使用市售商品,常用的电泳缓冲液为pH8.6的巴比妥缓冲液,浓度在0.05-0.09mol/L。
⒉操作要点
⑴膜的预处理:必须于电泳前将膜片浸泡于缓冲液,浸透后,取出膜片并用滤纸吸去多余的缓冲液,不可吸得过干。
⑵加样:样品用量依样品浓度、本身性质、染色方法及检测方法等因素决定。对血清蛋白质的常规电泳分析,每cm加样线不超过1μl,相当于60-80μg的蛋白质。
⑶电泳:可在室温下进行。电压为25V/cm,电流为0.4-0.6mA/cm宽。
⑷染色:一般蛋白质染色常使用氨基黑和丽春红,糖蛋白用甲苯胺蓝或过碘酸-Schiff试剂,脂蛋白则用苏丹黑或品红亚硫酸染色。
⑸脱色与透明:对水溶性染料最普遍应用的脱色剂是5%醋酸水溶液。为了长期保存或进行光吸收扫描测定,可浸入冰醋酸:无水乙醇=30:70(V/V)的透明液中。
(二)凝胶电泳
以淀粉胶、琼脂或琼脂糖凝胶、聚丙烯酰胺凝胶等作为支持介质的区带电泳法称为凝胶电泳。其中聚丙烯酰胺凝胶电泳(polyacrylamide gel electrophoresis,PAGE)普遍用于分离蛋白质及较小分子的核酸。琼脂糖凝胶孔径较大,对一般蛋白质不起分子筛作用,但适用于分离同工酶及其亚型,大分子核酸等应用较广,介绍如下:
⒈琼脂糖凝胶电泳的原理概述琼脂糖是由琼脂分离制备的链状多糖。其结构单元是D-半乳糖和3.6-脱水-L-半乳糖。许多琼脂糖链依氢键及其它力的作用使其互相盘绕形成绳状琼脂糖束,构成大网孔型凝胶。因此该凝胶适合于免疫复合物、核酸与核蛋白的分离、鉴定及纯化。在临床生化检验中常用于LDH、CK等同工酶的检测。
⒉琼脂糖凝胶电泳分离核酸的基本技术在一定浓度的琼脂糖凝胶介质中,DNA分子的电泳迁移率与其分子量的常用对数成反比;分子构型也对迁移率有影响,如共价闭环DNA>直线DNA>开环双链DNA。当凝胶浓度太高时,凝胶孔径变小,环状DNA(球形)不能进入胶中,相对迁移率为0,而同等大小的直线DNA(刚性棒状)可以按长轴方向前移,相对迁移率大于0。
⑴设备与试剂:琼脂糖凝胶电泳分为垂直及水平型两种。其中水平型可制备低浓度琼脂糖凝胶,而且制胶与加样都比较方便,故应用比较广泛。核酸分离一般用连续缓冲体系,常用的有TBE(0.08mol/l Tris·HCl,pH8.5,0.08mol/L硼酸,0.0024mol/lEDTA)和THE(0.04mol/l Tris·HCl。pH7.8,0.2mol/L醋酸钠,0.0018mol/lEDTA)。
⑵凝胶制备:用上述缓冲液配制0.5%-0.8%琼脂糖凝胶溶液,沸水浴或微波炉加热使之融化,冷至55℃时加入溴化乙锭(EB)至终浓度为0.5μg/ml,然后将其注入玻璃板或有机玻璃板组装好的模子中,厚度依样品浓度而定。注胶时,梳齿下端距玻璃板0.5-1.0mm,待脱凝固后,取出梳子,加入适量电极缓冲液使板胶浸没在缓冲液下1mm处。
⑶样品制备与加样:溶解于TBE或THE内的样品应含指示染料(0.025%溴酚蓝或桔黄橙)、蔗糖(10%-15%)或甘油(5%-10%),也可使用2.5%Fico Ⅱ增加比重,使样品集中,每齿孔可加样5-10μg。
⑷电泳:一般电压为5-15V/cm。对大分子的分离可用电压5V/cm。电泳过程最好在低温条件下进行。
⑸样品回收:电泳结束后在紫外灯下观察样品的分离情况,对需要的DNA分子或特殊片段可从电泳后的凝胶中以不同的方法进行回收,如电泳洗脱法:在紫外灯下切取含核酸区带的凝胶,将其装入透析袋(内含适量新鲜电泳缓冲液),扎紧透析袋后,平放在水平型电泳槽两电极之间的浅层缓冲液中,100V电泳2-3小时,然后正负电极交换,反向电泳2分钟,使透析袋上的DNA释放出来。吸出含DNA的溶液,进行酚抽提、乙醇沉淀等步骤即可完成样品的回收。
其它还有低融点琼脂糖法、醋酸铵溶液浸出法、冷冻挤压法等,但各种方法都仅仅有利于小分子量DNA片段(<1kb)的回收,随着DNA分子量的增大,回收量显著下降。
(三)等电聚焦电泳技术
等电聚焦(isoelectricfocusing,IEF)是60年代中期问世的一种利用有pH梯度的介质分离等电点不同的蛋白质的电泳技术。由于其分辨率可达0.01pH单位,因此特别适合于分离分子量相近而等电点不同的蛋白质组分。
⒈IEF的基本原理在IEF的电泳中,具有pH梯度的介质其分布是从阳极到阴极,pH值逐渐增大。如前所述,蛋白质分子具有两性解离及等电点的特征,这样在碱性区域蛋白质分子带负电荷向阳极移动,直至某一pH位点时失去电荷而停止移动,此处介质的pH恰好等于聚焦蛋白质分子的等电点(pl)。同理,位于酸性区域的蛋白质分子带正电荷向阴极移动,直到它们的等电点上聚焦为止。可见在该方法中,等电点是蛋白质组分的特性量度,将等电点不同的蛋白质混合物加入有pH梯度的凝胶介质中,在电场内经过一定时间后,各组分将分别聚焦在各自等电点相应的pH位置上,形成分离的蛋白质区带。
⒉pH梯度的组成pH梯度的组成方式有二种,一种是人工pH梯度,由于其不稳定,重复性差,现已不再使用。另一种是天然pH梯度。天然pH梯度的建立是在水平板或电泳管正负极间引入等电点彼此接近的一系列两性电解质的混合物,在正极端吸入酸液,如硫酸、磷酸或醋酸等,在负极端引入碱液,如氢氧化钠、氨水等。电泳开始前两性电解质的混合物pH为一均值,即各段介质中的pH相等,用pH表示。电泳开始后,混合物中pH最低的分子,带负电荷最多,pI1为其等电点,向正极移动速度最快,当移动到正极附近的酸液界面时,pH突然下降,甚至接近或稍低于PI1,这一分子不再向前移动而停留在此区域内。由于两性电解质具有一定的缓冲能力,使其周围一定的区域内介质的pH保持在它的等电点范围。pH稍高的第二种两性电解质,其等电点为pI2,也移向正极,由于pI2>pI1,因此定位于第一种两性电解质之后,这样,经过一定时间后,具有不同等电点的两性电解质按各自的等电点依次排列,形成了从正极到负极等电点递增,由低到高的线性pH梯度,如图16-3所示。
⒊两性电解质载体与支持介质理想的两性电解质载体应在pI处有足够的缓冲能力及电导,前者保证pH梯度的稳定,后者允许一定的电流通过。不同pI的两性电解质应有相似的电导系数从而使整个体系的电导均匀。两性电解质的分子量要小,易于应用分子筛或透析方法将其与被分离的高分子物质分开,而且不应与被分离物质发生反应或使之变性。
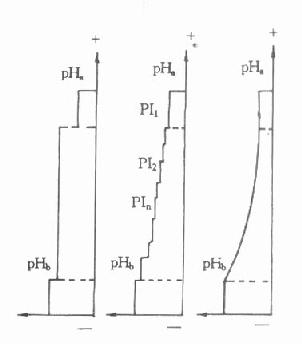
图16-3 pH梯度形成示意图
常用的pH梯度支持介质有聚丙烯酰胺凝胶、琼脂糖凝胶、葡聚糖凝胶等,其中聚丙烯酰胺凝胶为最常应用。
电泳后,不可用染色剂直接染色,因为常用的蛋白质染色剂也能和两性电解质结合,因此应先将凝胶浸泡在5%的三氯醋酸中去除两性电解质,然后再以适当的方法染色。
(四)其他电泳技术
⒈IEF/SDS-PAGE双向电泳法1975年O′Farrall等人根据不同组份之间的等电点差异和分子量差异建立了IEF/Sd S-PAGE双向电泳。其中IEF电泳(管柱状)为第一向,SDS-PAGE为第二向(平板)。在进行第一向IEF电泳时,电泳体系中应加入高浓度尿素、适量非离子型去污剂NP-40。蛋白质样品中除含有这两种物质外还应有二硫苏糖醇以促使蛋白质变性和肽链舒展。
IEF电泳结束后,将圆柱形凝胶在SDS-PAGE所应用的样品处理液(内含SDS、β-巯基乙醇)中振荡平衡,然后包埋在SDS-PAGE的凝胶板上端,即可进行第二向电泳。
IEF/SDS-PAGE双向电泳对蛋白质(包括核糖体蛋白、组蛋白等)的分离是极为精细的,因此特别适合于分离细菌或细胞中复杂的蛋白质组分。
⒉毛细管电泳 Neuhoff等人于1973年建立了毛细管均一浓度和梯度浓度凝胶用来分析微量蛋白质的方法,即微柱胶电泳,均一浓度的凝胶是将毛细管浸入凝胶混合液中,使凝胶充满总体积的2/3左右,然后将其揿入约厚2mm的代用粘土垫上,封闭管底,用一支直径比盛凝胶的毛细管更细的硬质玻璃毛细管吸水铺在凝胶上。聚合后,除去水层并用毛细管加蛋白质溶液(0.1-1.0μl,浓度为1-3mg/ml)于凝胶上,毛细管的空隙用电极缓冲液注满,切除插入粘土部分,即可电泳。
目前毛细管电泳分析仪的诞生,特别是美国生物系统公司的高效电泳色谱仪为DNA片段、蛋白质及多肽等生物大分子的分离、回收提供了快速、有效的途径。高效电泳色谱法是将凝胶电泳解析度和快速液相色谱技术溶为一体,在从凝胶中洗脱样品时,连续的洗脱液流载着分离好的成分,通过一个连机检测器,将结果显示并打印记录。高效电泳色谱法既具有凝胶电泳固有的高分辨率,生物相容性的优点,又可方便地连续洗脱样品。
第三节 离心技术的应用
离心技术(centrifugaltechnique)是根据颗粒在作匀速圆周运动时受到一个外向的离心力的行为而发展起来的一种分离技术。这项技术应用很广,诸如分离出化学反应后的沉淀物,天然的生物大分子、无机物、有机物,在生物化学以及其它的生物学领域常用来收集细胞、细胞器及生物大分子物质。
一、基本原理的分类
(一)基本原理
⒈离心力(centrifugal force,Fc)离心作用是根据在一定角度速度下作圆周运动的任何物体都受到一个向外的离心力进行的。离心力(Fc)的大小等于离心加速度ω2X与颗粒质量m的乘积,即:
![]()
其中ω是旋转角速度,以弧度/秒为单位;X是颗粒离开旋转中心的距离,以cm为单位;m是质量,以克为单位。
⒉相对离心力(relative centrifugal force,RCF)由于各种离心机转子的半径或者离心管至旋转轴中心的距离不同,离心力而受变化,因此在文献中常用“相对离心力”或“数字×g”表示离心力,只要RCF值不变,一个样品可以在不同的离心机上获得相同的结果。
RCF就是实际离心场转化为重力加速度的倍数。
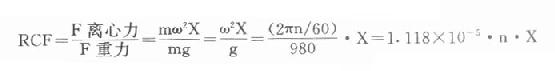
式中X为离心转子的半径距离,以cm为单位;g为地球重力加速度(980cm/sec2);n为转子每分钟的转数(rpm)。
在上式的基础上,Dole和Cotzias制作了与转子速度和半径相对应的离心力的转换列线图,见图16-4,在用图16-4将离心机转数换成相对离心力时,先在离心机半径标尺上取已知的离心机半径和在转数标尺上取已知的离心机转数,然后将这两点间划一条直线,在图中间RCF标尺上的交叉点,即为相应的离心力数值。例已知离心机转数为2500rpm,离心机的半径为7.7cm,将两点连接起来交于RCF标尺,此交点500×g即是RCF值。
⒊沉降系数(sedimentation coefficient,s)根据1924年Svedberg对沉降系数下的定义:颗粒在单位离心力场中粒子移动的速度。
![]()
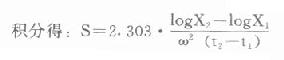
若ω用2πn/60表示,则
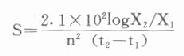
式中X1为离心前粒子离旋转轴的距离;X2为离心后粒子离旋转轴的距离。S实际上时常在10-13秒左右,故把沉降系数10-13秒称为一个Svedberg单位,简写S,量纲为秒。
⒋沉降速度(sedimentation velocity)沉降速度是指在强大离心力作用下,单位时间内物质运动的距离。
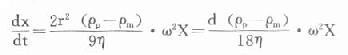
式中r为球形粒子半径;d为球形粒子直径;η为流体介质的粘度;ρP为粒子的密度;ρm为介质的密度。
从上式可知,粒子的沉降速度与粒子直径的平方、粒子的密度和介质密度之差成正比;离心力场增大,粒子的沉降速度也增加,将此式代入上项沉降系数公式中,则S的表示式也可表示为:
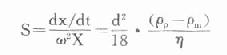
从该式中可看出,①当ρP>ρm,则S>0,粒子顺着离心方向沉降。②当ρP=ρm,则S=0,粒子到达某一位置后达到平衡。③当ρP<ρm,则S<0,粒子逆着离心方向上浮。
⒌沉降时间(sedimentation time,Ts)在实际工作中,常常遇到要求在已有的离心机上把某一种溶质从溶液中全部沉降分离出来的问题,这就必须首先知道用多大转速与多长时间可达到目的。如果转速已知,则需解决沉降时间来确定分离某粒子所需的时间。
根据沉降系数(S)式可得:


图16-4 离心力的转换列线图
将左侧r点与右侧n点连成一条直线,与中间RCF相交的
点即为相对离心力(×g)RCF=1.118×102×r×n2。
积分得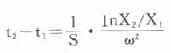
式中X2为离心转轴中心至离心管底内壁的距离;X1为离心转轴至样品溶液弯月面之间的距离,那么样品粒子完全沉降到底管内壁的时间(t2-t1)用Ts表示则式可改为:
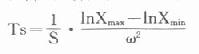
式中Ts以小时为单位,S以Svedberg为单位。
⒍K系数(k factor) K系数是用来描述在一个转子中,将粒子沉降下来的效率。也就是溶液恢复成澄清程度的一个指数,所以也叫“cleaning factor”。原则上,K系数愈小的,愈容易,也愈快将粒子沉降。
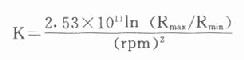
其中Rmax为转子最大半径;Rmin为转子最小半径。由其公式可知,K系数与离心转速及粒子沉降的路径有关。所以K系数是一个变数。当转速废,或者离心管的溶液量不同,即粒子沉降的路径改变时,K系数就改变了。通常,离心机的转子说明书中提供的K系数,都是根据最大路径及在最大转速下所计算出来的数值。如果已知粒子的沉降系数为80S的Polysome,采用的转子的K系数是323,那么预计沉降到管底所需的离心时间是T=k/S=4h,利用此公式预估的离心时间,对水平式转子最适合;对固定角式转子而言,实际时间将比预估的时间来得快些。
(二)分类
根据离心原理,按照实际工作的需要,目前已设计出许多离心方法,综合起来大致可分三类。
⒈平衡离心法根据粒子大小、形状不同进行分离,包括差速离心法(differential velocitycentrifugation)和速率区带离心法(rate zonal centrifugation)。
⒉等密度离心法(jsopycnic centrifugation)又称等比重离心法,依粒子密度差进行分离,等密度离心法和上述速率区带离心法合称为密度梯度离心法。
⒊经典式沉降平衡离心法用于对生物大分子分子量的测定、纯度估计、构象变化等。
二、离心分离方法
(一)差速离心法
它利用不同的粒子在离心力场中沉降的差别,在同一离心条件下,沉降速度不同,通过不断增加相对离心力,使一个非均匀混合液内的大小、形状不同的粒子分部沉淀。操作过程中一般是在离心后用倾倒的办法把上清液与沉淀分开,然后将上清液加高转速离心,分离出第二部分沉淀,如此往复加高转速,逐级分离出所需要的物质。
差速离心的分辨率不高,沉淀系数在同一个数量级内的各种粒子不容易分开,常用于其他分离手段之前的粗制品提取。
例如用差速离心法分离已破碎的细胞各组份
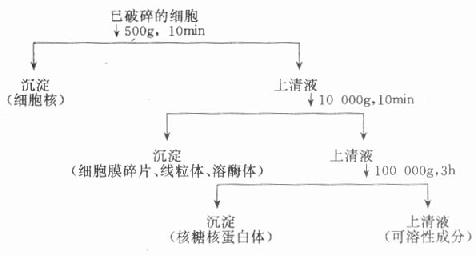
(二)速率区带离心法
速率区带离心法是在离心前于离心管内先装入密度梯度介质(如蔗糖、甘油、KBr、CsCl等),待分离的样品铺在梯度液的顶部、离心管底部或梯度层中间,同梯度液一起离心。离心后在近旋转轴处(X1)的介质密度最小,离旋转轴最远处(X2)介质的密度最大,但最大介质密度必须小于样品中粒子的最小密度,即ρP>ρm。这种方法是根据分离的粒子在梯度液中沉降速度的不同,使具有不同沉降速度的粒子处于不同的密度梯度层内分成一系列区带,达到彼此分离的目的。梯度液在离心过程中以及离心完毕后,取样时起着支持介质和稳定剂的作用,避免因机械振动而引起已分层的粒子再混合。
由于ρP>ρm可知S>0,因此该离心法的离心时间要严格控制,既有足够的时间使各种粒子在介质梯度中形成区带,又要控制在任一粒子达到沉淀前。如果离心时间过长,所有的样品可全部到达离心管底部;离心时间不足,样品还没有分离。由于此法是一种不完全的沉降,沉降受物质本身大小的影响较大,一般是应用在物质大小相异而密度相同的情况。常用的梯度液有Ficoll、Percoll及蔗糖。
(三)等密度离心法
等密度离心法是在离心前预先配制介质的密度梯度,此种密度梯度液包含了被分离样品中所有粒子的密度,待分离的样品铺在梯度液顶上或和梯度液先混合,离心开始后,当梯度液由于离心力的作用逐渐形成底浓而管顶稀的密度梯度,与此同时原来分布均匀的粒子也发生重新分布。当管底介质的密度大于粒子的密度,即ρm>ρP时粒子上浮;在弯顶处ρP>ρm时,则粒子沉降,最后粒子进入到一个它本身的密度位置即ρP=ρm,此时dx/dt为零粒子不再移动,粒子形成纯组份的区带,与样品粒子的密度有关,而与粒子的大小和其他参数无关,因此只要转速、温度不变,则延长离心时间也不能改变这些粒子的成带位置。
此法一般应用于物质的大小相近,而密度差异较大时。常用的梯度液是CsCl。
(四)梯度溶液的制备
⒈梯度材料的选择原则作为一种理想的梯度材料应具备以下几点:①与被分离的生物材料不发生反应即完全惰性,且易与所分离的生物粒子分开。②可达到要求的密度范围,且在所要求的密度范围内,粘度低,渗透压低,离子强度和pH变化较小。③不会对离心设备发生腐蚀作用。④容易纯化,价格便宜或容易回收。⑤浓度便于测定,如具有折光率。⑥对于超速离心分析工作来说,它的物理性质、热力学性质应该是已知的。这些条件是理想条件,完全符合每种性能的梯度材料几乎是没有的。下面介绍几种基本上符合上述原则的梯度材料:①糖类:蔗糖、甘油、聚蔗糖(Ficoll)、右旋糖酐、糖原。②无机盐类:CsCl(氯化铯)、RbCl(氯化铷)、NaCl、KBr等。③有机碘化物:三碘苯甲酰葡萄糖胺(matrizamide)等。④硅溶胶:如Percoll。⑤蛋白质:如牛血清白蛋白。⑥重水。⑦非水溶性有机物:如氟代碳等。
⒉梯度材料的应用范围
⑴蔗糖:水溶性大,性质稳定,渗透压较高,其最高密度可达1.33g/ml,且由于价格低容易制备,是现在实验室里常用于细胞器、病毒、RNA分离的梯度材料,但由于有较大的渗透压,不宜用于细胞的分离。
⑵聚蔗糖:商品名Ficoll,常采用Ficoll-400也就是相对分子重量为400000,Ficoll渗透压低,但它的粘度却特别高,为此常与泛影葡胺混合使用以降低粘度。主要用于分离各种细胞包括血细胞、成纤维细胞、肿瘤细胞、鼠肝细胞等。
⑶氯化铯:是一种离子性介质、水溶性大,最高密度可达1.91g/ml。由于它是重金属盐类,在离心时形成的梯度有较好的分辨率,被广泛地用于DNA、质粒、病毒和脂蛋白的分离,但价格较贵。
⑷卤化盐类:KBr和NaCl可用于脂蛋白分离,KI和NaI可用于RNA分离,其分辨率高于铯盐。NaCl梯度也可用于分离脂蛋白,NaI梯度可分离天然或变性的DNA。
⑸Percoll:是商品名,它是一种SiO2胶体外面包了一层聚乙烯吡咯酮(PVP),渗透压低,它对生物材料的影响小,而且颗粒稳定,在冷却和冻融情况下还是稳定的,其粘度高,且在酸性pH和高离子强度下不稳定。它可用于细胞、细胞器和病毒的分离。
三、分析性超速离心
与制备性超速离心不同的是:分析性超速离心主要是为了研究生物大分子的沉降特性和结构,而不是专门收集某一特定组份。因此它使用了特殊的转子和检测手段,以便连续监视物质在一个离心场中的沉降过程。
(一)分析性超速离心的工作原理
分析性超速离心机主要由一个椭圆形的转子、一套真空系统和一套光学系统所组成。该转子通过一个柔性的轴联接成一个高速的驱动装置,此轴可使转子在旋转时形成自己的轴。转子在一个冷冻的真空腔中旋转,其容纳二个小室:分析室和配衡室。配衡室是一个经过精密加工的金属块,作为分析室的平衡用。分析室的容量一般为1ml,呈扇形排列在转子中,其工作原理与一个普遍水平转子相同。分析室有上下二个平面的石英窗,离心机中装有的光学系统可保证在整个离心期间都能观察小室中正在沉降的物质,可以通过对紫外光的吸收(如对蛋白质和DNA)或折谢率的不同对沉降物进行监视。后一方法的原理是:当光线通过一个具有不同密度区的透明液,在这些区带的界面上产生光的折射。在分析室中物质沉降时重粒子和轻粒子之间形成的界面就象一个折射的透镜,结果在检测系统的照相底板上产出一“峰”。由于沉降不断进行,界面向前推进,故“峰”也在移动,从峰移动的速度可以得到物质沉降速度的指标。图16-5是分析性超速离心系统的示意图。
(二)分析性超速离心的应用
⒈测定生物大分子的相对分子重量测定相对分子重量主要有三种方法:沉降速度、沉降平衡和接近沉降平衡。其中应用最广的是沉降速度,超速离心在高速中进行,这个速度使得任意分布的粒子通过溶剂从旋转的中心辐射地向外移动,在清除了粒子的那部分溶剂和尚含有沉降物的那部分溶剂之间形成一个明显的界面,该界面随时间的移动而移动,这就是粒子沉降速度的一个指标,然后用照相记录,即可求出粒子的沉降系数。
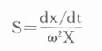
分子或粒子的相对分子重量则可从Svedberg方程式来确定:
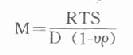
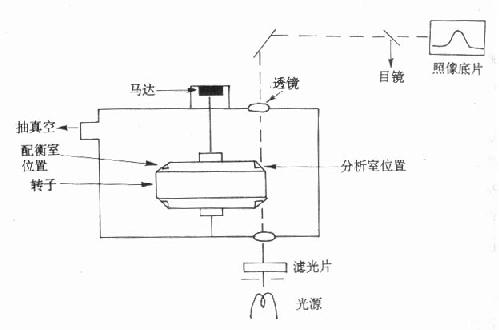
图16-5 分析性超速离心系统图示
式中M:该分子不含水的相对分子重量;R:气体常数;T:绝对温度;S:分子的沉降系数;ν:分子的微分比容(当一克溶质加到一个大体积的溶液中所占有的体积);ρ:溶剂的密度。
⒉生物大分子的纯度估计静分析性超速离心已广泛地应用于研究DNA制剂、病毒和蛋白质的纯度。用沉降速度的技术来分析沉降界面是测定制剂均质性的最常用方法之一,出现单一清晰的界面一般认为是均质的,如有杂质则在主峰的一侧或二侧出现小峰。
⒊分析生物大分子中的构象变化分析性超速离心已成功地用于检测大分子构象的变化,例如DNA可能以单股或双股出现,其中每一股在本质上可能是线性的,也可能是环状的,如果遇到某种因素(温度或有机溶剂)DNA分子可能发生一些构象上的变化,这些变化也许可逆、也许不可逆,这些构象上的变化可以通过检查样品在沉降速度上的差异来证实。
第四节 层析技术的应用
一、层析技术的原理和分类
(一)层析技术的原理
层析法是目前广泛应用的一种分离技术。本世纪初俄国植物学家M.Tswett发现并使用这一技术证明了植物的叶子中不仅有叶绿素还含有其它色素。现在层析法已成为生物化学、分子生物学及其它学科领域有效的分离分析工具之一。
层析法是利用不同物质理化性质的差异而建立起来的技术。所有的层析系统都由两个相组成:一是固定相,它或者是固体物质或者是固定于固体物质上的成分;另一是流动相,即可以流动的物质,如水和各种溶媒。当待分离的混合物随溶媒(流动相)通过固定相时,由于各组份的理化性质存在差异,与两相发生相互作用(吸附、溶解、结合等)的能力不同,在两相中的分配(含量对比)不同,而且随溶媒向前移动,各组份不断地在两相中进行再分配。与固定相相互作用力越弱的组份,随流动相移动时受到的阻滞作用小,向前移动的速度快。反之,与固定相相互作用越强的组份,向前移动速度越慢。分部收集流出液,可得到样品中所含的各单一组份,从而达到将各组份分离的目的。
(二)层析法分类见表16-5~7
(三)层析法的特点与应用
表16-5 按两相所处状态分类
| 流动相 | |||
| 液体 | 气体 | ||
| 液体 | 液-液层析法 | 气-液层析法 | |
| 固定相 | |||
| 固体 | 液-固层析法 | 气-固层析法 | |
层析法是根据物质的理化性质不同而建立的分离分析方法。根据层析峰的位置及峰高或峰面积,可以定性及定量。层析法与光学、电学或电化学仪器连用,可检测出层析后各组份的浓度或质量,同时绘出层析图。层析仪与电子计算机联用,可使操作及数据处理自动化,大大缩短分析时间。由于层析法具有分辨率高、灵敏度高、选择性好、速度快等特点,因此适用于杂质多、含量少的复杂样品分析,尤其适用于生物样品的分离分析。近年来,已成为生物化学及分子生物学常用的分析方法。在医药卫生、环境化学、高分子材料、石油化工等方面也得到了广泛的应用。
表16-6 按层析原理分类
| 名称 | 分离原理 |
| 吸附层析法 | 组份在吸附剂表面吸附固定相是固体吸附剂,各能力不同 |
| 分配层析法 | 各组份在流动相和静止液相(固相)中的分配系数不同 |
| 离子交换层析法 | 固定相是离子交换剂,各组份与离子交换剂亲和力不同 |
| 凝胶层析法 | 固定相是多孔凝胶,各组份的分子大小不同,因而在凝胶上受阻滞的程度不同 |
| 亲和层析法 | 固定相只能与一种待分离组份专一结合,以此和无亲和力的其它组份分离 |
表16-7 按操作形式不同分类
| 名称 | 操作形式 |
| 柱层析法 | 固定相装于柱内,使样品沿着一个方向前移而达分离 |
| 薄层层析法 | 将适当粘度的固定相均匀涂铺在薄板上,点样后用流动相展开,使各组份分离 |
| 纸层析法 | 用滤纸作液体的载体,点样后用流动相展开,使各组份分离 |
| 薄膜层析法 | 将适当的高分子有机吸附剂制成薄膜,以类似纸层析方法进行物质的分离 |
二、层析法实验技术
(一)凝胶层析法
凝胶层析又称分子筛过滤、排阻层析等。它的突出优点是层析所用的凝胶属于惰性载体,不带电荷,吸附力弱,操作条件比较温和,可在相当广的温度范围下进行,不需要有机溶剂,并且对分离成分理化性质的保持有独到之处。对于高分子物质有很好的分离效果。
⒈凝胶的选择根据实验目的不同选择不同型号的凝胶。如果实验目的是将样品中的大分子物质和小分子物质分开,由于它们在分配系数上有显着差异,这种分离又称组别分离,一般可选用Sephadex G-25和G-50,对于小肽和低分子量的物质(1000-5000)的脱盐可使用Sephadex G-10,G-15及Bio-Gel-p-2或4。如果实验目的是将样品中一些分子量比较近似的物质进行分离,这种分离又叫分级分离。一般选用排阻限度略大于样品中最高分子量物质的凝胶,层析过程中这些物质都能不同程度地深入到凝胶内部,由于Kd不同,最后得到分离。
⒉柱的直径与长度根据经验,组别分离时,大多采用2-30cm长的层析柱,分级分离时,一般需要100cm左右长的层析柱,其直径在1-5cm范围内,小于1cm产生管壁效应,大于5cm则稀释现象严重。长度L与直径D的比值L/D一般宜在7-10之间,但对移动慢的物质宜在30-40之间。
⒊凝胶柱的制备凝胶型号选定后,将干胶颗粒悬浮于5-10倍量的蒸馏水或洗脱液中充分溶胀,溶胀之后将极细的小颗粒倾泻出去。自然溶胀费时较长,加热可使溶胀加速,即在沸水浴中将湿凝胶浆逐渐升温至近沸,1-2小时即可达到凝胶的充分胀溶。加热法既可节省时间又可消毒。
凝胶的装填:将层析柱与地面垂直固定在架子上,下端流出口用夹子夹紧,柱顶可安装一个带有搅拌装置的较大容器,柱内充满洗脱液,将凝胶调成较稀薄的浆头液盛于柱顶的容器中,然后在微微地搅拌下使凝胶下沉于柱内,这样凝胶粒水平上升,直到所需高度为止,拆除柱顶装置,用相应的滤纸片轻轻盖在凝胶床表面。稍放置一段时间,再开始流动平衡,流速应低于层析时所需的流速。在平衡过程中逐渐增加到层析的流速,千万不能超过最终流速。平衡凝胶床过夜,使用前要检查层析床是否均匀,有无“纹路”或气泡,或加一些有色物质来观察色带的移动,如带狭窄、均匀平整说明层析柱的性能良好,色带出现歪曲、散乱、变宽时必须重新装柱。
⒋加样和洗脱凝胶床经过平衡后,在床顶部留下数亳升洗脱液使凝胶床饱和,再用滴管加入样品。一般样品体积不大于凝胶总床体积的5%-10%。样品浓度与分配系数无关,故样品浓度可以提高,但分子量较大的物质,溶液的粘度将随浓度增加而增大,使分子运动受限,故样品与洗脱液的相对粘度不得超过1.5-2。样品加入后打开流出口,使样品渗入凝胶床内,当样品液面恰与凝胶床表面相平时,再加入数毫升洗脱液中洗管壁,使其全部进入凝胶床后,将层析床与洗脱液贮瓶及收集器相连,预先设计好流速,然后分部收集洗脱液,并对每一馏份做定性、定量测定。
⒌凝胶柱的重复使用、凝胶回收与保存一次装柱后可以反复使用,不必特殊处理,并不影响分离效果。为了防止凝胶染菌,可在一次层析后加入0.02%的叠氮钠,在下次层析前应将抑菌剂除去,以免干扰洗脱液的测定。
如果不再使用可将其回收,一般方法是将凝胶用水冲洗干净滤干,依次用70%、90%、95%乙醇脱水平衡至乙醇浓度达90%以上,滤干,再用乙醚洗去乙醇、滤干、干燥保存。湿态保存方法是凝胶浆中加入抑菌剂或水冲洗到中性,密封后高压灭菌保存。
⒍凝胶层析的应用
⑴脱盐:高分子(如蛋白质、核酸、多糖等)溶液中的低分子量杂质,可以用凝胶层析法除去,这一操作称为脱盐。本法脱盐操作简便、快速、蛋白质和酶类等在脱盐过程中不易变性。适用的凝胶为SephadexG-10、15、25或Bio-Gel-p-2、4、6。柱长与直径之比为5-15,样品体积可达柱床体积的25%-30%,为了防止蛋白质脱盐后溶解度降低会形成沉淀吸附于柱上,一般用醋酸铵等挥发性盐类缓冲液使层析柱平衡,然后加入样品,再用同样缓冲液洗脱,收集的洗脱液用冷冻干燥法除去挥发性盐类。
⑵用于分离提纯:凝胶层析法已广泛用于酶、蛋白质、氨基酸、多糖、激素、生物碱等物质的分离提纯。凝胶对热原有较强的吸附力,可用来去除无离子水中的致热原制备注射用水。
⑶测定高分子物质的分子量:用一系列已知分子量的标准品放入同一凝胶柱内,在同一条件下层析,记录每一分钟成分的洗脱体积,并以洗脱体积对分子量的对数作图,在一定分子量范围内可得一直线,即分子量的标准曲线。测定未知物质的分子量时,可将此样品加在测定了标准曲线的凝胶柱内洗肿后,根据物质的洗脱体积,在标准曲线上查出它的分子量。
⑷高分子溶液的浓缩:通常将SephadexG-25或50干胶投入到稀的高分子溶液中,这时水分和低分子量的物质就会进入凝胶粒子内部的孔隙中,而高分子物质则排阻在凝胶颗粒之外,再经离心或过滤,将溶胀的凝胶分离出去,就得到了浓缩的高分子溶液。
(二)离子交换层析法
离子交换层析法是以具有离子交换性能的物质作固定相,利用它与流动相中的离子能进行可逆的交换性质来分离离子型化合物的一种方法。
⒈离子交换剂预处理和装柱对于离子交换纤维素要用流水洗去少量碎的不易沉淀的颗粒,以保证有较好的均匀度,对于已溶胀好的产品则不必经这一步骤。溶胀的交换剂使用前要用稀酸或稀碱处理,使之成为带H+或OH-的交换剂型。阴离子交换剂常用“碱-酸-碱”处理,使最终转为-OH-型或盐型交换剂;对于阳离子交换剂则用“酸-碱-酸”处理,使最终转为-H-型交换剂。洗涤好的纤维素使用前必须平衡至所需的pH和离子强度。已平衡的交换剂在装柱前还要减压除气泡。为了避免颗粒大小不等的交换剂在自然沉降时分层,要适当加压装柱,同时使柱床压紧,减少死体积,有利于分辨率的提高。柱子装好后再用起始缓冲液淋洗,直至达到充分平衡方可使用。
⒉加样与洗脱加样:层析所用的样品应与起始缓冲液有相同的pH和离子强度,所选定的pH值应落在交换剂与被结合物有相反电荷的范围,同时要注意离子强度应低,可用透析、凝胶过滤或稀释法达此目的。样品中的不溶物应在透析后或凝胶过滤前,以离心法除去。为了达到满意的分离效果,上样量要适当,不要超过柱的负荷能力。柱的负荷能力可用交换容量来推算,通常上样量为交换剂交换总量的1%-5%。
洗脱:已结合样品的离子交换前,可通过改变溶液的pH或改变离子强度的方法将结合物洗脱,也可同时改变pH与离子强度。为了使复杂的组份分离完全,往往需要逐步改变pH或离子强度,其中最简单的方法是阶段洗脱法,即分次将不同pH与离子强度的溶液加入,使不同成分逐步洗脱。由于这种洗脱pH与离子强度的变化大,使许多洗脱体积相近的成分同时洗脱,纯度较差,不适宜精细的分离。最好的洗脱方法是连续梯度洗脱,洗脱装置见图16-6。两个容器放于同一水平上,第一个容器盛有一定pH的缓冲液,第二个容器含有高盐浓度或不同pH的缓冲液,两容器连通,第一个容器与柱相连,当溶液由第一容器流入柱时,第二容器中的溶液就会自动来补充,经搅拌与第一容器的溶液相混合,这样流入柱中的缓冲液的洗脱能力即成梯度变化。第一容器中任何时间的浓度都可用下式进行计算:
C=C2-(C2-C1)(1-V)A2/A1
式中A1、A2分别代表两容器的截面积:C1、C2分别表示容器中溶液的浓度;V为流出体积对总体积之比。当A1=A2时为线性梯度,当A1>A2时为凹形梯度,A1>A2时为凸形梯度。
洗脱时应满足以下要求:①洗脱液体积应足够大,一般要几十倍于床体积,从而使分离的各峰不致于太拥挤。②梯度的上限要足够高,使紧密吸附的物质能被洗脱下来。③梯度不要上升太快,要恰好使移动的区带在快到柱末端时达到解吸状态。目的物的过早解吸,会引起区带扩散;而目的物的过晚解吸会使峰形过宽。
图16-6 梯度洗脱示意图
⒊洗脱馏份的分析 按一定体积(5-10ml/管)收集的洗脱液可逐管进行测定,得到层析图谱。依实验目的的不同,可采用适宜的检测方法(生物活性测定、免疫学测定等)确定图谱中目的物的位置,并回收目的物。
⒋离子交换剂的再生与保存离子交换剂可在柱上再生。如离子交换纤维素可用2mol/:NaCl淋洗柱,若有强吸附物则可用0.1mol/l NaOH洗柱;若有脂溶性物质则可用非离子型去污剂洗柱后再生,也可用乙醇洗涤,其顺序为:0.5mol/l NaOH-水-乙醇-水-20%NaOH-水。保存离子交换剂时要加防腐剂。对阴离子交换剂宜用0.002%氯已定(洗必泰),阳离子交换剂可用乙基硫柳汞(0.005%)。有些产品建立用0.02%叠氮钠。
⒌离子交换层析的应用离子交换层析技术已广泛用于各学科领域。在生物化学及临床生化检验中主要用于分离氨基酸、多肽及蛋白质,也可用于分离核酸、核苷酸及其它带电荷的生物分子。
(三)高效液相层析法
高效液相层析法(HPLC)是近二十年来发展起来的一项新颖快速的分离技术。它是在经典液相层析法基础上,引进了气相层析的理论具有气相层析的全部优点。由于HPLC分离能力强、测定灵敏度高,可在室温下进行,应用范围极广,无论是极性还是非极性,小分子还是大分子,热稳定还是不稳定的化合物均可用此法测定。对蛋白质、核酸、氨基酸、生物碱、类固醇和类脂等尤为有利。
高效液相层析法的基本概念和分离理论与经典的液相色谱法及气相色谱法一致,因而其塔板理论及动力学理论等都可用于高效液相层析。
⒈高效液相层析仪典型的高效液相层析仪包括输液系统、层析柱与检测系统三部分。流动相用一高压泵输入。这种高压泵应满足以下条件:①流量恒定,无脉动,并有较大的调节范围。②能抗溶剂腐蚀。③有较高的输出压力,一般要达到15~300kg/c,也有的高达800kg/c。④泵的死体积要小。梯度洗脱装置必须具备两台高压泵,一台输送强溶剂,一台输送弱溶剂,两泵运转速度用电脑控制,并可按一定的要求改变流动相的组成,以改善分离效果。一般用微量注射器直接进样,也可采用六通阀门进样。HPLC中所用的检测器最多应用的是紫外吸收检测,灵敏度可达ng水平。此外,还有荧光检测器、示差析光检测器、电化学检测器等。
⒉HPLC的类型与应用
⑴液-固吸附层析:固定相是具有吸附活性的吸附剂,常用的有硅胶、氧化铝、高分子有机酸或聚酰胺凝胶等。液-固吸附层析中的流动相依其所起的作用不同,分为“底剂”和洗脱剂两类,底剂起决定基本色谱的分离作用,洗脱剂起调节试样组份的滞留时间长短,并对试样中某几个组份具有选择性作用。流动相中底剂与洗脱剂成分的组合和选择,直接影响色谱的分离情况,一般底剂为极性较低的溶剂,如正已烷、环已烷、戊烷、石油醚等,洗脱剂则根据试样性质选用针对性溶剂,如醚、酯、酮、醇和酸等。本法可用于分离异构体、抗氧化剂与维生素等。
⑵液-液分配层析:固定相为单体固定液构成。将固定液的官能团结合在薄壳或多孔型硅胶上,经酸洗、中和、干燥活化、使表面保持一定的硅羟基。这种以化学键合相为固定相的液-液层析称为化学键合相层析。另一种利用离子对原理的液-液分配层析为离子对层析。化学键合层析分:①极性键合相层析:固定相为极性基团,氰基、氨基及双羟基三种。流动相为非极性或极性较小的溶剂。极性小的组份先出峰,极性大的后出峰,这称为正相层析法,适用于分离极性化合物。②非极性键合相层析:固定相为非极性基团,如十八烷基(C18)、辛烷基(C8)、甲基与苯基等,流动相用强极性溶剂,如水、醇、乙腈或无机盐缓冲液。最常用的是不同比例的水和甲醇配制的混合溶剂,水不仅起洗脱作用还可掩盖载体表面的硅羟基,防止因吸附而至的拖尾现象。极性大的组份先出峰,极性小的组份后出峰,恰好与正相法相反,故称反相层析。本法适用于小分子物质的分离,如肽、核苷酸、糖类、氨基酸的衍生物等。离子对分配层析分:①正相离子对层析:此法常以水吸附在硅胶上作为固定相,把与分离组份带相反电荷的配对离子以一定浓度溶于水或缓冲液涂渍在硅胶上。流动相为极性较低的有机溶剂。在层析过程中,待分离的离子与水相中配对离子形成中性离子对,在水相和有机相中进行分配,而达到分离。本法优点是流动相选择余地大,缺点是固定相易流失。②反向离子对层析:固定相是疏水性键合硅胶,如C18键合相,待分离离子和带相反电荷的配对离子同时存在于强极性的流动相中,生成的中性离子对在流动相和键合相之间进行分配,而得到分离。本法优点是固定相不存在流失问题、流动相含水或缓冲液更适用于电离性化合物的分离。
⑶离子交换层析:原理与普通离子交换相同。在离子交换HPLC中,固定相多用离子性键合相,故本法又称离子性键合相层析。流动相主要是水溶液,pH值最好在被分离酸、碱的pK值附近。
(四)亲和层析法
亲和层析是利用待分离物质和它的特异性配体间具有特异的亲和力,从而达到分离目的的一类特殊层析技术。
具有专一亲和力的生物分子对主要有:抗原与抗体、DNA与互补DNA或RNA、酶与它的底物或竞争性抑制剂、激素(或药物)与它们的受体、维生素和它的特异结合蛋白、糖蛋白与它相应的植物凝集素等。可亲和的一对分子中的一方以共价键形式与不溶性载体相连作为固定相吸附剂,当含有混合组份的样品通过此固定相时,只有和固定相分子有特异亲和力的物质,才能被固定相吸附结合,其它没有亲和力的无关组份就随流动相流出,然后改变流动组成份,将结合的亲和物洗脱下来。亲和层析中所用的载体称为基质,与基质共价连接的化合物称配基。
亲和层析纯化过程简单、迅速,且分离效率高。对分离含量极少又不稳定的活性物质尤为有效。但本法必须针对某一分离对象,制备专一的配基和寻求层析的稳定条件,因此亲和层析的应用范围受到了一定的限制。
亲和层析可用于纯化生物大分子:稀溶液的浓缩;不稳定蛋白质的贮藏;从纯化的分子中除去残余的污染物;用免疫吸附剂吸附纯化对此尚无互补配体的生物大分子;分离核酸是亲和层析应用的一个重要方面。
应用实例:2-胰凝乳蛋白酶的提纯
⒈亲和吸附剂(Σ-氨基-N-乙酰-D-色氨酸)的制备取一定体积的Sepharose4B加100mgCNBr。搅拌混合物,滴加2-8mol/l NaOH,使溶液pH维持在11.0。20℃左右,8-12分钟完成反应。在布氏漏斗中抽滤活化的琼脂糖,然后用20倍体积冷的0.1mol/l NaHCO3(pH9.0)溶液洗涤。将上述活化的Sepharose4B与等体积0.1mol/l NaHCO3(pH9.0并在冰箱中预冷)溶液混合,接着迅速加入α-胰蛋白酶抑制剂(Σ-氨基-N-乙酰-色氨酸甲酯)。抑制剂的最大用量为每毫升Sepharose 4B 65μmol/L。4℃缓慢搅拌约24小时,而后再用水和缓冲液反复地洗至没有游离的抑制剂为止。制得的亲和吸附剂在50mmol/l Tris-HCl缓冲液(pH8.0)中保存备用。
⒉分离 亲和吸附剂装柱后用50mmol/LTris-HCl(pH8.0)缓冲液平衡(室温)。样品溶于同样缓冲液中并上柱,再应用同样缓冲液淋洗柱子。流速为30-60ml/小时,直至280nm无光吸收时停止洗涤,然后改用0.2mol/L醋酸缓冲液(pH3.0)洗脱。
(五)聚焦层析法
聚焦层析是一种操作简单、廉价的层析技术。它的原理是根据各种蛋白质的等电点不同进行分离的过程,因此本方法具有高分辨、高度浓缩和高度专一等特点。聚焦层析所用的凝胶首先用高pH溶液平衡,然后用多元缓冲液进行洗脱,多元缓冲液pH呈梯度下降。
聚焦层析所用凝胶主要有两种:MONOP和多元缓冲液交换剂(PBE)。其中MONOP是带孔小珠,孔中被带正电荷的胺基填充,适用于高效聚焦层析。多元缓冲液交换剂是一种交换凝胶,适用作普通聚焦层析的介质。各种凝胶性质见表16-8。
表16-8 聚焦层析技术数据
| 凝胶名称 | pH范围 | 洗脱 |
| MONOP | 11-8 | Pharmalyte 8-10.5 |
| PBE | 11-8 | Pharmalyte 8-10.5 |
| MONOP | 9-6 | 多元缓冲液96 |
| PBE 94 | 7-4 | 多元缓冲液74 |
| PBE94 | 7-4 | 多元缓冲液74 |
NONOP可制成两种高效液相柱:MONOp HR5/5(1ml),和MONOp HR5/20(5ml)。使用时根据样品复杂性选择相应的柱。HR5/5分辨率为pI>0.2,HR5/20pI>0.02。
多元缓冲液交换剂PBE,属珠状交换剂凝胶,有PBe 118(pH11-8)和PBE(pH9-4)。在聚焦层析时,通过使用不同的洗脱液,可使交换容量发生改变,并且使pH达到更广的范围。
第五节 电化学分析技术的应用
一、概述
利用物质的电化学性质,测定化学电池的电位、电流或电量的变化进行分析的方法称为电化学分析法。
电化学分析法有多种,如测定原电池电动势以求物质含量的分析方法称为电位法(potential method)或电位分析法;通过对电阻的测定以求物质含量的分析法称为电导法;而借助某些物理量的突变作为滴定分析终点的指示,则称为电容量分析法等。
电位分析法是利用电极电位和浓度之间的关系来确定物质含量的分析方法,表示电极电位的基本公式是能斯特方程式。由于单个电极电位的绝对值无法测量,在大多数情况下,电位法是基于测量原电池的电动势,构成电池的两个电极,一个电极的电位随待测离子浓度而变化,能指示待测离子浓度,称为指示电极;另一个电极的电位则不受试液组成变化的影响,具有较恒定的数值,称为参比电极。指示电极和参化电极共同浸入试液中,构成一个原电池,通过测定原电池的电动势,便可求得待测离子的浓度,这一方法亦称为直接电位法。
离子选择电极分析法是电位分析法中发展最为迅速、最活跃的分支。对某些离子测定的灵敏度可达10-6数量级。在许多情况下不破坏试液或不用进行复杂的预处理,对有色、浑浊溶液都可进行分析。
二、离子选择性电极分类
基于离子选择电极绝大多数都是膜电极这一事实,依据膜电位响应机制、膜的组成和结构,其分类如下:
(一)玻璃膜电极
玻璃膜电极属于刚性基质电极。敏感膜由玻璃材料制成。由于玻璃的组成不同,可制成H+、Na+、K+、Li+和Ag+等离子选择性电极
第十七章 血清酶定量的检测技术
第一节 概述
酶在正常和病理代谢中起着重要作用,因此酶的测定是医学实验室的一项重要工作,在医学研究和医院常规工作中都要进行大量酶的测定。但要作好酶的测定不是一件很容易的事,因为它有很多与其它定量测定不同或独特之处,作好酶的测定除了必须掌握一般分析技术知识外,还须了解酶测定的特点、方法分类和各类方法的优缺点,然后根据各自实验室情况、研究目的和临床需要,才能设计和应用好各个具体的酶测定方法,创造性地作好酶的测定。
目前常用于酶定量测定的方法是测量酶的活性浓度,此方法并不直接测酶蛋白含量,实际上是测定酶催化反应的速度,并由此间接地推算出标本中酶浓度的高低,这是酶测定方法独特之处。
使用间接方法并不是因为该法比直接测定酶蛋白有很多优点,事实上间接方法有不少不足之处,在很大程度上采用间接法是出于无奈,因为在所测标本中酶蛋白浓度和其它蛋白相比明显偏低,例如在血清中大多数酶蛋白含量多在g/L水平,用一般化学方法从含量高达70g/L蛋白质的血清中将酶蛋白分离出来并直接测定其含量显然是很困难的。这样科学家才想到利用酶蛋白的催化特性来测定酶;微量酶蛋白可以明显加快所催化反应的速度,并在特定条件下,此反应速度(v)可和酶浓度(E)成正比例。可以下列二式表示之:
v=△P/△t=k·E
此式以单位时间(t)内产生物(P)生成量表示反应速度
v=-△S/△t=k·E
此式中以底物(S)减少量取代产物生成量。
以上二式是衡量测酶活性浓度方法是否准确可靠的基本标准。因为不是任何情况下酶的催化反应速度都能与酶浓度成正比例,当设计不当,所选择条件不合适时,反应速度v虽可能随酶浓度增加而加快,但不成正比例,即v≠k[E],所得结果出现误差,从表17-1可以清楚看到此问题。
方法1所测反应速度(v)和酶浓度间存在一个明显正比例关系,各标本μg/L和酶浓度间高度的一致,相反方法2各标本的U/L不能准确反应酶浓度(μg/L)间的比例关系,浓度愈高误差愈大,例如标本5酶浓度为标本1的5倍。但其反应速度结果却显示为3.5倍,在临床工作中这种误差有可能给医师诊断疾病判断病情带来困难,在科研工作中也有可能导致不恰当的结论。方法3的结果说明在标本1到标本3的酶浓度之间,测定结果是准确的,但在高浓度标本时可能导致误差,这是在实际工作中常能遇到的情况。
表17-1 三个测同一酶方法结果的比较
| 标本 | 酶浓度(μg/L) | 测定结果(U/L) | ||
| 方法1 | 方法2 | 方法3 | ||
| 1 | 1 | 100 | 50 | 10 |
| 2 | 2 | 200 | 90 | 20 |
| 3 | 3 | 300 | 125 | 30 |
| 4 | 4 | 400 | 155 | 38 |
| 5 | 5 | 500 | 175 | 45 |
表17-1还显示了酶活性浓度测定的另一个重要特点,即测定结果U/L的相对性,不同于用质量单位mol/L表示浓度时高度一致性,如用不同方法测5份不同浓度血糖标本。其绝对值(μmol/L)不会有太大差异。但在酶活性浓度测定中往往不存在这样高的一致性。如表17-1三个方法结果可在同一方法内进行比较。而绝不能在方法间进行比较,否则会得出荒谬的结论。这是因为这些方法测的是反应速度,只能用速度单位(一单位表示每一分钟有1μmol/L反应物的变化)来间接表示酶含量的高低,而速度快慢不仅受酶含量多少影响,还与其它很多因素有关,表17-1中三法之所以有这样大的结果差异,很可能是由于三种方法使用了不同种类的底物,由于酶与不同底物亲和性不一样,反应速度的绝对值(μmol min-1)出现差异是不足为奇的。
这些叙述说明了在考虑或设计酶活性浓度测定方法时,重要的是要选择好各种条件,使在尽可能宽的范围内所测的反应速率v和酶浓度E之间存在着正比例关系。
第二节 酶活性测定的常用技术和方法
从广义上说,任何一种可以测定反应物变化速度的分析技术,不论是经典的比色法、容量法,还是现代的仪器分析技术都可用来测定酶活性浓度,但从历史和发展来看,主要还是使用分光亮度法和量气法。
一、量气法
在封闭的反应系统中如有气体变化,通过测量变化后的气体体积或压力很容易计算出气体变化量,这是量气法的基本原理。曾在检验科广泛应用的Van-slyke测二氧化碳结合力的方法就是量气法的一个典型例子。Warburg进一步加以发展,设计出专用于测定酶活性的华勃呼吸仪。这种仪器特别适用于测定那些在反应中产生或消耗气体的酶,例如氧化酶反应涉及到O2的消耗,脱羧酶会产生CO2。但也不仅限于这些酶,科学家采用与CO2气体保持平衡过的重碳酸盐体系,可用来测定各种产生H+的酶反应,如各种还原酶,可使NADH变为NAD和H+,而H+会促使反应体系中重碳酸盐变为CO2气体。
二、比色法与分光亮度法
在20世纪上半个世纪华勃仪得到研究实验室广泛的应用,并在酶学上得到丰硕的成果。但此法操作烦琐,技术要求高而且灵敏度低。临床常规中很少使用。多使用简单易行的比色法测酶活性。在上半个世纪建立了一些适用于常规工作的测酶活性浓度的方法,如测定淀粉酶的Somogyi法,碱性磷酸酶的Bodansky法、King法等等。这些方法都是在酶和底物作用一段时间后停止酶反应,加入各种化学试剂与产物或基质反应呈色,用比色计在可见光处比色,同时将被测物质作标准管或标准曲线,比较后计算出在此段时间内产物生成量或底物消耗量,从而求得反应速率v。
比色法从50年代起逐步被分光光度法所取代。这是因为分光光度法有以下几个显著优点:一是测定范围不只局限在可见光,还可扩展到紫外和红外部分。这就为扩大测定酶范围提供了可能性。二是提供了寻找一类不需停止酶反应就可直接测定产物生成量或底物消耗量方法的可能性。例某一酶催化下列反应A->B+C,A、B、C三种物质用分光光度法的吸收光谱如图17-1所示。
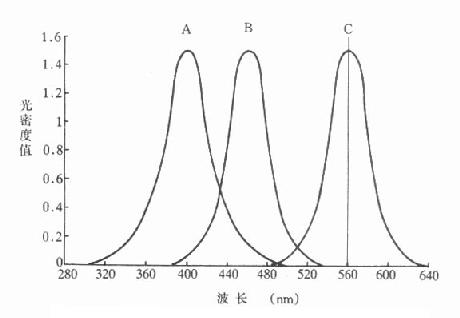
图17-1 A、B、C三种物质的吸收曲线
可以看到C在560nm处有一吸收峰,而A和B在此处无吸光度变化因此无需停止酶反应,只要在560nm处测定吸光度变化就很容易计算出C的变化速度,而且C物质比A、B二物质有更高的吸收峰,即灵敏度最高。
这类方法中最成功的是Warburg在50年代利用NAD(P)H和NAD(P)吸收光谱差异建立的测酶活性浓度方法。NAD(P)H在340nm处有一吸收峰,而NAD(P)在此波段却毫无吸光性。因此建立了一类和原来比色法截然不同方法。不需停止酶反应,在340nm根据吸光度变化,就可观察到酶反应变化全过程。
第三个优点是不需要如比色法那样,作标准管或标准曲线,因为分光光度计使用近似单色光的光源,在此条件下,某一特定物质的吸光度为常数,即人们所熟悉的摩尔吸光度(molar absorbance)。根据此值从吸光度△A/△T不难计算出酶催化反应速度v。
分光光度计的这些简便、准确等特点使它在近年来已逐步取代比色法而成为目前最流行的方法。其缺点是需要精确带恒温装置的分光光度计,在经济不发达地区尚难推广。
分光光度法的技术多样化。设计得当可用于各种酶的测定。表17-2是一些可用于分光光度法的氧化还原物质特性。
除了前述的NAD(P)H系统可用于脱氢酶测定外,可利用黄素单核苷酸(FMN)和黄素腺嘌呤二核苷酸(FAD)来测定各种含这二个辅基的酶。它们的氧化型在450nm有一很强吸收峰,而还原型的吸光度很低,同样细胞色素还原型在可见光有一个非常明显和很窄的吸收峰,都使人们很容易用分光光度法研究这些酶的作用。上述物质主要用于氧化还原酶的测定。
科学家还设计出一系列人工合成酶的底物,用于其它酶的分光光度法测定。例如合成了很多对硝基酚的衍生物,用于各种水解酶的测定。碱性磷酸酶底物磷酸对硝基酚就是一个成功例子。又如测芳香基硫酸酯酶,可使用人工合成的硫酸对硝基酚为底物,其分解产物的吸收峰由原来的278nm变为318nm,Webb成功地在330nm进行此酶监测。
表17-2 一些氧化还原物质的特性
| 物质 | 波长(nm) | 克分子吸光度 | |
| 还原型 | 氧化型 | ||
| NAD,NADP | 340 | 6300 | |
| FMN | 450 | 12200 | |
| FAD | 450 | 11300 | |
| 细胞色素C | 550 | 29500 | 8300 |
| 亚甲蓝(等消光点) | 610 | 41000 | |
| 二氢酚吲哚酚 | 600 | 21000 | |
| 吩嗪甲酯硫酸 | 388 | 1500 | 22000 |
| 抗坏血酸 | 265 | 15100 | |
| 连二亚硫酸盐 | 314 | 8000 | |
| 氰化铁(亚铁) | 420 | 1020 | |
分光光度法的上述原理还可以用于其它酶的测定,如烯醇化酶、延胡索酸水解酶的底物由于含不饱和键,在330nm处有很强吸收峰,而酶作用产物无此不饱和键。则不难在330nm处对这些酶进行直接测定。
此后在分光光度法的发展过程中又导入了酶偶联技术。使得分光光度法几乎能测定所有的酶。因此临床实验室工作者如不能很好掌握分光光度法的技术,不了解各种影响因素,要作好酶的测定是很困难的。
三、荧光法和同位素法
分光亮度法有一个缺点,即灵敏度较低。有些标本中酶浓度很低时往往测不出来。此时可考虑改用荧光法,可将测定灵敏度提高2-3个数量级。如科学家合成了一系列甲基伞形酮的衍生物,可取代对硝基酚衍生物做为一些水解酶的底物,由于水解产物甲基伞形酮有强烈荧光,大大提高测定的灵敏度,就是分光亮度法中最常用的NAD(P)H反应系统,也可改用荧光法,在340nm紫外线激发下NAD(P)H产生强烈的蓝色荧光,而NAD(P)不被激发。此外,还可使用在荧光法基础上发展起来的时间分辨荧光法,例如北京医院就曾利用此种灵敏度极高方法测定很难用其它方法检测的脑脊液中微量的烯醇化酶。荧光法不易掌握,对所用的试剂、容器和仪器都要求很高,否则易产生非特异荧光干扰测定,或者引起荧光的淬灭使测定不准,故此种方法多用于研究实验室,少用于常规实验室。为提高灵敏度,还可使用同位素标记的底物进行酶测定,例如,有人以C12标记的乙酰胆碱为底物测定胆碱酯酶,在酶作用后以离子交换法分离出含C14的乙酸。同位素方法由于对人体有害,操作麻烦,目前已很少使用。
四、其它方法
离子选择电极法,旋光法等有时用于测定特定的酶,当酶反应牵涉到有酸碱变化时,很容易用pH仪直接观察酶反应过程中H+的变化。直接用pH仪测酶反应有两个缺点:一是随pH变化,会偏离酶作用的最适pH值,不可避免地引起酶反应速度变慢。其二是如测定的标本不是纯酶时标本中其他蛋白及其它有缓衡能力的物质将会影响所测pH变化的程度。此时如改用电位滴定仪则更为适合。此仪器可在酶反应过程中不断向反应体系中加入酸或碱以维持反应体系pH的恒定,而加入的酸碱量只与体系中H+变化量相关,和反应体系中缓衡能力无关。
同样如酶反应中有O2变化,可使用氧电极来监测酶反应过程,这可用来测定葡萄糖氧化酶活性,Chappell还成功地将此技术用于测线粒体的氧化能力。还曾有人尝试用二氧化碳和氨电极测酶,由于这些电极反应时间较慢,不利于检测酶反应速度。有些酶的反应物为光学异构体,则可根据旋光度变化来追踪酶反应。某些反应物如羟基酸本身虽无旋光性,但与钼结合后产生很高的旋光性。根据此特性建立了测定延胡索酸水合酶的方法。还有个别酶的测定使用了极谱法、高效液相色谱法等。总之,实验室工作者完全可以根据实验室现有仪器和技术,创造性地建立一些新的测活性浓度的方法。
第三节 酶活性测定条件的选择和限定
引言中已强调了仅在一定条件下酶反应速度v才和酶量成正比例。一定条件是什么?这些条件之间有无主次关系?如何选择合适的测定条件?这些都是实验室工作者在设计或选择测酶活性浓度方法时必须面对的问题。
首先可从理论上探讨在什么特定情况下的反应速度才可能与酶量成正比例。酶的整个反应过程可简化如下:
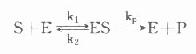
式中Kp可看成为ES的解离常数.Michaclis和Menten通过推导可得出下式方程式
![]()
此式中V为最大反应速度,对一定浓度的酶而言为一常数。Km为实验室工作所熟系的米氏常数。上式也可改写为:
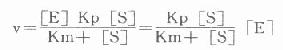
Kp和Km虽为常数,但[S]为变量,因此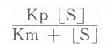 在一般情况下不可能为常数,即v≠k[E],从理论上很清楚说明反应速度v和酶量E之间在绝大多数条件下只存正比,即v∝[E]。而非正比例关系。但在底物[S]浓度很大,远远超过Km的特殊条件下Km值由于相对很小可忽略不计。此式可变为:
在一般情况下不可能为常数,即v≠k[E],从理论上很清楚说明反应速度v和酶量E之间在绝大多数条件下只存正比,即v∝[E]。而非正比例关系。但在底物[S]浓度很大,远远超过Km的特殊条件下Km值由于相对很小可忽略不计。此式可变为:
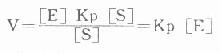
由于底物[S]浓度很大,使酶饱和。ES已达极限,反应速度不再增加,故此时反应速度为最大反应V。即从理论上说只有测定的是酶最大反应V。此时反应速度才和酶量E成正比例。也只有在此基础上建立起来的测定方法才是可靠的、准确的。
Kp是酶学中很重要的一个常数,即周转数(Turnover number),从上式可得出。

其含义为每分钟每分子酶转换底物分子数。酶的Kp数值约在50-105min-1之间。碳酸酐酶是目前已知最高酶变率的酶(36×10-6min-1)。Kp的倒数代表每一酶催化循环的时间,以碳酸酐酶为例
1/Kp=lmin/136×106=0.028×10-6min=1.7μs
即每隔1.7μs,一分子酶就和一个底物结合,反应一次。
国际临床化学联合会(IFCC)和不少国家包括我国的一些学会建立了或正在建立测定酶活性浓度的推荐或参考方法。都毫无例外地提出测酶活性浓度方法所选择的测定条件应是酶反应的“最适条件”以保证酶具有最大的催化活性。实质上就是基于以上理论考虑,要测定酶的最大反应速度V。
我国检验学会文件中对最适条件是这样提出:①选合适的底物、辅因子、活化剂、变构剂的种类和浓度。②指示酶和辅助酶的种类和浓度。③反应混合液的最适pH,缓冲液种类和浓度。④其它影响酶活性的因素,如去除各种抑制剂。并提出:在某些情况下,为了获得更好的测定重复性或更大的临床价值,可考虑对上述“最适条件”作适度的修改。
一、底物、辅因子、活化剂、变构剂的种类和浓度
这一项包括几种因子,其中最重要的是底物种类和浓度,这是每种测酶活性浓度方法中首先需要选择的项目,选择恰当与否,对该酶测定至关重要。后面几项则不是每种酶测定都会遇到的选择。
(一)选合适的底物种类和浓度
如所测的酶专一性不强,可作用于多种底物,首先就需决定选择哪一类底物作为测此酶的底物。如该项酶测定主要用于临床诊疗工作,则首先应考虑选有较高诊断价值的底物。例如临床上测定酸性磷酸酶主要用于诊断前列腺癌,所选的底物应对前列腺酸性磷酸酶有较高的特异性。不易被其它组织如红细胞、血小板中酸性磷酸酶所作用,在常用的底物中以麝香草酚磷酸盐最符合上述选择条件。但如进行酶基础研究,探讨其在体内作用时,则选择酶在体内的生理底物更为合适,一般而言在多种底物中,Km最小的底物往往是此酶的生理底物。
选择Km小的底物测定酶还有一个优点,就是在最大反应速度V的底物浓度也将最低。在实际工作中可能意味着试剂成本可能较低,不易出现底物难溶解的困难。
底物专一性强的酶虽然不面临上述选择,但只要所测酶催化的是可逆反应,则和专一性不强酶一样,都面临着是选择正向还是逆向反应来测定酶,不同反应方向的底物必然是不一样的。这方面的选择更多从技术和实用方面加以考虑往往选择速度较快的方向,因为这可以提高测定的灵敏度,如测定肌酸激酶(CK),由于磷酸肌酸价格昂贵且不稳定,早期多选用正向反应,底物为肌酸和ATP速度太慢,只是逆向反应的1/6,测定灵敏度太低,以致正常标本结果误差很大。近年来都改用逆向反应测定CK,明显提高CK测定的精密度和准确度。在此问题上,实用考虑也起了不小作用。例如测定乳酸脱氢酶(LD)时,如考虑到正向反应明显快于逆向反应,则应考虑以丙酮酸和NADH为底物,这正是IFCC和其它一些学会推荐的方法。但目前市售的测LD的试剂盒不少都以乳酸和NAD+为底物。因为NAD+价格明显低于NADH,并且稳定可长期储存。科研工作中如需测LD,还是最好采用IFCC的方法。
确定底物种类后,重要的问题是选择底物的合适浓度,在讨论此问题之前,有必要先简单复习一下底物浓度和酶反应速度之间的关系,因其不同于其它化学反应有其独特之处,图17-2很形象表示出这种差异。
反应物浓度[S]
图17-2a 反应物浓度对化学反应的影响
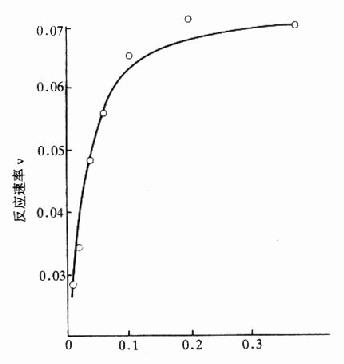
底物浓度[S]
图17-2b 底物浓度对酶反应的影响
图a表示在一般化学反应中遵循质量作用定律,反应速度随反应物质浓度增加成正比例增加,图b则表示酶反应中底物浓度对反应速度的影响,当底物浓度很低时,酶反应速度几乎随底物增加成正比例加快,进一步升高底物浓度,虽然反应速度也加快,但增加速度愈来愈慢,不成比例。到一定程度,反应速度趋于恒定为一常数,实际上就是趋向最大反应速度V,测定此处的反应速度V,最能准确地反映酶量多少。在此处底物浓度变化,对反应速度影响很小。
Michaclis和Menten二式首先在本世纪初对酶反应的此项独特现象从理论上加以合理解释。并推导出著名的米-门方程式:
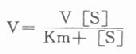
此公式对选择酶测定的底物浓度有重要的指导作用。Km对每一种酶而言在一定条件下为一常数,并可从上述方程式求出,在反应速度为最大反应速度V一半时,代入上式得:
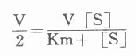
Vkm+V[S]=2V[S]
VKm=V[S]
[S]=Km
换言之,当酶促反应速度为最大反应速度一半时,此时的底物就相当于酶的米氏常数Km。以mol/L表示之,大多数酶Km在10-3至10-5mol/L之间。
当我们知道或计算出某一酶的Km后就可以计算出不同底物浓度和Km间的比值,化入米-门方程式就可计算出此时酶促反应速度相当于最大反应速度的百分比,见表:17-3
表17-3 底物浓度与Km比值相当的最大反应百分比
| [S]/Km | V/V×100 |
| 100.0 | 99.0 |
| 10.0 | 91.0 |
| 1.0 | 50.0 |
| 0.1 | 9.0 |
| 0.01 | 0.99 |
依据上表一般都认为酶测定时底物浓度最好为Km的20倍乃至100倍。在实际工作考虑到底物溶解度的限制,价格的昂贵,可将底物浓度降为Km的10倍。再低就不适合酶的测定,可能产生较大误差。
以上规律适用于大多数酶。一些酶还有其特殊情况,常能遇到的是当底物浓度增加到一定范围后,酶反应速度不仅不增加,反而下降。如乳酸脱氢酶,当丙酮酸浓度超过一定量时,酶活性反而下降,故丙酮酸浓度不能过高。
以上介绍的米-门方程式只适用于单一底物的酶,不少酶催化反应二底物乃至更多底物,有关二底物的米-门方程以及此时底物浓度的选择可参考一些有关书籍。实际工作中最简单的作法是先将其中底物之一的浓度选得很高,使酶饱和,然后求出另一底物的表观Km,反之亦然,最后按上述规律决定二底物的合适浓度。
(二)辅因子、活化剂的种类和浓度
从广义上说,凡能促进酶及反应物进入活化状态从而加速酶催化反应的物质都能称为辅因子,它包括种类很广的物质。英汉生化词典将辅因子(cofactors)定义为“一种酶的活性所需要的一种非蛋白质成分”。这种辅因子可能是一种金属离子激活剂或一种有机分子(辅酶)。它们或松或紧地与酶相结合;紧密接合的辅因子称为“辅基”。将激活剂(activator)定义为“一种金属离子,作为一种酶的辅助因子。”
在前面已谈到,测酶活性浓度时应该在最适条件下测酶反应的最大速度。如测定酶需要辅因子,活化剂时,应选择合适的种类和浓度加入到测定酶的体系中,在些过程中可能会涉及到下列四类物质。
首先是辅酶(Coenzyme)。很多酶反应都必须有辅酶参加。如相当部分的还原酶需要辅酶Ⅰ(NAD+)和辅酶Ⅱ(NADD+)参加。ATP是激酶(Kinase)反应中不可少的辅酶,这类物质一般是小的有机化合物,和辅基不同点是与酶蛋白结合很松弛,用透析和其它方法很易将它们与酶分开,尽管它们不用于酶的底物,特异性不强,往往参加一系列酶反应和代谢过程。但在作用方式上和底物类似,在酶反应过程中与酶结合、分离及反复循环。在方法学上,可将它们按底物处理,即可按米-门方程式求出其Km,按底物规律选择其浓度。
第二是辅基,辅基虽也是小的有机化合物,但却是酶蛋白不可分割部分,不含辅基的酶蛋白称为脱辅基酶蛋白(apoenzyme),没有催化活性,必须加入足量辅基,和它结合成为全酶(holoenzyme),才可能有催化活性。最典型的例子是各种转氨酶需要磷酸吡哆醛为其辅基,在反应过程中。氨基酸将其氨基交给吡哆醛变为吡哆胺,本身接受醛基成为酮酸,然后吡哆胺将氨基转交给酮酸生成氨基酸,本身又变回吡哆醛。从此机制不难理解为何脱辅基酶蛋白无催化活性。
辅基和辅酶不同点不仅表现在和酶蛋白结合紧密,不易为透析所除去,和酶作用方式也不同,不似辅酶迅速与酶结合,又迅速分解为酶和产物,辅基显著特点之一是与酶结合需要一定时间,因此在酶测定时,如按底物一样来处理辅基,在酶反应开始时才加入辅基,则开始阶段反应较慢,经过一段延滞期后,反应才达到应有速度。因此在酶测定时,往往是先加入足量辅基,作用一定时间如10分钟后,再加入底物开始酶反应。
很多辅酶和辅基来自维生素或结构中含有维生素,如NAD和NADP来自维生素尼克酸,转氨酶的辅基磷酸吡哆醛来自维生素B6(吡哆醇)。来自维生素B1的焦磷酸硫胺素是丙酮酸脱羧反应的辅羧化酶。从维生素B2(核黄素)形成的FAD,FMN是很多呼吸链上酶的辅基,维生素H(生物素)在羧化和脱羧作用中起辅基作用,叶酸衍生物参与一碳基团的转移。维生素B12的衍生物钴胺酰胺参与酶催化的异构反应。
第三类物质是离子,很多酶需要特定离子帮助才能使其反应达到最大速度。最常见的是二价金属离子如Mg2+、Zn2+、Mn2+、Ca2+、Fe2+等。所有转移磷酸的酶,如激酶类和碱性磷酸酶的反应都需要Mg2+的参加。所以如在反应体系中加入金属螯合剂如ED-TA或以他们为抗凝剂常抑制一些酶的活性。因此酶测定的标本多采用血清而不采用血浆,以外还有单价的K+。如丙酮酸激酶反应同时需要K+和Mg2+。淀粉酶催化的反应需要阴离子Cl-,5mmol/L氯离子可加速淀粉酶的反应三倍。
离子加速酶反应的机制是多种多样,Zn2+是碱性磷酸酶和羧基肽酶A整体结构的一部分,可稳定酶的三级和四级结构。Cl-加速淀粉酶反应的机制可能与它能与酶分子中某些带阳电荷基团结合有关,改变了在催化作用中起重要作用的基团的电离常数,有些离子可使酶分子带阳电荷,和带阴性电荷的底物结合,在酶反应中起了桥梁作用。必须注意的是,过量的离子往往抑制酶反应速度,在测定酶时要选择合适的浓度。
第四类是一些无法包括在前三类的其它加速酶反应物质,如巯基化合物可稳定酶的双硫键。在肌酸激酶反应体系中加入它将明显增高酶活性,并且随所加巯基化合物的不同,增加的速度也有差异,所以在测肌酸激酶时要选择好巯基化合物的种类与合适的浓度。
还有所谓的表观激活作用,该物质并不是真正加快反应速度,而是由于和抑制剂作用抵消了抑制作用,从表面上看似乎是加速了酶的反应作用。
(三)选变构剂的种类与浓度
有一类特殊的酶叫变构酶(allosteric enzyme),其反应速度和底物关系不同于一般酶的双曲线,而出现S形曲线,这是由于这种酶具有二个或更多个独特的部位-或是相互作用的催化部位,或是相互作用的催化部位与调节部位,其结果是酶与底物作用速度将受另一类物质的影响,这类物质命名为变构剂或效应物。根据作用不同又分为变构激活剂和变构抑制剂或者正负效应物,而不同浓度的变构剂会明显改变曲线形状。变构酶在物质代谢中有重要的调节作用。在测定变构酶活性浓度时,必须选择好变构剂种类和浓度。一般来说应选择在饱和浓度的变构剂的条件下测定构酶的活性浓度。
二、指示酶与辅助酶的种类和浓度
采用酶偶联法测定酶活性浓度时,反应体系必须加入指示酶,有些方法还加用辅助酶,在选择或设计此类方法时,必须考虑合适的酶和浓度,有关问题将在后面“连续监测法酶活性浓度”一节中详加讨论。
三、反应体系的最适pH、缓冲液的种类和浓度
固定酶反应的其它条件,在不同pH处测定酶反应速度,可得各种类型的酶活性与pH关系见图17-3A。
图17-3A 酶活性与pH的函数关系曲线可能具有的几种形状
最常见的是(a)的对称钟形曲线,少数的如(b)(c)在一侧即偏酸或偏碱处活性最高。生化学家将酶活性最高处的pH称为最适pH。一般来说,血清中大多数酶最适pH接近中性(pH6.5-7.5)。有些酶在最适pH处活性变化尖锐明显,也有些平坦宽广。测定酶活性浓度时一定要选择在最适pH处,不仅因为此处酶反应速度最大,测定灵敏度最高,还因为此处酶活性变化的斜率最小,如反应体系中出现pH变化时,对测定结果影响最小。
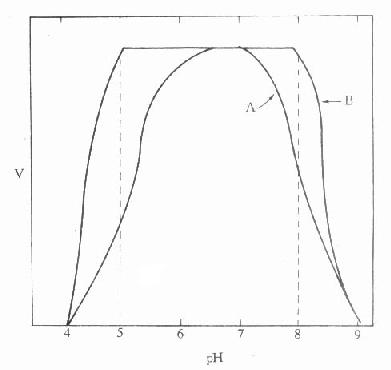
图17-3B pH对酶活性及稳定性的作用
曲线A:V对pH作图曲线B:酶先在pH5及pH8预孵育后,在pH6.8测活性
氢离子可以通过多种途径影响酶催化反应。如在脱氢酶反应中往往需要氢离子参加,也可能产生氢离子,从理论上可以将氢离子看成是该反应的底物和产物之一。此外,当pH变化时,可影响到底物、酶、酶-底物复合物的解离状态和构型,甚至还可能影响到各种辅因子,从而影响酶活性。
PH对酶还有一个重要的作用,就是影响酶的稳定性。图17-3B介绍一个有关实验。
图中曲线A是最适pH实验的结果,出现典型的钟形曲线,其最适pH为6.8,高于或低于6.8时,酶反应速度下降,下降原因可能与上述诸因素有关,但也可能由于酶稳定性下降失活所致,或者是二类原因之总和。通过曲线B的实验,可将上述二类原因分清。此时先将酶与不同pH底物缓冲液温育一段时间,此时间一般与测定时间相当,然后再将pH调回最适pH处测其活性。从曲线B可以说在pH5-6.8以及pH6.8-8.0之间酶活性的下降与酶灭活关系不大,酶在最适pH处储存常数稳定。但有些酶贮存在最适pH时,并不一定比在其它pH处更稳定。
最适pH并非是酶的特征性常数,易受多种因素影响而改变,如缓冲液的种类、底物浓度、温度等,在研究pH对酶稳定性影响还应注意到酶浓度高低,在低浓度时,酶易解离为单体,常比多聚体更易灭活。还应注意试剂中各种防腐剂和其它添加剂的影响。
最后必须强调的是本节讨论的pH并不是指底物缓冲液的pH,而是底物和标本混合后的pH对反应速度的影响,由于实验室所测的标本很少是纯酶样品,多为体液或组织粗提液。它们也是有一定pH值和缓冲能力的缓冲液,和底物缓冲液混后后,不一定维持住原来pH,特别是当二溶液的pH相差甚远时,变化更大。例如碱性磷酸酶最适pH为10.2,虽然底物pH也是10.2,但血清标本pH为中性,混合液的pH必然低于10.2,降低程度取决于血清的pH和缓冲能力。从而影响酶测定结果,临床观察证实:当血清标本放置过夜后用金氏法再测碱性磷酸酶时,结果常升高。有人认为这与血清放置过长,由于CO2逸出引起血清pH偏碱有关。
在下列情况极易引起混合液pH偏离底物缓冲液的pH:一是标本用量太大,如以前有些方法,为节省底物,底物缓冲液用量和血清用量相等,此时混合液pH可能在此二液pH之间,有可能引起较大测定误差。所以在文件中规定“标本在总体积中的比例应在10%以下”。就是要尽量减少标本中各种物质对测定结果的影响。二是底物缓冲液缓冲能力太低。如金氏法测碱性磷酸酶时使用的碳酸盐缓冲液的浓度为0.01mol/L,浓度这样低的缓冲液,必定要受标本pH的影响。
过去长期以来,认为缓冲液作用就是维持酶反应的最适pH,忽视了缓冲物质对酶反应的其它作用和影响,Howell等研究了酶活性在三种不同缓冲液的变化情况见图17-4。
图17-4 各种缓冲液的温度及pH的关系
TRIS:三羟甲基氨基甲烷0.1mol/L
TRA:三乙醇胺0.1mol/L
三种不同缓冲液不仅最适pH不一样,最大反应速度也有差异,从测定酶活性浓度角度,应该选用枸橼酸缓冲液,类似现象在大多数酶都能观察到,仅是程度不同,其中以碱性磷酸酶活性受到缓冲液种类影响最为显著,在二乙醇胺缓冲液所测酶活性约比在碳酸盐缓冲液所测的高2倍。
由于缓冲液含有大量离子,或影响酶蛋白的构型,或影响底物的解离程度等等,从而影响酶活性。Allert曾拟定了一种作用模式,并进行数学计算,得出相应方程式来说明反应体系中电解质会影响最大反应速度V,且不同浓度电解质的影响有差异。因此在方法设计时,选完缓冲物质后,还必须了解不同浓度缓冲液对酶活性的影响。一般而言,随缓冲液浓度增加,电解质干扰酶和底物结合,酶活性将逐步下降,所以选择缓冲液浓度时,常需在浓度较高以保持足够缓冲能力和浓度较低以免抑制酶活性两个相矛盾作用之间取得平衡。
在目前酶测定常用的一大类所谓生物缓冲物质,如图中的Tris、三乙醇胺受温度影响较大。因此在我国学会文件中指出“配制缓冲液时不仅要指出其pH值,还应说明配制温度,以避免因温度不同而引起的误差。”在此图中还可看到,即使是同一种缓冲液,随其它物质存在,也有可能改变温度的影响。因此在实际配制缓冲液时,应首先将配制溶液温度调节到规定温度,然后在pH计监测下加入相应的酸或碱将溶液pH调到要求范围。不控制温度,不用pH计盲目按一些书提供的量配制缓冲液是不可取的。
四、其它影响酶活性因素
除了上述主要因素外,一些其它因素也会影响酶活性,如反应体系中含有氧化剂,可氧化酶蛋白中甲硫氨酸、色氨酸和酪氨酸残基,使酶失活,试剂盒中常用的一些表面活性剂如Tween-80、Brig-35等会抑制一些酶的活性。
其它因素中,对酶测定而言,最重要的是抑制剂的作用,抑制作用是酶学中的一个重要研究内容。但如只涉及到设计和选择测定酶的方法,则比较简单,既然要测定的是“最适条件”下的最大反应速度,那么不论是可逆或不可逆抑制剂,也不论是竞争性、非竞争性、反竞争性抑制剂都应在反应体系中设法除去。此问题在测定尿液中尤为突出。尿中排出大量代谢产物,不少物质会影响酶,如脲可使酶解聚、磷酸盐抑制磷酸酶活性。在病理情况下尤其使用各种药物后,使情况更为复杂,此时尿中可出现不少正常情况下不出现物质,即可能抑制酶活性,也可能激活酶活性。为使脲酶测定结果有一定可比性,最好在测定脲酶前,通过透析、凝胶层析或超滤将小分子化合物与酶分开,这样较为可靠。
五、温度的控制
要了解此问题,应注意温度对酶活性影响的双重性。首先,化学速度随温度升高而加快,酶反应也不例外。Q10值即温度增加10℃,化学速度的变化率。酶的Q10值约在1.5-2.5之间。
温度与反应速度之间关系,可用Arrhenius方程式来表示:
Logk=-E/2.303RT+常数
式中k是反应常数,T为绝对温度,R为气体常数。E为活化能也是常数。在酶反应中V=K·[E]。所以在酶反应中,LogV和1/T作图为一直线,斜率为-E/2.303R。通过每一直线斜率不难计算出该酶反应的活化能E。
但是,测定酶时都需要酶和底物在一定温度下作用一段时间,在此期间,温度有可能使酶失活,不同酶在此方面差异很大,有些酶如胎盘碱性磷酸酶在56℃放置15分钟,活性也不下降,有些酶如酸性磷酸酶,37℃1小时后,失去50%酶活性。酶的稳定性还与其它因素,如作用时间、pH、有无底物、有无其它蛋白等有关。同为碱性磷酸酶,在肝提取液中远不如血清中稳定,37℃作用15分钟后肝中此酶活性将显著下降。又如将血清pH降低在pH6.0以下,血清中酸性磷酸酶将长期稳定。
图17-5 预温15分(37℃,pH9.9)对ALP活性的影响
○与●表示肝ALP活性不预温和预温的变化
△与▲表示血清ALP活性不预温和预温的变化
图17-5形象说明预温过程对酶活性的影响。
目前常规工作实验室越来越多的使用37℃,一些国家已明确推荐使用此温度。这更多地是从实际工作方便来考虑。因为温度升高,反应速度快,灵敏度高。同时延滞时间和测定时间都可能缩短。从而有利于提高工作效率。尤其目前在常规实验室中广泛使用全自动生化分析仪,有高效率的恒温系统,使反应系统很快升温并维持在37℃,可避免温度差异引起的误差。但在37℃时,不可避免地一些酶可能失活。
早期曾推荐使用25℃为酶测定温度,其优点为接近室温,反应体系温度很容易平衡到此温度,这对于当时使用无有效控温系统的分光光度计来测酶活化性有其特别方便之处。但温度低。反应太慢。在有些温热地区,当室温超过25℃时,还需使用降温系统很不方便。因此目前已很少有实验室采用此温度。
IFCC推荐的30℃,兼顾了上述二温度的优点,即保证了一定的反应速度。又无酶失活之扰,此外还有一个上述二温度没有的优点,即可以镓作为此温度的基准物质,纯镓的熔点为29.77℃。保证了测定系统计30℃的高度准确性,而37℃和25℃至今尚无一个最准确的确定方法。
30℃和37℃是目前使用最广泛的二种测定酶的使用的温度。在比较不同实验室测定结果时,一定要注意由此引起来的差异,否则将导致临床诊断的困难和误差。一些作者提出使用二温度间的转换系数使二温度测定结果的比较变为可比。虽然一些作者持怀疑态度,用一个系数能代表所有标本中的变化情况吗?最新研究证实只要所用方法合适,人类血清中酶对温度变化的反应差异不大,在无法统一温度情况下,这还不失为一权宜之计,常用酶的温度转换系数可参见表17-4:
表17-4 常见酶温度转化系数
| 25℃ | 30℃ | 37℃ | |
| CK | 0.64 | 1.00 | 1.56 |
| LD | 0.75 | 1.00 | 1.44 |
| ALT | 0.76 | 1.00 | 1.38 |
| AST | 0.73 | 1.00 | 1.52 |
| ALP | 0.78 | 1.00 | 1.30 |
| GGT | 0.73 | 1.00 | 1.31 |
| CHE | 0.81 | 1.00 | 1.26 |
| HBDH | 0.85 | 1.00 | 1.10 |
不论选用何种温度测酶,由于酶反应受温度影响很大,在测定时间内,反应体系的温度变化应控制在±0.1℃内。应用国产普通恒温水浴箱来测酶活性是不合适的,应使用带有搅拌器的高级恒温水浴。
第四节 测定酶活性浓度的两大类方法
一、固定时间法(取样法)
前面已经提到,早期临床实验室测酶活性时常使用比色法,先让酶与底物在一定温度下作用一段固定的时间,然后停止酶反应,加入试剂进行化学反应呈色测出底物和产物的变化。由于比色法灵敏度较低,或由于手工操作不易控制好,常定在30分钟左右。历史上这类方法常被命名为“终点法”、“二点法”、“固定时间法”和“取样法”。大多数文献使用“固定时间法”。此命名指出了用这类方法测酶活性浓度,必须保证酶和底物在所选定的温度下作用时间要很精确,否则将引起较大误差。但“取样法”的命名更具有特征性。因它指出此类方法和下面将提到的另一类连续监测法相比较,最基本一点的是此类方法停止反应后才测底物或物的变化。很难进行连续监测,而另一类方法则不需停止酶反应,就可测定反应物的变化,很容易观察到反应的整个过程。后一类方法开始于50年代,Warburg首先用分光光度计。在340nm处直接监测到反应物NADH的动态变化过程。由于不停止酶反应,不需添加其它呈色试剂。测定方法简单,结果准确,逐步取代了“取样法”成为目前测酶活性的主要方法。
由于经历了一个发展过程,命名比较混乱而且大部分不甚确切,IFCC建议命名为“连续监测法”,不用目前广为流行的“动态法”术语。其原因是目前测酶活性浓度方法都是基于化学反应。图17-6是一个典型的化学反应过程。
图17-6 动态和平衡概念的示意图
可以看出整个反应过程,可以大致分为二个时期。一开始为动态期,化学反应按一定速度进行,反应物浓度随时间而变化,且变化程度不断变小,到一定时间。化学反应或者达到平衡,或者反应达到终点,此时反应速度为零,反应物浓度不变。最近IFCC文件按所用方法所测定的反应是在达到平衡前还是平衡后。分别命名为动态法或平衡法。一般文献往往将后一类方法称为终点法。
二、连续监测法与反应进程的分析
从这个权威性的方法分类来看,前面所提到的二类测酶活性浓度方法都应归纳为动态法。目前广为流行的“动态法”命名显然是不确切的或者是不合适的。应该更多采用IFCC文件中建议的“连续监测法”命名。
从酶反应曲线来比较此二类方法。图17-7是一个典型酶反应过程。
![酶促反应中基质[S]、产物[P]](/lilunshuji/linchuangshengwuhuaxue/linchuangshengwuhuaxue177.jpg)
图17-7 酶促反应中基质[S]、产物[P]
和反应速率V在不同时间变化的模式图
在酶作用下,底物[S]浓度不断下降,随之有相应产物[P]产生。不少酶在反应一开始的阶段,由于各种因素影响,反应速度较慢,称为延滞期,随后在过量浓度的底物存在条件下,酶反应以恒定的速度进行,不受底物浓度变化的影响,这段反应称为线性反应期或零级反应期。随时间延长,反应速度除与酶量有关,还与底物浓度有关。即v=k(a-x),式中a为原来底物浓度,x为消耗的底物浓度,如反应物浓度项上指数为1,称为一级反应,假如反应速率受二种或二种以上物质浓度的影响,则各个反应物浓度项上指数总和作为反应级数,可以分别为一级、二级或多级反应,总的将这段反应时期称为非线性反应期。
很明显如测反应速度的目的是为了从此推算出酶含量的多少,则只有测线性反应期的反应速度才能作到此点。在延滞期、非线性期测的结果不准确,一般是偏低的。
取样法由于方法学上的限制,一般只测二管:一为没发生酶反应的对照管,另一管测酶作用一段时间后反应物的变化,实际上测得是平均速度,而且二管间隔时间都较长,在半小时左右,图17-8中t,t分别代表不同时间二管反应物的量,虽然从t到t反应物变化量是相同的。但实际反应过程是多种多样的。
只有当反应如曲线A时,用“固定时间法”才能测出真实的酶活性,实际工作中是很少见的,图中曲线B代表了最常见的反应情况,在一个很短的线性反应期后在大部分测定期间内主要为非线性反应期。曲线C说明在测定期间包括了延滞期,曲线D则不仅包括了延滞期,还包括非线性期,在这些反应中如用固定时间法来测定,结果是不够准确的,一般是偏低的,而且酶浓度愈高,偏离程度愈大。
假如从上节叙述中得出一个重要结论:即在设计和选择测定酶活性浓度方法时必须是在“最适条件”下测定最大反应速度。那么从这段叙述中可得出一个对上述结论一个很重要的补充,即所测的最大反应速度还应该是初速度即V。
理论上的V在实际工作中是不存在的,必须让酶和底物作用一段时间,消耗掉一定量的底物,才能测出反应速度,一般说,如消耗底物在5%内所测到的反应速度都可认为是初速度,如底物浓度很高时,底物消耗在20%以内的反应往往还在线性反应期。
连续监测法能满足上述重要要求。由于不需停止酶反应。很容易得到整个酶反应曲线,然后根据反应曲线情况,避开延滞期和非线性反应期,只在线性反应期测定最大反应初速度。同时由于分光光度法的高灵敏度,自动生化分析仪的高精密度,连续监测法能在很短时间,甚至在1分钟反应时间,准确测定反应速度,从而求出准确的酶活性浓度。“固定时间法”测定时间长达30分钟,很难满足上述方法学上测定初速度V的要求。
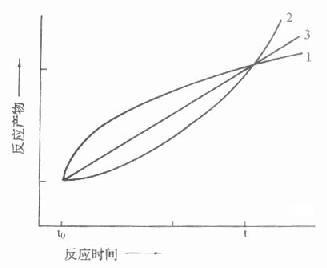
图17-8 二点测定法中可能引起的误差
从理论上说,只有当测定的是初速度,米氏方程式才能成立,并可能测出真正的Km。用“固定时间法”测出的速度受到逆反应速度的影响。可以下式表示有逆向反应存在时的酶反应过程。
k1 k2k3
E+S→ES→EP→E+P
← ← ←
K-1k-2k-3
存在着EP和P时,就会出现逆反应,此时所测的为一表观速度(Va)或反应的净速度。
Va=k2[ES]-k-2[EP]
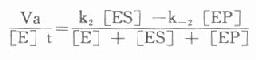
[E]t代表酶总量,在反应过程中包含了游离酶[E]、酶底物复合物[ES]和酶产物复合物[]EP]。
Ks为[ES]的解离常数,Kp为[EP]的解离常数,故:

代入上式得:
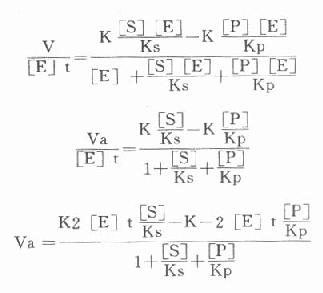
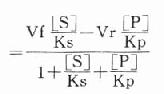
根据Maldanc公式Keq=VfKp/VrKs得:

在反应特定时间,产物[P]为一定值,上式得:
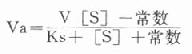
此公式从理论上说明,如所测反应有逆反应存在时,不存在原来的米-门方程式。用各种作图法很难求出准确的米氏常数。如果说所测酶反应不仅存在着逆反应,产物还抑制了酶反应,则动力学公式将更为复杂。这就是为什么不仅在建立方法时,就是在研究酶的动力学时,也总是希望所测量的都是酶反应的初速度。
以上这二类方法并不是概括了所有测酶活性浓度的方法,也并不排除在非线性反应期测酶活性浓度的可能性。如固定底物变化量,则不同标本达到此变化终点的时间,与酶量成反比例,Somogyi曾提出一种测淀粉酶的方法,酶与底物混合后。每隔半分钟取一滴出来加入碘液呈色,观察颜色由紫色变为红色时间,也就是淀粉酶全部水介变为糊精的时间,并由此时间的长短计算出酶含量高低,这类方法不需使用很高浓度的底物,有其独特之外,在自动生化分析仪中也曾使用类似的方法,值得人们重视。
三、连续监测法测酶活性浓度
连续监测法具有众多的优点,随着科学技术的发展,自动生化仪的使用,正在逐步取代“固定时间法”,发达国家从50年代到70年代末用了约20余年的时间,我国从80年代初开始使用连续监测法,虽然发展很快,但至少仍有不少地区医院实验室仍使用老的方法,就是已使用新法的实验室,也不一定掌握得很好,本节将比较详细地从分类,酶偶联反应,测定注意事项等方面对连续监测法加以介绍。
(一)连续监测法种类
⒈直接连续监测法这类方法是使用各种分析手段,如分光亮度法、荧光法、pH计、旋光计、电导仪等等,在不停止酶反应条件下直接测反应体系中底物或产物的变化,从而计算出酶活性浓度,其中尤以分光光度法应用最多,应用最广的有NAD(P)H反应系统,可以测定大部分的脱氢酶,还有的是所谓“色素原”底物,其本身为无色或微黄色,在酶作用下生成有色化合物,适用于测定水解酶和一些转移酶。
⒉间接连续监测法直接法试剂简单,操作方便,可惜的是只有当底物与产物之间,在光学性质或其它理化性质有显著差异时,才有可能使用此法。到目前为止,大部分酶都无法用直接法测定。
科学家还曾设法在原来反应体系中添加一些试剂,这些试剂必须不与酶作用也不影响酶活性,同时又能与酶反应物迅速作用,产生可以被直接测定的物质,典型的例子是Ellman的胆碱酯酶测定法,底物为硫代乙酰胆碱,酶水解产物硫代胆碱与添加的二硫代硝基苯甲酸(DTNB)作用立刻生成黄色化合物,可在412nm用分光光度法连续监测。
实际工作中,更多的是在反应体系中加入另一酶试剂。进行适当的酶促反应,完全可以胜任上述工作。目前酶偶联法已经成为应用最广、最频繁测酶活性浓度的方法。
最简单的酶偶联反应为以下模式:
![]()
Ex为所要测定的酶,A、B二物质分别为其底物和产物,对此二物质变化,无法直接监测,此时可加入其它酶,其底物为Ex的产物B,其反应产物C可直接监测,这样通过第二个酶反应有可能推测出第一个酶的活性浓度,外加的第二个酶称之为指示酶Ei。
如果一些酶反应找不到合适的指示酶与其直接偶联,此时往往可以加入另一种酶,将二者连接起来,模式如下:
![]()
将这种联接的酶称之为辅助酶(Ea)。个别情况可能使用二种乃至二种以上的辅助酶。
酶偶联反应与一般酶反应的一个重要区别处在于其有一个明显的延滞期。酶偶联反应一开始只存在着底物A,不存在指示酶的反应,随着产物B的出现和增加,指示酶反应随之加快,Ex和Ei反应速度相等,也就是达到稳态。从酶反应开始至稳态期间,指示酶反应较慢且不稳定,称为延滞期。显然这期间指示酶反应速度不能代表测定酶量多少。设计或选择酶测定方法时,如果用酶偶联法,延滞期越短越好,测定时间一定要避开此期。
是不是所有类型的酶偶联反应都可用来测酶活性浓度?回答是否定的。因为测酶的活性浓度是依据测定酶反应速度——△A/△t或△B/△t求出。在酶偶联法,此值无法直接求出,而是通过测定指示酶反应△C/△t间接求出,要使酶偶联法测得的酶活性浓度准确可靠,则Vind=Vx。换言之,指示酶的最大反应速度必须等于或接近测定酶的最大反应速度。
当测定酶的反应速度明显大于指示酶,此时A很快转变为B,由于指示酶反应慢,中间产物B大量推积,最终产物产生速度明显慢于底物A的消失速度。
当指示酶速度加大后,中间产物B堆积减小,指示酶反应速率偏差程度也变小。只有当指示酶大量存在时,出现产物B就能将其迅速变为C。
可以看出在延滞期后(即B达到高峰后的时期),C和A的变化速度非常一致。也就是只有在些情况下,才是测定酶浓度的理想条件。
从理论上说,用酶偶联反应测酶活性浓度时,最好条件应是测定酶反应为限速反应。动力学上为零级反应,而指示酶为一级反应,酶反应速度与指示酶底物浓度相关。
(二)指示酶、辅助酶的种类和浓度
指示酶、辅助酶的种类:常规化验中常用的酶偶联法中,多以脱氢酶为指示酶,在常规化验中的自动分析仪几乎无一例外都有340nm波长,通过NAD(P)H系统可以很方便地监测到指示酶反应。但从理论上说,往往可以有不止一种偶联方法,只要设法使偶联反应中最后一个是指示酶反应,前面已提到测CK可以正向逆向二个方向建立二种不同酶偶联的反应。又如在丙氨酸转氨基酶(ALT)测定法中,正向反应后产生丙酮酸和谷氨酶,目前最常用的是用乳酸脱氢酶与丙酮酸偶联反应,伴有NADH下降。但也可以用谷氨酸脱氢酶与谷氨酸作用,伴有NADH生成。
总之,酶偶联法为实验室工作者开辟了一个广阔前景。在设计和选择测定酶活性浓度方法时,应该创造更新的方法,而不应拘泥于书本上的方法。
在选择时首先应考虑方法的特异性。在ALT方法,测定丙酮酸显然优于测谷氨酸,因为多种氨基转移酶都能产生谷氨酸,而只有ALT产生丙酮酸。还有干扰反应或副反应,在ALT测定中,由于正常以及病理血清中含有一定量丙酮酸,将受到它的干扰,又由于底物中含有大量α-酮戊二酸,可被血中谷氨酸脱氢酶作用消耗NADH。
其次应考虑酶偶联反应的合理性,如能直接与指示酶偶联,就没有必要加入辅助酶,所选用的指示酶、辅助酶除了应选Km小的酶(这样容易使它们催化反应速度大大超过测定酶反应速度),还必须考虑指示酶、辅助酶是在测定酶的“最适条件”下工作,此时往往不是指示酶、辅助酶的最适条件,如二者差异太大不利于整个酶系统的反应,例如乳酸脱氢酶最适pH为7.4,而丙氨酸氨基转移酶最适pH也是7.4,乳酸脱氢酶作为指示酶比谷氨酸脱氢酶更为合适,因后者最适pH为8.4。
如最后制成试剂盒,则还需从经济角度选择一些价廉物美,又易取材、纯化得到的酶制剂。
(三)指示酶、辅助酶的浓度
从上节描述中可以知道建立一个合适的酶偶联反应体系,不是很容易的指示酶,辅助酶的反应要能准确反映出测定酶含量,中间产物必须低,延滞期必须短,要作到此点,这些工具酶用量很大,但用量过大经济上不合算,所以在酶偶联测定法中,选用适当量的指示酶是一个重要的问题。一般可用反复试验法,即试验性地选用不同量的指示酸直到偶联酶反应中指示酶反应速度不随工具酶的增加而升高,并在所选用的“最适条件”下,指示酶反应速度和酶活性浓度成正比例。
这种方法工作量大,而且不一定能得到最适结果,较好的办法是可先从理论上进行计算,得出一个大约结果,然后在此范围内进行试验,这样可以节约时间和精力。
最简单的方法是根据Vx/(Km)x=Vi/(Km)i的比值来选择指示酶的用量Vi,式中Vx为测定酶的测定上限,(Km)x和(Km)i分别是测定酶和指示酶的米氏常数;有人计算过当比值为1:100时,测定值低4%;如改为1:10时,指示酶的反应速度将比测定酶反应速度慢28%,如增加到1:1000,则误差只有0.7%。
这是因为,当我们用酶偶联反应体系测酶活性浓度时,测定酶的反应必须是体系中的限速反应,工具酶所催化的最大反应速度必须远远大于测定酶,由于工具酶所催化的反应必须在中间产物浓度很低条件下进行,并且将其很快转变为最终产物,反应体系中不应有中间产物堆积,否则将导致误差。
下面举一实例,在肌酸激酶测定法中,CK的Km为2.4mmol/L,指示酶6-磷酸葡萄糖脱氢酶的Km为0.27mmol/L,如我们将CK测定上限定为450μ/L(实际测定时标本稀释60倍,实际为7.5μ/L),如希望上述比例为1:100。代入上式:
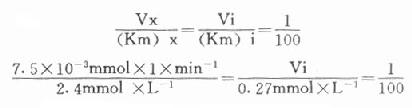
Vi=3.1×10-3×min-1×0.27mmol×1
=0.835×10-3×mmol×min-1
=0.835U
如反应体系为1ml,求出在此反应体系中只需0.835U的6-磷酸葡萄糖脱氢酶即可。
另一法可以根据米氏方程式来计算,在酶偶联反应中,指示酶催化速度(Vi)的米氏方程式为:
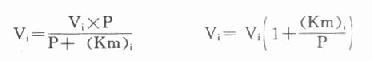
以天冬氨酸氨基转移酶(AST)为例希望中间产物P浓度很低,定为0.001mmol/L,指示酶苹果酸脱氢酶。Km为0.0165mmol/L,如设定AST上限为300U/L(标本为1:12稀释,实测上限为25U/L)。代入上式:
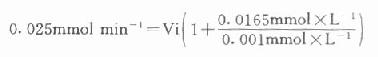
Vi=0.0014mmol min-1=1.4U
得1.4U,如反应体系为3ml,则试剂中苹果酸脱氢酶用量为466U/L,目前试剂中用量为600U/L。从以上计算来看,试剂中用量是足够的。
在酶偶联反应体系中,不可避免会出现延滞期,在测定酶时希望此延滞期愈短愈好,此时可利用Mcclure介绍的计算法计算出一定延滞期时所需指示酶的量。
Mcclure假定在下面酶偶联反应中:
![]()
第一个反应为零级反应,第二个反应为一级反应,且中间产物B浓度远小于指示酶的Km,即B<<(Km)i。
通过推导,可得
dB/dt=k1-k2B
上式积分得:
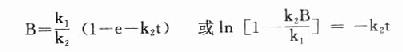
当t足够大时,e-k2t→0,此时B浓度不变达到稳态,此时B的浓度为Bss,代入上式:
首先可得 Bss=k1/k2
以及ln[1-B/Bss] =-k2t
同时在稳态下,k2是指示酶正向反应的常数,在一级反应中k2=Vi/(Km)i。
令F=B/Bss,此即在时间t时产物B为其稳态浓度的百分数,一般选用0.99。
最后可得公式:
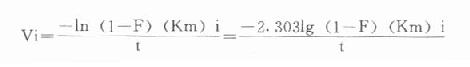 Vi=
Vi=
例如在CK测定中,指示酶6-磷酸葡萄糖脱氢酶Km为0.11mmol/L,我们希望延滞时间为1分钟,则指示酶用量为:
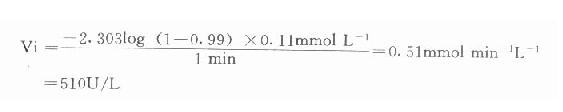
总之,从上式可清楚看到,延滞时间t与指示酶的Km成正比例,用量成反比。
四、酶活性的浓度单位
50年代以前酶活性浓度单位的命名混乱,常以方法提出者的姓氏来命名,例如淀粉酶的Somogyi单位,碱性磷酸酶的King单位等等,定义参差不齐,给临床医师带来很大不便,尤其在建立“连续监测法”测酶后,大量酶应用于临床,此混乱现象更为突出。1963年国际生化协会通过广泛讨论,提出一个国际单位定义来表示酶量的多少,即1分钟能转化1微摩尔底物(μmolmin-1)的酶量为一个国际单位,以IU表示之,由于意见不一致,至今尚未指定酶反应温度,而同一量的酶,在不同温度时间为1分钟所转化底物的量将有明显差异。为避免临床上误认为只要是同一国际单位的酶量在国际上无论何处所测结果都应一致,目前大多数实验工作者常省略国际二字(简写也由IU改为U)。
临床上测定的不是酶的绝对量而是浓度,1963年并未明确规定用ml或L表示体积。旧的文献中可见到mU/ml,或U/L。目前在临床化学中,几乎都习惯用U/L来表示体液中酶活性浓度。我国由于近年来大量用自动分析仪和连续监测法测酶,已逐步不再使用各种古老单位,而使用U/L来表示酶活性浓度。
近年来国际上大力推广SI制,我国已明确SI制为法定计量单位制,SI制中酶活性单位为Katal,即1秒中转化1个摩尔底物(mol s-1)的酶量,Katal对体液中酶量而言显然过大,常用单位为ukatal或nkatal。
上述国际单位和katal间关系如下:
1U=1μmol min-1=16.67nmol s-1=16.67nkatal
在我国不论实验室还是临床医师对katal都不太熟悉,如报告使用katal/L报告酶结果时,最好同时注明相应的U/L。
(一)连续监测法中酶活性浓度的计算
前面已提到连续监测法优点之一是计算方便,不需作标准曲线或标准管,用分光光度计监测酶反应过程时,很容易求出反应体系每分钟吸光度变化,根据摩尔吸光系数可求出△A相当测定物质变化的微摩尔数,由于临床医师需要知道的是标本中而不是反应体系中酶的浓度,计算中要考虑标本的稀释倍数,假如比色杯光径不是1cm,则还应考虑光径不同对△A的影响,这样整个计算公式应为:
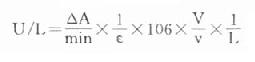
此中ε为摩尔(线性)吸光体系(mol-1·L·cm-1)
△A为吸光度变化
v为标本体积(ml)
V为反应体系体积(ml)
L为光径(cm)
在实际测定中后面几项皆为常数,所以上式常简化为:
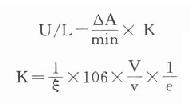
(二)常数K的意义和设置
在测酶时,常数K值的选择是很重要的。此值过高虽然测定的线性范围较宽,但重复性差,反之,虽然精密度好,但线性窄。
此问题与仪器测定的嗓音(noise)密切相关,自动分析仪吸光度读数嗓音一般都需控制在0.001,也就是仪器须保证对同一溶液反复进行测定时,吸光度误差最好控制0.001上下,虽然此值不大,但已可能使测定结果产生1/1000K值的误差,如K值为6000,代入上式,则每分钟测定吸光度如有0.001微小变化,结果将是出现上下6U/L的误差,对于一此参考值较低的酶,如转氨酶而言显然太大,临床医师无法容忍这么大差异。
K值设置的首先出发点应是测定酶的判断值或参考值上限,应保证这些值测定的可靠,所以转氨酶的常数K一般在3000左右,不少人宁愿在1500左右,另外还应考虑到测定时间,一些半自动分析仪测定时间短到0.5分,此时0.001嗓音对每分钟△A误差将是0.002,反之,测定时间延长到2分钟,误差也小一半,也就是说,如测定时间长,则K值可以设置大一些,如测定时间只有0.5分钟,K值一般不超过4000。
改变K值最方便的途径就是改变标本稀释度,稀释倍数愈大,K值愈大。
(三)常数K值的检验
摩尔吸光系数(ε)对一定物质而言常是一个定值,但在一些外界条件影响下也会有所变化。最明显例子是对硝基酚,随pH不同,颜色差异明显,当pH>10时,405nm处其ε为18500,在pH7.0,ε下降为9400,颜色下降几乎一半。温度变化时,也会对NAD(P)H的ε产生一些影响。当波长大于334nm时,随温度升高,同一波长的ε值会轻度下降。
同时ε值是指用分光光度法,也就是在近似单色光的光源条件下才能成立,实际上大多数自动分析仪使用的是干涉滤片,波峰值可能出现差异,个别的半波宽可能为10nm乃至更大。由于杂散光的存在,会明显改变ε值,假如再考虑到注加系统的误差,实测K值很可能与理论K值不一致。Onuki检测了三台Hitachi-7150型自动分析仪测AST的K值分别为-5466、-5421、-5559。而理论K值为-6111则结果约增加为1.12倍。
测定实际的K值不是一件困难的事,目前有些自动分析仪已有相应的软件和试剂可以进行检测。事实上对一些性质稳定的物质如对硝基酚、对硝基苯胺,可以用高纯试剂配成标准品或直接购买可靠的校准品,将它们作为标本进行酶的测定,根据已知标准品的浓度和吸光度变化就可计算出实际K值,但此法不适用于测与NAD(P)H反应有关酶测定的K值。因为NAD(P)H不稳定,此时可使用测葡萄糖的紫外分光光度法试剂盒,对已知浓度的葡萄糖标准品进行终点法测定,此法产生与葡萄糖相等摩尔数的NAD(P)H,故不难从吸光度变化求出K值,也有人用乳酸脱氢酶测已知量的丙酮酸来求K值。
五、连续监测法中的干扰因素及其控制
大多数测定标本不是纯酶制剂,而是体液或组织液。其中除了测定酶外,还存在着其它酶和各种物质,如使用酶偶联反应,在反应体系中又外添了大量的各种酶制剂,因此在反应体系中可能出现一些我们不希望的副反应或旁路反应,这些都有可能对测定反应产生干扰。
(一)其它酶和物质的干扰
如组织匀浆中往往含有NADH-细胞色素C还原酶,它将干扰各种还原酶的测定。临床酶测定中最典型的例子是血液中丙酮酸对丙氨酸氨基转移酶测定的干扰,由于反应体系中含有大量乳酸脱氢酶和NADH,可与丙酮酸反应消耗NADH,引起340nm处吸亮度下降,假如将此NADH下降也算为ALT活性将引起误差。其它如红细胞中腺苷酸激酶(AK)对CK测定的干扰,在CK的酶偶联体系中ADP是CK底物,但它又同时是AK的底物,二个酶反应都产生ATP,在工具酶(已糖激酶和6-磷酸葡萄糖脱氢酶)作用下都产生NADH引起340nm处吸光度上升,其结果是CK测定结果偏高,为避免此种干扰,所以在反应体系中加入AK的抑制剂AMP和二腺苷酸5′磷酸。
(二)工具酶的污染
目前试剂中所用的试剂酶多从动物组织或细菌中提取,不可避免地会污染有其它酶,如不注意此问题,会引起不正确结果,例如丙酮酸羧化酶催化下列反应:
![]()
可加入苹果酸脱氢酶和NADH进行测定,将草酰乙酸转变为苹果酸,并将NADH转变为NAP+。如果工具酶苹果酸脱氢酶不纯,含有乳酸脱氢酶,也可以作用底物之一的丙酮酸,同时消耗NADH,这样就无法准确测定丙酮酸羧化酶活性。
更有甚者,有些工具酶中就混有测定酶,这种情况下,将产生很高的本底。
所以所用的工具酶必须很纯,并检查污染酶的含量,例如测定天冬氨酸氨基转移酶所用的苹果酸脱氢酶中所含杂的AST时,按IFCC规定不应超过0.005%。
(三)非酶反应
有些底物不稳定,没有酶的作用就能自行反应,例如ALP的底物磷酸对硝基酚配成的底物溶液,室温放置过夜,即自行水解释放出对硝基成黄色。又如测醛缩酶时,其底物醛类化合物可以和NAD+起非酶反应产生一种具有类似NADH吸收光谱的化合物,给测定带来困难。
(四)分析容器的污染
如冲洗不当,分析容器和管道中混杂有各种物质,可能影响酶活性,如微量重金属可使酶失活,残留的表面活性剂可能抑制酶活性。
(五)沉淀形成
使用光学法监测酶反应时,如有沉淀形成或组织匀浆中颗粒的下沉都会引起吸光度变化,引起测定结果误差,此情况常见于底物溶解度低而反应体系中底物浓度偏高。可见于用γ-谷氨酰对硝基苯胺为底物测GGT时。
对上述的一些问题常可通过试剂空白管检出并加以校正。不同厂家的ALT,AST试剂盒由于杂酶存在,试剂空白管可以测出多少不等转氨酶活性,个别可达5U/L以上,这种试剂无法使用,较好的试剂盒也在2U/L左右。如不作试剂空白管对结果加以校正,所测结果将偏高,用半自动分析仪测定酶时特别要注意作试剂空白管,有时还需作标本对照管,例如测ALT时为除去GLD的干扰,可以在底物溶液中不加入丙氨酸,与标本中GLD作用引起NADH下降。
解决这些问题另一个有效措施就是不用单一试剂测酶活性,改用双试剂,先加的第一试剂常不含底物或底物之一,但含有所有其它成分,与标本作用一段时间待吸光度停止变化后,再加入底物开始测定酶的反应。
第十八章 诊断分子生物学基本技术
第一节 概述
现代医学是随着自然科学各基础学科的发展而发展。在临床观察的基础上,其检验、诊断、治疗和病理生理学的进展是与实验研究方法的创新与改进分不开的。分子生物学的迅速发展,全面地渗透到医学科学各个领域中,揭示了许多新现象,提出了不少新问题,是医学向分子水平迈进的一个重要依据和手段。使医学科学达到了一个新的阶段,医学分子生物学已逐渐形成了比较完整的理论体系。医学检验与诊断学也不例外,分子生物学技术在医学检验诊断中的广泛应用,逐步形成诊断分子生物学(diagnostic molecular biology)这一学科分支。诊断分子生物学的内容主要包括:
⒈内源基因异常的检验与诊断 在分子水平为疾病的发生机制,治疗和预后提供实验依据。自身基因结构异常导致基因功能异常而致病,或使其表达产物蛋白质的结构与功能异常而致病,或使基因调控失常而致病,如遗传性疾病、肿瘤等。
⒉外源基因侵入体内的检验与诊断 对多种病源微生物(如细菌、病毒、衣原体、支原体等)的检出,可望实现快速、灵敏、准确。在分子水平可更准确地分类亚种和变种。探明病源微生物致病的机制。
生物大分子指核酸和蛋白质分子,是生命物质的基本要素,如最简单的生命体病毒、噬菌体仅以核酸,蛋白质为主,却表现了生命的最基本的功能。核酸的表达产物是蛋白质,为因果关系。核酸是生命的存在形式,蛋白质是生命的表现形式。许多疾病的临床表现与某些蛋白质的结构和功能异常有关,但究其原因可能是基因的结构和功能异常导致基因表达产物蛋白质异常所为。
蛋白质结构与功能的检验和临床诊断应用方法已日趋完善,而核酸结构与功能的研究历史短,研究前景广,是目前分子生物学研究的主要内容。特别是在临床检验、诊断、预后、治疗方面的开发前景受到医学工作者关注。
核酸(nucleicacid)具有储存和表达遗传信息的功能,前者为脱氧核糖核酸(deoxyri-bonucleic acid,DNA)承担,后者由核糖核酸(ribonucleic acid,RAN)参与。碱基(base)、脱氧核糖(deoxyribose)或核糖(ribose)、磷酸(phosphate)三种分子组成核苷酸(nucleotide),核苷酸之间以磷酸二酯键在纵向按一定的排列顺序组成核酸的一级结构,是链状结构。在此基础上形成二级或更高级的空间结构,方能显示其生物功能。
组成DNA的碱基有腺嘌呤(adenine A)、鸟嘌呤(guanine G)、胞嘧啶(cytosine C)和胸腺嘧啶(thymine T),RNA的碱基为A、G、C和尿嘧啶(uracil U)。碱基在紫外光260nm波长附近有较强吸收峰,该特性常用于核酸分析。碱基(嘌呤环的N-9或嘧啶环的N-1)与核糖(C-1′)以糖苷键结合形成核苷,核苷(C-5′)与磷酸以磷酸酯键结合形成核苷酸。核苷可与1-3个磷酸结合,如腺苷与一个磷酸结合为腺苷酸(AMP),与二个磷酸结合为二磷酸腺苷(ADP),与三个磷酸结合为三磷酸腺苷(ATP)。第一个核苷酸的C-3′位羟基与第二个核苷酸的C-5′位磷酸以磷酸二酯键结合,以此类推成百上万的核苷酸结合可形成一条大分子核酸单链,单链之间的碱基再以氢键结合可形成较稳定的核酸大分子双链结构(二级结构)。
核酸的简写方式为5′AGCTGTAC3′,此外的英语字母已不代表碱基,而是指核苷酸。核酸的书写方向与合成方向一致,都是5′→3′。习惯上碱基原子次序不用‘′’而核糖环原子标以‘′’,故5′表示核糖的C-5,并与磷酸基团连接,3′为核糖的C-3,并带有-OH基团。可写作5′pAGCTGTAC-OH3′。
核酸的高级结构以1953年Watson和Crick发表的DNA双螺旋结构最著名。DNA分子由两条核苷酸组成的单链,按右手螺旋盘旋,两条单链走向相反。两条单链之间的横向碱基A=T以二个氢键配对,G≡C以三个氢键配对,使DNA分子的双链之间有严格准确的碱基配对关系,形成DNA二级结构。在此基础上可形成超螺旋,即三级结构。
DNA分子的一个重要物理性质是在过酸、过碱或加热的溶液中,双链间的氢键可分开成单链,称为变性。变性是可逆的,改变条件,解开的单链可重新按碱基配对规则,A=T、G≡C严格配对形成双链,形成的双链与变性前的结构完全相同,称为复性。解链的DNA溶液在260nm处吸光值A260增大,即核苷酸>单链DNA>双链DNA,称为高色效应,反之为低色效应。以50μg/ml为例,A260分别为:双链DNA=1.00,单链DNA=1.34自由碱基=1.60;或A260=1.00时双链DNA=50μg/ml,单链DNA=37μg/ml,mRNA=40μg/ml。一般同时在A280测定蛋白质含量,纯化的DNa A260/A280≥1.8;纯化的mRNaA260/A280≥2.0,即蛋白质含量少。实验室常用加热使DNA变性,因为G≡C之间有三个氢键,而A=T之间只有二个氢键,解链温度取决于DNA分子中G≡C配对多少。以加热温度对A260作图可以得到一条解链曲线,解链开始到完全解链的温度范围的中点温度称为解链温度(meltingtemperature,Tm)又称熔链温度。Tm值大小与DNA的G+C含量百分数成正比。虽有推算Tm值的经验公式Tm=4(G+C)+2(A+T),但只能用于20个核苷酸以下的DNA小分子。而且实验条件可影响Tm。如三氯醋酸钠存在使Tm变小,氯化钠存在可使Tm升高。变性的DNA溶液经处理后,使单链的碱基重新配对形成双链的过程叫复性或退火。同源DNA复性叫复性DNA,不同源DNA复性的过程叫核酸杂交,形成的双链叫杂化双链。杂交可以发生在DNA分子之间,也可以发生在DNA与RNA分子之间,如DNA-DNA′;DNA-mRNA。影响复性的因素很多,如离子强度、温度、DNA浓度和长度等,一般采用0.15-0.5mol/l NaCl;温度低于Tm20-25℃,过高不利于复性,过低易发生单链内碱基随机配对或非特异性结合。复性的开始是碰撞,因此DNA浓度高时,碰撞机会多,复性速度快。
DNA的生物功能是携带、传递和表达遗传信息。基因是DNA大分子中一段有生物功能的片段。即在自然界可表达一分子具有生物活性的肽链,也可携带和传递给子代表达相同的肽链的信息。基因组是所有基因的集合。
DNA碱基严格的配对,稳定的结构可将携带的遗传信息保留在生命体内。当细胞分裂时,DNA双螺旋结构解开分为两条单链,暴露碱基。在DNA合成酶作用下,两条链各自为模板(母链),按碱基配对原则合成与母链严格互补的互补链。子代细胞的DNA分子一条链来自亲代,一条是按碱基配对原则合成的新链,这一过程为复制,这种复制的方式称为半保留复制。半保留复制使子代与亲代的DNA分子双链碱基序列完全一致。遗传信息就这样准确地传递给子代。复制过程以母链为模板,游离的脱氧三磷酸核苷为合成新链的原料,即dATP、dGTP、dCTP、dTTP,总称dNTP。催化新链合成反应的酶是以DNA为模板的DNA聚合酶(DNA-dependentDNA polymerase)。DNA聚合酶需要一段寡核苷酸作为引物(primer)引导,以母链为模板,按碱基配对原则,将dNTP逐个由5′→3′方向合成新链,合成的新链走向5′→3′与母链3′→5′走向相反。1958年首先在大肠埃希菌发现DNA聚合酶Ⅰ(polⅠ),继而发现原核生物有DNA聚合酶Ⅰ、Ⅱ、Ⅲ。聚合酶Ⅲ主要功能是复制,聚合酶Ⅰ的主要功能是在DNA碱基配对发生错误或DNA损伤时起修复作用,聚合酶Ⅱ的功能尚不清楚。三种聚合酶均有从5′向3′或3′向5′逐一把核苷酸从核酸链上水解下来的核酸外切酶(exonuclease)作用。聚合酶Ⅰ外切酶活性最强,便于切除错误和修复。用枯草菌溶素(蛋白酶)水解聚合酶Ⅰ后产生大、小片段,其中大自然切去了3′→5′的外切酶活性,保留5′→3′方向的聚合酶和外切酶活性,称为Klenow片段(Klenow fragment),是分子生物学研究中常用的工具酶。
DNA的另一个生物功能是通过转录和翻译,将遗传信息表达为具有生物功能的产物-基因产物,如肽链、酶、蛋白质等。使遗传得到表达,生命得以表现。
RNA有信使RNA(mRNA)、转运RNA(tRNA)和核糖体RNA(rRNA),结构相对简单,生物功能主要转录DNA的遗传信息,翻译DNA的遗传信息,表达DNA的遗传信息。
合成RNA的过程为转录。转录酶是依赖DNA的RNA聚合酶(DNA-dependentRNA polymerase)。转录与复制有相似之外,均需依赖DNA的聚合酶,都以DNA为模板;均需核苷酸作原料,都为5′向3′延长;且均遵从碱基配对原则。但也有明显的不同之处,复制是把作为模板的DNA两条母链均复制,而转录只转录DNA双链中的有意义链;合成原料前者利用脱氧核苷酸mRNA、tRNA、rRNA三种RNA,且RNA中无胸腺嘧啶T,而以尿嘧啶U代替T,碱基配对也由A=T转为A=U;参与复制的DNA聚合酶需引物,而用于转录的RNA聚合酶无需引物,自身有识别转录起点的功能。与复制不同的是转录只需以一条DNA链为模板,因此把可作为转录模板的一条DNA链称为有意义链(sense strand or Watson strand),与此对应的一条不参与转录的链称为反意义链(antisense strand or Crick strand)。有意义链并不固定在DNA双链的某一条链上,而是在两条链上交替出现,使转录也是交替进行,这一现象称为不对称转录。转录的重要产物是mRNA,因为mRNA可作为肽链合成的模板,指导蛋白质合成。由此把可转录产生mRNA并指导蛋白质合成的一段DNA称为结构基因(structural gene),其余的DNA可转录tRNA和rRNA,还有许多起调节作用的调节基因,但有相当一部分的DNA功能尚不清楚,既不承担转录功能也无调节作用。真核生物的基因往往是一种断裂基因(splitegene)即一个结构基因,一条有意义链中的DNA碱基序列并不都能表达为蛋白质。可表达为蛋白质的区域称编码区,反之为非编码区,编码区与非编码区在基因中相间排列。1978年,Gilbert把可编码蛋白质的DNA序列称外显子(exon),把非编码称内含子(intron)。真核细胞刚完成转录的mRNA含外显子和内含子,称杂化核RNA(heteroge-neous nuclear RNA,hnRNA),这种初级mRNA的分子量往往比成熟的mRNA大几倍。hnRNA在细胞核内经剪接加工,切去内含子,并在5′端加上7-甲基鸟嘌呤和三磷酸鸟苷的帽子结构(capstructure)在3′端加上多聚脱氧腺苷酸(poly A)后,进入细胞质为成熟的mRNA。
基因表达在复制、转录、翻译水平及各水平表达后加工均可进行调控。目前对原核生物的转录水平调控研究较多,较为清楚。原核生物中,功能相关的几个结构基因往往排列在一起,转录出一段较长的mRNA,称为多顺反之(polycistron),并指导合成(翻译)一套功能相关的蛋白质。这样的一组基因及调节部分称为操纵子(operon)。乳糖操纵子(Lac operon)就是由调控区的启动子(promoter,p)、操纵区(operator,o)和表达β-半乳糖苷酶(Z)、β-半乳糖苷透性酶(Y)、β-半乳糖苷乙酰转移酶(a)三种功能相关酶的相关基因组成。乳糖操纵子是可诱导的负调控型操纵子,在高效诱导剂异丙基硫代半乳糖苷(isopropyl-β-D-thiogalactoside,IPTG)诱导下,产生相应的酶并作用底物5-溴-4-氯-3-吲哚-半乳糖苷(5-bromo-4-chloro-3-indolyl-β-D-galactoside,x-gal)产生蓝色,在重组基因筛选中被广泛应用。有名的操纵子还有可诱导的正调控操纵子-阿拉伯糖操纵子(Ara operon),可阻遏操纵子-色氨酸操纵子(Trp operon)。可诱导指关闭的基因在诱导剂存在下可开启表达,可阻遏指开放的基因在阻遏物存在下可关闭表达。
由转录把DNA上基因的信息(脱氧核苷酸序列)变为mRNA核苷酸序列,生物体合成mRNA并把转录的信息(核苷酸序列)转变为蛋白质的氨基酸序列的过程称翻译。复制、转录在细胞核完成,翻译在细胞质进行。
翻译从mRNA5′开始,向3′方向每三个碱基为一个密码(codon),称三联体密码。4种碱基可组合64种密码,代表了20种氨基酸和翻译的起点与终止密码,如5′-CGUGG AUAA-3′代表精氨酸甘氨酸和终止密码。密码之间无标点符号,当多一个或少一个核苷酸时,可使密码重排列移码(frame shiff),由此引起的变异称移码突变。翻译过程中以mRNA为模板,rRNA为场所,tRNA搬运与密码对应的氨基酸,自mRNA5′端开始合成多肽链,多肽链合成从氨基(N)端开始,第一个氨基酸的羧基与第二个氨基酸的氨基形成肽键,在羧基(C)端结束。故肽链合成方向是N→C。经翻译后加工形成有活性的蛋白质。
1958年,Crick把复制、转录、翻译的基因表达(geneexpression)过程总结为分子生物学的中心法则(central dogma)。以后在病毒学研究中发现了依赖RNA的DNA聚合酶能进行逆转录,即反转录酶(reverse transcriptase,逆转录酶),可以RNA为模板复制DNA,由此得到的DNA称cDNA(complementary DNA,互补DNA,反转录DNA)。后来还发现了RNA复制RNA,M13噬菌体的DNA可以单链存在,有时DNA还能以三链、四链体形式存在,大大丰富了中心法则。
第二节 分子生物学实验基础
分子生物学是在生物化学基础上发展起来的,以研究核酸和蛋白质结构、功能等生命本质的学科,在核酸、蛋白质分子水平研究发病、诊断、治疗和预后的机制。其中基因工程(基因技术,基因重组)是目前分子生物学研究热点,这些技术可以改造或扩增基因和基因产物,使微量的研究对象达到分析水平,是研究基因调控和表达的方法,也是分子水平研究疾病发生机制、基因诊断和基因治疗的方法。转化(transformation)、转染、转导、转位等是自然界基因重组存在的方式,也是人工基因重组常采用的手段。基因重组的目的之一是基因克隆(gene clone),基因克隆可理解为以一分子基因为模板扩增得到的与模板分子结构完全相同的基因。使需要分析研究的微量、混杂的目的基因易于纯化,得以增量,便于分析。
外来基因引起细胞生物性状改变的过程叫转化(transformation),以噬菌体把外源基因导入细菌的过程叫转染(transfection)。利用载体(噬菌体或病毒)把遗传物质从一种宿主传给另一种宿主的过程叫转导(transduction)。一个或一组基因从一处转移到基因组另一处的过程叫转位(transposition),这些游动的基因叫转位子。
一、基因工程的常用工具
(一)载体
载体(Vector)是把外源DNA(目的基因)导入宿主细胞,使之传代、扩增、表达的工具。载体有质粒(plasmid)、噬菌体、单链丝状噬菌体和粘性末端质粒(粘粒)、病毒等。载体具有能自我复制;有可选择的,便于筛选、鉴定的遗传标记;有供外源DNA插入的位点;本身体积小等特征。
质粒存在于多种细菌,是染色体(核)以外的独立遗传因子,由双链环状DNA组成,几乎完全裸露,很少有蛋白质结合。质粒有严紧型和松弛型之分。严紧型由DNA多聚酶Ⅲ复制,一个细胞可复制1-5个质粒。而松弛型由DNA多聚酶Ⅰ复制,一个细胞可复制30-50个质粒,如果用氯霉素可阻止蛋白质合成,使质粒有效利用原料,复制更多的质粒。质粒经过改造品种繁多,常用的有pBR322、pUC系列等。这些质粒都含有多个基本基因,如复制起动区(复制原点Ori),便于复制扩增;抗抗生素标记(抗氨芐青霉素Apr、抗四环素Tcr等)或大肠埃希菌部分乳糖操纵子(E.coli LacZ)等,便于基因重组体的筛选;基因发动子(乳糖操纵子Lac、色氨酸操纵子Trp等)和转录终止序列,便于插入的外源基因转录、翻译表达。质粒上还有许多限制性内切酶的切点,即基因插入位点,又叫基因重组位点,基因克隆位点。
常用噬菌体载体有单链噬菌体M13系统;双链噬菌体系统。噬菌体应和相应的宿主细胞配合使用。以上载体各有特点,便于选择,灵活应用。
(二)工具酶
工具酶是基因重组技术不可缺少的工具。主要有限制性内切酶、连接酶、核酸聚合酶、逆转录酶、核酸酶等。
限制性内切酶有Ⅰ型和Ⅱ型限制性DNA内切酶之分,Ⅱ型能严格识别核酸序列,并在识别区内特定的核苷酸处切开DNA双链。故通常所指都是Ⅱ型限制性DNA内切酶。识别分四核苷酸和六核苷酸,其序列旋转对称。切口分开端和粘端,产生3′-OH和5′-P末端。内切酶品种多,使用时应注意温度、缓冲液用量(一般1μg DNA/2-5单位酶)等反应条件。
| 酶 | 识别序列 | 切口 | |
| Alu Ⅰ | …AGCT… | …AG CT… | 四核苷酸 |
| …TCGA… | …TC GA… | 平端切口 | |
| Eco R1 | …GAATTC… | …G AATTC… | 六核苷酸 |
| …CTTAAG… | …CTTAA G… | 粘端切口 |
连接酶有T4噬菌体DNA连接酶、T4噬菌体RNA连接酶、大肠埃希菌DNA连接酶等。DNA连接酶可连接平端,也连接粘端。反应需有Mg2+和ATP存在,pH7.5-7.6。最适温度37℃,30℃以下活性明显下降,但考虑到被连接DNa 的稳定性和粘性末端的退火温度,一般平端连接用20-25℃,粘端连接用12℃左右。
聚合酶有DNA聚合酶(以DNA为模板合成DNA大肠埃希菌DNA聚合酶Ⅰ,大肠埃希菌DNA聚合酶Ⅰ大片段(Klenow大片段),T4或T7噬菌体DNA聚合酶等);RNA聚合酶(以DNA为模板合成RNA,T7或T3噬菌体RNA聚合酶);逆转录酶(以RNA为模板合成DNA,除RNA病毒中发现外,发现大肠埃希菌DNA聚合酶Ⅰ和Taq DNA聚合酶都有逆转录活性)。
大肠埃希菌DNA聚合酶Ⅰ具有5′→3′聚合酶活性和5′→3′,3′→5′外切酶活性。Klenow片段是DNA聚合酶Ⅰ被枯草杆菌酶作用产生的一个大片段,有5′→3′聚合酶和5′→3′外切酶活性,无3′→5′外切酶活性。可用于缺口翻译(Nick translation)法标记核酸,也可用于DNA序列测定,修补DNA链等。
核酸酶有DNase、RNase、核酸酶S1等,可水解相应的DNA和RNA,核酸酶S1可降解单链DNA和RNA,用量增大也可降解双链核酸。它可用于切去ds-cDNA合成中产生的发夹环。
末端转移酶在Mg2+存在下,选择3′-OH端单链DNA为引物加成核苷酸,在Co2+存在下,选择3′-OH端双链DNA为引物加成核苷酸,形成多聚核苷酸尾。常用于核酸末端标记和核酸连接的互补多聚尾(连接器)。
碱性磷酸酶去除5′-P,可防止二分子DNA片段5′端P基团自身空间障碍,影响DNA分子之间的连接,一般用碱性磷酸酶处理载体DNA除去5′端P基团,在连接酶作用下目的基因的5′端P先与载体3′端OH连接,再通过复制修复另一条链,使二条链完全连接。该方法大大提高了连接效率(图18-1)。
图18-1 经碱性磷酸酶处理后载体DNA与目的基因DNA的连接
二、目的基因
常把需研究的基因称为目的基因,需分析的基因称靶基因,在基因克隆过程中有时两者均称为插入基因,有时三者含义相近。简单的原核生物目的基因可从细胞核中直接分离得到,但人类的基因分布在23对染色体上,较难从直接法得到。简短的目的基因可在了解一级结构或通过了解多肽链一级结构氨基酸编码的核苷酸序列基础上人工合成。但多数的目的基因由mRNA合成cDNA(complementaryDNA,反转录DNA)得到。CDNA通过各种方式与载体连接,克隆可得到全长cDNA或片段,用于探针制备、序列分析、基因表达等研究。因此以cDNA为研究材料反映了mRNA的转录及对以后翻译的影响情况,即反映某一基因(DNA)外显子的情况。(图18-2)
图18-2 目的基因cDNA的合成
三、基因库的建立
建立基因库的目的是为了便于目的基因的保存、扩增和纯化。基因库是利用工具酶,通过基因重组技术和转化、筛选而建立,也是基因克隆的过程。实际上是利用低等生物如细菌、病毒、噬菌体等生长快,繁殖力强的特点,而作为一种核酸扩增筛选的载体,把人类等高等生物的基因或其片段用工具酶插入,重组于其中,经筛选,克隆得到目的基因与载体重组的重组基因,成为便于保存,取用方便的基因库。基因库交流是研究室之间交流目的基因的常用方式。
(一)基因重组
基因重组方案很多,简单的可用同一种限制性内切酶分别在载体和目的基因切出相对应的切口,便于连接。也可用末端连接酶合成连接器(图18-3)。近年,Okayama方案为实验室普遍采用,该方法可得到全长的cDNA。
(二)转化
转化目的是把有复制能力,但在细胞外无复制活性的目的基因-载体重组体装入细胞(大肠埃希菌),使目的基因随载体在细胞内复制、扩增。实验步骤介绍如下:
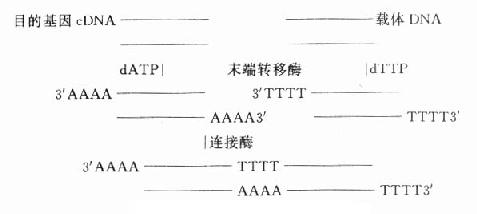
图18-3 基因重组方案之一
⒈大肠埃希菌的处理(增加细胞膜通透性,便于基因进入细胞)大肠埃希菌(E.Coli cells)接种于Lb agar培养基,37℃培养一晚。挑选3~5个大菌落,接种于50ml LB培养基,37℃,振摇培养一晚后,在A550测定,要求细菌繁殖一定量(一般为细菌对数生长期),野生型(rec+,5x107cells/ml)为0.2-0.3,缺陷型(rec-,5x107cells/ml)为0.5-0.6。离心弃去上清液,用20ml,50mmol/L,CaCl2悬浮菌体。冰浴20分钟,低温离心。弃上清液,用2.5ml冰50mmol/l CaCl2悬浮。4℃可保存48小时,用于转化,称competent cells。
⒉质粒(如经基因重组建立的重组质粒,基因库等)的转化 将competent cells(以美国BRL公司DH10B菌株为例)置冰水浴。同时将Falcon2059(传热系数稳定)塑料试管置冰水浴。取100μl菌液(DH10B)于2059试管中,加10倍稀释的巯基乙醇1μl,冰浴轻摇2分钟,放置10分钟。取0.1-50ng质粒加入试管,轻轻摇匀,冰浴30分钟。此间备好42℃水浴。并将SOC培养基置42℃备用。将试管置于42℃,轻摇45秒钟,马上置冰浴2分钟。使competent cells热胀冷缩,把质粒导入菌体。加42℃SOC培养基0.9ml于试管,37℃培养1小时。1000rpm离心10分钟,弃上清液,用200μlSOC培养基悬浮菌体。接种于LB-氨芐青霉素(100U/ml)培养皿,37℃培养一晚。已转化的菌体可形成菌落(因质粒上有抗氨芐青霉素基因)。在了解目的基因或其产物的基础上对菌落进行鉴定。并在LB培养液中扩大培养。基因库的建立、转化、筛选、扩增、纯化的过程就是基因克隆的过程。
LB培养基(1L):10g蛋白胨,5g酵母膏,10gNaCl,高压灭菌,pH7.5。
SOB培养基(1L):20g蛋白胨,5g酵母膏,0.5gNaCl,高压灭菌。另外准备2mol/L镁溶液(1mol/L氯化镁与1mol/L硫酸镁等量混匀,过滤灭菌),临用前加1ml于100ml培养基。
SOC培养基(100ml):临用前加入1ml过滤灭菌的2mol/L葡萄糖。
四、核酸的分离与纯化
核酸存在于多种细胞,如病毒、细菌、寄生虫、动植物细胞;多种标本中,如血液、组织、唾液、尿液等其它来源的标本。因此分离方法复杂而多样。又因各种DNA、RNA的丰度差异,各种分析方法对核酸的纯度与量的要求有差异,因此在实验前应对采用的方法有所了解和选择。总的说来核酸的分离与纯化是在溶解细胞的基础上,利用苯酚等有机溶剂抽提(核酸溶于缓冲液,即水相),分离,纯化;乙醇、丙酮等有机溶剂沉淀,收获。溶解细胞的方法因标本不同而不同,有用SDS加NaOH,有用蛋白酶,有的用超声波破碎等方法,苯酚提取主要使蛋白质变性沉淀于有机相,而核酸保留在水相,达到分离核酸的目的。实验上生物标本中含量最多的就是蛋白质。为了除去分离过程中残留的有机溶剂,常用的方法是加冷乙醇和盐沉淀核酸,通过离心回收核酸,然后用70%-80%乙醇洗涤沉淀,除去多余的盐,以免影响核酸溶解和抑制后续步骤的酶促反应。为了得到纯的核酸可用蛋白酶除去蛋白,用RNA酶除去RNA,得到纯的DNA,用DNA酶除去DNA而获得RNA。目前开发了许多商品化的核酸分离柱,可简单、快速地分离得到纯度很高的DNA或RNA。其分离原理有的利用核酸的分子量差异,有的利用需分离核酸的特点与其特异性结合达到分离、回收的目的。
(一)质粒的分离与纯化
含质粒的E. Coli cells经LB培养液250ml扩大培养,倒入50ml离心管,4℃,3000rpm,离心15分钟。弃去上清液。(弃去液丢弃前应作消毒处理,以免污染环境)。加lysis buffer(50mmol/L Glucose,10mmol/l EDTA,25mmol/l Tris-HCl,pH8.0)1-2ml使之悬浮。放置室温5分钟,加3.5-7ml新配制SDS/NaOH溶液(1mol/l NaOH 8ml,20%SDs2ml,蒸馏水30ml)上下摇匀,置冰水浴5分钟。禁用混匀器,以免DNA分子断裂),加2.5mol/L醋酸钾缓冲液(pH4.8)2.6-5.2ml,上下摇匀,冰浴5分钟。4℃,3000rpm,离心15分钟。沉淀蛋白质。取上清液,加无水乙醇12-24ml(上清液的二倍)放室温15分钟后,10000rpm离心,弃上清液。加约为沉淀物体积的0.4倍TE(10mmol/lTris-Hcl pH8.0,10mmol/l EDTA)溶解沉淀后,加0.2倍的7.5mol/L醋酸铵。可用混匀器混匀,置冰浴10分钟。4℃,10000rpm离心5min,弃上清液。加沉淀物0.4倍的10mmol/lTris-HCl pH7.5,10mmol/l EDTA缓冲液,1/10体积的5mg/ml RNase,37℃,30分钟保温。加等量的phenol(酚)/CIAA(480ml氯仿,20ml异戊醇),混匀。4℃,10000rpm,离心5分钟。取上层液加酚/CIAA重复一次。取上层液加1/10体积5mol/l NaCl和2倍无水乙醇,―20℃过夜或―70℃2小时以上。4℃,10000rpm,离心10分钟,弃上清液。加80%乙醇不摇动,直接10000rpm离心,弃上清液,真空干燥。加适量(约200μl)TE溶解。测定含量后备用。
(二)重组质粒中目的基因的分离与纯化
先取少量纯化的重组质粒,用限制性内切酶切出目的基因,经电泳分离,确认。以200μl含2mg DNA(重组质粒)为例:取10μl含100μg DNA,加90μl TE。按1μgDNA加2-5U限制性内切酶,1/10体积缓冲液,1-2小时保温。缓冲液种类和酶反应温度因内切酶种类而异。1/10体积3mol/l NaAc,2倍无水乙醇,-70℃,2小时(-20℃过夜),10000rpm,10分钟离心,弃上清液(乙醇沉淀)。适量TE溶解沉淀(切断的DNA质粒与目的基因混合物),电泳分离(用相应分子量DNA标准同时电泳),在紫外光下观察结果,与标准DNA分子量比较,确认目的基因和内切酶的切割效果。
⒈电泳条件:
| 凝胶制备: | 1%琼脂糖凝胶 | agarose(mg) | X50TAE(ml) | EB(溴化乙锭μl) |
| (agarose) | 1000 | 2.0 | 4.0 | |
| 总体积(ml) | ||||
| 100 |
| 胶浓度(%): | 0.3 | 0.6 | 0.7 | 0.9 | 1.2 | 1.5 | 2.0 |
| DNA长度(kb): | 60-5 | 20-1 | 10-0.8 | 7-0.5 | 6-0.4 | 4-0.2 | 3-0.1 |
| 电泳缓冲液: | X50TAE(ml) | EB(μl) | 总体积(ml) | ||||
| 20 | 30 | 1000 |
X50TAE:Trismabase 54g;乙酸57.1ml;0.5m EDTApH8.0 20ml;蒸馏水至1000ml。
EB:10mg/mlethidium bromide。(EB有致癌性,操作应小心)。
经电泳确认后,取适量DNA(重组质粒)同上条件切开,用1.5%低熔点(<65℃熔解)琼脂糖凝胶,电泳分离。在紫外光下,切出含目的基因区带(凝胶),放入离心管中,提取纯化。
⒉从低熔点凝胶提取,纯化DNA片段 加与凝胶体积相等的TE(10mmol/lTris-HCl pH8.0,0.1mmol/l EDTA),置65℃水浴5分钟保温,使凝胶完全溶解。待放至室温,加等量酚(TE饱和,TE封在上层,取下层酚),轻轻混匀(不用混匀),12000rpm,3分钟离心。反复1-2次。取上层液,加0.1体积3mol/L醋酸钠(pH5.2)和2.5倍体积无水乙醇,进行乙醇沉淀。将纯化的DNA加适量TE溶解,测定含量,备用(可用于目的基因结构分析,探针制备等)。除低熔点凝胶回收法外,如用一般凝胶可用透析带短暂电泳,离心管低部加玻璃棉,高速离心等方法回收DNA片段。
(三)标本DNA的分离与纯化
用5-10ml 1xNTE(NaCl 100mmol/L ,Tris-HCl10mmol/l pH7.4,EDTa 1mmol/L pH8.0)调整细胞为2×107个(组织应切碎置液氮冰冻,高速搅切成粉末后,加入缓冲液)。加1/10-1/20体积(V)10mg/ml蛋白酶(Sigma Ⅷ型),1/20v 10%SDS,37℃2小时。加等量酚/3xNTE(3xNTE上封)混匀(至少7分钟)。4℃,3000rpm,10分钟。取上清液,加2mlTE(Tris-HCl 10mmol/L pH7.5,EDTa 1mmol/L pH8.0),充分混匀。乙醇沉淀。2mlTE溶解沉淀。加1/30xNTE,蛋白酶K(10mg/ml)代替蛋白酶重复2-4步骤。测定含量。
(四)样品RNA的分离,纯化
含106个细胞液加等量纯化溶液[4mol/L胍基硫氰酸盐,25mmol/L柠檬酸pH7.0,0.5%肌氨酸(sarcosyl),0.1mol/l 2-巯基乙醇]。总体积为细胞沉淀4-5倍,混匀。加0.1体积2ml/L乙酸钠(pH4.1),1体积酚(重蒸水上封),0.2体积CIAA,混匀器剧烈混合10秒钟。冰水浴15分钟。10000xg4℃,20分钟。离心(不用刹车)。小心取出上清液。加等量丙酮(或2倍乙醇),-20℃,60分钟以上。10000xg,4℃,20分钟离心。弃上清液。用1.5ml离心管约加0.3ml纯化溶液,重复3)步骤,离心10分钟,弃上清液。加75%乙醇,10000xg,4℃,10分钟离心。弃上清液。真空干燥15分钟,备用。
五、探针制备
探针制备就是目的基因的标记,核酸标记方法常用的有缺口平移法、随机引物法、末端标记法等。标记物质有放射性元素(如32P等)和非放射性物质(如生物素、地高辛等)。32P是最常用的核苷酸标记同位素,被标记的dNTP本身就带有磷酸基团,便于标记,特点是比活性高,可达9000Ci/mmol;发射的β射线能量高,可达1.70MeV,用它标记的探针自显影时间短,灵敏度高。32P的半寿期为14天,应随标随用,一般标记后,在一周内使用,它虽带来不便,但给使用后废弃物处理减轻了压力。使用32P标记物应注意防护,操作时应有1-1.5cm厚聚甲基丙烯酸甲酯有机玻璃隔离保护,避免直接照射。操作人员胸前应佩带个人计量器,定期检测。结束实验后,应用专用探测器(盖革-米勒检测器),检查工作区域、手、衣服等,以免污染发生。其它同位素标记物,同样应注意放射线防护。
非放射性标记有酶标和化学物标记法。酶标方法与免疫测定ELISA方法相似,只是被标记的核酸代替了被标记的抗体,事实上被标记的抗体也称为探针,阅读文献时应加以注意。现有许多商品是生物素(biotin)、地高辛标记的,如生物素-dUTP、生物素-dATP、地高辛-dUTP等。血凝素(avidin)与生物素有非常高的亲和性,当血凝素标记上过氧化物酶或碱性磷酸酶(血凝素-酶),经杂交反应最终形成:探针-生物素-血凝素-酶复合物(ABC法)。酶催化底物显色,观察结果。与一般的酶反应底物不同,ABC法底物显色后为不溶物,以便观测结果。酶标记法复杂、重复性差、成本高,但便于运输保存,灵敏度与放射物标记相当。化学物标记有的也是利用相应酶标抗体形成特异复合物,与上述方法相当,有的则可自发光。化学标记法简单、成本低,但灵敏度相对较低。(表18-1)
表18-1 探针标记及特性
| 标记物 | 检测方法 | 特点 |
| 32P | β射线,自显影 | 灵敏度最高,半寿期14天,放射强度1.7MeV |
| 35S | β射线,自显影 | 灵敏度高,分辨率好,半寿期87天,0.17MeV |
| 3H | β射线,自显影 | 低敏度高,分辨率高,半寿期12年,0.01MeV |
| 125I | γ射线,自显影 | 灵敏度高,分辨率高,半寿期60天,0.04MeV |
| 生物素 | 酶标血凝素,显色 | 灵敏度好,避免有内源性生物素标本 |
| 地高辛 | 酶标地高辛配体,显色 | 灵敏度好,分辨率一般 |
| T-T二聚体 | 酶标抗二聚体抗体,显色 | 灵敏度好,紫外照射形成二聚体而标记 |
| BrdU | 酶标抗BrdU抗体,显色 | 灵敏度好,细菌内含质粒复制掺入 |
| 铕-补骨脂素 | 荧光 | 灵敏度一般 |
缺口平移标记法先由DNase Ⅰ在DNA双链上切出随机切口,DNA聚合酶Ⅰ沿缺口水解5′端核苷酸和3′端修复加入被标记核苷酸同时进行,切口平行推移,可均一标记DNA链。(图18-4、5)
缺口平移法快速、简便、成本相对较低、比活性较高、标记均一。主要利用了DNA聚合酶Ⅰ修复DNA的功能,用于大分子DNA标记(>1000bp最好),单链DNA、RNA不能用该法标记。随机引物法具有上述优点,可代替缺口平移法。此外大小、单双DNA均可标记,标记均匀,标记率高,但不能标记环状DNA。利用末端转移酶可进行“尾标”,尾标适用于寡核苷酸探针标记,用于大分子核酸标记因尾巴短而标记比活性降低。尾标不能用dTTP标记,因mRNA的多聚(A)尾而影响杂交特异性。寡核苷酸探针多用于核酸“点”突变检测,该探针可用核酸合成仪人工合成。克隆探针一般较长,特异性
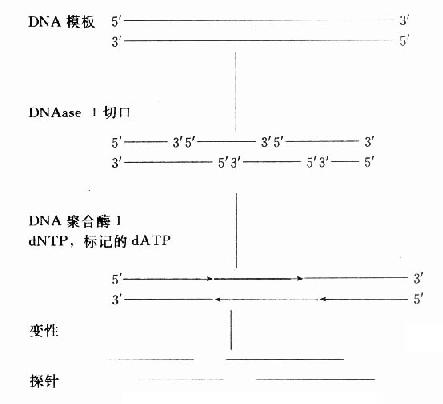
图18-5 DNA随机引物标记法
好,标记量大,杂交检出信号强。合成探针长度短,一般在20-50核苷酸之间,过长合成成本高,且易出现聚合酶合成错误,杂交时间长。太短则特异性下降。合成探针设计还应注意碱基组成G≡C含40%-60%,一种碱基连续重复不超过4个,以免非特异性杂交产生。还要注意探针自身序列内应无互补区域以免产生“发夹”结构,影响杂交。一个好的探针最终在实践中才能加以确认。另外,还有利用M13噬菌体载体可复制单链DNA的特点,复制合成DNA探针,利用转录载体(DNA),转录合成RNA探针。下面就GIBCoBRL公司(Bethesda Research Laboratories美国)的缺口平移法标记生物素试剂盒(bionick labeling system)为例介绍如下:
| 试剂: | 10x dNTP Mix.250μl | 10x Enzyme Mix 250μl |
| 0.2mmol/L each dCTP,dCTP,dTTP | 0.5 units/μl DNA Polymerase Ⅰ | |
| 0.1mmol/L dATP | 0.0075 units/μl DNase Ⅰ | |
| 0.1mmol/L biotin-14-dATP | 50mmol/L Tris-HCl(pH7.5) | |
| 500mmol/L Tris-HCl(pH7.8) | 5mmol/L magnesium acetate | |
| 50mmol/L MgCl2 | 1mmol/L β-mercaptoethanol | |
| 100mmol/L β-mercaptoethanol | 0.1mmol/L phenylmethyl- | |
| sulfonyl fluoride | ||
| 100μg/ml nuclease-free BSA | 50%(v/v)glycerol | |
| 100μg/ml nuclease-free BSA | ||
| Control DNA:5μg pBR322 in TE | ||
| stop buffer 300 mM EDTA | ||
| autoclaved H2O |
操作步骤:
将5μl 10x dNTP Mix.;-μl DNA(相当1μg需标记的DNA量);-μl灭菌蒸馏水加至45μl;再加5μl10x Enzyme Mix.依次加入1.5-2ml离心管后。15000xg离心15秒钟。16℃保温1小时。加5μl Stop Buffer终止反应。乙醇沉淀,备用。
第三节 分子生物学实验诊断技术
一、核酸杂交技术
检验方法建立的基本要素是特异性和灵敏度,在复杂的物体中,使无法感觉的特定物质进入人类的观察范围。核酸杂交检测技术就是利用核酸碱基严格配对的特异性,核酸标记物的灵敏度而建立的检测核酸结构与功能的方法。该法建立以来已有二十年,目前研究实验室用得多,临床实验室用得较少,除成本高外,关键是操作复杂,重复性差,仅能半定量。成本高可随经济发展而缓和,但其它缺点尚需努力克服。
杂交实验的探针应具有特异性,对应不同的杂交方法靶基因(被检基因)应有一定的纯度与丰度。杂交反应的开始是碰撞,探针浓度高则速率增加,一般32P标记探记探针5-100ng/ml,非放射性探针25-1000ng/ml,原位杂交无论放射还是非放射物标记,一般都需0.1-5.0μg/ml。当探针过量时,杂交速率取决于探针长度,如一个100ng/ml20个核苷酸的合成寡核苷酸探针与1-100pg 1kb长的靶基因杂交,10分钟能达到最大杂交率。而相同条件下2kb的克隆探针需160小时。主要原因是这里所指浓度是重量浓度而非摩尔浓度。长探针因标记量大,小的摩尔浓度已足以达到需要的检测灵敏度。为了促进长探针(>250个核苷酸)的杂交速率,加入杂交促进剂是非常必要的。最常用的是硫酸葡聚糖,使用浓度为5%-10%,浓度过高会增加杂交液粘度。聚乙二醇也作为促进剂,价廉且粘度低,但检测本底太高。另外杂交的温度、盐浓度、甲酰胺浓度可调节杂交分子形成的稳定性。理论选用DNA:DNA杂交温度T=Tm-25℃,此时杂交分子最易形成。但具体实验中影响Tm值的因素很多,一般杂交温度低,形成的杂交分子较稳定。RNA:DNA杂交分子的Tm大10-15℃,RNA:RNA杂交分子的Tm大20-25℃,使得杂交温度提高,此时需调节甲酰胺浓度来调节杂交温度。好的实验条件最终应在实践中摸索建立。
(一)斑点杂交
将少量核酸样品点样在硝酸纤维素滤膜上,80℃烘烤后可牢固地固定在膜上,再用探针进行杂交。尼龙膜,特别是聚偏氟乙烯膜(PVDF)与DNA结合力更高,坚韧、易操作。点样可手工,也可用真空点样品。检测可用放射性探针自显影或非放射性探针显色。可用于DNA或RNA分析。下面以非放射性探针分析DNA为例,结果是显色。
取硝酸纤维素膜和滤纸在2xSSC(NaCl 300mmol/L,柠檬酸钠30mmol/L),pH7.0浸15分钟,平铺滤纸在点样抽滤器上,再铺上硝酸纤维滤膜,真空抽气使点样器减压,膜显出凹面。点样50μl(5-10μgDNA,可直接点血清样品)于凹面,撤去真空,把膜晾干。取滤纸浸0.5mol/l NaOH,1.0mol/l NaCl,把膜平铺于滤纸上20分钟,变性。取滤纸二张分别浸在0.5mol/l Tris-Hcl pH7.5和1.0mol/l Tris-HCl,0.6mol/l NaCl pH7.5,分别依次把膜平铺于滤纸上,各中和15分钟,中和后膜的pH为7.2-7.5。于80℃,30-45分钟烘干。(RNA分析点样缓冲液系统不同,无需变性,中和,余大致相同)用塑料封口机把膜和2.5ml预杂交缓冲液封入塑料袋中,42℃,水浴30分钟。
| 预杂交缓冲液: | 65℃6xSSC | (原液:20xSSPE) |
| 5xDenhardt | (50xDenhardt) | |
| 0.5%SDS | (10%SDS) | |
| 100μg/ml变性鲑精DNA片段 | (1mg/ml) | |
| 42℃增加50%甲酰胺 | (原液),配成20ml. |
50xDenhardt:5g聚蔗糖(Ficoll,400型,Parmacia),5g聚乙烯吡咯烷酮和5g牛血清白蛋白(组份Ⅴ,Sigma)加水至500ml。过滤后-20℃保存。
探针2ml(50-100ng/2ml预杂交缓冲液)于100℃,10分钟,马上置冰浴5分钟(变性)。加入袋封口。42℃水浴过夜。去杂交液(可回收)。以2xSSC,0.1%SDS15ml,50℃5分钟洗二次。0.1SSC,0.1%SDS洗二次。封闭:另取袋封入膜和封闭液(蛋白质50mg/ml,100mmol/l Tris-HCl(pH7.5)/150mmol/LNaCl)2.5ml,37℃,30分钟。去封闭液(可回收)。100mmol/lTris-HCl(pH7.5)/150mmol/L NaCl ,15ml,5分钟洗二次。亲和反应:酶标抗体3ml(酶有碱性磷酸酶、过氧化物酶等,标记物有抗地高辛、生物素等,现以地高辛标碱性磷酸酶为例4μg/3ml 100mmol/L Tris-HCl,150mmol/l MgCl2,10mg/ml牛血清白蛋白),封入袋中37℃,30分钟。100mmol/l Tris-Hcl,100mmol/l NaCl,50mmol/l MgCl2,pH9.5,10ml,1分钟洗一次。取硝基四氮唑兰(NBT)75mg/ml 70%二甲基甲酰胺25μl,4-溴-5-氯-3-吲哚磷酸50mg/ml二甲基甲酰胺20μl(底物)和100mmol/l Tris-HCl,100mmol/l NaCl,50mmol/l MgCl2,pH9.56ml混匀,37℃,避光反应三小时,显色。蒸馏水洗膜,晾干。观察结果。
(二)Southern blot
1975年E.Southern发明了将DNA切开,电泳分离,再变性,印迹到硝酸纤维(Nc)滤膜上,用探针进行杂交,检测DNA的方法。自显影或显色用于DNA分析。以32P标记放射性探针为例,放射自显影观察结果。(图18-6)。
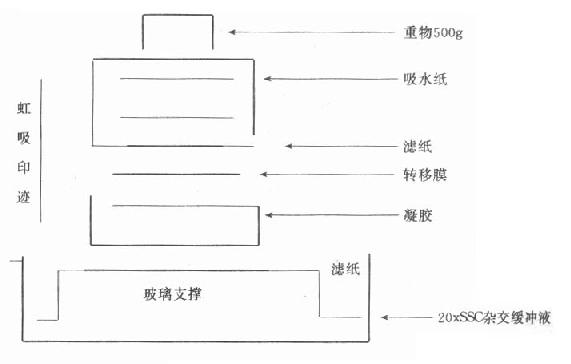
图18-6 Southernblot体系
前述分离、纯化的DNA样品5-10μg/100μlTE,加限制性内切酶3Unit/1μgDNA和内切酶相应缓冲液,在相应温度保温1-3小时(不同的酶所用的缓冲液和温度不同),乙醇沉淀,15-20μl TE溶解DNA断片。前述电泳条件,与DNA分子量标准品(Marker,Ladder)一起,3-4V/cm电泳(30V12-15小时)。在紫外光下观察Marker充分分离,结束电泳。将胶放入0.5mol/l NaCl,0.5mol/L NaOH液体中30分钟,使DNA变性。同时将膜用蒸馏水浸润,在0.5mol/L NaCl,0.5mol/L NaOH液体的方盘皿上,放上滤纸两头浸在液体中,依次放上凝胶、膜、滤纸、一尺厚的吸水纸,压力1kg的重物。转移(印迹,blot)过夜。翌日,把膜放在0.5mol/l Tris-HCl,pH7.0-7.5,1mol/L NaCl液中,中和膜上碱性变性液15-30分钟。戴上手套,用手指压在膜上来回充分洗膜。室温干燥一小时。用塑料封口机,把膜和预杂交缓冲液封在塑料口袋中,注意驱除袋内汽泡。热水浴过夜。探针变性后,加入袋中再封口。热水浴过夜。
| 洗膜: | 2xSSPE,0.5%SDS,总体积200ml,室温30分钟。 |
| 0.4%xSSPE,0.5%SDS,总体积200ml,60℃,30分钟。 | |
| 将膜包入塑料袋,在暗室贴在X光底片上。-70℃,自显影1-2天。 | |
| 冲片显影,观察结果(背景不清晰可重新洗膜再显影)。 |
(三)Northern blot
用于RNA分析,电泳条件与转膜方法与Southern blot不同外,RNA不必变性与中和,电泳时加电醛防止RNA发夹结构形成。其它步骤相同。
为了防止RNase水解需分析的mRNA,尽可能将器皿在160-180℃干热灭菌8小时以上,也可加0.1%焦碳酸二乙酯(DEPC)泡2小时,灭菌水淋洗干净,100℃干烤15分钟。不能干热灭菌的试剂等,也可加1%DEPC处理12小时(但不能用于Tris)后,100℃。加热15分钟(或高压灭菌15分钟)以抑制、分解RNase。1.0g琼脂糖,灭菌水80ml加热溶解。加x50TAE2ml,EB25μl,灭菌水至1000ml。样品缓冲液,x50TAE20μl,50%去离子甲酰胺500μl,甲醛180μl,混匀。约10-30μg/10μl纯化的RNA样品,加二倍(20μl)样品缓冲液(可点样一个凝胶孔lane)。60℃水浴15min,马上置冰浴。加1/10电泳指示剂。点样电泳,75-80V电泳4-5小时(指示剂泳动7~8cm),结束电泳。电泳期间正负极缓冲液要不断交换(可用蠕动泵),以免pH改变。电泳结束,将凝胶放在紫外光下可观察到人rRNA大亚基28S(5.1kb),小亚基18S(2.0kb),记下大小亚基移动距离(或拍照),可作为被测mRNA分子量分析的参照物。凝胶在50mmol/l NaOH中变性30分钟(低浓度碱,此步可省)。膜在10xSSPE中浸20分钟。用10xSSPE转移过夜(同Southern blot)。80℃1-2小时干燥。用探针杂交后,显影或显色。
(四)原位杂交(insituhybridization)
在保持细胞形状条件下,进行细胞内杂交,显影或显色。用于DNA或RNA分析。
细胞用离心涂片机涂片或组织切片置于载玻片上。4%多聚甲醛(PFA)/PBS固定(固定时间因标本而异10-20分钟,也可用含2%甲醛,0.05%戊二醛,2.5mmol/lCaCl2的0.1mol/L磷酸缓冲液pH7.3,500W微波炉中照射10-20秒,固定)。马上用PBS洗标本三次,2.5μg/ml蛋白酶K,37℃,5-20分钟(因标本和固定条件而定)处理。磷酸盐类缓冲液(PBs NaCl 137mmol/l,KCl 2.7mmol/L,Na2HPO48.1mmol/L,KH2PO41.5mmol/L)洗涤。4%PFA/PBS后固定,PBS洗一次,0.2%甘氨酸/PBS洗二次,每次15分钟。预杂交:42℃,1小时。预杂交缓冲液:10%硫酸葡聚糖,10%Denhardt液,0.5%吐温-20,250μg/ml鲑鱼精子DNA,500μg/ml酵母tRNA。杂交:42℃,4小时。用2x杂交缓冲液(4xSSC,0.2mol/L磷酸钠pH6.5,2xDenhardt)溶解已标记探针,使其终浓度为0.1-1.0μg/ml。DNA检测时把探针覆盖在标本上,置100℃5分钟(变性)取出,杂交。mRNA检测则先把探针置95℃水浴3分钟,立即置冰水浴,再覆盖标本上进行杂交。覆盖标本用液量100-200μl洗涤:2xSSC,50℃过夜。0.2xSSC,室温1小时。载玻片浸在缓冲液中。显色(试剂、方法参照斑点杂交的封闭-显色部分)20分钟-2小时,显微镜下观察结果。
二、核酸扩增技术
通过大肠埃希菌繁殖而增殖重组质粒,也是扩增核酸的手段。但在试管中有效扩增DNA的方法,是在90年代才开展起来的多聚酶链反应(polymerase chain reaction,PCR)新技术。由于PCR灵敏度高、特异性好、操作方便,在我国发展很快。目前,PCR技术结合分子生物学实验技术(如逆转录反应杂交技术等)开发了许多PCR新技术(reverse transcriptase PCR;PCR-single strandconformation polymorphism,PCR-SSCP;巢式PCR(二次PCR);热启动PCR等)。RT-PCR用于RNA分析;PCR-SSCP分辨率高,可用于点突变分析;二次PCR特异性好,灵敏度高;热启动PCR可提高特异性。
PCR技术原理是在了解DNA一级结构基础上设计引物(primer或sense,人工合成),DNA多聚酶可识别并结合引物,以单链DNA为模板,dNTP为底物,合成其互补链。通过反复变性,反复合成互补链的过程,达到DNA扩增的目的。人的DNA扩增引物长度多为20个核甘酸。(图18-7)

图18-7 PCR技术原理
PCR反应:DNA样品0.1-1μg,引物1,2各25-100pmol,dNTP各200μmol/L,Tris-HCl(pH8.3)10mmol/L,KCl 50mmol/L,MgCl21.5mmol/L,gelatin(明胶)0.01%,Taq多聚酶2.5-5U,矿物油一滴。93℃7分钟→50-55℃2分钟→70-72℃3-1.5分钟→93℃2分钟。
![]()
将反应物和Marker点样于凝胶一起电泳,紫外光下观察结果,拍照保存结果。反应物也可用于印迹(杂交)实验、多态分析、探针制备等。
注意事项:①DNA样品纯度高,Taq酶(和Klenow酶)有逆转录酶活性,DNA和RNA不能混在一起。②设计的引物分子内(特别3′端)和引物,1,2分子间不形成双链。选择性好,不增幅目的DNA以外区域的DNA。引物的G+C含量为40%-60%。③dNTP浓度过高易使Taq酶合成错误,一般在200μmol/L,有人建议40-50μmol/L。④KCl>75mmol/L对酶有抑制作用。Mg2+浓度过高,NDA不易变性,>10mmol/L可抑制酶活性40%-50%。⑤多聚酶:Taq多聚酶从嗜热菌分离得到(1989年,在此以前1985年利用Klenow酶因不耐热每经过一次热变性要补加一次酶),现在经基因克隆的重组体产物Taq酶最适pH8.3~8.8(室温8.3-8.4),反应温度75-80℃,95℃以上失活明显。无3′→5′校读活性,对SDS敏感(<0.01%活性也受到抑制,加吐温-20可抵制SDS抑制)。该酶有逆转录酶活性。⑥退火温度一般设定为Tm-5℃,但实际上与引物一级结构序列有关,设计不当可造成非特异性退火,而造成非特异扩增,此时应调整温度和相应的时间。⑦循环次数受到的影响因素较多,增加次数可提高灵敏度,但易出现非特异扩增。⑧PCR灵敏高,被检样品极易被污染,样品纯化,扩增,检测区域应分开。房间应经常用紫外光照射。扩增物应定点丢弃,处理。
三、核酸限制性片段长度多态性(RFLP)分析
RFLP分析是限制性内切酶、核酸电泳、印迹(blot)技术、探针-杂交技术的综合应用,多用于临床遗传性疾病的基因诊断。基本原理是遗传性疾病多有基因的缺陷,如基因缺失、基因插入、编码区小范围核苷酸序列改变或点突变等。前二者可将纯化的正常人和病人DNA同时用限制性内切酶切成片段,经电泳分离、印迹、探针杂交等找到目的基因。由于患者基因的缺失或插入,造成被限制性内切酶切开的片段(分子量)大小与正常人发生差异(限制性片段长度多态),使其电泳迁移率发生差异,而达到基因诊断的目的。小区域或点突变,可选用一个限制性内切酶的切点正好在这一变异位置,使切割作用和正常人发生差异(如正常人基因可切断而患者不能,或反之),达到诊断目的。现在PCR产物经变性为DNA单链,用聚丙烯酰胺凝胶电泳分离,只要有一个核苷酸发生变异,其电泳迁移率就会改变,进行多态分析(PCR-SSCP)。实际上把基因的异常位置设计在PCR扩增区域内,就能进行多态分析。(图18-8)
核酸纯化→内切酶作用→电泳分离→Southern blot→探针-杂交→显影,显色
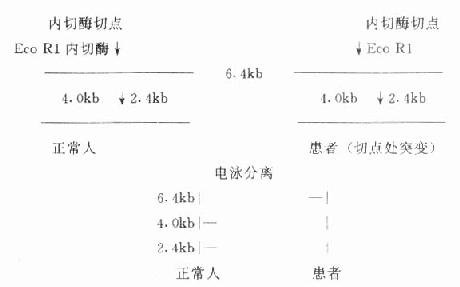
图18-8 点突变Eco R1RFL
四、核酸一级结构(核苷酸序列sequence)分析
核酸一级结构分析是基因分析最直接的第一手资料。目前DNA、RNA以及蛋白质的一级结构分析中,DNA的碱基序列分析方法多样、简单、快速。下面就M13噬菌体-双脱氧核苷酸(ddNTP)的DNA测序法介绍如下。
M13是单链DNA噬菌体,转染宿主菌体复制为双链增殖,又以单链形式透出菌体。把待测DNA重组插入双链M13复制型DNA中,经培养扩增,可分离得到单链M13DNA(含待测单链DNA的碱基序列),即复制模板。在待测DNA的3′端人工合成引物,在DNA聚合酶Ⅰ作用下,复制链延长。在ddNTP(2′,3′双脱氧核糖核苷三磷酸)存在下,复制延长随机受阻,形成分子量(长度)大小不等的片段。电泳分离,自显影,读图18-9分析碱基序列。

图18-9 核酸碱基序列分析
0.2-0.3mm厚度聚丙烯酰胺-尿素凝胶,高压(1600V)电泳,可分辨一个核苷酸的差异。电泳后干燥凝胶,自显影,自下往上读图,得到5′→3′的合成链碱基序列,其互补链是待测DNA的3′→5′碱基序列。
第四节 诊断分子生物学技术的临床应用
一种检验方法从建立到临床应用涉及许多因素,关键是临床需要和质量可靠,结果可信、实用二个方面。随着对发病机制研究的不断深入,在分子水平诊断、治疗、预后疾病已为人们所认识。分子生物学技术一般操作较复杂,成本较高,质控较差,但它在基因分子水平揭示发病的机制及本质、诊断、治疗疾病的媚力不可抗拒,人们正在努力改进技术以便符合临床要求。如PCR技术的出现,立即得到临床的响应。
一、诊断分子生物学技术应用范围
外源基因侵入致病,如病毒、细菌、支原体、衣原体、寄生虫等的传统检验、分类方法效率低,往往不能满足临床要求。而外源致病生物侵入体内发病时,拷贝数较高,样品取材、核酸提取相对容易,加之诊断分子生物学技术的灵敏度与特异性,使临床应用价值提高。特别是病毒致病日益猖獗,临床诊断的不断需要,及应用试剂盒的不断开发,操作手续的不断简化,外源基因的检验方法在临床不断得到应用。该领域的发展趋势很可能成为诊断分子生物学技术在临床应用的突破口,应用前景广阔。
内源性基因诊断目前在遗传性疾病诊断方面应用较多,不断向肿瘤(癌基因)及其它领域深入,尚在数据积累,研究阶段。
二、诊断分子生物学技术质量控制
对检验技术的具体要求首先是检验诊断学的质量保证,由完善、合理的实验设计来完成。
⒈特异性杂交技术是通过基因克隆制备探针来完成,PCR是通过引物来完成,RFLP是利用限制内切酶来完成等。即探针、引物、限制性内切酶在反应中都具有特异性。
⒉灵敏度 杂交技术用同位素或酶标记探针,PCR反复扩增20-30次都是提高灵敏度的手段。二次PCR、PCR与Southern blot合用都是典型的例子。
⒊操作简单 杂交技术因操作复杂且费时(需1-2天以上出结果),在临床实验室难以推广。而PCR方法相对简单、快速,故推广迅速。
⒋实验成本分子生物学实验因试剂多数依赖进口,成本高,给临床推广造成一定的困难。试剂国产化是今后的努力方向。
重复性和稳定性也是我们关心的问题,但报告较少。
本世纪生命科学飞跃发展,尤其是近年来分子生物学新技术日新月异,使过去难以想象的实验正在进入常规实验室。以基因结构、基因功能研究为基础,已深入到医学研究各领域,在神经分子生物学、肿瘤分子生物学、基因治疗等医学领域做出了引人注目的成绩。分子生物学技术、诊断分子生物学(基因诊断)、基因治疗已经是医学工作者不可回避的课题。分子生物学技术理论深,内容多,应用广。本章只介绍一些基本知识的技术作为入门,更详尽的内容可参考有关专著和资料。
第十九章 临床生物化学分析仪的性能与应用
临床生化自动分析仪,就是把生化分析中的取样、加试剂、去干扰、混合、保温反应、检测、结果计算、显示和打印,以及清洗等步骤自动化的仪器。它完全模仿并代替了手工操作。不仅提高了工作效率,而且减少了主观误差,稳定了检验质量。由于这类仪器一般都具有灵敏、准确、快速、节省和标准化等优点,使得生化自动分析仪在临床生化分析中得到了广泛应用。
第一节 临床生化自动分析仪的类型
一、概述
自50年代Skeggs首次介绍一种临床生化分析仪的原理以来,随着科学技术尤其是医学科学的发展,各种生化自动分析仪和诊断试剂均有了很大发展,根据仪器的结构原理不同,可分为连续流动式(管道式)、分立式、离心式和干片式四类。新型的自动分析仪常装有:样品识别、自动加样、自动检测、数据处理、打印报告和自动报警等装置。
(一)管道式分析仪
管道式分析仪的特点是测定项目相同的各待测样品与试剂混合后的化学反应,是在同一管道中经流动过程完成的。这类仪器一般可分为空气分段系统式和非分段系统式。所谓空气分段系统是指在吸入管道的每一个样品、试剂以及混合后的反应液之间,均由一小段空气间隔开;而非分段系统是靠试剂空白或缓冲液来间隔每个样品的反应液。在管道式分析仪中,以空气分段系统式最多,且较典型,整套仪器是由样品盘、比例泵、混合管、透析器、恒温器、比色计和记录器几个部件所组成(图19-1)。管道内的圆圈表示气泡,气泡可将样品及试剂分隔为许多液柱,并起一定的搅拌作用,但气泡影响比色,必须在比色前除去。
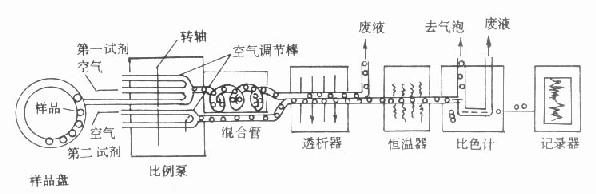
图19-1 单通道管道式自动分析仪结构示意图
将几个单通道管道式分析仪结合起来,对一个样品同时测定几个项目。著名的有12通道分析仪,命名为顺序多项自动分析仪(sequential multiple autoanalyzer),简称SMA12/60。同时发展为SMAC,C为计算机(computer)的缩写。这类大型分析仪至今仍有使用者。它对每个样品可同时分析12个项目,每小时可分析60个样品。后来发现不加入空气泡,更有利于结果的检测,发展为流动注射分析法(flowinjection analysis)。试剂的流动仍用比例泵驱动,样品用转动阀加入液流,反应后比色检测。
(二)分立式分析仪
所谓分立式,是指按手工操作的方式编排程序,并以有节奏的机械操作代替手工,各环节用转送带连接起来,按顺序依次操作。分立式分析仪与管道式分析仪在结构上的主要区别为:前者各个样品和试剂在各自的试管中起反应,而后者是在同一管道中起反应;前者采用由采样器和加液器组成的稀释器来取样和加试剂,而不用比例泵;前者一般没有透析器,如要除蛋白质等干扰,需另行处理。恒温器必须能容纳需保温的试管和试管架,所以比管道式分析仪的体积要大。除此以外,其它部件与管道式的基本相似。分立式分析仪的基本结构如图19-2。Dupout公司还推出一种袋式分析仪,即试管变为塑料袋,反应在袋内进行,也以袋作比色杯。这种袋只能一次性使用。
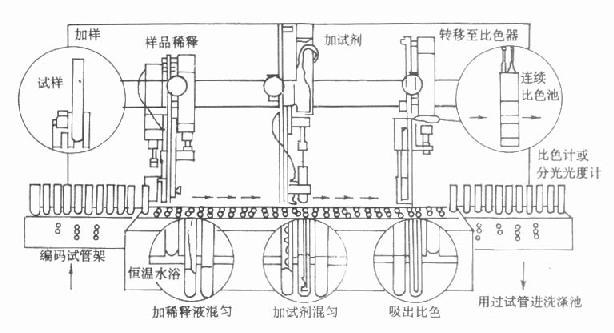
图19-2 分立式自动分析仪结构示意图
(三)离心式分析仪
离心式分析仪是1969年以后发展起来的一种分析仪,由Anderson设计,其特点是化学反应器装在离心机的转子位置,该圆形反应器称为转头,先将样品和试剂分别置于转头内,当离心机开动后,圆盘内的样品和试剂受离心力的作用而相互混合发生反应,最后流入圆盘外圈的比色槽内,通过比色计进行检测(图19-3)。这类分析仪特点是①在整个分析过程中,各样品与试剂的混合、反应和检测等每一步骤,几乎都是同时完成的,不同于管道式和分立式分析仪的“顺序分析”,而是基于“同步分析”的原理而设计。②样品量和试剂量均为微量级(样品用1-50μl,试剂120-300μl),快速分析(每小时可分析600个样品以上)。③转头是这类分析仪的特殊结构。早期的转头由转移盘、比色槽、上下玻璃卷和上下套壳六个部件组成,现已被一次成型的塑料制品代替。转头转动时,各比色槽被轮流连续监测,如转速为960r/min,转头上有20个比色槽,则每分钟可接受19200个电信号,配有微机进行控制和数据处理。
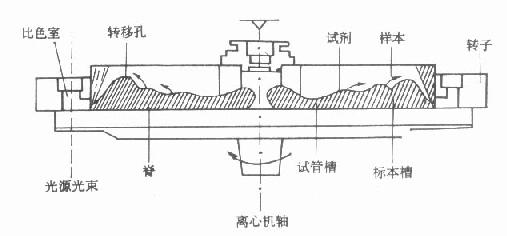
图19-3 离心式自动分析仪工作原理示意图
离心式分析仪主要由两部分组成,一为加样部分,二为分析部分。加样部分包括样品盘、试剂盘、吸样臂(或管)、试剂臂(加液器)和电子控制部分(键盘和显示器等)。加样时转头置于加样部分。加样完毕后将转头移至离心机上。分析部分,除安装转头的离心机外,还有温控和光学检测系统,并有微机信息处理和显示系统。
(四)干片式分析仪
干片式分析仪是80年代问世的。首先Eastman Kodak公司以其精湛的化学工艺造出了测定血清中血糖、尿素、蛋白质、胆固醇等的干式试剂片。当加上定量的血清后,在干片的前面产生颜色反应,用反射光度计检测即可进行定量。这类方法完全革除了液体试剂,故称干化学法。Boehringer Mannheim公司推出了用全血的干征试剂。即将血细胞排除于滤膜之外,而血浆与试剂发生反应后显色检测。干片试剂结构示意图见图19-4。
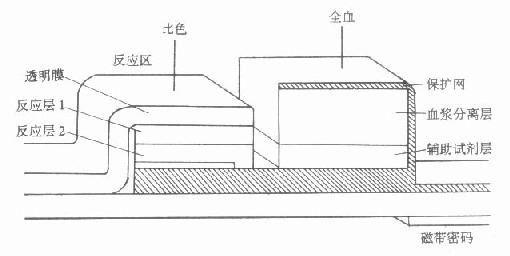
图19-4 平片试剂结构示意图
干片不仅包括试剂,也可由电极构成,所以这类分析仪也可进行电解质的测定。这类干片均为一次性使用,故成本较贵。
二、半自动生化分析仪
半自动生化分析仪指在分析过程中的部分操作(如加样、保温、吸入比色、结果记录等某一步骤)需手工完成,而另一部分操作则可由仪器自动完成。这类仪器的特点是体积小,结构简单,灵活性大,既可分开单独使用,又可与其他仪器配套使用,价格便宜,一般在分立式分析仪中多常见。
荷兰VitalScientific生产的ISP-M半自动生化分析仪,是一种典型且性能比较好的半自动生化分析仪,能应用于动力学法、两点法及终点法的检测,可以采用人机对话的方式把要检测的30个项目的各参数输入仪器进行贮存,也可以随试验项目、方法、试剂等条件的改变而随时进行修改。它有一个微量流动比色杯,因此每次检测所需反应液很少,一般用400μl左右即可。还有一个非常敏感的能控制三种温度(25℃、30℃、37℃)的恒温器,能在数秒钟内迅速达到要求的温度。所有的检测步骤及试剂结果,可由显示窗显示及由打印机自动打印出来。
这类仪器一般不受试剂、方法的限制,国产试剂、自己配制的试剂及自行设计的方法均可在仪器上进行检测,而且由于仪器的体积小、重量轻、操作方便、反应迅速,尤其适合中、小型检验单位作为日常生化检测的主要仪器,也适用于作急诊生化检测及外出执行任务时使用。
这类仪器使用时应注意:①大多使用固定的比色池,结果的重复性比较好,但由于检测方法、所加试剂及样品量都很少,因此要特别注意取样和加试剂的准确性。②如反复出现非线性(NL)大于10%,应考虑试剂是否变质及所编程序是否合适。③在作动力学方法检测时,要注意试验的反应原理,如果是吸光度下降的应在浓度因数(F)值前面加负号,否则结果不准。做酶动力学方法检测时,还要注意开始的吸光度(Ai),如果特别低,可能由于样品中酶活力较高,在延迟时间内基质已被消耗完。此时,必须把样品进行稀释后再进行检测。④出现仪器不吸液体,常常由于仪器长时间不用,吸液管被压扁而堵塞,只要稍加揉擦即可畅通,如管道由于不及时清洗而造成的堵塞,这就要进行清洗或更换。⑤在使用过程中,严格吸入强酸试剂,否则将会损坏吸管的金属头,每次使用完毕,都必须用蒸馏水冲洗比色池和管道,如果在反复多次吸入蒸馏水后,不能调整零点,应用清洁液浸泡比色池。具体操作是将温度放在37℃,用10%清洁液(dete-rgent)充满比色池,浸泡15分钟,再用水冲洗。仪器长时间不用,应在一个月内通电一次,否则仪器内程序的记忆即消失。
三、全自动生化分析仪
全自动生化分析仪,从加样至出结果的全过程完全由仪器自动完成。操作者只需把样品放在分析仪的特定位置上,选用程序开动仪器即可等取检验报告。由于分析中没有手工操作步骤,故主观误差很少,且由于该类仪器一般都具有自动报告异常情况,自动校正自身工作状态的功能,因此系统误差也较小,给使用者带来很大的便利。
自美国Technicon公司于1957年成功地生产了世界上第一台全自动生化分析仪后,各种型号和功能不同的全自动生化分析仪不断涌现,为医院临床生化检验的自动化迈出了十分重要的一步。这类仪器的功能全,灵活性大,易于操作,大多采用吸光度、荧光、光散、浊度测定技术和离子选择电极系统,用于常规生化、特殊蛋白和药物监测等检测,可随机安排程序,既可根据需要输入单一项目成批分析,又可按临床医生根据患者病情不同要求,选择性的多项组合分析;结果报告既能打印出每个项目的报告,也能按人打印出所做全部项目的累积报告。在测试过程中可随时加入急诊项目,优先分析,打印出报告后,仪器仍按原输入程序继续进行检测。仪器以微机控制,采用人机对话方式来安排程序操作,可自由编排和清除程序,可贮存100个以上分析方法,可进行统计学处理,求均值、标准差、变异系数、相关系数等。有的分析仪采用了化学惰性“液囊式技术”(capsulechemistry technology),可将不同的分析标本或各种试验项目严格地分立开,互不掺杂,无需用水作为冲洗系统去防止交叉污染,更有利于随时任选项目检测。
这类仪器适应在大、中型检测单位用于临床生化项目的检测。
纵观临床生化自动分析仪的发展史,也是临床生化检测方法学的发展史,最初出现的管道式分析仪适应了化学分析的需要,设计了透析器等排除蛋白质干扰的装置,但是随着表面活性剂的应用,消除了蛋白质的沉淀干扰,已无必要进行透析分离。随着酶试剂的生产,许多以特异性酶促反应的测定方法,消除了干扰物的影响。这是分立式和离心式分析仪发展的原因之一。干片式分析仪更是试剂生产工艺革新的产物。随着方法学的改进,分析的反应部分的体积逐渐缩小,而检测、监控和信息处理部分,多数由微机担任,体积则逐渐扩展,直至可将测定结果通过网络系统,输送至病房。有人预言,随着电化学分析法的进展,各种特异性电极的研制成功,将来的自动分析仪有可能由许多精细的电极组成,则其结构将比现有的分析仪更简单化。
第二节 临床生化自动分析仪的性能评价与合理选用
目前,临床生化自动分析仪的规格型号很多,生产厂也很多,随着技术的革新和应用的要求,在性能和结构上都有了很大改进和发展。正确评价与合理选用这些仪器十分重要。
一、自动化程度
对一台生化分析仪来说,自动化程度越高,说明仪器功能越强。仪器的自动化程度高低取决于仪器所使用微处理机的功能大小,根据仪器计算机功能的不同,自动化分析仪一般可分为全自动和半自动两种。
全自动分析仪比较适合于样品量多,化验项目多,人力相对不足的综合性大医院的临床化验室应用。而对于样品数量、化验项目少的小医院或专科医院使用半自动分析仪则更为合适。
二、分析效率
这里的分析效率,系指在测定方法相同的情况下不同分析仪的分析速度。显然,分析速度决定于一次测定中可测样品多少和可测项目的多少。对不同类型的分析仪,由于其结构和设计原理的不同,微机应用程序不同造成自动化程度的差异,也直接影响到分析仪的分析效率。
离心式分析仪采用同步分析原理设计,测定中所有样品的混合、反应及比色几乎同时进行。另外,其加样部分与分析部分又可各自独立工作,在一批样品分析的同时,可进行另一批样品的加样,节约了时间。因此,这类分析仪的分析速度快于其它类型。当然这也不是绝对的,当样品很少时,甚至只有一个样品,但为了保持离心机的平衡,也必须在转头的空槽中用蒸馏水一一加入方可。这样分析显然还不如用半自动的分析仪更为快一些。
三、应用范围
自动生化分析仪的应用范围包括可测试的生化项目、其它项目、反应的类型及分析方法的种类等。应用范围广的分析仪不仅能测多种临床生化检验指标,而且还可进行药物监测和各种特异蛋白的分析、微量元素测定等。分析方法除了分光光度法外,还能进行浊度比色法、离子选择性电极法、荧光法等测定。既能用终点法,又可用动态法测定。它可使酶免疫技术、固相酶技术得以应用,从而在科研和技术生产开发工作中,发挥更大的效益。有些分析仪采用了独特的双波长光路设计,可消除“背景噪声”,排除样品中溶血、脂血及胆红素等成分的干扰,精确灵敏,在一些特殊的测量分析中很有用。通常小型的、程序固定式的半自动化的分析仪,其工作应用范围相对较窄,选择时应予注意。
四、分析准确度
生化分析仪测量的准确度愈高愈好。准确度取决于各部件(加液、温控、波长、记时等)的加工精确度及其精确的工作状态。如有的分析仪采用样品液体感应探针,准确吸样,使样品携带率低于0.5%,不仅能准确地吸取微量样品,而且还能充分混合样品及试剂,使测量结果精确度提高。另外,克服分析仪中交叉污染,也是保证测定结果准确的重要环节。生化分析仪一般对取样探针有自动清洗装置,从而保证测试结果的准确率。
除此之外,仪器的寿命、仪器的维修保养方式和途径、测定所需样品和试剂的数量,以及配套试剂盒的供应等,特别是仪器的性能价格比,在选用时都应一并考虑,使选用的分析仪能够物尽其用,且又经济实惠,能够取得最大的效益。
第三节 临床生化自动分析的方法
自动分析仪的应用决不仅仅是操作方法的变化,由于引进了一系列高精技术,所用的方法测定原理都有了迅速的发展。因此,用好自动分析仪的一个重要前提,必须对自动分析仪可以提供的测试方法类型、主要实验室参数和仪器室条件及仪器操作有所了解。
一、分析方法的分类
临床生化常用方法根据其测定的原理可作如图19-5法分类。一类方法是物理方法,测定是物质固有的物理特性。另一类是物理化学方法,也就是将所测定物质进行一些化学转化后再进行测定。动态法是在所测定的物质浓度在不断变化中,由传感器获得相应的改变的信号进行计算的方法。平衡法是由传感器得到相对不变的信号进行计算的方法。现在越来越多被临床生化应用的酶试剂方法,无论是用自动分析仪记录或分光亮度计或普通比色计都包括在动态法的范围内。
(一)平衡法(终点法)
这类方法的特点是被测物质(酶反应的底物)在酶反应过程中应完全被转化或消耗掉,即达到反应的终点。通常这类方法操作简单,一般不需要作标准管,通过一些已知的理化常数,例如克分子吸光系数等不难将结果计算出来。其缺点是反应时间较长,特别不适合于离心式自动分析仪。还有少数酶反应,底物并未完全消耗掉,只是达到一个动态平衡,此时,往往需要同时作标准管。
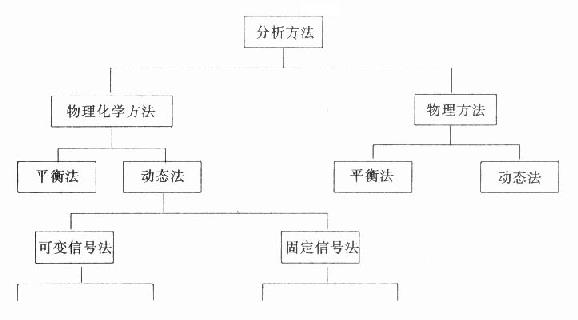
图19-5 临床生化方法的分类
⒈一步法 试剂酶的底物(S)就是所测的物质,在试剂酶作用于S后完全转化为产物(P),由于S和P具有完全不同的理化性质,从而可根据对S或P的浓度变化进行连续监测。使用最多的还是光度法,尤其是在340nm处测定NAD(P)H的生成或消耗量。在实际应用上最多的仍是和NAD(P)H有关的脱氢反应。如用乙醇脱氢酶测乙醇的含量或用乳酸脱氢酶测丙酮酸等。
⒉酶偶联反应和测定酶活性方法一样,有时单用一个酶(辅助酶,Ea)不行,因为S和P之间往往无明显差异,不能直接测定,此时可以加入另外的酶(指示酶,Ei),将反应偶联起来,通过测定指示酶反应间接推算出所测物质,即第一个酶作用底物的浓度。
反应通式如下:

若Ei是一个脱氢酶,在反应过程中同时伴有NAD(P)H和NAD(P)间的转化,故不难在340nm处进行连续监测。另一常用的酶是过氧化物酶,可以通过新生态氧和一些色素原作用显色而进行测定。例如用已糖激酶(HK)法和葡萄糖氧化酶(GOD)法来测定葡萄糖等。
另外,还有一类少用的偶联酶反应,指示酶反应在辅助酶反应之前,而不是在后。
![]()
式中S1是欲测物质,往往另一底物S2不稳定,通过先行的指示酶产生S2,如乙酰CoA的测定。

其中草酰乙酸很不稳定,可通过苹果酸脱氢酶作用生成。
从理论上说酶催化反应都是可逆的反应,除水解酶外,大多数酶往往都不易将底物完全转换或消耗掉。常可通过增加不需测定的另一底物浓度,改变pH,使用捕获剂(trap-ping agent)等改变平衡点,使反应偏向一侧,达到或接近反应完全。
(二)动态法
动态法在自动分析仪中的应用十分广泛,但是动态法的分类一直比较混乱。一般可分为可变信号法和固定信号法两类。
⒈可变信号法
⑴直接法即根据所得到的吸光度变化直接计算结果:由于对所收集数据多少有很大意义,所以又区分为一点法、二点法、多点法。一点法是在一个预先设置时间测定各个标本吸光度。二点法则是在一点法基础上多测一点,即在酶反应仍未进行时的吸光度,和一点法相比可以通过空白测定扣除这部分误差,但是仍无法了解反应过程,避免不了延缓期或非线性反应所引起的误差。多点法如使用确当,对标本空白和反应过程都有详细了解,可用以下两法:
一法是求出不同测定点间吸光度的差异即所谓delta法。各点吸光度差异可用б=△A=An-An-1计算出。根据这些数据可以观察反应是否符合线性,并计算出被测物质浓度。偏离线性的数据在计算结果时应摒弃。很多早期自动生化仪都使用此法。另一法则是使用回归法,根据一定公式如线性公式y=a+bx对获得的多个数据进行计算,求出斜率b和截距a。这类方法也可用于浓度计算。和delta方法相比,不仅可适用于线性反应,也可适用于其它非线性反应。
⑵导数法在此法中使用电子线路计算出反应瞬间的一阶和二阶导数:bekman system TR就使用了这样方法,它可以同时计算出一阶和二阶导数,用二阶导数来观察哪一段反应为线性反应,并用线性反应段上的一阶导数计算出物质浓度。此法单用导数方法并不记录吸光度变化(△A),所以此法所提供的信息往往少于回归法,例如无法观察到反应中底物的消耗情况。
⑶积分法此法原理是运算放大器对反应曲线二个节段的吸光度进行积分。并计算出部分面积之差。Vitatron分析仪用此差值来考虑线性段,但仍用△A法计算结果。
⒉固定信号法测定原理是随着反应进行,通过不断添加试剂使吸光度变化在很窄范围内,此时所测的是加试剂的量,并依此来计算物质浓度。最典型例子是以对硝基酚作为指示剂的乙酰胆碱酯酶方法,在反应过程中不断加入碱以中和酶水解产物乙酸以维持pH不变,然后根据所加碱量计算酶含量。这一类方法由于操作费时麻烦,目前应用很少,但不应忘记这类方法能维持反应条件如pH的恒定性,其它如在NADH参与的反应中不断加入NADH有助于避免底物的消耗,在一些特殊情况下可能还是有用的。
由此可见,假如从方便简单不需复杂仪器而言,则一点和二点法应为首选。而回归法、积分和导数法不易进行。如从可靠性来进行比较,则多点回归法应居榜首,一点和二点法最不准确。
回归法的缺点是此法可以不考虑收集的数据是否可靠,不管各数据是否偏离曲线或明显不呈线性,也可求得一个线性方程式。所以,一个好的回归方程式还应给出标准差和有关参数,这样即可用来评价用回归法所得结果的可靠性。根据Pardue意见,使用较多数据且设计良好的回归方法是首选的。
(三)固定时间法(二点法)
固定时间法在自动分析仪中的应用,有助于我们解决反应的特异性问题,最明显的例子就是苦味酸法测肌酐和溴甲酚绿法测白蛋白。人们早就知道很多物质如维生素C、乙酰醋酸、葡萄糖、果糖以及某些药物也能和苦味酸呈色。近年来发现这些干扰物质呈色较慢,提出用自动生化仪只测定开始一分钟的反应,明显提高了苦味酸法测肌酐的特异性。用溴甲酚绿测白蛋白法,此法由于简单易行,在70年代取代了经典的盐析法,但随即发现一部分球蛋白如α球蛋白也能和溴甲酚绿结合,但这类反应比较迟,所以目前更多地使用自动分析仪测定开始一分钟的反应来计算白蛋白量。
固定时间法之所以受到愈来愈多注意,更重要因素是酶试剂的应用(详见有关章节)。
(四)空白对照方法
由于使用了各种高新技术,在自动生化分析仪中人们可以根据实际情况,使用各种空白和对照方法。
⒈固定法空白①试剂;②标本+盐水;③标本+空白试剂;④标本+灭能试剂;⑤标本+完全试剂(时间不同);
⒉动态法空白①试剂空白;②标本+空白试剂。
从理论上说固定空白法用“标本+完全试剂”是最好的办法,此法可以扣除各种因素的误差,如试剂、比色杯等,这也是在自动生化仪应用较多的方法,而用手工法是不易做到的。动态空白仅用于少数试剂不稳定方法,如以固红B测天门冬氨酸转氨酶。
二、主要实验参数的选择(设置)
一般自动分析仪有12个主要数据,即波长、温度、标本及试剂量、空白时间(Tb)、开始收集实验数据时间(Ti)、实验数据收集窗时间(TW)、实验数据收集完成时间(Td)和最后时间(Tf)、速率时间(Tr)、线性限度(L)、异常吸收率(ABN)、曲线拟合、浓度因素(F)。这些参数都是通过方法学的研究之后,根据反应的具体情况加以确定的。
(一)波长
根据分光亮度计的光吸收曲线或者一个比色杯不同波长的反应曲线选择分析波长和空白波长。对快速反应,以光吸收曲线的低凹区波长为空白波长,光吸收曲线中吸亮度最大而较平坦区的波长为分析波长。
(二)温度
一般均选用30℃。
(三)样品量及试剂量
可根据手工法按比例缩减或者重新设计。要考虑到检测灵敏度、线性范围,尽可能将样品稀释倍数大些,以降低样品中其它成分的影响。
(四)分析时间的确定
用高、中、低浓度的标准液进行实验用研究模式,进行酶促反应时间曲线或反应速度时间曲线的观察,根据吸收率动态变化的具体数据,确定有关的分析时间。
⒈Tb 终点法,吸光度上升型,一般选择反应尚示开始而试剂和样品已充分混合均匀,一般定为4秒。动态法的Tb=Ti。
⒉Ti 终点法选择反应接近平衡期,动态法则选择接近进入线性期。这个参数不很严格。如果Ti定的太早,过多的不符合要求的数据点将舍弃。Ti定的太晚,数据收集时间就要延长,在动态法还可能使底物耗尽的发生机会增多。在终点法Ti大致定于Tf的70%或接近于反应完全时,动态法确定Ti时,主要考虑避开酶反应的延迟期以及试剂和样品混合后温度不平衡期。
⒊TW 根据高、中、低浓度标准液反应速度时间曲线加以确定。要求各标准液的吸光度变化在该时间内均在线性判断标准范围内。终点法一般为4-10秒。在动态法中,为了使反应不出现未反应(NR)的标志,在Tb与Tf之间至少有8mA的吸收率变化。在快速反应时,TW从30秒开始,按10秒向上递增。在慢反应可达120秒。TW不应太长,否则可以偏离线性,也减低工作效率。
⒋Tr Tr值的大小对方法的精密度有较显著的影响。为了保证方法的精密度,使△A>1.0mA是必要的。如Tr时间短,△A太小,测定方法的精密度就降低。一般情况下,Tr定为20-60秒。在现有程序中,是以正常低值标本的△A>1mA来决定Tr值。如ALT测定,正常低值为7u/L,Tr=7u/L/2.57=2.7mA/60秒,30秒=1.35mA,故选30秒为Tr时间。
(五)线性判断标准(线性限L)
指在数据收集窗时间内吸收率变化的允许范围。作为终点法,从理论上讲,化学反应达到平衡(终点),吸光度应该稳定不变,也就是说,在TW内吸收率变化应该是零,即线性判断标准应该定为零。在实际中,由于信号有一定的飘移,更主要是测定要求快速,是在反应还没真正到达终点时进行测定,加上自动分析仪检测的灵敏度大多较高,精度可达到0.1mA,所以在TW期间的吸收率会有一定的波动。不同的化学反应或酶促反应,吸光度变化程度不一。要根据实验结果选择一个适当的吸光度变化范围作为线性判断标准,以此来判定反应是否达到了平衡。这个标准值如果定的太小,反应也很难达到要求的标准,出现非线性的机会太多,或者要延长观测时间,降低工作效率,这就不符合快速分析的要求。相反,这个线性判断标准定的太大,就失去了判断线性的意义,出现反应根本没有达到终点就错误地判为达到标准,影响测定结果的可靠性。在终点法中,L单位是mA/2秒,大多选用1.0mA/2秒。如出现反应不完全,可按0.5mA顺序增加。在动态法中,L单位是mA/秒,一般从0.1mA/秒开始,如果试验结果出现非线性(NL)情况多,可以增大,按0.01mA递增,但不可超过0.1mA。
(六)异常吸收率限度
这一参数主要根据每个方法实验测定的线性范围来确定。即用吸光率作为纵轴,标准液浓度为横轴,绘制标准曲线。这个线性段向非线性段转折拐点的相应吸收率值即为异常吸收率限度。终点法吸收率超过此值被标志为超出限度(XL)。在动态法中,试剂空白的吸收率超过ABN,ABN被标志为试剂吸收率异常(AB),测定杯的吸收率超过ABN时,ABN被标志为底物耗尽(EX)。
三、仪器室条件及仪器操作
(一)仪器室条件
⒈接入仪器室的电源线应大于仪器及室内电器所指定的安培容量。
⒉电压不稳定或低于额定电压的地区需加装大于指定功率的电压稳压器。
⒊经常停电地区可装大于指定功率的不间断电源,电源的工作时间任选。
⒋应安装空调器以保持恒温与无尘。空调器的制冷量应大于仪器的产热量,室温波动每分钟应不大于1.33℃。试验温度对室温是有要求的,各型号仪器要求不一,一般30℃试验的室温是18-25℃,37℃试验的室温要求是18-30℃。
(二)仪器操作
各种型号的仪器都有规定的日常操作程序,应按照说明书要求进行。
第二十章 临床生物化学方法的选择、建立和评价
临床生化方法的选择、建立和评价,是临床生化质量控制的重要基础工作,它们之间的关系可用图20-1表示。
本章内容主要指光谱分析方法的选择、建立和评价,对其它分析方法也有参考价值。其中所述评价系统适用于常规分析方法的评价,在评价试剂盒时也可供参考。
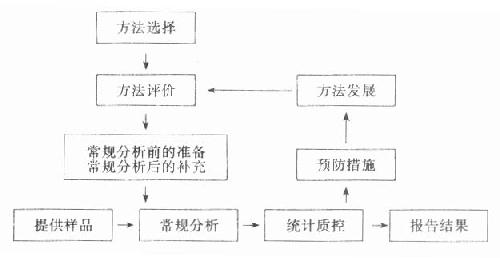
图20-1 方法选择、建立与评价之间的关系
第一节 临床生化方法的选择
一、方法和标准品的分级
(一)方法的分级
临床生化成分的测定方法很多,根据其准确度与精密度的不同可以分为三级。
⒈决定性方法(definitive method)是指准确度最高,系统误差最小,经过详细的研究,没有发现产生误差的原因或在某些方面不够明确的方法,测定结果为“确定值”,与“真值”最接近。由于技术要求太高,费用昂贵,不直接用于鉴定常规方法的准确性,只用于发展及评价参考方法和标准品。
⒉参考方法(reference method)是指准确度与精密度已经充分证实的分析方法,干扰因素少,系统误差很小,与重复测定的随机误差相比可以忽略不计,有适当的灵敏度、特异性、直线性及较宽的分析范围。这类方法应在条件优越的实验室中应经常使用。但它主要应用于鉴定常规方法,评价其误差大小,干扰因素,并决定其是否可以被接受。它也用于鉴定二级标准品,为质控血清定值,也可用于评价商品试剂盒等。
⒊常规方法(routine method)常规方法的性能指标符合临床或其它目的需要,有适当的分析范围,而且经济实用。所谓偏差已知的方法是指准确度已经确定的方法,其准确度不明者称为偏差未知的方法。常规方法在作出评价以后,经有关学术组织认可,可以作为推荐方法。
临床化学方法之间关系可见图20-2。
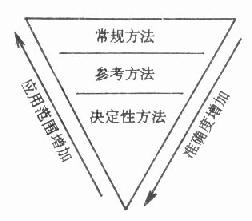
图20-2 临床化学方法的关系图
(二)标准品的分级
国际标准化委员会将标准品(参考物)的定义暂定为:它的一种或几种物理或化学性质已经充分确定,被用于校正仪器或证实一种测定方法的物质,并将其分成三级。
⒈一级标准品一级标准品(原级参考物)是已经确定的稳定而均一的物质,它的数值已由决定性方法确定,或由高度准确的若干方法确定,所含杂质也已经定量。它可用于校正决定性方法,评价及校正参考方法以及为“二级标准品”定值。一级标准品都有证书,在美国由国家标准局(NBS)发给合格证书,并指明它的性质和有关数据。
⒉二级标准品二级标准品(次级标准品)或是纯溶液(水或有机溶剂的溶液),或存在于相似基质中。这类标准品可由实验室自己配制或为商品,其中有关物质的量由参考方法定值或用一级标准品比较而确定,主要用于常规方法的标化或为控制物定值。
⒊控制物控制物有冻干的或溶液,可以用适当的标准品(一级或二级)以参考方法定值,用于质量控制,不用于标化(除经准确定值者)。
二、方法选择的要求
国际临床化学协会(IFCC)提出:常规方法应具有实用性和可靠性两方面的性能指标,至于某一项具体分析方法所应具有的性能标准,可由临床化学家根据采用这一试验的目的决定。
(一)实用性
一般应具备:①微量快速能便于急诊,适合成套项目分析。②费用低廉,包括劳动力与试剂、设备及一般管理费用等。③方法简便。④应用安全可靠。
(二)可靠性
一般具有较高的精密度和准确度,以及较大的检测能力。①精密度,变异系数(CV%)一般应小于5%。②准确度与特异性,准确度是指测定值与“真值”的符合程度。一般偏差系数(CB)应小于5%。特异性是指只对待测物质起化学反应,不与其它结构类似的化合物发生反应。③检测能力,即“检测限度”,或“检出限”。检出限通常是指能与适当的“空白”读数相区别的待测物的最小值。
三、方法选择的步骤
(一)广泛查阅文献
根据方法选择的要求对已发表的各种方法进行比较与检验,确定哪些方法有充分的科学根据及真实的使用价值。必须仔细分析各种方法的特点,选择适合于本实验室具体条件的方法,并且要判断所选方法能否满足本室工作的需要。如果文章中没有写上方法的有关性能资料或引用文献中的有关论述,则不应轻易采用。
(二)选定候选方法
经初步选定的方法称为候选方法。候选方法确定后,应该写出该法的原理与参考文献,所用仪器、试剂的来源、牌号、纯度(或配套试剂盒),标本收集要求,详细的操作步骤,标准曲线和结果计算,文献中列出的性能指标,参考值与参考范围,以及其它应注意的事项与安全防护措施等。
(三)进行初步试验
为了实用的目的,初试应该包括①标准曲线及其重复性;②质控血清或新鲜标本的重复试验;③分析待测物浓度不同的标本,与公认的参考方法的测定值作对比;④要有足够的试验证明仪器与试剂能符合要求。这些初试工作有希望揭露明显的问题。例如未曾预料的困难或误差来源等。初试的特殊目的是使分析工作者熟悉有关技术;掌握各分析步骤的特性;操作是否可以改进或简化;初试所得到的一切资料用于确定是否有必要作进一步研究,如需要和技术上的某些修改,应在评价实验之前作好。
第二节 临床生化方法的建立
一、方法建立的原理确定
临床生化方法按其传统分类可分为光谱法、层析法、电泳法、酶法、电化学法、分子生物学和免疫化学法等,这些方法的建立首先要根据其技术的原理(详见前面有关章节)和被测物质的物理化学性质来确定其方法建立的原理,例如亮度法测定的对象是各种有色物质,有色物质溶液的颜色深浅与其浓度有一定的关系,浓度大时色深,浓度小时色浅,比较颜色的深浅即可测知待测物质的浓度。为此,首先要知道待测物质本身是否有色,若本身无色,能与何种物质在什么条件下呈色,弄清后即可与一已知浓度的标准品作比较,然后分别测出两者的吸亮度,从郎伯-比耳定律可得知:
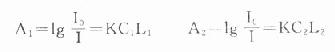
由于分析的物质相同,处理也一样,故K值相同,再因为测定时用同样比色皿(L1=L2),所以光度法中测得未知液与标准液吸光度的比值就等于其浓度的比值。(A1/A2=C1/C2)。如C1为未知物浓度,则可得到计算公式如下:
C1=A1×C2/A2
只要再乘以测定时标本的稀释倍数,即为通常光度法测定的通用的计算公式,但此公式只有在测定时空白管为零的条件下才适用。
二、方法建立时的条件选择
在方法建立的原理确定后,尚需根据其原理来选择合适的条件,以保证所建的方法可靠。下面以亮度法为例介绍其条件选择的主要环节。颜色反应是亮度分析的基础,因此,用作亮度测定的颜色反应至少要具备以下几条:反应有较高的特异性,从而干扰小;反应产生的有色物质吸光系数要大,大者灵敏度高;有色产物的离解度要小,小则稳定;此外,还要求有色产物有恒定的组成成分,使产生的颜色稳定。由于影响颜色反应的因素很多,如温度、pH、试剂浓度、反应的时间等等。这些因素在建立新方法时都要逐项试验,确定最佳实验条件。
(一)选择合适的吸收光谱
实际上就是测定颜色反应产生的光吸收曲线。方法是在整个可见光区(400-700nm)以10nm(在接近最大吸收波长范围还应再细分)间隔。分为十几个点测定其在不同波长时的吸光度,然后以吸光度为纵坐标,波长为横坐标绘成光吸收曲线。在条件许可的情况下,应采用双光束分光光度计在可见光区进行波长扫描,结果较为可靠。同时,还应以同法分别测定其空白液和标准物颜色产物的吸收曲线。测定光吸收曲线的目的是找出最大吸收波长,作为光度法波长选择的依据。因为在最大吸收波长测定,可获得最大灵敏度,且能更符合比耳定律。然而在实际使用中,不能单纯考虑灵敏度高,还要考虑在测定浓度范围能否符合比耳定律,并能在光度计准确区域内(吸光度0.2-0.85之间)读出读数。有时由于测定的最大吸收波长并不是所要测定物质的特性吸收波长,即同时存在具有相似最大吸收的干扰物质,为了保证实验的特异性,常改用特有而非最大吸收波长进行测定。例如用磷钼酸测定血糖,选用波长420nm,溶液对此波长的吸收比其它波长光线的吸收都低,其目的是使参考值有一个较低的吸收,这样可允许在相同情况下对高出参考值的血糖也能得到准确的读数。因为在这项测定中,高血糖是常见的变化方向。
(二)测定方法适用的浓度范围
根据光吸收定律,溶液的吸光度应与溶液的浓度成正比。但由于有色物质的电离、水解、缔合等原因当溶液稀释或增浓时,有色物质颜色的深浅并不按比例降低或增高,因而也就不符合光吸收定律。测定方法适用的浓度范围是指分析的浓度范围在直线线性段,浓度与吸光度之比为一常数,此时直线上任何一点的斜率tanθ相等,即:
tanθ=A1/C1=A2/C2=A3/C3=……=An/Cn=K
此处K即为校正常数,可用于结果计算。
通常稀溶液都是符合比耳定律的,但在浓度过高和过低时往往都不呈直线,说明低浓度部分亦有仪器的灵敏度和方法的检测能力限制,所以,具体的测定就宜选择在直线段浓度范围内进行。以此还可用于确定标本的用量,使具有临床意义的标本测定结果都落在直线范围内。
(三)观察影响颜色反应的因素
影响颜色反应的主要因素有:溶液pH的影响、杂质的影响、反应的温度和时间、以及颜色的稳定性等。这些都是光度法测定误差的主要来源。都需要通过实验来观察并控制在一个适宜的范围内。从而保证方法的准确性和精密性。
⒈溶液pH的影响有些溶液的颜色对pH值的改变非常灵敏。例如白蛋白在pH4左右可与溴甲酚绿结合后由黄色变成绿色,绿色的深浅与白蛋白浓度成正比,但是溴甲酚绿本身是一种pH指示剂,当pH5.4时即由黄色转变成绿色,在pH3.8时又由绿色变为黄色。在白蛋白与溴甲酚绿结合颜色反应中,如pH控制不好就很容易出现误差,又如每种酶都有各自的最适pH,酶促反应中,只有在其最适pH时才能发挥充分的催化活性。因此,在建立方法时可在不同pH值条件下(其它条件不变)令其反应,以观察颜色反应的吸光度变化,从而选择合适的pH值范围来保证颜色反应,以求较高的灵敏度和较好的线性。有时还需用相应的pH缓冲液来维持反应的pH,以利颜色反应的正常进行。
⒉杂质的影响由于临床生化标本中除含待测物质外,还含有许多非测定物质,成分十分复杂,它们有的可与有色产物作用,使产生的颜色逐渐退去,有的与待测物质相类似,也能缓慢地与显色试剂生成有色化合物或沉淀,有的本身是有色的等,都会影响被测物溶液的颜色。试验方法是将各种可疑物质的纯溶液作标本单独进行测定,如产生颜色反应,这说明该方法的特异性不高。如果将待测物质混入可疑物质作标本测定,改变了被测物质与试剂间的反应,这是干扰(具体方法见第三节)。为此就需要进一步改进方法,或设立标本空白,或将标本作适当处理,除去干扰物,或加入合适的干扰物掩蔽剂等,以提高方法的特异性。
⒊反应的温度和时间有些有色物质能迅速反应生成,另一些有色物质须经过相当长时间后反应才能完全。提高反应的温度可以加速化学反应,缩短反应的时间。因此,如果在不恰当的温度和时间内进行比色,将会造成相当大的误差。这可以在不同的温度下(如设定25℃,30℃,37℃)令其反应,通过检测吸光度来观察各自反应终点时间(一般达到反应终点,随后测定的吸光度不再变化),以选定适宜临床应用的反应最佳温度和时间。
⒋稳定性试验待测物质经显色反应产生的有色物稳定与否,关系到测定结果的可靠性。某些有色物质虽然生成很快,但却不太稳定,放置后色泽会逐步消退或起变化,有些干扰物虽不能与被测物质同时发生颜色反应,但当放置一段时间后也能缓慢地与试剂显色,使溶液颜色加深。因此,在方法建立的同时作稳定性试验很有必要。试验的方法是用被测标本、标准溶液、空白溶液按方法要求进行显色反应后,即刻及室温放置不同时间,分别测定其吸光度,根据其吸光度的变化确定方法稳定的时间范围。一般要求有色溶液能稳定二小时以上就能满足临床应用的需要。必要时还要加以注明。
三、方法建立后的临床观察
方法建立的目的是为了更好地应用于临床,为疾病的诊断和治疗提供可靠的依据。因此,在方法建立后必须作临床观察试验。
(一)正常参考值试验
制定参考值(referencevalues)时,首先要阅读有关资料,依照经典的文献作为研究设计的依据,使设计尽量合理,结果令人信服,有实用价值。1978年,国际临床化学委员会下属的“参考值”理论专家组发表专门文件,推荐出一套有关参考值的概念、定义、建立和使用方案。参考值的建立应包括参考个体、参考人群、参考标本、参考值、参考分布、参考限和参考区间。它们之间的关系如下:

具体试验是对选定的一定人群中的健康人,按性别、年龄等分组,每组至少100人,应用所建立方法测定,如所得某体液成分的结果呈正态分析,经统计处理求出均值(X)和标准差(S),然后将X±2S定为正常参考范围。但如胆红素、铁、尿素及碱性磷酸酶等,常呈不对称的偏态分布,需经适当的数据处理,使之接近正态分布的形式,这样就可参照正态分析数据一样来对待。常用的有对数转换法,使每一个数值转换为相应的对数值,用其对数值作图,若呈正态分布,则利用此对数值计算X和S值,最后将结果转换回为相应的自然对数值,求得均值及参考值的上限和下限。确定的参考值要求符合以前的调查报告。
(二)临床病例观察试验
临床病例观察的目的是指出所建方法的某项试验,对特定疾病的预示值(PV)。预示值为一项诊断试验确定或排队某疾病存在与否的诊断概率。预示值尚可分为阳性预示值(PV+)和阴性预示值(PV-)它们分别表示确定诊断或排除诊断的把握有多大,是诊断试验的评价指标。诊断试验评价指标除预示值外,还有诊断特异性、诊断灵敏度和准确度。这些指标都与疾病的发生率有关。现将有关名词的概念,作简要地介绍。
⒈真阳性者(TP)经试验而被正确分类的患者的数目。
⒉假阳性者(FP)经试验而被错误分类的无病试者的数目。
⒊真阴性者(TN)经试验而被正确分类的无病试者的数目。
⒋假阴性者(FN)经试验而被错误分类的患病试者的数目。
⒌诊断灵敏度 患病试者的阳性百分率。

⒍诊断特异性 无病试者的阴性百分率。
![]()
⒎阳性结果的预示值(PV+)阳性结果中真阳性者的百分数。
![]()
⒏阴性结果的预示值(PV-)阴性结果中真阴性者的百分数。
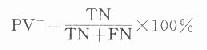
⒐实验的总有效率所有实验结果中正确结果的百分数。
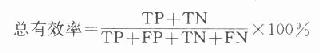
在临床应用中,主要以阳性结果的预示值观察实验结果的使用价值。但在固定参考范围的条件下,对不同发病率的人群进行相同指标检测时,所得到的预示值会有很大变化。例如用某方法对某专科病房检测100个标本,发病率为50%,方法诊断灵敏度为95%,诊断特异性为95%。由此数据可知:
TP=50×95%=47.5人;FN=50-47.5=2.5人;
TN=50×95%=47.5人;FP=50-47.5=2.5人;
TP+FP=50人,TN+FN=50人
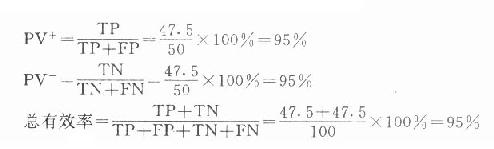
临床对该检测方法的运用满意。经同样方法用于普查,调查某地区发病率为5%,检测1000例中,950人为非病人,50人为病人。
TP=50×95%=47.5人;FN=50-47.5=2.5人;
TN=950×95%=902.5人;FP=950-902.5=47.5人;
PV+=47.5/95×100%=50%
PV-=902.5/905×100%=99.7%
总有效率=(47.5+902.5)/(95+905)×100%=95%
临床认为这个检验方法使用价值不高,因为在普查中假阳性率占50%,给进一步诊断带来困难。如可能可调整参数值范围来提高方法的使用价值。一般认为,用于过筛实验的分析方法,希望有最高的灵敏度;用于确诊实验的分析方法,必须有高特异性。
(三)质量控制观察
方法是用控制物随临床常规标本一起用所建方法测定,连续测定一个月。以观察在实验室常规应用中的准确性和精密性,判断是否满足临床应用的要求,具体做法见第二十一章。
第三节 临床生化方法学的评价
一、评价实验与分析误差类型的关系
方法学评价(evalution of methodology)的基本内容是通过实验途径,测定并评价方法的精密度与准确度,在实验中测定的是不精密度与不准确度,不伦精密度还是准确度,强调的都是误差,评价实验的过程就是对误差的测定。因此,方法评价的各项实验是配合检测各种类型的分析误差而设计的,表20-1表示了它们之间的关系。
表20-1 评价实验与分析误差类型的关系
| 分析误差的类型 | 评价实验 | |
| 初步试验 | 最后试验 | |
| 偶然误差 | 批内(或天内)重复性 | 天间重复性 |
| 恒定误差 | 干扰 | 与比较方法对比 |
| 比例误差 | 回收 | |
二、实验方法的评价
(一)重复性试验
目的在于考察候选方法的随机误差,重复性试验的方法是对同一材料分成数份试验样品,进行多次分析测定。
⒈试验形式与方法 ①批内重复性试验:可以用同一份标本在相当短的时间内,按规定的操作方法,在较稳定的条件下,作多次(一般为20次)重复性测定。计算其X、S与CV。不同性质的分析对CV值的要求不同,一般应小于5%,精密的分析或多次平行测定,要求小于2%。②天内重复性试验:在一天内对一个或数个标本作数批重复测定,其它条件和批内相同,但因在一天内重复测定几批,所受影响因素要比批内多,所得的CV值可能比批内大一些。③天间重复性试验:将同一标本每天一次随机插入常规标本中测定,连续20个工作日,这样得到的CV值比上述批内与天内大,能反映实际工作的情况。④病人标本的平行双份测定:如不易得到含有合适基质的稳定样品(如作血液pH的重复性测定的标本),可采用病人标本的双份测定来代替重复性试验,选择一批病人标本(20至100份),待测物浓度应在实验方法的分析范围内,有高有低;记录标本的特性,如有某种药物、混浊、溶血或有尿毒症等,可影响其准确性。可作一批或几批分析,但每一标本都平行做两份,用双份分析结果之差(d)来计算S(∑d/2N)作为不精密度的指标。
⒉分析样品的选择作重复性试验的样品,应用标准液、控制物溶液、病人标本或混合血清均可,视其用途而定。作批内重复性试验,用标准液可以得到各种不同浓度,较简便,它代表了最好的重复性性能。若结果良好,说明方法操作是适宜的,再进一步用其它样品进行测定。在进行天间重复性试验时,用冻干质控血清较好,因其稳定、方便。其它样品进行测定。在进行天间重复性试验时,用冻干质控血清较好,因其稳定、方便。
⒊分析物浓度的选择进行重复性试验时被测物浓度,宜选择在医学上具有决定性意义的浓度进行试验,因为这种浓度在临床分析实验结果时最有用处。
(二)回收试验
所谓回收即分析方法正确测定加入常规分析样品中的纯分析物的能力。目的是测定比例系统误差,以衡量候选方法的准确度。
⒈方法将被分析的纯品标准液加入病人样品中,成为分析样品,原病人样品加入相同量的无分析物的溶液作基础样品,然后用实验方法分析,两者测定结果的差值为回收量。
⒉回收率计算 以测定血清钙为例说明如下:
⑴样品制备
1)基础样品:血清2ml+蒸馏水0.1ml;
2)分析样品:血清2ml+4mmol/L钙标准液0.1ml;
3)分析样品:血清2ml+25mmol/L钙标准液0.1ml;
⑵计算公式及计算结果

故本例的②③管的加入量分别为:

回收浓度=分析样品测得浓度-基础样品测得浓度
回收率(%)=回收浓度/加入浓度×100%
本例结果见表20-2
表20-2 血清钙回收试验结果
| 测得浓度(mmol/L) | 加入浓度(mmol/L) | 回收浓度(mmol/L) | 回收率% |
| ①1.70 | ― | ― | ― |
| ②1.88 | 0.19 | 0.18 | 94.7 |
| ③2.85 | 1.19 | 1.15 | 96.6 |
| 平均 | 95.7 |
理想的结果期望回收率达到100%,现在平均回收率为95.7%,有4.3%的比例误差,它表明一个含钙真值为2.5mmol/L的标本,若用该方法测定,约为2.39mmol/L(2.5×95.79%),误差为0.11mmol/L(2.5×4.3%)。
⒊注意事项
①准确吸量,因为被分析物的理论值是根据加入标准液体积及原样品的体积计算所得,吸量稍有不准,就会影响结果。因此,选择经过校准的吸管,严格地清洗与干燥,以及按正规要求吸量均很重要。②样品中加入标准液后,总的浓度必须在测定方法的分析范围内,一般须作加入高、中、低不同浓度的回收试验,计算平均回收率。③加入标准液后,最好使实验样品中被测浓度达到医学上具有决定意义的浓度。④加入标准液的体积在整个样品中要少,一般在10%以内。若稀释过大,误差将发生改变,甚至消失。
(三)干扰试验
干扰试验的目的也是用来衡量候选方法的准确度的。在加入一定浓度的干扰物的条件下,形成的是恒定系统误差。干扰物浓度不同,误差大小也不同。
⒈方法基本与回收试验一样,但是加入的是疑有干扰或非特异性反应的物质而不是标准液。以肌酐对葡萄糖测定的干扰试验方法及结果简述如下:
样品制备
1)基础样品:0.9ml血清+0.1ml蒸馏水。
2)分析样品:0.9ml血清+0.1ml 442μmol/L肌酐溶液。
3) 分析样品:0.9ml血清+0.1ml 884μmol/L肌酐溶液。
所有各样品均作双份葡萄糖测定取平均值。加入值的计算同回收试验。分析样品与基础样品两值之差为干扰值。结果如下:
| 葡萄糖测得值 | 加入肌酐值 | 干扰值 |
| 5.55mmol/L | - | - |
| 5.77mmol/L | 442μmol/L | 0.22mmol/L |
| 6.16mmol/L | 884μmol/L | 0.61mmol/L |
结果说明:每88.4μmol/L肌酐约使葡萄糖测定增加0.056mmol/L。在正常人,肌酐约为132μmol/L,将使葡萄糖测定带入约0.11mmol/L的误差,在临床上并无意义。但肌酐病理值可达884-1786μmol/L,对葡萄糖测定引入约0.56-1.11mmol/L误差,就会影响临床应用。
⒉可疑干扰物的浓度加入可疑干扰物的浓度须达到有价值的范围,有可能应达到病理标本的最高浓度值,在确证有影响后应测定在何浓度时,产生的误差在临床上无意义,即确定使分析结果影响临床应用价值的最低可疑物浓度值。
⒊被试验的物质可根据该方法的反应原理,提示出可能的干扰物。一般应考虑的是用胆红素、溶血(血红蛋白)、脂血、防腐剂、抗凝剂等进行试验。但干扰试验有一定的局限性,因为人们只能试验某些物质的影响,还有许多药物和食物成分未经试验,亦不能认为无关。干扰试验也可用比较方法进行,比较方法若为常规方法更应如此,若两个方法具有相同干扰,这种干扰不应作为否定试验方法的理由,试验方法应在其它性能方面较原常规方法为好,从而对检验质量有所提高。
⒋消除干扰的常用方法 ①作空白(对照)试验。一是试剂空白,可以校正标本读数中的试剂部分;另一种是标本空白,用以补偿标本中被测物以外的其它物质的影响。②采用各种物理、化学的方法、分离除去干扰物。③近年来出现的由计算机控制的双波长或多波长检测仪器,在排除干扰因素方面既有效又简便(详见仪器分析有关内容)。
(四)方法比较实验
对比试验用于检测候选方法的系统分析误差,包括比例和恒定两种系统误差在内。如果对适当的数据进行分析,也可提供系统误差的性质究竟是属恒定误差还是比例误差。
⒈方法对一组病人的标本用候选方法和对比方法同时进行分析测定,最后观察两者之间的差异。这是考验候选方法是否可被采用的重要措施。
⒉比较方法的选择在临床生化常规方法评价时,最好选择参考方法作为对比方法。这样在解释结果时,就可把方法间的任何分析误差都可归于候选方法。
⒊注意事项 ①试验样品数:一般作40-100例,包括各种疾病的样品,即包括在常规工作中可能遇到的整个分析范围。选择合适的样品比增加试验样品的数目更为重要。②重复分析:一般每个样品用两种方法各测定一次,最好各测定两次,不是平行测定,而是分两批进行。两种方法及两批测定,须在同一天内,最好是在4小时内进行。③时间间隔:每天测定2-5个样品,约测定20天。④结果的图示:根据实验结果作坐标图,比较方法所得数值为横坐标,候选方法所得数值为纵坐标,用以定性地估计分析误差。进行双份测定时,候选方法的第一次结果与对比方法的第一次结果作图;第二次结果也如此。溶血、脂血、黄疸标本,可在相应的散点上标以H、L、I等符号。每天测定的结果应及时绘于图上,以便及时检查结果,发现问题,对结果有矛盾的样品,可取原样重新测定。
⒋统计处理方法比较试验所得的结果是配对资料,可进行配对资料的统计处理,包括配对t检验、相关和回归分析等。通常都选用相关回归分析方法,因为此法可以计算出所研究浓度范围内任一浓度的系统误差。从回归方程y=a+bx中,可显示出候选方法有无系统性误差。这种误差分三类:恒定系统误差与比例系统误差,或两者兼有。回归线的截距a代表恒定误差,回归系数(回归线的斜率)b代表比例误差,相关系数r代表两种方法的相关性是否密切。
实际情况并不简单,X与Y值都有偶然误差存在,b与a只是估计值,需要确定其可信限。在评价一种候选方法时,如果a=0,b的数值稍偏离1.0,那么这种偏离程度是否小到可以忽略不计,因而使候选方法可以接受。这就需要参考a及b值的变动范围,根据经验判断是否可以适用于临床检验的目的。在方法比较试验中的相关系数r可作为候选方法可否接受的一种指针,但r值随病人标本测定范围的增加而增大,因此,在配对资料分析中,不应片面地根据相关系数r来判断两种方法分析结果的符合程度。
第四节 临床生化方法学性能判断
候选方法可否被接受,最后根据评价实验中的误差结果进行归纳,作出判断。West-gard曾经对医学决定水平上的分析误差,采用统计学方法制定出一套判断指标,首先是制定“可允许误差的95%限度”,然后计算各项误差并与其比较,任何一项指标大于可允许误差都不能被接受。
一、方法学性能标准及其制定
(一)性能标准
性能标准(performance standards,PS)也称分析目标,应根据不同的应用目的(筛选、诊断、预后、监测)而异。由允许分析误差(allowableanalyticalerror)和医学决定水平(medical decision level)这两项内容决定。
⒈允许分析误差 用EA表示,它被规定为95%样品的允许误差限度,即95%的病人样品其误差应小于这个限度。
⒉医学决定水平 用Xc表示,临床判断结果具有意义的分析物浓度。
EA和Xc两项内容,就是一个测定方法的性能指标。但对于每一医学决定水平都应规定相应的性能标准,即在一定Xc值下的EA值。以血清葡萄糖测定为例。在Xcl=2.8mmol/L,Xc2=6.7mmol/L,Xc3=8.9mmol/L时,其相应的EA均为0.56mmol/L,而在Xc4=16.8mmol/L,其EA为1.4mmol/L。
这表示当葡萄糖浓度在2.8mmol/L、6.7mmol/L和8.9mmol/L时,95%的样品具有的误差不得大于0.56mmol/L,而浓度在16.8mmol/L时,95%的样品所具有的误差不得大于1.4mmol/L。这里指的是总误差。
(二)性能标准的制定
制定的性能标准,既应反映临床应用与解释结果的要求,称为医学效用限度(medicalusefulness limits),又应基本符合实验室所能达到的技能状态(state of art)。因此,需要由临床医学家和临床化学家共同研究制定。
⒈根据参考值与参考范围而定的标准这是Tonks于1963年提出的,其公式是:

最大的可允许误差定为±10%,但对某些物质(如酶)则提高到±20%。由于其允许误差以参考范围表示,有临床实用意义,但缺点是参考范围的宽度与试验本身的不精密度有关,一项不精密的方法由于测出的参考范围较宽,以致可制定出较宽的可允许误差限度,就会使一项性能不够好的分析方法的应用合法化。
⒉根据临床观察制定的标准这往往反映参与与制定的医生的经验,综合临床及实验室的意见,提出在医学决定水平上的不精密度标准。还有不准确度及综合不精密度与不准确度的标准。一般说来,在参考范围内的中间及很不正常的水平时,医生允许有较大的变异系数。特别是用于诊断,而不是用于监视疗效的。但在医学决定水平需要最严格的指标。这种方法的缺点是主观成分较大,而且收集临床的意见也很困难,它依赖于临床医生对实验室工作的充分了解。
⒊根据生物学变异制定的不精密度标准生物学变异或称生理变异(CVB)包括个体内变异(CVP)及个体间变异(CVg),也就是通常所说的生理波动。
美国病理学会建议,为了在人群中筛选某些疾病,不精密度(CV)应等于或小于个体内及个体间变异的二分之一(即≤1/2CVB)。对于个别试验,目的在于辅助诊断或监测治疗效果,则CV应小于或等于1/2CVP。但目前文献报告的生物学变异数据不一致,由于这种方法比较好,将来可能会有更多的研究。
⒋根据实验室技能状态制定的标准技能状态是根据技术熟练而且误差最小的一组实验室的数据,计算出在参考范围高限的变异系数作为标准的,这实际上是指当前技术水平所可能达到的技能状态。当然,技能状态有继续进步的趋势,在没有更好的标准以前,技能状态还会继续用作实验室工作性能的标准。对于生物变异相当大的项目,以技能状态作为不精密度的标准更为合适。
二、使用单值判断指标判断
单值判断指标(single-value criteria)较简单,在评价过程中用于初步估量。
⒈计算公式 单值判断指标的计算公式见表20-3
表20-3 单值判断指标
| 误差类别 | 判断指标 | 备注 |
| 随机误差(RE) | 1.96TM<EA Xc |
STM=重复试验的标准差 |
| 比例误差(PE) | (|R-100|)(棧?/FONT>EA 100 |
R=平均回收率 |
| 恒定误差(CE) | |偏差|<EA | 由干扰试验测出 |
| 系统误差(SE) | |(a+bXc)-Xc|<EA | 对比试验回归方程 |
| 总误差(TE=RE+SE) | 1.96STM+|(a+bXc)|<EA | 包括偶然及系统误差 |
⒉结果判断单值判断指标是可接受性能的估计指标。对前述各项实验
结果经计算得到各项误差值后,分别与EA值比较,必须都比EA小,该方法才初步判断为可接受,否则为不可接受,可改进分析方法减少误差或排除该方法。
判断举例:用单值判断指标判断某法测定血肌酐的结果:
设Xc=176.8μmol/L时,EA=35.4μmol/L,
⑴偶然误差
重复性误差:n=20,X=176.8μmol/L,STM=3.536μmol/L
判断:1.96STM=1.96×3.563μmol/L=6.93μmol/L,因RE<EA,偶然误差可以接受。
⑵系统误差:回归方程:n=50,a=-0.98,b=0.925,Y=-0.98+0.925X。
SE=|(a+bXc)-Xc|=|(-0.98+0.925×176.8)-176.8|=14.24μmol/L
判断:|(a+bXc)Xc|<EA,系统误差可以接受。
⑶总误差:RE=6.93μmol/L,SE=14.24μmol/L
判断:RE+SE=6.93+14.24=21.17μmol/L<EA,总误差可以接受,故该方法为可接受。
对于一个判断为不能接受的方法,为了减少某一误差而作了改进,则各个评价试验都须重新进行做。
单值判断指标虽简单,但主要问题是各项试验的样品数都较小,使测定值极可能是分析误差的不可靠测量,最后使实验估计发生错误。因此,只有在假设所有实验结果是绝对正确的前提下,才能进行上述计算。为了在适当的样品数下,能以最小的代价取得实验误差测定的最大可靠性,可用可信区间判断指标。
三、使用可信区间判断指标判断
可信区间判断指标比较复杂,但能对方法性能提供更客观的决定,起最后判断作用。
⒈90%可信区间,可信上限及可信下限统计学的规律说明,每种测定结果的可靠性与测定次数有关,次数愈多,结果反映真实性愈强;但实际上,不可能进行大量的测定。在统计学中为了估量分析误差的不确定性,对于每一误差可计算其可信区间,用可信上限与可信下限代替单值的估量,EU为误差的可信上限,EL为误差的可信下限。West-gard推荐用90%的可信区间,这样,EU将是误差单侧的95%上限,用此判断候选方法的可接受性比较可靠。假如,EU<EA,则方法性能为可接受;假如EL>EA,则方法必须改进,以减少误差,否则排除。假如EU>EA,但EL<EA,说明仅有的数据不足以作出任何有关可接受性的结论,还需继续实验以收集更多的数据,以便对分析误差作出较好的估量。可信区间判断指标见表20-4
表20-4 可信区间判断指标

这些指标在形式上与表20-3的单值判断指标相似,最明显的差别是对每一类型误差用两个判断指标,其一是判断可接受性,其二是判断排除。对RE、PE及CE的判断指标,仅用了误差估量的上限和下限。SE和TE的判断指标较为复杂,引入了一个新的术语“W”。
W是回归线可信区间的宽度(与给定的Xc相对应的Yc值范围),对于一给定的Xc,Yc的上下可信限由方程(a+bXc)±W计算得到。W计算式如下:
W=t(SY/X)〔1/N+(Xc-X)2/∑(Xi-X)2〕1/2
W的大小取决于选择的百分区间(这里是90%),即和选择的值有关(这里选双侧)。W也和回归线标准差SY/x成正比关系,SY/x直接反应方法对比数据的不确定性。中括号内的式子表明,在N很大,Xc=X,W很小,若Xc无论在哪一方向逐渐偏高X,则(Xc-X)之差增大,W也增大。如图20-3表示。
图20-3 回归线的可信区间
⒉计算举例这里介绍某血清钙测定方法的各种评价实验数据,用五个例子概括说明。需指出的是,作为误差估量的上下限,必须具有相同的代数符号,否则下限应取作零。
例一用可信区间判断指标确定偶然误差(RE)是否为可接受。
由重复性试验获得:N=21,Y=11.0mmol/L,STM=0.08mmol/L
假设Xc=11.0mmol/L时,EA=0.5mmol/L,
⑴计算STN的95%可信限
STMU=STM×fu STML=STM×fL
由表20-5查自由度
df=(N-1)fU、fL的值(f为计算因素)
现N=21,df=20,fU=1.358,fL=0.7979
则:STMU=0.08×1.358=0.11mmol/L
STML=0.08×0.7979=0.06mmol/L
表20-5计算单侧可信限(95%)的f值
| df | fu | fl |
| 10 | 1.593 | 0.7391 |
| 20 | 1.358 | 0.7979 |
| 30 | 1.274 | 0.8279 |
| 40 | 1.228 | 0.8480 |
| 60 | 1.179 | 0.8710 |
| 80 | 1.151 | 0.8860 |
| 100 | 1.133 | 0.8968 |
⑵计算REU和REL
REU=1.96STMU=1.96×0.11=0.22mmol/L
REL=1.96STML=1.96×0.06=0.12mmol/L
⑶将以上估量值与EA比较
REU<EA即0.22<0.5。因此,由重复性试验所得的RE是在可接受的低水平上。
例二由可信区间判断指标判断比例误差(PE)是否为可接受。
回收试验的数据:N=9,R=99、98、98、99、100、98、99、100(%)
设:Xc=11.0mmol/L,EA=0.5mmol/L
⑴计算平均回收率R及标准误SR:
R=98.9% SR=0.78%
⑵计算平均回收率的标准误SR:
![]()
表20-6 90%可信区间或95%可信限的t值
| df | T | df | t |
| 3 | 2.35 | 12 | 1.78 |
| 4 | 2.13 | 14 | 1.76 |
| 5 | 2.02 | 16 | 1.75 |
| 6 | 1.94 | 20 | 1.72 |
| 7 | 1.90 | 25 | 1.71 |
| 8 | 1.86 | 30 | 1.70 |
| 9 | 1.83 | 40 | 1.68 |
| 10 | 1.81 | 120 | 1.66 |
⑶计算R的95%可信限
RU=R+t×SR RL=R-t×SR
从表20-6 t值表中查t值
现N=9,df=8,t0.01=1.86
则:RU=98.9+1.86×0.26
98.9+0.48=99.4%
RL=98.9-1.86×0.26
=98.9-0.484=98.42%
⑷计算%PE的上下可信限:
%PEU=|RU或L-100|U
%PEL=|RU或L-100|L
%PEU=|98.4-100|=1.6%
%PEL=|99.4-100|=0.6%
⑸将%PEU及%PEL与Xc相乘,换算成浓度单位%PEU及%PEL。
PEU=%PEU·Xc=1.6%×11.0mmol/L=0.18mmol/L
PEL=%PEL×Xc=0.6×11.0mmol/L=0.07mmol/L
⑹将以上测得的PRU值与EA作比较
PEU<EA即0.18<0.5。因此,作为回收试验测得的PE值是在可接受的低水平上。
例三用可信区间判断指标确定恒定误差(CE)是否为可接受。
由干扰试验得到下列数据表20-7
表20-7 干扰试验结果
| 标本号 | 测得浓度mmol/L | 干扰值 Mmol/L |
|
| 未加干扰物 | 加干扰物 | ||
| 1 | 10.72 | 10.69 | -0.03 |
| 2 | 8.56 | 8.51 | -0.05 |
| 3 | 11.60 | 11.58 | -0.02 |
| 4 | 6.45 | 6.41 | -0.04 |
| 5 | 14.12 | 14.08 | -0.04 |
| 6 | 12.30 | 12.27 | -0.03 |
| 7 | 9.75 | 9.72 | -0.03 |
| 8 | 5.84 | 5.82 | -0.02 |
| 9 | 10.41 | 10.38 | -0.03 |
设:Xc=11.0mmol/L,EA=0.5mmol/L
⑴计算平均干扰值d及标准差Sd:
d=0.032mmol/L,Sd=0.0097mmol/L
⑵计算CE的95%可信限:
![]()
从表20-6查得当N=9,df=8,t0.01=1.86
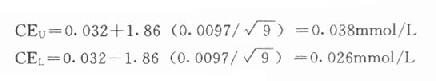
⑶将以上CE值与EA作比较
CEU<EA即0.038<0.5,因此,通过试验测得的CE值,在可接受的水平。
例四用可信区间判断指标确定系统误差(SE)是否为可接受。
由方法对比试验得到下列数据:
N=109,a=0.25mmol/L,b=0.981,SY/x=0.351mmol/L,X=8.42mmol/L,Sx=5.42mmol/L
设:Xc=11.0mmol/L时,EA=0.5mmol/L
⑴计算相当于Xc的候选方法Yc值:
Yc=a+bXc=0.25+0.981×11.0=11.04mmol/L
⑵计算Yc的可信区间宽度W:
W=t(SY/x)〔1/N+(Xc-X)2/∑(Xi-X)2〕1/2〕
查表20-6,N=109,df用120,t=1.66
从Sx式中计算∑(Xi-X)2,即![]()
则∑(Xi-X)2=Sx2(N-1),因此
∑(Xi-X)2=(5.42)2(109-1)=3172.65
将各数据代入得
W=1.66(0.351)〔1/109+(11.0-8.42)2/3172.65〕1/2=0.062mmol/L
⑶计算SEU及SEL:
SEU=|(Yc±W)-Xc|U=|(11.04+0.062)-11.0|U=0.102mmol/L
SEL=|(Yc±W)-Xc|L=|(11.04-0.062)-11.0|L=0.022mmol/L
注意:用+W还是用-W,需视能获得较大的SEU值而定。
⑷将以上SEU值与EA值作比较
SEU<EA,即0.102<0.5,因此,从方法对比试验中测得的系统误差在可接受的低水平。
例五 用可信区间判断指标确定总误差(TE)是否不为可接受。
用例一重复试验和例四方法比较试验所得RE及SE计算总误差。
设:Xc=11.0mmol/L时,EA=0.5mmol/L,
⑴计算TEU:
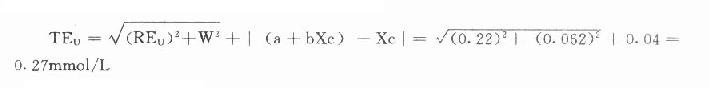
⑵计算TEL:
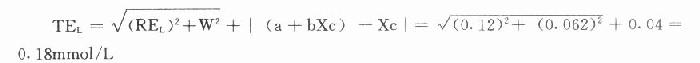
⑶将以上TE值和EA值作比较
TEU<EA,即0.27<0.5,因此,TE值已足以小到可接受的水平。
如果候选方法被得出可接受性的结论,那么接着就要进行评价后实施;最后进入方法应用阶段。不要以为一经评价合格的方法就可产生高质量的结果,还须建立质控系统,以便随时发现合格的方法在实施过程中出现的问题。要善于发现其中还存在的不足并进一步研究解决使其日臻完善。
评价实验完成以后,应该写出书面报告,报告中除了叙述操作方法(包括试剂配制与所需器材)以外,应着重写出所选方法的各项性能指标,要特别重视实验数据的科学整理与客观陈述。评价实验的结果也可整理成论文在专业杂志上发表。
第二十一章 临床生物化学检验质量控制
医学检验结果的可靠性直接影响医疗质量。质量控制或质量管理(quality control,QC)的目的,就是检测分析过程中的误差,控制与分析有关的各个环节,防止得出不可靠的结果。1947年Belk和Sundeman首先调查了不同临床实验室的分析结果,发现有惊人的差异。1950年Levy和Jennings首先将工业质量管理上的控制图移植到医学检验中来,采用每天随常规标本分析控制血清,将结果作成控制图。此后,质控图逐渐成为质控中被广泛接受的方法,此法实际上是一种统计质量控制。至50年代后期60年代初期,统计质量控制由只作为质量控制方法的一部分,发展为更趋完备的全面质量控制(total quality control TQC)。现在检验质控已遍及各个领域,并已成为国际性的活动。
第一节 全面质量控制的内容
临床生化检验要得到良好的分析结果,应认真实行全面的质量控制程序。它包括影响分析结果可靠性的各方面因素或各个环节,以及检验的全过程,即以预防性质量控制为主,回顾性质量控制为辅的科学管理方法。
一、预防性质量控制
预防性质量控制至少包括以下几项内容。
⒈领导重视、组织健全、措施落实、经费与人员有保证,确保质量控制经常化,真正起到保证质量的作用。
⒉重视检验人员的素质提高,尤其要加强在实践中坚持不断的培训教育,工作量与人员数要有适当的比例,负荷过大或过小都不适宜。
⒊要保证实验室中仪器、水、试剂、标准品及控制品的质量(参考分析化学)。
⒋选择并评价各种分析方法(见19章),各种分析方法须有良好的精密性和准确性,且有详尽的操作规程。
⒌实验室的组织管理要落实,如环境、设备、规章制度等,各级人员的职责要明确。
⒍做好标本分析前的正确处理工作,如病人准备、标本收集方法及初步处理方法等。
二、回顾性质量控制
包括做好室内质量控制和参加室间质量评价等工作(详见第二、三节)。
第二节 临床生化检验室内质量控制
室内质量控制系各实验室为了监测和评价本室工作质量,以决定常规检验报告能否发出所采取的一系列检查、控制手段,包括实验室工作的全过程,旨在检测和控制本室常规工作的精密度,并检测其准确度的改变,提高本室常规工作中批间和日间标本检测的一致性,室内质量控制的内容。见表21-1,本节主要介绍分析后的质控知识,其它内容参见有关章节。
表21-1 室内质量控制内容
| 分析前的质控 | 分析中的质控 | 分析后的质控 |
| 病人准备 | 量具与测量 | 计算结果 |
| 标志收集 | 试剂质量 | 标准曲线 |
| 标本处理 | 反应条件 | 控制图绘制 |
| 标本贮存与转运 | 比色测定 | 结果统计分析 |
一、控制物的种类及其使用
控制物是一个广义名词,它包括标准控制物溶液及控制血清等,目前国内外主要使用未定值的控制血清,随常规标本做室内质控。
(一)控制血清的种类
⒈液态控制血清 有动物血和人血制备的控制血清。目前液态控制血清多为实验室自己制备,其中各种成分的含量经实验室多次测定后确定。
⒉冻干控制血清 多由动物血清(猪、牛、马等)制备,分定值与未定值两种。未定值控制血清多用于室内质控,其成分含量由该实验室多次测定后确定。定值血清多由厂家测定或由邀请某些设备及技术水平较好的实验室协助共同测定,此种血清多供室内核对数值使用。
⒊参考血清 其成分经几种参考方法测定。用冻干血清时其瓶间变异小于0.25%,一般是先经透析,除去某些成分,再加入已知量的所需纯品。可用于校正仪器及评价试验方法。
(二)使用控制血清注意事项
⒈使用自制冰冻血清 分装后应注意冰冻保存或低温-10℃~-20℃。如果在一天内连续使用,应注意加盖,避免蒸发。液体血清的优点是不存在分装的问题。但液体血清容易发生细菌污染,细菌污染会改变许多生化成分。
⒉使用冰冻混合血清 由低温情况下取出后,应放置使全溶解,注意混匀后再使用。
⒊使用冻干血清 应严格按照说明书规定操作,注意冻干血清的均一性。即各瓶间差异应在允许范围内,如有怀疑,可随机抽取二三瓶互作测定,观察瓶间差异。如怀疑血清变质或不能较长时间保存,投入使用前应做稳定性试验。一般采用-10或-20℃,室温及37℃三种温度保存,连续定期做测定,将所得数值做比较,观察差异情况。
⒋定值及定限问题 目前均有市售定值控制血清供应。用户往往容易盲目采用产品商所给的数值,但各实验室所用的实验方法不同,因此,在使用前,还应作校对。
二、室内质量控制的主要方法
(一)常规质控图(X-S图)
X-S图法由于它只需要单一浓度的未定值血清,绘图方法简单易懂,有较为成熟的理论和实际经验。因此,成为国内外目前采用最广泛的一种常规室内质量控制方法。一般步骤和具体做法如下:
⒈最佳条件和常规条件下的变异的测定及计算 选择含量均匀、稳定性良好的未定值质控血清,在“最佳条件”,对该批血清反复测定(至少20份),计算出20个结果的X、S和CV,此CV即为最佳条件下的变异(optimalcondition variance,OCV)。取测过OCV的质量控制血清,每天随病人标本测定一瓶,20天后,计算X、S和CV,此CV即为常规条件下的变异(routinecondition variance,RCV)。OCV的测定方法是日间、批间或批内进行,尚无统一意见。但应在尽可能保证最佳条件的前提下,使OCV与RCV有较好的对应性。以便在室内质量控制工作中作对比分析。因此,可每天作4、5批测定,每批测一瓶血清,这样在4、5天内即可得到20个数值,所测得的OCV综合表达了批间和日间精密度。RCV是常规条件下日间精密度的表达指标,因此,测定时必须保证和标本同样的处理条件,而且每天要重新打开一瓶,并只测一份,20个数据要来自20天测定。在测定过程中,如有特殊情况发生,应做详细记录,并将该数据删除,补做一个数据。求出X、S和CV之后,应观察有无超出X±3S范围的数据,在OCV测定中,如有某个数据超出X±3S范围,则应废除全部数据,重新测定OCV。在RCV测定中,有一个数据超出X±3S范围,则删除此数据,用剩余的19个数据计算RCV:如有一个以上数据超出X±3S范围,则应废除该批数据,重新测定RCV。计算结果:通常RCV比OCV大,但一般不超过2倍。且对同一批号质量控制血清OCV和RCV测定中所得X应十分一致,否则应查找原因。OCV过大,往往提示检测方法本身有问题,或测定时未处于最佳条件。RCV过大,则说明常规工作中控制过松,没有达到应有的精密度水平,或由于检测方法不够稳定,难于掌握。
⒉OCV和RCV图的绘制 OCV和RCV的作图十分相似,当使用全国统一印发的质控图纸时,基本步骤如下:
⑴在纵坐标上标出X、X+2S、X-2S、X+3S、X-3S的标志,并将其具体值标在左侧标尺上。
⑵用红笔画出X±2S,用蓝笔画出X±3S线:即成为一张OCV(或RCV)的“空图”。还应填齐图纸上方的各项,如测定项目,测定单位,血清来源及批号,起止日期,主要仪器及使用波长等。测定过程中的特殊情况应在备注栏内记录。
⑶在图纸下方“日期”、“测定值”和“操作者”栏内按原始记录填入相应内容,边填写边核对:注意数据顺序,应严格按实际操作情况,不得颠倒。日期一栏内,在OCV测定中,应改为检测批次(或序号)。在RCV测定中应改为填写实际日期,未做测定的节假日、星期天在图上做出空格似更为合理。因为这样可以真实反映客观情况,便于分析误差原因。
⑷画出每个检测值所对应的图点:每个图点在图上的横坐标为其日期(或序号)而纵坐标(即与X轴相距的小格数目)可按下式计算:

计算结果为正数,则点在X轴上方,反之,则点在X轴下方。各点均画在图上后,用直线将各点顺序连接,以便于观察。OCV和RCV图中各点应呈正态分布,如失去正态分布特点。应进行分析。
⒊X-S图在常规工作质量控制中的应用
⑴按本节前面介绍的RCV“空图”绘制方法:在每个月第一天根据所要用的质量控制血清在RCV测定中所得的X、S绘制一张“空图”。图中X线为靶直线,X±2S线为警告线,X±3S为失控线。
⑵每天将该批号质量控制血清按有关规定复溶(液体的血清则融化),随同各项目病人标本的测定同时测定一份:并按上述方法画出图上的对应点,用直线将该点与前一天的点连接。如果点在X±3S线以外,则为失控。应立即报告有关负责人,迅速查找原因,必要时复测标本,然后才可发出化验报告,并应对失控情况查找的过程及结果处理等详细记录。如果点在X±2S线以外,或出现连续5点以上在X一侧等规律性变化,均应及时向有关负责人反映,并积极查找原因。但当天的化验报告一般可以发出。
⑶月底计算当月全部质量控制血清检测结果的X、S和CV,并进行图形分析和小结,将质量控制图存入质量控制资料档案。
⒋X-S图失控的表现及其处理(图21-1)
⑴测定值的分布规律:按统计学规律,控制血清的数值应依据下列规律分布:①95%的结果应落在X±2S范围内。②有5%的结果可在X±2S外,但在X±3S内;③均值两侧的数据分布几乎相同,不能有连续5次结果在均值的同一侧,或5次数值渐升或渐降,不能有连续有2次结果在X±2S以外;④没有数值落在X±3S以外,结果违反上述规律时,称为失控。
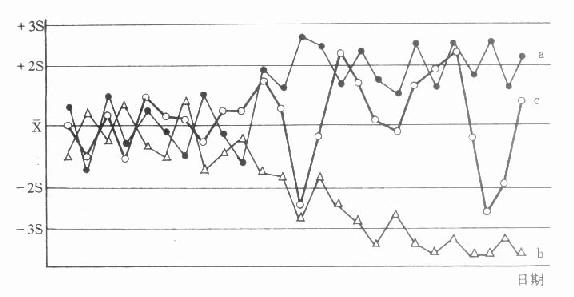
图21-1 X-S质控图的几种失控表现
a.曲线发生漂移;b.曲线趋势性变化;c.曲线精密度出现变化
⑵质控图的几种失控表现:①曲线漂移:“漂移”现象提示存在系统误差,准确度发生了一次性的向上或向下的改变。这种变化往往是由于一个突然出现的新的情况引起的。如更换标准品的生产厂家及批号;重新配制试剂及操作人员的变换等。在寻找原因时,应重点注意“漂移”现象的前后发生了哪些变动因素(图21-1a)。②趋势性变化:向上或向下的趋势性变化,表明检测的准确度发生了逐渐的变化(图21-1b)。这种变化往往是由于一个逐渐改变的因素造成的,如试剂的挥发、吸水、沉淀析出、分光光度计的波长渐渐偏移、光电池老化及质控血清本身的变质等。而更换标准品、试剂或操作者等一次性变化的因素则不大可能造成趋势性变化。③精密度的变化:指常规测定中出现日间差异较大的情况(图21-1c)。从图21-1中可见准确性无明显变化,但精密度从第26天起开始发生变化。
⑶失控后的处理:一般可按以下步骤处理:①迅速回顾、检查整个操作过程,是否有发生错误的环节,如计算错误、吸管及波长选择是否用错等。②如操作步骤均无问题,可重复试验一次,如有改进,说明误差很可能为操作错误所致,如加标本量或试剂量的错误,波长或滤光片选择的错误等偶然误差所致。③如重作后仍不能更正,可取一份新鲜的控制血清重作,观察是否能更正;④如仍不能解决问题,取一份定值血清,用同样方法测定,如结果良好,可能为原血清变质。⑤如仍未得到更正,应仔细检查仪器的各种性能是否正常。⑥如仍未得到纠正,应重新配制试剂或标准液,重新在操作中查找原因。
(二)改良Monica质控图
改良Monica质控图用于室内常规项目的质控,能同时监测精密度和准确度,及时指示存在的误差大小和性质,制作简便,应用及时,形象直观,能将室内质控和室间质评有机地直接联系起来,从质控图上就可基本预测参加室间质评的变异指数得分情况。
⒈Monica质控图绘制
⑴用质量可靠的定值质控血清的测定结果绘图。
⑵该图与X-S图基本相似。其模式如图21-2,在纵轴居中绘出平行于横轴的靶值(target value,T)线,并绘出上下警告值线(warning line)和最大允许值线(max error)4条平行于靶值线的直线。警告值线和最大允许值线用一定范围的选定的变异系数(chosen coefficient variation,CCV)来确定。
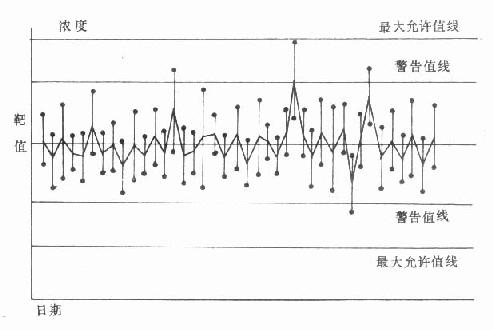
图21-2 Monica质控图模式图
⑶分别以T±0.8CCV×T和T±1.5CCV×T作为警告值和最大允许值。例如:定值质控血清的氯的靶值为115mmol/L,CCV=2.2%,则警告值定为115±0.8×2.2%×115,即113.0-117.0mmol/L,最大允许值定为115±1.5×2.2%×15,即111.2-118.8mmol/L。
在临床生化常规项目的室内质控中,制作Monica质控图时,采用0.8CCV和1.5CCV作为误差界限的目的是试图将室内质控和室间质评更为有机地直接联系起来。根据变异指数(Variance Index,VI)的计算公式:
VI=〔(|X-T|)×100/T〕×100/CCV
式中(|X-T|)/T是测定值的相对误差,用d表示,则:VI=d×100/CCV,若设定VI=80时,则d=0.8CCV。
因此,对一个测定结果,当VI≤80时,则d≤0.8CCV;当80<VI≤150时,则0.8CCV<d≤1.5CCV;当VI>150时,则d<1.5CCV;同理,当d≤0.8CCV时,则VI≤80,即测定结果的相对误差不大于0.8CCV时,VI值也不会大于80,则VIS也不会大于80,在室间质评中成绩为优秀;当0.8CCV<d≤1.5CCV时,则80<VI≤150,即测定结果的相对误差不大于1.5CCV时,VIS也不会大于150,在室间质评中为及格;如果d>1.5CCV,则VI>150,即测定结果的相对误差大于1.5CCV时,VIS就在150以上。在室间质评中不及格。
由此可见,用Monica质控图进行室内质控,如能做到全部测定结果落在警告线内,参加室间质评可获得优秀成绩;如果有些点子分布在警告线外,但在最大允许线内,参加室间质评可望及格;如果经瓽出现超出最大允许线的点子,参加室间质评很难达到及格。
⑷在进行病人标本常规测定中,插入两份质控血清同时操作。将此两个测定值点在质控图上,用垂线将此两点连接,再用红点标出垂线的中点(代表双份测定的均值),然后用细线将相邻的红点连起来见图21-2。
⒉结果分析与判断标准 按照Monica原判断标准,最理想的结果是质控血清的全部测定值(不是双份测定的均值)都落在警告线内。如果超出警告线,但仍在最大允许线内,该批病人标本测定结果仍然有效,即“在控”,但指示测定误差较大,须仔细检查可能产生误差的各个环节。出现任何落在最大允许线外的点都表示“失控”,该批测定结果无效,应查明误差原因并予消除后重新测定。
在Monica质控图中,由于相邻各红点连线与X-S质控图相似,因此,X-S质控图的分析判断标准也基本适用Monica质控图。
在Monica质控图中,双份重复测定值的垂线的长短可作为精密度的指示,愈短精密度愈好,愈长则愈差。红点离靶值线的远近可作为准确度的指示,离得愈近,准确度愈好,愈远则愈差。可根据垂线的长短和红点的位置,分析判断该批测定结果的精密度和准确度及其误差的大小和性质。
⒊Monica质控图的优点
⑴制作简单,应用方便:只要有质量可靠的定值质控血清,就可立即制图应用。而X-S质控图需进行20天定值确定X与S后方可绘图。
⑵Monica质控图与X-S质控图很相似:只是使用定值质控血清,作双份重复测定,用双份测定值的垂直线的长短来监测偶然误差,用垂线中点的位置来监测准确度,既形象直观,又容易理解和掌握。
⑶Monica质控图采用统一CCV作为允许误差来确定警告线和最大允许线的界限值,能将临床生化常规项目的室内质控与室间质评有机地联系起来:从室内质控图上,既可基本预测参加室间质评可能获得的变异指数得分情况,更能充分发挥室内质控与室间质评二者之间相互补充,相互促进的作用。
除以上介绍的两种室内质控方法外,尚有均数和范围质控图,累加与质控图和病人标本测定结果均值图等,一般应用较少,必要时参考有关文献。
第三节 临床生化检验室间质量评价
室间评价(external quality assessment)应在室内质控的基础上进行,方法是组织若干实验室,共同在规定的时间内,测定同一批血清,收集测定结果作出统计学分析,目的在于调查各参加实验室的工作质量,观察试验的准确性;比较各实验室间的数值,并采取相应措施,使各实验室结果渐趋一致。
一、室间质量评价应具备的条件
(一)要有一支素质较高的质控技术队伍
他们已掌握质控的有关知识,能统计分析质控的测定结果,组织研究存在的问题及采取相应的措施。
(二)参加室间质控的各实验室要有室内质控的基础
要先建立室内质控制度,如果在参加前未实行室内质控,室间的质控工作,只能起调查了解情况的作用。
(三)要有良好的质控血清作为调查样本
在一个小范围进行室间评价,可以采用自制的液体质控血清,但在范围大的地区进行,最好用高低两个浓度的冻干血清,一个浓度在正常范围内,另一个应在病理情况下可能出现的高浓度。室间质控冻干血清应具有含量准确,均一性好及成分稳定的特点。使用时应注意全部溶解,有的血清溶解后有些混浊,但不影响测定结果,控制血清应注意避免强光照射,不用时宜放暗处。而且使用控制血清时,均应按照肝炎抗原阳性标本处理。
(四)调查样本定值的方法更可靠
目前调查样本的定值方法主要有:①使用可靠的决定性方法或参考方法定值;②采用生产质控血清时加入纯物质定值,血清先经透析除去欲测成分,再加入定量的纯物质;③几个参考实验室常规方法测定,取均值作为该血清的定值;④使用参考实验室报告结果的均值为质控血清的定值。
国内外多采用第④种方法。依据是①在正常情况下,参考实验室所得数值往往和质控结果的均值相似。②对某一成分用不同的测定方法测定结果,数值往往相近似。③国外多年使用的试验,认为均值可以代表该血清真正的近似值。④参加的实验室越多,所得结果的分布越趋向常态分布,均值越接近真值。
(五)确定好参考实验室
参考实验室应具有良好的仪器设备,较强的技术力量,还要热心于质控工作。参考实验室除提供各成分的测定数值外,还应对质控过程中的问题,提出分析意见和解决问题的建议。因此,在进行室间评价中,必须有几个参考实验室作参谋和顾问。
(六)统一测定方法及测定标准
室间质评结果的统计分析,要在相同方法的基础上进行,例如血糖测定中,用测“真糖”的方法和有测还原物质的方法,结果要分开统计,否则不好比较。如果能把测定方法统一在推荐的方法上,更为理想。室间质控分发控制血清的同时,应发送标准液。
二、室间质量评价的组织方法
室间质控的组织,应从实际出发,由小到大,先在较小的范围内进行,取得经验教训,再扩大到全地区或全国。方法是:①首先确定负责分发和收集、统计结果的单位。分发血清单位,定期发送血清,并制定表格,要求逐项填写。②组织各参与实验室,进行编号,测定结果的分布用不记名方式,只有承担统计分析的单位掌握实验室的名称。③按规定日期进行测定,并要求在指定的日期前将结果报送统计单位,逾期未报者不参加统计。
三、室间质量控制的统计方法
主要以测定值与靶值的离散程度为评价依据。常用的方法有以下几种:
(一)计算均值法
把相同测定方法的实验室的测定结果归在一起,计算均值及标准差,就均值为靶值,观察各实验室结果是否满意。如用血清的定值作为靶值,则靶值的确定方法应告知参与单位,评价时结果与靶值相关在1S以内者为满意结果,2S为警戒线;大于3S者为逾限。把统计所得结果列表或作图发给参与单位。
(二)变异指数得分(VIS)
这种评分方法是由英国全国性质量评价活动倡导并被WHO推荐的方法,原计算公式为:
![]()
VI=V×100/CCV
其中V为变异百分数,X为各室测定结果,X为各实验室结果均值(剔除X+3S以外的结果)VI为变异指数,CCV为选定的变异系数。鉴于我国多数地区的实际情况在1985年全国临床检验质量控制会议上通过的提案建议将上述公式改为:
![]()
VI=V×100/CCV
其中T为靶值,其它符号含义与原公式相同。
VIS即变异指数得分。VI<400时,VIS=VI;当VI>40时,VIS=400,其主要目的在于防止由于个别过大的偶然差错造成对检测水平全面评价的假象。有些国家对VI<50的情况,取VIS=0,以示鼓励,我国未采取此做法。
例如:某次室间质量评价活动中,血清氯的靶值为98mmol/L,实验室A测得结果为101mmol/L,CCV为2.2,该室血清氯检测得分为:
V=(101-98)×100/98=3.06
VI=3.06×100/2.2=139
此时VI<400,VIS=VI=139。该室即得分的139。
一般情况下,VIS只计整数位,并不带正负符号。
上面的公式和例子说明VIS越低越好,当测定结果与靶值完全一样时,VIS=0。WHO对发展中国家在国际质量评价中的标准为VIS<50为优秀;VIS<100为良好;VIS<150为及格。我国根据具体情况确定对参加全国质量评价活动的实验室VIS<80为优良;VIS<150为及格。一般认为,VIS>200,表明结果中有临床上不允许的误差,而VIS=400的测定结果则会造成临床的严重失误,是绝对不许可的。
目前国内和国际推荐的CV值见表21-2
表21-2 国内国际推荐使用的CV值
| 测定项目 | 我国推荐的 | WHO推荐的 | WHO推荐的 |
| RCV(%) | CCV(%) | RCV(%) | |
| 钾 | 3.5 | 2.9 | 3.0 |
| 钠 | 2.0 | 1.6 | 2.0 |
| 氯 | 2.5 | 2.2 | 3.0 |
| 钙 | 4.0 | 4.0 | 4.0 |
| 磷 | 7.0 | 7.8 | ― |
| 血糖 | 5.0 | 7.7 | 6.0 |
| 尿素氮 | 6.0 | 5.7 | 6.0 |
| 尿酸 | 7.5 | 7.7 | 10.0 |
| 肌酐 | 8.0 | 8.9 | 8.0 |
| 总蛋白 | 4.0 | 3.9 | 4.0 |
| 白蛋白 | 5.0 | 7.5 | 6.0 |
| 胆红素 | ― | 9.6 | 12.0 |
(三)变异指数移动总均值(overall mean runningvariance index score,OMRVIS)
OMRVIS是动态反映实验室工作质量的一个指标,表示实验室工作质量提高或下降的总趋势。假设在一次室间质量评价活动中,某实验室得到12个项目的VIS,其平均值则代表了这次活动时该室工作的质量水平。但这个水平并非偶然的和孤立的,它既是前一阶段工作质量情况发展的必然结果,又是下一阶段工作质量的基础。因此,它只是一个连续的、不断发展变化过程中的一个环节。为了真实生动地反映这一工作质量连续变化的客观过程,采取移动平均值的表达方式,它较少受偶然因素的影响而对总的发展趋势有较好的代表性。
OMRVIS原定义为最近30个VIS的平均值。为了使OMRVIS的变化与测定时间有更明确的关联,在全国临床检验质量控制会议的提案中,建议全国统一以最近三次质评活动的VIS的平均值为OMRVIS。
四、室间质量评价的作图法
(一)改良Youden作图法
此图是以S为单位的离散图,它是用两种不同浓度的控制物发给各参加单位同时进行测定后,收集结果,所有参加单位的结果均值的相交点为靶值(图中以“+”表示),纵坐标和横坐标等距离以标准差为单位表示。在+2S处作与轴垂直的平行线,即可成正方形的方框,反映试验结果的离散程度。并作45°对角线,以评价各单位的操作技术。将各参与单位的结果按其在图中的位置标出,凡在方框内的为在允许范围之内;凡超出方框外的各点,为超出2S范围的点(图21-3)。
①、②号实验室在对角线上分别成比例地降低和增高,属系统误差。③号实验室低浓度血清测出值偏高,高浓度血清测出值偏低;④号实验室高浓度血清测出值偏高。低浓度血清测出值偏低,均是偶然误差。这种图较醒目,易于了解,不需要复杂的统计学计算,但需要有不同浓度血清。
图21-3 改良Youden作图法
(二)结果分析
不论被测物质是高浓度或低浓度的,实验室测定结果总量持续偏高或偏低,说明有系统误差。表示某测定结果的点将落在对角线上。也可由仪器的性能问题或吸管刻度不准等因素引起误差。方框外的各个接近对角线的点,就代表这样情况。位于其它部位的点则属于随机误差所致,如测定时的温度突然升高或降低,标本弄错等原因。
第二十二章 临床生物化学实验室数据的作用和有效使用
临床医生要求临床生物化学实验室对所进行的检测项目提供可靠的数据,同时又要求能够尽快地发出报告,以便为临床诊断和治疗及时提供信息。此外,临床医生也要求实验室能提供相当范围的可测项目,以适应不断发展的临床需要。因此一个好的临床生物化学实验室要不断地研究开发新项目、新技术,更新旧项目,淘汰陈旧的技术和方法,建立质量控制的保证制度。
为了充分有效地利用实验室,必须强调临床医生与临床生物化学家的密切合作,及时交流,以确保实验结果的可靠性,并能运用现代生物化学和医学两个方面知识来理解这些数据,对它们进行合理的评价,并把它转化为更高层次的临床生物语言和概念,对诊断、治疗疾病作出决策和指导。
为此,一个现代化的临床检验科室应当把①数据可靠②不断发展、更新、补充新项目和③提供临床咨询,作为服务质量的三大管理目标。
为了充分发挥临床生物化学实验室的作用,能对分析的结果正确地评价和利用,应当了解以下几个问题。
第一节 临床生物化学实验室的检验项目
根据临床需要,各项检验项目包括:①监测病情和治疗效果;②协助疾病的诊断。前者在临床生物化学实验室工作中占很大分量。经常可通过检测某几项特定的血液生物化学指标对病情作出有效判断,例如糖尿病病人的血糖浓度和肾功能衰竭病人的肌酐测定,治疗药物的血浓度等,这些监测项目有足够的分析精密度。
依靠生物化学检测项目本身即可对疾病作出诊断,以先天性代谢异常、内分泌功能紊乱等为最有效。这一类疾病的临床初步诊断及其它检查结果提示的诊断往往可以通过生物化学检测的结果加以修正或作出进一步诊断,并对病情判断提供参考根据。但在通常情况下,医生对病情获得初步印象后,期望通过生物化学检测证实他们的设想,如通过血清酶活力测定对疑诊心肌梗死或急性肝炎的病人加以证实,对急腹症的病人和黄疸病人作出鉴别诊断等。但对一些症状不典型和较轻的或早期病人,合理选择项目更为重要。在这类项目中,化学实验室的正常参考值范围和方法的准确性往往是很重要的基础。它特别要求医生和临床生物化学家的密切配合,才能有效评价和使用实验结果。为了方法的准确性,有时需要加测正常对照。此外,为了诊断目的,常常采用成套试验,即同时测定一组相关试验,这种方法可以增加诊断的灵敏度和特异性。
建立合理的成套项目,最好由医生和临床生物化学家共同设计,以避免不必要的项目,它不仅浪费时间、精力和经济,有时甚至带来混乱和干扰。组合这些项目应对所选项目的分析误差和临床意义(正常参考值及其范围)均有可靠的评价。成套试验特别适用于对一些疾病的筛选诊断,或对一些无前期症状的疾病,或是一些疾病的危险因素作预防性监测。
为了有效地进行工作,实验室一般分常规标本、急症标本和特约标本几种类型。
常规标本化验一般指每日规定好程序的一系列化验项目(有些项目可规定隔日进行或每周哪几天进行),这样标本的采集和运送保存均应有效配合。
急症标本包括病情急需以及标本应立即处理两种情况。为了节省精力、时间和试剂,对急症标本数量应适当控制。一般实验室可将以下标本列入急症标本:血清钾、钠、氯、CO2结合力、血糖、淀粉酶、尿素氮、肌酐、钙、转氨酶、肌酸激酶、血pH、PO2、PCO2等,以及某些治疗性药物的监测。
特约标本系指在临床或化验室方面需要作特别工作的试验,需先预约于双方共同认为合适的特定时间内收集标本,进行分析。
此外,化验单要求填写清楚,必要的内容必须填全。除了避免张冠李戴的严重错误外,有些项目如年龄、性别、饮食、用药情况等因素对实验室结果的解释有重要影响,应予写明。登记必须可靠,发出报告必须核对。
第二节 标本的正确收集和保存
正确地收集、运送和保存标本是十分重要的环节,不恰当的标本收集与保存,往往是导致实验结果不可靠的主要原因,且又往往不易被检测者发现,因而必须予以重视。
⒈标本收集前应考虑的因素 包括病人的饮食、药物与采集的时间等。有些化学组成容易受到饮食成分的干扰,有时还可受到近期食谱的影响。许多药物可干扰测定结果,应加以注明,并尽可能在试验前停药。有些药物可能对某一种测定方法发生干扰,而对另一种方法却无影响,因此医生和检验人员之间的密切联系更显得必要。某些血浆组成的浓度可有昼夜节律性变化(如铁、皮质醇等),则应规定恰当的标本收集时间。
⒉标本收集时应考虑的因素 最常见的是病人的体位、皮肤消毒剂的选择;采血部位;有无瘀血与溶血、抗凝剂的成分和用量等。有资料表明,人体从平卧状态转为直立状态15分钟内,静脉血中血浆蛋白及某些与血浆蛋白结合的成分的浓度可增高10%-13%,这是由于细胞外液的重新分布所致。
当患者处于静脉输液时,不应在输液部位静脉采取标本。
⒊标本收集后应考虑的因素 一般应即时送化验室。有些检查必须立即进行,必要时可冰箱保存,或分离血浆(血清)后保存。
第三节 分析数据的正确评价与合理使用
临床医生申请一个(组)生化实验室检验项目,不外乎要求提供以下的信息:
⒈对疾病的诊断;
⒉判断病情的严重程度及其发展;
⒊监测疾病的进程,对治疗的反映提供指导,掌握药物的浓度与副作用;
⒋并发症的监测;
⒌筛选病例-职业病、危险人群、流行病区疾病的普查或其他特定人群中进行;
⒍研究比较;
⒎为今后判断健康状态或病情发展建立一个基础值。
为了正确评价与合理使用临床生物化学实验室各项分析的数据和结果,各个实验室必须做到:
⑴对各个检测项目分析方法的准确度、精密度、特异性、灵敏度与最小检出限作出评价。
⑵提供适应自己实验室的正常参考值。
前者涉及分析方法学的评价,后者牵涉有关正常值、参考值的基本概念。如参考值的建立、“健康”参考人群的选择、参考范围以及临床判断性水平(或限值)等一系列问题的正确理解和使用。这些已在第二十章中详述和讨论。此处仅作概括性的提示。
绝大多数临床生物化学实验室的报告为一系列测定数据,如何看待这些数据,合理地评价和有效地利用这些数据呢?
通常临床医生要求报告能回答两个重要问题:①报告的结果是否属正常范围?②本次报告结果和前一次报告之间有无显著意义?为此,首先要区分这一差异是否在分析变异范围内,进一步要区分这一差异是否在生理变异或生物学变异范围内,这样,才能判断有无临床意义。
要回答第一个问题,首先要将测定结果与正常参考值作比较。判断一个化验结果是正常还是异常,最理想的方法是将病人在发病前(健康状态时)的一系列基础数据与发病后的数据相比较。这种用病人自身作为对照的方法,常可用于作选择期手术的病人(如测定手术前后血浆尿素含量,血清酶的水平)等。在未建立这一制度前,只有采用从“健康”参考人群获得的数据作参考值来进行比较。
在人群中,许多化学成分含量是常态分布的。但也有一些组分是呈偏态分布的,如血清胆红素、铁、尿素及碱性磷酸酶等。由正常“健康”人群获得的化学分析结果常用其平均数(X)或M值及其离散状态标准差(SD)来表示之。数据的分布状态见表22-1。
表22-1 健康人群中测定数据的正常分布情况
| 测定结果在平均值(X)±SD的范围 | 结果出现的频率(%) |
| X±1SD | 68 |
| X±2SD | 95 |
| X±3SD | 99 |
| X±4SD | 99.9 |
| 测出结果(R)与平均值(X)之差R’ | 正常人获得在此范围结果的机率 |
| 2SD<R’<3SD | 5% |
| 3SD<R’<4SD | 1% |
| 4SD<R’<5SD以上 | 1‰或更小 |
应当理解,在此SD值包括了分析方法和生物学两个方面的变异因素。
一般把“正常范围”定在95%健康人群所分布的数值内(即在一常态分布的变量中,取X±2SD)。由于健康人群的结果尚有5%机率可以落在这一“范围”之外,因此,“正常范围”这一词,严格来说是不太妥当的,容易引起误解。为此,近年一些作者建议该用“参考值”、“参考范围”来代替“正常值”及“正常范围”。这种表示法避免了机械地把落在“正常范围”的数值理解为一定是正常的,而所有在这一范围之外的结果就一定不正常。
对于呈偏态分布的数据,可以经过适当的数字处理使之接近于正态分布形式后,参照常态分布数据来对待。常用的有对数转换法,即将每一个数值转换为相应的对数值来作图,使之呈常态分布,利用此对数值计算出X及SD值。最后又将数值转换为相应的平均值,定为其“参考值”并定其上限和下限。
对参考值的建立和应用,还应注意几点:①以上统计学处理是基于假定数据的分布是呈一定规律的。在多数情况下,也确是如此。假如对数据的分布状态无法作出判断时,可求助于其它统计方法的处理,其细节可参考专著。②有些数值会受不同年龄、性别和其它特征的影响而有差异,依次对这些化学组分测定的结果,应和相似人群(同一年龄组、性别组及其它特征相同)所测得的参考值比较。③参考值的上、下限可人为地加以选择和判断。选择的依据和目的是为了尽量减少正常健康人群和疾病人群间的数值重叠。④为了临床判断病情和采取相应治疗措施的需要,可以对某项目测定结果的数值制定一个“判断有无临床意义”的上限和下限,有人建议称之为“临床判断水平”,这一限值可用以除外或确定某一临床情况的存在与否,或提示将出现某一病理生理变化。“临床判断水平”(或限值)与“正常参考范围”不是同一概念,但是在临床使用中可以相互补充。
在实际工作中,每一个实验室都有责任与临床医生密切合作,建立一个既实用又可靠的“参考值”和必要的“临床判断限值”。由于方法学和个体的生理差异,各个实验室应制定适用于自己的参考值,并将这些参考值汇印成册,供临床医生分析结果时使用。
现在,再来回答第二个问题:即两次化验的结果之间有无差异,以及这种差异有无显著意义?
任何测定方法,不论操作如何严格细心,仍会有一定差异。例如,测定血清标本钾浓度由同一人操作,使用同一来源的标本和相同的分析方法,反复进行5次,测定结果可获得如下数值:3.6、3.4、3.6、3.5、3.6mmol/L,统计处理得其平均值X为3.53,其标准差SD为0.089。
因此,临床生物化学实验室要有一定的质量保证制度,以便确定日常工作中分析结果的误差程度。在进行质控标本分析时,要避免检测者的主观性。通过质控标本分析或方法评价实验的数据可以获得分析精密度的指标,通常以标准差(SD)来表示之。必须注意,这里所用的SD值指仅由分析方法学带来的变异。所以和前面提到的由于标本来自不同个体的人群所测得的化学成分结果的平均值及各个数值间的差异,是不同范畴的两个概念。后者是用以建立参考值时用的平均值和SD值。参考值中的标准差主要来自被研究人群的生物差异(也包括分析方法带来的精密度不足的差异)。可以简单地认为:如果两次标本的测定结果的差异落在分析精密度的3个SD之内,这种变化是没有生物学意义的。因此,各个实验室应通过质量控制保证制度,掌握本实验室各个测定项目的分析精密度。
临床医生应了解实验室分析方法中的精密度(即分析精密度)。通常以分析方法的变异系数(coefficient variation,CV值)来表示之。这样,不致使自己对由于分析精密度不足带来的化验报告中无价值的变化而作出错误的判断。
血清中某些化学成分的参考值分布范围较宽,这部分地反映了:①个体间存在的生物学差异;②由于方法学的缺陷导致分析精密度的不足。
在建立一个参考值及参考范围的过程中,可通过,①改进分析技术;②标本收集条件的标准化,而使参考范围减小到最理想的程度。这样才有可能减出最微小,但又是“显著区别于正常标准”的变化。
第四节 临床生物化学实验室中计算机的应用
微处理机在临床生物化学实验室全面管理中的作用及其进展已有专着介绍。这里仅就有关实验数据的处理与报告方面的应用作一概括性的提示。
应用计算机系统处理实验数据并打印报告等具有多方面的应用,如通过编定的程序,可从大量实验数据中筛选出最有价值的数据,以提供参比;也可将原始数据进行分析、加工,做出更高层次的图解和评论、判断、解释,供临床医生参考。例如:
(一)选出最适参考值
某些化学成分的参考值随年龄或性别差异而有不同。计算机辅助系统可很方便地根据申请单中列出的要求,参照测定标本的性别、年龄及其它特征,自动而迅速地处理这些变数,列出最适参考值及其范围,供判断比较。
(二)用标准差(SD)为单位分析测定值与正常参考值的偏离状态
通过编制计算机程序,可将某一项目的参考值(即根据正常人群测定的数据及其分布状态求得平均值)及其参考范围(通常以SD形式表示)储存于计算机系统,以后每一项分析结果与参考值之间的差值均可用SD值来表示。表22-2中列出一系列生物化学测定结果,均以三种不同方式来表示,即国际单位、惯用单位和SD单位。可以看出,使用SD系统表示可协助临床医生很快地识别异常结果。对使用不同计算单位的分析方法均可用这种统一的方式来标志其偏离状态(或异常程度)的级别。
表22-2 几种血浆化学成分采血不同方式来表示测定结果
| 测定项目 | 国际单位 | 惯用单位 | SD单位 | 正常参考值 |
| 钠 | 141mmol/L | 141mEq/L | -0.5 | 142.5±3mEq/l (136.5-148.5) |
| HCO3- | 18mmol/L | 18mEq/L | -6.0 | 27±1.5mEq/l (24-30) |
| 钙 | 2.75mmol/L | 11mg/dL | +3.0 | 9.5±0.5mg/dl (8.5-10.5) |
| 铁 | 25μmol/L | 140μg/dl | +0.4 | 130±25μg/dl (80-180) |
| 胆固醇 | 5.2mmol/L | 200mg/dl | 200±300mg/dl (140-260) |
|
| 白蛋白 | 43g/L | 4.3g/dl | +0.5 | 4.15±0.3g/dl (3.6-4.7) |
将检测的结果用SD单位表示,将更有利于数据的进一步统计处理,诸如进行多因素分析等。
(三)测定结果的储存与调用
需要时可以在几秒钟内调阅任何病例,过去任何一天的任一项结果,并将最近一次的检查结果和过去一系列结果进行比较,进行评论,并对异常结果作出特殊标记。
(四)资料的分析与筛选
在一系列筛选试验中,临床医生可能面临一大堆数据而发现困扰。使用计算机报告技术,可帮助我们将注意力集中在一些重要数据上。例如,通过采用选择性报告程序,要求计算机显示和打印出我们要求的项目,并加上那些测定结果是偏离正常参考值过大的数据(在计算机程序指令中提出)。
(五)排除病人使用药物的影响
当病人正在接受某种药物治疗时,可能影响某项实验结果。据调查,近1万种药物在一定条件下会影响某项成分的测定结果。如在实验申请单中标明有关药物的信息,通过储存于计算机的信息分析,可查出有无药物影响因素的存在,并能在报告中给予提示。
(六)计算机辅助诊断
用计算机辅助分析处理实验数据,可获得更高层次的评价意见和诊断报告。例如,目前已有不少程序可将血清蛋白电泳图谱的数据、同工酶谱的数据和成套的肝功能实验结果以及综合血液电解质酸碱平衡状态的检测数据,经计算机处理后,作出相应的诊断意见。这项工作正在进一步发展中。例如,用计算机程序辅助对电解质、酸碱平衡紊乱病人的处理,在治疗性药物监测中,几次血药浓度的数据分析可以辅助医生及时调整给药的剂量和间隔时间等。
计算机技术也正在被用于研究血清诸化学成分之间的统计学关系及其规律,这意味着将一组实验结果的数据作为整体在分析考虑时,其诊断意义往往更为显著。这种组合的实验项目分析有可能提供更灵敏而特异的信息,今后将有可能发展成为有前途的一种诊断程序。
第五节 临床生化检验医师的岗位、职责与作用
随着检验医学的迅速发展、新的检测项目及检测技术日新月异地引进到临床实验中来,为了有效地发挥实验室数据的作用,不少医院的检验科室均建立了临床咨询岗位,建立了多专业的临床实验室咨询的制度和职责,大大加强了实验室专家与临床医师之间的联系,活跃了临床与实验室两大部门的交流和共同提高,使实验室数据更好地被利用,更及时地被理解,转化为更高层次的诊断信息。
根据几大医院的实践,有关生化检验医师的职责可参考以下各项:
⒈能运用完整的实验设备,为临床医师提供诊断咨询服务;
⒉在充分考虑到该项测定的价/效益的情况能适应临床需要的(在速度上及时在分析数据方面可靠的)分析结果;
⒊提供对实验结果的解释,分析和提供进一步检查需要的建议;
⒋对个别病人进行连续监测,为控制调整其治疗方案提供实验依据;
⒌协助临床共同创建完善新的诊断和治疗方法;
⒍进行基础的与应用的临床观察和研究;
⒎参加医院和社会的科研项目;
⒏协助医院对职工的培训与继续教育。
以上职责和作用又可以归纳为以下几个方面的活动并形成一定制度。
⒈日常为病人服务的有关行政和技术管理工作,包括生化专家帮助临床医师在每天处理病人中遇到的问题咨询,为了胜任此项工作,称职的临床实验室生化专家必需具备有关疾病病理生理的基础知识,这将使他们可以适当地选择和解释生化实验及其结果。咨询中还包括多种内分泌和代谢病诊断中功能实验的设计实施,药物监测实验中多种标本的适当收集时间。结合临床解释阴离子间隙和容积渗分子间隙,合理选择组合试验并对结果作出评价。
为了给临床医生提供方便有效的参考指导,检验科要为实验的选择和对样品的要求提供手册和简易操作说明。
实验报告的解释是实验室诊断中的不可缺少的组成部分,部分实验包括多数功能试验和组合试验应附有解释性的评论和描述分析。
检验科咨询人员也可提供直接与病人处理有关的问题,如联合的脂质代谢门诊部、甲状腺门诊部、遗传代谢门诊部等。
⒉建立临床联系会诊与病例讨论会制度实验室的有效作用,很大程度上依赖于他们与临床医生及病人之间的交流,定期的行政交流和学术性讨论,都将促使临床实验室人员与医生之间的相互理解和提高。不少临床生化专家进行内分泌病例讨论会,也有外科、内科、传染病专题研讨会等。
⒊坚持分析质量的保证制度生化专家对分析结果的质量负有主要的责任,总体的质量控制包括分析前变化的控制,分析过程质量的保证,分析过程中干扰的识别,实验室管理的各个环节,新实验的开发,要有方法学评价实验,分析有效价值的报告。
⒋重视研究开发工作
⑴正确组合配套实验,进行有计划的多个专题的组合评价讨论会,并加以总结提高,这是目前临床生化实验室研究工作的一个方面,对于提高诊断率,判断病理进程有重要意义。
⑵不断开发新试验,推出新项目,主动对临床医师进行专题综述介绍,并在时间中紧密配合临床进行个案随访,以总结经验。