糖分解代谢途径先天代谢异常可有丙酮酸激酶缺乏病,丙酮酸脱氢酶缺乏症和磷酸果糖代谢异常所致恶性发烧。
(一)丙酮酸激酶(PK)缺乏病
在糖酵解过程中,丙酮酸激酶催化磷酸烯醇式丙酮酸生成烯醇式丙酮酸,同时产生ATP,是酵解途径产生ATP的反应之一,PK缺乏将导致成熟红细胞缺乏ATP,进而发生溶血。
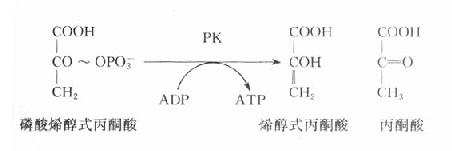
表3-6 各型糖原贮积病的特征
| 类型 | Ⅰ | Ⅱ | Ⅲ | Ⅳ | Ⅴ | Ⅵ | Ⅶ | Ⅷ | Ⅸa | Ⅸb | Ⅹ | ||
| 受害器官 | 肝肾 | 所有组积 | 肝、肌、心 | 肝单核吞噬细包系统 | 肌肉 | 肝 | 肌肉 | 脑 | 肝 | 肝 | 肝、肌肉 | 肝 | |
| 糖原结构 | N | N | 短枝异常 | 长枝异常 | N | N | N | 巨大颗粒 | N | N | |||
| 酶缺陷 | G-6-P酶 | α葡萄糖 苷酶 | 脱枝酶 | 分枝酶 | 肌磷酸化酶 | 肝磷酸化酶 | 磷酸果粒糠酶 | 肝磷酸化酶 | 同左 | 糖原合成酶 | |||
| 空腹低血糖 | ++ | + | ± | ± | ++ | ||||||||
| 对胰高血 糖素反应 |
血糖 | ↑N | 0↑ | ↑ | ↑ | 0± | ↑ | ± | N进餐0空腹 | ||||
| 血乳酸 | ↑↑ | ↑ | ↑ | ||||||||||
| 半乳糖 果糖试验 |
血糖 | ↑ | ↑ | ↑ | ↑ | ↑ | ↑ | ↑ | |||||
| 血乳酸 | ↑↑ | ↑ | ↑ | ||||||||||
| 剧烈运动后血乳酸 | ↑ | ↑ | ↑ | ↑ | ↑ | ||||||||
| 脂质代谢 | ↑↑ | N | 饿后 FFa ↑ |
运动后肌摄取 FA↑ |
TG↑ Ch↑ |
TG↑ Ch↑ |
TG↑ Ch↑ |
||||||
| 其它诊断性试验 | 肝活检 | 红细胞抗原 ↑ | 红细胞糖原结构异常 | 红细胞磷酸化酶↓ | |||||||||
| 临床特征 | 生长停滞,肝大 | 心衰 | 似Ⅰ型但不明显 | 肝硬化腹水 | 肌无力 | 肝大 | 肌无力 | 意识障碍 | 肝大,常染色体隐性遗传 | 肝大伴性遗传 | 肝大 | 轻度肝大 | |
注:N-正常;O-阴性;↑-升高;↓降低;±-可疑
网织红细胞中含有线粒体,故可通过糖有氧氧化产生足量的ATP。而成熟红细胞中不含线粒体,完全依赖糖酵解供能。红细胞内生成的ATP主要用于维持细胞内外的离子梯度,特别是通过Na+-K+-ATP酶维持细胞内外Na+、K+浓度梯度。这对于维持红细胞双凹形状十分重要。若缺乏ATP,红细胞将发生肿胀,易发生溶血,实验室检查可以见到自身溶血试验阳性。PK的遗传缺陷是糖酵解途径中遗传性缺陷导致溶血性贫血的最多见原因。PK缺陷时,细胞中PK活性仅为正常细胞的5%-25%,故虽然PK缺陷少见,但其造成的溶血性贫血却对机体危害甚大。
(二)丙酮酸脱氢酶复合物缺乏症
丙酮酸脱氢酶复合物由丙铜酸脱氢酶、二氢硫辛酸转乙酰基酶、二氢硫辛酸脱氢酶及NAD+、FAD、CoASH、焦磷酸硫胺素、硫辛酸三个酶、五个辅助因子组成,其氧化的丙酮酸氧化脱羧生成乙酰辅酶A的反应是糖进入三羧酸循环、彻底氧化成CO2和水、产生大量ATP的关键。
在儿童中发现有多种丙酮酸代谢异常的疾病,其中有些是由于丙酮酸脱氢酶复合物中某些组份先天性缺陷所致。该酶复合物中各种亚基(催化亚基和调节亚基)都可能发生先天性缺陷。这些缺陷都可使丙酮酸不能继续氧化产生ATP,使脑组织不能有效地利用葡萄糖供能,进而影响了儿童大脑的发育和功能,严重者可导致死亡。
丙酮酸不能进一步氧化,致使患儿血液中乳酸、丙酮酸和丙氨酸的浓度显著升高,出现慢性乳酸酸中毒。丙酮酸脱氢酶的缺陷可以通过皮肤成纤维细胞培养并进行酶学测定予以测定。此类病人在一定程度上可通过进食生酮食物和限制糖的摄入使病情缓解或得到控制。
(三)磷酸果糖代谢异常
磷酸果糖激酶与果糖-1,6-二磷酸酶是作用相反的一对酶,它们所催化的化学反应是糖代谢途径中的一处无效循环(又称底物循环)。

由于酶的遗传性缺陷,以上无效循环得不到控制,造成ATP大量分解产热:ATP+H2O→ADP+Pi+热。临床上可因服用氟烷而诱发恶性发烧。
恶性发烧是一种罕见的遗传缺陷性疾病,其发病率占儿童的1/15000,成人的1/50000-1/100000。病人常因服用某种药物,如吸入氟烷而在几分钟内突然发病,表现为体温骤然升高、代谢性和呼吸性酸中毒,以及高血钾症和肌肉强直。人们认为,氟烷可以促进肌肉中上述两个酶所催化的耗能无效循环,诱发恶性发烧的产生。