一、血清碱性蛋白质的分离及其氨基酸的组成分析
最初在培养离体成纤维细胞和脂肪细胞时,偶然发现培养基中加入血清可使细胞甘油三酯(TG)的合成量增加,后来证明这是因为血清中含有一种呈碱性的蛋白质分子,根据这一特性,1988年,Cianflone采用层析法将这种蛋白质分离出来,并依据其生理功能命名为酰化作用剌激蛋白,该蛋白质分子量为14.0kD,等电点为9.10。1989年Kwiterovich采用IEF和SDS/PAGE技术从人血浆中分离纯化三种碱性蛋白质分子,并测得其分子量和等电点分别为:碱性蛋白Ⅰ(BPⅠ)14.0kD,9.10;碱性蛋白Ⅱ(BPⅡ)27.5kD,8.48;碱性蛋白Ⅲ(BPⅢ)55.0kD.8.73。分析这三种碱性蛋白质,发现他们分别由不同的氨基酸组成:BPⅠ精氨酸的含量约为BPⅡ、BPⅢ的两倍,半胱氨酸含量为BPⅢ的三倍;BPⅡ不含半胱氨酸而富含脯氨酸;BPⅢ蛋氨酸的含量是BPⅠ、BPⅡ的2~3倍,而组氨酸的含量只有BPⅠ、BPⅡ的一半,BPⅢ还富含丝氨酸。另外,这三种蛋白质都含有较丰富的非极性氨基酸,因此当用葡聚糖或聚丙烯酰胺对其进行层析法分离时,观察到的相反的现象可能是这些氨基酸在水相中相互作用的结果。而组成该蛋白质分子的Asn/Asp和Gln/Glu都以酰胺基团形式存在,保证了这些蛋白质分子的等电点呈碱性。
Kwiterovich采用免疫印迹分析表明:BPⅠ只能同抗ASP免疫血清发生特异性反应,与免疫前血清无反应。而BPⅡ和BPⅢ都能与抗ASP免疫血清和免疫前血清发生反应,因此没有理由相信BPⅡ和BPⅢ与ASP是同一种物质,但Kwiterovich认为BPⅠ和ASP为同一种物质,因为许多实验结果也显示BPⅠ和ASP具有相似的等电点、分子量和氨基酸组成,而且都具有酰化作用激活性,但目前还没有BPⅠ和ASP分子克隆的结果证实这一结论,见表7-1所示。
表7-1 碱性蛋白Ⅰ和酰化作用剌激蛋白氨基酸组成分析
| 残 基 | 碱性蛋白Ⅰ | 酰化作用剌激蛋白 | ||
| 摩尔组分 | 整 数 | 摩尔组分 | 整 数 | |
| Asp/Asn | 9.47 | 12 | 10.3 | 14 |
| Clu/Gln | 11.76 | 15 | 14.1 | 19 |
| Ser | 5.75 | 7 | 6.6 | 9 |
| Gly | 9.11 | 12 | 11.7 | 16 |
| Arg | 8.25 | 10 | 5.8 | 8 |
| Cys | 3.01 | 4 | 5.4 | 7 |
| Thr | 5.16 | 7 | 4.9 | 7 |
| Ala | 7.54 | 10 | 8.2 | 11 |
| Val | 5.23 | 7 | 4.8 | 6 |
| Met | 1.59 | 2 | 1.5 | 2 |
| Ile | 3.61 | 5 | 2.7 | 4 |
| Leu | 8.03 | 10 | 6.2 | 8 |
| Phe | 3.61 | 5 | 3.2 | 4 |
| His | 2.95 | 4 | 1.4 | 2 |
| Lys | 6.75 | 9 | 5.2 | 7 |
| Pro | 5.02 | 6 | 5.2 | 7 |
| Trp | ND | ND | ||
| 残基总数 | 129 | 135 | ||
| 分子量 | 14335Da | 14514Da | ||
| 等电点 | 9.10 | 9.10 | ||
氨基酸组成分析也证实BPⅠ和ASP都含有丰富的半胱氨酸,可以凭借其形成二硫键产生广泛的生物学效应,一般这些半胱氨酸残基都以氧化的形式存在,当采用含β-巯基乙醇的缓冲液来纯化ASP时,其生物活性会完全丧失,可见半胱氨酸的存在对维持其酰化作用剌激活性很有必要.现已证明,BPⅠ、BPⅡ、BPⅢ的生理功能不同于胆固醇转运蛋白(sterol carrier protein ,SCP)和脂肪酸连结蛋白(fatty-acidbinding protein ,FABP),也不同于其他碱性蛋白如髓鞘碱性蛋白(MBP)。
二、血浆碱性蛋白对细胞脂质代谢的影响
碱性蛋白Ⅰ、Ⅱ、Ⅲ都具有酰化剌激活性,能剌激成纤维细胞合成TG,胆固醇酯(ChE)和磷脂(PL)。1994年,Peter分别采集了6个健康成人和6个家族性高载脂蛋白β脂蛋白血症患者的成纤维细胞进行细胞脂质代谢的研究:在正常人培养的成纤维细胞中,BPⅠ、BPⅡ和BPⅢ分别使其细胞内甘油三酯(TG)的含量增加2、1.5和1.4倍;但在HyperApoB患者成纤维细胞中,BPⅠ的作用下降了50%,而BPⅡ和BPⅢ仍能发挥其有效的酰化作用剌激活性。
在正常成纤维细胞,BPⅠ也有剌激胆固醇酯合成的能力,而在HyperApoB患者成纤维细胞,BPⅠ的这种作用基本上消失了。但BPⅡ能使HyperApoB患者成纤维细胞内胆固醇酯的含量异常升高至正常细胞含量的六倍,而且BPⅡ对胆固醇酯和总胆固醇的影响是平行的,游离胆固醇未发现有何变化。BPⅠ还可使正常细胞总磷脂含量升高约两倍,而在HyperApoB患者成纤维细胞,BPⅠ对磷脂的合成代谢影响甚微,只相当于正常细胞约三分之一的能力。
已有研究表明,未用碱性蛋白(BPs)处理的正常细胞和HyperApoB患者细胞,其脂质代谢无显著性差异,表明碱性蛋白(BPs)的作用机制可能是加速细胞合成脂质或抑制细胞内脂质的水解。Teng等发现在正常人脂肪细胞,ASP(或BPⅠ)的作用是促使甘油三酯(TG)合成增加而不是抑制了细胞内甘油三酯的水解,同样,在HyperApoB患者脂肪细胞内甘油三酯的减少不是由于(BPⅠ)使甘油三酯水解增加而是使其合成减少。因此Kwiterovich认为BPⅠ促进正常细胞合成甘油三酯,而HyperApoB由于存在某些缺陷限制了BPⅠ的酰化剌激活性。
三.不同脂质、脂蛋白和LDL-ApoB浓度下碱性蛋白对细胞代谢的影响
Kwiterovich 选择正常人和家族性高载脂蛋白β脂蛋白血症患者成纤维细胞进行培养,分别观察碱性蛋白Ⅰ、Ⅱ在不同脂质、脂蛋白和LDL-ApoB浓度下对细胞油酸代谢的影响,结果发现BPⅠ剌激油酸转化为甘油三酯的比率与血浆总胆固醇、甘油三酯、LDL-C和LDL-ApoB水平呈显着性相关(P<0.01),同血浆高密度脂蛋白浓度正相关(P<0.05),相似的关系也见于油酸转化为胆固醇酯的过程中。因此认为BPⅠ可能影响HyperApoB的病理生理过程,对各种表型的形成起重要作用。如总胆固醇、甘油三酯、LDL-ApoB浓度越高,则高密度脂蛋白浓度越低。而Cianflone则认为,除了家族性高胆固醇血症,酰化作用剌激蛋白(ASP)诱导成纤维细胞合成甘油三酯的量与血浆低密度脂蛋白水平呈负相关,与血浆高密度脂蛋白和极低密度脂蛋白水平无关。
同BPⅠ相反,BPⅡ诱导胆固醇酯合成的量同血浆总胆固醇、LDL-C和LDL-ApoB的浓度呈显著性正相关(P<0.01),同血浆高密度脂蛋白水平呈负相关(P=0.06),同血浆甘油三酯的浓度有正相关趋势(P=0.01)。同样,BPⅡ剌激高载脂蛋白β脂蛋白血症患者成纤维细胞合成胆固醇酯的能力与血浆高水平的LDL-C和LDL-ApoB密切相关,血浆高水平的LDL-C和LDLApoB为BPⅡ促进胆固醇的大量酯化创造了良好的条件。碱性蛋白Ⅱ(BPⅡ)可能以不同于碱性蛋白Ⅰ(BPⅠ)的方式影响血浆LDL浓度:BPⅠ主要通过影响外周细胞(如脂肪细胞)而发挥生物效应,因为在HyperApoB患者外周细胞对游离脂肪酸的转化降低;而BPⅡ则促进肝组织对游离脂肪酸的吸收并剌激肝细胞合成分泌载脂蛋白B。
碱性蛋白Ⅲ(BPⅢ)能显著剌激单核细胞源性巨噬细胞合成胆固醇酯,而对甘油三酯的代谢无影响。BPⅠ和BPⅡ对单核源性巨噬细胞的甘油三酯和胆固醇的代谢没有影响,表明碱性蛋白生物效应可能具有细胞特异性。
四、酰化作用剌激蛋白与细胞的连结
同正常人成纤维细胞一样,酰化作用剌激蛋白(ASP)能明显促进家族性高胆固醇血症和LDL-ApoB水平正常的IV型高脂蛋白血症患者的成纤维细胞合成甘油三酯,并且ASP与成纤维细胞结合具有一定的特异性,结合曲线可呈饱和状态,表明家族性高胆固醇血症和LDLApoB水平正常的IV型高脂蛋白血症患者,同正常细胞一样具有同一类ASP受体,而且该受体的最大亲和力与正常对照组基本一致。
研究表明,ASP与正常细胞和HyperApoB细胞连结的典型曲线为矩形双曲线。Scatcharel线性回归分析表明,正常细胞的斜率为-0.068±0.01(μg/ml)-1而HyperApoB患者细胞的斜率为-0.08±0.02(μg/ml)-1统计学无显著性差异,而HyperApoB细胞x轴截距只相当于正常细胞的一半这些结果表明,ASP与正常细胞和HyperApoB患者细胞表面受体的连结都呈单级式,kD值为1.05×10-6M,只是HyperApoB细胞表面受体下降了一半。但有研究表明,只有约50%的HyperApoB患者细胞存在这种缺陷,且此种缺陷与高脂血症的严重程度无关。Babirak则认为脂蛋白脂肪酶的活性缺失也可能导致高载脂蛋白β脂蛋白血症,而并未发现该患者的外周细胞表面的酰化剌激蛋白受体有所减少。
同胰岛素相比,ASP可能更有促进脂肪组织合成甘油三酯的潜力,对餐后脂肪酸的分布发挥其重要作用。ASP调节外周脂肪组织吸收脂肪酸合成甘油三酯,迅速解除游离脂肪酸对脂蛋白脂肪酶(LPL)活性的抑制,促使LPL更有效地水解餐后升高的甘油三酯,并改善LPL对富含甘油三酯的脂蛋白的清除,而HyperApoB患者由于细胞膜ASP受体数目降低,外周组织不能有效地利用血浆中的脂肪酸和葡萄糖,致使大量脂肪酸重新分布到肝组织,剌激肝细胞合成并分泌大量ApoB,装配VLDL和LDL。
五、碱性蛋白的信使传递途径
碱性蛋白Ⅰ和碱性蛋白Ⅱ可能是通过第二信使系统介导而影响细胞甘油三酯和胆固醇酯的代谢。Peter选择蛋白激酶C(PKC)的激活剂和抑制剂来研究BPⅠ、BPⅡ的作用途径,H-7是PKC较为有效的抑制剂,其作用是参与ATP竞争而不是与Ca2+或磷脂相互作用,实验结果表明,H-7抵消了HyperApoB患者细胞对BPⅠ的反应性,而且HyperApoB患者细胞对BPⅡ异常反应也被高浓度的H-7所遏制。在缺乏BPs时,PKC的激活剂C:8能同样有效地剌激正常细胞和HyperApoB患者细胞,表明在HyperApoB细胞,经甘油二酯(DG)途径激活PKC的过程无异常。但当BPs存在时,HyperApoB细胞的代谢缺陷并不因为PKC的激活剂C:8的出现而有所改善,所以HyperApoB的代谢缺陷不是在DG-PKC途径,而在第二信使系统的其他环节上。
Baldo也认为ASP是通过蛋白激酶C(PKC)途径介导而发挥生物效应的,因为PKC的激活剂豆寇酰佛波乙酯(PMA)和1-油酰基-2-乙酰-rar-甘油(OAG)能模拟ASP的生物效应,当成纤维细胞对PMA和OAG的反应性处于饱和状态时,改变培养基中的ASP浓度,并不能使甘油三酯的合成增加;PKC特异性抑制剂星形孢菌素Cal-phostinC和GF109203x也可完全抑制ASP的生物活性;ASP可使细胞内DG合成增加,而DG是PKC的有效激活剂;ASP激活PKC从胞浆转位到细胞膜,使细胞膜对脂肪酸的吸收和葡萄糖的转运增加。
为了进一步明确HyperApoB患者的细胞变异环节,1995年,Kwiterovich采用染料木黄酮(genistein)作为探针来研究正常细胞和HyperApoB患者细胞对BPⅠ的反应性。结果显示木黄酮存在时正常细胞和HyperApoB患者成纤维细胞的脂质代谢存在明显差异,可以推测酪氨酸蛋白激酶(TPK)的磷酸化在HyperApoB病理生理过程中扮演重要角色,且这一现象也被其他两种TPK抑制作用所证实(herbimycinA和tryphostinA47)。
在缺乏BPⅠ的F-12培养基中补充木黄酮(92.5nmol/ml),正常细胞甘油三酯含量下降10%,而HyperApoB患者成纤维细胞中总胆固醇和非酯化胆固醇含量下降25%。正常成纤维细胞和HyperApoB患者成纤维细胞中总胆固醇和非酯化胆固醇的量都显著减少,且HyperApoB患者成纤维细胞这两种脂质下降更显著,还没有发现成纤维细胞内胆固醇酯有何变化。当用BPⅠ和木黄酮(或者herbimycinA、tryphostinA47)处理培养基后,正常细胞合成甘油三酯的量与只用BPⅠ处理HyperApoB细胞合成甘油合成甘油三酯的量一致。这些现象表明,BPⅠ的生物活性,是通过位于细胞膜上TPK所介导的HyperApoB细胞,对BPⅠ反应性缺失,由于TPK的磷酸化为一些TPK的抑制剂所阻断,经木黄酮处理的正常细胞和HyperApoB细胞仍对BPⅠ有一些残余反应,其机理有待进一步探讨。
非受体型TPK包括SH2和SH3两个功能域,通过磷酸化作用,激活跨膜受体TPK,实际上SH2和(或)SH3蛋白是磷脂酶C-r(PLC-r)、ras-GTP酶活化蛋白(ras GAP)和磷脂酰肌醇3激酶(pI-3K)的同系物,其功能是裂解4,5二磷脂酰肌醇为DG和三磷酸肌醇(IP3),因此这些SH2和SH3蛋白也具有TPK活性,并放大TPK的信号,最终将其传到核内的转录机构,以调节特定基因的表达或通过改变细胞内蛋白的功能状态,使细胞的代谢水平发生变化,已证实HerbimycinA直接抑制PLC-r。因此,HyperApoB的细胞缺陷可能存在于跨膜受体TPK,也可能存在于原型细胞质信号蛋白的识别和结合方式。
从目前的资料来看,木黄酮对BPⅠ生物活性的抑制不是通过降低HMGCoA还原酶的活性而实现的,因为木黄酮能抑制甲羟戊酸内酯转变成非酯化胆固醇,而不能抑制HMGCoA还原酶的磷酸化。Sato已经阐明HMGCoA还原酶的磷酸化可降低其催化能力,如果木黄酮抑制了HMGCoA还原酶的磷酸化,那么将使胆固醇的生物合成增加而不是降低,但是这样无法解释经木黄酮处理的正常成纤维细胞和HyperApoB细胞内总胆固醇和非酯化胆固醇的量显著减少而胆固醇酯的变化不大的现象。
六、脂肪信号Adipsin-ASP系统
1983年,Spiegelman用梯度凝胶电泳和Northern杂交技术检测培养的鼠3T3脂肪细胞和前脂肪细胞总mRNA水平时,发现脂肪细胞较前脂肪细胞多三条特异的mRNA带,其中adipsin mRNA是分化的脂肪细胞所特有的,尤其在鼠的白色和褐色脂肪组织adipsinmRNA水平分别下降75%和68%,在胰岛素缺失的实验模型中,adipsin mRNA水平增加2~3倍,而在遗传性肥胖实验模型中,脂肪组织adipsin mRNA水平降低96%~99%。许多研究表明,adipsin是一种丝氨酸蛋白酶,由脂肪组织和坐骨神经产生,存在于循环血液中。Rosen用酶分析认为adipsin具有补体因子D的活性,而抗体中和试验也强烈提示鼠adipsin是人补体因子D仅有的同系物,White采用氨基酸序列分析表明adipsin同丝氨酸蛋白酶家族中主要成员有30%同源性,而与人补体因子D有60%同源性,而且经重组的adipsin可能替代补体因子D激活补体旁路途径。另外还发现,在肥胖型老鼠的血液中,补体因子D的活性也随着adipsin mRNA的水平下降而降低。最近White对人adipsin mRNA的成功克隆最终证实了adipsin和补体因子D为同一种物质。
在脊椎动物,补体系统是主要免疫防御体系之一。外来微生物的溶解是通过激活了抗体依赖性补体激活途径或抗体非依赖性补体激活途径来实现的,这两种激活途径都可形成裂解成孔复合体(lytic pore complex),破坏感染的细胞膜成分,导致细胞的裂解。我们知道,大多数补体蛋白都是由肝细胞和免疫细胞合成的,而脂肪组织特异性蛋白酶adipsin是人补体因子D的同系物,说明补体旁路激活与脂肪细胞的生物学行为间存在某种联系,受这一结果的启发,Lisa终于取得令人满意的结果:脂肪细胞和脂肪组织,还可合成补体旁路激活途径中的另外两个关键补体因子C3和B,表明脂肪组织在局部的免疫防御反应中起有重要的作用。SDSPAGE分析表明,补体因子B和D由一条多肽链组成,而C3由两条链组成,C3α(110kD)和C3β(70kD)之间由二硫键相连。将因子C3、B和adipsin/D放在一起孵育发现,adipsin/D将因子B酶解为Ba和Bb,因子Bb与C3相连形成C3转化酶,将C3裂解为C3α(110kD)和C3α(9kD)。过敏毒素C3α可能改变血管的通透性和收缩性,是一种趋化性细胞因子,能诱导单核细胞释放IL-1,Ba和Bb,调节淋巴细胞的生长,Bb还诱导人单核细胞的扩展,抑制单核细胞的迁移。
ASP是一个更具潜力的脂肪酸酰化剌激因子,他诱导TG合成具有双重效应:其一,ASP诱导葡萄糖经细胞器运输到细胞表面,使细胞膜对葡萄糖特异性转运增加,尽管细胞膜对脂肪酸的转运是由ASP间接引起的,但细胞对脂肪酸净吸收量的增加激活了甘油三酯合成的限速酶-甘油二酯酰基转移酶:其二,ASP实际上是补体C3的一个片段,命名为C3αdesArg,他具有调节细胞内甘油三酯合成的功能。
1993年,Baldo对ASP进行了氨基酸组成、分子量和N-端氨基酸序列分析发现,ASP和C3α的氨基酸组成非常接近,只是C3α比ASP多一个精氨酸,且N-端氨基酸序列均为N-Ser-Val-Gln-Leu-Thr-Glu-Lys-Arg-Met-ASP,离子喷射电离技术也证实ASP的质量单位(8933±0.3)与C3αdesArg(8932.5)一致,而不同于C3α(9088.7).
在血浆羧肽酶的作用下,C3α羧基未端Arg很容易被水解掉而形成C3αdesArg,尽管C3αdesArg曾被认为是C3的非活性产物,只有过敏毒素的作用,然而目前的资料已显示C3αdesArg确实能够促进细胞甘油三酯的合成。
已经明确,ASP能剌激成纤维细胞、脂肪细胞和HepG2细胞合成TG,且对分化的脂肪细胞影响最大,胆仍没有证据表明ASP对脂质水解有何影响,因为HyperApoB细胞TG的含量下降是由于该细胞对ASP的反应性下降而非细胞内TG的水解增加所致。但Cianflone在分子水平利用RT-PCR技术检测了甘油醛-3-磷酸脱氢酶(GAP)、脂蛋白脂肪酶、adipsin、因子C3和因子b mRNA水平与ASP(C3α-desArg)之间的关系后发现,成纤维细胞和未分化的前脂肪细胞只能合成微量ASP,而分化的脂肪细胞在相同时间内产生大量ASP(是前者的8倍);成纤维细胞和分化的脂肪细胞内GAp mRNA水平无差异,而LDL mRNA在分化的脂肪细胞内明显增加,其意义有待进一步探索。另外,分化的脂肪细胞内因子C3和adipsin mRNA水平异常升高,因子B mRNA水平只略有升高,这些变化与ASP(C3α-desArg)大量增加平行。
因此,adipsin/D-ASP/C3α-desArg系统可能具有非常重要的生理意义。当脂肪组织对脂肪酸的摄取能力下降时,大量脂肪酸滞留于血浆中,LPL的活性则受到严重抑制,不能有效水解血浆中的TG,而Adipisn-ASP系统可以上调脂肪组织对血浆游离脂肪酸的吸收率,使血浆中游离脂肪酸的水平不致于上升到有害的程度,LPL也能充分发挥其有效的生物活性,水解血浆中的TG和富含TG的脂蛋白,有效地调节甘油三酯的水解和合成。
七、HyperApoB的病理生理
HyperApoB是Sniderman分析了大量临床资料后于1980年首先提出的,该患者脂代谢紊乱的主要表现是血ApoB和LDL水平升高,LDL-C同LDL-ApoB之比明显降低。据Montreal心脏研究中心对100例HyperApoB合并冠状动脉粥样硬化患者实施主动脉-冠状动脉搭桥术后,十年随访结果显示:血清LDL-ApoB水平升高和HDL-ch水平降低是冠状动脉粥样硬化病变进展的两个最有价值的指标,不管是发生在原位冠状血管,还是在隐静脉旁路移植块。Brown等对血清LDL≥72.5μmol/L、有冠状动脉搭桥术史和早发冠心病的遗传背景的患者采用控制饮食和降低脂质相结合的治疗方案。结果表明:当血清LDL-ApoB的浓度下降时,冠状动脉内的粥样斑块也随之消退。
正常人的LDL是一类非均质的大分子物质,密度为1.019~1.063kg/L,HyperApoB患者LDL的分子量小于2.0×106D,直径仅有23nm,Sf值也小于5.0,而密度则高达1.055kg/L,该患者LDL组成分析表明,胆固醇酯的含量明显降低,ApoB含量明显升高,而甘油三酯、磷脂和非酯化胆固醇含量基本不变。另外,HyperApoB患者往往合并血清Lp(α)水平升高,而PROCAM研究中心则认为血清Lp(α)水平与ApoB或ApoA-Ⅰ的浓度变化无关。
大量研究显示HyperApoB患者体内LDL受体活性是正常的,并认为LDL-ApoB的增加是大量VLDL-ApoB的继发性改变。于是,许多实验室开始研究HyperApoB患者ApoB的分子或基因变异情况,Gavish应用ApoB的单克隆抗体封闭ApoB的某种抗原点位,发现LDL-ApoB的浓度。但大规模遗传连锁分析还没有确认HyperApoB患者同ApoB基因所在染色单体之间的关系,而且Johns Hopkins冠心病研究小组也没有发现ApoB的影响变异与HyperApoB之间存在某种密切关系。
从目前资料来看,ApoB的基因变异或缺失似乎对HyperApoB的影响甚微,无法解释HyperApoB的各种表型。已发现HyperApoB每一种表型都存在于两种代谢障碍。其一,肝组织大量合成VLDL,致使致密的LDL水平升高,Teng用ApoB的单克隆抗体分别封闭致密和疏松的LDL微粒,发现致密的LDL与LDL之间向乎无反应,而疏松的LDL微粒与LDL受体之间的高亲和力下降,致使LDL表面的ApoB重新分布。来自动物体内和体外的实验结果也证实疏松的LDL很容易从血液循环中清除,而致密LDL常需要很长时间。Kwiterovich认为致密LDL可能通过清道夫受体途径从血液中清除,这种代谢方式将促成动脉粥样硬化的形成和胆固醇酯在血管壁的沉积,但研究结果出乎意料,来源于HyperApoB患者体内的LDL并不能促进正常人或HyperApoB患者单核源性巨噬细胞大量合成胆固醇酯。而且Knight等也没有发现单核源性巨噬细胞优先吸收和降解HyperApoB患者的致密LDL。其二,从饮食中摄入的脂肪经乳糜途径延迟,血中游离脂肪酸的浓度明显升高。Sniderman等的研究表明脂肪细胞和成纤维细胞摄取游离脂肪酸合成甘油三酯的能力下降,可能是血中游离脂肪酸水平升高的主要原因,而游离脂肪酸则诱导肝细胞合成和分泌装配VLDL的载脂蛋白B。
由于血清碱性蛋白与脂代谢密切相关,而HyperApoB的发病机制又无法从ApoB的基因变异、LDL受体和清道夫受体水平上找到合理的解释,许多实验室已将目光转入血清碱性蛋白质的研究上,并试图解开HyperApoB的发病机制。尽管目前的研究水平仍停留在对碱性蛋白质结构和功能的探讨上,但Kwiterovich已根据现有的研究成果,对HyperApoB的生化特征从碱性蛋白质及其受体的角度作出了大胆的假设,如图7-3所示。
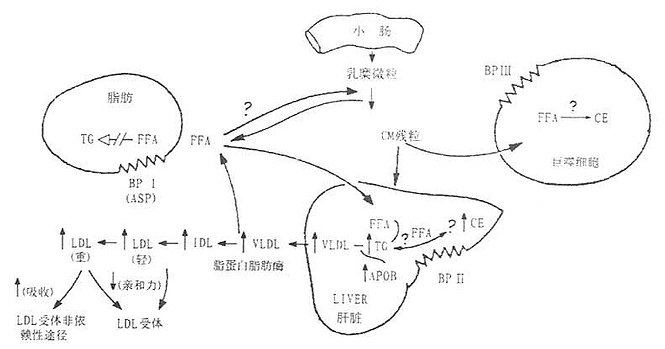
图7-3 HyperApoB病理假说图解
综上所述,三种血清碱性蛋白质所具有酰化剌激活性确实为我们进一步认识血浆脂质、脂蛋白和游离脂肪酸的代谢及HyperApoB的致病机制提供了新的视野,但其确切的生化机制有待进一步阐明。血清碱性蛋白质与补体系统之间的关系。以及碱性蛋白的组织特异性仍将是今后研究的重点。