第一章 绪论
本章介绍病理生理学的概念、内容、学科性质及其在医学中的地位,发展简史,学习和研究时所应具有的指导思想,以期对本课程有一个概括的基本认识。
病理生理学(PathologicPhysiology或Pathophysiology)是基础医学理论学科之一,它同时还肩负着基础医学课程到临床课程之间的桥梁作用。它的任务是研究疾病发生的原因和条件,研究整个疾病过程中的患病机体的机能、代谢的动态变化及其发生机理,从而揭示疾病发生、发展和转归的规律,阐明疾病的本质,为疾病的防治提供理论基础。
人体患病是一个复杂过程。我们愈能明了其规律便愈能更准确地诊断、治病疾病。同时也能运用疾病这个客观事物的认识去预防疾病。
第一节 病理生理学的内容
就基础医学兼桥梁的一个学科而言,病理生理学的范围非常广泛,临床各科的任何疾病以及在实验动物上复制的任何疾病,都有病理生理学的问题。在此应特别指出的是尽管疾病种类繁多,但是所有的疾病,或者是定位于不同器官的许多疾病,都可发生一些共同的变化,都具有一些共同规律。而同一器官的疾病以至每一种具体的疾病,又各有其特殊的变化和特殊的规律。因此病理生理学的内容可以分成三个部分,即疾病概论、基本病理过程以及各系统病理生理学。疾病概论所包含的内容是关于各种疾病的普遍规律性问题,如疾病发生的原因和条件,疾病时稳态(homeostasis)调节的紊乱及其规律,疾病的转归等。基本病理过程是指不同器官、系统在许多不同疾病中可能出现的共同的、成套的病理变化,如水、电解质和酸碱平衡紊乱、缺氧、发热、炎症、播散性血管内凝血、休克等。各系统病理生理学的主要内容之一是各系统的许多疾病在其发展过程中可能出现的一些常见的共同的病理生理变化,例如心血管系统的心力衰竭,呼吸系统的呼吸衰竭,肝胆系统的肝性脑病和黄疸,泌尿系统的肾功能衰竭等。人体系统很多,如神经系统、血液系统、免疫系统、骨骼关节系统等等,病理生理学将不能一一叙述。至于每一种疾病的特殊变化和特殊规律,虽然也属于各系统病理生理学的范围,但病种过多,学时有限,故许多具体疾病的病理生理学问题,将分别在有关学科特别是临床学科教材中论及,或者参考详为论述的专著。
对于医学教育来说,我们的计划和大纲只规定了必要的病理生理学部分内容。包括疾病概论、基本病理过程和各系统理生理学等共十六章,这都是重要的内容,属于病理生理学的基本理论,也是进一步深造的基础。
能过本门课程的教学,目的是使学员对所学的内容,掌握基本概念、各种病理生理过程是怎样发生和发展的,在其发展过程中有哪些主要的功能和代谢变化,这些变化是怎样产生的,并能运用这些基本理论对具体疾病的病理生理学问题进行分析综合,为临床医学、军事医学的学习和实践打下必要的理论基础。
第二节 病理生理学的学科性质及其在医学中的地位
当前,医学科学的各个学科,既各有专业范围,各有本身特点,又愈来明显地互相依赖、互相渗透、互相促进;而且,医学科学与数学、物理学、化学、生物学等一般自然科学的关系也日益密切。正因为如此,现代医学才能前所未有的速度蓬勃发展。病理生理学是从机能角度提示疾病本质的学科。与不少其他基础学科一样,病理生理学也是一门与多学科密切相关的综合性边缘学科。为了研究患病机体复杂的机能、代谢变化及其发生发展的机制,必须运用有关基础学科的理论和方法。因此,病理生理学与生物学、遗传学、免疫学、生理学、生理物理学和生物化学等有密切的关系。这些基础学科的每一重大进展,都有力地促进了病理生理学的发展。对于医学来说,熟悉这些基础学科的有关理论和方法,也是学好病理生理学的先决条件之一。
另一方面,病理生理学又与临床各科密切相关。在各科的临床实践中,往往都有或者都会不断出现迫切需要解决的病理生理学问题,诸如疾病原因和条件的探索,发病机制的阐明,诊疗和预防措施的改进等等。病理生理学专业工作者以及其他学科特别是临床各科从事病理生理学研究的人员,就必须对这些问题进行深入的研究,使人们对疾病的认识,不断有所提高,有所深化。病理生理在病因和发病机制方面的研究成果,常常使疾病的防治不断地改进,甚至发生重大的变革。例如,从上世纪末至本世纪中叶,人们一直认为许多休克病人的共同发病环节是小动脉、微动脉等小血管因血管运动中枢麻痹而扩张所引起的动脉血压下降,因而临床上曾经广泛采用的治疗措施之一是用血管收缩药来使微动脉等小血管收缩并从而血压回升。但是,这种疗法对不少病人的疗效并不理想,有时甚至反而会使病情恶化。到本世纪六十年代,人们对休克进行了深入的病理生理学研究,发现多数休克动物或休克病人的共同发病环节不是微动脉等小血管的扩张而是小动脉、微动脉、后微动脉、毛细血管前括约肌的痉挛性收缩,特别是持续较久的微静脉痉挛性收缩,从而使组织的动脉血液灌流量急剧减少。这就是休克时微循环衰竭学说的基本观点。根据这个学说,目前临床上比较广泛采用的治疗措施之一是结合补液应用血管扩张药。实践证明,这种疗法效果要好得多。有人认为,血管扩张药的作用主要在于解除微静脉的痉挛,因为休克发展到一定时期后,许多病人的微动脉、后微动脉、毛细血管前括约肌等可能已经发生扩张。然而,血管活性药物的正确使用,微循环的改善,组织动脉血液灌流量的正常化,仍然未能挽救所有休克的病人。随着近年来对休克进行的细胞水平、亚细胞水平和分子水平的病理生理研究,人们发现在休克时,除了由于微循环衰竭而发生的缺血、缺氧可以继发地损害组织,细胞以外,休克动因还可直接损害细胞,使细胞膜电位降低,线粒体、溶酶体受损,从而导致ATP生成减少等一系列严重的代谢变化直至发生细胞坏死崩解,并进而引起严重的全身性变化如内脏出血和心力衰竭等等(参阅第十章)。因此,在休克的现代治疗中,已经开始采用ATP以纠正细胞能量代谢障碍,用糖皮质激素以稳定溶酶体等措施,并已经取得一定效果。由此可见,病理生理学的研究成果,往往能促进临床医学不断发展,对于医生来说,学好病理生理学,也是学习临床学科的重要条件。可以认为,病理生理学是沟通基础医学和临床医学的桥梁,起着承前启后的的作用。
病理生理学是一门理论性较强的学科,必须认真学习本学科和复习有关邻近学科的基础理论,并且应用这些基本理论,通过科学思维来正确认识疾病中出现的各种变化,不断提高分析综合和解决问题的能力。
病理生理学又是一门实践性较强的学科。为了探索疾病发生的原因和条件,病理生理学工作者有时需要作一定的流行病学调查;为了研究疾病时机能代谢的动态变化及其发生机制,除了必须作周密的临床观察之外,还应当在不损害病人的前提下,进行一些必要的临床实验研究。但是,大部分实验研究是不容许在人身上进行的。这就需要在动物身上复制人类疾病的模型,人为的控制各种条件,以便从各个方面对机能、代谢变化进行深入的动态观察,并且对复制的疾病进行治疗并探索疗效的机制。这就是实验病理学和实验治疗学。动物实验的结果往往可以成为临床医学的重要借鉴和参考。病理生理的大量研究结果,主要是来自实验研究,特别是来自动物实验研究。因此,病理生理学又是一门实验性科学。在病理生理学的教学内容中,也安排了一些动物实验,其目的在于通过具体操作和实验设计,通过对所得结果的分析综合,提高独立思考和独立工作的能力,为将来进行科学研究工作打下一定的基础。此外,通过实验,也可以印证理论,使感性认识与理性认识更好地结合。
第三节 学习和研究病理生理学的指导思想
学习和研究病理生理学,如同学习和研究其他学科一样,必须自觉地以马克思主义哲学即辩证唯物论的宇宙观和方法论作为指导思想,运用唯物辩证法的最根本的法则,即对立统一的法则或矛盾的法则,去研究疾病中的各种问题。唯有这样,才能更客观、更全面的认识疾病,才能避免唯心主义和形而上学,才能避免机械地片面地理解疾病。
结合本学科的特点,应当特别注意下述几方面的问题:
(一)必须辩证地认识病因中原因和条件的应用
例如,就肺炎球菌性肺炎的病因而言,在肺炎双球菌侵入呼吸道后,还不一定能引起肺炎。只有当机体因受寒、饥饿、疲劳或醉洒等因素使呼吸道的防御屏障包括免疫功能遭到削弱,以致机体在与病菌的相互作用中不能排除或消灭病菌时,才会发生肺炎。在这里,肺炎双球菌是疾病的原因,而受寒、饥饿、疲劳或醉洒等因素则属于疾病的条件。可以看出,原因是引起相应疾病并决定疾病特异性的必不可少的因素,而某些条件则是在原因作用于机体或侵入机体的前提下,决定某些疾病是否发生的因素。正确认识和区分原因和条件在疾病发生发展中的作用,对于正确进行疾病的防治,有很重要的意义(参阅第二章)。
(二)必须认识疾病是一个运动、发展的过程
例如,在肺炎双球菌侵入肺泡引起肺炎的最初阶段,患部肺泡壁毛细血管显著扩张,血管内液体进入肺泡而形成水肿液,继而红细胞和纤维蛋白原亦从血管内渗出而进入肺泡,纤维蛋白原在肺泡内转变为纤维蛋白而使肺泡陷于实变;最后,由于大量白细胞实变肺泡并释出蛋白水解酶而使纤维蛋白溶解,肺泡内液体继而被吸收或咳出,患部肺泡的呼吸功能乃得以恢复。与在许多其他疾病时一样,推动肺炎不断运动发展的动力是损害同机体的抗损害反应之间的矛盾斗争:肺泡内纤维蛋白的形成可以阻止肺炎双球菌继续蔓延,白细胞大量进入肺泡,可以消灭病原菌并有助于肺泡恢复通气,这些,再加上特异性抗体的形成等等,无疑都是重要的抗损害反应。正确认识疾病的运动发展,正确区分许多疾病中推动疾病运动发展的损害与抗损害的反应及其相互斗争的规律,往往是正确处理疾病的重要基础。此外,也应当认识,正确而及时的治疗措施也可以能动地改变疾病的自然发展过程。例如,在肺炎的早期,可以通过抗菌素的抑菌作用和杀菌作用来消灭病原菌,通过一般支持疗法来协助和加强机体的抗损害反应,从而可以使肺炎的发展过程大大缩短,患者乃得以更快地恢复健康。
(三)必须正确认识形态、机能和代谢变化的辩证关系,正确认识全身和局部变化的辩证关系
辩证唯物论的基本观点之一就是各种事物都是互相联系、互相制约而不是彼此孤立的。正常器官、组织、细胞的形态结构、机能和代谢是密切相关不可分割的统一体。没有正常的形态结构和代谢过程,便不可能有正常的机能;反之,没有正常的代谢过程和机能活动,细胞和组织将不可能不断地自我更新,其正常结构也将不能维持。结构破坏或代谢异常的肌肉固然不能正常地收缩,而缺氧所致的代谢障碍也将引起肌肉组织的结构破坏;长期瘫痪的肌肉也将发生萎缩。在所有疾病中,形态、机能和代谢的异常变化是互相联系、互相制约的。例如,肺炎球菌性肺炎时,患部肺泡由于充满纤维蛋而无气实变,这种形态结构的变化就使患部肺泡丧失通气功能,其结果可引起全身缺氧和代谢变化。
无论是在正常或患病时,机能的任一局部与全身之间,以及各个局部之间,都是通过神经和体液的途径紧密地联系在一起的,孤立地看待疾病时的局部变化或全身变化,否认它们之间的相互影响相互制约的观点,都是错误的。前文已经提到,肺炎时患部的肺泡实变,可以引起全身缺氧,炎症区代谢产物吸收后对骨髓的刺激,是使全身血液中的白细胞增多的重要因素,而全身血液中白细胞的增多又有利于白细胞更多地进入肺部炎症区,从而起吞噬、杀菌、溶解肺泡内纤维蛋白等作用,使病变趋向愈复。
第四节 病理生理学的发展简史
在整个医学的漫长发展史中,病理生理学是一门比较年轻的学科,是科学发展和实践需要的必然产物。十九世纪中叶,人们开始认识到,仅仅用临床观察和尸体解剖的方法,还不足以使人们对疾病的本质有全面的、深刻的认识。于是有人开始在动物身上用实验的方法来研究疾病时机能、代谢的动态变化,为以后病理生理学的发展,奠定了基础。二十世纪以来,特别是最近一、二十年以来,随着一般自然科学和医学基础科学的飞跃发展以及各种先进技术的广泛采用,病理生理学也在自己的领域中取得了重大的进展,使人们对许多医学基础理论问题和许多疾病机制的认识,提高到一个新的水平,即亚细胞水平和分子水平,而病理生理学研究的这些新成就又迅速应用于临床实践,使临床医学也不断得到新的发展。
我国在解放前,没有病理生理学这个专业,少数学者在实验病理学方面做过一定的工作,实际上也就是病理生理学的早期研究工作。
解放以来,在党中央的亲切关怀和正确领导下,我国的病理生理学,作为一门新兴的学科,有了较大的发展。在医学教育方面,自从1955年以来,全国医学院校普遍开设了病理生理学这门新的课程。广大病理生理学教师,患诚党的教育事业,把“自力更生”和“洋为中用”的结合起来,在教材建设、教学改革等方面,通过辛勤的劳动,经过反复曲折,终于走上了具有自己特色的病理生理学教学的发展道路。在科学研究方面,我国的病理生理学工作者在医学遗传学、肿瘤病因学、肿瘤发病学、免疫病理学、移植免疫学、冻伤、烧伤、休克、微循环障碍、缺氧、高山病、发热、炎症、放射病、心血管系统疾病、血液病、内分泌系统疾病、中西医结合以及某些传染病、地方病如钩端螺旋体病、克山病、黑热病、低血钾麻痹等等各个方面,都作出了可喜的成绩。
自从1980年在广州召开的全国病理生理学第三次学术会议上成立了中国生理科学会病理生理学专业委员会以来,全国各地病理生理学教学和科学研究工作有了更快的发展,我们深信,我国的病理生理学工作者,今后一定能为社会主义祖国的现代化建设,特别是为医学科学的现代化,作出更大贡献。
第二章 疾病概论
第一节 疾病的概念
根据目前对疾病(disease)的认识,可将其概念归纳如下:
疾病是机体在一定病因的损害性作用下,因自稳调节(homeostatic control)紊乱而发生的异常生命活动过程。在多数疾病,机体对病因所引起的损害发生一系列抗损害反应。自稳调节的紊乱,损害和抗损害反应,表现为疾病过程中各种复杂的机能、代谢和形态结构的异常变化,而这些变化又可使机体各器官系统之间以及机体与外界环境之间的协调关系发生障碍,从而引起各种症状、体征和行为异常,特别是对环境适应能力和劳动能力的减弱甚至丧失。
上述概念、概括了疾病如下的基本特征:
第一,疾病是有原因的。疾病的原因简称病因,它包括致病因子和条件。目前虽然有些疾病的原因还不清楚,但随着医学科学的发展,迟早总会被阐明的。疾病的发生必须有一定的原因,但往往不单纯是致病因子直接作用的结果,与机体的反应特征和诱发疾病的条件也有密切关系。因此研究疾病的发生,应从致病因子、条件、机体反应性三个方面来考虑。
第二,疾病是一个有规律的发展过程。在其发展的不同阶段,有不同的变化,这些变化之间往往有一定的因果联系。掌握了疾病发展变化的规律,不仅可以了解当时所发生的变化,而且可以预计它可能的发展和转归,及早采取有效的预防和治疗措施。
第三,疾病时,体内发生一系列的功能、代谢和形态结构的变化,并由此而产生各种症状和体征,这是我们认识疾病的基础。这些变化往往是相互联系和相互影响的,但就其性质来说,可以分为两类,一类变化是疾病过程中造成的损害性变化,另一种是机体对抗害而产生的防御代偿适应性变化。
第四,疾病是完整机体的反应,但不同的疾病又在一定部位(器官或系统)有它特殊的变化。局部的变化往往是受神经和体液因素调节的影响的,同时又通过神经和体液因素而影响到全身,引起全身功能和代谢变化。所以认识疾病和治疗疾病,应从整体观念出发,辩证地处理好疾病过程中局部和全身的相互关系。
第五,疾病时,机体内各器官系统之间的平衡关系和机体与外界环境之间的平衡关系受到破坏,机体对外界环境适应能力降低,劳动力减弱或丧失,是疾病的又一个重要特征。治疗的着眼点应放在重新建立机体内外环境的平衡关系,恢复劳动力。
所谓病理过程(pathologicalprocess)是指存在于不同疾病中的共同的、成套的机能、代谢和形态结构的异常变化。例如阑尾炎、肺炎以及所有其他炎性疾病都有炎症这个病理过程,包括变质、渗出和增生等基本病理变化。病理过程可以局部变化为主,如血栓形成、栓塞、梗死、炎症等,也可以全身反应为主,如发热、休克等,一种疾病可以包含几种病理过程,如肺炎球菌性肺炎时有炎症、发热、缺氧甚至休克等病理过程。
第二节 病因学概论
一般来说,所谓病因,应当包括致病因子和条件(包括通常所谓诱因)两方面的因素,它们在疾病的发生发展中,起着不同的作用。
致病因子是指能够引起某一疾病的某种特定因素而言。例如伤寒杆菌能引起伤寒、疟原虫能引起疟疾,等等,因此,伤寒杆菌就是伤寒的原因,疟原虫就是疟疾的原因。原因是引起疾病的必不可少的、决定疾病特异性的因素;没有这个因素,相应的疾病就不可能发生。然而,在许多情况下,仅有致病因子对机体的作用,往往还不足以使疾病发生。例如,与同一感冒患者密切相处的许多人,虽然都可能受到感冒病毒的侵袭,但其中可能只有少数人发生感冒而大多数人并不发生。这里,感冒是否发生,就取决于某些条件是否具备。条件是指在疾病的致病因子作用于机体的前提下,决定疾病发生发展的因素而言。有些条件可使机体的抵抗力(resistance)降低或易感性(susceptibility)、敏感性(sensitivity)增高,从而使机体的在相应致病因子的作用下易于发病;有些条件则可使相应的致病因子能以更多的机会、更大的强度作用于机体而引起疾病;例如,免疫功能不足、过劳、月经期、过敏性鼻炎等等条件能使机体对感冒病毒的抵抗力降低或易感性增高;因此,具备其中一个或一个以上条件的机体在接触感冒病毒后就易于发病,而不具备上述条件的机体(这是大多数)即使受到感冒病毒的侵袭,一般也不致发病。促使感冒发病的条件还有年龄因素(学龄前儿童感冒发病率较高)、季节因素(寒冷季节中感冒发病率较高)与感冒患者相处特别密切而持续又较久等等。
许多条件是一些自然因素,包括气象条件、地理环境等等。例如,夏季和初秋天气炎热有利于肠道致病菌(伤寒杆菌、痢疾杆菌等)在外界环境中繁殖,也有利于苍蝇的孳生,从而使肠道致病菌易于传播;同时,炎热天气可能使人体消化液分泌减少和肠蠕动减弱,消化道的抵抗力可因而降低,而且炎热季节中人们爱吃生冷食物,与肠道致病菌接触的机会可能增多。因此,炎热季节中容易发生消化道传染病如痢疾、伤寒等等。冬春季天气寒冷,人们在室内停留时间较长,如通风不良,居住拥挤就有利于呼吸道致病微生物的传播。因而容易发生呼吸道传染病如麻疹、白喉、流行性脑脊髓膜炎等等。我国吸血虫病主要见于长江两岸和南方湖沼水网地区,是由于这些地区适宜于中间宿主的大量繁殖,而水源又易被含有血吸虫卵的人畜粪便所污染的缘故。
也有许多条件属于社会因素。大量事实表明,社会制度、社会环境对人类疾病的发生发展有重大影响。在我国解放前,统治阶级残酷地剥削和压迫劳动人民;人民精神苦闷,生活贫困,营养不足,又加上过度劳累,因而对疾病的抵抗力很弱。同时,恶劣的卫生条件又使各种致病微生物、寄生虫得以大量繁殖孳生,各种劳动保护措施又十分欠缺。这些条件(社会因素)就决定了旧社会中各种传染病、寄生虫病的猖獗流行和工伤事故、职业病的大量发生、而娼妓制度的存在,又使性病广泛传播。新中国成立以后,我国人民在党中央的正确领导下生活水平、劳动条件和卫生条件都逐步有所提高和改善,休质也不断有所增强。多年来,我国在党的卫生方针指引下,通过一系列强有力的有效措施,取得了卫生保健事业各方面的伟大成就。例如,解放前在我国危害严重的烈性传染病鼠疫、天花等已经绝迹,黑热病早在五十年代末就已基本消灭,血吸虫病也逐步得到控制,而娼妓制度废除,也使性病逐步趋于消灭等等。但是,由于极左路线的干扰,卫生管理制度的不够完善以及医疗卫生设施的不足,某些社会因素尚未得到应有的控制,因而在疾病防治方面也还有不少问题有待解决,例如,随着工业发展而出现的废气、废水、废渣对环境的污染,饮食卫生管理不善以致病毒性肝炎和一些常见的消化道传染病如痢疾、伤寒等尚未得到充分的控制,等等,都是值得重视的问题。
区分致病因子和条件的作用和意义,对于许多疾病的防治,具有重要的实际意义。根据不同情况,我们既可以侧重于采取在体内外消灭致病因子或防止其侵入机体的各种措施,也可以侧重于采取排除相应各种条件的措施,或者采取两者并重的办法,来达到防治疾病的目的。例如,目前对防止疟疾的流行来说,消灭致病因子(疟原虫)是主要的,因而采取的主要措施应当是彻底治疗疟疾现症患者,疟疾流行区居民普遍预防服药,消灭蚊虫和防止蚊虫叮咬等。对于消灭天花来说,全民接种牛痘疫苗,以排除对天花的免疫能力不足这个条件,却是最有效的措施。对于防止结核病的流行而言,则针对致病因子(结核杆菌)的措施如隔离和治疗开放性肺结核患者,乳牛结核病的防和牛乳的消毒等等,以及排除发病条件的措施如不断改善营养和居住条件,合理安排工作以及接种卡介苗以增强特异免疫等等,都有十分重要意义,应当尽可能兼顾并重而不能有所偏废。
条件对于许多疾病的发生发展的重要意义,已如上述,但是,也有许多疾病的发生,似乎并不需要相应条件的存在,例如机械暴力、高温、氰化物等剧毒化学制剂作用于机体时,毋须任何条件,即可分别引起创伤、烧伤和中毒。
还应当注意的是,同一因素,对一种疾病来说是条件,而对另一种疾病却可以是原因,因而应当作具体分析。例如营养不足使机体抵抗力降低,可以是结核病发生的条件,而长期严重的营养不足本身又是营养不良症的致病因子。
所谓诱因或诱发因素(precipitatingfactor)是指能够加强某一疾病或病理过程的原因的作用,从而促进疾病或病理过程发生的因素。例如,昏迷病人容易发生上呼吸道带菌分泌物的吸入,因而昏迷可以成为肺炎的诱因。又如,肝性脑病发生的重要致病因子之一是氨中毒,而食管静脉破裂出血是肝性脑病的重要诱因,因为大量血液进入肠道后,其中蛋白质分解产物氨基酸,经肠内细菌作用可产生大量的氨,因而可使血氨水平突然显著增高而诱发肝性脑病。
致病因子的种类很多。根据习惯,可以基本上仍按致病因子分类,但是考虑致病因子的同时,必须注意条件所起的作用:
一、生物性因素
各种致病性微生物(如病毒、支原体、立克次体、细菌、螺旋体、真菌等)和寄生虫(如原虫、蠕虫等)是很常见的致病因子。这些因素致病力量的强弱,除了与其入侵机体的数量有关以外,还取决于它们的侵袭力(invasiveness)和毒力。所谓侵袭力是指这些因素穿过机体的屏障以及在体内散布、蔓延的能力。梅毒螺旋体能穿过完整的皮肤和粘膜,某些链球菌能产生透明质酸酶(hyaluronidase)以水解而破坏结缔组织的完整性,因而都有较强的侵袭力。所谓毒力主要是指致病微生物产生外毒素或内毒素的能力。例如,白喉杆菌的侵袭力虽然不强,但因产生毒性很强的外毒素,故而是致病性很强的细菌。
致病微生物作用于机体后是否引起发病以及发病后的病情轻重,往往取决于一系列条件,其中,机体免疫功能低下是促使许多感染性疾病发生的特别重要条件,应当引起足够的重视。
二、化学性因素
许多无机和有机化学物质具有毒性,称为毒物(poison)。一定剂量的毒物被摄入机体后即可引起中毒或死亡。毒性(toxicity)极强的毒物如氰化物、有机磷农药等,即使剂量很小,也可导致严重的损害或死亡。不少毒物对机体的某些器官系统有选择性的损害作用。例如,一氧化碳与血红蛋白有很强的亲和力,因而能选择性地作用于红细胞,形成碳氧血红蛋白而导致缺氧;升汞主要引起肾脏损害;四氯化碳主要损害肝脏;巴比妥类药物主要作用于中枢神经系统,等等。熟悉毒物的选择性毒性作用,对于理解中毒性疾病的发病机制和采取正确治疗措施,都有重要的意义。
某些条件对于中毒性疾病的发生发展,也起一定作用。例如,毒物对机体的影响,在一定程度上取决于机体对该毒物的排泄速度:阿托品可被机体较快地随尿排出,故一般不致发生蓄积作用;而机体排泄铅的速度很慢,因而长期食入非中毒剂量的铅可导致铅在体内蓄积而发生铅中毒。如果机体的排泄功能发生障碍,毒物在体内停留时间就将延长,机体受到的损害也将更为严重。由于正常的肝脏有强大的解毒功能,能使许多毒物减弱或解除毒性,因而肝脏功能的损害,将减低机体对毒物的耐受能力。
三、物理性因素
能损害机体的物理因素主要有机械暴力(引起创伤、震荡、骨折、脱臼等)、高温(引起烧伤或中署)低温(引起冻伤或全身过冷)、电流(引起电击伤)、激光(高能量激光由于热的作用可引起蛋白质变性和酶的失活)、大气压的改变(引起减压病等)、电离辐射(引起放射病)等。
物理因素是否引起疾病以及引起疾病的严重程度,主要取决于这些因素的强度、作用部位和范围、作用的持续时间等。例如,温度愈高,作用面积愈大,则引起的烧伤愈严重;同样强度的交流电通过肢体时,可只引起烧伤;但如通过心脏,则可引起心室纤维颤动而致死。然而,在有些情况下,某些条件在发病中也起一定作用。例如,在空气干燥,风速较大而利于发汗散热的条件下,人体可以经受得住50~60℃的环境高温,而在空气湿度大、风速小、不利于蒸发对流散热的条件下,30~35℃的气温就可能引起中署。
四、营养性因素
营养过多和和营养不足都可引起疾病。长期摄入热量过多可以引起肥胖病,摄入某些雄生素特别维生素A和D过多也可引起中毒。营养不足可以由营养物质摄入不足或消化、吸收不良所引起,也可以是需要增加而供应相对不足的结果。,例如,生长发育旺盛的儿童和少年,孕妇和甲状腺能亢进或长期发热的患者等,营养需要或营养物质的消耗显著增加,如不相应的增补,就易发生营养不足。营养不足常见类型是总热量不足,蛋白质不足,各种维生素、必需氨基酸和必需脂肪酸的不足。此外,其它营养素如水和无机物包括钠、钾、钙、镁、磷、氯和微量元素如铁、氟、锌、铜、钼、锰、硒、碘、铬、钴等的缺乏都可以成为疾病的原因,而其中许多物质如水、钠、钾、钙、镁、铁、铜、氟、硒等的过多,也可引起疾病。
氧虽然一般不列为营养因素,但比起所有营养因素来,氧更是机体绝不可缺的物质。缺氧可引起极严重的后果,严重的缺氧可在数分钟内导致死亡。然而,缺氧对机体的影响也取决于一些条件。例如,中枢神经系统的抑制,代谢率的降低,长期锻炼和适应等都能提高机体对缺氧的耐受性。氧吸入过多时,可以发生氧中毒,多见于高压氧或常压高浓度氧持续吸入时(参阅第三章)
五、遗传性因素
遗传物质的改变可以直接引起遗传性疾病,例如某种染色体畸变可以引起先天愚型(mongolism)或Down综合征,某种基因突变可以引起血友病(hemophilia)等。遗传因素的改变也可使机体获得遗传易感性(genetic predisposition),必须加上一定的环境因素的作用才能使机体发生相应的疾病。例如,某种基因突变可使红细胞葡萄糖-6-磷酸脱氢酶(glucose-6-phosphatedehydrogenase)发生缺陷,以致红细胞还原型谷胱甘肽的含量较低,而还原型谷胱甘肽又为维持红细胞膜的稳定性所必需。这样的个体,在通常情况下还不致发生溶血;但当他们吃了过多的蚕豆或服用伯氨喹啉、磺胺等具有氧化作用的药物时,就可发生溶血。
遗传物质改变的机制尚不十分明了。但有资料表明,某些外界环境可以引起遗传物质的改变,例如电离辐射可以引起染色体损害。
六、先天性因素
与遗传因素不同,先天性因素不是指遗传物质的改变,而是指那些能够损害正在发育的胎儿的有害因素。例如,孕妇如患风疹,则风疹病毒可能损害胎儿而引起先天性心脏病。
七、免疫性因素
在某些个体,主要可能是由于遗传因素的影响,免疫系统对一些抗原的刺激常发生异常强烈的反应并从而导致组织、细胞的损害和生理功能的障碍。这种异常的免疫反应称为变态反应(allergy)或超敏反应(hypersensitivity),异种血清蛋白,一些致病微生物等都可引起变态反应;甚至某些食物(如虾、牛乳、蛋类等)、某些花粉,某些药物(如青霉素等)在某些个体也可引起诸如荨麻疹、支气管哮喘甚至过敏性休克等变态反应性疾病。有些个体能对自身抗原发生免疫反应并引起自身组织损害,称为自身免疫性疾病(autoimmune disease)。自身免疫性疾病的发生与遗传有密切关系。一些自身免疫疾病如全身性红斑狼疮(systemiclupus erythematosis, SLE)等多见于女性,因而其发生与女性激素的作用可能有一定的关系。各种原因引起的免疫缺陷病(immunodeficiency disease)的共同特点是容易发生致病微生物的感染,细胞免疫缺陷的另一后果是容易发生恶性肿瘤(参阅第十六章)。
八、精神性因素
长期的忧虑、悲伤、恐惧等不良情绪和强烈的精神创伤在某些疾病的发生中可能起重要作用。例如,有人认为,某些人之所以发生高血压病或消化性溃疡,可能与长期的精神过度紧张有关;长期的思想冲突或精神负担可使某些人发生神经衰弱,等等。在这方面,个体特点(条件)是非常重要的。同样的精神刺激,对有些人并无显著影响,而对另一些人却可造成长期的不良情绪,并可进而引起某些疾病。
综合以上所介绍的八类因素来看,一类疾病只能由某一类致病因子所引起。一种疾病只能由一种致病因子所引起。就多数疾病而言,情况确是如此。但是,也应当注意,同一类疾病,也可以由不同类的致病因子所引起。例如,恶性肿瘤就可以由许多致病因子引起。所谓致癌因素,就是指引起恶性肿瘤各种致病因子,而一系列条件则在恶性肿瘤的发生发展中也起重要的作用。
很多种类的化学物质有致癌作用,称为化学致癌物,包括无机化合物如砷化合物、铬酸盐、镍化合物以及复杂的有机化合物如多环芳烃、芳香胺、亚硝胺、偶氮染料、烷化剂、肼、氯乙烯、某些激素如雄二醇、不少药物如氮芥、非那西丁等。某些黄曲霉菌产生的黄曲霉素(aflatoxin)也是一种作用强烈的化学致癌物。据目前所知,有些化学致癌物进入体内后毋须经过代谢即能致癌,如氮芥、环氧化物等称为直接致癌物质;但许多化学致癌物活性并不高,要在进入体内后在代谢过程中得到活化而转变为高度活性的亲电子物质以后,才能与细胞中的大分子亲核基团形成共价键结合而发挥其致癌作用,此种致癌物称为间接致癌物。例如,间接致癌物苯并芘进入机体后,可被细胞内质网中的一组酶(p-450系统或芳烃羟化酶系统)经过一系列反应而活化成高度活性的亲电子物质,这一组酶的活性水平受基因调节控制。不同的个体,由于遗传差异而致这些酶的活性水平有所不同,因而对苯并芘等间接致癌物的敏感性不同。在化学致癌的机制方面,还有许多问题尚待解决。
此外,重要的致癌因素还有病毒和一些物理性因素如电离辐射、长期的慢性机械性刺激等等。这些因素致癌的机制也是十分复杂的。
遗传因素对恶性肿瘤发生的影响,不仅表现在上述不同个体细胞内质网中活化致癌物的某些酶系统的活性水平不同,因而对致癌物的敏感性有个体差异,而且还表现在人类恶性肿瘤的发生与人类白细胞抗原(human leukocyte antigen HLA)和ABO血型抗原有关系。国外有一些群体调查表明,乳腺癌、肠癌、直肠癌、前列腺癌与HLA-A1、A3、A7、B8、B12有正关联;胃癌与ABO血型的B抗原有正关联。
细胞免疫功能的不足,内分泌失调以致某些激素(如雌激素等)持续作用于靶器官,也是恶性肿瘤发生的重要因素。年龄因素在肿瘤的发生中也有一定的意义,如癌多发在40岁以上的人,而肉瘤则多见于青年人,根据一般材料,女性患肿瘤疾病者略多于男性,可能与女性生殖系统和乳腺肿瘤发生率较高有关。
应当着重指出,性别和年龄因素除了在恶性肿瘤的发生中起一定作用以外,还作为重要条件而影响许多其他疾病的发生发展。
就性别而言,现知妇女易患胆石病、癔病和甲状腺机能亢进等疾患,而男人则易患动脉粥样硬化、胃癌等疾病。至于性别如何影响疾病的发生,有些问题尚未充分阐明。
在年龄因素方面,目前国内外特别重视老年性因素在许多疾病的发生发展中作为条件所起的作用。众所周知动脉粥样硬化及其所引起的心血管病是最重要的老年人疾病;这种疾病的病死率欧美占第一位,本病在我国也相当常见。尽管动脉粥样硬化的发病因素很多,但其中作为条件的老年性因素无疑占有重要位置。首先,老年人动脉内膜往往发生纤维性增厚。据研究,这种老年性变化就有利于胆固醇在内膜的沉积而引起动脉粥样硬化。其次,老年人由性激素生理性减少等原因,容易发生肥胖,肥胖本身和肥胖的常见合并症如高血压、糖尿病等都有利于动脉粥样硬化的发生。此外由于年龄关系以及由于肥胖而加重骨、关节负荷的缘故,老年人易发生关节和骨骼的退行性变化,从而往往导致退行性骨、关节炎。老年人骨骼比较疏松,易发生骨折,骨折也较难愈合。老年人大脑也发生退行性变化。对环境变动的适应能力减弱。老年免疫功能减退,故对感染的抵抗力降低。前文已经提到,老年人较易发生癌症,这可能一方面与胸腺功能减退有关,另一方面与某些致癌因素长时间的蓄积作用有关。
除了老年以外,其他的年龄阶段也可作为条件而影响疾病的发生发展;例如,小儿易患呼吸道及消化道传染病,可能与小儿的解剖生理特点和防御机能不够完善有关。
第三节 发病学概论
病因作用于机体使疾病发生以后,疾病便作为一个运动发展的过程不断向前演变、推移,经过一定的时间或阶段后,最终趋于结束,这便是发病学(Pathogenesis)所要研究的问题。因此,疾病发展、经过的全过程应当包括它的最终的结局,即疾病的转归,包括恢复健康和死亡。但为了便于理解,疾病转归问题将在第五节中专门论述,而本节则着重讨论疾病发展经过中某些重要的问题——疾病时自稳调节的紊乱,疾病过程中的因果转化和疾病过程中的损害和抗损害反应。
一、疾病时自稳调节的紊乱
正常机体主要在神经和体液的调节下,在不断变动的内外环境因素作用下能够维持各器官系统机能和代谢的正常进行,维持内环境的相对的动态稳定性,这就是自稳调节控制下的自稳态或称内环境稳定(homeostasis)。正常机体的血压、心率、体温、代谢强度、腺体分泌,神经系统和免疫功能状态以及内环境中各种有机物质和无机盐类的浓度、体液的pH等等,往往有赖于两类互相拮抗而又互相协调的自稳调节的影响而被控制在一个狭隘的正常波动范围。这是整个机体的正常生命活动所必不可少的。
疾病发生发展的基本环节就是病因通过其对机体的损害性作用而使体内自稳调节的某一个方面发生紊乱,而自稳调节任何一个方面的紊乱,不仅会使相应的机能或代谢活动发生障碍,而且往往会通过连锁反应,牵动其他环节,使自稳调节的其他方面也相继发生紊乱,从而引起更为广泛而严重的生命活动障碍。以糖代谢和血糖水平的调节为例,交感神经兴奋,肾上腺素,胰高血糖素,糖皮质激素,腺垂体生长激素等可分别间接或直接地通过促进肝糖原分解和糖的异生等环节使血糖升高,而迷走神经兴奋和胰岛素则可分别间接或直接地促进肝糖原合成,抑制糖的异生以及促进组织摄取利用糖而使血糖降低。正常血糖水平,有赖于上述两方面因素相反相成的作用而得以维持。当某些病因因素使胰岛素受损或使腺垂体功能亢进以致胰岛素分泌不足或生长素分泌过多时,均可使糖代谢发生紊乱,血糖水平显著增高,而糖代谢紊乱的进一步发展将导致脂类代谢自稳调节的紊乱,表现为脂肪酸的分解占优势而发生酮症酸中毒,说明酸碱平衡的自稳调节也继之发生紊乱。
在自稳态的维持中,反馈调节起着重要作用。例如当糖皮质激素分泌过多时,可反馈地抑制下丘脑和腺垂体,从而使促肾上肾皮质激素释放激素(corticotropoin-releasing hormone, CRH)和 ACTH的分泌减少,这样就可使糖皮质激素的分泌降至正常水平。反之,当血浆中糖皮质激素减少时,上述的反馈抑制作用就有所减弱,CRH和ACTH的分泌随即增加而使糖皮质激素在血浆中又升至正常水平。这样,上述反馈调节就能使正常人血浆和糖皮质激素浓度维持在一个相对恒定的水平。当反馈调节发生障碍时,自稳态就会发生紊乱而引起一系列异常变化。例如,肾上腺一性腺综合征(adrenogenital syndrome)患者可能因遗传缺陷而致肾上腺皮质11-羟化酶缺乏,因而皮质醇(cortisol)和皮质酮(corticosterone)生成不足,故对CRH和ACTH的反馈抑制失效,腺垂体乃不断分泌更多的ACTH,肾上腺皮质性激素的生成就因而增多,故患者血中和组织中ACTH,17-酮类醇、雄激素明显增多,女性患者可出现男性化症状。
二、疾病过程中的因果转化
在各种自稳调节的控制下,正常机体各器官系统的机能和代谢活动互相依赖,互相制约,体现了极为完善的协调关系。由此理解,当某一器官系统的一个部分受到病因的损害作用而发生机能代谢紊乱,自稳态不能维持时,就有可能通过连锁反应而引起本器官系统其他部分或者其他器官系统机能代谢的变化。这就是疾病中的因果转化,即原始病因使机体某一部分发生损害后,这种损害又可以作为发病学原因(pathogenetic cause)而引起另一些变化,而后者又可作为新的发病学原因而引起新的变化。如此,原因和结果交替不已,疾病就不断发展起来。前述的糖尿病时糖代谢、脂类代谢和酸碱平衡相继发生紊乱,便是疾病时因果转化的一个例子。又如,原始病因机械暴力短暂地作用于机体,可使组织受损,血管破裂而导致大出血,大出血使心输出量减少和动脉血压下降,血压下降可反射性地使交感神经兴奋,皮肤、腹腔内脏的小动脉、微动脉等乃因而收缩,这种血管收缩虽可引起外周组织缺氧,但却可减少出血,在一定时间内又可维持动脉血压于一定水平,故有利于心、脑的动脉血液供应。外周组织(主要是皮肤和腹腔内脏)持续的缺血缺氧将导致大量血液淤积在毛细血管和微静脉内(参阅第十章),其结果是回心血量锐减,心输出量进一步减少和动脉血压进一步降低,组织缺氧就更严重,于是就有更多的血液淤积在循环中,回心血量又随之而更加减少。可见,组织缺血缺氧,毛细血管和微静脉内大量血液的淤积。回心血量减少,动脉血压降低等几个环节互为因果,循环不已,而每一次因果循环都能使病情更加恶化,故这种循环称为恶性循环(vicious circle)。实际上,严重外伤时机体内的因果转化情况还要复杂得多,上面所述,仅仅是一个概略的轮廓而已。
认识疾病发展过程中的因素转化以及在某些疾病某些情况下可能出现的恶性循环,对于正确地治疗疾病和防止疾病的恶化,具有重要意义。在上述的严重外伤发展过程中,如能及时采取有效的止血措施和输血输液,就可以阻断上述连锁反应的发展,从而防止病情的恶化。如果恶性循环已经出现,则可通过输血补液,正确使用血管活性药物,纠正酸中毒等措施来打断恶性循环,使病情向着有利于机体的方向发展。
随着因果转化的不断向前推移,一些疾病就可以呈现出比较明显的阶段性。例如,在上述的严重外伤引起出血性休克的过程中,机体可经历休克初期(微循环缺血期)、休克期(微循环淤血期)和休克期晚期(难治期)第三期(参阅第十章);严重大面积烧伤患者往往要经历休克,感染,肾功能不全等几个阶段;各种传染病则一般要经历潜伏期,前驱期,显明期和转归期等几个阶段;在伤寒病历时数周的显明期中,患者的临床表现。回肠病变和免疫反应等等,每周都不相同。具体分析疾病各阶段中的因果转化和可能出现的恶性循环,显然是正确处理疾病的重要基础。
三、疾病时的损害和抗损害反应
分析许多疾病中因果转化的连锁反应,可以看出其中两类变化:其一是原始病因引起的以及在以后连锁反应中继发出现的损害性变化,其二则是对抗这些损害的各种反应,包括各种生理性防御适应性反应和代偿作用。损害和抗损害反应之间相互依存又相互斗争的复杂关系是推动很多疾病不断发展演变,推动因果连锁反应不断向前推移的能基本动力。前述的机械暴力作用于机体例子中,组织破坏、血管破裂、出血、缺氧等属于损害性变化。而动脉压的初步下降所致的反射性交感神经兴奋以及因而发生的血管收缩,由于可减少出血并在一定时间内有助于维持动脉血压于一定水平从而有利于心、脑的动脉血液供应,故属抗损害反应。此外,同时发生的率加快、心缩加强可以增加心输出量,血液凝固过程加速又有利于止血,因而也属抗损害反应。如果损害较轻,则通过上述抗损害反应和适当的及时治疗,机体便可恢复健康;如损害严重,抗损害反应不足以抗衡损害性变化,又无适当的治疗,则病人可因创伤性或失血性休克而死亡。可见,损害和抗损害反应之间的对比往往影响着疾病的发展方向和转归。应当注意的是有些变化可以既有抗损害意义又有损害作用;而且,随着条件的改变和时间的推移,原来以抗损害为主的变化可以转化为损害性变化。例如,上述的创伤时的血管收缩有抗损害意义,但血管收缩同时也有使外周组织缺氧的损害作用,而持续的组织缺血缺氧,将导致微循环障碍而使回心血量锐减(参阅第十章),这就说明原来有抗损害意义的血管收缩,此时已转化成为对机体有严重损害作用的变化。正确区分疾病过程中的损害性变化和抗损害性反应,有重要的实践意义。在临床实践中,原则上应当尽可能支持和保持抗损害性反应而排除或减轻损害性变化,但当抗损害性反应转化为损害性变化时,就应当排除或减轻这种变化。目前,休克治疗中常应用血管扩张药来改善组织的动脉血液灌流以减轻或消除组织缺氧,并且获得较好效果,其理论基础就在于此。
对不同损害所发生的抗损害反应往往各有特点。例如,创伤时的反应已如上述,而在炎症性疾病时,机体的局部反应往往是渗出和增生,全身反应则可有发热、白细胞数目的变化等。然而,不同的损害也可引起某些共同的反应。例如,各种强烈因素如麻醉、感染,中毒、出血、创伤、烧伤、休克、过冷等,都能引起机体的应激反应(stress reaction),即通过下丘脑一腺垂体引起肾上腺皮质激素大量分泌,从而使机体的防御适应能力在短期内有所加强。这是常见于各种急性危重疾病的一种非特异性损害反应,对机体适应各种强烈因素的刺激起着重要作用。
疾病时抗损害反应的一个重要方面是各种代偿和适应反应。例如一侧肾功能完全丧失后对侧健康肾可加强活动而维持正常的泌尿功能;组织缺氧时,糖酵解过程加强,氧合血红蛋白释放氧的能力和组织利用氧的能力增强;某些组织和细胞坏死后发生的再生等等。
疾病时机体内发生的反应,主要是对抗损害的,但又不是所有疾病时的所有反应都针对损害的。例如,在许多致病微生物引起的疾病,机体的反应不是仅仅针对微生物所造成的损害,而十分重要的正是机体的免疫反应也是针对微生物本身和/或其他代谢产物的。
损害和抗损害的斗争,诚然是许多疾病时的一个重要问题。但是,在红绿色盲、唇裂、腭裂、多指症、先天愚型、睾丸女性化(testicular feminization)、先天性睾丸发育不全(Klinefelter’s syndrome)以及由遗传缺陷所引起的种种严重畸形的患者,有明显的机能、代谢和形态结构上的异常变化,但在他们身上似还很难找出令人信服的损害与抗损害反应的斗争;即使有这种斗争,但能否作为决定疾病发展方向的主要矛盾,亦尚属疑问。
第四节 疾病时的症状、体征和社会行为异常
疾病过程中机体内的一系列机能、代谢和形态结构异常变化所引起的病人主观上的异常感觉称为症状(symptom),如疼痛,不适,畏寒等;异常变化引起的现象如能用体格检查的方法检出,就称为体征(sign),例如心脏杂音,肺部罗音,血压升高,反射异常等。但是也应当注意,有的疾病,特别是在某些疾病的早期,也可以不伴有症状和体征。据调查,成年人大多都有动脉粥硬化,但其中只有少数人出现临床症状;许多早期癌症的患者也可以毫无主观症状和容易察见的体征。但如果对这些无症状患者进行相应的实验或特殊检查,往往能够发现异常变化。因此,对某些疾病如恶性肿瘤、动脉粥硬化、血吸虫病等在一范围内进行普查,以求早期诊断和早期治疗,是非常重要的。
所谓社会行为(socialbehavior),就是指劳动、人与人之间的交往等一切作为社会成员的活动,其中,劳动是最重要的。疾病时的各种异常变化,可在不同程度上影响体力劳动和脑力劳动的能力,从劳动力减退直至完全丧失劳动力,从暂时的缺勤直至长期或终生不能参加工作。在劳动力受影响的同时,其他社会行为也将受到影响,如不能参加正常的社交活动、文娱活动和旅游活动等,甚至不能自理生活。某些精神病患者除了丧失劳动和不能进行正常的社交活动以外,还可能进行犯罪活动,这也是社会行为异常的一种表现。
第五节 疾病的转归
一般疾病发展过程常可分为四期:潜伏期、前驱期、症状明显期、转归期。但也不尽然。例如,有些疾病如红绿色盲、先天愚型、先天性睾丸发育不全等遗传疾病,一旦发生以后,在患者一生中很少发生明显变化和发展,或者变化、发展的很慢。这些疾病既不直接引起患者死亡,迄今也无法使之治愈或改善。患者最后往往因其他疾病而死亡。但是大多数疾病在经历一定时间或若干阶段以后,终将趋于结束,这就是疾病的转归。当然,诊断和治疗是否及时与正确,对疾病的转归起着极为重要的作用。本节专论疾病的转归。
疾病的转归有完全恢复健康,不完全恢复健康和死亡三种情况。
一、完全恢复健康
完全恢复健康或痊愈(completerecovery)是指致病因素以及疾病时发生的各种损害性变化完全消除或得到控制,机体的机能、代谢活动完全恢复正常,形态结构破坏得到充分的修复,一切症状体征均先后消失,机体的自稳调节以及机体对外界环境的适应能力,社会行为包括劳动力也完全恢复正常。完全恢复健康说明机体的防御、代偿等反应取得绝对的优势。完全恢复健康是常见的。不少传染病痊愈以后,机体还能获得特异的免疫性。
二、不完全恢复健康
不完全恢复健康(incompleterecovery)是指损害性变化得到了控制,主要症状已经消失,但体内仍存在着某些病理变化,只是通过代偿反应才能维持着相对正常的生命活动。如果过分地增加机体的功能负荷,就可因代偿失调而致疾病再现。例如心瓣膜病引起的心力衰竭经内科治疗后,患者的主要症状可以消失,但心瓣膜的病变依然存在,只是由于心脏及心外的各种代偿反应,才能维持“正常”的血液循环。如果不适当地增加体力负荷,则又可导致代偿失调而重新出现心力衰竭时的血液循环障碍。严格地说,这种所谓不完全恢复健康的人,实际上并不健康,而仍然应当被看成是病人,并应受到恰当的保护和照顾。因外伤或其他疾病引起的各种残废,如肢体截除,肢体瘫痪等,也应归入不完全恢复健康的范畴。
三、死亡
如果疾病时的各种严重损害占优势而防御、代偿等抗损害反应相对不足,或者自稳调节的紊乱十分严重,不能建立新的平衡,又无及时和正确治疗,病人就可发生死亡(death)。当然,有些疾病,即使经过迄今为止最好的及时治疗,仍将导致患者死亡。
死亡的原因可以是生命重要器官(如心、脑、肝、肾、肺、肾上腺等)发生严重的不可恢复的损伤,也可以是慢性消耗性疾病(如严重的结核病、恶性肿瘤等)引起的全身极度衰竭,也可以是由于失血、休克、窒息、中毒等因素使各器官系统之间的协调发生严重的障碍。
近年来,人们对死亡的认识,正在经历着某些重要的变化。
按照传统的概念,死亡被认为是一个经历下述三个阶段的过程:
1、濒死期机体各系统的机能产生严重的障碍,中枢神经系统脑干以上的部分处于深度抑制状态,表现为意识模糊或消失,反射迟钝,心跳减弱,血压降低,呼吸微弱,或出现周期性呼吸。由于缺氧,糖酵解过程占优势,乳酸等酸性中间代谢产物增多;同时,ATP形成不足,能量供应锐减,各种机能活动乃愈益减弱。凝死期的持续时间因病而异,例如因心跳或呼吸骤停的病人,可以不经过或无明显的濒死期而直接进入临床死亡期,称为猝死(sudden death)。因慢性疾病死亡的病人,其濒死期一般较长。可持续数小时至2~3昼夜。
2、临床死亡期主要标志为心跳和呼吸完全停止。此时反射消失,延髓处于深度抑制状态,但各种组织中仍然进行着微弱的代谢过程。动物实验证明,在一般条件下,临床死亡期的持续时间约为5~6分钟,即血液供应完全停止后,大脑所能耐受缺氧的时间。超过这个时间,大脑将发生不可恢复的变化。
在濒死期或临床死亡期,重要器官的代谢过程尚未停止。如果这种情况是由于失血、窒息、触电等原因引起,则及时采取一系列紧急抢救措施,就可能起复苏或复活的作用。
3、生物学死亡期是死亡过程的最后阶段。此时从脑皮质开始到整个神经系统以及其他各器官系统的新陈代谢相继停止并出现不可逆的变化;整个机体已不可能复活,但某些组织在一定时间内仍可有极为微弱的代谢活动。此期中逐渐出理尸斑、尸冷、尸僵,最后尸体开始腐败。
六十年代末七十年代初以来,由于社会、法律、医学本身特别是器官移植方面的需要,人们对死亡进行了大量研究,就死亡问题,提出了如下一些新概念:
(一)死亡的定义
Bernat 认为,死亡应当是指机体作为一个整体(orgaism as a whole)的机能的永久性停止。所谓机体作为一个整体的机能的意义,就是指主要在神经体液的控制调节下,各器官系统复杂而又相互协调的活动所共同体现出来的综合或整合的机能活动;同时,这种整合的机能活动还保证着机体对环境因素的作用能有所反应,对环境有一定的适应能力。例如,体温调节就是机体作为一个整体的机能之一。体温调节是一种复杂的过程,它的调节中枢在下丘脑。体温调节对于维持一切细胞的生命活动是十分重要的。体温调节机能的丧失是机体作为一个整体停止机能活动的重要标志之一。
机体作为一个整体的机能的永久性停止是指整体的死亡而并不意味着各器官组织同时都发生死亡。在整体死亡以后一定时间内,有些器官、系统和某些组织、细胞还能继续进行机能活动。例如,当一个病人的作为一个整体的机能停止,即作为整体已经死亡以后,如果继续使用人工呼吸,则此人的血液循环还可能维持2周左右。
(二)死亡的标志
机体作为一个整体的机能永远停止的标志是全脑机能的永久性消失。简言之,整体死亡的标志就是脑死亡(brain death)。
对于机体的各种复杂的生命活动,脑起着形成、联系、整合和调节的作用,在一定时间内,一个全脑彻底破坏而继续进行人工呼吸的人,只不过是一组人为地维持着功能的某些器官、系统而已,因为作为一个整体的机体,已经停止机能活动。
脑死亡以后,尽管采取人工呼吸等一切抢救措施,各器官、系统的机能仍将在一定时间内先后停止,机能的各个部分将不可避免地先后发生死亡。例如,正常时脑干呼吸中枢发放呼吸的冲动,延髓的血压调节中枢协助调血液循环;因此,脑的破坏将导致呼吸停止或全身血管扩张。尽管此时采用人工吸收等一切积极的措施,心脏仍将在数周以内停止跳动。
除了属于大脑新皮质机能活动的高级神经活动如思维、语言、定向等以外,机体作为一个整体的机能还包括属于脑干各部分的若干机能,例如神经内分泌调节、体温调节、觅食行为等等。因此,作为死亡的标志应当是全脑的死亡,即全脑机能的完全的、不可逆的丧失,而不能只是大脑新皮质机能的丧失。
(三)判断死亡的根据
判定死亡,即判定脑死亡——全脑机能不可逆的停止的根据应当是:各种有关检查的结果都一致表明,脑干和大脑两半球的机能已全部、永远地消失。根据近年研究,判定脑死亡的主要根据可大致归纳如下:
1、不可逆昏迷和大脑无反应性不可逆昏迷(irreversible coma)是不能逆转的意识丧失状态。所谓大脑无反应性(cerebral unresponsivity)是指深度昏迷的患者对施加的外界刺激不发生有目的的反应,不听从指挥,不自动地发声,在给予疼痛性刺激时也不反应发声。
2、呼吸停止无自动呼吸,表现为至少需要进行15分钟的人呼吸后,仍无自动呼吸。
3、瞳孔散大是重要根据,但非绝对必需,有的患者可无瞳孔散大,但瞳孔固定(对光反应消失)是必有的。
4、颅神经反射消失包括瞳孔反射,角膜反射,视听反射(oculoauditoryreflex眼向拍手处转动),咳嗽反射,恶心反射,吞咽反射等的消失。
5、脑电波消失应当注意的是过量的中枢神经系统抑制药中毒和冬眠状态时,脑电波也处于零电位,但这种状态不一定是脑死亡的表现。除此以外,零电位脑电图是表示脑死亡的重要根据之一。
如果可能,再加用动脉造影等方法证明脑血液循环停止,则可进一步肯定脑死亡的诊断。至于确诊脑死亡所需的时间,一般认为上述5项检查结果持续存在24小时而无逆转倾向时,即可宣告死亡。近来也有人认为这些结果只需持续存在6小时就可发出死亡通知。而且,如果有一次脑血管造影证明脑血管灌流完全停止,就可以立刻宣告死亡。
有没有条件做脑血管造影和脑电图,没有条件用人工呼吸机进行抢救时,一般就可以根据心跳和呼吸的永久性停止来诊断脑死亡,因为已经证明,心跳和呼吸的不可逆停止如不作抢救,很快就会导致全脑机能的永久性地丧失。
脑死亡等新概念的提出,对于器官移植来说,有非常重要的实践意义。器官移植能否成功,长期效果是否良好,在很大程度上取决于移植器官从供者身上摘除时和摘除前一定时间内血液的灌流情况。从血液循环已经停止的供者,特别是血液循环停止以前有持续低血压的供者取下的器官的移植效果,一般要比摘除前仍有较好血液灌流的器官的效果为差。实践证明,已经确诊脑死亡借助人工呼吸在一定时间内维持着血液循环的患者(实际上是死者)是器官移植的良好供者,用他们的器官移植给适当的受者,可获得好效果。国外已有法律规定,只要医生确诊病人已经发生脑死亡,就可以取其器官进行移值。
脑死亡概念的提出,使人们对复苏的概念也应作出新的考虑,因为一旦医生明确宣告脑死亡,复苏或复活就完全不能实现。复苏成功,必须表明机体尚未发生脑死亡。
脑死亡概念的提出,使医生们能精确地判定死亡时间的发生,对于解决可能牵涉到的一些法律问题,也是有利的。
对比关于死亡的新旧概念,可以看出传统概念中有一些不妥之处。
1、将涉死期归属于死亡过程是不妥的。涉死期中,病人只是垂死(dying)而并未死亡,亦即尚未发生脑死亡。
2、将临床死亡的标志简单地概括为心跳,呼吸完全停止和反射消失,是不够全面的。延髓型小儿麻痹症病人的自动呼吸可以完全停止;某些心跳病患者的心跳也可以完全停止,但他们可分别借助于人工呼吸和起搏器而继续存活。可见,在上述情况下,对自动呼吸或心跳完全停止甚至永久性停止的某些病人,如果及时采用适当措施,可使病人非但不发生临床死亡,而且还可能长期存活。反之,心跳的存在也并不一定意味着生命的存在;如前所述,确诊脑死亡继续人工呼吸的某些患者(实际上是死者),其血液循环还可能持续数周。
3、整体死亡时,尽管各器官组织的死亡和崩解并不同时发生,而是在一段相当长的时间中相继出现,但是把整体死亡看成是经历濒死期,临床死亡期和生物学死亡期三个阶段的一个过程,也未必恰当。因为第一,机体各组织的崩解破坏,即所谓生物死亡期的变化,虽然一般都在心跳、呼吸停止以后发生,但有些也可在心跳、呼吸停止以前出现,因此,生物学死亡期和前面两个时期就难于截然划分。第二,把死亡看成是一个过程,对于准确地宣布死亡的时间也会造成一定的困难,而准确宣布死亡时间,往往正是社会和法律所需要的。
把死亡看成是一个事变(event)而不看成是象前文所述那样一个过程,似比较合理。这个事变就是脑死亡。医师们可以准确地诊断死亡,并通知死亡的时间,尽管死者体内的许多器官、组织还要经历一段时间才相继发生死亡。由此可见,用关于死亡的新概念来取代和补充传统概念,不论在理论上或实践上都有重要的意义。
第三章 缺氧
第一节 缺氧的概念
氧参与生物氧化,是正常生命活动不可缺少的物质。成人在静息状态下,每分钟耗氧量约250毫升;活动时,耗氧量增加。但人体内氧储量极少,有赖于外界环境氧的供给和通过呼吸、血液、血液循环不断地完成氧的摄取和运输,以保证细胞生物氧化的地需要。
当组织得不到充足的氧,或不能充分利用氧时,组织的代谢、机能、甚至形态结构都可能发生异常变化,这一病理过程称为缺氧(hypoxia)。
缺氧是许多疾病所共有的一个基本病理过程。例如休克、呼吸功能不全、心功能不全、贫血等,都可以引起缺氧。缺氧在军事医学中也是个非常重要的课题,例如高原适应不全症主要是个缺氧的问题;高空飞行、潜水作业、密闭舱或坑道内作业,如果处理不当或发生意外,都可发生缺氧。所以研究缺氧发生和发展的规律以及缺氧所引起的病理生理变化,对缺氧的防治,保障部队战斗力,具有重要的意义。
氧的获得和利用是个复杂的过程。组织的供氧量=动脉血氧含量×血流量;组织的耗氧量=(动脉血氧含量-静脉血氧含量)×血流量。故血氧是反映组织的供氧与耗氧的重要指标。
常用的血氧指标及其意义
氧分压(PO2)是指以物理状态溶解在血浆内的氧分子所产生的张力(故又称氧张力)。在100毫升37℃的血液内、以物理状态溶解的氧,每0.003毫升可产生0.133kPa(1mmHg)的氧分压。正常人在静息状态,呼吸海平面空气,以物理状态溶解在动脉血内的氧约0.3毫升%,动脉血氧分压(PaO2)约13.3kPa(100mmHg);静脉血氧分压(PvO2)正常约5.32kPa(40mmHg)。
PaO2主要取决于肺泡氧分压(PAO2)的高低、氧通过肺泡膜弥散入血的量、肺泡通气量与肺血流量的比例。如果外界空气氧分压低或肺泡通气减少,使肺泡氧分压降低,或弥散障碍、通气/血流比例失调,使肺动-静脉血功能性或解剖性分流增加,都可使PaO2降低。
氧含量 是指100毫升血液内所含的氧毫升数,包括实际与血红蛋白结合的氧和溶解在血浆内的氧。正常动脉血氧含量约19.3毫升%,混合静脉血氧含量约12毫升%。
血液氧含量主要取决于PaO2与血红蛋白的质和量。PaO2明显降低或血红蛋白结合氧的能力降低,使血红蛋白饱和度降低,或单位容积血液内血红蛋白量减少,都可使氧含量减少。
氧容量 指氧分压为19.95kPa(150mmHg),二氧化碳分压为5.32kPa(40mmHg),湿度38℃,在体外100毫升血液内血红蛋白所结合的氧量。正常血红蛋白在上述条件下,每克能结合氧1.34~1.36毫升。若按每100毫升血液含量含血红蛋白15克计算,动脉血和静脉血氧容量约20毫升%。
氧含量取决于单位容积血液内血红蛋白的量和血红蛋白结合氧的能力。如果血红蛋白含量减少(贫血)或血红蛋结合氧的能力降低(如高铁血红蛋白、碳氧血红蛋白),则氧容量减少,氧含量也随之减少。如果单位容积血液内血红蛋白的量和性质正常,只是由于氧分压降低使血红蛋白氧饱和度降低。此时氧含量减少,但氧容量是正常的。
氧饱和度 是指血红蛋白与氧结合达到饱和程度的百分数。1克血红蛋白最多能与1.36毫升的氧结合,氧饱和度达到100%。氧饱和度可以下列公式表示:
氧饱和度(%)=实际1克血红蛋白结合的氧(毫升)/1.36(毫升)×100
正常动脉血氧饱和度约95~97%,混合静脉血氧饱和度约75%。
氧饱和度高低主要取决于氧分压的高低,氧分压与氧饱和度之间的关系,可用氧离曲线来表示(图3-1)。由于血红蛋白的生理特点,氧离曲线呈S形,PO27.98kPa(60mmHg)以下,才会使氧饱和度明显降低,氧含量明显减少,从而引起缺氧。
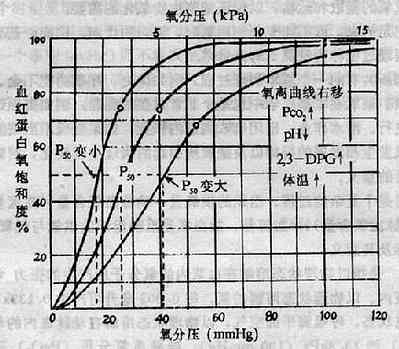
图3-1 氧离曲线
中间曲线为标准状态下(38℃、PCO2 5.32kPa(40mmIIg)、pH7.4)的氧离曲线,P50约3.59kPa (27mmHg)
动脉血氧分压和氧饱和度
混合静脉血氧分压和氧饱和度
kPa: 千帕斯卡(Kilo-Pascal),1mmHg=0.133kPa
血红蛋白与氧亲和力高低,常用P50表示。P50是指血液在38℃,pH7.4,PCO2 5.32kPa(400Hg)的条件下,使氧饱和度达到50%时的氧分压。正常成人P50约为3.59kPa(27mmHg)。血液PCO2升高、pH降低、湿度升高或红细胞内2,3-DPG含量缯加,都可使血红蛋白氧亲和力降低,氧离曲线右移,P50增大(图3-1);反之,使血红蛋白与氧亲和力升高,氧离曲线左移,P50变小。血红蛋白的结构与功能异常,不易与氧结合或不易解离氧,对P50也有影响。
动静脉血氧差 即动脉血氧含量减去静脉血氧含量所得的毫升,说明组织对氧消耗量。由于各组织器官耗氧量不同,各器官动静脉血氧差很不一样。正常动脉血与混合静脉血氧差约6~8毫升%。
动静脉血氧差变化取决于组织从单位容积血液内摄取氧的多少。PaO2明显降低,动脉血与组织氧分压梯差变小;微循环动静脉吻合支开放,使流经真毛血管的血量减少;红细胞变形能力降低或红细胞聚集,使血液流变性发生改变;细胞受损,利用氧的能力降低,都可使组织细胞从血液中的摄取的减少,动静脉血氧减少变小。淤血,血流缓慢,虽然单位时间动脉血灌流减少,但由于血流缓慢和氧离曲线右移,组织从单位容积血液内摄取的氧增多,动静脉血氧差加大。各型缺氧时动静脉血氧差的变化,要对具体情况作具体的分析。
第二节 缺氧的原因和类型
根据缺氧发生的速度,有急性缺氧和慢性缺氧。根据缺氧时PaO2的变化,有低张性低氧血症和等张性低氧血症。根据缺氧的原因,有乏氧性缺氧、血液性缺氧化、循环性缺氧、组织性缺氧。
一、乏氧性缺氧(Hypoxic Anoxia)
乏氧性缺氧是指由于肺泡氧分压降低,或静脉血分流入动脉,血液从肺摄取的氧减少,以致动脉血氧含量减少,PaO2降低。属于低张性低氧血症(hypotonic hypoxemia)。
(一)原因
1、吸入气氧分压低 例如高原或高空,大气压低;通风不好的矿井、坑道内;吸入低氧的混合气体(如吸入气摄入高浓度的氮、氢或笑气)。由于吸入气氧分压低,PAO2和PaO2随之降低。
2、外呼吸功能障碍 呼吸运动减弱或肺的疾患(如窒息、慢性阻塞性肺疾患、肺水肿、肺炎等),致肺泡通气量减少,PACO2升高,PAO2降低,结果血液通过肺摄取的氧减少,动脉血氧含量和PAO2降低。由呼吸功能障碍而引起的缺氧,又称呼吸性缺氧(respiratoryanoxia)。
3、静脉血分流入动脉(静脉血掺杂)增多正常掺杂到动脉的静脉血约占心输出量的2-3%。如果心房或心室中隔缺损,伴有肺动脉狭窄或肺动脉高压,右心的静脉血可部分经缺损处流入左心。又如肺的疾患,引起弥散障碍或通气/血流比例失调,或肺动脉吻合支开放,可致肺动静脉血功能性或解剖性分流增加。静脉血分流入动脉增多,达到心输出量的50%,PaO2可降到6.65kPa(50mmHg)以下。如果此时肺泡气通气量正常,则肺泡与动脉血氧分压差加大。
(二)、乏氧性缺氧的特点(图3-2)
1、动脉血氧分压、氧饱和度和氧含量都降低,静脉血氧分压、氧饱和度和氧含量亦随之降低。
2、动脉血和静脉血氧容量正常。如果由于慢性缺氧,使单位容积血液内红细胞数和血红蛋白量增多,氧容量增加。
3、动脉血氧差接近正常。如果PaO2太低,动脉血与组织氧分压差明显变小,血氧弥散到组织内减少,可使动静脉血氧差降低。
4、除血氧变化外,根据肺泡通气量,PaO2有不同的变化,例如严重的肺气功能障碍,CO2排出少,PaCO2升高;如果过度换气,CO2排出多,则PaCO2降低。
二、血液性缺氧(Hemic Anoxia)
血液性缺氧是指由于血红蛋白含量减少或性质发生改变,致血液携带的氧减少,血氧含量降低,或血红蛋白结合的氧不易释出所引起的缺氧。由于以物理状态溶解在血液内的氧不受血红蛋白的影响,这型缺氧的PaO2正常,属于等张性低氧血症(isotonic hypoxemia)。
(一)原因
1、贫血(anemia)各种原因引起的贫血,单位容积血液内红细胞数和血红蛋白量减少,虽然PaO2和氧饱和度正常,但氧容量降低,氧含量随之减少。虽然由血红蛋白携带的氧减少,但由于单位容积的红细胞数减少,血液粘度降低,血流加快,运输氧的能力提高(单位时间内血液给组织运输的氧量以血细胞压积为30%时最高),一般当贫血使血细胞压积低于20%,才会引起组织对氧供给不足。
2、高铁血红蛋白血症(methemoglobinemia)血红蛋白的二价铁,在氧化剂的作用下,可氧化成三价铁,形成高铁血红蛋白(methemoglobin,HbFe3+OH),也称变性血红蛋白或羟化血红蛋白。高铁血红蛋白的三价铁因与羟基牢固结合而丧失携带氧的能力,加上血红蛋白分子的四个二价铁中有一部分氧化为三价铁后还能使剩余的Fe2+与氧的亲和力增高,导致氧离曲线左移,使组织缺氧。生理情况下,血液中不断形成极少量的高铁血红蛋白,又不断地被液中的还原剂如NADH,抗坏血酸,还原型谷胱甘肽等还原为二价铁的血红蛋白,使正常血液中高铁血红蛋白含量占血红蛋白的1.7%以下。当亚硝酸盐、过氯酸盐、磺胺等氧化剂中毒时,如使血中高铁血红蛋白含量增加至20~50%,就可出现头疼、衰弱、昏迷、呼吸困难和心动过速等症状。较常见的是食用大量含硝酸盐的腌菜后,经肠道细菌将硝酸盐还原为亚硝酸盐,吸收后形成高铁血红蛋白血症,称为“肠源性发绀”(enterogenouscyanosis)。
3、碳氧血红蛋白血症(carboxyemoglobinemia)碳氧血红蛋白血症是由于CO中毒引起的,CO与Hb的亲和力为O2与Hb亲和力的218倍(37℃),Hb与CO结合后就不能与O2结合。另一方面,CO还能抑制红细胞内糖酵解,使其2,3-DPG生成减少,氧离曲线左移,HbO2中的氧不易释出,从而加重组织缺氧,血液HbCO含量达到Hb总量的10~20%,就可引起轻度缺氧;当吸入气中有0.1%的CO时,血液中的血红蛋白可能有50%为HbCO,则可发生极为严重的缺氧。
(二)血液性缺氧的特点(图3-2)
1.PaO2正常,氧容量和氧含量减少。
2.血红蛋白氧饱和度,贫血性缺氧正常,高铁血红蛋白血症和碳氧血红蛋白血症降低。
3.动静脉血氧差常小于正常。
4.由于PaO2正常,一般不引起肺通气增加。严重贫血不出现紫绀。高铁血红蛋白呈咖啡色(皮肤、粘膜青紫),碳氧血红蛋白呈樱桃红色。
三、循环性缺氧(Circulatory Anoxia)
循环性缺氧是指由于血液循环障碍,供给组织的血液减少而引起的缺氧,又称低血流性缺氧(hypokinetic anoxia)。循环性缺氧可以是局部的(如血管狭窄或阻塞);也可以是全身性的(如心力衰竭、休克)。由于动脉狭窄或阻塞,致动脉血灌流不足而引起的缺氧,又称缺血性缺氧(ischemic anoxia);由于静脉血回流受阻,血流缓慢,微循环淤血,导致动脉血灌流减少而引起的缺氧,称淤血性缺氧(stagnant anoxia)。
(一)原因
1、血管的狭窄或阻塞 可见于血管的栓塞、受压、血管的病变如动脉粥样硬化或脉管炎与血栓形成等。
2、心力衰竭 由于心输出量减少和静脉血回流受阻,而引起组织淤血和缺氧。
3、休克 由于微循环缺血、淤血和微血栓的形成,动脉血灌流急剧减少,而引起缺氧。
(二)循环性缺氧的特点(图3-2)
1.动脉血氧分压、氧饱和度和氧含量正常。氧容量一般是正常的。
2、由于血流缓慢和氧离曲线右移,组织从单位容积血液内摄取的氧增多,静脉血氧分压、氧饱和度和氧含量降低,动静脉血氧差别加大。休克时,如果微循环动静脉吻合支开放,或细胞利用氧的能力降低,动静脉血氧差也可以变小。
3.不仅组织缺氧,组织内代谢产物也不能及时运出,所以低血流性缺氧比乏氧性缺氧对组织细胞损害更为严重。
四、组织性缺氧(Histogenous Anoxia)
由组织细胞利用氧异常所引起的缺氧称为组织性缺氧。
(一)原因
1、组织中毒如氰化物、硫化氢、磷等可引起组织中毒性缺氧(histotoxic anoxia)。最典型的是氰化物中毒。各种氰化物,如HCN、KCN、NaCN、NH4CN等可由消化道、呼吸道或皮肤进入体内,迅速与氧化型细胞色素氧化酶的三价铁结合为氰化高铁细胞色素氧化酶,使之不能还原成还原型细胞色素氧化酶,以致呼吸链中断,组织不能利用氧。0.06克的HCN即可使人死亡。硫化氢、砷化物等中毒也主要由于抑制细胞色素氧化酶等而影响了细胞的氧化过程。细菌毒素、放射线等也可能损伤线粒体的呼吸功能而引起氧的利用障碍。
2、组织水肿组织间液和细胞内液的异常增多,使气体弥散距离增大,引起内呼吸障碍。
3、组织需氧过多如冠状动脉硬化的病人运动或情绪激动时,心肌耗氧量增加可诱发心绞痛。
(二)组织性缺氧的特点(图3-2)
1.动脉血氧分压、氧饱和度和氧含量正常。
2.静脉血氧分压、氧饱和度和氧含量高于正常,动脉血氧差变小。
因组织需氧过多引起缺氧时,组织耗氧量是增加的,静脉血氧含量与氧分压较低,使动静脉血氧增大。
表3-1 各型缺氧的血氧变化
| 缺氧类型 | 动脉血氧分压 | 动脉血氧饱和度 | 血氧容量 | 动脉血氧含量 | 动-静 脉氧差 |
| 乏氧性缺氧 | ↓ | ↓ | N | ↓ | ↓和N |
| 血液性缺氧 | N | N | ↓或N | ↓或N | ↓ |
| 循环性缺氧 | N | N | N | N | ↑ |
| 组织性缺氧 | N | N | N | N | ↑或↓ |
↓降低 ↑升高N正常
缺氧虽分为上述四型,但在实际情况中所见的,往往是混合型。例如失血性休克,既有血红蛋白减少所致的血液性缺氧,又有有微循环障碍所致的循环性缺氧。又如心力衰竭,既有循环障碍引起的循环性缺氧,又可继发肺淤血、水肿而引起呼吸性缺氧。因此,对具体病人,要作全面地具体分析。
第三节 缺氧时机体的机能代谢变化
缺氧时机体的机能代谢变化,包括机体对缺氧的代偿性反应和由缺氧引起的代谢与机能障碍。轻度缺氧主要引起机体代偿性反应;严重缺氧而机体代偿不全时,出现的变化以代谢机能障碍为主。机体在急性缺氧时与慢性缺氧时的代偿性反应也有区别。急性缺氧是由于机体来不及代偿而较易发生代谢的机能障碍。各种类型的缺氧所引起的变化,既有相似之处,又各具特点,以下主要以低张性缺氧为例,说明缺氧对机体的影响。
一、代偿性反应
动脉血氧分压一般要降至8kPa(60mmHg)以下,才会使组织缺氧,才引起机体的代偿反应,包括增强呼吸血液循环,增加血液运送氧和组织利用氧的功能等。
(一)呼吸系统
PaO2降低(低于8kPa)可刺激颈动脉体和主动脉体化学感受器。反射性地引起呼吸加深加快,从而使肺泡通气量增加,肺泡气氧分压升高,PaO2也随之升高。吸入10%氧时,通气量可增加50%;吸入5%氧可使通气量增加3倍。胸廓呼吸运动的增强使胸内负压增大,还可促进静脉回流,增加心输出量和肺血流量,有利于氧的摄取和运输。但过度通气使PaO2降低,减低了CO2对延髓的中枢化学感受器的刺激,可限制肺通气的增强。
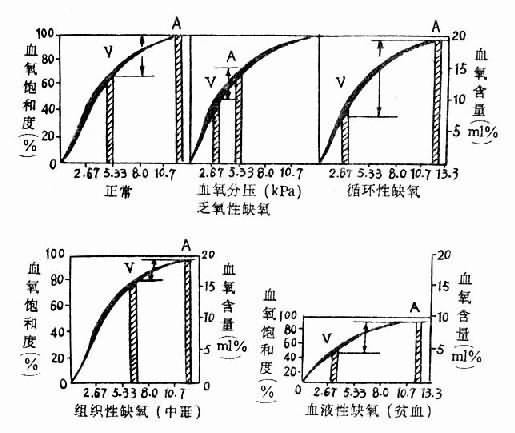
图3-2 各型缺氧的血氧变化特点
A动脉V静脉图中kPa相当于mmHg的数值
| kPa | mmHg |
| 2.67 | 20 |
| 5.33 | 40 |
| 8.0 | 60 |
| 10.7 | 80 |
低张性缺氧所引起的肺通气变化与缺氧持续的时间有关。如人达到400m高原后,肺通气量立即增加,但仅比在海平面高65%。数日后,肺通气量可高达在海平面的5~7倍。但久居高原,肺通气量逐渐回降,至仅比海平面者高15%左右。在急性缺氧早期肺通气增加较少,可能因过度通气形成的低碳酸血症和呼吸性碱中毒对呼吸中枢的抑制作用,使肺通气的增加受限。2~3日后,通过肾脏代偿性地排出HCO3-,脑脊液内的HCO3-也逐渐通过血脑屏障进入血液,使脑组织中pH逐渐恢复正常,此时方能充分显示缺氧兴奋缺氧的作用。久居高原肺气量回降,可能与外周化学感受器对缺氧的敏感性降低有关。据观察,世居高原者之颈动脉体的平均体积比世居海平面者大6.7倍,患慢性阻塞性肺病的病人的颈动脉比正常人大一倍以上。电镜观察表明,在慢性低张性缺氧的早期,颈动脉体增大,其中I型细胞增多,因I型细胞中嗜锇体含儿茶酚胺类神经介质,其增多可能具代偿意义。但在缺氧晚期,在增大的颈动脉体中嗜锇体的中心(core)缩小、晕轮(halo)加宽,有时整个嗜锇体为空泡所取代。这可能是颈动脉化学感受器敏感性降低的原因。长期缺氧使肺通气反应减弱,这也是一种慢性适应性反应。因为肺通气每增加1L,呼吸肌耗氧增加0.5ml,可能加剧机体氧的供求矛盾,故长期呼吸运动增强显然是对机体不利的。
肺通气量增加是对急性低张性缺氧最重要的代偿性反应。此反应的强弱存在显著的个体差异,代偿良好者肺通气量增加较多,PaO2比代偿不良者高。PaCO2也较低。
血液性缺氧和组织性缺氧因PaO2不低,故呼吸一般不增强;循环性缺氧如累及肺循环,如心力衰竭引起肺淤血、水肿时,可使呼吸加快。
(二)循环系统
低张性缺氧引起的代偿性心血管反应,主要表现为心输出量增加、血流分布改变、肺血管收缩与毛细血管增生。
1、心输出量增加 有报道进入高原(6100m)30天的人,其心输出量比平原居民高2~3倍。在高原久住后,心输出量逐渐减少。心输出量增加可提高全身组织的供氧量,故对急性缺氧有一定的代偿意义。心输出量增加主要是由于:
(1)心率加快:过去认为心率加快是颈动脉体和主动脉体化学感受器刺激反射性地引起。但有人实验,在控制呼吸不变的情况下,缺氧刺激血管化学感受器却使心率变慢。因此缺氧时心率加快很可能是通气增加所致肺膨胀对肺牵张感受器的刺激,反射性地通过交感神经引起的。
然而呼吸运动过深反而通过反射使心率减慢,外周血管扩张和血压下降。
(2)心收缩性增强:缺氧作为一种应激原,可引起交感神经兴奋,作用于心脏β—肾上腺素能受体,使心收缩性增强。
(3)静脉回流量增加:胸廓呼吸运动及心脏活动增强,可导致静脉回流量增加和心输出量增多。
2、血流分布改变器官血流量取决于血液灌注的压力(即动、静脉压差)和器官血流的阻力。后者主要取决于开放的血管数量与内径大小。缺氧时,一方面交感神经兴奋引起的血管收缩;另一方面局部组织因缺氧产生的乳酸、腺苷等代谢产物则使血管扩张。这两种作用的平衡关系决定器官的血管是收缩或扩张,以及血流量是减少或增多。急性缺氧时,皮肤、腹腔内脏交感神经兴奋,缩血管作用占优势,故血管收缩;而心、脑血管因以局部组织代谢的产物的扩血管作用为主,故血管扩张,血流增加。这种血流分布的改变显然对于保证生命重要器官缺氧的供应是有利的。
心肌活动消耗的能量主要来自有氧代谢。心脏重量约占体重之0.4~0.5%,静息时冠脉流量约占心输出量之4~5%,其动—静脉血氧含量差约为12ml%,表明心肌耗氧量大,由单位容积血液摄取的氧量多。心肌缺氧时,进一步提高对单位容积血液中氧的摄取率很有限,主要依靠扩张冠状血管以增加心肌的供氧。冠脉扩张由局部代谢产物(腺苷、H+、K+、PGI2等)与冠脉平滑肌中β—肾上腺能受体占优势所致,其中腺苷的作用最为重要。当心肌细胞缺氧时,由ATP、ADP生成的AMP增多,AMP在5—核苷酸酶的作用下,脱去磷酸,形成腺苷。腺苷易透过细胞膜进入组织液,作用于冠状血管,使之扩张。通常组织液中的腺苷大部分进入细胞,重新磷酸化生成AMP,一部分被腺苷脱氨酶灭活。缺氧时,腺苷脱氨酶活性可能降低,这也是局部腺苷增多的一个原因。
3、肺血管收缩肺血管直接对缺氧的反应与体血管相反。肺泡缺氧及混合静脉血的氧分压降低都引起肺小动脉收缩,从而使缺氧的肺泡的血流量减少。如果是由肺泡通气量减少引起的肺泡缺氧,则肺血管的收缩反应有利于维持肺泡通气与血流的适当比例,使流经这部分肺泡的血液仍能获得较充分的氧,从而可维持较高的PaO2。此外,正常情况下由于重力作用,通过肺尖部的肺泡通气量与血流量的比值过大,肺泡气中氧不能充分地被血液运走。当缺氧引起较广泛的肺血管收缩,导致肺动脉压升高时,肺上部的血流增加,肺上部的肺泡通气能得到更充分的利用。
缺氧引起肺血管收缩的机制较复杂,尚未完全阐明,研究结果也有矛盾。当前具倾向性的观点:①交感神经作用:缺氧所致交感神经兴奋可作用于肺血管的α受体引起血管收缩反应。②体液因素作用:缺氧可促使肺组织内肥大细胞、肺泡巨噬细胞、血管内皮细胞等释放组胺、前列腺素和白三烯等血管活性物质,其中有的能收缩肺血管,如白三烯(leukotriene,LTs)、血栓素A2(thromboxane A2、TXA2)、前列腺素F2a(prostaglandin F2a,PGF2a)等,有的扩张血管,如前列环素(prostacyclin,PGI2)、前列腺素E1(prostaglandin E1 PGE1)等。在肺血管收缩反应中,缩血管物质生成与释放增加,起介导作用;扩血管物质的生成与释放也可增加,起调节作用。两者力量对比决定肺血管收缩反应的强度。组胺作用于H1受体使肺血管收缩,作用于H2受体则使之扩张。在缺氧性肺血管收缩反应中,组胺释放增多,主要作用于H2受体以限制肺血管的收缩。③缺氧直接对血管平滑肌作用:缺氧使平滑肌细胞膜对Na+、Ca2+的通透性增高,促使Na+、Ca2+的通透性增高,促使Na+、Ca2+内流,导致肌细胞兴奋性与收缩性增高。这一观点还有待进一步证实。看来缺氧性肺血管收缩反应是多因素综合作用的结果。
4、毛细血管增生长期慢性缺氧可促使毛细血管增生。尤其是脑、心脏和骨骼肌的毛细血管增生更显著。毛细血管的密度增加可缩短血氧弥散至细胞的距离,增加对细胞的供氧量。
(三)血液系统
缺氧可使骨髓造血增强及氧合血红蛋白解离曲线右移,从而增加氧的运输和释放。
1、红细胞增多 移居到3600m高原的男性居民红细胞计数通常约为6×1012/L(6×106/mm3),Hb为210g/L(21g/dl)左右。慢性缺氧所致红细胞增多主要是骨髓造血增强所致。当低氧血流经肾脏近球小体时,能刺激近球细胞,使其中颗粒增多,生成并释放促红细胞生成素(erythropoietin),促红细胞生成素能促使红细胞系单向干细胞分化为原红细胞,并促进其分化、增殖和成熟,加速Hb的合成和使骨髓内的网织红细胞和红细胞释放入血液。当血浆中促红细胞生成素增高到一定水平时,可因红细胞增多使缺氧缓解,肾脏促红细胞生成素的产生因而减少,通过这种反馈机制控制着血浆促红细胞生成素的含量。红细胞增多可增加血液的氧容量和氧含量,从而增加组织的供氧量。
2、氧合血红蛋白解离曲线右移缺氧时,红细胞内2,3—DPG增加,导致氧离曲线右移,即血红蛋白与氧的亲和力降低,易于将结合的氧释出供组织利用。但是,如果PaO2低于8kPa,则氧离曲线的右移将使血液通过肺泡时结合的氧量减少,使之失去代偿意义。
2,3—DPG是红细胞内糖酵解过程的中间产物。缺氧时红细胞中生成的2,3—DPG增多是因为:①低张性缺氧者氧合血红蛋白(HbO2)减少,脱氧血红蛋白(Hb)增多,前者中央孔穴小,不能结合2,3—DPG;后者中央孔穴较大,可结合2,3—DPG。故当脱氧血红蛋白增多,红细胞内游离的2,3—DPG减少,使2,3—DPG对二磷酸甘油酶变位酶(diphosphoglycerate mutase, DPGM)及磷酸果糖激酶的抑制作用减弱,从而使糖酵解增强及2,3—DPG的生成增多;②低张性缺氧时出现的代偿性肺过度通气所致呼吸性碱中毒,以及由于脱氧血红蛋白稍偏碱性,致使pH增高,pH增高能激活磷酸果糖激酶使糖酵解增强,2,3—DPG合成增加,另一方面,pH增高还能抑制2,3—DPG磷酸酶(2,3—DPg phosphatase, 2,3—DPG)的活性,使2,3—DPG的分解减少(图3-3,图3-4)。
2,3—DPG增多使氧离曲线右移,是因为:①2,3—DPG与脱氧血红蛋白结合,可稳定后者的空间构型,使之不易与氧结合;②2,3—DPG是一种不能透出红细胞的有机酸,增多时能降低红细胞内pH,而pH下降通过Bohr效应可使血红蛋白与氧的亲和力降低。(Bohr效应系指H+和Pco2对Hb与O2亲和力的影响,当H+浓度或Pco2增高时,Hb与O2的亲和力降低,氧离曲线右移)。
P50为反映Hb与O2的亲和力的指标,指的是血红蛋白氧饱和度为50%时的氧分压,正常为3.47~3.6kPa(26~27mmHg)。红细胞内2,3—DPg 浓度每增高1μm/gHb,P50将升高约0.1kPa。
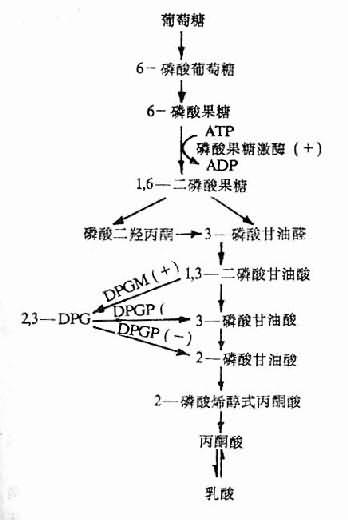
图3-3,2,3—DPG的生成与分解
DPGM二磷酸甘油酸变位酶;
DPGP二磷酸甘油酸磷酸酶;
(+)pH增高时促进反应;
(-)PH增高时抑制反应
图3-4 2,3-DPG结合于HHb分子的中央空穴示意图
(四)组织细胞的适应
在供氧不足的情况下,组织细胞可通过增强利用氧的能力和增强无氧酵解过以获取维持生命活动所必须的能量。
1、组织细胞利用氧的能力增强 慢性缺氧时,细胞内线粒体的数目和膜的表面积均增加,呼吸链中的酶如琥珀酸脱氢酶、细胞色素氧化酶可增加,使细胞的内呼吸功能增强。如胎儿在母体内处于相对缺氧的环境,其细胞线粒体的呼吸功能为成年动物的3倍,至出生后10~14天,线粒体呼吸功能才降至成年动物水平。
2、无氧酵解增强发 严重缺氧时,ATP生成减少,ATP/ADP比值下降,以致磷酸果糖激酶活性增强,该酶是控制糖酵解过程最主要的限速酶,其活性增强可促使糖酵解过程加强,在一定的程度上可补偿能量的不足。
3、肌红蛋白增加 慢性缺氧可使肌肉中肌红细胞蛋白含量增多。肌红蛋白和氧的亲和力较大,当氧分压为1.33kPa(10mmHg)时,血红蛋白的氧饱和度约为10%,而肌红蛋白的氧饱和度可达70%,当氧分压进一步降低时,肌红蛋白可释出大量的氧供细胞利用。肌红蛋白的增加可能具有储存氧的作用。
肺通气及心脏活动的增强可在缺氧时立即发生。但这些代偿功能活动本身消耗能量和氧,红细胞的增生和组织利用氧能力的增强需较长的时间,但为较经济的代偿方式。急性缺氧时以呼吸系统和循环系统的代偿反应为主;慢性缺氧者,如世居高原的居民,主要靠增加组织利用氧和血液运送氧的能力以适应慢性缺氧。其肺通气量、心率及输出量并不多于居住海平面者。
二、缺氧时机体的机能代谢障碍
严重缺氧,如低张性缺氧者PaO2低于4kPa(30mmHg)时,组织细胞可发生严重的缺氧性损伤,器官可发生功能障碍甚而功能衰竭。
(一)缺氧性细胞损伤
缺氧性细损伤(hypoxic cell damage)主要为细胞膜、线粒体溶酶体的变化。
1、细胞膜的变化 在细胞内ATP含量减少以前,细胞膜电位已开始下降。其原因为细胞膜对离子的通透性增高,导致离子顺浓度差透过细胞膜。
(1)钠离子内流:Na+内流使细胞内Na+浓度增加,可激活Na+-K+泵以泵出Na+,从而消耗ATP。ATP消耗量增多可促使线粒体氧化磷酸化过程增强,严重缺氧时,线粒体呼吸功能降低使ATP生成减少,以至Na+-K+泵不能充分运转,进一步使细胞内Na+增多。细胞内Na+的增多促使水进入细胞,导致细胞水肿。血管内皮细胞肿胀可堵塞微血管,加重微循环缺氧。
(2)钾离子外流:K+外流使细胞内缺K+。而K+为蛋白质包括酶等合成代谢所必需。细胞内缺钾将导致合成代谢障碍,酶的生成减少,将进一步影响ATP的生成和离子泵的功能。
(3)钙离子的内流:细胞外Ca2+浓度比胞浆中游离Ca2+高1000倍以上。细胞内Ca2+逆浓度外流和肌浆网、线粒体逆浓度差摄Ca2+均为耗能过程。当严重缺氧时使细胞膜对Ca2+的对通透性增高量Ca2+内流将增加;ATP减少将影响Ca2+的外流和摄取,使胞浆Ca2+浓度增高。Ca2+增多可抑制线粒体的呼吸功能;可激活磷脂酶,使膜磷脂分解,引起溶酶体的损伤及其水解酶释出;还可激活一种蛋白酶,使黄嘌呤脱氢酶(D型)转变为黄嘌呤氧化酶(O型)。由此增加自由基的形成,加重细胞的损伤。
2、线粒体的变化 细胞内的氧约有80-90%在线粒体内用于氧化磷酸化生成ATP,仅10~20%在线粒体外用于生物合成、降解及生物转化(解毒)作用等。轻度缺氧或缺氧早期线粒体呼吸功能是增强的。严重缺氧首先影响线粒体外氧的作用,使神经介质的生成和生物转化过程等降低,当线粒体部位氧分压降到监界点0.1kPa(<1mmHg)时,可降低线粒休的呼吸功能,使ATP生成减少。呼吸功能降低主要因脱氢酶活性下降,严重时线粒体可出现肿胀、嵴崩解、外膜破裂和基质外溢等病变。
3、溶酶体的变化缺氧时因糖酵解增强,乳酸生成增多,和脂肪氧化不全使其中间代谢产物酮体增多。导致酸中毒。pH降低可引起磷脂酶活性增高,使溶酶体膜磷脂被分解,膜通透性增高,结果使溶酶体肿胀、破裂,和大量溶酶体酶的释出,进而导致细胞本身及其周围组织的溶解、坏死。
(二)中枢神经系统的机能障碍
脑重仅为体重为2%左右,而脑血流量约占心输出量之15%,脑耗氧量约为总耗氧量的23%,所以脑对缺氧十分敏感。脑灰质比白质的耗氧量多5倍,对缺氧的耐受性更差。急性缺氧可引起头痛、情绪激动、思维力、记忆力、判断力降低或丧失以及运动不协调等。慢性缺氧者则有易疲劳、思睡、注意力不集中及精神抑郁等症状。严重缺氧可导致烦躁不安、惊厥、昏迷甚而死亡。正常人脑静脉血氧分压约为4.53kPa(34mmHg),当降至3.73kPa(28mmHg)以下可出现神经错乱等;降至2.53kPa(19mmHg)以下时可出现意识丧失;低达1.6kPa(12mmHg)时将危及生命。缺氧引起脑组织的形态学变化主要是脑细胞变性、坏死、脑细胞肿胀及脑水肿。
缺氧引起中枢神经系统机能障碍的机制较复杂。神经细胞膜电位的降低、神经介质的合成减少、ATP的生成不足、酸中毒、细胞内游离Ca2+增多、溶酶体酶的释放以及细胞水肿等,均可导致神经系统的功能障碍,甚而神经细胞结构的破坏、当PaO2低于6.67kPa(50mmHg)时,可使脑血管扩张。缺氧与酸中毒还使脑微血管通透性增高,从而导致脑水肿(图3-5)。脑血管扩张、脑细胞及脑间质水肿可使颅内压升高,由此引起头痛、呕吐等症状。
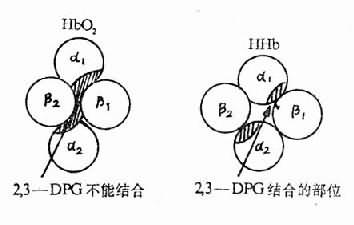
图3-5 缺氧时脑水肿发生机理
(三)外呼吸功能障碍
急性低张性缺氧,如快速登上4000m以上的高原时,可在1-4天内发生肺水肿,表现为呼吸困难、咳嗽、咳出血性泡沫痰、肺部有湿性罗音、皮肤粘膜发绀等。因高原肺水肿的动物模型难以复制成功,故其发病机制至今尚不清楚。因为肺水肿与肺动脉高压呈正相关,故有人强调肺毛细血管压力增高的作用。可能缺氧所致外周血管收缩使回心血量增加。和肺血量增多;加上缺氧性肺血管收缩反应使肺血流阻力增加,导致肺动脉高压。由于肺血管收缩强度不一,致使肺血流分布不均,在肺血管收缩较轻或不收缩的部位肺泡毛细血管血流增加,毛细血管压力增高,从而引起压力性肺水肿。也有人强调肺微血管通透性增高的作用。因为患者支气管肺泡洗出液中蛋白质含量较高,并有大量肺泡巨噬细胞,可测得补体C3a、LTB4、TXB2等;尸检可见肺泡水肿、炎性细胞浸润及透明膜形成。但高原性肺水肿不同于其它原因引起的成人呼吸窘迫综合征,前者经休息、氧疗或下山后短期内即可痊愈;而成人呼吸窘迫综合征经治疗往往要数月后才能痊愈。肺内血压高和流速对微血管的切应力(流动的血液作用于血管壁的力与管壁平等方向的分力)可能是导致微血管内皮损伤和血管通透性增高的一个因素。肺水肿影响肺的换气功能,可使PaO2进一步下降。
PaO2过低可直接抑制呼吸中枢,使呼吸抑制,肺通气量减少,导致中枢性呼吸衰竭。
(四)循环功能障碍
严重的全身性缺氧时,心脏可受累,如高原性心脏病、肺原性心脏病、贫血性心脏病等,甚而发生心力衰竭。今以高原性心脏病为例说明缺氧引起循环障碍的机制。
1、肺动脉高压 肺泡缺氧所致肺血管收缩反应可增加肺循环阻力,可导致严重的肺动脉高压。慢性缺氧使肺小动脉长期处于收缩状态,可引起肺血管中膜平滑肌肥大,血管硬化,形成稳定的肺动脉高压。肺动脉高压增加右室射血的阻力。另外,缺氧所致红细胞增多,使血液粘度增高,也可增加肺循环阻力。肺动脉高压可导致右心室肥大,甚至心力衰竭。
2、心肌的收缩与舒张功能降低心肌缺氧可降低心肌的舒缩功能,甚而使心肌发生变性、坏死。(参阅第十四章心血管系统病理生理学)
3、心律失常 严重缺氧可引起窦性心动过缓、期前收缩、甚至发生心室纤颤致死。心动过缓可能为严重的PaO2降低对颈动脉体化学感受器的刺激,反射性地兴奋迷走神经所致。此外,持久缺氧也往往显示副交感优势使心率变慢。期前收缩与室颤的发生与心肌细胞内K+减少、Na+增加使静息膜电位降低、心肌兴奋性增高、和传导性降低有关。缺氧部位的心肌静息膜电位降低,使其与相邻较完好的心肌之间形成电位差,从而产生“损伤电流”,可成为异位激动的起源,严重的心肌受损可导致完全的传导阻滞。
4、静脉回流减少 脑严重缺氧时,呼吸中枢的抑制使胸廓运动减弱,可导致静脉回流减少,全身性极严重而持久的缺氧使体内产生大量乳酸、腺苷等代谢产物,后者可直接扩张外周血管,使外周血管床扩大,大量血液淤积在外周,回心血量减少,使心输出量减少,而引起循环循衰竭。
除以上所述神经系统、呼吸与循环系统机能障碍外,肝、肾、消化道、内分泌等各系统的功能均可因严重缺氧而受损害。
第四节 影响机体缺氧耐受性的因素
年龄、机体的机能状态、营养、锻炼、气候等许多因素都可影响机体对缺氧的耐受性,这些因素可以归纳为两点,即代谢耗氧率与机能的代偿能力。
一、代谢耗氧率
基础代谢高者, 如发热、机体过热、或甲状腺机能亢进的病人,由于耗氧多,故对缺氧的耐受性较低。寒冷、体力活动、情绪激动等可增加机体耗氧量,也使对缺氧的耐受性降低。体温降低、神经系统的抑制则因能降低机能耗氧率使对缺氧的耐受性升高。故低温麻醉可用于心脏外科手术,以延长手术所必需阻断血流的时间。
二、机体的代偿能力
机体通过呼吸、循环和血液系统的代偿性反应能增加组织的供氧。通过组织细胞的代偿性反应能提高利用氧的能力。这些代偿性反应存在着显著的个体差异,因而各人对缺氧的耐受性也很不相同。有心、肺疾病及血液病者对缺氧耐受性低,老年人因为肺和心脏的功能储备降低、骨髓的造血干细胞减少、外周血液红细胞数减少,以及细胞某些呼吸酶活性降低等原因,均可导致对缺氧的适应能力下降。另外,代偿能力是可以通过锻炼提高的。轻度的缺氧刺激可调动机体的代偿能力。如登高山者如采取缓慢的队梯性的上升要比快速上升者能更好地适应。慢性贫血的病人血经蛋白即使很低仍能维持正常活动,而急性失血使血红蛋白减少到同等程度就可能引起严重的代谢机能障碍。
第五节 缺疗与氧中毒
一、氧疗
各类缺氧的治疗,除了消除引起缺氧的原因以外,均可给病人吸氧。但氧疗的效果因缺氧的类型而异。
氧疗对低张性缺氧的效果最好。由于病人PaO2及SaO2明显低于正常。吸氧可提高肺泡气氧分压,使PaO2及SaO2增高,血氧含量增多,因而对组织的供氧增加。但由静脉血分流入动脉引起的低张性缺氧,因分流的血液未经肺泡直接掺入动脉血,故吸氧对改善缺氧的作用不大。
血液性缺氧、循环性缺氧和组织缺氧者PaO2及SaO2正常,因为可结合氧的血红蛋白已达95%左右的饱和度,故吸氧虽然可明显提高PaO2,而SaO2的增加却很有限,但吸氧可增加血浆内溶解的氧。通常在海平面吸入空气时,100ml血液中血浆内溶解的氧仅为0.31ml;吸入纯氧时,要达1.7ml%;吸入3个大气压的纯氧时,溶解的氧可增至6ml%。而通常组织从100ml血液中摄氧量平均约为5ml。可见,吸入高浓度氧或高压氧使血浆中溶解氧量增加能改善组织的供氧。组织性缺氧时,供氧一般虽无障碍,而是组织利用氧的能力降低;通过氧疗提高血浆与组织之间的氧分压梯度,以促进氧的弥散,也可能有一定治疗作用。一氧化碳中毒者吸入纯氧,使血液的氧分压升高,氧可与CO竞争与血红蛋白结合,从而加速HbCO的解离,促进CO的排出,故氧疗效果较好。
二、氧中毒
O2虽为生命活动所必需,但0.5个大气压以上的氧却对任何细胞都有毒性作用,可引起氧中毒(oxygen intoxication)。
氧中毒时细胞受损的机制一般认为与活性氧的毒性作用有关(参阅第十三章缺血与再灌注损伤)。
氧中毒的发生取决于氧分压而不是氧浓度。吸入气的氧分压(PiO2)与氧浓度(FiO2)的关系如公式:PiO2=(PB-6.27)×FiO2,式中PB为吸入气压力(kPa)。6.27(kPa,即47mmHg)为水蒸汽压。潜水员在深50m的海水下作业(PB约为608kPa 即4560mmHg)时,虽然吸入气的氧浓度正常(FiO2=0.21),氧分压(FiO2)却高达126.4kPa(948mmHg),从而可导致氧中毒;相反,宇航员在1/3大气压环境中工作,即使吸入纯氧(FiO2=1),PiO2也仅27.5kPa(206mmHg),不易出现氧中毒。当吸入气的氧分压过高时,因肺泡气及动脉血的氧分压随着增高,使血液与组织细胞之间的氧分压差增大,氧的弥散加速,组织细胞因获得过多氧而中毒。
人类氧中毒有两型:肺型与脑型。
肺型氧中毒 发生于吸入一个大气压左右的氧8小时以后,出现胸骨后疼痛、咳嗽、呼吸困难、肺活量减少、PaO2下降。肺部呈炎性病变,有炎性细胞浸润、充血、水肿、出血和肺不张。氧疗的病人如发生氧中毒,吸氧反而使PaO2下降,加重缺氧,造成难以调和的治疗矛盾,故氧疗时应控制吸氧的浓度和时间,严防氧中毒的发生。
脑型氧中毒 吸入2-3个大气压以上的氧,可在短时内引起脑型氧中毒(6个大气压的氧数分种;4个大气压氧数十分钟),病人主要出现视觉、听觉障碍、恶心、抽搐、晕厥等神经症状,严重者可昏迷、死亡。高压氧疗时,病人出现神经症状,应区分“脑型氧中毒”与由缺氧引起的“缺氧性脑病”。前者病人先抽搐以后才昏迷,抽搐时病人是清醒的;后者则先昏迷后抽搐。对氧中毒者应控制吸氧,但对缺氧病脑病者则应加强氧疗。
附 高原适应不全症(高山病)
Syndrome of Insufficient AltitudeAdaptation (Mountain Sickness)
人未经适应就迅速进入3000米以上高原,或由海拔较低的高原进入海拔更高的地区,加上寒冷气候的影响,或体力负荷过重,而使机体对低氧环境耐受性降低,以致个体适应能力不足,一部分可出现一系列症状和机能代谢变化,称为高原适应不全症。国外习惯用急性高山病和慢性高山病。
我军对高原适应不全症提出了下列分型:
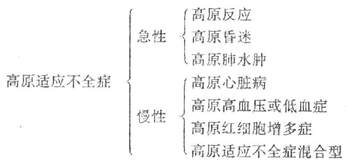
上述分型简明,有利于诊断治疗。但对具体病人来说,常常是混合型,很少是单一的,但在一定阶段可能以某一型为主。
我国幅员辽阔,海拔3000米以上的高原、高山地区,约占全国总面积的六分之一。这些地区大多分布在边疆省区,具有重要的国防意义。高原地带气候多变,寒冷、风大、空气稀薄,对人体构成了一个特殊的自然环境。其中空气稀薄,大气压和氧分压降低,是高原环境对机体影响的主要因素。
在高原地区世居的少数民族,对高原环境已经适应,但一般人口稀少,对这些地区的经济建设需要内地支援。我军有守卫边疆的任务,内地人员进入高原地区日渐增多,因此如何保证进入高原的人员健康,我是军卫生工作的重要任务。
在海平地区,空气在每平方厘米上所形成的压力为101.3kPa(760毫米汞柱),在干燥空气中氧占20.40%,故氧分压为21.15kPa(159毫米汞柱)。空气中氧所占比例基本不受高原影响,当大气压力因海拔增高而降低时,则氧分压按比例降低。下面选择几个不同高度的大气压和氧分压的改变列表如下(表3-2)。
初抵3000米以上高原地区,由于大气压中氧分降低,肺泡气和动脉血氧分压也相应的降低,毛细血管血液与细胞线粒体间氧分压梯度差缩小,从而引起缺氧。如果逐渐登高,有一个锻炼适应过程,在低氧分压环境中,机体可发生一系列代偿适应性变化,如通气加强,肺泡膜的弥散能力提高;循环功能加强,输送氧的能力增加;红细胞和血红蛋白含量增加,红细胞中2,3-二磷酸甘油酸增多,氧离曲线右移,通过这些代偿作用,以便使组织可利用氧达到或接近正常水平。机体具有一定的适应能力,可以较长期居住高原地区。一般地说,长期居住可适应的最大高度为5000米。但有人适应能力较弱,在5000米以下一定高度就失去了适应能力,而出现高原适应不全症。
在高原地区除了大气压降低对机体的主要作用,还有气候的影响,如寒冷、大风、雨雪以及紫外线照射等。这些因素降低机体适应能力,往往是高原适应不全症的诱发和加重因素。因此在相同高度的不同地区,由于气候不同,因而引起高原反应的发病率也不一样。
表3-2 不同的海拔高度大气压和氧分压的改变
| 高度 | 大气压 kPa | 氧分压 kPa | |
| 英尺 | 米 | ||
| 101.31(760.0mmHg) | 21.15(159.0mmHg) | ||
| 10,000 | 3,048 | 69.51 (522.6mmHg) | 14.55(109.4mmHg) |
| 17,000 | 5,182 | 52.59(395.4mmHg) | 11.01(82.8mmHg) |
| 20,000 | 6,097 | 46.44(349.2mmHg) | 9.72(73.1mmHg) |
| 23,000 | 7,010 | 40.88(307.4mmHg) | 8.57(64.4mmHg) |
| 26,000 | 7,925 | 35.88(269.8mmHg) | 7.51(56.5mmHg) |
| 28,000 | 8,534 | 32.82(246.8mmHg) | 6.88(51.7mmHg) |
| 33,000 | 10,058 | 26.12(196.4mmHg) | 5.47(41.7mmHg) |
三、急性高原适应不全症
(一)高原反应
从海平面地区进入高原地区以后,在不同的高度,人群中有些可出现一系列不适反应,如头痛、头昏、失眠、乏力、四肢发麻、眼花、耳鸣;重的可发生食欲不振、恶心、呕吐、胸闷、呼吸困难、心慌、浮肿等症状,称为高原反应。多数人在两周内由于代偿适应功能的建立,症状可自行消失。
这些人发病较快,如无合并症恢复也较快,一般3~5天即可恢复。也有少数人持续数月而不恢复的。
初入高原的人到海拔1000米以上就有人发病,随着高度增加,发病率增高。下面是一部分人群从海拔1000米处出发到5000米高度,高原反应发病率的情况(图3-6)。一般在3500米以下发病率占37~51%,3600~5000米发病率占50%。有一个单位在3000米居留一段时间,再登高,抵达4500~5000米时才有一些人发生高原反应。
急性高原反应的发生率与个体代偿适应能力和预先有无锻炼有关。
在代偿适应反应中主要是通过肺泡能气量增加,以增加肺泡气氧分压;并通过加强血液对氧的输送和增加组织对氧的利用。高原反应的人主要是这几个环节的代偿能力低下或发生障碍所致。
因肺泡有水蒸气和较高的CO2分压,再加上氧的弥散入血,肺泡气与大气氧分压有一个很大的梯度差(表3-3)。
在海平地区,大气→肺泡气→动脉血→混合静脉血的氧分压之间的梯度差较大。而进入高原,抵达5791米高度时,它们之间的梯度差变小。例如,在海平大气氧分压为21.15kPa(159毫米汞柱),肺泡氧分压为13.83kPa(104毫米汞柱),相差7.32kPa(55毫米汞柱);而登高抵达5791米处,大气氧分压为10.64kPa(80毫米汞柱),肺泡氧分压为5.99kPa(45毫米汞柱),只相差4.66kPa(35毫米汞柱)(图3-7)。此时大气与肺泡气氧分压差变小,主要是由于代偿性通气增加的结果。
通气增加是机体在高原的代偿适应的重要环节,主要通过呼吸加深来实现的,呼吸频率增加不明显(表3-4)。由于呼吸加深,每分肺泡通气量增加,以提高肺泡气分压。
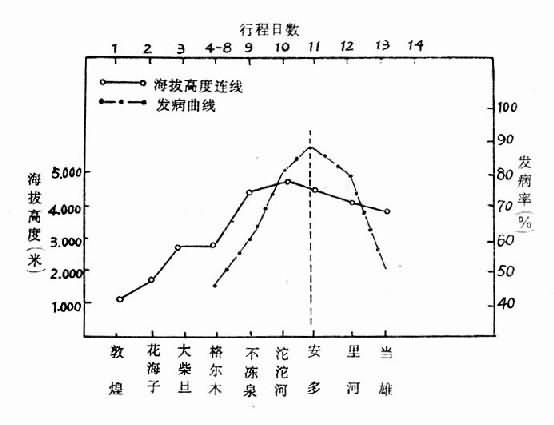
图3-6 高原反应发病率与行程
─·海拔高度·─·─·发病率
表3-3 在海平地区呼吸过程中各部分气体分压变化
| 气体分压(kPa) | 大气 | 吸入气 | 肺泡气 | 呼出气 | 动脉血 | 混合静脉血 |
| 氮 | 79.40 | 74.93 | 75.68 | 75.28 | 76.21 | 76.21 |
| 氧 | 21.15 | 19.86 | 13.83 | 15.96 | 13.30 | 5.32 |
| 二氧化碳 | 0.04 | 0.04 | 5.32 | 3.59 | 5.32 | 6.12 |
| 水蒸气 | 0.49 | 6.25 | 6.25 | 6.25 | 6.25 | 6.25 |
| 总 量 | 101.08 | 101.08 | 101.08 | 101.08 | 101.08 | 93.90 |
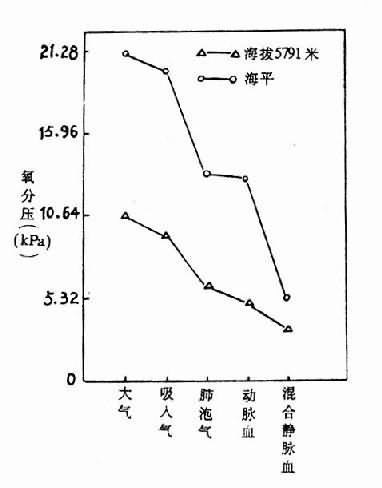
图3-7 在海平和5791米高度时氧分压的变化
表3-4 在不同高度每分通气量和呼吸频率的变化
| 高度(米) | 每分通气量(升) | 每分呼吸次数 |
| 海平地区 | 8.85 | 12 |
| 3658 | 9.71 | 12 |
| 5486 | 11.06 | 12 |
| 6706 | 15.31 | 15 |
发生高原反应的人,常常对低氧环境代偿反应较弱,没有明显的呼吸加深,因此每分通气量增加不多。由于这些人代偿适应反应弱,因此登高到一定高度,缺氧症状比较严重。目前有人认为,可用减压舱测定人群每分通气量变化,作为预测人群地高度适应的能力。
呼吸加深,可提高肺泡膜弥散功能,使肺泡氧进入肺泡壁毛细血管增加。我国健康成人静息时氧的弥散量为23毫升/0.133千帕(1毫米汞柱)/分,每分钟约有250毫升的氧弥弥散入血,供组织代谢需要。缺氧时,由于呼吸加强,参与气体交换的肺泡数增多和肺泡容量增加,同时开放的肺毛细血管数也增多和毛细血管扩张,这样就增加了肺泡—毛细血管膜弥散面积,血液从肺泡摄取较多的氧。如果这种代偿能力弱,可促使高原反应发生。
缺氧时,向组织输送氧的能力提高。正常人在海平地区安静状态下,平均每分种100毫升血给组织输送约为5毫升氧,正常心脏每分输出量约为5000毫升,那么每分钟就有250毫升氧输送给组织。在高原地区,一方面心脏活动增强,每分输出量增加,组织血流量增多;另一方面,红细胞数和血红蛋白增多,增强携带氧的能力。这样在一定限度内可以补偿组织代谢对氧的需要。如果这种代偿适应能力降低或尚未建立起来,也易发生高原反应。
初进高原,由于呼吸加强,二氧化碳排出过多,还可引起低碳酸血症和呼吸硷中毒。PaCO2降低,可引起脑血管收缩,部分抵销缺氧引起脑血管扩张的反应,容易发生意识丧失。
(二)高原昏迷
进入高原地区较高高度,有少数人可发生昏迷。在昏迷发生前,常有头痛、头晕、呕吐等症状。昏迷发生后常出现阵发性抽搐,大小便失禁,病人瞳孔常缩小而固定,或忽大忽小;少数病例有肢体强直或肢体弛缓性瘫痪。1/5的病例眼底有小动脉痉挛,静脉扩张,视网膜乳头水肿。
高原昏迷常在3500米以上的高度发生。根据67例发病高度统计,3500~4000米发病11.9%,4000~4500米发病28.4%,4500~5110米发病59.7%。一般进入高原后1~10天内发病。
高原昏迷发生的机理,主要是由于缺氧(见缺氧时中枢神经系统的变化)。早期可能由于缺氧,氧化过程障碍,能量供应减少,而致脑细胞功能降低。正常脑内ATP贮存量很少。ATP是推动“钠泵”的能源。ATP减少。“钠泵”作用减弱,则钾与钠离子在细胞内、外的浓度差减小,膜电位降低。人吸入含8%氧的混合气体3~5分钟,脑电就出现每分钟2~7次的慢波。高山运动员在减压舱内,当减压到相当于9000米高度大气压力时,脑电α波减少,慢波增多,出现神经细胞功能抑制现象。
缺氧进一步加重,ATP形成更加减少,不能维持细胞内外离子浓度差,细胞内钠离子增多,氯也进入细胞内,水随之进入增多,发生细胞内水肿。大白鼠实验性缺氧,有4~6%的大白鼠发生细胞内水肿。1962年印度士兵急性高山病死亡率病例中,3/4有脑水肿病变。
除神经细胞水肿外,神经胶质细胞和血管内皮细胞也发生水肿。毛细血管和小静脉周围神经胶质细胞水肿,可以压迫血管;血管内皮细胞水肿,有的呈疱疹状向管腔突出或脱落,使血管腔狭窄或堵塞,造成脑微循环障碍,缺氧和脑水肿进一步加重。
严重缺氧,还可引起呼吸中枢兴奋性降低,二氧化碳排出减少,动脉血二氧化碳分压升高。二氧化碳分压升高和缺氧,可使脑血管失去自家调节,而引起脑血管扩张,血流量增加,毛细血管流体静压和通透性升高,组织间液和脑脊液生成增多。有的实验证明,动脉血二氧化碳分压升高到6.65kPa(50mmHg),脑脊液平均增加55%,脑脊液压力由正常0.69~1.78kPa(70~180毫米水柱)升高到2.82kPa(285毫米水柱)。在西藏地区高原昏迷病人,有人测定过10例脑脊液,其中7例脑脊液压力升高。
脑水肿(包括细胞水肿和间质水肿)和脑脊液增多,使颅内压升高超过3.96kPa(400毫米水柱),可压迫脑的小血管,动脉血灌流明显减少。高原昏迷的许多临床症状是由于缺氧和颅内压升高所引起的。
(三)高原肺水肿
高原肺水肿发病率较高。一般由海平地区初入高原或重返高原一周内发病。据218例高原肺水肿发病与高度的关系统计,3500~4000米,占28.3%;4000~4500米,占34.4%;4500~5100米,占37.3%。
高原肺水肿发病急骤,临床症状除有一般高原反应症状外,所有患者均有不同程度的咳嗽,开始为干咳或有少量痰,以后即咳出均匀混合、稀薄的粉红色或白色泡沫痰。呼吸急促,有时每分钟高达30~40次。病人惊恐不安,心慌、胸闷、紫绀、两肺满布湿罗音。
高原肺水肿的主要病理变化是广泛的呈片块状分布的肺泡水肿,偶而可见透明膜形成(这是肺泡水肿液中的纤维蛋白沉积所致)。肺水肿最早在血管周围间隙形成(间质水肿),当这些间隙扩张,压力超过了肺泡压,则液体进入肺泡。微动脉和毛细血管充血,偶而可见血管周围有出血。
高原肺水肿发生机理目前还不清楚。以往推测可能是肺静脉压升高,肺毛细血管通透性增加。近年来应用心导管直接测定肺动脉压、楔压(wedge pressure)、肺静脉压和左心房压力,发现高原肺水肿病人动脉压升高,楔压正常或降低,肺静脉压和左心房压力正常。因此可以排除左心衰竭或肺静脉收缩而引起高原肺水肿的论点。
高原性缺氧引起肺动脉压升高,已为许多研究所证实,随着高度增加,肺动脉压也增加(图3-8)。在单纯性缺氧,肺泡氧分压降到7.98kPa(60mmHg)以下,也引起肺动脉压升高。曾进高原患过高原的肺水的人,肺动脉对缺氧敏感性升高,则很快发生肺动脉高压。Hultgren观察了5例曾患过高原肺水肿的人,在海平地区,安静时的肺动脉压平均为1.84kPa(13.8mmHg),肺泡气氧分压降到5.99kPa(45mmHg),肺动脉压平均为3.06kPa(23mmHg),有两例高出正常的范围(图3-9)。高原肺水肿的发生与肺动脉压升高有关,但是肺动脉压升高是如何引起肺水肿的,现在还不清楚。有人推测肺动脉收缩是区域性的,高山缺氧时可能引起一部分肺动脉收缩,另一些小动脉尚未收缩,则肺的血流大量进入肺动脉尚未收缩的区域,因此这些局部的微循环中血量增多,流体静压增加,液体向血管周围间隙转移增多,当组织间液生成增多超过淋巴液回流量,则引起肺水肿。所以高原肺水肿在X线下看到的往往呈片块状分布。
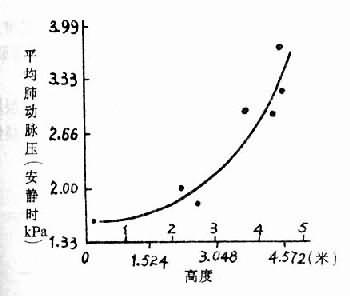
图3-8 不同高度肺动脉压的变化
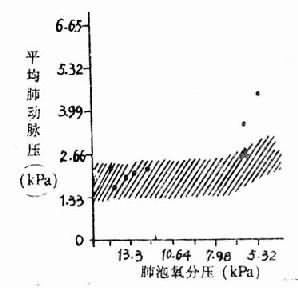
图3-9 人缺氧时肺动脉压的变化
肺微循环有旁路,从小动脉发出分枝直接进入毛细血管静脉端(图3-10),称为前终末微动脉(preterminal arteriole),正常是闭合的。当肺微动脉收缩时,则此旁路开放,血液从小动脉直接进入毛细血管静脉端,血量增加,压力增高,液体渗出增多。由于这些血液没有进行气体交换,故动脉血氧分压急剧下降。
高山缺氧还引起外周静脉收缩,回心血量增加,肺血量增加,Wood测定前臂肢体容积,推测静脉容量。在海平地区前臂肢体容积测定溢出水量平均为4.4毫升;登山到3596米时,第三下降到3.4毫升,表明有静脉收缩。而高原肺水肿病人降到2.5毫升。周围静脉收缩,中心血量增多,可能是促进肺水肿发生的一个因素。
此外,缺氧可使肺毛细血管通透性升高,也是引起肺水肿的重要因素。
四、慢性高原适应不全症
慢性高原适应不全症包括高原心脏病、高原血压或低血压和高原红细胞增多症。有的是高原心脏病、高原高血压和高原红细胞增多症三型中有两型以上并存的混合型。
(一)高原心脏病
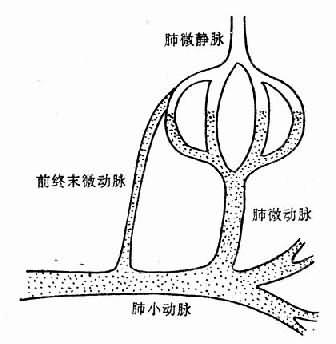
图3-10 肺小动脉与肺毛细血管静脉端的交通支—前 终末微动脉
久居高原地区的人,有少数(特别是儿童)逐渐出现心悸、气喘、胸闷、浮肿、右心室增大等现象,可考虑有高原心脏病的可能。X线检查:肺动脉圆锥突出,右肺下动脉增宽。心电图图形;右心肥厚,电轴右偏,有时出现肺型P波。
高原心脏病的发生,多数人认为是由于高原缺氧,肺小动脉持续收缩,引起肺小动脉肌层肥厚,管壁增厚,管腔狭窄,阻力增加,而使肺动脉压持续升高(表3-5),加重右心室负担。慢性缺氧,还使红细胞生成增多,血液粘滞性增加,又加重心脏的负担。
表3-5 慢性高原适应不全症人与高原居民右心和肺动脉压比较
| 慢性高原适应不全症(Monge氏病) | 高原世居者 | 海平正常人 | |
| 右心房平均压(kPa) | 0.52(3.9mmHg) | 0.39(2.9mmHg) | 0.35(2.6mmHg) |
| 右心室平均压(kPa) | 3.86(29mmHg) | 2.00(15mmHg) | 1.20(9mmHg) |
| 肺动脉(收缩压、舒张压、平均压)kPa | 8.51(64mmHg) | 4.52(34mmHg) | 2.93(9mmHg) |
| 4.39(33mmHg) | 1.73(13mmHg) | 0.80(6mmHg) | |
| 6.25(47mmHg) | 3.06(23mmHg) | 1.60(12mmHg) | |
| 肺动脉楔压(kPa) | 0.76(5.7mmHg) | 0.92(6.9mmHg) | 0.82(6.2mmHg) |
有人比较高原习服的人和高原心脏病患者的右心室功和心脏指数,证明高原心脏病患者心脏负担加重(表3-6)。
表3-6 高原习服的人和高原心脏病患者的右心室功
和心脏指数比较(五例的平均数)
| 高原习服者 | 高原心脏病者 | |
| 心脏指数(升/分/米2) | 3.83 | 4.01 |
| 右心室功(公斤-米/分/米2) | 1.08 | 2.47 |
长期肺动脉压升高,心脏负担加重,将引起右心室代偿性肥大,最后可发展为右心衰竭。但也有少数病例是以右心衰竭为主的全心衰竭。
(二)高原高血压和高原低血压
进入高原以后,部分人发生头痛、头昏、失眠等症状,血压持续在18.62/11.97kPa(140/90毫米汞柱)以上,返回平原,血压又恢复,称为高原低高血压。高原高血压的特点是以舒张压升高为主,超过11.97kPa(90毫米柱),收缩压略有升高或在正常范围内,因而脉压变小。
高原高血压的发生机理,可能是由于缺氧使大脑皮层对皮层下枢的调节功能减弱,血管运动中枢兴奋性升高,通过交感神经兴奋和肾上腺素分泌增多,引起小动脉收缩;另外,红细胞增多,血液粘滞性升高,也是引起外周阻力增加的一个因素。
进入高原后,还有少数人血压降到11.97/7.98kPa(90/60毫米汞柱)以下,并伴有头昏、眩晕、乏力等症状,称为高原低血压。高原低血压以收缩压降低为主。其发生机理不清楚,可能因缺氧,植物神经功能紊乱,迷走神经张力增加,引起心动缓慢和外周阻力降低;也有人认为,缺氧通过某些生理活动性物质的作用,使小动脉平滑肌紧张性降低。
(三)高原红细胞增多症
进入高原后,红细胞数超过650万/立方毫米,血红蛋白超过20克%,血红细胞压积超过65%,并伴有紫绀、头痛、头昏、乏力、呼吸困难等,称为高原红细胞增多症。
红细胞和血红蛋白增多,是机体对缺氧的代偿适应性变化,进入高原后可以很快出现,以进入高原后6~14天最明显,一般持续6~8个月,开始血量不增加,以后血量有所增加(图3-11)。组织缺氧,肾脏产生促红细胞生成因子(erythrogenin)增多,使血浆中促红细胞生成素原(erythropoietinogen)变为促红细胞生成素(erythropoietin),刺激骨髓红细胞生成增多,结果单位容积血液内红细胞数和血红蛋白量增加。
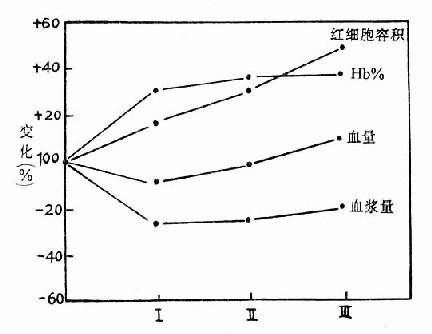
图3-11 登山以后不同时期血液的变化
Ⅰ在4000和5791米处停留8周后
Ⅱ在5791米处停留3~6周后
Ⅲ在5791米处停留9~14周后
高原红细胞增多症的发生机理,可能是:
1、在慢性缺氧条件下,中枢化学感受器对二氧化碳的敏感性和主动脉体与颈动脉体对缺氧的敏感性降低,不能充分发挥呼吸的代偿功能,肺泡通气量增加不明显,肺泡氧分压降低。
2、血中二氧化碳分压和红细胞内2,3-二磷酸甘油酸高于同一高度的健康居民,氧离曲线右移(图3-11),血红蛋白对氧亲和力降低,血液从肺摄取的氧减少,更加重缺氧,使得红细胞生成更多。
第四章 发热
第一节 发热的概念
发热(fever)是临床常见的疾病症状之一,也是许多疾病所共有的病理过程。临床上常把体温上升超过正常值的0.5℃,通称为发热,这种概念不够精确。许多情况可使体温超出正常0.5℃,但其本质并非发热。根据体温调节调定点的理论,发热是在致热原的作用下使体温调节中枢的调定点上移而引起的调节性体温升高。多数病理性体温升高(如传染性或炎症性发热)均属这样。但少数病理性体温升高是因体温调节机构失调控或调节障碍而产生,其本质不同于发热,应称之为过热(hyperthermia)。如皮肤有广泛鱼鳞癣或是先天性汗腺缺陷,因散热障碍,夏季可出现体温升高;甲状腺机能亢进造成异常产热而致体温升高,以及环境高温(中暑)引起的体温升高,均属此类。
此外,在剧烈运动时,妇女月经前期、妊娠期等体温也可上升高于0.5℃,但它们属于生理性体温升高,也不宜称为发热。
现把这些概念归纳如图4-1。
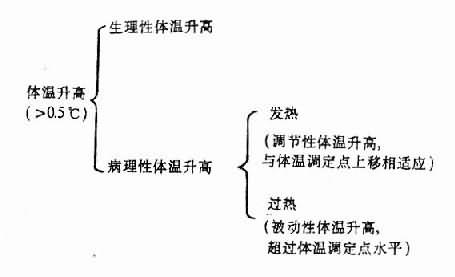
图4-1体温升高的分类
发热通常不是独立疾病,而是发热性疾病的重要病理过程和临床表现。体温升高不超过38℃为低热;38-39℃为中等热;39-40℃为高热;超过41℃为过高热。许多疾病常是由于早期出现发热而被察觉的,因而它是疾病的重要信号,甚至是潜在恶性病灶(肿瘤)的信号。在整个病程中,体温曲线变化往往反映病情变化,对判断病情、评价疗效和估计预后。均有重要参考价值。
第二节 发热的原因和机制
一、致热原和激活物的概念
传统上把能引起人体或动物发热的物质,通称为致热原(pyrogen)。根据来源又把致热原划分为外源性致热原和内生致热原,用以表示来自体外或体内。近年来不少学者认为,许多外源性致热原(传染原或致炎剌激物)可能主要是激活产内生致热原细胞,使后者产生和释放内生致热原,再通过某种途径引起发热。因此,外源性致热原用乃是体内产生内生致热原细胞的激活物(activators),或称为发热激活物。此概念并不排除一些外源性致热原与机体相作用,在体内引起激活物的产生,因而体内某些产物,也可成为产内生致热原细胞的激活物。当然也不排除有些激活物或其成分,如能通过血脑屏障,也可能以一定方式作用于体温调节中枢,而发挥双重作用(即既可促使内生致热原的产生,又可作用于中枢),或还可能通过内生致热原以外的中介物从外周进入脑内,参与发热的机制。
在第一种内生致热原(白细胞致热原)被发现后,最近几年又相继发现三种新的内生致热原,这些新发现的内生致热原还来不及系统深入的研究,而白细胞致热原则已积累大量系统研究资料。关于发热激活物的系列资料,都是围绕白细胞致热原所进行的实验研究所取得的。
二、发热激活物的主要种类和性质
有许多物质(包括外源性致热原和体内某些产物)能够激活产内生致热原细胞而使其产生和释放白细胞致热原。下面介绍的仅是几种常见或重要的激活物。
(一)微生物
革兰氏阴性细菌的菌壁含有内毒素(endotoxin,ET),后者是一种有代表性的细菌致热原(bacterial pyrogen)。给家兔微量静脉内或更微量脑内(视前区-前下丘脑)注射,均可引起明显发热。ET的活性成分是脂多糖,它有三个组成部分,即O-特异侧链、核心多糖和脂质A(lipid A)。脂质A是决定致热性的主要成分。
临床上输液或输血过程中所产生的发热反应,多数就是由于污染ET所致,因其耐热性很高,需干热160℃2小时才能灭活,一般灭菌方法不能清除。目前多数学者认为,ET性发热是由于ET激活了产内生致热原细胞,使其释放白细胞致热原所致体外实验证明。用微量ET与白细胞培育,可使后者产生释放白细胞致热原;给家兔或狗静脉内注射ET,在引起发热的同时,血清中出现大量循环白细胞致热原。最近的一些研究证明,ET还能激活单核细胞产生其它内生致热原。此外,ET在外周还可能引起其它代学介质的生成,后者经血脑屏障进入脑内而参与中枢机制。但出不能完全排除ET本身或其降解产物进入脑内发挥致热作用的可能性。ET的分子量很大,达1,000-2,000KD,一般剂量静脉内注射,显然难以通过血脑屏障并进入脑内。但是,大剂量注射ET,有无可能削弱血脑屏障而致小量ET通过,或者由于某些生理过程(包括传染、毒血症或高热)提高了脑毛细血管的通透性,导致ET或其降解产物得以自由通过,这个可能性还不能完全排除。有的学者报道,单独注射ET不能通过血脑屏障,但联合使用A型链球菌致热性外毒素时,ET就能够通过血脑屏障。
革兰氏阳性细菌(如肺炎球菌、白色葡萄球菌、溶血性链球菌等)感染也能引起发热。给家兔静脉内注射活的或加热杀死的葡萄球菌,均能引起发热,因而其效应不取决于传染是否成立,而可能是细菌颗粒本身起的作用。加热杀死的葡萄球菌在体外与白细胞培育,能激活产内生致热原细胞,使其产生释放白细胞致热原。在剂量—效应关系上,取决于细菌颗粒数与细胞数的比例。
从革兰氏阳性细菌体内能分离出有致热性的外毒素,例如从葡萄球菌分离出的肠毒素,和从A型溶血性链球菌分离出的红疹毒素(erythrogenic toxin),都是强激活物,微量给动物静脉内注射即可引起发热。体外实验证明,红疹毒素与家兔白细胞培养,能使后者产生释放白细胞致热原。
病毒感染,例如把流感病毒、麻疹病毒或者Coxsackie病毒注入家兔静脉内,都可引起动物发热。在发热的同时,血清中出现循环白细胞致热原。实验证明,病毒也可能过激活产内生致热原细胞产生释放白细胞致热原引起发热,其激活作用可能与血细胞凝集素(hemagglutinin)有关。在体外用副流感病毒与家兔血白细胞培育,能激活后者释放白细胞致热原。
此外,螺旋体(回归热的病原体疏螺旋体属Borrelia,钩端螺旋体等)及真菌引入体内也可引起发热。在体外把赫姆斯氏包柔氏螺旋体(Borrelia hermsii)或酵母分别与人体白细胞培育,都能激活后者产生释放白细胞致热原。
(二)致炎物和炎症灶激活物
有些致炎物如硅酸结晶,尿酸结晶等,在体内不但可引起炎症反应,其本身还具有激活产内生致热原细胞的作用,并已证明,尿酸结晶或硅酸结晶的激活作用,不取决于细胞对它们的吞噬,因为用细胞松弛素B或秋水仙素制止吞噬,不影响白细胞致热原的产生和释放。
除某些非传染性致炎物以及传染原有激活作用之外,非传染性炎性渗出液中还含有激活物。实验证明,给家兔腹腔灌注生理盐水后,把从腹腔收集到的渗出白细胞置于生理盐水中培育。能释放白细胞致热原;但把正常血白细胞置于生理盐水中培育,则无白细胞致热原释放。若先加入从腹腔收集到的无细胞渗出液到正常血白细胞中培育,然后再置于生理盐水中培育,则血白细胞也能释放白细胞致热原,表明在渗出液中含有激活物。
(三)抗原-抗体复合物。
抗原-抗体复合物对产内生致热原细胞也有激活作用,举下述实验为证;
皮内注射青霉素-佐剂乳状物或静脉内注射水溶性青霉素或肌肉内注射普鲁卡因青霉素,使家兔致敏,然后给这种致敏兔静脉内注入青霉素-血清蛋白结合物,可引起动物发热;在有致敏兔的含抗体血清的参与下,把致敏兔的血细胞与上述结合物作体外培育,能释放白细胞致热原,表明抗原-抗体复合物起了激活作用。用牛血清蛋白使家兔致敏。然后把致敏动物的血浆或血清转移给正常家兔,再用特异抗原攻击受血动物,可以引起后者发热。但牛血清蛋白对正常家兔却无致热作用,表明是由于抗原-抗体复合物起了激活作用。已证明,用人体血清蛋白给家兔致敏后,再用人体血清蛋白攻击,约5分钟后就可在循环血中出现抗原-抗体复合物。
(四)淋巴因子
淋巴细胞不产生和释放内生致热原,但抗原或外凝集素能剌激淋巴细胞产生淋巴因子(lymphokine)后者对产内生致热原细胞有激活作用。实验证明,用卡介苗(bacille Calmette-Guerin,BCG)给家兔致敏,然后用旧结核菌素攻击可引起发热。这种反应可通过致敏的脾和淋巴结细胞被动转移给正常家兔。把致敏或未致敏的家兔血白细胞在体外与特异抗原培育时,不能释放白细胞致热原,如果同时加入致敏的淋巴细胞一起培育,则能使白细胞释放白细胞致热原。这是因为致敏淋巴细胞-抗原混合物所形成的一种可溶性产物起激活作用,这种产物就是淋巴因子,它可能主要来自T淋巴细胞。
(五)类固醇
体内某些类固醇(steroid)产物对人体有明显的致热性,睾丸酮的中间代谢产物本胆烷醇酮(etiocholanolone)是其典型代表。石胆酸也有类似作用。但实验证明,本胆烷醇酮的种系特异性很强,给狗、猫、大鼠、小鼠、豚鼠、家兔和猴作肌肉内注射,均不引起发热,只有当给人体肌肉内注射时,才引起明显发热。体外实验证明,人体血白细胞与本胆烷醇酮培育,经几小时激活能产生释放白细胞致热原。
已证明,本胆烷醇酮的致热性取决于类固醇的5-β-H构型,因为5-α-本胆烷醇酮不具致热性。同样,它在体外对白细胞的激活作用,也取决于5-β-H构型。
某些周期性发热病人,常找不到发热的原因,而血浆中的本胆烷醇酮的浓度有所增高。另一种类固醇,如糖皮质激素和雌激素,则能抑制白细胞致热原的产生释放。因此有人用类固醇代谢失调来解释某些周期性发热。例如一些肝硬变的发热病人,伴有血浆中本胆烷醇酮浓度升高,被怀疑是类固醇代谢失调所致,但仍有争议。
三、内生致热原
(一)白细胞致热原
1.细胞来源 1984年Beeson等首先发现家兔腹腔无菌性渗出白细胞培育于无菌生理盐液中,能产生释放致热原,并称之为白细胞致热原(leucocytic pyrogen,LP)。为表示其来自体内,又称之为内生致热原(endogenous pyrogen,EP)。现在已经证明,白细胞中的单核细胞是产生LP的主要细胞。此外,组织巨噬细胞,包括肝星状细胞、肺泡巨噬细胞、腹腔巨噬细胞和脾巨噬细胞等,以及某些肿瘤细胞,均可产生并释放LP。
近年来对LP的系统研究中,发现它除引起发热外,还引起许多疾病急性期反应,表明其生物活性与白细胞介素-1(interleukin-1,IL-1)一致,现已公认LP就是IL-1。由于从不同侧面研究IL-1或LP,对它的细胞来源有了更多了解。根据目前所知,能产生IL-1(LP)的细胞种类,可列表如下(表4-1)。
表4-1 LP(IL-1)的细胞来源
| 巨噬细胞 | 肿瘤细胞 | 其它细胞 |
| 血单核细胞 | 骨髓单核细胞性肿瘤细胞 | 表皮角化细胞 |
| 肺泡巨噬细胞 | 白血病细胞 | 郎罕氏细胞 |
| 肝星状细胞 | 何杰金氏淋巴肉瘤细胞 | 角膜上皮细胞 |
| 脾巨噬细胞 | 牙龈渗出细胞 | |
| 腹腔巨噬细胞 | 肾细胞癌细胞 | 神经胶质细胞 |
| 滑膜巨噬细胞 | 肾小球膜细胞 | |
| 骨髓巨噬细胞 |
2.产生和释放 产LP细胞如何产生和释放LP,根据对人体白细胞体外培育实验的观察,这一过程包括三个阶段,即激活、产生和释放。
激活过程可能从激活物的有效成分与产LP细胞膜的特异受体结合开始,然后发生吞噬(以及消化细菌)。此时细胞产生一系列的代谢反应,包括耗氧量增多、糖酵解增强,以及各种水解酶的释放等。曾有人认为激活过程就是吞噬过程,但进一步的研究表明,事实不尽如此,当吞噬被事先制止时,某些激活物颗粒仍然能发挥激活作用。
一般在激活后(指体外培育)1~2小时,可能是LP生成的初期。在此期,事先加入到培养基中的同位素标记氨基酸能掺入到新生成的LP中;若加入蛋白质和核糖核酸的合成抑制物,则可抑制LP的生成,表明此期需要有新合成的核糖核酸(mRNA)和蛋白质来保证。已证明LP合成是需能过程。给氟化钠能阻止合成。
过了此期,即在激活2小时之后,似乎不再需要蛋白质的新合成了。因为在此之后加入蛋白质合成抑制物,已不再影响LP的生成和释放了。
血白细胞合成的LP,在3~16小时内释放。在此期LP可能由非活化型经酶的作用,转化为活化型。不像合成是需能的,释放LP是不需能的过程;阻断细胞呼吸不干扰LP的释出只有细胞死亡或破裂才中止释放。因此LP可能是通过细胞膜而释出的。
3.化学性质据目前所知,LP大致是一种较小分子的蛋白质,共耐热性低,加热70℃20分钟即可破坏其致热活性。蛋白酶如胃蛋白酶、胰蛋白酶或链霉蛋白酶,都能破坏其致热性。
LP在体内的动力学尚欠系统研究,但已知它主要由肾脏清除。
LP的分子量各家报道不一,多数认为人或兔的LP约13~15KD;也有人报道,人体提纯的LP有15KD和38KD两种,后者同样不耐热,也可为胰蛋白酶所破坏。后来的资料表明,较大分子的LP可能是分子量为15KD的LP的三聚体。据报道,人体和小鼠肿瘤细胞也可释出不同分子量的LP,可能是二聚体或三聚体。
LP的等电点有两型,即p17和p15这两型都有相同的致热性和其它生物活性。
4.抗原性和致热性交叉反应LP表现高度的抗原特异性。在兔体内对人体LP产生的抗体,只能破坏人体LP的致热性,而对家兔、豚鼠和猴的LP的致热性不能破坏。根据这种抗原特异性,有的实验室已建立对人体LP的放射免疫检测方法。
虽然LP有高度抗原特异性,但其致热性则在某些种系动物中可呈交叉反应。例如人体LP可引起家兔或小鼠发热;家兔LP能引起蜥蜴发热,大鼠LP可引起家兔发热。这种交叉致热性表明,上述不同种系动物产生的LP,必然有共同的有效部分,能为其靶细胞的特异受体所接受。
5.生物学效应 LP有明显的致热性。从家兔渗出白细胞制备的LP,其粗提物的致热性约为300~400μg蛋白质引起家兔(约2.5~3.0kg体重)平均发热1℃;经滤膜过滤和层析滤过而获得的较纯的LP,致热性约为0.1~0.2μg引起家兔发热1℃。表明LP的致热性很强。
除发热效应之外,LP还能引起疾病急性期的多种反应,包括中性粒细胞增多、低铁血症、低锌血症、高铜血症、肝脏急性期蛋白合成增多。后者包括纤维蛋白原、结合珠蛋白、血浆铜蓝蛋白、C-反应蛋白、α1-抗胰蛋白酶、血清淀粉样物质A及某些补体成分等。与此同时还出现肌肉蛋白水解增多和氨基酸血症,以保证急性期蛋白合成增多的需要。因此,LP是疾病急性期反应的一种中介分子或系列中介分子之一。
(二)新发现的内生致热原
除LP外,近年来又发现三种内生致热原。
1.干扰素干扰素(interferon,IFN)是细胞对病毒感染的反应产物,这种糖蛋白物质去糖后仍具活性。由人类白细胞诱生的hIFN,已应用于临床,有抗病毒,抑制细胞尤其肿瘤细胞生长的作用。1984年Dinarello等证明,给家兔静脉内注射hIFN。能引起单相热,其致热性不是由于污染ET。对ET产生耐受性的小鼠,注射hIFN仍引起发热且不减弱;其致热性也不是由于LP的作用。在家兔hIFN性发热期间,循环血未出现LP;体外培育单核细胞加入适量hIFN不引起LP的释放。给猫脑室内(intracerebroventricular,ICV)注射hIFN照例引起发热,表明它本身具有致热性。
2.肿瘤坏死因子肿瘤坏死因子(tumor necrosis factor,TNF)也是巨噬细胞分泌的一种蛋白质,ET能诱生之。重组TNF(rTNF)已用于临床1期治疗肿瘤,有非特异杀伤肿瘤细胞的作用,给人注射能引起发热反应。Dinarello等(1986)用家兔实验验证其致热性:静脉内注射1μg/kg迅速引起单相热,10μg/kg引起双相热,在第二热相血浆中出现循环LP。体外实验证明。rTNF能激活单核细胞产生LP。TNF在70℃中加热30分钟,失去致热性50%。加热的10μg/kg只引起单相热。但LP加热70℃30分钟,则失去全部致热性。TNF不同于ET,每天注射不出现耐受性。Dinarello等认为TNF双相热的第一热峰是TNF直接作用于体温调节中枢所致,第二热相是通过LP而引起的。
3.巨噬细胞炎症蛋白-1 最近Wolpe等(1988)新发现一种单核细胞因子,是一种肝素-结合蛋白质,对人体多形核白细胞有化学促活作用(chemokinesis),在体外能引起中性粒细胞产生H2O2,皮下注射此因子能引起炎症反应,故称之为巨噬细胞炎症蛋白-1(macrophageinflammatory protein-1,MIP-1)。进一步研究(Davatelis等,1989)发现,MIP-1给家兔静脉内注射引起剂量依赖性发热反应,热型呈单相。其致热性既不是由于污染ET,也不是由于含有LP或TNF,也不依赖于PGE,表明它是另一种具有致热性的EP。
四、致热原的作用部位
哺乳类动物和人类的体温相对恒定,是依赖体温调节中枢调控产热和散热的平衡来维持的。视前区-前下脑(preopticanterior hypothalamus,POAH)是体温调节中枢的高级部分,次级部分是延脑、桥脑、中脑和脊髓等。当POAH进行正常活动时,次级中枢退居次要或备用地位。而当POAH失去活动(如被病灶或人工破坏)时,次级中枢可能取代之而发挥积极作用。无论对体温调节或致热原的反应,可能都是如此。
至于致热原(包括ET或LP)的作用部位,迄今尚难确定。许多实验证明,在脑内存在着对ET或LP起反应的敏感区。用直接微量注射的方法显示,这种敏感区正好集中于下丘脑体温调节中枢,其它中枢部位的敏感性较低或不敏感。因此,只要有小量ET或LP通过血脑屏障进入脑内,就有可能作用于敏感区而引起发热效应。目前还未有证据可以表明,ET或LP能作用于外周温度感受器或其它外周调温结构而引起发热。
由于ET的分子量很大,LP的分子量较小,因此多数学者认为,循环ET不能通过血脑屏障而作用于POAH,LP则能通过血脑屏障而作用于POAH。其实至今对此仍不能最后肯定或否定。关于ET能否通过血脑屏障,前文已有论述。关于LP,近年来有的学者提出其作用部位可能位于血脑屏障外的脑血管区。这个特殊部位,称为下丘脑终板血管器(organum vasculosum laminae terminalis,OVLT)位于第三脑室壁的视上隐窝处(图4-2)。这里的毛细血管属于有孔毛细血管,LP可能通过这种毛细血管而作用于血管外周间隙中的巨噬细胞,由后者释放介质再作用于OVLT区神通元(与POAH相联系)或弥散通过室管膜血脑屏障的紧密连接,而作用于POAH的神经元。这种主张也有待进一步验证。
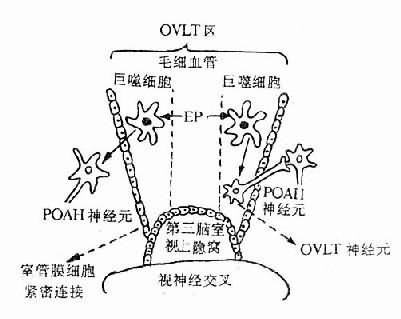
图4-2 OVLT在发热病学中的作用示意图(引自Stitt,1986)
近年来新发现的三种内生致热原的作用部位尚待确定。
五、内生致热原的作用方式
无论EP是否通过血脑屏障,它在给动物静脉内注射后,总要经过一段潜伏期才引起发热。因而它很可能要通过某种或多个中间环节,导致调定点上移,再通过调温反应而引起发热。

许多学者推测有某种或某些中枢介质(也称中枢发热介质)参与发热的中枢机制。先后被研究的有单胺(去甲肾上腺素、5-羟色胺)、前列腺素E(PGE)、花生四烯酸的其它衍生物,cAMP和Na+/Ca2+比值等。而最受重视的是PGE、cAMP和Na+/Ca2+比值。
(一)前列腺素
主张PGE是EP引起发热的主要介质的现行假说的最重要依据是:①脑内(下丘脑)或ICV注射PGE引起发热;②LP静脉内注射或IFn ICV注射引起发热是时,CSF中PGE2明显增多;③下脑组织分别与LP、IFN或TNF在体外培育时,都使PGE2合成增多;④阻断PGE合成的药物,对LP、IFN或TNF性发热都能解热。
鉴于目前仍无直接证据以示LP能从外周进入脑内,因而有的学者修改了此假说,提出PGE的释放部位是在OVLT区孔性毛细血管外周的巨噬细胞(参阅图4-2)。LP激活后者释出PGE,作用于OVLT区的神经元或弥散过室管膜细胞紧密连接而作用于POAH的神经元。
此外,Dinarello等(1986)认为TNF引起的双相热的第一相,是TNF引起下丘脑PGE增多的效应。
但是许多资料不支持PGE作为发热介质,其根据是:(1)PGE的两种特异拮抗物SC19220和HR546能抑止PGE性发热,但不能抑制LP性发热;(2)小剂量阿司匹林在抑制LP引起的CSF PGE增多的同时,可不抑制体温上升;(3)家兔两侧POAH摘除或损伤后,向该处或ICV注入PGE均不引起发热,但ICV注入LP仍能引起发热,表明不需PGE参与;(4)LP注入家兔POAH,使大部分热敏神经元敏感性受抑制,大部分冷敏神经元的敏感性提高,但PGE注入POAH,大部分热敏神经元不受影响,约1/2冷敏神经元也不受影响;(5)MIP-1的致热性与PGE无关。
因此,目前还难肯定PGE是EP性发热的主要介质。
(二)cAMP
脑内有较高cAMP,也有丰富的cAMP合成降解酶系。它又是脑内多种介质的信使和突触传递的重要介质,故当PGE作为发热介质有争议的同时,cAMP能否作为发热介质参与中枢机制,倍受重视。十多年前国外学者积累了一些资料,支持cAMP参与发热中枢机制,主要是:①把二丁酰cAMP给猫、兔、大鼠脑内注射,迅速引起发热;②家兔静脉内注射LP引起发热时,CSF中cAMP浓度明显增高,而环境高温引起的体温升高,不伴有CSF中cAMP增多。③注射茶碱(磷酸二酯酶抑制物)在增高脑内cAMP浓度的同时,增强LP性发热;而注射尼克酸(磷酸二酯酶激活物)则在降低cAMP浓度的同时,使LP性发热减弱。
至于LP如何引起脑内cAMP增多,最新研究资料表明,LP可能通过提高Na+/Ca2+比值,再引起脑内cAMP增多。
(三)Na+/Ca2+比值
实验表明,用生理盐水替换人工脑脊液作动物脑室灌注时,引起了猫的体温明显上升,而加入CaCl2则可防止体温上升。用等渗蔗糖溶液灌注后下丘脑,体温无变化;若加入Na+,就引起体温上升;若加入Ca2+,则可降温。因而提出体温调定点受Na+/Ca2+比值所调控,强调Ca2+浓度是调定点的生理学基础,Na+/Ca2+比值上升可致调定点上移,并确定其敏感区位于后下丘脑。
进一步实验证明,静脉内注射LP引起发热时,增加灌注脑室的人工脑脊液中的Ca2+浓度,可抑制发热效应。若把灌注液改为等渗蔗糖溶液,则静脉内注射LP不引起发热,表明LP可能通过提高下丘脑Na+/Ca2+比值,使调定点上移而启动调温反应,引起体温上升。在应用放射性同位素钠和钙的实验中发现,发热时下丘脑组织内Na+/Ca2+比值上升。
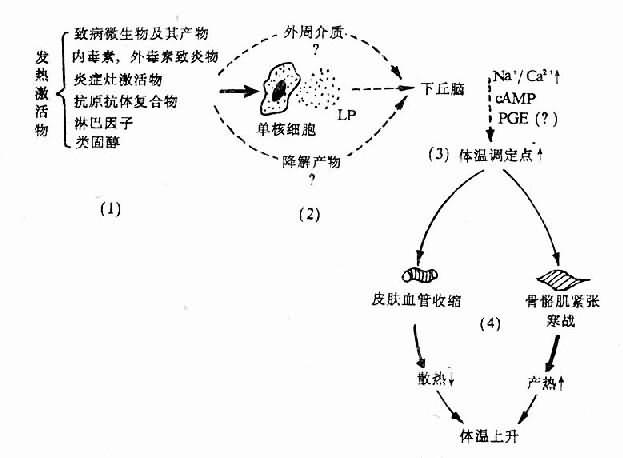
图4-3 发热发病学基本环节示意图
(未包括新发现的EP)
lP如何引起Na+/Ca2+比值上升,Na+/Ca2+比值上升又如何引起调定点上移,尚缺乏深入研究。但最近国内学孝者的研究证明,用降钙剂(EGTA)灌注侧脑室引起发热时,CSF的cAMP明显增多;若事先灌注CaCl2,可使EGTA性体温升高被制止,而且CSF中cAMP的增多也明显受抑制,体温变化与cAMP浓度变化呈明显正相关。继而又发现事先给家兔侧脑室灌注CaCl2,不但抑制静脉内注射LP引起的体温上升,而且抑制了LP引起的CSF中cAMP的增多,体温变化也与cAMP浓度变化呈明显正相关。因此提出:《LP→下丘脑Na+/Ca2+↑→cAMP↑》可能是多种致热原引起发热的重要共同途径。
总之,发热的发生机制比较复杂,有不少细节仍未查明,但主要的或基本的环节已比较清楚。概括起来,多数发热发病学的第一环节是激活物的作用,但至今其作用方式所知不多;第二环节,即共同的中介环节主要是EP。后者有多种,它们可能以不同给合或先后作用于POAH,或作用于外周靶细胞,再通过发热介质参与作用;第三环节是中枢机制,无论EP是否直接进入脑内,很可能要在下丘脑通过中枢介质才引起体温调定点上移,也不排除激活物的降解产物或外周介质到达下丘脑参与作用;第四环节是调定点上移后引起调温效应器的反应。此时由于中心温度低于体温调定点的新水平,从体温调节中枢发出调温指令抵达产热器官和散热器官,一方面通过运动神经引起骨骼肌的紧张度增高或寒战,使产热增多;另一方面经交感神经系统引起皮肤血管收缩,使散热减少;由于产热大于散热,体温乃相应上升直至与调定点新高度相适应。这些基本环节可用下列模式图加以表示(图4-3)。
第三节 发热的时相及其热代谢特点
多数发热尤其急性传染病和急性炎症的发热,其临床经过大致可分三个时相,每个时相有各自的临床和热代谢特点。
一、体温上升期
发热的第一时相是中心体温开始迅速或逐渐上升,快者约几小时或一昼夜就达高峰;慢者需几天才达高峰,称为体温上升期(stadium incrementi)。此期许多病人自感发冷或恶寒,并可出现“鸡皮”和寒战、皮肤苍白等现象。皮肤苍白是皮肤血管收缩使血流减少所致。由于浅层血液减少,皮温下降并剌激冷感受器,信息传入中枢时自感发冷,严重时出现恶寒。在此同时经交感神经传出的冲动又引起皮肤竖毛肌的收缩,故出现“鸡皮”。寒战则是骨骼肌的不随意周期性收缩,是下丘脑发出的冲动,经脊髓侧系的网状脊髓束和红核脊髓束,通过运动神经传递到运动终板而引起的。皮肤温度下降由冷感受器传入信息也是引起寒战的一个因素。故此期又可称寒战期。此期是因体温调定点上移,中心温度低于调定点唤起的调温反应,故热代谢的特点是散热减少和产和产热增多,产热大于散热,体温因而上升。是故当病人感到发冷或恶寒时,中心温度其实已上升了。
寒战在诊断上有参考意义。反复寒战超过一天可能是疟疾或菌血症。在传染病过程中,再次发生寒战,是传染原侵入血流的信号,但寒战不限于传染病。
二、高峰期
当体温上升到与新的调定点水平相适应的高度后。就波动于较高的水平上,称为高峰期或热稽留期(fastigium)。此期病人的皮肤颜色发红,自觉酷热和皮肤干燥,其中心体温已达到或略高于体温调定点的新水平,故下丘脑不再发出引起“冷反应”的冲动。除寒战及“鸡皮”现象消失外,皮肤血管由收缩转为舒张;血温上升也有舒血管作用;浅层血管舒张使皮肤血流增多,因而皮肤发红,散热也因而增加。由于温度较高的血液灌注提高了皮肤温度,热感受器将信息传入中枢,故产生酷热感。高热使皮肤水分蒸发较多,因而皮肤和口唇比较干燥。高峰期持续时间不一,从几小时(如疟疾)、几天(如大叶性肺炎)至一周以上(如伤寒)。本期的热代谢特点是中心体温与上升的调定点水平相适应,产热与散热在较高水平上保持相对平衡,波动也可较大。
三、退热期
退热期(stadium decrementi或defervescence)中因发热激活物在体内被控制或消失,EP及增多的发热介质也被清除(LP主要自肾脏清除),上升的体温调定点乃回降到正常水平。由于调定点水平低于中心体温,故从下丘脑发出降温指令,不仅引起皮肤血管舒张,还可引起大量出汗,故又称出汗期,由于皮肤比较潮湿。
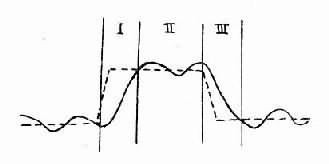
图4-4 发热三个时相体温与调定点的关系示意图
I体温上升期;II高峰期;III退热期……调定点动态曲线;~体温曲线。
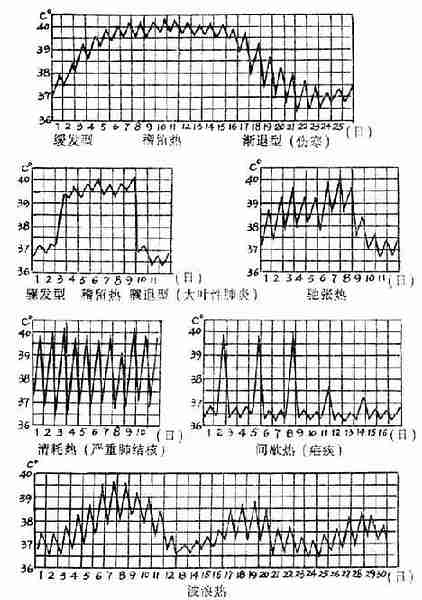
图4-5 常见的发热热型
出汗是一种速效的散热反应,但大量出汗可造成脱水,甚至循环衰竭,应注意监护,补充水和电解质,尤其是在心肌劳损患者,更应密切注意。本期的热代谢特点是散热多于产热,故体温下降,直至与已回降的调定点相适应。热的消退可快可慢,快者几小时或24小时内降至正常,称为热的骤退(crisis),慢者需几天才降至正常,称热的渐退(lysis)。
在这三个时相中,体温与调定点的关系见图4-4。
[附]常见的发热热型
在许多疾病过程中,发热过程持续时间与体温升高水平是不完全相同的。将这些病人的体温按一定时间记录,绘制成曲线图(即所谓热型),可以发现有不同热型(图4-5)。为什么许多发热疾病热型不一样,至今尚无满意的解释,可能与致病微生物的特异性和机体反应性有关。长期积累的资料表明,一定的疾病具有其特殊热型,了解这些热型,有助于鉴别诊断。
第四节 发热机体的主要机能和代谢改变
一、代谢改变
发热机体的代谢改变包含两个方面,一方面是在致热原作用后,体温调节中枢对产热进行调节,提高骨骼肌的物质代谢,使调节性产热增多;另一方面是体温升高本身的作用,一般公认,体温升高1℃,基础代谢率提高13%,例如伤寒病人体温上升并保持于39~40℃,其基础代谢率约增高30~40%(低热量饮食条件下)。因此持久发热使物质消耗明显增多。如果营养物质摄入不足,就会消耗自身物质,并易出现维生素C和B的缺乏,故必须保证有足够能量供应,包括补充足量维生素。
(一)蛋白质代谢
高热传染病人的蛋白分解加强,尿氮比正常人增加2~3倍,可出现负氮平衡,即摄入未能补足消耗。蛋白质分解加强除与体温升高有关外,与LP的作用关系重大。已经证明LP通过PGE合成增多而使骨骼肌蛋白质大量分解,后者是疾病急性期反应之一,除保证能量需求之外,还保证提供肝脏大量氨基酸。用于急性期反应蛋白的合成和组织修复等的需要。
(二)糖和脂肪代谢
发热时糖代谢加强,肝糖原和肌糖原分解增多,血糖因而增多,糖原储备减少。由于葡萄糖的无氧酵解也增强。组织内乳酸因而增加。发热时脂肪分解也显着加强,由于糖代谢加强使糖原储备不足,摄入相对减少,乃动员储备脂肪,后者大量消耗而致消瘦。由于脂肪分解加强和氧化不全,有的病人可出现酮血症酮尿。
(三)水盐代谢
发热时水盐代谢有变化。在发热高峰期,尿量常明显减少,出现少尿和尿色加深,氯化钠排出随而减少,Na+和Cl-滞留于体内;而在退热期,随着尿量增多和大量排汗,钠盐的排出也相应增多。
在高峰期,高热使皮肤和呼吸道水分蒸发增多。加上出汗和饮水不足,可引起脱水,脱水又可加重发热。因此要注意持久高热者的饮食情况,确定合理摄水量,尤其是在退热期,大量排汗可加重脱水。必须补足水分。
二、生理机能改变
发热时有一系列生理机能改变,有的是体温升高引起,有的不是,有的则未确定。
(一)心血管机能改变
体温上升1℃,心率每分钟平均增加18(12~27)次,若按华氏温度计算,则上升1°F,每分钟约增加10次。这是血温升高剌激窦房结及交感神经—肾上腺髓质系统活动增强所致。心率加快一般使心输出量增多,但对心肌劳损或心肌有潜在病灶的病人,则加重了心肌负担,可诱发心力衰竭。在寒战期动脉血压可轻度上升,是外周血管收缩和心率加快的结果;在高峰期由于外周血管舒张,动脉血压轻度下降,高血压病人下降较为明显。体温骤退,特别是用解热药引起体温骤退时,可因大量出汗而导致休克。
(二)呼吸机能改变
发热时呼吸加快,是上升的血温剌激呼吸中枢以及提高呼吸中枢对CO2的敏感性所致。传统上把此看作一种加强散热的反应。
(三)消化机能改变
发热时出现食欲不振和唾液分泌减少。前者使饮食减退,后者使口腔粘膜干燥,当然后者与水分蒸发过多也有关。动物实验证明,IL-1能引起食欲不振。
有些发热病人还有胃液和胃酸分必减少,胃肠道蠕动减弱(并可鼓肠)。这些变化只部分与发热有关。实验证明,注射ET可在引起发热的同时,导致胃肠蠕动减弱和分泌减少。给解热药抑制体温上升,这些变化未能完全消失。
(四)中枢神经系统机能改变
高热时对中枢神经系统的影响较大,突出表现是头痛,机制未明。有的病人有谵语和幻觉。实验证明,注射LP能诱导睡眠,这可能对传染病人睡眠较多作出部分解释。
小儿在高热中可出现搐搦,常见于出生后6月~6岁之间的儿童,称热惊厥。多为全身搐搦,发作时间较短,称单纯性热惊厥。这种儿童的脑本来正常,无既往脑病史;而有些原来有既往脑病史的儿童,其热惊厥则表现为局部搐搦,发作持续时间也较长。热惊厥的发作,可能与体温上升的高度和上升的速度都有一定关系。对原来有脑病史的儿童,发热可能降低搐搦发作的剌激阈。
第五节 发热的生物学意义
发热是不是防御反应,是否有利于传染病康复?至今资料和观点矛盾。现把正反两类资料摘要对照如下表:
不难看出,上列双方资料相互矛盾。故目前对发热的生物学意义,未能做一般结论。只能具体情况作具体分析和判断。自临床广泛使用解热药以来,除个别外,未见对普通传染病的病情或终局产生明显影响。一般既不使病情恶化,又未能促进传染病康复。虽然发热疗法曾用于治疗淋菌性尿道炎和神经梅素等,有一定效果。但这些疾病本身并无发热。故不能据此判断发热能促进传染病康复。另外许多无菌性炎症性发热,则谈不上想象中的好处。全身性红斑狼疮和类风湿关节炎是自身免疫性疾病,发热促进免疫反应必然会带来更大的伤害。
| 提高抵抗力 | 降低抵抗力 |
| 1.体温升高提高传染生存率 · 蜥蜴或金鱼感染嗜水性产气单胞菌,体温升高者生存率较高。 · 过热动物感染炭疽杆菌、痢疾杆菌和霍乱弧菌,比常温延缓死亡或提高生存率。 · 发热提高动物对痢疾中毒的抵抗力,延缓中毒死亡或提高生存率。 2.体温升高增强某些防御反应 · 激活白细胞吞噬活力,促进白细胞游走,有利于消灭局部细菌。 · 促进干扰素的产生,有抗病毒、抗细菌和抗癌效应。 · 有利于T淋巴细胞增殖和增加抗体产生 · IL-1引起的低铁血症不利细菌生长,发热对此有协同作用 |
1.体温升高降低传染生存率。 · 大鼠感染肠炎沙门氏菌,冷却视前区致发热增高1℃,使生存率由77%降至0% · 常温小鼠能忍受的破伤风芽胞感染剂量,可使发热小鼠感染致死。 · 发热或过热提高动物对细菌内毒素的毒血症敏感性,显著降低半致死量。提高中毒死亡率 2.体温升高抑制某些防御反应 (1)明显降低自然杀伤细胞(NK)的活性。 (2)抑制IL-1的释放,从而抑制IL-1引 起的免疫反应 (3)使血中补体和细菌凝集素减少甚至消失 (4)使家兔伤寒被动免疫的抗体效价下降,抗体消耗加快 |
一般认为发热>40.5℃对病体有不良效应,包括脱水、谵妄,心肺负荷增加,负营养平衡等。至于中度发热对病体的利弊如何?尤其是在哺乳类动物和人体,还必须进行系统深入研究,特别要把发热和非发热因素严格区分开,才能得出精确的答案。
第六节 发热的处理原则
基于对发热发病学的新认识和解热药作用原理的了解,对发热病人的处理,提出下述原则。
一、对一般发热不急于解热
由于热型和热程变化,可反映病情变化,并可作为诊断、评价疗效和估计预后的重要参考,而发热不过高或不太持久又不至有多大的危害,故在疾病未得到有效治疗时,不必强行解热。解热本身不能导致疾病康复,且药效短暂,药效一过,体温又会上升。相反,疾病一经确诊而治疗奏效,则热自退。急于解热使热程被干扰,就失去参考价值,有弊无益。
二、下列情况应及时解热
1.体温过高(如40℃以上)使患者明显不适、头痛、意识障碍和惊厥者。
2.恶性肿瘤患者(持续发热加重病体消耗)。
3.心肌梗塞或心肌劳损者(发热加重心肌负荷)。
三、选用适宜的解热措施
1.针对发热病因传染病的根本治疗方法是消除传染原和传染灶。当抗感染奏效时,随着传染灶(包括炎症灶)的消退,便出现退热。为促进退热,解热药可与抗感染疗法合并使用。
2.针对发热机制中心环节 根据发热机制及现有解热药的药理作用,可针对下列三个环节采取措施以达到解热:(1)干扰或阻止EP的合成和释放,包括制止或减少激活物的产生或发挥作用;(2)妨碍或对抗EP对体温调节中枢的作用;(3)阻断发热介质的合成。这些措施可导致上升的调定点下降而退热。目前临床上采用的解热药包括化学解热药和类固醇解热药。前者以水杨酸盐为代表,对其解热原理有以下解释:①作用于POAH及附近,以某种方式使中枢神经元的机能复原;②阻断PGE的合成(通过抑制环加氧酶),但PGE作为发热介质仍有争议。以糖皮质激素(抗炎激素)为代表的类固醇解热剂的解热作用也有下列解释:①抑制产LP细胞合成和释放LP;②抑制免疫反应;③抑制炎症反应(包括降低微血管通透性、抑制白细胞游出和抗渗出等),使炎灶EP和激活物减少;④中枢效应:小量注入POAH有解热作用,但方式不清楚。
3.针剌解热疗法,有一定效果,机制未明。
四、加强对高热或持久发热病人的护理
1.注意水盐代谢,补足水分,预防脱水。
2.保证充足易消化的营养食物。包括维生素。
3.监护心血管功能,对心肌能劳损者,在退热期或用解热药致大量排汗时,要防止休克的发生
第五章 水和电解质代谢紊乱
人和高等动物机体内的细胞也象水中的单细胞生物一样是在液体环境之中的。和单细胞生物不同的是人体大量细胞拥挤在相对来说很少量的细胞外液中,这是进化的结果。但人具有精确的调节机构,能不断更新并保持细胞外液化学成分、理化特性和容量方面的相对恒定,这就是对生命活动具有十分重要意义的内环境。
水、电解质代谢紊乱在临床上十分常见。许多器官系统的疾病,一些全身性的病理过程,都可以引起或伴有水、电解质代谢紊乱;外界环境的某些变化,某些变化,某些医原性因素如药物使用不当,也常可导致水、电解质代谢紊乱。如果得不到及时的纠正,水、电解质代谢紊乱本身又可使全身各器管系统特别是心血管系统、神经系统的生理功能和机体的物质代谢发生相应的障碍,严重时常可导致死亡。因此,水、电解质代谢紊乱的问题,是医学科学中极为重要的问题之一,受到了医学科学工作者的普遍重视。
内环境的相对恒定主要是在神经一内分泌调节下实现的。故本章在简述水、电解质平衡调节的基础上,着重讨论水、钠、钾、镁的代谢紊乱。应当指出,水与电解质代谢紊乱之间,某一电解质与其它电解质的代谢紊乱之间,水、电解质与酸硷平衡紊乱之间关系密切,它们互相联系互相影响,一旦发生紊乱往往是综合的,即一种障碍往往可以伴有或引起另一种或另一些障碍。
第一节 水、电解质平衡的调节
水、电解质的平衡,受神经系统和某些激素的调节,而这种调节又主要是通过神经特别是一些激素对肾处理水和电解质的影响而得以实现的。
一、渴感的作用
下丘脑视上核侧面有口渴中枢。使这个中枢兴奋的主要剌激是血浆晶体渗透压的升高,因为这可使口渴中枢的神经细胞脱水而引起渴感。渴则思饮寻水,饮水后血浆渗透压回降,渴感乃消失。此外有效血容量的减少和血管紧张素Ⅱ的增多也可以引起渴感。
二、抗利尿激素的作用
抗利尿激素(antidiuretic hormone,ADH)主要是下丘脑视上核神经细胞所分泌并在神经垂体贮存的激素。ADH能提高肾远曲小管和集合管对水的通透性,从而使水的重吸收增加(图5-1)。
促使ADH释放的主要剌激是血浆晶体渗透压的增高和循环血量的减少。当机体失去大量水分而使血浆晶体渗透压增高时,便可剌激下丘脑视上核或其周围区的渗透压感受器而使ADH释放增多。血浆渗透压乃可因肾重吸收水分增多而有所回降。大量饮水时的情况正好相反。由于ADH释放减少,肾排水增多,血浆渗透压乃得以回升。血量过多时,可剌激左心房和胸腔内大静脉的容量感受器。反射性地引起ADH释放减少,结果引起利尿而使血量回降。反之,当失血等原因使血量减少时,ADH乃可因容量感受器所受剌激减弱而释放增加,尿量因而减少而有助于血量的恢复。
此外,动脉血压升高可通过剌激颈动脉窦压力感受器而反射性地抑制AKH的释放;疼痛剌激和情绪紧张可使ADH释放增多;血管紧张素Ⅰ增多也可剌激ADH的分泌。
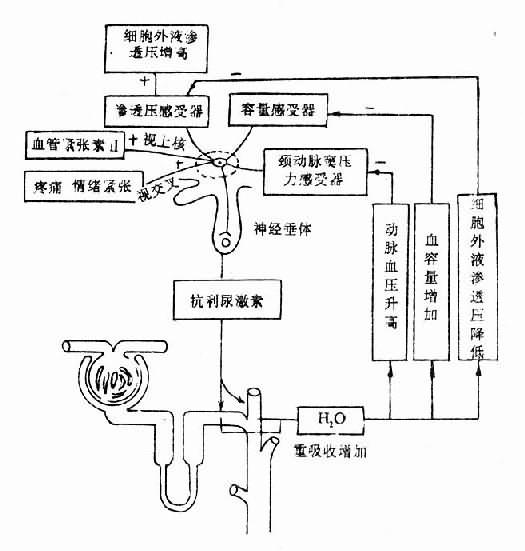
图5-1 抗利尿激素的调节及其作用示意图
三、醛固酮的作用
醛固酮(aldosterone)是肾上腺皮质球状带分泌的盐皮质激素。醛固酮的主要作用是促进肾远曲小管和集合管对Na+的主动重吸收,同时通过Na+、-K+和Na+-H交换而促进K+和H+的排出,所以说醛固酮有排钾、排氢、保钠的作用。随着Na+主动重吸收的增加,Cl-和水的重吸收也增多,可见醛固酮也有保水作用(图5-2)。
醛固酮的分泌主要受肾素—血管紧张素系统和血浆Na+、K+浓度的调节。当失血等原因使血容量减少,动脉血压降低时,肾入球小动脉管壁的牵张感受器就因入球小动脉血压下降和血容量减少而受到剌激,近球细胞的肾素分泌乃增多。同时由于肾小球滤过率也相应减少,流经致密斑的Na+亦因而减少,这也可使近球细胞的肾素分泌增多。(另一种完全相反的见解是,远曲小管起始部分肾小管液Na+浓度的增加,可剌激致密斑而使近球细胞分泌肾素增多。目前这两种看法尚未能统一。)肾素增多后,血管紧张素Ⅰ、Ⅱ、Ⅲ便相继增多,血管紧张素Ⅱ和Ⅲ都能剌激肾上腺皮质球状带使醛固酮的合成和分泌增多。
此外,近球细胞处的小动脉管内有交感神经末梢支配,肾交感神经兴奋时能使肾素的释放量增加。肾上腺素和去甲肾上腺素也可直接剌激近球细胞,使肾素释放增加。
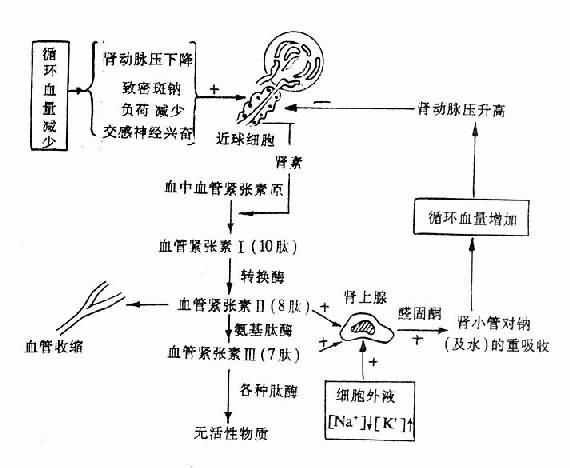
图5-2 醛固酮分泌的调节及其作用示意图
血浆K+浓度升高或Na+浓度降低,可直接剌激肾上腺皮质球状带使醛固酮分泌增多;反之,当血浆K+浓度降低或Na+浓度升高时,醛固酮的分泌减少。
四、“第三因子”的作用
有人在用狗作的实验中观察到,当细胞外液容量增加时,血浆中出现一种抑制肾小管重吸收Na+从而导致尿钠排出增多的性质未明的物质,称为“称钠激素”(natriuretic hormone)或“第三因子”。但这方面还有许多问题有待阐明。有些资料也未能证实这种物质的存在。
五、心房利钠因子的作用
80年代初以来,哺乳动物心房中心房利钠因子(atrial natriuretic factor,ANF)的发现和随后一系列的研究,为人们理解体液容量和血压的调节开辟了一个新的时代,也是医学和生理学研究的一个重大进展。ANF后来也被称为心房利钠多肽(atrial natriureticpolypeptide,ANP)因为已经证明它是一种多肽。
ANP主要存在于哺乳动物其中也包括人的心房肌细胞的细胞浆中。ANP已经分离提纯,并且已能人工合成,其氨基酸序列亦已确定。从动物心房肌获得的这类多肽称为心钠素(cardionatrin)或心房肽(atriopeptin)而从人类心房肌所得者称为人心房利钠多肽(human atrial natriuretic polypeptide,hANP)而ANP 则是它们的通称。
动物实验证明,急性的血容量增加可使ANP释放入血,从而引起强大的利钠和利尿作用。血容量增加可能是通过增高右心房压力,牵张心房肌而使ANP释放的。反之,限制钠、水摄入或减少静脉回心血量则能减少ANP的释放。
已经证明,一些动物的动脉、肾、肾上腺皮质球状带等有ANP的特异受体,ANP是通过这些受体作用于细胞膜上的鸟苷酸环化酶,以细胞内的环鸟苷酸(cGMP)作为第二信使而发挥其效应的。
ANP对水、电解质代谢有如下的重要影响:
(一)强大的利钠、利尿作用其机制在于抑制肾髓质集合管对Na+的重吸收。ANP也可能通过改变肾内血流分布、增加肾小球滤过率而发挥利钠、利尿的作用。
(二)拮抗肾素一醛固酮系统的作用实验证明,ANP能抑制体外培养的肾上腺皮质球状带细胞合成和分泌醛固酮;体内试验又证明ANP能使血浆肾素活性下降,有人认为ANP可能直接抑制近球细胞分泌肾素。
(三)ANP能显著减轻失水或失血后血浆中ADH水平增高的程度ANP及其与肾素—醛固酮系统以及ADH之间的相互作用,对于精密地调节水、电解质平衡起着重要作用。ANP还有舒张血管,降低血压的作用。
根据其释放、对远隔器官的作用以及以后在肝、肾、肺等器官中降解等特点,已公认ANP为一种新的激素,因而心脏除了是泵血器官以外,同时也是一个内分泌器官,这是内分泌学的一个新的突破。
六、甲状旁腺激素的作用
甲状旁腺激素是甲状旁腺分泌的激素。它能促进肾远曲小管的集合管对Ca2+的重吸收,抑制近曲小管对磷酸盐的重吸收,抑制近曲小管对Na+、K+和HCO3-的重吸收。甲状旁腺激素还能促进肾小管对Mg2+的重吸收。关于Mg2+重吸收的部位,尚无一致的看法。有人报道Mg2+在近曲小管和髓袢升支被重吸收,而加一些报道则认为Mg2+主要在髓袢特别是髓袢升支的粗段被重吸收,而近曲和远曲小管基本上不能重吸收Mg2+。甲状旁腺激素的分泌主要受血浆Ca2+浓度的调节:Ca2+浓度下降可使甲状旁腺激素的分泌增加,反之则甲状旁腺激素的分泌减少。
第二节 水、钠代谢紊乱
水、钠代谢紊乱常同时或先后发生,关系密切,通常一起讨论,紊乱有各种分类方法,为了便于理解,根据临床上通常采用的方法分为脱水(包括失钠)和水中毒进行讨论。
一、脱水
脱水(dehydration)系指体液容量的明显减少。脱水按细胞外液的渗透压不同可分为三种类型。以失水为主者,称为高渗(原发)性脱水;以失钠为主者,称为低渗(继发)性脱水;水、钠各按其在血浆中的含量成比例丢失者,称为等渗性脱水。
(一)高渗性脱水
高渗性脱水(hypertonic dehydration)以失水多于失钠、血清钠浓度>150mmol/L(150mEq/L)、血浆渗透压>310mOsm /L为主要特征。
1.原因和机制
(1).单纯失水
①经肺失水:任何原因引起的过度通气都可使呼吸道粘膜的不感蒸发加强以致大量失水;
②经皮肤失水:例如在发热或甲状腺机能亢进时,通过皮肤的不感蒸发每日可失水数升;
③经肾失水:中枢性尿崩症时因ADH产生和释放不足,肾性尿崩症时因肾远曲小管和集合管对ADH的反应缺乏,故肾脏可排出大量水分。失水发生在肾单位的最远侧部分,亦即在这个部分以前,大部分钠离子已经被重吸收。因此,病人可排出10~15L的稀释尿而其中只含几个mmol的钠。
单纯失水时机体的总钠含量可以正常。
⑵失水大于失钠:即低渗液的丧失,见于:
①胃肠道失液:呕吐和腹泻时可能丧失含钠量低的消化液,如部分婴幼儿腹泻的病儿,粪便钠浓度在60mmol/L以下;
②大量出汗:汗为低渗液;大汗时每小时可丢失水分800ml左右。
③经肾丧失低渗尿:如反复静脉内输注甘露醇、尿素、高渗葡萄糖等时,可因肾小管液渗透压增高而引起渗透性利尿,排水多于排钠。
在这些情况下,机体既失水,又失钠,但失水不成比例地多于失钠。
⑶饮水不足
上述原因在渴感正常的人,在可以得到水喝和能够喝水的情况下,很少引起高渗性脱水,因为在水分丧失的早期,血浆渗透压稍有增高时,就会剌激口渴中枢。在喝水后,血浆渗透压即可恢复。因此,只有在下述情况才会发生明显的高渗性脱水:①水源断绝:如沙漠迷路;②不能或不会饮水:如频繁呕吐的病人、昏迷病人、极度衰弱的病人等;③渴感障碍:下丘脑病变可损害口渴中枢;在有些并不引起失语症的大脑皮质脑血管意外的老年病人,也可发生渴感障碍。
在临床实践中,高渗性脱水的原因常是综合性的,如婴幼儿腹泻时高渗性脱水的原因除了丢失肠液、入水不足外,还有发热出汗,呼吸增快等因素引起的失水过多。
2.对机体的影响
⑴因失水多于失钠,细胞外液渗透压增高,剌激口渴中枢(渴感障碍者除外),促使患者找水喝。
⑵除尿崩症患者外,细胞外液渗透压增高剌激下丘脑渗透压感受器而使ADH释放增多,从而使肾重吸收水增多,尿量减少而比重增高。
⑶细胞外液渗透压增高可使渗透压相对较低的细胞内液中的水向细胞外转移。以上三点都能使细胞外液得到水分补充,使渗透压倾向于回降。
可见,高渗性脱水时细胞内、外液都有所减少,但因细胞外液可能从几方面得到补充,故细胞外液和血容量的减少不如低渗性脱水时明显,发生休克者也较少。
⑷早期或轻症患者,由于血容量减少不明显,醛固酮分泌不增多,故尿中仍有钠排出,其浓度还可因水重吸收增多而增高;在晚期和重症病例,可因血容量减少、醛固酮分泌增多而致尿钠含量减少。
⑸细胞外液渗透压增高使脑细胞脱水时可引起一系列中枢神经系统功能障碍的症状,包括嗜睡、肌肉抽搐、昏迷,甚至导致死亡。脑体积因脱水而显著缩小时,颅骨与脑皮质之间的血管张力增大,因而可导致静脉破裂而出现局部脑内出血和蛛网膜下出血。
⑹脱水严重的病例,尤其是小儿,由于从皮肤蒸发的水分减少,散热受到影响,因而可以发生脱水热。
根据脱水程度可将高渗性脱水分为轻度,中度和重度三级。①轻度:失水量相当于体重的2~5%,患者粘膜干燥,汗少,皮肤弹性减低,口渴,尿量少,尿渗透压通常>600mOsm/L,尿比重>1.020(肾脏浓缩功能障碍者如尿崩症患者等除外),可出现酸中毒,但不发生休克,婴幼儿患者啼哭无泪,前囱凹陷,眼球张力低下。②中度:失水量相当于体重的5~10%。临床表现有严重口渴,恶心,腋窝和腹股沟干燥,皮肤弹性缺乏,血液浓缩,心动过速,体位性低血压,中心静脉压下降,表情淡漠,肾功能低下,少尿,血浆肌酐和尿素素氮水平增高,血清钾浓度可在正常范围的上限或稍高,尿渗透压通常大于800mOsm/L,尿比重>1.025(肾脏浓缩功能障碍者如尿崩症患者等除外),发生酸中毒。③重度:失水量相当于体重的10~15%。患者经常发生休克,临床主要表现有少尿或无尿,血压下降,脉搏快而弱。肾脏功能受损害,血浆肌酐和尿素氮上升;血清[K+]升高。代谢性酸中毒通常严重。重度脱水常可导致死亡。脱水程度超过此界限时,很少人能够耐受。
3.防治原则
首先应防治原发疾病,防止某些原因的作用。高渗性脱水时因血钠浓度高,故应给予5%葡萄糖溶液。高钠血症严重者可静脉内注射2.5%或3%葡萄糖溶液。应当注意,高渗性脱水时血钠浓度高,但患者仍有钠丢失,故还应补充一定量的含钠溶液,以免发生细胞外液低渗。
(二)低渗性脱水
低渗性脱水(hypotonic dehydration)以失钠多于失水,血清钠浓度<130mmol/L(<130mEq/L),血浆渗透压280mOsm/L为主要特征。
1.原因和机制
⑴丧失大量消化液而只补充水分:这是最常见的原因。大多是因呕吐、腹泻,部分是因胃、肠吸引术丢失体液而只补充水分或输注葡萄糖溶液。
⑵大汗后只补充水分:汗虽为低渗液,但大量出汗也可伴有明显的钠丢失(每小时可丢失30~40mEq左右的钠),若只补充水分则可造成细胞外液低渗。
⑶大面积烧伤:烧伤面积大,大量体液丢失而只补充水时,可发生低渗性脱水。
⑷肾性失钠:可见于以下情况:①水肿患者长期连续使用排钠性利尿剂(如氯噻嗪类、速尿及利尿酸等)时,由于肾单位稀释段对钠的重吸收被抑制,故钠从尿中大量丢失。如再限制钠盐摄入,则钠的缺乏更为明显;②急性肾功能衰竭多尿时期,主要是肾小管液中尿素等溶质浓度增高,故可通过渗透性利尿作用使肾小管上皮细胞对钠、水重吸收减少;③在所谓“失盐性肾炎”的患者,由于受损的肾小管上皮细胞对醛固酮的反应性降低,故远侧肾小管(近年有人认为是集合管)细胞对钠重吸收障碍;④Addison病时,主要是因为醛固酮分泌减少,故肾小管对钠重吸收减少。对上述些经肾失钠的病人,如果只补充水分而忽略了补钠盐,就可能引起低渗性脱水。
由此可见,低渗性脱水的发生,往往与措施不当(失钠后只补水而不补充钠)有关。这一点应当引起充分的注意。但是,也必须指出,即使没有这些不适当的措施,大量体液丢失本身也可以使有些患者发生低渗性脱水。这是因为大量体液丢失所致的细胞外液容量的显著减少,可通过对容量感受器的剌激而引起ADH分泌增多,结果是肾脏重吸收水分增加,因而引起细胞外液低渗(低渗性脱水)。
2.对机体的影响
在细胞外液容量尚末减少时,由于细胞外液渗透压降低,ADH分泌减少,故肾小管上皮细胞对水重吸收减少而导致肾脏排出的水分增多。因此,早期患者可排出较多的低渗尿。水分排出的增多一方面可使细胞外液容量进一步减缩,因而可使患者倾向于发生休克,另一方面可使细胞外液渗透压得到一定程度的恢复,因而又具有一定的代偿意义。如果细胞外液的渗透压仍然得不到恢复,则细胞外液可向渗透压相对较高的细胞内转移,故细胞内液并无丢失而细胞外液量则显著减少,患者易发生休克,这是本型脱水的主要特点。此外由于血钠浓度低,致密斑(位于远曲小管起始部)的钠负荷减轻。故肾素—血管紧张素—醛固酮系统的活性增强,醛酮分泌增多,因而可使肾小管上皮细胞对钠的重吸收增强,尿中Na+或Cl-排出减少。肾素—血管紧张素—醛固酮系统活性增强也与细胞外液特别是有效循环血量减少,以致肾脏入球小动脉压力降低、牵张感受器被兴奋,从而使肾素释放增多有关。
当脱水进一步发展以致细胞外液容量严重不足时,又可因容量感受器受剌激而使ADH分泌增多,从而使肾脏重吸收水分增多,其结果是一方面在一定程度上维持细胞外液容量,使之不致过分减少,另一方面则又可使细胞外液渗透压降低,从而促使水分向细胞内转移。
在临床上,伴随着休克倾向的出现,患者往往有静脉塌陷、动脉血压降低、脉搏细速、四肢厥冷、尿量减少,氮质血症等表现。由于细胞外液特别是细胞间液显著减少,因而患者皮肤弹性丧失,眼窝和婴儿囟门内陷。
根据缺钠程度和临床症状,也可将低渗性脱水分为三度:①轻度:相当于成人每公斤体重缺失氯化钠0.5g。患者常感疲乏、头晕,直立时可发生昏倒(昏厥),尿中氯化钠很少或缺如;②中度:每公斤体重缺失氯化钠0.5g~0.75g。此时患者可有厌食、恶心呕吐、视力模糊、收缩压轻度降低、起立时昏倒、心率加快、脉搏细弱、皮肤弹性减弱、面容消瘦等表现;③重度:每公斤体重缺失氯化钠0.75g~1.25g,患者可有表情淡漠、木僵等神经症状。最后发生昏迷,关有严重休克。
3.防治原则
除去除原因(如停用利尿药)、防治原发疾病外,一般应用等渗氯化钠溶液及时补足血管内容量即可达到治疗目的。如已发生休克,要及时积极抢救。
(三)等渗性脱水
水与钠按其在正常血浆中的浓度成比例丢失时,可引起等渗性脱水(isotonicdehydration)。即使是不按比例丢失,但脱水后经过机体调节。血钠浓度仍维持在130~145mmol/L,渗透压仍保持在280~310mOsm/L者,亦属等渗性脱水。
1.原因及机制
⑴小肠液丧失:从十二指肠到回盲部的所有小肠分泌液以及胆汁和胰液的钠浓度都在120~140mmol/L之间。因此,小肠炎所致的腹泻、小肠瘘、小肠梗阻等可引起等渗体液的丧失。
⑵大量胸水和腹水形成等。
2.对机体的影响
细胞外液容量减少而渗透压在正常范围,故细胞内外液之间维持了水的平衡,细胞内液容量无明显变化。血容量减少又可通过醛固酮和ADH的增多而使肾对钠、水的重吸收增加,因而细胞外液得到一定的补充,同时尿钠含量减少,尿比重增高。如血容量减少得迅速而严重,患者也可发生休克。
如不予及时处理,则可通过不感蒸发继续丧失水分而转变为高渗性脱水;如只补充水分而不补钠盐,又可转变为低渗性脱水。(三型脱水的比较见表5-1)。
3.防治原则
防治原发病,输注渗透压偏低的氯化钠溶液,其渗透压以等渗溶液渗透压的1/2~2/3为宜。
表5-1 三 型 脱 水 的 比 较
| 高渗必脱水 | 低渗性脱水 | 等渗性脱水 | |
| 发病原理 | 水摄入不足或丧失过多 | 体液丧失而单纯补水 | 水和钠等比例丧失而未予补充 |
| 发病原因 | 细胞外液高渗,细胞内液丧失为主 | 细胞外液低渗,细胞外液丧失为主 | 细胞外液等渗,以后高渗,细胞内外液均有丧失 |
| 主要表现和影响 | 口渴、尿少、脑细胞脱水 | 脱水体征、休克、脑细胞水肿 | 口渴、尿少、脱水体征、休克 |
| 化验 1.血清钠毫当量/升 2.尿氯化钠 |
150以上 有 |
130以下 减少或无 |
130~150 减少,但有 |
| 治疗 | 补充水分为主 | 补充生理盐水或3%氯化钠溶液 | 补充偏低渗的氯化钠溶液 |
二、水中毒
正常人摄入较多的水时,由于神经—内分泌系统和肾脏的调节作用。可将体内多余的水很快经由肾脏排出,故不致发生水潴留,更不会发生水中毒(Water intoxication)。但给处在ADH分泌过多或肾脏排水功能低下的患者输入过多的水分时,则可引起水在体内潴留,并伴有包括低钠血症在内的一系列症状和体征,即出现所谓水中毒。
(一)原因
1.ADH分泌过多 由于ADH是具有促进肾脏远曲小管和集合管上皮细胞重吸收水的作用,故各种原因引起的ADH分泌过多,均可使水分经肾排出减少,从而使机体易于发生水中毒。
ADH分泌过多的原因可归纳为下列几个方面;
⑴ADH分泌异常增多综合征(Syndrome lf inappropriate ADH secretion,SIADH):见于以下疾病的某些病例:①恶性肿瘤:如肺燕麦细胞癌、胰腺癌、何杰金氏病以及淋巴肉瘤等;②中枢神经系统疾病:如脑肿瘤、脑脓肿、硬脑膜下出血、蛛网膜下腔出血、脑血管血栓形成、病毒性或细菌性脑炎、细菌性或结核性脑膜炎以及早老性痴呆等;③肺疾患:如肺结核、肺脓肿、病毒性及细菌性肺炎等。
经生物鉴定或放射免疫法测定,发现上述疾病患者血清、尿及癌组织(尤其是肺燕麦细胞癌)提取液中ADH或ADH样物质增多。SIADH可能是由于这些肿瘤形成并释出较多的类似ADH的多肽类物质。或者是由于某些病变直接剌激下丘脑,使之分泌ADH过多所致。
⑵药物:促进ADH释放和/或使其作用增强的药物有异丙肾上腺素、吗啡和扑热息痛等。抗利尿但其机制未明者有环磷酰胺,阿米替林(amitriptyline)和氟奋乃静等。
⑶各种原因所致的应激:见于手术,创伤及强烈精神剌激等时。正常状态下,副交感神经兴奋有抑制ADH分泌的作用;应激时交感神经兴奋而副交感神经受抑制从而解除副交感神经对ADH分泌的抑制,结果是ADH分泌增多而易引致水中毒。
⑷有效循环血容量减少:左心房内膜下有容量(牵张)感受器,经迷走神经与下丘脑联系。血容量过多时左心房扩张,这种感受器就受剌激而兴奋,传入冲动沿迷走神经到达下丘脑后,可使ADH释放减少。有效循环血容量减少(如休克等)时,从左心房传至下丘脑抑制ADH释放的冲动减少,故ADH分泌增多;如果此时输液过快过多可导致水中毒。
⑸肾上腺皮质功能低下:肾上腺皮质激素对下丘脑分泌ADH具有抑制作用。肾上腺皮质功能低下时,由于肾上腺皮质激素分泌减少,对下丘脑分泌ADH的抑制作用也就减弱,因而ADH分泌增多。如果此时大量进水可发生水中毒。
2.肾排水功能不足 在急慢性肾功能不全少尿期,因肾脏排水功能急剧降低,如果入水量不加限制,则可引起水在体内潴留;严重心力衰竭或肝硬变时,由于有效循环血量和肾血流量减少,肾脏排水也明显减少,若增加水负荷亦易引起水中毒。
3.低渗性脱水晚期由于胞外液低渗,细胞外液向细胞内转移。可造成细胞内水肿,如此时输入大量水分就可引起水中毒。
(二)对机体的影响
细胞外液因水过多而被稀释,故血钠浓度降低,渗透压下降。加之肾脏不能将过多的水分及时排出,水分乃向渗透压相对高的细胞内转移而引起细胞水肿,结果是细胞内、外液容量均增多而渗透压都降低。由于细胞内液的容量大于细胞外液的容量,所以潴留的水分大部分积聚在细胞内,因此在轻度水中毒患者,组织间隙中水潴留的程度尚不足以引起明显的凹隐性水肿。
急性水中毒时,由于脑神经细胞水肿和颅内压增高,故脑症状出现最早而且突出,可发生各种神经精神症状,如凝视、失语、精神错乱、定向失常、嗜睡、烦躁等并可有视神经乳头水肿,严重者可因发生脑疝而致呼吸心跳骤停,轻度或慢性水中毒患者,发病缓慢,症状常不明显,多被原发病的症状、体征所掩盖,可有嗜睡、头痛、恶心、呕吐、软弱无力及肌肉挛痛等症状。
有人报道,水中毒时因细胞外液容量扩大,醛固酮分泌抑制(尽管细胞外液钠浓度降低),故远曲小管对钠重吸收减弱,从而引起肾持续性排钠增多,机体出现钠负平衡。但也有资料表明水中毒时醛固酮并不减少,反而可以增多,这可能与病因不同有关。至于尿比重,则主要因ADH增多而较高。
(三)防治原则
首先应防治原发疾患,防止引起水中毒原因作用。轻症患者在暂停给水后即可自行恢复。对于重症急性水中毒患者,则应立即静脉内输注甘露醇、山梨醇等渗性利尿剂或速尿等强利尿剂以减轻脑细胞水肿和促进体内水分的排出。3~5%高渗氯化钠溶液静脉滴注可迅速缓解体液的低渗状态,但须密切注意,因钠离子过多可使细胞外液容量增大而加重心脏负荷。
第三节 钾代谢紊乱
钾代谢乱主要是指细胞外液中钾离子浓度的异常变化,包括低钾血症(hypokalemia)和高钾血症(hyperkalemia)。关于在病理情况下细胞内钾离子浓度的改变及其对机体的影响等问题,迄今还知之不多。
一、低钾血症
血清钾浓度低于3.5mmol/L(3.5mEq/L,正常人血清钾浓度的范围为3.5~5.5mmol/L)称为低钾血症。低钾血症时,机体的含钾总量不一定减少,细胞外钾向细胞内转移时,情况就是如此。但是,在大多数情况下,低钾血症的患者也伴有体钾总量的减少——缺钾(potassium deficit)。
(一)原因和机制
1.钾摄入减少一般饮食含钾都比较丰富。故只要能正常进食,机体就不致缺钾。消化道梗阻、昏迷、手术后较长时间禁食的患者,不能进食。如果给这些患者静脉内输入营养时没有同时补钾或补钾不够,就可导致缺钾和低钾血症。然而,如果摄入不足是唯一原因,则在一定时间内缺钾程度可以因为肾的保钾功能而不十分严重。当钾摄入不足时,在4~7天内可将尿钾排泄量减少到20mmol/L以下,在7~10天内则可降至5~10mmol/L(正常时尿钾排泄量为38~150mmol/L)。
2.钾排出过多
⑴经胃肠道失钾:这是小儿失钾最重要的原因,常见于严重腹泻呕吐等伴有大量消化液丧失的患者。腹泻时粪便中K+的浓度可达30~50mmol/L。此时随粪丢失的钾可比正常时多10~20倍。粪钾含量之所以增多,一方面是因为腹泻而使钾在小肠的吸收减少,另一方面是由于腹泻所致的血容量减少可使醛固酮分泌增多,而醛固酮不仅可使尿钾排出增多,也可使结肠分泌钾的作用加强。由于胃液含钾量只有5~10mmol/L,故剧烈呕吐时,胃液的丧失并非失钾的主要原因,而大量的钾是经肾随尿丧失的,因为呕吐所引起的代谢性碱中毒可使肾排钾增多(详后文),呕吐引起的血容量减少也可通过继发性醛固酮增多而促进肾排钾。
⑵经肾失钾:这是成人失钾最重要的原因。引起肾排钾增多的常见因素有:
①利尿药的长期连续使用或用量过多:例如,抑制近曲小管钠、水重吸收的利尿药(碳酸酐酶抑制药乙酰唑胺),抑制髓袢升支粗段Cl-和Na+重吸收的利尿药(速尿、利尿酸、噻嗪类等)都能使到达远侧肾小管的原尿流量增加,而此处的流量增加是促进肾小管钾分泌增多的重要原因。上述利尿药还能使到达远曲小管的Na+量增多,从而通过Na+-K+交换加强而导致失钾。许多利尿药还有一个引起肾排钾增多的共同机制:通过血容量的减少而导致醛固酮分泌增多。速尿、利尿酸、噻嗪类的作用在于抑制髓袢升支粗段对Cl-的重吸收从而也抑制了Na+的重吸收。所以,这些药物的长期使用既可导致低钠血症,又可导致低氯血症。已经证明,任何原因引起的低氯血症均可使肾排钾增多。其可能机制之一是低氯血症似能直接剌激远侧肾小管的泌钾功能。
②某些肾脏疾病:如远侧肾小管性酸中毒时,由于远曲小管泌氢功能障碍,因而H+-Na+交换减少而K+-Na+交换增多而导致失钾。近侧肾小管性酸中毒时,近曲小管HCO3-的重吸收减少,到达远曲小管的HCO3-增多是促进远曲小管排钾增多的重要原因(详后文)。急性肾小管坏死的多尿期,由于肾小管液中尿素增多所致的渗透性利尿,以及新生肾小管上皮对水、电解质重吸收的功能不足,故可发生排钾增多。
③肾上腺皮质激素过多:原性和继发怀醛固酮增多时,肾远曲小管和集合管Na+-K+交换增加,因而起排钾保钠的作用。Cushing综合征时,糖皮质激素皮质醇的分泌大量增多。皮质醇也有一定的盐皮质激素样的作用。大量、长期的皮质醇增多也能促进远曲小管和集合管的Na+-K+交换而导致肾排钾增多。
④远曲小管中不易重吸收的阴离子增多:HCO3-、SO42-、HPO42-、NO3-、β-羟丁酸、乙酰乙酸、青霉素等均属此。它们在远曲小管液中增多时,由于不能被重吸收而增大原尿的负电荷,因而K+易从肾小管上皮细胞进入管腔液而随尿丧失。
⑤镁缺失:镁缺失常常引起低钾血症。髓袢升支的钾重吸收有赖于肾小管上皮细胞中的Na+-K+-ATR酶,而这种酶又需Mg2+的激活。缺镁时,可能因为细胞内Mg2+缺失而使此酶失活,因而该处钾重吸收发生障碍而致失钾。动物实验还证明,镁缺失还可引起醛固酮增多,这也可能是导致失钾的原因。
⑥碱中毒:碱中毒时,肾小管上皮细胞排H+减少,故H+-Na+交换加强,故随尿排钾增多。
(3)经皮肤失钾:汗液含钾只有9mmol/L。在一般情况下,出汗不致引起低钾血症。但在高温环境中进行重体力劳动时,大量出汗亦可导致钾的丧失。
3.细胞外钾向细胞内转移细胞外钾向细胞内转移时,可发生低钾血症,但在机体的含钾总量并不因而减少。
⑴低钾性周期性麻痹:发作时细胞外钾向细胞内转移,是一种家族性疾病。
⑵碱中毒:细胞内H+移至细胞外以起代偿作用,同时细胞外K+进入细胞。
⑶过量胰岛素:用大剂量胰岛素治疗糖尿病酮症酸中毒时,发生低钾血症的机制有二:
①胰岛素促进细胞糖原合成,糖原合成需要钾,血浆钾乃随葡萄糖进入细胞以合成糖原。
②胰岛素有可能直接剌激骨骼肌细胞膜上的Na+-K+-ATP酶,从而使肌细胞内Na+排出增多而细胞外K+进入肌细胞增多。
⑷钡中毒:抗日战争时期四川某地发生大批“趴病”病例,临床表现主要是肌肉软弱无力和瘫痪,严重者常因呼吸肌麻痹而死亡。经我国学者杜公振等研究,确定该病的原因是钡中毒。但当时钡中毒引起瘫痪的机制尚未阐明。现已确证,钡中毒引起瘫痪的机制在于钡中毒引起了低钾血症。钡中毒时,细胞膜上的Na+-K+-ATP酶继续活动。故细胞外液中的钾不断进入细胞。但钾从细胞内流出的孔道却被特异地阻断,因而发生低钾血症。引起钡中毒的是一些溶于酸的钡盐如醋酸钡、碳酸钡、氯化钡、氢氧化钡、硝酸钡和硫化钡等。
4.粗制生棉油中毒近二三十年来,在我国某些棉产区出现一种低血钾麻痹症,在一些省内又被称为“软病”。其临床主要特征是四肢肌肉极度软弱或发生弛缓性麻痹,严重者常因呼吸肌麻痹而死亡,血清钾浓度明显降低。往往在同一地区有许多人发病。病因与食用粗制生棉籽油有密切关系。粗制生棉油是农村一些小型油厂和榨坊生产的。这些厂的生产工艺不合规格。棉籽未经充分蒸炒甚至未曾脱壳就用来榨油,榨出的油又未按规定进行加碱精炼。因此棉籽中的许多毒性物质存于油中。与“软病”的发生和随后的一系列研究,都是棉酚(gossypol)。“软病”时低钾血症的发生机制尚未阐明。“软病”的发现和随后的一系列研究,都是我国学者进行的。迄今为止,国外的书刊中,尚无该病的记载。
(二)对机体的影响
低钾血症对机体的影响,在不同的个体有很大的差别。低钾血症的临床表现也常被原发病和钠水代谢紊乱所掩盖。低钾血症的症状取决于失钾的快慢和血钾降低的程度。失钾快则症状出现快,而且也较严重;失钾慢则缺钾虽已较重,症状也不一定显著。一般说来,血清钾浓度愈低,症状愈严重。但有一点应当强调指出,在可兴奋的组织内,兴奋性不仅与血清钾降低的程度有关。而更重要的还取决于细胞内钾浓度与细胞外钾浓度之比([K+]i/[K+]e)。比值大则兴奋性减低,比值小则兴奋性增高。
虽然细胞内的许多酶需要钾激活,但是细胞内钾浓度的轻度降低(例如从160降至130mmol/L)是否会明显地影响这些酶的活性,尚不清楚。
动物实验证明,缺钾时细胞内外发生离子交换。即细胞内K+逸出而细胞外Na+和H+进入细胞。缺钾比较严重时,细胞内Na+和H+的积聚可达到足以影响酶活性的程度。因此,缺钾引起的细胞功能障碍很可能是细胞内钠离子浓度和pH改变的结果。
低钾血症对机体的影响如下:
1.对骨骼肌的影响主要是超极化阻滞。低钾血症时[K+]i/[K+]e的比值增大,因而肌细胞静息电位负值增大。静息电位与阈电位的距离增大,细胞兴奋性于是降低,严重时甚至不能兴奋,亦即细胞处于超极化阻滞状态。临床上先是出现肌肉无力。继而可发生弛缓性麻痹。这种变化在四肢肌肉最为明显,严重者可发生呼吸肌麻痹,这是低钾血症患者的主要死亡原因之一(图5-3)。
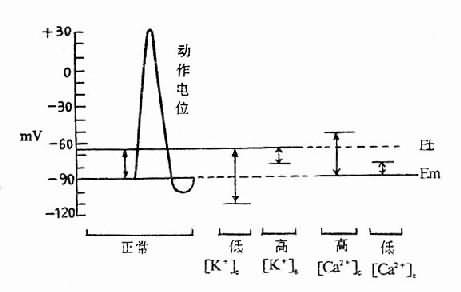
图5-3 细胞外液K+、Ca2+浓度和正常骨骼肌静息膜电位(Em)与阈电位(Et)的关系
肌肉兴奋性的这种变化在急性缺钾要比在慢性缺钾时严重得多。因为在急性缺钾时,细胞外钾浓度已经显著降低而细胞内钾在短时间内尚来不及较多地外逸,故细胞内外钾的浓度差明显增大,[K+]i/[K+]e比值显著增大。在慢性缺钾时,随着时间的推移。细胞内钾释出也较多,因而[K+]i/[K+]e比值变化可以不大。因此,同一水平的低钾血症,在急性缺钾患者可引起严重的肌肉麻痹而在慢性缺钾患者却可无明显的肌肉症状。
2.对心脏的影响
⑴对兴奋性的影响:按理论推测,细胞外液钾浓度降低时,由于细胞膜内外K+浓度差增大,细胞内K+外流应当增多而使心肌细胞静息电位负值增大而呈超极化状态。但实际上当血清钾浓度降低特别是明显降低(如低于3mmol/L)时,静息电位负值反而减少,这可能是由于细胞外液钾浓度降低时,心肌细胞膜的钾电导(potassium conductance)降低,从而使细胞内钾外流减少,而基础的内向钠电流使膜部分去极化所致。静息电位负值的减少使静息电位与阈电位的距离减小,因而引起兴奋所需的剌激也较小,所以心肌的兴奋性增高。细胞外液钾浓度降低时对钙内流的抑制作用减小,故钙内流加速而使复极化2期(坪期)缩短,心肌的有效不应期也随之而缩短。心肌细胞膜的钾电导降低所致的钾外流减小,又使3期复极的时间延长。近年有人从低钾血症病人的右心室尖部所记录的心肌细胞动作电位中也观察到3期复极时间的延长。3期复极时间的延长也就说明心肌超常期延长。上述变化使整个动作电位的时间延长,因而后一次0期除极化波可在前一次复极化完华之前到达。在心电图上可见反映2期复极的S-T段压低。相当于3期复极的T波压低和增宽,并可在其末期出现明显的U波,相当于心室动作电位时间的Q-T间期延长。
⑵对自律性的影响:在心房传导组织、房室束-浦肯野纤维网的快反应自律细胞,当3期复极末达到最大复极电位(-90mV)后,由于膜上Ik通道通透性进行性衰减使细胞内钾的外流逐渐减少,而钠离子又从细胞外缓慢而不断地进入细胞(背景电流),故进入细胞的正电荷量逐渐超过逸出细胞的正电荷量,膜就逐渐去极化,当到达阈电位时就发生0期去极化。这就是快反应细胞的自动去极化。在低钾血症时钾电导降低,故在到达最大复极电位后,细胞内钾的外流比正常减慢而钠内流相对加速。因而这些快反应自律细胞的自动去极化加速,自律性增高。
⑶对传导性的影响:低钾血症时因心肌静息电位负值变小,去极化时钠内流速度减慢。故0期膜内电位上升的速度减慢,幅度减小,兴奋的扩布因而减慢,心肌传导性降低。在心电图上,可见P-R间期延长,说明去极化波由心房传导到心室所需的时间延长,QRS综合波增宽,说明心室内传导性降低(图5-4)。
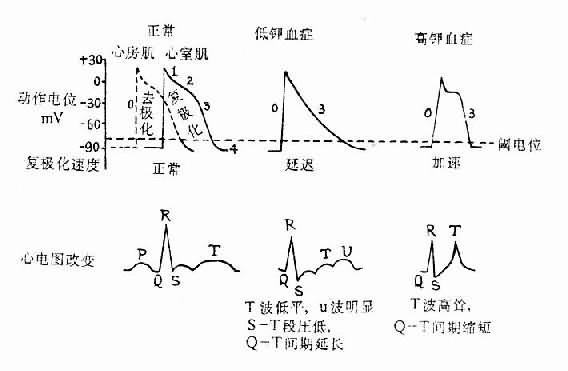
图5-4 血浆钾浓度对心肌细胞膜电位及心电图的影响
由上述可见,低钾血症时由于心肌兴奋性增高、超常期延长和异位起搏点自律性增高等原因,容易发生心律失常。传导性降低所致的传导缓慢和单向传导阻滞,加上有效不应期的缩短有助于兴奋折返,因而也可引起包括心室纤维颤动在内的心律失常。
(4)对收缩性的影响:如前所述,细胞外液钾浓度降低时对钙内流的抑制作用减小,故在2期复极时钙内流加速,心肌细胞内Ca2+浓度增高,兴奋-收缩偶联过程加强,心肌收缩性增强。然而,低钾血症对心肌收缩性的影响因缺钾的程度和持续时间而异:在早期或轻度低钾血症时,心肌收缩性增强;但在严重的慢性缺钾时,心肌收缩性减弱。与此相应的组织学变化是:在实验动物的心肌中可见横纹的消失、间质细胞浸润、不同程度的心肌坏死和瘢痕形成。由此也可以理解,有些严重慢性缺钾的狗,可因心力衰竭而发生肺水肿。然而在临床上,缺钾很少成为心力衰竭的原因。
3.对肾的影响
⑴尿浓缩功能障碍:在慢性缺钾伴有低钾血症时,常出现尿浓缩的障碍。由此可以理解,慢性缺钾的病人常有多尿和低比重尿的临床表现。尿浓缩功能障碍的发生机制在于:
①远曲小管对ADH的反应性不足;
②低钾血症时髓袢升支NaCL的重吸收不足以致髓质渗透压梯度的形成发生障碍。
⑵肾血流量减少:人和动物缺钾时都可发生肾血管收缩,从而引起肾血流量减少。引起肾血管收缩的因素有:
①肾内血管收缩性的前列腺素的生成不成比例地增多;
②血管紧张素Ⅱ的水平增高。
⑶肾小球滤过率减少:在实验动物,肾小球滤过率的减少似与肾血流量的减少平行。在病人,严重而持续的缺钾也可使肾小球滤过率明显减少。时间久后,可导致肾的器质性损害。
⑷肾形态结构的变化:在大鼠,缺钾引起的病变主要见于髓质集合管,表现为增殖性反应包括上皮细胞肿胀、增生和胞质内显著的颗粒形成。持久的缺钾可导致间质瘢痕形成、肾小球硬化和肾小管扩张等器质性变化。在人,慢性缺钾主要引起近曲小管上皮细胞的空泡形成,也可发生间质瘢痕形成、间质淋巴细胞浸润和肾小管萎缩等变化。
以上的变化中,除了显著的纤维化和肾组织的丧失以外,一般都是可复性的。
4.对胃肠的影响
钾缺乏可引起胃肠运动减弱。患者常发生恶心、呕吐和厌食,严重缺钾可致难以忍受的腹胀甚至麻痹性肠梗阻。
5.对代谢的影响
⑴糖代谢:血浆钾浓度的降低可抑制胰腺分泌胰岛素,因而低钾血症患者的糖原合成发生障碍,对葡萄糖的耐量不足,易发生高血糖。应当看到,这时的胰岛素分泌减少也有一定的代偿意义,因为胰岛素可通过保养进细胞内糖原合成和直接剌激骨骼肌细胞膜上的Na+K+-ATP酶而使细胞外钾向组织内转移。可见低钾血症时的胰岛素分泌减少,有助于防止血浆钾浓度的进一步降低。
⑵蛋白代谢:缺钾可以引起负氮平衡,因为钾是蛋白合成所必需。在儿童,钾缺乏可以成为生长障碍的原因之一。
⑶水、电解质和酸碱平衡:
①醛固酮分泌减少;血浆钾浓度降低能直接抑制肾上腺皮质球带合成醛固酮。血浆醛固酮水平的降低能减少肾远曲小管等对钾的排泄,因而也有一定的代偿意义;
②肾产氨增加:低钾血症时可能通过细胞内酸中毒而使肾脏远曲小管产氨增加,氨排出的增多可使远曲小管排钾减少,因而也有代偿意义;
③多尿多饮:慢性缺钾时,尿浓缩功能减退,因而排出大量低比重尿。水分的丧失引起渴感。动物实验证明缺钾也能剌激渴感,从而引起多饮。
④肾排氯增多:缺钾时,全部肾小管特别是其远侧部分对氯的重吸收减少。
⑤酸碱平衡:低钾血症患者的酸碱平衡状态与原发疾病或引起低钾血症的原因有密切关系。例如,当原发疾病为肾小管酸中毒,或引起缺钾的原因为腹泻时,患者就可伴有代谢性酸中毒。当引起缺钾的原因是长时间应用高效能利尿药如速尿、利尿酸时,患者就有代谢性碱中毒。但是,缺钾和低钾血症本身却往往倾向于引起代谢性碱中毒。这是因为,第一,低钾血症时,远曲小管内K+-Na+交换减少,故H+-Na+交换增多,因而排H+增多;而且,如前所述,低钾血症时肾远曲小管产氨和排氨增多,氨又可与H+增多结合成NH4+而排出;第二,低钾血症时(原因为细胞外钾向细胞内转移者除外),细胞内K+向细胞外释出,细胞外的H+进入细胞,从而使细胞外液H+浓度降低;第三,如前所述,缺钾时肾排氯增多,而机体缺氯可引起代谢性碱中毒(参阅《酸碱平衡紊乱》)。可见在一个具体的低钾血症患者,酸碱平衡的状态是由原发疾病、缺钾原因和低钾血症本身的影响来共同决定的。
(三)防治原则
1.防治原发疾病,去除引起缺钾的原因如停用某些利尿药等。
2.补钾如果低钾血症较重(血清钾低于2.5~3.0mmol/L)或者还有显著的临床表现如心律失常、肌肉瘫痪等,则应及时补钾。
补钾最好口服,每天以40~120mmol为宜。只有当情况危急,缺钾即将引起威胁生命的并发症时,或者因恶心、呕吐等原因使患者不能口服时才应静脉内补钾。而且,只有当每日尿量在500ml以上才容许静脉内补钾。输入液的钾浓度不得超过40mmol/L,每小时滴入的量一般不应超过10mmol。静脉内补钾时要定时测定血钾浓度,作心电图描记以进行监护。
细胞内缺钾恢复较慢,有时需补钾4~6日后细胞内外的钾才能达到平衡,有的严重的慢性缺钾患者需补钾10~15日以上。
如低钾血症伴有代谢性碱中毒或酸碱状态无明显变化,宜用KCL。KCL对各种原因引起的低钾血症实际上也都适用,因为低钾血症本身就可以引起缺氯。如低钾血症伴有酸中毒,则可用KHCO3或柠檬酸钾,以同时纠正低钾血症和酸中毒。
3.纠正水和其它电解质代谢紊乱引起低钾血症的原因中,有不少可以同时引起水和其他电解质如钠、镁等的丧失,因此应当及时检查,一经发现就必须积极处理。如前所述,如果低钾血症是由缺镁引起,则如不补镁,单纯补钾是无效的。
二、高钾血症
血清钾浓度高于55mmol/L称为高钾血症(hyperkalemia)。体内钾过多在理论上可以引起细胞内钾含量增高。但在实际上,高钾血症极少伴有可测知的细胞内钾含量的增高。这可能是因为,只要有相对小量的钾在体内贮留,就会引起威胁生命的高钾血症,而且这也说明,细胞内容纳钾积聚的余地是很小的。
应当指出,高钾血症也未必总是伴有体内钾过多。例如,在未经治疗的糖尿病酮症酸中毒病人,可因渗透性利尿(因高血糖所致)而使尿钾的排出大量增加,机体因而可处于缺钾状态。但是,大量失水所致的肾血流量减少和肾小球滤过率减少等原因,又可导致高钾血症。
(一)原因和机制
1.钾潴留
⑴钾摄入过多:在肾功能正常时,因钾摄入过多而引起高钾血症是罕见的。当然,静脉内过多过快地输入钾盐是有可能起高钾血症的。口服钾不足以引起威胁生命的高钾血症,因为胃肠道对钾的吸收有限,而且在大量口服钾盐时还会引起呕吐或腹泻。
⑵肾排钾减少:这是引起高钾血症的主要原因。临床上高钾血症最常见于不论何种原因引起的急性而严重的肾小球滤过率减少。任何原因引起的少尿也常伴有高钾血症。这些情况,主要见于急性肾功能衰竭。在慢性肾功能衰竭的发展过程中,病人逐渐对排钾进行适应,表现为每一肾单位排钾量增加。据估计,肾正常的成人每天可以摄入大约每公斤体重10mmol的钾而不发生明显的高钾血症。如果一个病人的肾小球滤过率减至每分钟10ml(正常的1/10弱),他还能每天随尿排钾60mmol而使血清钾水平保持正常;如果肾小球滤过率降至每分钟5ml,则病人还能每天随尿排钾30mmol;在这些病人,肠道排钾也随着慢性肾功能衰竭的发展而适应性地增加,因而又可有10~20mmol的钾每天随粪排出,血清钾仍得以保持于正常范围。然而,肾小球滤过率显著减少的病人显然不能排出额外的大的钾负荷。当然,慢性肾功能衰竭晚期患者尿量过少时,也可因钾的排出过少而发生高钾血症。
⑶肾小管分泌钾的功能缺陷:在间质性肾炎患者,肾小管和肾间质受损,故肾小管泌钾功能障碍;全身性红斑狼疮、肾的淀粉样变等也可损害肾小管而使其泌钾功能受损。
⑷盐皮质激素缺乏:醛固酮主要作用于肾远曲小管和集合管,促进其对钠的重吸收和钾氢的排泌。此外,对结肠粘膜、涎腺及汗腺等也有同样的作用。
醛固酮的不足较常见于Addison病。患者盐皮质激素和糖皮质激素都不足。最常见的电解质紊乱是低钠血症。如果食盐摄入充分,还不致发生高钾血症。如限制食盐就会导致高钾血症,因为限制食盐摄入时,到达远曲小管的Na+减少,Na+-K+交换因而减少,再加上因醛固酮和皮质醇的不足而使肾排钾减少,因而易于导致高钾血症。
⑸留钾利尿药的大量使用:安体舒通是醛固酮的对抗药,能抵消醛固酮的排钾保钠作用,长时间大量应用时可导致钾在体内潴留而导致高钾血症。氨苯蝶啶能抑制远曲小管和集合管对钾的分泌,长时间大量应用也可引起钾在体内潴留和高钾血症。
2.细胞内钾释出过多
⑴酸中毒:酸中毒可伴有高钾血症,因为酸中毒时细胞外液的H+进入细胞而细胞内的K+释出至细胞外。
⑵缺氧:缺氧时细胞内ATP生成不足,细胞膜上钠-钾泵运转发生障碍,故钠离子潴留于细胞内,细胞外液中的K+不易进入细胞。
⑶高钾性周期性麻痹:发作时细胞内钾向细胞外转移,是一种家族性疾病。
⑷细胞和组织的损伤和破坏。
①血管内溶血:重度溶血如血型不合输血时,红细胞的破坏使大量理K+进入血浆。此时如肾功能正常,则过多的钾还可随尿排出,若伴有肾功能损害,即可发生明显的高钾血症。
②严重创伤特别是在挤压综合征(crush syndrome)伴有肌肉组织大量损伤时,从损伤组织可释出大量的K+。特别是挤压综合征常伴有急性肾功能衰竭,因而易发生威胁生命的高钾血症。
(二)对机体的影响
1.对骨骼肌的影响轻度高钾血症(血清钾5.5~7mmol/L)时,细胞外液钾浓度的增高使[K+]i/[K+]e的比值减小,静息期细胞内K+外流减少,因而静息电位负值减小,与阈电位的距离减小,引起兴奋所需的阈剌激也较小,即肌肉的兴奋性增高。临床上可出现肢体感觉异常、剌痛、肌肉震颤等症状。在严重高钾血症(血清钾7~9mmol/L)时骨骼肌细胞的静息电位过小,因而快钠孔道失活,细胞处于去极化阻滞状态而不能被兴奋。临床上可出现肌肉软弱甚至驰缓性麻痹等症状。肌肉症状常先出现于四肢,然后向躯干发展,也可波及呼吸肌。
高钾血症对骨骼肌的影响比较次要,因为在骨骼肌完全麻痹以前,病人往往已因致命性的心律紊乱或心搏骤停而死亡。
2.对心脏的影响
⑴对兴奋性的影响:与对骨骼肌的影响相似,在轻度高钾血症时,[K+]i/[K+]e比值减小,静息期细胞内K+外流减少,静息电位负值减小,故心肌兴奋性增高。静息电位减小说明细胞膜处于部分去极化状态,因而在动作电位的0期,膜内电位上升的速度较慢,幅度较小。这是因为在部分去极化的状态下,膜的快钠孔道部分失活,所以在0期钠的快速内流减少。当血清钾显著升高时,由于静息电位过小,心肌兴奋性也将降低甚至消失,因为这时快钠孔道大部或全部都已失活,心搏可因而停止。
高钾血症时携带复极化钾电流的Ix孔道在开放的速度与程度上都加大,故钾外流加速,复极化3期加速,因此动作电位时间和有效不应期均缩短。Ix孔道开放的加速与加大,虽然也倾向于使复极化2期(坪)缩短,但由于细胞外液中K+浓度的增高抑制了Ca2+在2期的内流,故坪实际上有所延长。心电图上相当于心室复极化的T波狭窄而高耸,相当于心室动作电位时间的Q-T间期缩短。
⑵对自律性的影响:在高钾血症时,心房传导组织、房室束-浦肯野纤维网的快反应自律细胞膜上的钾电导增高,故在到达最大复极电位后,细胞内钾的外流比正常时加快而钠内流相对减慢,因而自动去极化减慢,自律性降低。
⑶对传导性的影响:如前文所述,高钾血症时动作电位0期膜内电位上升的速度减慢,幅度减小,因而兴奋的扩布减慢,传导性降低,故心房内,房室间或心室内均可发生传导延缓或阻滞。心电图上可见代表心房去极化的P波压低、增宽或消失,代表房-室传导的P-R间期延长,代表心室去极化的R波降低,代表心室内传导的QRS综合波增宽。
高钾血症时心肌传导性的降低也可引起传导缓慢和单向阻滞,同时有效不应期又缩短,因而也易形成兴奋折返并进而引起包括心室纤维颤动在内的心律失常。严重的高钾血症时可因心肌兴奋性消失或严重的传导阻滞而导致心搏骤停。
⑷对收缩性的影响:如前所述,高钾血症时细胞外液K+浓度的增高抑制了心肌复极2期时Ca2+的内流,故心肌细胞同内Ca2+浓度降低,兴奋-收缩偶联减弱,收缩性降低。
应当提到,无论是对于骨骼肌还是对于心脏,血钾升高的速度愈快,影响也愈严重。
3.对内分泌和电解质、酸碱平衡的影响
⑴胰岛素和高血糖素;血浆K+浓度上升1.0mmol/L以上时便能直接剌激胰岛素释放。胰岛素的增多可促进骨骼肌细胞摄取细胞外液中的K+,因而在高钾血症时有代偿意义。与此同时,高钾血症还直接剌激胰高血糖素的分泌,后者与增多的胰岛素共同维持血糖的调节。
⑵儿茶酚胺:大鼠实验证明,血浆钾浓度的显著增高能使血浆肾上腺素水平升高。肾上腺素对α受体和β受体都有剌激作用。静脉内注射肾上腺素后最初3分钟内引起血钾浓度升高。这是肾上腺素作用于α受体使肝释放K+的结果。随后,由于肾上腺素剌激骨骼肌细胞的β受体,从而使骨骼肌加速摄取细胞外钾,故也有代偿意义。
⑶电解质和酸碱平衡:高钾血症似能减少肾产氨,从而使H+排出减少而倾向于发生代谢性酸中毒。酸中毒的另一原因是高钾血症时细胞外液K+移入细胞内而细胞内的H+移向细胞外。高钾血症还有利钠作用。但高钾血症作用于肾单位的哪一部分而引起Na+的重吸收减少,尚不清楚。高钾血症又能直接剌激肾上腺皮质球状带,使醛固酮的分泌增多,醛固酮的增多能促进钾的排出,故有代偿意义。而且,醛固酮的增多还能抵消高钾血症的利钠作用。从而减少机体失钠。
(三)防治原则
1.防治原发疾病,去除引起高钾血症的原因包括严禁静脉内推注钾溶液等。
2.降低血钾 如果心电图上除了T波高耸还有其他变化,如果血清K+浓度高于6.5mmol/L,必须迅速采取强有力的措施来降低血钾。
⑴使钾向细胞内转移:葡萄糖和胰岛素同时静脉内注射。可使细胞外钾向细胞内转移。应用碳酸氢钠(不能与钙剂一起注射)不仅能通过提高血浆pH,并且还能通过对K+的直接作用而促使K+进入细胞内。
⑵使钾排出体外:阳离子交换树脂聚苯乙烯磺酸钠(sodium polystyrene sulfonate)经口服或灌肠应用后,能在胃肠道内进行Na+-K+交换而促进体钾排出。对于严重高钾血症患者,可用腹膜透析或血液透析来移除体内过多的钾。
3.注射钙剂和钠盐 Ca2+能使阈电位上移(负值减小),使静息电位与阈电位间的距离稍为拉开,因而心肌细胞的兴奋性也倾向于有所恢复。此外,细胞外液Ca2+浓度的增高,可使复极化2期钙内流增加,从而使心肌细胞内Ca2+浓度增高,心肌收缩性增强。输入钠盐后细胞外液Na+浓度增高,心肌细胞内外电化学梯度增大,故在去极化时钠内流加快,动作电位0期上升速度可有所加快,幅度也有所增大,故可改善心肌的传导性。
4.纠正其他电解质代谢紊乱在引起高钾血症的原因中,有些也可以同时引起高镁血症,故应及时检查并给予相应的处理(见后文)。
第四节 镁代谢紊乱
镁在含量上是机体内第四位的阳离子,仅次于钙、钠、钾;在细胞内,镁的含量仅次于钾而占第二位。
镁代谢紊乱主要是指细胞外液中镁浓度的变化,包括低镁血症(hypomagnesemia)和高镁血症(hypermagnesemia)。
一、低镁血症
(一)原因和机制
1.镁摄入不足一般饮食含镁也比较丰富,故只要能正常进食,机体就不致缺镁。成人每天镁的摄入量约为10mmol(20mEq)。营养不良、长期禁食、厌食、长期经静脉营养未注意镁的补充均可导致镁摄入不足,而小量的镁仍继续随尿排出,故可发生低镁血症。
2.镁排出过多
⑴经胃肠道排出过多;正常时饮食中镁的40~70%随粪便排出体外。严重的腹泻和持续的胃肠吸引可使镁经消化道吸收减少而排出过多。
⑵经肾排镁过多:正常肾小球滤过的镁约有25%在近曲小管被重吸收,60~70%在髓袢升支和远曲小管重吸收。随尿排出的镁,大约相当于摄入镁量的30~60%。在下列情况下,肾排镁增多:
①利尿药:特别是髓袢利尿药如加速尿、利尿酸等可抑制髓袢对镁的重吸收而致镁丧失,长期使用时可引起低镁血症。由甘露醇、尿素或葡萄糖所致的渗透性利尿亦可引起镁随尿排出过多。
②高钙血症:钙与镁在肾小管中被重吸收时有相互竟争的作用,因而任何原因引起的高钙血症(如甲状旁腺功能亢进、维生素D中毒时)均可使肾小管重吸收镁减少。甲状旁腺激素(parathyroid hormone,PTH)有促进肾小管重吸收镁的作用。甲状旁腺功能亢进时,过多的PTH本应使更多的镁在肾小管内重吸收,但这种作用被高钙血症所完全对消。
③严重的甲状旁腺功能减退:由于PTH减少,肾小管中镁的重吸收减少。
④醛固酮增多:醛固酮也能抑制肾小管重吸收镁,故原发性醛固酮增多症和各种原因引起的继发性醛固酮增多症均可能引起低镁血症。
⑤糖尿病酮症酸中毒:酸中毒能明显地妨碍肾小管对镁的重吸收,高血糖又可通过渗透性利尿而使镁随尿排出增多。
⑥酒精中毒:急慢性酒精中毒常伴有低镁血症,其机制是多因素性的:血中酒精浓度增高能增加肾脏排镁,可能乙醇能抑制肾小管对镁的重吸收;慢性洒精中毒者往往伴有营养不良和腹泻,等等。
⑦强心甙:洋地黄类药物也有促进肾排镁的作用。
⑧庆大霉素和二氨二氯络铂(cisplatin)引起肾小管损害时能使肾保镁的功能发生可复性的缺陷。
⑨肾疾患:急性肾小管坏死多尿期、慢性肾盂肾炎、肾小管酸中毒等疾病分别因渗透性利尿和肾小管功能受损而导致镁随尿排出增多。
3.细胞外液镁转入细胞过多用胰岛素治疗糖尿病酮症酸中毒时,因糖原合成需要镁,故细胞外液中的镁过多地转向细胞内液,故有助于引起低镁血症。
(二)对机体的影响
1.对神经-肌肉的影响在正常情况下,运动神经末梢在动作电位去极相的影响下,大量含乙酰胆碱的襄泡向轴突膜移动。通过出泡作用,大量乙酰胆碱得以释出至神经-肌肉接头的间隙。襄泡的释放除受轴突膜电位变化的影响外,还与细胞间液中的Ca2+和Mg2+的浓度有关。动作电位的去极相可引起膜上的Ca2+通道开放,而Ca2+的进入量也决定着襄泡释放的数量;Mg2+则能竟争性地进入轴突,对抗Ca2+的作用。低镁血症时,Ca2+的进入增多,故乙酰胆碱的释放量也增多。此外,Mg2+还能抑制终板膜上乙酰胆碱受体对乙酰胆碱的敏感性。低镁血症时,这种抑制减弱。因此,神经-肌肉接头处兴奋传递加强。而且,Mg2+还能抑制神经纤维和骨骼肌的应激性;低镁血症时,神经纤维和骨骼肌的应激性就增高,故在临床上可出现一系列神经-肌肉应激性增高的表现如小束肌纤维收缩、震颤、阳性的Chvostek征和Trousseau征和手足搐搦。Mg2+还有抑制中枢神经系统的作用;低镁血症时这种抑制减弱,故可出现反射亢进,对声、光反应的过强、焦虑、易激动等症状。Mg2+对平滑肌也有抑制作用,低镁血症时平滑肌的兴奋可导致呕吐或腹泻。
2.对代谢的影响
⑴低钙血症:中度至重度低镁血症常可引起低钙血症,其机制涉及到甲状旁腺机能的障碍。有人发现,在低镁血症时,病人循环血液中的免疫反应性甲状旁腺激素(immunoreactive PTH,IPTH)减少。如果给这种病人静脉内注射镁剂,则IPTH浓度在数分钟内即可明显升高,提示PTH的分泌有障碍而不象是合成有障碍。血钙降低剌激PTH分泌是通过甲状旁腺腺体细胞膜结合的腺苷酸环化酶介导的。此酶需Mg2+激活,而此时血浆Mg2+浓度降低,故不易激活此酶。因此,虽然血钙已有初步降低,也不能剌激甲状旁腺分泌PTH,血钙乃进一步降低而导致低钙血症。
此时,PTH的靶器官如骨骼系统和肾小管上皮等对PTH的反应也减弱。这是因为PTH也必须通过腺苷酸环化酶的介导才能促进靶器官的功能活动。低镁血症时靶器官上的腺苷酸环化酶同样也不能激活,因而骨钙的动员和钙在肾小管的重吸收发生障碍,血钙得不到补充。这也是低钙血症发生的重要原因。
⑵低钾血症:镁缺乏时常可出现低钾血症。实验证明,限制大鼠饮食中的镁含量可使尿钾排出增加,骨骼肌含量减少。如只补钾而不及时补镁,则血钾亦难以恢复。可见,低镁可使低钾难以纠正。临床上也可以看到,在某些病例,低镁是持续的难治性低钾的原因。在这些病例,如只补钾而不补镁,低钾血症同样也得不到纠正。关于低镁时肾保钾功能减退的机制,可参阅本章第三节:低钾血症的原因和机制。
3.对心脏的影响体外灌流实验证明,镁对于离体动物的心肌组织,有稳定其生物电活动的作用。去除灌流液中的Mg2+可使心肌细胞静息电位负值显著变小,说明缺镁可使心肌的兴奋性增高。此外,对于浦肯野细胞等快反应自律细胞的缓慢而恒定的钠内流,镁也有阻断作用,而这种内向电流,又是这些细胞自动去极化的一个基础。低镁血症时,这种阻断作用减弱,钠离子内流相对加速,因而心肌快反应自律细胞的自动去极化加速,自律性增高。由于缺镁时心肌的兴奋性和自律性均升高,故易发生心律失常。
除了直接作用外,缺镁也可通过引起低钾血症而导致心律失常,因为低钾血症也可使心肌的兴奋性和自律性增高,而且还能使有效不应期缩短,超常期延长。
低镁血症时的心律失常可以很严重,甚至也可发生心室纤维颤动。
除此以外,缺镁也可以引起心肌形态结构的变化。例如,因为镁是许多酶系所必需的辅因子,故严重缺镁可引起心肌细胞的代谢障碍从而导致心肌坏死,并可能过缺钾而引起心肌细胞完整性的破坏。动物实验中,缺镁饮食引起的心肌坏死可能与低镁血症使冠状血管痉挛有关。
(三)防治原则
1.防治原发疾病,防止或排除引起低镁血症的原因的作用。
2.补镁 严重低镁血症且有症状特别是各种类型的心律失常时必须及时补镁。对于缺镁引起的严重心律失常,其他疗法往往都无效果。只有静脉内缓慢注射或滴注镁盐(一般是用硫酸镁)才能奏效。静脉内补镁要谨慎,如患者肾功能受损,则更要格外小心。在补镁过程中要常常测定血清镁浓度,必须防止因补镁过快而转变为高镁血症。小儿静脉内补镁时还应特别注意防止低血压的发生,因为镁可使外周小动脉等血管扩张。对于较轻的低镁血症,也可通过肌肉内注射的途径补镁。补镁的剂量须视缺镁的程度和症状的轻重而定。
3.纠正水和其他电解质代谢紊乱 包括补水,特别是补钾和补钙,因为低镁血症常伴有失水、低钾血症和低钙血症。
二、高镁血症
血清镁浓度高于1.25mmol/L(2.5mEq/L)时为高镁血症。
(一)原因和机制
1.镁摄入过多见于静脉内补镁过快过多时。这种情况在肾功能受损的病人更易发生。
2.肾排镁过少正常时肾有很大的排镁能力,故口服或注射较多的镁盐在肾功能正常的人不致引起高镁血症。肾排镁减少是高镁血症最重要的原因,见于:
⑴肾功能衰竭:急性或慢性肾功能衰竭伴有少尿或无尿时,由于肾小球滤过功能减弱等原因,肾排镁减少,故易发生高镁血症。此时如果不适当地给病人应用含镁药物,更将促进和加重高镁血症。
⑵严重脱水伴有少尿:随着尿量减少,镁的排出也减少,故易发生高镁血症。
糖尿病酮症酸中毒昏迷患者在治疗前,往往因为多尿、呕吐、入水减少而发生严重的脱水和少尿,因而血清镁可以升高。此外,在胰岛素治疗前,细胞内分解代谢占优势,故细胞内镁向细胞外释出,这也是引起高镁血症的一个原因。
⑶甲状腺功能减退:甲状腺素有抑制肾小管重吸收镁,促进尿镁排出的作用,故某些粘液水肿的病人可能发生高镁血症。
⑷醛固酮减少:醛固酮也有抑制肾小管重吸收镁。促进尿镁排出的作用,故某些Addison病患者可发生高镁血症。
(二)对机体的影响
在血清镁浓度不超过2mmol/L(4mEq/L)时,临床上很难觉察高镁血症对机体的影响。只有当血清镁浓度升至3mmol/L(6mEq/L)或更高时,才可看到高镁血症所引起的临床症状:
1.对神经-肌肉接头处的影响镁能抑制神经-肌肉接头处的兴奋传递,高浓度的镁有箭毒样的作用。故高镁血症病人可发生显著的肌无力甚至也弛缓性麻痹,四肢、吞咽和呼吸肌都可以被波及,因而可导致驰缓性四瘫,吞咽和说话困难,严重者可因呼吸肌麻痹而死亡。
2.对中枢神经系统的影响镁能抑制中枢神经系统的突触传递,抑制中枢神经系统的功能活动。高镁血症因而也可以引起深腱反射减弱或消失,有的病人还可发生嗜睡或昏迷。
3.对心脏的影响高浓度的镁能抑制房室和心室内传导,并降低心肌兴奋性,故可引起传导阻滞和心动过缓。心电图上可见P-R间期延长和QRS综合波增宽。
4.对平滑肌的影响 镁对平滑肌亦有抑制作用。高镁血症时血管平滑肌的抑制可使小动脉、微动脉等扩张,从而导致外周阻力降低和动脉血压下降。对内脏平滑肌的抑制可引起暧气、呕吐、便秘、尿潴留等症状。
(三)防治原则
1.防治原发疾病,尽可能改善肾功能,包括纠正脱水。
2.静脉内注射葡萄糖酸钙,因为Ca2+在某些方面能与Mg2+相拮抗。
3.使镁排出体外 可用透析疗法以去除体内过多的镁。如肾功能尚好,也可以适当使用利尿药使肾排镁增多。
4.人工呼吸用于抢救呼吸肌麻痹患者。
5.治疗其他电解质紊乱引起高镁血症的原因往往也会引起高钾血症,因此应当及时检查血清钾,发现高钾血症后应积极治疗。
第六章 酸碱平衡紊乱
第一节 正常平衡的调节
在正常情况下,人体血浆pH值平均为7.4,变动范围很小(pH7.35~7.45)。而机体每日代谢产酸量是很大的,例如,非挥发酸可达50~100毫克当量,CO2可达400升。这些酸性物质必须及时处理,否则血浆pH值不能保持正常。这就靠一整套调节机构密切协同来完成。
一、化学缓冲物质的作用(化学反应可以瞬间完成)
血液中有一系列缓冲物质。根据化学上的缓冲作用原理,我们人为地把它们归纳为四个主要的缓冲对,即NaHCO3/H2CO3,Na2HPO4/NaH2PO4,B·血浆蛋白/H·血浆蛋白,B·Hb/H·Hb。它们具有很强且很迅速的缓冲酸碱度改变的能力。例如,我们将10mmol(毫克分子)的HC1加1000毫升中性蒸馏水中,其pH值可从7降至2,但同量酸加于1000毫升血浆中,其pH值的变化却很小,以致一般几乎测不出来。
每一对缓冲物质既能缓冲酯也能缓冲碱,其中以NaHCO3/H2CO3这一对最为重要,因为它的量最大。它对酸的缓冲区反应如下:
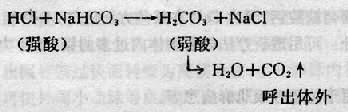
从上面的反应可以看出,经NaHCO3缓冲,解离度大的强酸HC1转变为解离度小的弱酸H2CO3,后者在体液中的解离度仅约为前者的1/1500。因此使[H+]大为减小。而且H2CO3还能分解为H2O和CO2,CO2又能呼出体外。医学实践中将血浆中NaHCO3称为碱贮备,以二氧化碳结合力表示之。正常值为27mmol(或毫克当量)/升或60容积%。
二、离子转移(一般在2~4小时完成)
这也是缓冲方式之一。当能改变酸碱平衡的离子如H+、HCO3-等在细胞外液中升高时,它们能进入细胞内(或进入骨内)换出与其符号相同的离子,以保持细胞外液之pH值。其变化可简单表示如下:
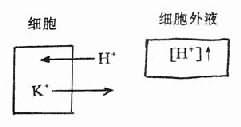
进入细胞内的H+可与细胞内的化学缓冲物质起化学反应而被缓冲。根据实验研究估算,酸或碱在细胞外液增加后,一般约经2~4小时,将有1/2的量进入细胞内。所以尽管细胞内液量大于细胞外液量一倍,但二者化学缓冲总能力却大致相等,可见细胞外液中缓冲物质的浓度是高于细胞内液的。
这个事实对治疗有关。举一代谢性酸中毒病人用NaHCO3治疗时的情况为例:
病人体重=60公斤
细胞外液量=0.20×60=12升
治疗前血浆[HCO3-]=13mmo1(或毫克当量)/升
给NaHCO3量=100mmo1

病人治疗后实测血浆[HCO3-]=16.7mmol/升
实践证明,上述规律是较为可靠的。因此在纠正酸碱中毒时应考虑离子转移这一变化。计算输液量时要将这一变化估计在内。
三、呼吸调节
呼吸调节(一般10~30分钟即可将轻度的一次性变动代偿过来)。[H+]增高和[CO2]增高,均能剌激呼吸中枢;H+还对颈动脉体和主动脉体的化学感受器起刺激作用,这都可引起呼吸加深加快,使CO2排出增加。从上面介绍的缓冲反应来看,每排出一个CO2分子,也就等于清除了一个H+离子,如:
H++HCO3-→+H2CO→H2C+CO2↑(增多) 碱贮 (呼出)
四、肾脏调节
肾脏调节(数小时至数日完成):肾脏是酸碱平衡调节的最终保证。因为只有CO2可以通过呼吸排出体外(CO2即可代表可挥发酸H2CO3,因为CO2+H2O→H2CO3),而其它如乳酸、丙酮酸、β-羟丁酸、乙酰乙酸、硫酸、磷酸、尿酸、草酸等均为非挥发酸,最终均需通过肾脏把前面三项调节所造成的变动调整过来。把过多的排出,把不足的保留下来。因此肾脏调节机体酸碱平衡的功能正常与否,关系重大。
肾脏排酸有三种形式:
(一)自由H+:虽然它决定着尿液pH值,但是其量却很少(每日只1~2毫克当量)。尿pH为5时,[H+]为0.00001克分子,pH为8时,[H+]为0.0000000克分子。尽管它确是一种存在形式,它只是尿中大量酸性物质的一个很小部分。而肾脏排酸主要是以下二种形式。
(二)NH4-:是H+与NH3作用形成的。NH3由肾小管产生,在体内是很强的碱性物,与H+的亲和力极大。它弥散至尿中与H+结合。示意如下:
人体每日由代射所产生的非挥发酸总量约50~100mmol,其中一半以上(1/2~2/3)是以此种形式排出的。
(三)可滴定酸:是由H+与缓冲物质(主要是HPO4=作用形成的。用NaOH把尿滴定至与血浆相同之pH,即可测知可滴定酸量。可滴定酸的形式约占肾排酸总量的少一半(1/3~1/2)。
肾小管上皮细胞分泌的H+,在近曲小管几乎全部用于重吸收HCO3-;在远曲小管则几乎全部用于生成可滴定酸和NH4+。这与肾脏疾病的代谢性酸中毒如肾小管性酸中毒的关系密切。可滴定酸的形成示意如下:
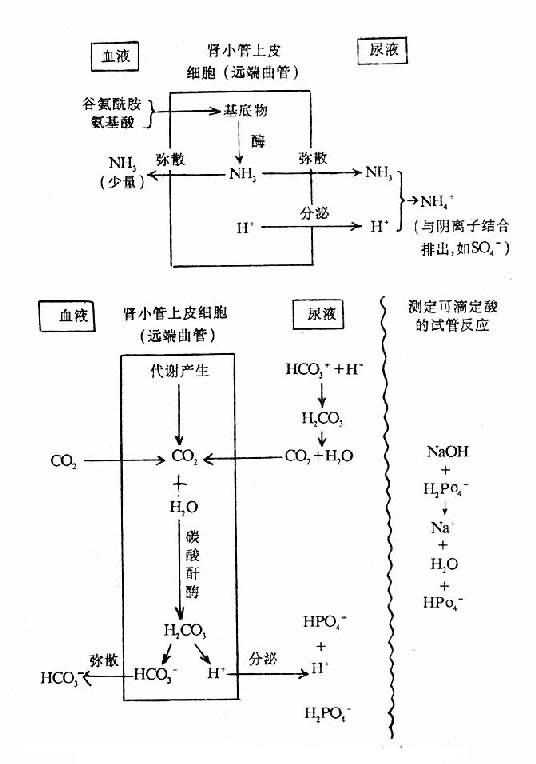
第二节 平衡失调的检测指标
一、pH值
血液pH值易于测定,是酸碱平衡障碍的一个很有用的指标。它反映体液中的氢离子浓度[H+],其值是以[H+]的负对数表示。婴幼儿低于儿童,儿童低于成人。例如新生儿血浆pH值为7.3~7.35,就处于成人pH值7.35~7.45的下限以下。这是因为年龄越小血浆二氧化碳分压越高,乃属正常生理范围。正常成人动脉血液pH值比静脉血液者高约0.02~0.1,组织间液pH值与血浆者近似。细胞内液pH比细胞外液者低,视细胞代谢旺盛程度而不同,其范围约6.0~7.4,平均7.0。
血浆的pH值主要取决于血浆中[HCO3-]与[H2CO3]的比值,其间的相互关系可从Henderson-Hasselbalch方程式中看得很清楚。式中pKa是H2CO3解离常数的负对数值,
![]()
在37℃环境中为6.1,此数值被视为一个比较恒定的数字。[HCO3-]主要由肾脏调节,动脉血浆中的浓度平均为24mmol/L。[H2CO3]由于可以分解为水和CO2,因此可以由呼吸调节。而且血浆中溶解的CO2只有很小部分呈H2CO3形式,CO2与H2CO3之比仅约800:1。由于实用上的方便我们不把这两者分开,而是由血浆CO2的溶解系数计出[H2CO3]。即当Pco2为0.133kPa(1mmHg)时,血浆中溶解的CO2和H2CO3的浓度为0.03mmol/L。Pco2的正常值平均为5.33kPa(40mmHg),计算[H2CO3]=40×=1.2mmol/L。代入上式,则正常的血浆pH=6.1+log(24/1.2)=6.1+log20=6.1+1.3=7.4。
血浆pH低于正常表明有酸中毒,高于正常表明有碱中毒。但只看pH的变化还不能区分是代射性还是呼吸性酸碱中毒。要区分代谢性和呼吸性酸碱中毒,还需要知道[HCO3-]和[H2CO3]何者是原发性变化者,即谁是先起变化的。当血浆[H2CO3]原发性上升或[HCO3-]原发性降低,以致pH小于7.35时,即为失代偿性酸中毒,前者称为呼吸性酸中毒,后者称为代谢性酸中毒。当血浆[H2CO3]原发性降低或[HCO3-]原发性增高以致pH大于7.45时即为失代偿性碱中毒,前者称为呼吸性碱中毒,后者称为代谢性碱中毒。
pH值处于正常范围内,也可能存在酸碱平衡障碍。因为在酸碱中毒时,通过机体的上述调节作用,尽管[HCO3-]和[H2CO3]的绝对值已经发生改变,但二者的比值仍维持在20:1附近,pH值则可保持于正常范围内。这类情况则称为代偿性酸中毒或碱中毒。代偿性酸中毒时血浆pH值在正常范围的近下限处,代偿性碱性酸中毒时血浆pH值在正常范围的近上限处。但需要注意,在某些类型的混合型酸碱平衡障碍时,血浆pH值可以是正常的。这在后面将予以介绍。
二、二氧化碳分压
二氧化碳分压(Pco2)是指物理溶解于动脉血浆中的CO2分子所产生的压力(即张力)。其正常平均值为5.33kPa(40mmHg,范围为33~47mmHg)。Pco2是反应呼吸性酸碱平衡障碍的重要指标。由于通气过度,CO2排出过多,其值低于正常,是为呼吸性碱中毒的变化。由于通气不足,CO2排出过少而在体内潴留,其值高于正常,是为呼吸性酸中毒的变化。在代谢性酸中毒时,由于呼吸加深加快的代偿反应,可使病人Pco2值下降而低于正常。也就是说当[HCO3-]原发性下降时,H2CO3-有代偿性继发的下降以使NaHCO3/H2CO3比值变动尽量减少或仍能维持20:1。在代射性碱中毒时,则与此相反,Pco2值可上升而高于正常。
三、二氧化碳结合力
二氧化碳结合力(CO2Combining Power,CO2C.P.)是指静脉血浆HCO3-中的CO2含量,亦即呈化学结合状态的CO2量。正常值平均为27mmol/l 。也常用容积%来表示这个指标的值,正常平均为60容积%,范围为50~70容积%。
血浆二氧化碳结合力可反应血浆中NaHCO3的含量。二氧化碳结合力增高时可能是代谢性碱中毒,也可能是有代偿反应的呼吸性酸中毒;因为后者是H2CO3原发性升高,NaHCO3便会有代偿性的继发增高。二氧化碳结合力降低时可能是代谢性酸中毒,也可能是有代偿反应的呼吸性碱中毒。二氧化碳结合力的测定方法建立于1917年,它在测定血浆NaHCO3(碱贮)上起过重要作用,现因测定pH值、Pco2等方便可靠,故二氧化碳结合力的测定方法已日渐少用。
四、标准碳酸氢盐和实际碳酸氢盐
标准碳酸氢盐(Standard bicarbonate,S.B.)是指动脉血液标本在38℃和血红蛋白完全氧合的条件下,用Pco2为5.33kPa的气体平衡后所测得的血浆[HCO3-]。因为这种方法已排除呼吸因素的影响,故为判断代谢性酸碱中毒的指标。正常平均值为24mmo1/L,范围为22~27mmol/L。代谢性酸中毒病人S.B.降低,代谢性碱中毒时则S.B.升高。但在慢性呼吸性酸中毒中或碱中毒时,由于代偿作用(肾脏长时间调节)也可在前者有所升高,后者有所降低。
实际碳酸氢盐(Actual bicarbonate,A.B.)是指隔绝空气的血液标本,在保持其原有Pco2和血氧饱和度不变的条件下测得的血浆碳酸氢盐浓度。因此A.B.受代谢和呼吸两方面因素的影响。它和二氧化碳结合力的差别主要是A.B.反映动脉血浆的值,二氧化碳结合力是用静脉血浆测定的。A.B.的正常值同S.B.,因为正常人的条件和测定S.B.的人工条件是相同的。但A.B.与S.B.的差值能反映呼吸因素对酸碱平衡的影响。正常人,A.B.=S.B.。病人有CO2贮积时,A.B>S.B.,指示呼吸性酸中毒;病人有CO2呼出过多即通气过度时,A.B.<S.B.,指示呼吸性碱中毒。两者数值均高于正常指示有代谢性碱中毒(或慢性呼吸性酸中毒有代偿变化)。
五、缓冲碱
缓冲碱(buffer base,B.B)是指动脉血液中具有缓冲作用的碱性物质的总和。也就是人体血液中具有缓冲作用的负离子的总和。这些负离子有HCO3-,HPO4=,Hb-,Pr-等,它们都能结合H+。通常用氧饱和的全血测定,这样测出的称全血缓冲碱(buffer base of blood, BBb)正常值为50±5mEq/L,其中:
HCO3- =20mEq/L
HPO4= =7mEq/L
Pr- =8mEq/L
Hb- =15mEq/L
如果离出血浆测定其缓冲碱总量,则称为血浆缓冲碱(buffer baseof plasma, BBp),正常值为40~48mmol/L,平均45mEq/L,其中:
HCO3- =22~27mEq/L
HPO4= =2~3mEq/L
Pr- =16~18mEq/L
一般使用血液缓冲碱总量一词均指全血缓冲碱而言,它是反映代谢性因素的指标,呼吸因素所造成Pco2升高或降低对它没有明显影响。B.B.降低指示代谢性酸中毒,B.B.升高指示代谢性碱中毒。
六、碱过剩和碱缺失
碱过剩(base excess, B.E.)是指在标准条件下,即在38℃,Pco25.33kPa,Hb为15g%,100%氧饱和的情况下,用酸或碱将人体1升全血滴定至正常pH7.4时所用的酸或碱的mmol数。如需用酸滴定,显然指示血中碱量多于正常,即称为碱过剩。它代表全血缓冲碱总量的变化。但表示法不同,是以用去酸量的mEq数表示,意即多出多少mEq缓冲碱,此种情况用正值即+B.E.表示,见于代谢性酸中毒。反之,如需用碱滴定,则表示碱缺失(basedeficit),指示血中缓冲碱减少,此种情况用负值表示即-B.E.。见于代谢性酸中毒。但在慢性呼吸性酸中毒或碱中毒时,由于肾脏的长时间代偿作用,B.E.也可以分别增加或减少。正常人的B.E.值在0附近,正常范围为0±3mEq/L。
碱过剩也分全血与血浆(代表细胞外液)二种测法。细胞外液碱过剩(B.E.ecf)是反映代谢性因素的较好指标;因为测定时血浆或其它细胞外液需经Pco2为5.33kPa的气体平衡,可以排出血液中Pco2升降的影响。
全血缓冲碱(B.E.b)由于包括Hb在内,故受病人Hb的量的影响,Hb是全血缓冲碱的重要成份,所以测定后要视病人Hb浓度按照方便的公式予以校正。
七、负离子间隙
负离子间隙(Anion Gap, AG)是指从血浆中的未测定负离子(Undetermined Anion, UA)包括:

减去未测定的正离子(Undertermined Cation, UC)包括:

二者之量的差值。即AG=UA-UC,亦即我们通常测定的[Na+]-([Cl-]+[HCO3-])。其关系如图6-1。
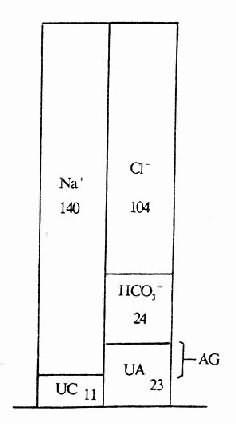
图 6-1 血浆负离子间隙示意图(数字为mEq/L)
AG正常值为12±2mEq/L。AG对于区别代谢性酸中毒的原因有重要作用。这在代谢性酸中毒中部份将予以介绍。
小结:上面介绍了七项常用的反映酸碱平衡障碍的指标,是当前临床上经常使用的,要熟悉其含义及在诊断上的意义。尽管如此,现在有不少临床工作者认为测定pH、Pco2和HCO3-三个指标即可区别呼吸性或代谢性酸、碱中毒,并且也可以区别代偿性或失代偿性酸、碱中毒。
至于S.B.、B.B.、B.E.等指标,由于排除了呼吸因素的影响,且抽血不需隔绝空气,有其优点,临床工作者也非常重视。它们对于诊断有特殊的帮助。不过需注意的是这些指标都是在体外用全血测定,滴定结果与体内的情况有些差别。例如,在急性Pco2改变时,同样的Pco2水平在体外作用于全血后可形成较多的HCO3-,而在体内则是作用于全血后可形成较多的HCO3-,而在体内则是作用于全身细胞,形成的HCO3-就较少,这是由于红细胞比体内的其它细胞具有更大的缓冲CO2增加的能力,故能形成较多HCO3-。不过这点差别还不至影响临床实用所要求的精确性,故而目前普遍在实际中应用。
第三节 酸中毒
一、代谢性酸中毒
代谢性酸中毒(Metabolic Acidosis)的特征是血浆[HCO-]原发性减少。
代谢性酸中毒又可根据AG是否增加分为二类:AG增加类代谢性酸中毒,病人血浆[Cl-]水平正常,亦即文献上经常提到的正常血氯性代谢性酸在毒。AG正常类代谢性酸中毒,病人血浆[Cl-]水平却升高,亦即文献上经常提到高血氯性代谢性酸中毒(见图6-2)。这其间的关系我们在本章未尾部分介绍清楚。
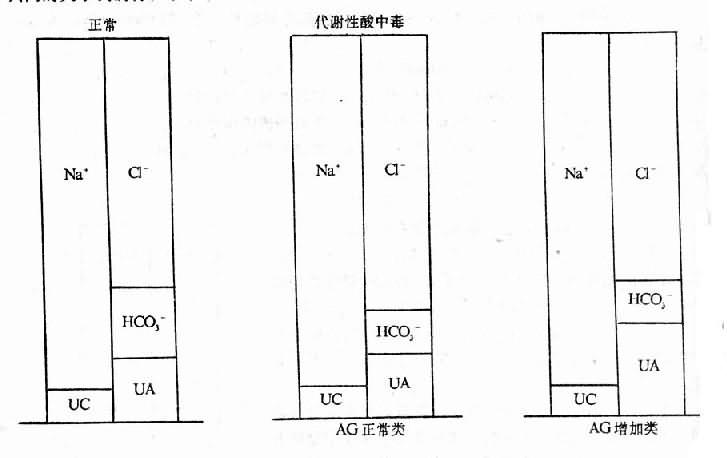
图6-2 正常和代谢性酸中毒时的负离子间隙改变类型
(一)原因和机制
1.酸性物质产生过多
(1)乳酸酸中毒:乳酸酸中毒(Lactic Acidosis)可见于各种原因引起的缺氧,其发病机制是缺氧时糖酵解过程加强,乳酸生成增加,因氧化过程不足而积累,导致血乳酸水平升高。这种酸中毒很常见。临床上伴有缺氧的病人休克、严重贫血、呼吸暂停、心脏停搏、CO中毒、氰化物中毒、癫痫发作及过于剧烈的运动、洒精中毒时的心脏呼吸抑制、严重肝病时肝脏对乳酸代谢障碍、糖尿病病人的糖氧化障碍、白血病时可能出现的恶性细胞糖酵解和加强等等均经常遇到。
乳酸酸中毒的特点:
血液中乳酸浓度升高,例如严重休克病人动脉血乳酸水平升高10倍以上。
血液中[乳酸-]/[丙酮酸-]比值增大(正常血浆乳酸浓度约1mmol/L,丙酮酸浓度约0.1mmol/L,二者比值为10:1)。
AG增大,血氯正常。故属于AG增加类正常血氯性代表谢性酸中毒。此种酸中毒血浆乳酸浓度常可超过6mmol/L,高者可达12mmol/L。[乳酸根-]是未测定负离子之一,其增加当使负离子间隙增加。这种病人丙酮酸也有增加。
(2)酮症酸中毒:酮症酸中毒(Ketoacidosis)是本体脂大量动用的情况下,如糖尿病、饥饿、妊娠反应较长时间有呕吐症状者、酒精中毒呕吐并数日少进食物者,脂肪酸在肝内氧化加强,酮体生成增加并超过了肝外利用量,因而出现酮血症。酮体包括丙酮、β-羟丁酸、乙酰乙酸,后两者是有机酸,导致代谢性酸中毒。这种酸中毒也是AG增加类正常血氯性代谢性酸中毒。
因胰岛素缺乏而发生糖尿病的病人,可以出现严重的酮症酸中毒,甚而致死。因为正常时人体胰岛素对抗脂解激素,使指解维持常量。当胰岛素缺乏时,脂解激素如ACTH、皮质醇、胰高血糖素及生长激素等的作用加强,大量激活脂肪细胞内的脂肪酶,使甘油三酯分解为甘油和脂肪酸的过程加强,脂肪酸大量进入肝脏,肝脏则生酮显著增加。
肝脏生酮增加与肉毒碱酰基转移酶(Acylcarnitine transferase)活性升高有关。因为正常时胰岛素对比酶具有抑制性调节作用,当胰岛毒缺乏时此酶活性显著增强。这时进入肝脏的脂肪酸形成脂肪酰辅酶A(Fatty acyl- CoA)之后,在此酶作用下大量进入线粒体,经β-氧化而生成大量的乙酰辅酶A,乙酰辅酶A是合成酮体的基础物质。正常情况下,乙酰辅酶A经柠檬酸合成酶的催化与草酰乙酸缩合成柠檬酸而进入三羧酸循环,或经乙酰辅酶A羧化酶的作用生成丙二酰辅酶A而合成脂肪酸,因此乙酰辅酶A合成酮体的量是很少的,肝外完全可以利用。此外,糖尿病病人肝细胞中增多的脂肪酰辅酶A还能抑制柠檬酸合成酶和乙酰辅酶A羧化酶的活性,使乙酰辅酶A进入三羧酸循环的通路不畅,同时也不易合成脂肪酸。这样就使大量乙酰辅酶a 肝内缩合成酮体。
非糖尿病病人的酮症酸中毒是糖原消耗补充不足,机体进而大量动用脂肪所致,如饥饿等。
图6-3 糖尿病酮症酸中毒的可能机制
图6-4 糖尿病时酮体生成增多的机制
2.肾脏排酸保碱功能障碍
不论肾小管上皮细胞H+排泌减少和碳酸氢盐生成减少还是肾小球滤过率严重下降,不论急性或慢性肾功能衰竭,均能引起肾性代谢性酸中毒。由于肾脏是机体酸碱平衡调节的最终保证,故肾衰的酸中毒更为严重,也是不得不采取血液透析措施的临床危重情况之一。
(1)肾功能衰竭:肾功能衰竭如果主要是由于肾小管功能障碍所引起时,则此时的代谢性酸中毒主要是因小管上皮细胞产NH3及排H+减少所致。正常肾小管上皮细胞内谷氨酰胺及氨基酸由血液供应,在谷氨酰胺酶及氨基酸化酶的催化作用下不断生成NH3,NH3弥散入管腔与肾小管上皮细胞分泌的H+结合形成NH4+,使尿液pH值升高,这就能使H+不断分泌入管腔,完成排酸过程。原尿中的Na+被NH4+不断换回,与HCO3-相伴而重新入血成为NaHCO3。这就是肾小管的主要排酸保碱功能。当肾小管发生病变从而引起此功能严重障碍时,即可发生酸中毒。此类酸中毒因肾小球滤过功能无大变化,并无酸类的阴离子因滤过障碍而在体内潴留,其特点为AG正常类高血氯性代谢性酸中毒。也就是说HPO4=、SO4=等阴离子没有潴留,故AG不增加,而HCO3-重吸收不足,则由另一种容易调节的阴离子Cl-代替,从而血氯上升。
肾功能衰竭如果主要是肾小球病变而使滤过功能障碍,则一般当肾小球滤过率不足正常的20%时,血浆中未测定阴离子HPO3=、SO4=和一些有机酸均可因潴留而增多。这时的特点是AG增加类正常血氯性代谢性酸中毒。HPO4=滤出减少,可以使可滴定酸排出减少,从而导致H+在体内潴留。
图6-3 糖尿病酮症酸中毒的可能机制
(2)碳酸酐酶抑制剂:例如使用乙酰唑胺作为利尿时,由于该药物抑制了肾小管上皮细胞中的碳酸酐酶活性,使CO2+H2O→H2CO3→H++HCO3-反应减弱,H+分泌减少,HCO3-重吸收减少,从而导致AG正常类高血氯性酸中毒。此时Na+、K+、HCO3-从尿中排出高于正常,可起利尿作用,用药时间长要注意上述类型酸中毒。
(3)肾小管性酸中毒:肾小管性酸中毒(Renal Tubular Acidosis, RTA)是肾脏酸化尿液的功能障碍而引起的AG正常类高血氯性代谢性酸中毒。目前按其发病机理可分四型。
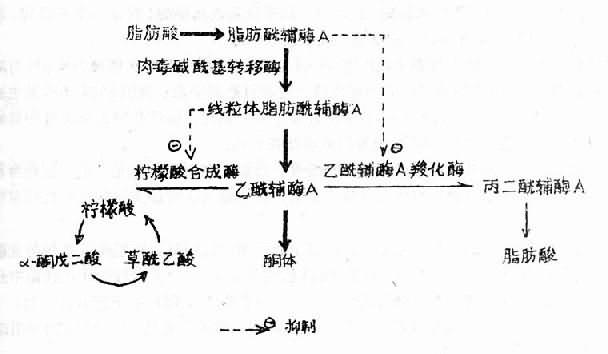
图6-4 糖尿病时酮体生成增多的机制
Ⅰ型-远端肾小管性酸中毒(Distal RTA)。是远端小管排H+障碍引起的。此时远端小管不能形成并维持正常管内与管周液的H+陡峭浓度差。小管上皮细胞形成H2CO3障碍,且管腔内H+还可弥散回管周液。它可能是肾小管上皮细胞排H+的一系列结构、功能和代谢的不正常引起的。其病因有原发性、自身免疫性、肾钙化、药物中毒(两性霉素B、甲苯、锂化合物、某些镇痛剂及麻醉剂)、肾盂肾炎、尿路阻塞、肾移植、麻疯、遗传性疾病、肝硬化等。
Ⅱ型—近端肾小管性酸中毒(Proximal RTA)。是近端小管重吸收HCO3-障碍引起的。此时尿中有大量HCO3-排出,血浆HCO3-降低。如果我们人为地将这类病人的血浆HCO3-升至正常水平并维持之,即可到肾丢失HCO3-超过滤过量的15%,这是一个很大的量。因此可导致严重酸中毒。当血浆HCO3-显著下降,酸中毒严重时,病人尿中HCO3-也就很少了,用上述办法方可观测到其障碍之所在。此型RTA的发病机理可能系主动转运的能量不足所致,多系遗传性的代谢障碍。
Ⅲ型-即Ⅰ-Ⅱ混合型,既有远端小管酸化尿的功能障碍,也有近端曲管重吸收HCO3-的障碍。
Ⅳ型-据目前资料认为系远端曲管阳离子交换障碍所致。此时管腔膜对H+通过有障碍。病人有低肾素性低醛固酮血症,高血钾。K+高时,与H+竞争,也使肾NH4+排出下降,H+潴留。常见于醛固酮缺乏症、肾脏对醛固酮反应性降低或其他如Ⅰ型或Ⅱ型的一些原因引起。
(4)肾上腺皮质功能低下(阿狄森氏病):一方面由于肾血流量下降,缓冲物质滤过减少,形成可滴定酸少;另一方面由于Na+重吸收减少,NH3和H+的排出也就减少,因为Na+的重吸收与NH3及H+的排出之间存在着一个交换关系。
3.肾外失碱肠液、胰液和胆汁中的[HCO3-]均高于血浆中的[HCO3-]水平。故当腹泻、肠瘘、肠道减压吸引等时,可因大量丢失[HCO3-]而引起AG正常类高血氯性代谢性酸中毒。输尿管乙状结肠吻合术后亦可丢失大量HCO3-而导致此类型酸中毒,其机理可能是Cl-被动重吸收而HCO3-大量排出,即Cl--HCO3-交换所致。
4.酸或成酸性药物摄入或输入过多氯化铵在肝脏内能分解生成氨和盐酸,用此祛痰剂日久量大可引起酸中毒。NH4Cl→NH3+H++Cl-。为AG正常类高血氯性代谢性酸中毒。氯化钙使用日久量大亦能导致此类酸中毒,其机制是Ca++在肠中吸收少,而Cl-与H+相伴随而被吸收,其量多于Ca++,Ca++能在肠内与缓冲碱之一的HPO4=相结合,使HPO4=吸收减少。Ca++也能与H2PO4-相结合生成不吸收的Ca3(PO4)2和H+,而H+伴随Cl-而被吸收。
水杨酸制剂如阿斯匹林(乙酰水杨酸)在体内可迅速分解成水杨酸,它是一个有机酸,消耗血浆的HCO3-,引起AG增加类正常血氯性代谢性酸中毒。
甲醇中毒时由于甲醇在体内代谢生成甲酸,可引起严重酸中毒,有的病例报告血pH可降至6.8。误饮含甲醇的工业酒精或将甲醇当作酒精饮用者可造成中毒。我国1987年曾发生过大批中毒病例。除甲醇的其它中毒危害外,AG增加类正常血氯性代谢性酸中毒是急性中毒的重要死亡原因之一。积极作用NaHCO3抢救的道理就在于此。
酸性食物如蛋白质代谢最终可形成硫酸、酮酸等,当然,在正常人并无问题。但是当肾功能低下时,高蛋白饮食是可能导致代谢性酸中毒的。这也是AG增加类正常血氯性代谢性酸中毒。
输注氨基酸溶液或水解蛋白溶液过多时,亦可引起代谢性酸中毒,特别是氨基酸的盐酸盐,在代谢中会分解出HCl来。这些溶液制备时pH值均调至7.4,但其盐酸盐能在代谢中分解出盐酸这一点仍需注意。临床上根据情况给病人补充一定量NaHCO3的道理就在于此。
5.稀释性酸中毒大量输入生理盐水,可以稀释体内的HCO3-并使Cl-增加,因而引起AG正常类高血氯性代谢性酸中毒。
(二)机体的代偿调节
机体发生代谢性酸中毒时,前面所提到的一整套调节机构将发挥代偿调节作用。如能保持pH值在正常范围内则称代偿性代谢性酸中毒,pH值低于正常下限则为失代偿性代谢性酸中毒。
1.细胞外液缓冲酸中毒时细胞外液[H+]升高,立即引起缓冲化学反应。以缓冲碱中HCO3-这一数量最多的为例,反应如下:
H++HCO3-→H2CO3→H2O+CO2↑
CO2通过呼吸加强而排出,HCO3-减少。
2.呼吸代偿 [H+]升高时,剌激延脑呼吸中枢、颈动脉体和主动脉体化学感受器,引起呼吸加深加快,肺泡通气量加大,排出更多CO2。
3.细胞外离子交换 H+进入细胞,K+出至细胞外。H+离子在细胞内与缓冲物质Pr-、HPO4=、Hb-等结合而被缓冲。H+亦能与骨内阳离子交换而缓冲。
4.肾脏代偿代谢性酸中毒非因肾脏功能障碍引起者,可由肾脏代偿。肾脏排酸的三种形式均加强。
(1)排H+增加,HCO3-重吸收加强:酸中毒时肾小管上皮细胞的碳酸酐酶活性增高,生成H+及HCO3-增多,H+分泌入管腔,换回Na+与HCO3-相伴而重吸收。显然这是一种排酸保碱过程。
(2)NH4+排出增多:酸中毒时肾小管上皮细胞产生NH3增多,可能是产NH3的底物如谷氨酰胺此时易于进入线粒体进行代谢的缘故。NH3弥散入管腔与H+结合生成NH4+,再结合阴离子从尿排出。这是肾脏排H+的主要方式,故代偿作用大。此过程伴有NaHCO3重吸收的增多。
(3)可滴定酸排出增加:酸中毒时肾小管上皮细胞H+分泌增多,能形成更多的酸性磷酸盐。
Na2HPO4+H+→NH2PO4+Na+(排出) (伴HCO3-重吸收)
Na2HPO4多带一个H+排出,同时也有碳酸氢钠重吸收的增加。Na2HPO4即是可滴定其量的酸性物质。
失代偿性代谢性酸中毒时反映酸碱平衡的指标变化如下:
pH↓ CO2C.P.↓
S.B. ↓ B.B.↓
A.B.↓ B.E.负值增大
A.G.未测定负离子增多者A.G.增加
未测定负离子不增者B.G.不增加
(三)对机体的影响
代谢性酸中毒对心血管和神经系统的功能有影响。特别是严重的酸中毒,发展急速时可由于这两大重要系统的功能障碍而导致死亡。慢性酸中毒还能影响骨骼系统。
1.心血管系统功能障碍:H+离子浓度升高时,心血管系统可发生下述变化:
(1)毛细血管前括约肌在[H+]升高时,对儿茶酚胺类的反应性降低,因而松弛扩张;但微静脉、小静脉都不如此敏感,因而仍能在一定[H+]限度内保持原口径。这种前松后不松的微循环血管状态,导致毛细血管容量不断扩大,回心血量减少,血压下降,严重时可发生休克。
(2)心脏收缩力减弱,搏出量减少。正常时Ca++与肌钙蛋白的钙受体结合是心肌收缩的重要步骤,但在酸中毒H+与Ca++竞争而抑制了Ca++的这种结合,故心肌收缩性减弱。既可加重微循环障碍,也可因供氧不足而加重已存在的酸中毒。
(3)心律失常:当细胞外液[H+]升高时,H+进入细胞内换出K+,使血钾浓度升高而出现高钾血症,从而引起心律失常。此外酸中毒时肾小管上皮细胞排H+增多,竞争性地抑制排K+,也是高钾血症的机制之一。再就是肾功能衰竭引起的酸中毒,高钾血症更为严重。此种心律失常表现为心脏传导阻滞和心室纤维性颤动。
2.神经系统功能障碍;代谢性酸中毒时神经系统功能障碍主要表现为抑制,严重者可发生嗜睡或昏迷。其发病机制可能与下列因素有关:(1)酸中毒时脑组织中谷氨酸脱羧酶活性增强,故γ-氨基丁酸生成增多,该物质对中枢神经系统有抑制作用:(2)酸中毒时生物氧化酶类的活性减弱,氧化磷酸化过程也因而减弱,ATP生成也就减少,因而脑组织能量供应不足。
3.骨骼系统的变化:慢性代谢性酸中毒如慢性肾功能衰竭、肾小管性酸中毒均可长时间存在达数年之久,由于不断从骨骼释放出钙盐,影响小儿骨骼的生长发育并可引起纤维性骨炎和佝偻病。在成人则可发生骨质软化病。
除以上三个主要方面的影响外,其它如呼吸功能也有改变。在代谢方面因许多酶的活性受抑制而有代谢紊乱。
(四)防治原则
1.积极防治引起代谢性酸中毒的原发病,纠正水、电解质紊乱,恢复有效循环血量,改善组织血液灌流状况,改善肾功能等。
2.给碱纠正代谢性酸中毒:严重酸中毒危及生命,则要及时给碱纠正。一般多用NaHCO3以补充HCO3-,去缓冲H+。乳酸钠也可用,不过在肝功能不全或乳酸酸中毒时不用,因为乳酸钠经肝代谢方能生成NaHCO3。三羟甲基氨基甲烷(Tris-hydroxymethylAminomethane THAM或Tris)近来常用。它不含Na+、HCO3-或CO2。其分子结构式为(CH2OH)3CNH2,它是以其OH-去中和H+的
例如:H2CO3+OH-→H2O+HCO3-;HCl+OH-→H2O+Cl-。可挥发酸均能中和。因此它可以用于代谢性酸中毒、呼吸性酸中毒,也可用于混合性酸中毒病人。
它的缺点是用得过多过快,病人呼吸抑制能导致缺氧及CO2重新积累。因为它能同时迅速降低H+和Pco2之故。此外,此药输注时不可漏出血管外,因为剌激性强能引起组织坏死。这些均应在使用中加以注意。
3.处理酸中毒时的高钾血症和病人失钾时的低钾血症:酸中毒常伴有高钾血症,在给碱纠正酸中毒时,H+从细胞内移至细胞外不断被缓冲,K+则从细胞外重新移向细胞内从而使血钾回降。但需注意,有的代谢性酸中毒病人因有失钾情况存在,虽有酸中毒但伴随着低血钾。纠正其酸中毒时血清钾浓度更会进一步下降引起严重甚至致命的低血钾。这种情况见于糖尿病人渗透性利尿而失钾,腹泻病人失钾等。纠正其酸中毒时需要依据血清钾下降程度适当补钾。
严重肾功能衰竭引起的酸中毒,则需进行腹膜透析或血液透析方能纠正其水、电解质、酸碱平衡以及代谢尾产物潴留等紊乱。
二、呼吸性酸中毒
呼吸性中毒(Respiratory Acidosis)的特征是血浆[H2CO3]原发性增高。
(一)原因和机制
1.呼吸中枢抑制一些中枢神经系统的病变如延脑肿瘤、延脑型脊髓灰质炎、脑炎、脑膜炎、椎动脉栓塞或血栓形成、颅内压升高、颅脑外伤等时,呼吸中枢活动可受抑制,使通气减少而CO2蓄积。此外,一些药物如麻醉剂、镇静剂、镇静剂(吗啡、巴比妥钠等)均有抑制呼吸的作用,剂量过大亦可引起通气不足。碳酸酐酶抑制剂如乙酰唑胺能引起代谢性酸中毒前已述及。它也能抑制红细胞中的碳酸酐酶而使CO2在肺内从红细胞中释放减少,从而引起动脉血Pco2升高。有酸中毒倾向的伤病员应慎用此药。
2.呼吸神经、肌肉功能障碍 见于脊髓灰质炎、急性感染性多发性神经炎(Guillain-barre综合征)肉毒中毒,重症肌无力,低钾血症或家族性周期性麻痹,高位脊髓损伤等。严重者呼吸肌可麻痹。
3.胸廓异常胸廓异常影响呼吸运动常见的有脊柱后、侧凸,连枷胸(Flail Chest),关系强直性脊柱炎(Ankylosing Spondylitis),心肺性肥胖综合征(Picwick综合征)等。
4.气道阻塞常见的有异物阻塞、喉头水肿和呕吐物的吸入等。
5.广泛性肺疾病是呼吸性酸中毒的最常见的原因。它包括慢性阻塞性肺疾病、支气管哮喘、严重间质性肺疾病等。这些病变均能严重妨碍肺泡通气。
6.CO2吸入过多指吸入气中CO2浓度过高,如坑道、坦克等空间狭小通风不良之环境中。此时肺泡通气量并不减少。
(二)机体的代偿调节
由于呼吸性酸中毒是由呼吸障碍引起,故呼吸代偿难以发挥。H2CO3增加可由非碳酸氢盐缓冲系统进行缓冲,并生成HCO3-。但这种缓冲是有限度的。
1.细胞内外离子交换和细胞内缓冲这是急性呼吸性酸中毒的主要代偿调节。
细胞内外离子交换是指细胞外液[H+]升高时,H+进入细胞内,换出同符号的K+等。这样可以缓解细胞外液[H+]的升高。这与代谢性酸中毒时的离子交换一样。
所胃细胞内缓冲,是指进入细胞内的H+为细胞内缓冲物质如蛋白质(Pr-)等所缓冲,以及CO2弥散进入红细胞内的反应。此时由于CO2的蓄积而使Pco2急速升高,CO2通过红细胞膜进入红细胞内的正常过程加强。CO2与H2O在红细胞碳酸酐酶的催化下生成H2CO3,H2CO3解离为H+与H2CO3-。H+由血红蛋白缓冲,HCO3-转移至血浆中,使血浆HCO3-呈代偿性增加,如能使NaHCO3/H2CO3保持在正常范围内,则为代偿性呼吸性酸中毒。由于急性呼吸性酸中毒常发展迅速,肾脏代偿缓慢,故常为失代偿性的。
以CO2弥散入红细胞的量增加为例,其反应如下:

急性呼吸酸中毒时Pco2升高,H2CO3增多,[HCO3-]升高代偿,Pco2与[HCO3-]二者关系如下式所示:
△[HCO3-]=△Pco2×0.07±1.5
例如Pco2从5.33kPa(40mmHg)迅速升高至10.7kPa(80mmHg)时,血浆[HCO3-]仅上升约3mmol(mEq)/L。且[HCO3-]的代偿性增加还有一定限度,在急性呼吸性酸中毒时,AB增加也不会超过30mmol(mEq)/L,因此常为失代偿性者。
2.肾脏代偿是慢性呼吸性酸中毒的主要代偿措施。肾脏是酸碱平衡调节的最终保证,但它的调节活动却比较缓慢,约6-12小时显示其作用,3-5日达最大效应。慢性呼吸性酸中毒指持续24小时以上的CO2蓄积,它也有急性呼吸性酸中毒时的离子交换和细胞内缓冲,但肾脏却可发挥其产NH3↑、排H+↑及重吸收NaHCO3↑的功能,使代偿更为有效。
慢性呼吸性酸中毒血浆[HCO3-]与Pco2之间的关系,如下式所示:
△[HCO3-]=△Pco2×0.4±3
例如,Pco2每升高1.33kPa(10mmHg),血浆[HCO3-]浓度可升高4mmol(mEq)/L左右。这比急性呼吸性酸中毒的代偿要有效得多,能在一定时期内维持pH值于正常范围呈代偿性呼吸性酸中毒。
失代偿性呼吸性酸中毒血浆反映酸碱平衡的指标变化如下:
pH↓ CO2C.P↑
S.B. 不变(有低偿性增加时可略↑)
A.B.↑
B.B.不变(有代偿性增加时可略↑)
B.E.不变(有代偿性增加时可呈正值↑)
此种酸中毒由于Cl-进入红细胞增多(HCO3-自红细胞转移至细胞外),故血浆CL-降低。
(三)对机体的影响
呼吸性酸中毒对机体的影响,就其体液[H+]升高的危害而言,与低谢性酸中毒是一致的。但呼吸性酸中毒特别是急性者因肾脏的代偿性调节比较缓慢,故常呈失代偿而更显严重。
呼吸性酸中毒可有CO2麻醉现象。这就使其神经系统症状更为严重。CO2麻醉的初期症状是头痛、视觉模糊、疲乏无力,进一步加重则病人表现精神错乱、震颤、谵妄、嗜睡直至昏迷。高浓度CO2麻醉时病人颅内压升高,视神经乳头可有水肿,这是由于CO2扩张脑血管所致。此外病人脑脊液pH值下降较其它细胞外液更多。这是由于CO2为脂溶性,极易通过血脑屏障;HCO3-为水溶性,通过此屏障缓慢之故。
呼吸性酸中毒时心血管方面的变化和代谢性酸中毒一致。也有微循环容量增大、血压下降,心肌收缩力减弱、心输出量下降和心律失常。因为这两类酸中毒时[H+]升高并能导致高钾血症是一致的。
呼吸性酸中毒病人可能伴有缺氧,这也是使病情加重的一个因素。
(四)防治原则
1.积极防治引起的呼吸性酸中毒的原发病。
2.改善肺泡通气,排出过多的CO2。根据情况可行气管切开,人工呼吸,解除支气管痉挛,祛痰,给氧等措施。
人工呼吸要适度,因为呼吸性酸中毒时NaHCO3/H2CO3中H2CO3原发性升高,NaH2CO3呈代偿性继发性升高。如果通气过度则血浆Pco2迅速下降,而NaHCO3仍在高水平,则病人转化为细胞外液碱中毒,脑脊液的情况也如此。可以引起低钾血症、血浆Ca++下降、中枢神经系统细胞外液碱中毒、昏迷甚至死亡。
3.酸中毒严重时如病人昏迷、心律失常,可给THAM治疗以中和过高的[H+]。NaHCO3溶液亦可使用,不过必须保证在有充分的肺泡通气的条件下才可作用。因为给NaHCO3纠正呼吸性酸中毒体液中过高的[H+],能生成CO2,如不能充分排出,会使CO2深度升高。
![]()
第四节 碱中毒
一、代谢性碱中毒
代谢性碱中毒(Metabolic Alkalosis)的特征是血浆[HCO3-]原发性增多。
(一)原因和机制
1.氢离子丢失过多
(1)胃液丢失:常见于幽门梗阻或高位肠梗阻时的剧烈呕吐,直接丢失胃酸(HCl)。胃腺壁细胞生成HCl,H+是胃腺壁细胞由CO2+H2O→H2CO3→H++HCO3-反应而来,Cl-则来自血浆。壁细胞中有碳酸酐酶促进此反应能迅速进行。H+与Cl-在胃腺腔内形成HCl分泌入胃内。进入小肠后HCl与肠液、胰液、胆汁等碱性消化液中的NaHCO3中和。碱性液的分泌是受H+入肠的剌激引起的。因此,如果HCl因呕吐而丢失,则肠液中NaHCO3分泌减少,体内将有潴留;再者,已分泌入肠的NaHCO3不被HCl中和,势必引起肠液中[HCO3-]升高而使其重吸收增加。这就使血中[HCO3-]上升而导致代谢性碱中毒。
胃液大量丢失时可伴有Cl+、K+的丢失和细胞外液容量减少,这些因素也与此时的低谢性碱中毒发生有关。低血Cl-时,同符号负离子HCO3-增多以补偿之,低血K+时由于离子转移而H+移入细胞内,细胞外液容量减少时由于醛固酮分泌增多而促进Na+重吸收而促使H+和K+排出,这些均能引起代谢性碱中毒。
(2)肾脏排H+过多:肾脏排出H+过多主要是由于醛固酮分泌增加引起的。醛固酮能促进远曲小管和集合管排出H+及K+,而加强Na+的重吸收。H+排出增多则由于H2COH3→H++HCO3-的反应,HCO3-生成多,与Na+相伴而重吸收也增加,从而引起代谢性碱中毒,同时也伴有低钾血症。
醛固酮分泌增加见于下列情况:①原发性醛固酮增多症。②柯兴综合征(Cushint’s Syndrome)—常由垂体分泌ACTH的肿瘤、原发性肾上腺皮质增生或肿瘤等所引起。皮质醇等激素的生成和释放增多,皮质醇也有盐皮质激素的活性,故亦能导致代谢性碱中毒。③先天性肾上腺皮质增生—可分为两型,17-羟化酶缺乏型(非男性化)和11-羟化酶缺乏型(男性化)。因为这些酶缺乏而导致皮质醇合成减少,血中皮质醇水平下降反馈地引起垂体分泌过多ACTH,促进肾上腺皮质合成并分泌更多脱氧皮质酮(Deoxycorticorticosterone,DOC)和皮质酮(Corticosterone)。DOC则具有明显的盐皮质激素活性。④Bartter综合征—这是以近球装置增生而肾素分泌增多为特点的综合征。通过肾素→血管紧张素→醛固酮系统引起醛固酮分泌增多,病人无高血压是因为其血管对血管紧张素Ⅱ的反应性降低。由于病人前列腺素分泌增多,故近年也提出交感神经兴奋而使前列腺素增多从而导致肾素分泌增多的机制。例如使用消炎痛抑制前列腺素合成,可以降低病人肾素及醛固酮水平,并使代谢性碱中毒及Na+、K+恢复正常。⑤近球装置肿瘤,其细胞能分泌大量肾素,引起高血压及代谢性碱中毒。⑥甘草及其制剂长期大量使用时,由于甘草酸(Glycyrrhizic Acid)具有盐皮质激素活性,故能引起类似醛固酮增多症时的代谢性碱中毒。⑦细胞外液容量减少时引起醛固酮分泌增多以加强Na+重吸收而保容量,可引起代谢性碱中毒。常见于速尿、利尿酸等髓袢利尿剂尿时或大量胃液丧失时。此种情况下,细胞外液每减少1升,血浆[HCO3-]约增加1.4mmol/L。速尿和利尿酸除可使细胞外液减少外,其抑制肾小管髓袢升支对Cl-、Na+的重吸收能导致到达远端曲管的Na+增多而使远端曲管排H+换Na+过程加强,这也与代谢性碱中毒的发生有关。⑧创伤和手术时的应激反应时有肾上腺皮质激素分泌增多,常伴以代谢性碱中毒。
2.碱性物质摄入过多
(1)碳酸氢盐摄入过多:例如溃疡病人服用过量的碳酸氢钠,中和胃酸后导致肠内NaHCO3明显升高时,特别是肾功能有障碍的病人由于肾脏调节HCO3-的能力下降可导致碱中毒。此外,在纠正酸中毒时,输入碳酸氢钠过量也同样会导致碱中毒。
(2)乳酸钠摄入过多:经肝脏代谢生成HCO3-。见于纠正酸中毒时输乳酸钠溶液过量。
(3)柠檬酸钠摄入过多:输血时所用液多用柠檬酸钠抗凝。每500毫升血液中有柠檬酸钠16.8mEq,经肝代谢性可生成HCO3-。故大量输血时(例如快速输入3000~4000毫升)可发生代谢性碱中毒。
3.缺钾
各种原因引起的血清钾减少,可引起血浆NaHCO3增多而发生代谢性碱中毒。其机理有①血清K+下降时,肾小管上皮细胞排K+相应减少而排H+增加,换回Na+、HCO3-增加。此时的代谢性碱中毒,不像一般碱中毒时排碱性尿,它却排酸性尿,称为反常酸性尿。②血清钾下降时,由于离子交换,K+移至细胞外以补充细胞外液的K+,而H+则进入细胞内以维持电中性,故导致代谢性碱中毒(此时细胞内却是酸中毒,当然细胞内冲物质可以缓冲进入细胞内的H+)。
4.缺氯:由于Cl-是肾小管中唯一的容易与Na+相继重吸收的阴离子,当原尿中[Cl-]降低时,肾小管便加强H+、K+的排出以换回Na+,HCO3-的重吸收增加,从而生成NaHCO3。因此低氯血症时由于失H+、K+而NaHCO3重吸收有增加,故能导致代谢性碱中毒。此时病人尿Cl-是降低的。另外,前述之速尿及利尿酸能抑制髓袢升支粗段对Cl-的主动重吸收从而造成缺Cl-。此时远端曲管加强排H+、K+以换回到达远端曲管过多的Na+。故同样可导致代谢性碱中毒。此时病人尿Cl-是升高的。
呕吐失去HCl,就是失Cl-,血浆及尿中Cl-下降,通过上述原尿中Cl-降低机制促使代谢性碱中毒发生。
(二)机体的代偿调节
1.细胞外液缓冲 代谢性碱中毒时体液[H+]降低,[OH-]升高,则OH-+H2CO3→HCO3-+H2O,OH-+HPr-→Pr-+H2O,以缓冲而减弱其碱性。体液中NaHCO3升高可与其它缓冲对起缓冲反应虽可使NaHCO3有所降低,但其它缓冲碱却有所增加。NaHCO3+Hpr→NaPr+H2CO3,NaHCO3+Na2HPO4→Na2HPO4+H2CO3。因而其缓冲效果不明显,因为缓冲碱总量并不能减少,还是要靠肾脏的代偿调节。
2.离子交换 此时细胞内H+向细胞外移动,K+则移向细胞内,故代谢性碱中毒能引起低血钾。
3.呼吸代偿 代谢性碱中毒时,由于细胞外液[HCO3-]升高,[H+]下降,导致呼吸中枢(延髓CO2敏感细胞即中枢化学感受器)及主动脉体、颈动脉体化学感受器兴奋性降低,出现呼吸抑制,肺泡通气减少,从而使血液中H2CO3上升。但这种代偿有其限度,因为通气减少将同时引起缺氧,缺氧对呼吸的剌激将限制此种代偿。
4.肾脏代偿代谢性碱中毒时肾脏的代偿是最主要的,它是代偿调节的最终保证。与代谢性酸中毒时的代偿反应正好相反,此时肾小管上皮细胞排H+减少,产NH3形成NH4+和可滴定酸排出均减少,对HCO3-的重吸收减少而使之排出增多。这是对代谢性碱中毒最为有效的代偿,其它三种代偿均是次要的。
需注意者,代谢性碱中毒一般均为碱性尿。但在低血钾引起的碱中毒却反常酸性尿。此因离子交换机制,H+进入肾小管上皮细胞换出K+以补充低血钾,小管内[H+]此时是高的。因此排出也多,虽为碱中毒却是酸性尿。
通过代偿调节如能使HCO3-/H2CO3的比值保持在正常范围内,则为代偿性代谢性碱中毒;否则,称为失代偿性代谢性碱中毒,其血浆中反映酸碱平衡的指标变化如下:
pH ↑ CO2C.P.↑
S.B. ↑ B.B. ↑
A.B.↑ B.E.正值增大
A.G.不变
(三)对机体的影响
1.神经肌肉功能障碍急性代谢性碱中毒病人常有神经肌肉应激性增高和手足搐搦症.是血浆[Ca++]下降所致,其机制是血浆pH值升高时血浆结合钙增多而游离钙减少。当代谢性碱中毒导致明显低血钾时,病人可发生肌肉无力或麻痹,腹胀甚至肠麻痹。
2.中枢神经系统功能障碍病人可有烦燥不安、精神错乱及谵妄等症状。其机制目前认为可能与中枢神经系统中γ-氨基丁酸减少有关。γ-氨基丁酸是中枢神经系统的一种起抑制作用的物质,参与维持中枢兴奋抑制的平衡。当[H+]下降时,谷氨酸脱羧酶活性降低,γ-氨基丁酸的生成便减少;同时γ-氨基丁酸转氨酶活性增加,使γ-氨基丁酸分解增加。这样就引起抑制的减弱而表现中枢的神经系统的兴奋症状。
严重代谢性碱中毒也有组织供氧不足的作用参与中枢神经系统功能障碍的发生。因pH值升高时氨离曲线左移,氧合血红蛋白在组织解离释氧减少,故病人也有缺氧的表现,症状可有兴奋直至昏迷。
3.低钾血症代谢性碱中毒经常伴有低钾血症。其机制是离子转移造成的。碱中毒时细胞内H+移向细胞外以平衡细胞外减少的H+,同时K+移向细胞内以维持电中性。此过程同样发生在肾小管上皮细胞,当细胞内K+增加时,肾排K+也就增加。故能引起低钾血症。从前面所介绍的全部情况看,低钾血症是代谢性碱中毒的原因之一,而代谢性碱中毒又能导致低钾血症,二者可以互为因果,这一点是要注意的。
(四)防治原则
1.积极防治引起代谢性碱中毒的原发病。
2.纠正低血钾症或低氯血症,如补充KCl,NaCl、CaCl2、NH4Cl等。其中NH4Cl既能纠正碱中毒也能补充Cl-,不过肝功能障碍患者不宜使用,因NH4Cl需经肝代谢。
3.纠正碱中毒 可使用碳酸肝酶抑制剂如乙酰唑胺以抑制肾小管上皮细胞中H2CO3的合成,从而减少H+的排出和HCO3-的重吸收。也可使用稀HCl以中和体液中过多的NaHCO3。大约是1mEq的酸可降低血浆[HCO3-]5mEq/L左右。醛固酮拮抗剂可减少H+、K+从肾脏排出,也有一定疗效。
二、呼吸性碱中毒
呼吸性碱中毒(Respiratory Alkalosis)的特征是血浆[H2CO3]原发性减少。
(一)原因和机制
1.精神性过度通气 这是呼吸性碱中毒的常见原因,但一般均不严重。严重者可以有头晕、感觉异常,偶尔有搐搦。常见于癔病发作患者。
2.代谢性过程异常 甲状腺机能亢进及发热等时,通气可明显增加超过了应排出的CO2量。可导致呼吸性碱中毒,但一般也不严重。但都说明通气量并非单单取决于体液中[H+]和Pco2,也与代谢强度和需氧情况有关。此时的通气过度可能是肺血流量增多通过反射性反应引起的。
3.乏氧性缺氧 乏氧性缺氧时的通气过度是对乏氧的代偿,但同时可以造成CO2排出过多而发生呼吸性碱中毒。常见于进入高原、高山或高空的人;胸廓及肺病变如肺炎、肺栓塞、气胸、肺淤血等引起胸廓、肺血管或肺组织传入神经受剌激而反射性通气增加的病人;此外,有些先天性心脏病患者,由于右至左分流增加而导致低张性低氧血症也能出现过度通气。这些均引起血浆H2CO3下降而出现呼吸性碱中毒。
4.中枢神经系统疾患 脑炎、脑膜炎、脑肿瘤、脑血管意外及颅脑损伤病人中有的呼吸中枢受到剌激而兴奋,出现通气过度。
5.水杨酸中毒 水杨酸能直接剌激呼吸中枢使其兴奋性升高,对正常剌激的敏感性也升高。因而出现过度通气。
6.革兰氏阴性杆菌败血症 革兰氏阴性杆菌进入血路而繁殖的病人,在体温血压还没有发生变化时即可出现明显的通气过度。Pco2有低至17mmHg者。此变化非常有助于诊断。其机理尚不清楚,因为动物实验中未能成功复制此一现象。
7.人工呼吸过度。
8.肝硬化 肝硬化有腹水及血NH3升高者可出现过度通气。可能系NH3对呼吸中枢的剌激作用引起的。当然,腹水上抬横隔也有剌激呼吸的作用,但是非肝硬化的腹水病人却无过度通气的反应。
9.代谢性酸中毒突然被纠正例如使用NaHCO3纠正代谢性酸中毒,细胞外液[HCO3-]浓度迅速升至正常,但通过血脑浆屏障很慢,约12~24小时,此时脑内仍为代谢性酸中毒,故过度通气仍持续存在。这就造成H2CO3过低的呼吸性碱中毒。
10.妊娠 有中等程度的通气增加,它超过CO2产量,目前认为系黄体酮对呼吸中枢的剌激作用,一些合成的黄体酮制剂也有此作用。妊娠反应期因呕吐、饮食不足可发生酮症酸中毒,妊娠反应期过后则可发生呼吸性碱中毒,有时引起手足搐搦。
(二)机体的代偿调节
1.细胞内外离子交换和细胞内缓冲急性呼吸性碱中毒细胞内外离子交换是H+自细胞内液转移至细胞外液以补充减少了的H+,而K+及Na+则自细胞外液转移至细胞内液。H++HCO3-→H2CO3,补充细胞外液的HCO3-使之有所升高,而HCO3-则有所降低。
细胞内缓冲是当血浆Pco2降低时,H2CO3下降,HCO3-/H2CO3比值增大,H2CO3-相对增多,它可进入细胞内进行缓冲,以进入红细胞为例其反应如下:
HCO3-自浆向红细胞转移,同时Cl-自红细胞向血浆转移。血浆NaHCO3有所减少以维持pH值不致明显升高。H+可由细胞内缓冲物质如酸性磷酸盐或HPr、HHb提供以补充细胞外液下降了的水平。这种细胞内缓冲大约红细胞担负1/3,其它细胞担负2/3。
急性呼吸性碱中毒时血浆Pco2与[HCO3-]二者的关系如下式所示:
△[HCO3-]=△Pco2×0.2±2.5
例如血浆Pco2迅速下降1.33kPa(10mmHg)(即从正常的5.33kPa(40mmHg)降至4.00kPa(300mmHg)),则[HCO3-]降低2mmol(mEq)/L左右。且[HCO3-]降低也有一定代偿限度,一般急性患者下降不超过6mmol(mEq)/L,即A.B.不低于18mmol(mEq)/L。因此常为失代偿者。
2.肾脏代偿是慢性呼吸性碱中毒的主要代偿措施。当血液Pco2下降HCO3-减少时,肾小管上皮细胞碳酸酐酶活性降低,H+的生成和排出下降。NH3→NH4+也因排出的H+下降而减少。与此同时HCO3-重吸收减少而排出增多。
呼吸性碱中毒不能依靠呼吸代偿。当然,如精神性过度通气,当碱中毒发展至Pco2明显降低,[H+]亦随之下降时,呼吸中枢的兴奋性将受抑制而使呼吸减慢变浅,这是发病环节中的反馈性自我调控,不属代偿。如癔病的发作此时可能就过去了。
在慢性呼吸性碱中毒时,血浆[HCO3-]与Pco2之间的关系如下式所示:
△[HCO3-]=△Pco2×0.5±2.5mmol(mEq)/L
例如血浆Pco2每降低1.33kPa(10mmHg),[HCO3-]可下降约5mmol(mEq)/L。这比急性者的代偿反应明显,概因慢性者有较充分的代偿时间。但其代偿也有限度,一般病人血浆[HCO3-]不会低于12mmol(mEq)/L。
失代偿性呼吸性碱中毒反映血浆酸碱平衡的指标变化如下:
pH ↑ CO2C.P.↓
S.B. 不变(有代偿性减少时可略↓)
A.B. ↓
B.B.. 不变(有代偿性减少时可略↓)
B.E. 不变(有代偿性减少时可呈负值增大)
此种碱中毒由于Cl-向红细胞外转移增加,故血浆Cl-升高。
(三)对机体的影响:
急性呼吸性碱中毒病人临床表现较为明显,常有诸如窒息感,气促,眩晕,易激动,四肢及口周围感觉异常等。严重者有意识障碍,其机理可能系Pco2↓导致脑血管收缩而缺血,手足搐搦是血浆[Ca++]下降所致。
慢性呼吸性碱中毒症状轻。因代偿发挥较好。
呼吸性碱中毒与代谢性碱中毒一样,也能导致低钾血症和组织缺氧。
(四)防治原则
1.防治原发病。
2.降低病人的通气过度,如精神性通气过度可用镇静剂。
3.吸入含5%CO2的混合气体,以提高血浆H2CO3浓度。
4.手足搐搦者可静脉适量补给钙剂以增加血浆[Ca++]。
第五节 混合型酸碱平衡障碍
混合型酸碱平衡障碍(Mixed Acid-BaseDisturbances)是指两种或两种以上的原发性酸碱平衡障碍同时并存。当两种原发性障碍使pH向同一方向变动时,则pH偏离正常更为显着,例如代谢性酸中毒合并呼吸性酸中毒的病人其pH值比单纯一种障碍更低。当两种障碍使pH向相反的方向变动时,血浆pH值取决于占优势的一种障碍,其变动幅度因受另外一种抵消而不及单纯一种障碍那样大。如果两种障碍引起pH相反的变动正好互相抵消,则病人血浆pH值可以正常,例如代谢性酸中毒合并呼吸性碱中毒。
一、常见种类
混合型酸碱平衡障碍常见的有下列五种。
(一)呼吸性酸中毒合并代谢性酸中毒
呼吸性酸中毒合并代谢性酸中毒见于:①慢性呼吸性酸中毒如阻塞性肺疾病同时发生中毒性休克伴有乳酸酸中毒;②心跳呼吸骤停发生急性呼吸性酸中毒和因缺氧发生乳酸酸中毒。此种混合型酸碱平衡障碍可使血浆pH值显着下降,血浆[HCO3-]可下降,Pco2可上升。例如患者血浆pH为7.0,Pco2为11.3kPa(85mmHg),[HCO3-]为14.4mmol(mEq)/L,B.E.为-12mmol(mEq)/L。
(二)呼吸性酸中毒合并代谢性碱中毒
呼吸性酸中毒合并代谢性碱中毒见于慢性阻塞性肺疾患发生高碳酸血症,又因肺源性心脏病心力衰竭而使用利尿剂如速尿、利尿酸等引起代谢性碱中毒的患者。这也是呼吸、心肾科室中常遇到的情况。病人血浆pH可以正常或轻度上升或下降,但[HCO3-]和Pco2均显著升高。[HCO3-]升高是代谢性碱中毒的特点而Pco2升高是呼吸性酸中毒的特点,二者比值却可保持不变或变动不大。例如患者血浆pH为7.4,Pco2为60mmHg,血浆[HCO3-]为34mEq/L,B.E.+14mEq/L。
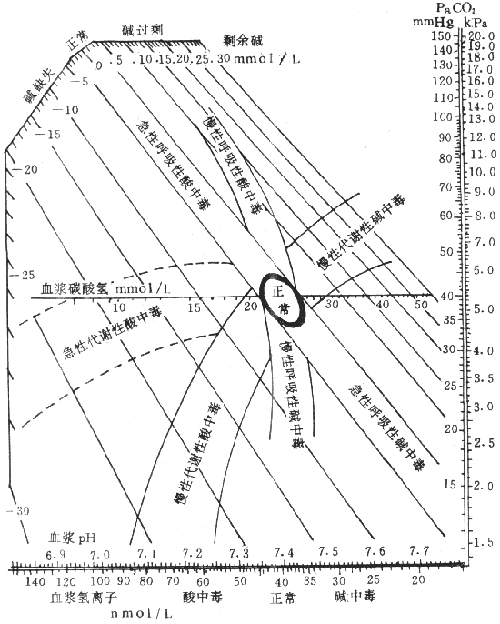
图6-5 各种类型酸碱平衡紊乱时血浆pH、Pco2、HCO3-变化示意图
(三)呼吸性碱中毒合并代谢性酸中毒
此种混合型酸碱平衡障碍可见于:①肾功能不全患者有代谢性酸中毒,又因发热而过度通气引起呼吸性碱中毒,如革兰氏阴性杆菌败血症引起的急性肾功能衰竭并伴有高热。②肝功不全患者可因NH3的剌激而过度通气,同时又因代谢障碍致乳酸酸中毒。③水杨酸剂量过大引起代谢性酸中毒,同时剌激呼吸中枢而导致过度通气。其血浆pH值可以正常、轻度上升或下降,但血浆[HCO3-]和Pco2均显著下降。[HCO3-]下降是代谢性酸中毒的特点,Pco2是呼吸性碱中毒的特点。二者比值却可保持不变或变动不大。例如患者血浆pH为7.36,Pco2为20mmHg,血浆[HCO3-]为14mEq/L,B.E.为-12mEq/L。
(四)呼吸性碱中毒合并代谢性碱中毒
此种混合型酸碱平衡障碍可见于:①发热呕吐患者,有过度通气引起的呼吸性碱中毒和呕吐引起的代谢性碱中毒。②肝硬变患者有腹水,因NH3的的剌激而通气过度,同时使用利尿剂或有呕吐时,此型血浆pH值明显升高,血浆[HCO3-]可升高,Pco2可降低。[HCO3-]升高是代谢性碱中毒的特点,Pco2降低是呼吸性碱中毒的特点。例如病人血浆pH为7.68,Pco2为29mmHg,血浆[HCO3-]为38mEq/L,B.E为+14mEq/L。
(五)代谢性酸中毒合并代谢性碱中毒
呼吸性酸碱中毒不能同时存在,但代谢性酸碱中毒却可并存。例如急性肾功衰患者有呕吐或行胃吸引术时,则可既有代谢性酸中毒也有代谢性碱中毒的病理过程存在。但血浆pH,[HCO3-],Pco2都可在正常范围内或稍偏高或偏低。
二、常用判断方法
混合型酸碱平衡障碍情况比较复杂,必须在充分研究分析疾病发生发展过程的基础上才能做出判断。尽管如此,有少数混合型酸碱平衡障碍仍然难以确定。目前国内设备先进的医院,临床不能作出肯定判断的仍有达2.2%者。可见在判断酸碱平衡障碍的原理和技术方面还需进一步研究。
为了临床实用方便,一些研究酸碱平衡障碍的学者依据血浆pH、Pco2和[HCO3-]的数值之间的关系,设计了一系列线算图(Nomogram),知道其中两个值便可从图上查出有关的其它指标的数值,并可迅速判断酸碱平衡障碍的性质。其中Siggaard-Anderson氏所设计的列线算图比较常用,附后以供参考。见图6-5。
第六节 需要明确的几个基本概念
由于医学对人体酸碱障碍研究已有百年历史,问题比较复杂,揭示其变化及规律是逐步的。因而存在着一些不同的看法和概念。在学习酸碱平衡障碍问题时,有些基本概念需要澄清,否则文献上的一些不同的命名和定义将给初学者带来困艰甚至混乱。在这里将介绍一些学习关系重大的基本概念。
一、什么是酸?什么是碱?
(一)定义
在探讨人体酸碱平衡障碍的文献中,对于酸和碱有三种不同的定义。我们在书籍、杂志中,经常可以见到三种定义混用,甚至并用,容易导致混乱。三种定义如下:
1.酸是阴离子,碱是阳离子
| 碱(mEq/L血浆) | 酸(mEq/L血浆) | ||
| Na+ | 142 | HCO3- | 27 |
| K+ | 4(5) | Cl- | 101(103) |
| Ca++ | 5 | HPO4= | 2 |
| Mg++ | 2 | SO4- | 1 |
| 153(154) | RCOO- | 6(5) | |
| 蛋白- | 16 | ||
| 153(154) | |||
2.酸是固定阴离子,碱是固定阳离子
| 碱 | 酸 |
| Na+ | Cl- |
| K+ | HPO4= |
| Ca++ | SO4- |
| Mg++ | RCOO- |
| 蛋白- |
“固定”是与“可挥发”相对应而言的,意即不能挥发的意思。如HCO3-可以生成CO2而从呼吸排出,即为可挥发的,因而不列入酸这一项中。其实这个定义是为了纠正前一个定义中把HCO3-作为酸这一明显错误的。HCO3-在体液pH条件下系一碱性物质,它能结合H+以减少[H+]的浓度。
3.酸是H+供给者,碱是接受者或OH-供给者
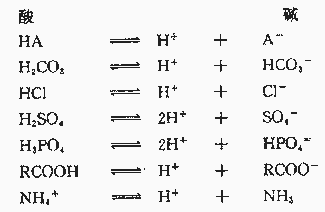
上述三种定义中的第三种是合理的,它与化学上的概念一致,也真正反映体内离子的化学作用。即在体内能解离生成H+者为酸,能接受H+者为碱。体液pH值就是反映体液中H+离子浓度的。酸碱平衡障碍所研究的基本变化就是[H+]在体液中的升降。
根据第三种定义则Na+、K+、Ca++、Mg++非酸亦非碱,所有表中所列的阴离子在体内均起碱的作用。近年在酸碱平衡障碍的有关文献中已逐步使用第三种概念以代替前二种。
(二)为什么会产生上述混乱?
这是因为过去很长时间内没有直接测定血液pH值的临床上适用的方便而准确的方法。所使用的方法是比较烦琐而且不够精确。过去一直从其它离子的变动间接判断血液pH,所以上述混乱是历史发展的原因造成的。
酸是阴离子、碱是阳离子的这种定义是医学引用化学概念发生偏差造成的。我们知道在电解槽中通直流电后,盐类电解质的阳离子去至阴极附近,它们再吸引OH-,因而使此处溶液变碱;阴离子则去至阳极附近,再吸收H+而使此处溶液变酸。以NaCl水溶液电解为例,阴极附近有NaOH,阴极附近有HCl。最初化学上称这些阳离子如Na+等为“形成碱的离子(Base Formers)”,而称Cl-等阴离子o “形成酸的离子(Acid Formers)”。使用中就习以为常把这些阳离子称为碱离子,阴离子称为酸离子。须知,这是不正确的。
(三)如何把这三个定义联系起来?
上述三种定义看起来是不同的,甚至截然相反,但是又都在使用。它们繁简不同地都可解释酸碱平衡障碍,其间的关系如何,便需说明。下列一个重要的化学缓冲反应式可以帮助说明:
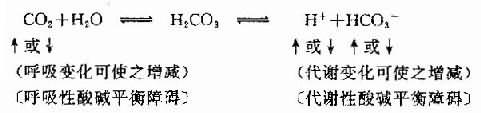
阴离子(除HCO3-外)增多为何酸中毒?——阴离子不是酸,但又有高血氯性酸中毒的说法(而Cl-是碱),这如何理解?这是因为除HCO3-以外的阴离子增加时,在维持离子平衡的过程中,最容易变动的HCO3-就减少(例如肾脏对它的重吸收减少,排出量就增加)。根据质量作用定律,上面的化学反应式右侧的HCO3-不断减少,反应向右进行,生成的H+就多,体液(H+)升高,pH值下降而趋于酸中毒。
阴离子(除HCO3-外)减少如何碱中毒?——与上面者相反,此时最容易变动的HCO3-就增多,上面化学反应式的反应向左进行,(H+)降低,pH值上升,趋于碱中毒。
阳离子增多如何碱中毒?——通常阳离子增多是伴以最易变动的HCO3-相应增多的,而HCO3-增多能使上述反应式的反应向左进行,H+被消耗而减少,pH值上升而趋于碱中毒。但有时阳离子增多并不伴有HCO3-增多,而是伴以其它阴离子如Cl-增加。此时HCO3-不增加,因而不发生碱中毒。所以说,“阳离子为碱,它的增加就会引起碱中毒”是不正确的。例如高钠血症、高钾血症并不与碱中毒等同。
阳离子减少为何酸中毒?——通常阳离子减少是伴以HCO3-相应减少的,而HCO3-减少能使反应向右进行,[H+]升高,pH降低而趋于酸中毒,但有时阳离子减少并不伴以HCO3-减少,因此阳离子减少不一定酸中毒的。同时,也可看出第三种定义比较精确,接触了问题的根本。使用第三种定义,也使酸碱中毒的概念明确。
按照第三种定义,上面所列体内阴离子均为碱,但它们的强弱不同。其强弱标准是以其接受H+的能力而定的。例如HCO3-比Cl-强的多,HCO3-是缓冲系统的重要缓冲碱,而Cl-则否。HCO3-接受H+生成H2CO3-,其在体内的解离度仅为Cl-接受H+生成的HCl的1/1500。换言之HCO3-“抓住”H+抓得更牢固,而Cl-则“抓”得很不牢,因为体内HCl几乎全部是呈解离的。
二、什么是固定酸(Fixed Acid)?什么是固定碱(Fixed Base)?
(一)两种定义
1.任何非挥发性的阴离子或阳离子称固定酸或固定碱,是与可挥发性酸H2CO3(可生成CO2)相区别的。
| 固定碱 | 固定酸 |
| Na+ K+ Ca++ Mg++ |
Cl- SO4= HPO4= RCCOO- 蛋白- |
2.不参加缓冲体系起作用的阴离子或阳离子称固定酸或固定碱。
| 固定碱 | 固定酸 |
| Na+ K+ Ca++ Mg++ |
CI- SO4- RCOO- |
(二)如何评价?
首先,上述两个定义中的酸与碱仍是与“酸是阴离子,碱是阳离子”的概念相联系的。因此,改称固定酸为固定阴离子,固定碱为固定阳离子,比较妥当,目前已逐渐采用。其次固定阴离子分两部分为妥,即固定阴离子和缓冲阴离子。因为在实际病例中,固定阴离子增多可能是AG增大类代谢性酸中毒,而缓冲阴离子增多则可能是代谢性碱中毒。这就是说,上述两种定义中,以第二种定义的规定比较更有实用意义,虽然它还不完善。
最后要说明的是阳离子全是“固定的”。因此“固定”二字实无必要,以用阳离子总量这一名词为宜。有的文献称为总碱量,这也是与“阳离子为碱”的旧概念相联系的名称,我们清楚它的含义就不会有什么误解了。
三、酸碱平衡障碍类型命名上的混乱
长期以来,文献上有一种以血氯高低来命名酸碱平衡障碍的方法,归结起来它是这样的:

仍以前述之化学缓冲反应式予以说明。
代谢性酸中毒时体液主要缓冲变化:

呼吸性酸中毒时体液主要缓冲变化:
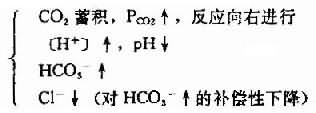
代谢性碱中毒时体液主要缓冲变化:

呼吸性碱中毒时体液主要缓冲变化:
归纳成表如下:
| pH | HCO3- | Pco2 | CI- | |
| 代谢性酸中毒 呼吸性酸中毒 代谢性碱中毒 呼吸性碱中毒 |
↓ ↓ ↑ ↑ |
↓ ↑ ↑ ↓ |
呼吸代偿可正常 ↑ 呼吸代偿可正常 ↓ |
↑ ↓ ↓ ↑ |
以氯命名的酸碱中毒,文献中并不少见。其主要缺点有二:(1)易使人误解,以为酸碱平衡障碍可由于Cl-的原发变动而引起。实际上Cl-的变动,在大多数情况下只是HCO3-变动的一个反映罢了,它是继发的。(2)有的情况Cl-并不象上述那样变动。例如代谢性酸中毒并非都是高血氯性酸中毒,前面已经讲到。例如有机酸生成过多的代谢性酸中毒如乳酸酸中毒、酮症酸中毒、肾小球滤过功能严重损害而使酸性物质在体内潴留以及水杨酸中毒等均是负离子间隙(AG)增大类代谢性酸中毒,血氯并不增高,而是其它阴离子增加。这类代谢性酸中毒显然不能称之为高血氯性酸中毒。
四、K[SB]+[/SB]代谢与酸碱平衡障碍之间的关系
K+代谢与酸碱平衡互相影响。有时K+代谢障碍在先,作为原发的改变,引起酸碱平衡障碍。有时则相反,是酸碱平衡障碍引起了K+代谢的变化。这就出现了诸如“低血钾性碱中毒”,“碱中毒性低血钾”等一些容易混淆的名词。现以肾小管上皮细胞为例,以图解表示如下:
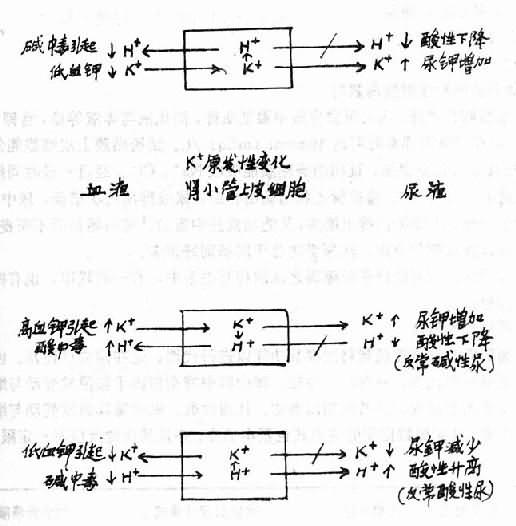
以上是K+代谢与酸碱平衡障碍之间的基本关系。但也应注意疾病发生发展过程中的变化是错综复杂的。因此在特殊情况下,也有特殊的变化出现。例如严重腹泻的病人,由于腹泻失钾而发生低血钾,同时又丧失HCO3-。这就使低血钾所能引起的碱中毒不能出现,而是一种代谢性酸中毒伴以低血钾。
又如绝大部分肾功能不全的代谢性酸中毒病人,其K+的问题是高血钾。但也有低血钾的。如果是低血钾,主要有下列原因:
1.钾进量不足
2.肾外途径丧失,如呕吐、腹泻
3.使用肾上腺皮质激素
4.纠正酸中毒时
5.给人输入葡萄糖同时注射胰岛素时
再如输尿管乙状结肠吻合术后,病人可发生酸中毒低血钾。消化液与体液等渗,含钾5~20mmol(mEq)/L,但在结肠下部有时可达30mmol(mEq)/L。结肠粘膜上皮细胞能分泌HCO3-和K+。用生理盐水做保留灌肠,证明结肠粘膜能吸收Na+、Cl-。经过一段时间抽出后,见抽出液中含多量K+、HCO3-。输尿管乙状结所吻合后,尿液排出乙状结肠,尿中H+可被吸收入血而引起酸中毒,且HCO3-排出增多,又能加重酸中毒。K+可因稀便而不断丧失,这就是此时酸中毒而伴以低血钾的原因。输尿管吻合于回肠则好得多。
以上所述,可以说明钾代谢与酸硷平衡障碍之间的相互关系中,有一般规律,也有特殊情况。具体病情要具体分析。
五、酸碱平衡障碍的代偿限度
机体发生酸碱平衡障碍时,调控机制将发挥其功能以进行代偿,这在前文已述及。由于急性呼吸性酸、碱中毒更易失代偿,故在呼吸性酸、碱中毒中特别指出了其原发变动与继发变动的数量关系,并以实测数据找出关系式加以表达。代谢性酸、碱中毒其原发变动与继发变动同样具有数量关系,其实测数据间的关系式也基本确立。并且其代偿反应有一定限度,现将其归纳如下表:
| 原发变化 | 代偿反应 | 代偿反应计算式 | 代偿升降限度 | |
| 代谢性酸中毒 | HCO3-↓↓↓ | Pco2↓↓ | △Pco2=△HCO3-×1.5+8±2 | 1.33kPa(10mmolHg) |
| 代谢性碱中毒 | HCO3-↑↑↑ | Pco2↑↑ | △Pco2=△HCO3-×0.9+9±2 | 7.33kPa(55mmHg) |
| 急性呼吸性酸中毒 | Pco2↑↑↑ | HCO3-↑↑ | △HCO3-=△Pco2×0.07±1.5 | 30mmol(mEq)/L |
| 慢性呼吸性酸中毒 | Pco2↑↑↑ | HCO3-↑ | △HCO3-=△Pco2×0.4±3 | 42mmol(mEq)/L |
| 急性呼吸性碱中毒 | Pco2↓↓↓ | HCO3-↓ | △HCO3-=△Pco2×0.2±2.5 | 18mmol(mEq)/L |
| 慢性呼吸性碱中毒 | PCO2↓↓↓ | HCO3-↓↓ | △HCO3-=△Pco2×0.5±2.5 | 12mmol(mEq)/L |
六、混合型酸碱平衡障碍分析概要
第七章 水肿
第一节 概述
一、水肿的概念
过多的液体在组织间隙或体腔中积聚,称为水肿(edema)。此定义把水肿局限于局部或全身血管外的细胞外液(指组织间液)容量的增多。其实,细胞内液的过多积聚,有时也称细胞水肿,如细胞中毒性脑水肿。后者未为上述定义所包括,许多水肿可不伴有细胞内液量的明显变化。为了把水肿这个术语限于细胞外,有人主张把细胞水肿称为细胞水化(cellularhydration)。
水肿按分布范围可分全身水肿(anasarca)和局部水肿(local edema)。也可按发生部位命名,如脑水肿、肺水肿、皮下水肿等。
正常体腔内只有少量液体,当体腔内液过多积聚时,称为积水(hydrops),如心包积水、胸腔积水(胸水)、腹腔积水(腹水)、脑室积水、阴囊积水等等。
水肿常按其原因而命名,如肾性水肿、肝性水肿、心性水肿、营养性水肿、静脉阻塞性水肿、淋巴水肿、炎症性水肿等。可见水肿并非独立疾病,而是疾病时的一种重要病理过程或体征。
二、水肿发病的基本因素
各类水肿的原因和发生机制虽不全一致,但基本因素不外两大类。
(一)全身水分进出平衡失调―钠水潴留
细胞外液量增多是因钠水摄入超过排出以致滞留。钠水能自由弥散或滤过毛细血管壁,故当钠水滞留引起血管内液增多时,必然引起血管外的细胞外液增多。增多的组织间液不能及时移走,积聚到一定程度就出现水肿。若事先已有组织间液积聚,则钠水滞留会加重水肿的发展。钠水滞留的基本机制是球-管失平衡而导致肾排钠和排水的减少。正常人能摄入比较大量的钠[例如每天0.34mol(20g)食盐]而不致发生钠滞留和水肿。但若排钠功能不足,则通常摄钠量就足以造成钠水滞留。平常由肾小管滤过的钠水总量中,只有0.5-1%左右被排出,而有99-99.5%被肾小管重吸收,其中约60-70%由近曲小管重吸收,这里钠的重吸收属主动过程即需能转运;钠水在远曲小管及集合管的重吸收,则主要受刺激所控制。这些调节因素保证了球-管平衡。如果肾小球滤过率下降不伴有相应的重吸收减少,肾排钠水就要减少;如果肾小球滤过量正常,而肾小管重吸收增多,肾排钠水量也会减少;如果肾小管球滤过率下降而肾小管重吸收增多,则肾排钠水就更加减少。这三种情况都可以引起球-管失平衡(图7-1),导致钠水滞留和细胞外液量增多。
图7-1 球-管失衡基本形式示意图
1.球管平衡;2~4.球管失衡;2.滤过↓,重吸收钠水正常;3.滤过正常,重吸收钠水↓;4.滤过↓重吸收钠水↓
球-管失平衡导致肾排钠水减少的原因,有原发和继发两类。
1.原发性肾排出钠水量减少肾原发疾病使肾小球滤过总量下降,而肾小管的重吸收没有相应减少,故引起肾排钠水量减少。这是急性肾小球肾炎时发生水肿的基本机制。
2.继发性肾排钠水量减少
(1)肾小球滤过钠水减少:任何原因使有效循环血量减少时,分布到肾的血流量就相应减少,加上动脉血压的相应降低导致颈动脉窦和主动脉弓压力感受器的牵张度减弱,反射地使肾血管收缩,肾血流就更减少。后者还能激活肾素-血管紧张素系统,血管紧张素Ⅱ浓度增加又可引起入球小动脉收缩,使肾小球血流量进一步减少。所有这些因素都导致肾小球滤过率下降。
(2)肾小管重吸收钠水增多:不同节段肾小管吸收钠水增多的机制不尽相同:
1)近曲小管重吸收钠水增多:当有效循环血量减少时,可引起近曲小管重吸收钠水增多,导致肾排钠水量减少。其机制有两种解释:①利钠激素分泌减少(详下);②肾内物理因素的作用。后者是指肾小球滤过分数(filtration fraction, FF)的增加。滤过分数=肾小球滤过率/肾血浆流量。正常约有20%的肾血流量由肾小球渡过。当充血性心力衰竭或肾病综合征等使有效循环血量减少从而引起肾血流量减少时,往往由于出球小动脉的收缩比入球小动脉的收缩更为明显,因而肾小球滤过率的下降也就不如肾血流量下降为明显,流入肾小管周围毛细血管的血液中,血浆蛋白的浓度也就相对增高,而管周毛细血管的流体静压则下降,这两个因素都促进近曲小管对钠水的重吸收。
2)远曲小管和集合管重吸收钠水增多:肾小管的这两段的重吸收钠水的功能,主要受下述肾外激素的调控:
①醛固酮增多:有效循环血量减少常引起醛固酮增多。因为有效循环血量减少使肾小动脉灌注压和肾小球滤过率下降,结果入球小动脉牵张感受器的牵张度减弱,致密斑也因到达的钠量减少而受剌激,从而激活了肾素-血管紧张素系统,使血管紧张素Ⅱ和Ⅲ增多,后两者剌激肾上腺皮质球状带,使之分泌较多的醛固酮,故血中醛固酮浓度增高。此外,肝功能严重损害可致醛固酮灭活减少,也是引起血浆醛固酮增多的附加因素。
醛固酮增多与水肿形成的关系并不恒定,多数进行性钠水滞留的病人,血浆醛固酮浓度往往增高,而处于平稳状态的水肿病人,则血浆醛固酮可在正常范围内。一些事实表明,单独醛固酮增多不一定导致持久滞钠和水肿。连续每天使用醛固酮使细胞外容量扩大时,开始时排钠减少,但几天后排钠回升到对照水平。此现象被称为“钠逃逸”或“醛固酮逃逸”。其本质仍未清楚,有人认为是第三因子(第一因子是肾小球滤过率,第二因子是醛固酮)的作用。可能在细胞外液容量扩大到一定程度后,第三因子分泌增多,近曲小管重吸收钠就减少,直至与醛固酮的作用相平衡为止。目前不少学者认为第三因子就是利钠激素(详下)。
②抗利尿激素:在全身水肿形成中,抗利尿激素(ADH)增多的滞水作用也有一定意义。有效循环血量或心排血量下降,使左心房壁和胸腔大血管壁的容量感受器所受的剌激减弱;加上有效循环血量下降激活了肾素-血管紧张素系统,以致血管紧张素Ⅱ生成增多,均可导致下丘脑-神经垂体分泌和释放ADH增多。此外,有些水肿(肝有损害)时,ADH增多部分地与肝灭活减少有关。
一些事实表明,ADH可参与某些全身水肿的机制,但可能不是钠水滞留所必需。把实验性腹水狗的神经垂体破坏,虽能造成尿崩症,但不能削弱钠滞留和腹水;事先破坏狗的神经垂体,然后造成下腔静脉(肝上方)狭窄,仍产生钠滞留和腹水。
③利钠激素或心房肽分泌减少:有些学者主张当血容量或有效循环血量下降时,可引起利钠激素(natriuretec hormone)减少(相反则增多)。此激素有抑制近曲小管重吸收钠的作用,故称利钠激素,并认为此即第三因子。另一些学者则对其存在有怀疑。但近年来一些报道,不仅承认其存在,而且认为是一种低分子物质,其作用至少有一部分是在近曲小管,并且认为上述“醛固酮逃逸”可能是利钠激素增多所致。因而利钠激素分泌减少就有利于醛固酮发挥滞钠作用和水肿的发生。
正当利钠激素的来源尚未解决之际,一些学者已从大鼠及人体心房组织提取纯化了心房肽(atriopeptin)或心房利钠多肽(atrial natriureticpolypeptide ,ANP),后者给大鼠静脉内注射能引起迅速而强烈的排钠利尿作用。近期资料表明,细胞外液容量变化能影响心房肌组织释放肽,后者到达靶器官与特异受体结合,可能通过cGMP而发挥利钠、利尿和扩血管作用,并能抑制醛固酮和ADH的释放。因此可以理解,心房肽的减少也可导致钠水储留而促成水肿的发生。至于心房肽是否就是上述的利钠激素,以及它们与水肿形成的关系,很有进一步研究的价值。
7
(二)血管内外液体交换失衡导致组织间液增多
组织间液生成和回收的平衡,受血管内外诸因素的调控。这些因素之一的失常或两个以上同时或先后失常,就可使血管内外液体交换失衡,引起组织间液生成过多或回收过少,或两者兼有,其结果都可使组织间液过多积聚而形成水肿。这些基本因素是:
1.毛细血管有效流体静压增高 毛细血管内的流体静压大于组织间液的流体静压,前者减去后者的值就是有效流体静压。因而毛细血管流体静压增高,可导致有效滤过压增高(有效滤过压等于有效流体静压减去有效胶体渗透压),它有利于毛细血管血浆的滤出而不利于组织间液的回收。全身或局部的静脉压升高,是有效流体静压增高的主要成因。静脉压升高可逆向转递到微静脉和毛细血管静脉端,使后者的流体静压增高,有效流体静压便随之增高。
局部静脉压增高的常见原因是血栓阻塞静脉腔,肿瘤或瘢痕压迫静脉壁等;全身体循环静脉压增高的常见原因是右心衰竭;而肺静脉压增高的常见原因则是左心衰竭。
2.有效胶体渗透压下降 血浆和组织间液中都含有能产生渗透作用的物质,故血管内外都有渗透压,渗透压有晶体渗透压与胶体渗透压之分,前者产自晶体物质尤其电解质;后者主要产自蛋白质。由于晶体物质能自由通过毛细血管壁,故对血管内外液体交换影响不大。在血管内外液体交换中,限制血浆液体由毛血细管向外滤出的主要力量,是有效胶体渗透压,它是血浆胶体渗透压减去组织间液体渗透压的差值。
血浆胶体渗透压主要取决于血浆蛋白尤其白蛋白的浓度,白蛋白比球蛋白有较大的渗透压,每克可形成0.73kPa(5.5mmHg)的胶体渗透压,而每克球蛋白则仅形成0.19kPa(1.4mmHg)的胶体渗透压。据以往统计,人体血浆胶体渗透压约为3.33kPa(25mmHg),而后来Cuyton所用的数据则按3.72kPa(28mmHg)计算。因组织间液的蛋白质含量很少,其胶体渗透压仅约0.67kPa(5mmHg),故有效胶体渗透压约等于3.72-0.67=3.05kPa(28-5=23mmHg)。这既是对抗血浆液体毛细血管滤出的主要力量,也是促进组织间液向毛细血管回收的力量。因而有效胶体渗透压下降将导致毛细血管动脉端滤出增多和静脉端回收减少,有利于液体在组织间隙积聚。以下三种基本情况可引起有效胶体渗透压下降。
(1)血浆蛋白浓度降低:当血浆蛋白尤其是白蛋白浓度下降时,因血浆胶体渗透压相应下降,有效胶体渗透压也随之下降,严重时可引起水肿。引起水肿的血浆白蛋白临界浓度,有人认为大约是2.0g%。但一般难确定,因往往不是单因素引起水肿。据报道,有的人即使血浆白蛋白浓度降至0.5g%,也不出现水肿。血浆蛋白浓度下降的主要原因是:①蛋白质丢失:肾病综合征时大量蛋白质从尿中丢失;蛋白质丢失性肠病时蛋白丢失于肠腔中而随粪排出;②合成障碍:见于肝实质严重损害(如肝硬变)或营养不良;③大量钠水滞留或输入大量非胶体溶液时使蛋白稀释。
(2)微血管壁通透性增高:正常毛细血管只容许微量血浆蛋白滤出,平均不超过5%,其它微血管则完全不容许蛋白滤过,因而毛细血管内外胶体渗透压梯度很大。但当微血管壁通透性增高时,血浆蛋白不仅可随液体从毛细血管壁滤出,也可从其它微血管尤其微静脉壁滤出,其结果是毛细血管静脉端和微静脉内的胶体渗透压下降,而组织间液的胶体渗透压则上升,使有效胶体渗透压下降。此时如淋巴回流不足以阻止组织间液积聚,就出现水肿。渗出性炎症时,炎症区的微血管壁通透性增高最曲型,故其水肿液所含蛋白质浓度较高,可达3~6g%。
(3)组织间液中蛋白积聚:正常组织间液只含小量蛋白质,平均0.4~0.6g%,但各器官组织有较大差别。这些蛋白质通常由淋巴携带经淋巴管排入体静脉,故不致在组织间隙中积聚,而且当淋巴加速时这种运输还不能加强。因而蛋白质在组织间液中积聚的原因,主要是微血管滤出增多并超过淋巴引流速度,以及淋巴回流受阻(详下)。
3.淋巴回流受阻平常淋巴管畅通,不仅能把滤出略多于回收而剩余的液体及所含小量蛋白质,输送回到血液循环中,而且在组织间液生成增多时,还能代偿地加强回流,把增多的组织间液排流出去,以防止液体在组织间隙中过多积聚,故可把它看成一种重要的抗水肿因素。但是,在某些病理情况下,当淋巴干道有了阻塞,使淋巴回流受阻或不能代偿地加强回流时,含蛋白质的淋巴液就可在组织间隙中积聚而形成淋巴水肿(lymphedema)。发生这种水肿时,非蛋白液体可由毛细血管回收,但蛋白质却可滞积,浓度可过3~5g%。
三、水肿的表现特征
(一)水肿液的性状
水肿液来自血浆液体成分,含有蛋白质、无机盐、葡萄糖、肌酐、尿素、氨基酸及其它可溶性物质。但蛋白质量及比例,则视水肿的原因而异,主要取决于微血管通透性是否增高及增高程度。通透性越高,蛋白质渗出越多,含量就越多,故水肿液的比重也越大;相反,当通透性不高时,则蛋白质含量较低(常<2g%),水肿液比重也较低。临床上习惯把比重低于1.015的水肿液称漏出液(transudate),比重高于1.018的称渗出液(exudate),后者即指炎症性渗出液。但也有例外,淋巴水肿时虽微血管通透性不增高,水肿液比重可不低于渗出液,原因已如上述。
(二)水肿器官和组织的特点
水肿器官的体积增大,重量也增加,包膜被牵引而紧张发亮。此外,在组织学上水肿部的间质纤维可被分隔而稀疏。
(三)体重变化
全身水肿时,体重能敏感地反映细胞外液容量的变化。因而动态检测体重的增减,是观察水肿消长的最有价值的指标,它比观察皮肤凹陷体征更敏感。
(四)皮下水肿的皮肤特征
皮下水肿是全身或躯体局部水肿的重要体征。当皮下组织有过多体液积聚时。皮肤肿胀,皱纹变浅,平滑而松软。临床上为验证有无水肿,常用手指按压内踝或胫前区皮肤,观察解压后有无留下凹陷,如留下压痕,表明已有显性水肿(frank edema),也称凹陷性水肿(pitting edema)。但此法不敏感,因显性水肿出现前已有隐性水肿(recessive edema)。为何组织间隙已有过量液体积聚,而不出现凹陷体征?这可用组织间隙中的凝胶体网状物的吸附力来解释。后者对液体有强大吸附力和膨胀性(图7-2)。液体被吸附呈凝胶态就不能自由移动,受到压力时也不易移动;只有当积聚的液量超过凝胶体结构的吸附力和膨胀度后,过多的液体才能呈游离状态。液离液体在组织间隙中则有高度移动性,故在有足量游离液积聚后,用手指按压该部皮肤,液离液乃从按压点向周围散开,于是出现凹陷(压痕)。解压后约经数秒到1分钟左右,才流回原处而平复。
图7-2 毛细血管、毛细淋巴管和凝胶体结构在水肿时的液体交换示意图
1.毛细血管;2.毛细淋巴管;3.组织间隙凝胶体网状物
(五)全身水肿的分布特点
常见的全身水肿是心性、肾性和肝性水肿,它们的分布各有特点,后者有助于鉴别诊断。右心衰竭时水肿先出现于低垂部,立位时以下肢尤其足踝部最早出现且较明显,然后向上扩展;肾性水肿先出现于面部,尤以眼睑部明显,然后向下扩展;肝性水肿多以腹水最显著,躯体部不明显。这些不同分布特点主要取决于:①组织结构特点:组织致密度和伸展性在一定程度上影响水肿液积聚的早晚和程度。有些部位(如眼睑部)的皮下组织很疏松,皮肤伸展性大,容易容纳水肿液积聚,在不受重力影响尤其平卧时,水肿较早在这些部位显露而易被发觉,故肾性水肿病人晨起时眼睑水肿比较明显;另一些部位(如手指、足趾尤其掌侧)因皮下组织比较致密,皮肤较厚而伸展性小,故不易容纳水肿液,因而水肿不易显露和被发觉;②重力和体位:毛细血管流体静压受重力效应的影响,故离心脏水平面向下垂直距离越远的部位,外周静脉血压及毛细血管流体静压就越高,因而立位或坐位的低垂部比平卧时的同部位,存在明显的差别。例如手部静脉血压的这种差别,可达0.49~0.98kPa(50~100mmH2O)。这种重力效应在全身体循环静脉於血时就更明显。因此,右心衰竭的水肿病人,低垂部比较容易和较早出现水肿。③局部血液动力因素:如有特定的局部因素,使某一体部或器官的毛细血管流体静压增高的程度,比重力效应更为显著,以致该部毛细血管流体静压增高明显高于低垂部,则该部水肿液的积聚,可比低垂部更早出现和更明显,因而肝性水肿时,由于肝静脉回流受阻,腹水往往比下肢水肿明显得多。
四、水肿对机体的影响
(一)水肿的有利效应
1.因为当血容量迅速增长时,大量液体及时转移到组织间隙中,可防止循环系统压力急剧上升,从而减免引起血管破裂和急性心力衰竭的危险。故可把水肿看成人体调节血容量的一种重要“安全阀”。
2.炎症性水肿的有利效应 炎症性水肿至少有下列保护效应:①水肿液能稀释毒素;②水肿液的大分子物质能吸附有害物质,阻碍其入血;③水肿液中纤维蛋白原形成纤维蛋白之后,在组织间隙中形成网状物或堵塞淋巴管腔,能阻碍细菌扩散,又有利于吞噬细胞游走;④通过渗出液可把抗体或药物运输至炎症灶。
3.水肿对某些病灶的可能有利效应传统上认为水肿液的积聚可引起组织、细胞的营养不足。但在特定条件下,例如对缺血(因血管内血栓形成)的组织(例如在冻伤时),水肿液的短时间积聚,在某种程度上起着营养液的作用,可能延缓组织坏死和有利于细胞修复。
(二)水肿的有害效应
1.水肿造成细胞组织的营养不良水肿液大量积聚使组织间隙扩大,可致细胞与毛细血管的距离延长,增加了营养物质向细胞弥散的距离。受骨壳或坚实包膜限制的器官或组织,急速发展的重度水肿可压迫微血管,使营养血流减少;慢性水肿促进水肿区纤维化,对血管也有压迫作用,可引起水肿区细胞营养不良,以致皮肤容易发生溃疡,伤口难以修复。水肿区对感染的抵抗力下降,易合并感染。
2.水肿对器官组织机能活动的影响 水肿对器官组织机能活动的影响,视水肿发展速度及程度而定。急速发展的重度水肿,因来有及适应或代偿,故比绶慢发展的水肿引起更加严重的机能障碍。更重要的决定因素是器官组织对生命活动的重要性。例如严重肢体水肿对整个生命活动无大妨碍;但咽部尤其声门的水肿,则可引起气道阻塞甚至窒息致死。此外,各种器官组织发生水肿时,将引起各自的特殊机能的活动紊乱或减弱。例如肠粘膜水肿引起消化吸收障碍和腹泻;脑水肿引起颅内压升高、脑疝及脑功能紊乱。
第二节 常见水肿类型的特点和发病机制
一、心性水肿
心性水肿的分布与心力衰竭发生的部位有关,左心衰竭主要引起肺水肿(心源性肺水肿),右心衰竭主要引起全身水肿。肺水肿有专节论述(参阅心血管系统病理生理学)。
(一)临床特点
右心衰竭时水肿的曲型表现是皮下水肿,常先出现于低垂部。在立、坐位时,一般以内踝和胫前区较明显;若卧床日久,则以骶部最明显。水肿可波及躯体各部,严重时还可有腹水、胸水和心包积水。
(二)发病机制
右心衰竭时水肿的发生与多因素有关,最重要的首推钠水滞留和毛细血管流体静压增高。
1.钠水滞留右心衰竭时体液总量明显增多,严重时可超过原体重的1/2。体液增多是由于钠水滞留。但血清钠的浓度可不增高,甚或偏低,这是因为过多体液的稀释作用、低钠饮食或服用利尿药造成利钠的缘故。当病人摄入较大量钠盐时体液滞留迅速加快,临床症状加重;而当控制钠盐摄入时水肿就明显消减。
心力衰竭时钠水滞留的基本机能是肾排出减少,主要成因是肾小球滤过率减少和肾小管重吸收钠水增多。心力衰竭时肾血流减少可超过心排血量减少的程度,表明肾血管发生收缩,入球小动脉收缩可致滤过降低,肾小球滤过率因而下降。肾血管收缩可能与交感神经活动加强以及肾素-血管紧张素系统活性加强有关。
肾小管滤过率下降不一定能单独引起钠水滞留。肾小管重吸收钠水增多是更重要的因素。肾小管重吸收钠水增多的机制是多方面的,首先是由于肾血流减少激活素-血管紧张素系统而使肾上腺皮质球状带分泌醛固酮增多。此外,肝淤血导致肝代谢减弱,以致对醛固酮的灭活减慢,也是使醛固酮增多的一个附加因素。
肾血流量减少时,往往是肾小球的出球小动脉比入球小动脉收缩得更为明显,从而使肾小球滤过分数增加,近曲小管对钠水的重吸收乃因而增加。
肾小管重吸收增多还与ADH增多有关,后者起滞水作用。ADH的增多则是由于有效循环血量减少和血管紧张素Ⅱ增多。至于利钠激素或心房肽是否参与,尚待研究。
2.体静脉血压和毛细血管流体静压增高心力衰竭时体静脉血压增高由下述三个因素所引起:①心收缩力减弱致排血量减少,不能适应静脉回流;②静脉紧张度增高:心排血量减少通过颈动脉窦压力感受器反射地引起静脉壁紧张度升高,小静脉收缩使回心血量增加和静脉血管容量减少,从而导致静脉血压升高。因此用血管扩张药能使静脉血压下降和改善心力衰竭症状;③钠水滞留使血容量增多。
上述三因素的作用引起静脉血压升高,后者又引起毛细管流体静压增高。
3.其它次要因素
(1)血浆胶体渗透压下降:病人血浆蛋白浓度偏低,但不明显,只个别病例较明显。可能与食欲不振、蛋白质摄入少、呕吐、肠粘膜淤血(吸收减少),以及少量蛋白质丢失于腹水及胸水有关,更重要的是钠水滞留引起的血浆稀释。
(2)淋巴回流减少:体静脉压增高可能使淋巴排入静脉系统遇到阻力,也许在一定程度上限制淋巴回流的代偿作用。
总之,心性水肿的发病机制是综合性的,钠水滞留和静脉压增高是不可缺少的基本因素。现用示意图(图7-3)表示如下:
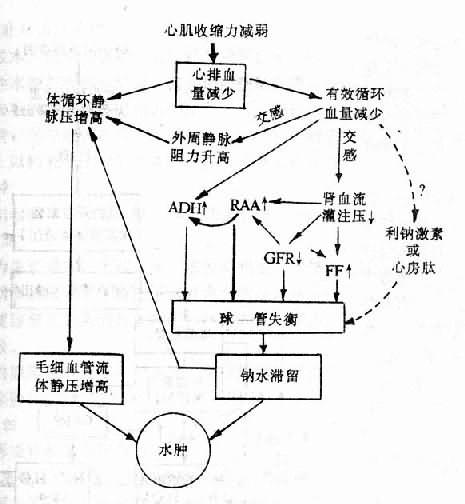
图7-3 心性水肿发生机制示意图
ADH抗利尿激素;RAA肾素-血管紧张素-醛固酮;GFR肾小球滤过率;FF滤过分数
(三)治疗原则
包括病因学疗法和发病学疗法两个方面。前者针对心力衰竭原因及其原发疾病,后者针对发病机制的主要环节。
1.改善心肌收缩力以提高心排血量。
2.消除滞积的钠水给利尿药清除过多积聚的钠水,这不仅能减轻水肿,还可减少静脉回心血量,从而解除衰竭心肌的额外负荷。
3.适当限制钠水摄入 钠水滞留是摄入超过排出的结果,如果不限制摄入,必会增加体液滞积并抵消利尿药的效应,故在应用利尿药的同时,要限制钠水摄入。
二、肾性水肿
肾原发功能障碍引起的全身水肿,称为肾性水肿(renaledema),是肾疾病的重要体征。肾性水肿起始时,低垂部的水肿不及眼睑和面部显着。往往是晨起先见眼睑或面部浮肿,随后才扩展到其它部位。这是因为体静脉压及外周毛细血管流体静压无明显增高,肺循环没有淤血,病人尚能平卧,故大量积滞的液体首先分布于组织间压较低和皮下组织疏松的部位。
肾性水肿可分两类,即以蛋白尿导致低蛋白血症为主的肾病性水肿,和以肾小球滤过率明显下降为主的炎性水肿。
(一)肾病性水肿
肾病性水肿(nephrotic edema)是肾病综合征的四大特征之一。后者除全身水肿外,还有蛋白尿、低蛋白血症和高脂血症。凡引起肾病综合征的原因,包括脂性肾病、膜性肾小球肾病、膜性增生性肾小球肾炎、肾淀粉样变性病、肾小球硬化等,都能引起肾病性水肿。
肾病性水肿发病机制的中心环节是低蛋白血症及因而起的血浆胶体渗透压下降,它是造成组织间液积聚的原发因素。低蛋白血症的原因是血浆蛋白(主要是白蛋白)大量随尿丢失,白蛋白的丢失量每天可达10~20g,大大超过蛋白合成的能力。继发钠水滞留也是重要因素,它是球-管失衡的结果。由于低蛋白血症的胶体渗透压下降,全身毛细血管的滤出增加,在引起组织间液增多的同时,也造成血浆容量的减少和有效循环血量下降,后者导致肾素-血管紧张素-醛固酮系统被激活;同时有效循环血量减少又激活下丘脑-神经垂体引起ADH释放增多;血管紧张素Ⅱ也引起ADH分泌增多。这些因素导致钠水滞留,本是对血浆容量和有效循环血量减少的代偿反应,但水肿活动期由于低蛋白血症未消除,钠水滞留又稀释了血浆蛋白,因而补充到血管内的液体,又成为水肿液的来源。
在钠水滞留的发展中,也可能有利钠激素或心房肽的释放减少参加作用,但肾内物理因素可能不起作用,因患者有明显的低蛋白血症,当血浆经过肾小球时,不断有白蛋白滤出,故血流到达肾小管周围毛细血管时,血浆蛋白浓度实际上低于正常,不可能促进近曲小管对钠水的重吸收。现把肾病性水肿的基本发生机制归纳如图7-4。
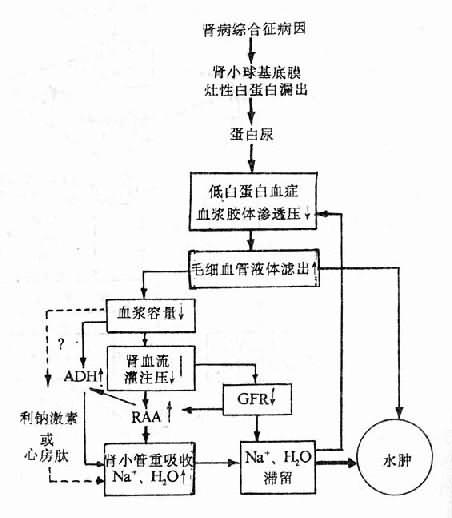
图7-4 肾病性水肿发生机制示意图
ADH抗利尿激素;RAA肾素-血管紧张素-醛固酮;GFR肾小球滤过率
(二)肾炎性水肿
主要见于急性肾小球肾炎患者。本病多由循环血中的免疫复合物所引起。临床表现尿的变化(血尿、蛋白尿、红细胞管型、少尿等、高血压和水肿。急性期过后水肿可消退。
早期认为这类水肿发生机制,是“毒素”导致全身毛细血管通透性增高。其根据是水肿液含蛋白质较多(>1g%),但进一步的研究证明,水肿液蛋白质含量不超过0.5g%。可能个别病例由于合并局部炎症反应,渗出液才会含较多蛋白质。目前认为这种全身水肿主要是球-管失衡导致钠水滞留所致。其实质是肾小球滤过率明显下降而不伴有肾小管重吸收的相应减少。由于肾小球血管内皮细胞和间质细胞发生肿胀和增生。炎性细胞渗出和纤维蛋白的堆积和充塞囊腔,使后者变得狭窄,以致通过肾小球的血流大为减少,肾小球的有效滤过面积又明显下降,其结果是肾小球钠水滤过显著下降。但此时完整的肾小管仍以正常速度重吸收钠和水,故产生高渗性少尿甚至无尿。大量钠水滞积于体内,引起血浆容量和血管外细胞外液量的明显增多,组织间液因而增多而不能被淋巴回流所代偿,于是出现全身水肿。肾小球毛细血管壁因炎症而通透性增高,故可出现蛋白尿,但低蛋白血症不明显。当然钠水滞留可稀释血浆蛋白,但此因素引起次要作用。
慢性肾小球炎有时也可伴有水肿,但不及急性肾小球肾炎明显。因残存肾单位能在一定程度上代偿。如出现水肿,其发生机制与下述因素有关;①正常肾单位明显减少使滤过总面积明显下降;②持续的肾性高血压加重左心负担,严重时导致心力衰竭;③长期蛋白尿所致的低蛋白血症。
(三)肾性水肿的治疗原则
病因治疗是根本,但秦效缓慢,必须针对发病机制及时治疗,原则如下:①限制钠盐报入:肾炎或肾病性水肿都有钠水滞留,都必须限制钠盐摄入,但要适当,长期禁钠可致低钠血症;②利尿:必要时在限钠同时投以利尿药,可促进钠水排出而缓解水肿,并可缓解高血压和减轻心脏负荷;③控制蛋白尿:对肾病性水肿必须控制蛋白尿,可用免疫抑制药(地塞米松、泼尼松等)以恢复肾小球的正常通透性;④补充血浆蛋白。
三、肝性水肿
肝原发疾病引起的体液异常积聚,称为肝性水肿(hepaticedema)。
(一)临床特点
往往以腹水为主要表现,下肢及皮下水肿不明显。若患者长期保持坐或立位,或因其它原因下肢静脉是明显淤血,则下肢皮下水肿也会明显。腹水病人因腹腔积液的牵张作用,加上肠道积气,可使腹部尤其两侧显着鼓胀;脐部外翻,腹腔内压过高易致肠疝,还可妨碍膈肌运动而影响呼吸。
腹水最常见的原因是肝硬变,但多见于失代偿期,代偿期很少有水肿,严重者还伴有胸水,也可致阴囊水肿。
(二)发病机制
肝性腹水的形成机制是多因素的:
1.肝静脉回流受阻和肝淋巴生成增多 肝血流1/3来自肝动脉,2/3来自门静脉,两路汇合于肝血窦,再至肝小叶的中央静脉,又汇集为小叶下静脉,最后联合成肝静脉,把来自肝的血送入下腔静脉。实验证明,肝静脉回流受阻可致肝淋巴生成增多并在腹腔积聚。用铝片环套在健康狗的下腔静脉胸段上,造成1/2(直径)环形狭窄,数天后出现腹水,水肿液由肝表面滴入腹腔,若先把肝移入胸腔再造成狭窄,则引起胸水而无腹水,表明水肿液来自肝。
窦后性肝硬变时肝静脉回流受阻是因广泛结缔组织增生和收缩,以及再生的肝细胞结节压迫肝静脉分支和肝血窦,致血管偏位,扭曲、闭塞或消失。回流受阻使肝静脉压升高,逆向传递至肝血窦,加上肝动脉压的向前传递,肝血窦内压乃明显上升而引起过多液体滤出,淋巴回流加速也未能充分排引,滞积的液体乃经包膜渗出,由于表面滴入腹腔而形成腹水。
2.门静脉高压和肠系膜淋巴生成增多当门静脉高压时,肠系膜区的毛细血管流体静压随而增高,液体由毛细血管滤出明显增多,肠淋巴生成增多超过淋巴回流的代偿,导致壁水肿并滤入腹腔参与腹水的形成。
3.钠水滞留上述两原发因素造成腹水后,血浆容量随之下降,钠水滞留是对后者的代偿反应。若原发因素未消除,则钠水滞留就成为腹水进一步发展的重要因素。过量钠水滞留加剧门静脉高压并使肝窦内压进一步升高,加速肝和肠系膜淋巴的生成,从而促进腹水发展。钠水滞留是因肾排钠水减少,后者又是由于醛固酮增多,包括分泌增多和肝灭活减少,此外ADH也参与钠水滞留的机制,至于利钠激素或心房肽有无分泌减少,尚待确定。
4.有关“有效胶体渗透压的下降”肝硬变时因肝合成白蛋白减少,可导致低蛋白血症。肝淋巴带走大量白蛋白丢失于腹腔,以及钠水滞留的稀释作用,也使低蛋白血症进一步加重。低蛋白血症导致血浆胶体渗透压下降,一直被认为是腹水基本成因之一。但近来有人提出相反主张,认为消化道的毛细血管壁尤其肝窦壁对血浆蛋白有较大通透性,因而平时该区组织间液的蛋白含量相当高,但在肝硬变时则可随着低蛋白血症的发展而相应下降。测算的结果表明的确如此,尤其是门静脉区组织的情况更是如此,以致肝窦和肠毛细血管有效胶体渗透压实际是增高了,而不是下降,对腹水形成并无促进作用。
可见,肝硬变时腹水形成机制是综合的,原发因素是肝内血管阻塞导致肝窦和肝外门静脉区毛细血管床流体静压增高;继发因素是钠水滞留,低蛋白血症的作用尚待确定。现把其综合机制用图解(图7-5)表示如下:
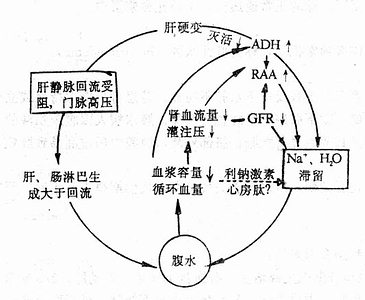
图7-5 肝硬变时腹水发生发展综合机制示意图
(三)肝性水肿的治疗原则
除改善肝功能和控制原发疾病之外,可根据发病机制采取下列措施:①限制钠盐和水摄入;②利尿:在限制钠水的同时投以利尿药很有效果;③腹腔穿剌:一般不采用,但腹水过多妨碍呼吸和病人明显不适、投利尿药无效或有副反应时,可慎用。速度不宜过快,反复小量抽放,效果尚佳;④门-腔静脉吻合分流术;把门静脉血液直接分流到下腔静脉中,使门静脉减压,一般以侧-侧门腔静脉吻合术较为优越。
四、肺水肿
肺间质有过量液体积聚和/或溢入肺泡腔内,称为肺水肿,无论哪一种肺水肿,其动力发展有一定顺序,取水肿液先在组织间隙中积聚,形成间质性肺水肿(interstial edema),然后发展为肺泡水肿(alveolar edema)。
(一)临床特点
急性肺水肿常突然发生甚至呈暴发性,表现严重呼吸困难、端坐呼吸、响亮吸气和呼气性喘鸣,听诊有水泡音,咳嗽时痰多,严重时分泌物从鼻腔或口腔流出,泡沫状痰,无色或粉红色(带血红染),痰中含大量蛋白质。慢性肺水肿症状和体征往往不严重,水肿液主要在肺间质中积聚,也有一定程度肺泡水肿,偶尔出现急性肺水肿发作。
(二)分类和原因
以发病机制为基础分为:
1.流体静压性肺水肿此类肺水肿是毛细血管流体静压增高所引起,故又称血液动力性肺水肿。它又分心源性肺水肿和非心源性血液动力性肺水肿。前者见于左心衰竭或二尖瓣狭窄;后者见于肺静脉阻塞或狭窄、过量输液或体循环血转移致肺循环等。
2.通透性肺水肿此类肺水肿是由肺泡上皮和/或微血管内皮通透性增高所引起。见于吸入毒气、细菌性病毒性肺炎、吸入性肺炎、吸入火灾烟雾、成人呼吸窘迫综合征、免疫反应(如药物特应性)等,也称肺泡中毒性水肿。
此外还有一些肺水肿的分类不够明确,如神经源性肺水肿、高原性肺水肿、肺栓塞、低血糖,子痫,呼吸道烧伤等引起的肺水肿。
(三)发病机制
1.通透性增高通透性肺水肿的发生,主要是由于肺毛细血管
■[此处缺少一些内容]■
才有可能出现这种情况。
3.血浆胶体渗透压下降 狗的实验证明,当血浆胶体渗透压降至2.00kPa(15mmHg)就可发生肺水肿;无左心衰竭的病人,在血浆蛋白减少的条件下,只需中度体液负荷(大量输液)就足以引起肺水肿;临床临床给休克病人输入大量晶体溶液容易引起肺水肿,与血浆蛋白被稀释很有关系。
4.淋巴管功能不全肺淋巴管病变导致排流不全,可致肺液体交换失衡。给狗造成轻度二尖瓣关闭不全或狭窄,左房压升达2.00kPa(15mmHg)时可无肺水肿但如同时不全结扎肺淋巴管后就出现水肿;全结扎肺淋巴管,虽无心瓣膜病也可引起肺水肿。已知当肺毛细血管滤出增多时,肺淋巴管能增加回流达3~10倍(慢性间质性肺水肿时可达25~100倍)以代偿之。故毛细血管流体静压如非急速上升至>4.00kPa(30mmHg),不易发生肺水肿。但当肺淋巴管功能不全时,这种代偿就受限制。例如在矽肺时由于慢性闭塞性淋巴管炎,阻碍了淋巴回流,在这种条件下,肺毛细血管压力只需升到2.00~3.33kPa(15~25mmHg),就足以引起肺水肿。
(四)治疗原则
1.一般疗法①改善通气和氧的供应以改善低氧血症;②纠正酸碱平衡紊乱;③利尿或在输入高分子溶液时加给利尿药,以减轻或解除肺水肿。
2.特殊疗法在准确判断基础上针对发病机制采取措施:①对抗肺毛细血管高压:针对病因解除流体静压增高。例如对心源性肺水肿病人改善心肌收缩力;对高血压引起的左心衰竭予以降压;对血容量过多的肺充血者解除肺充血(如给外周血管舒张药使血液转移到体循环);②对抗通透性增高:除解除病因外,选用降低通透性的药物,如抗炎或免疫抑制药(类固醇或非类固醇抗炎药物等)。
五、脑水肿
脑组织的液体含量增多引起脑容积增大,称为脑水肿。早期有人提出脑肿胀(brainswelling)和脑水肿是不同概念。脑水肿是指脑组织间隙的液体积聚过多,脑肿胀则指细胞液体积聚过多。其实两者并无本质区别,都是由于水肿液积聚而致脑容量增大,所不同的只是水肿液的分布和蛋白质含量。但脑容量增大不等于就有水肿,非水肿过程如脑静脉明显扩张(如CO2分压增高)时也可致脑容量增大,称为脑扩大(brainenlargement)。因而脑水肿不等于脑扩大,但脑扩大(如脑静脉阻塞或扩张)可引起脑水肿,脑水肿又可引起脑扩大。
(一)分类和特点
Klatzo把脑水肿分为两种基本类型:
1.血管源性脑水肿是最常见的一类。见于脑的外伤、肿瘤、出血、梗塞、脓肿,化脓性脑膜炎、铅中毒脑病及实验性脑冻伤等。血管源性脑水肿(vasogenic brain edema)的主要发病机制是毛细血管通透性增高,其主要特点是白质的细胞间隙有大量液体积聚,且富含蛋白质,灰质无此变化。灰质主要出现血管和神经元周围胶质成分的肿胀(胶质细胞水肿)。
2.细胞中毒性脑水肿临床多种原因引起的急性缺氧如心脏停跳、窒息、脑循不中断(缺血)等均可引起细胞中毒性脑水肿(cytotoxic brain edema),也称细胞性脑水肿。某些内源性中毒(尿毒症、糖尿病)、急性低钠血症(水中毒)、化脓性脑膜炎等也可引起这种水肿。动物实验中,局部涂搽或灌淳毒毛旋花子苷(G-strophanthin),或用二硝基酚、三乙基锡(triethyl tin)或3-乙酰吡啶(3-acetylpyridine)等代谢抑制物注射或涂擦,也可引起这种水肿。本类脑水肿的主要特点,是水肿液主要分布于细胞内,包括神经细胞、神经胶质细胞和血管内皮细胞等,细胞外间隙不但不扩大,反而缩小。灰质虽有弥漫性病变分布,但主要变化见于白质。
除上述两大类之外,Fishman又提出第三类脑水肿,称间质性脑水肿(interstitial brain edema),主要发生于阻塞性脑室积水时。当肿瘤、炎症或胶质增生堵塞了导水管或脑室孔道时,便可引起脑积水和相应脑室周围白质的间质性水肿。
脑水肿的临床表现视发展速度和严重程度而异,轻者无明显症状和体征,重者引起一系列功能紊乱:①颅内压增高引起的综合征;如头痛、头晕、呕吐、视神经乳头水肿,血压升高、心动过缓及意识障碍等;②局灶性脑体征:如一时性麻痹、半身轻瘫、单或双侧椎体性体征等;③脑疝引起的继发体征:脑扩大和颅内高压达临界点时,某些脑部因压力作用可脱位进入底池(basal cistern),出现压迫性脑疝。可表现中脑或延髓急性压迫综合征,后者可致呕吐、头晕、高血压、颈强直、角弓反张、意识丧失、呼吸间断甚至停止。
(二)发病机制
血管源性脑水肿的基本发病机制是微血管通透性增高。正常血脑屏障只容许一些小分子溶质通过,因脑毛细血管通透性很低,基外周几乎被星形胶质细胞终足所包围,后者被视为血脑屏障的组成部分(第二道屏障)。故平时组织间液几乎不含蛋白,但血管源性脑水肿时的水肿液含较多蛋白质表明微血管通透性已增高。实验观察发现水肿中心区毛细血管内皮细胞的大小吞饮泡囊增多;用铁蛋白作示踪剂,发现该颗粒出现于吞饮泡囊中,游离于胞浆和基底膜内,停留于细胞间隙中,和出现于水肿组织中,从而判定水肿液是经内皮细胞内和细胞之间的通道渗出并扩展的。通透性增高的机制尚不详知,可能与一些化学介质的作用有关。有人发现水肿白质中5-羟色胺明显增多,后者经脑脊髓液引入脑实质,可致微血管通透性增高;近期资料表明,自由基损伤内皮细胞的可能性很大,肌肉内注射自由基清除剂对苯二胺(DPPD),可减轻实验性冻伤性脑水肿。
在细胞中毒性脑水肿的发展中,微血管通透性不增高。目前认为这类水肿是脑细胞摄水增多而致肿胀。前文所述的各种代谢抑制物及急性缺氧可能都使ATP生成减少,致依赖于ATP提供能量的钠泵活动衰减,Na+不能向细胞外主动运转,水分乃进入细胞内以恢复平衡,故造成过量Na+和水在脑细胞内积聚。至于急性低钠血症时,则是因细胞外低渗,故水分转移到细胞内。新的资料表明,脑细胞膜含较多的多价不饱和脂肪酸,其不饱和双键易受自由基的影响而发生脂质过氧化反应,从而损伤膜结构和功能。此因素在细胞中中毒性脑水肿的发病机制中可能起重要作用。自由基损伤线粒体膜,后者功能受损又导致ATP生成减少。
间质性脑水肿液来自脑脊髓液,当脑脊髓液生成和回流的通路受阻(如导水管被肿瘤,或炎性增生所堵塞)时,它就在脑室中积聚,过多积聚使室内压上升,以致脑室管膜通透性增高甚至破裂,而溢入附近间质引起周围白质的间质性脑水肿(图7-6)。
(三)治疗原则
除针对病因外,主要对症治疗,原则是消肿,缩小脑容量或外科减压。
1.糖皮质激素疗法大剂量糖皮质激素尤其地塞米松对解除血管源性脑水肿有明显效果、对细胞中毒性脑水肿也有良好效果。其作用是抑制炎症反应、降低微血管通透性(抗渗出)、稳定细胞膜并恢复钠泵功能,改善线粒体功能,防止或减弱自由基引起的脂质过氧化反应,对炎症引起的间质性脑水肿也有效。
2.脱水疗法 ①渗透疗法:目的是使水分由脑组织转移到血液中,引起脑容积缩小和颅内降压,可作为应急措施。被选用的药物有尿素、甘露醇和甘油等,前两者静脉输注,后者口服;②利尿疗法:目的是增加钠水排出,减少细胞外积液。
3.外科减压疗法是解除脑肿胀和颅内高压的急救措施,不是常规治疗,但对严重的血肿和脓肿等是较好的治疗手段。
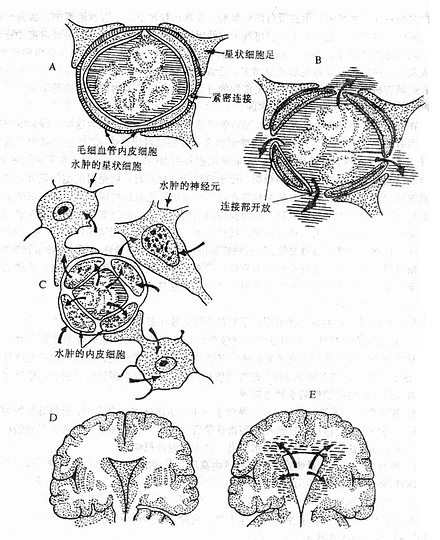
图7-6 各类脑水肿发生机制示意图
A 正常脑毛细血管;B 血管源性脑水肿;C 细胞中毒性脑水肿;d 正常脑矢状面;E间质性脑水肿
六、营养性水肿
营养不足引起的全身水肿称营养性水肿,也称营养不良性水肿。
(一)原因和临床特点
营养性水肿有原发和继发两类。原发营养不良见于战争或灾茺所致食物缺乏;继发营养不良见于:①因病摄入不足;②消化吸收障碍;③排泄或丢失过多。
水肿分布从组织疏松处开始,然后扩展至全身皮下,以低垂部为着,立位时下肢明显。
(二)发生机制
传统上认为主要成因是低蛋白血症,但实验表明,维持6个月营养不良可出现水肿,而血浆蛋白浓度却在正常范围内,故低蛋白血症不是唯一成因。另一重要因素是本来为肌肉和储备脂肪所占据的空间,因组织分解消耗而遗下空隙,使组织间隙有更大负压,易为液体所代替,在低垂部这种液体下坠而表现水肿。据检测,在半饥饿期间细胞外容量与细胞容量的比值增大,固实的组织被水样液所代替。还有资料表明这肿水肿的发生,可能与血中醛固酮和ADH增多也有关系。
(三)治疗原则
首先应解除病因和补充营养,使自身组织的消耗得以补充。补充食物要逐渐进行,使消化道逐渐适应。肠道有病则采用非经口补充。此外还应限制钠摄入或投利尿药。
七、特发性水肿
是病因尚未查明的一种全身性水肿。
(一)临床特点
几乎只发生于妇女,中年占多数。晨起眼睑浮肿,鼻梁变厚,面和手指发紧,随后乳房发胀不适,腹部膨胀,继而移行到下半身,足、战踝和小腿明显水肿,到傍晚或夜间活动(如上夜班)终了时最明显。一昼夜体重增减超过1.4kg。患者可有精神症状如情绪不安、抑郁甚至错乱,天气炎热可加重水肿。
(二)发病机制
仍不详知,早期认为与性激素或精神、情绪不稳定有关,但缺乏依据。近期资料表明可能与下列因素有关。
1.体位因素据研究,这种水肿病人微血管滤出过多与立位有关,称立位性水肿,并分为立位性钠滞留者(排钠减少)和立位性水滞留者(排水减少)。
2.微血管床异常据电镜观察,患者皮肤微血管壁的基底膜增厚,与肾病综合征时肾小球基底膜的改变相似,故提出原发微血管病导致水肿发生的假说。
3.体液因素据报道,参与特发性水肿发生的体液因素可能有醛固酮、ADH、多巴胺和缓激肽等。有人设想立位性钠滞留可能与醛固酮分泌增多有关,而立位性水滞留可能与ADH分泌增多有关;有人注意到卧位病人尿中多巴胺排出量明显减少,而多巴胺又有利钠作用。故提出多巴胺产生减少引起排钠减少的主张;还有人根据病人立位时的许多表现与缓激肽过多的表现很相似,推想缓缴肽参与其发生机制。
(三)治疗原则
原则上要解除病人的精神紧张并使之与医师取得合作,适当限钠,减少站立,晚饭后尽量躺卧于睡椅或脚板椅上(带脚板的椅子,脚板可以升降)午后7~8小时投利尿药,然的卧床(立位给药效果差)。有人试用高弹性袜和弹性压迫外套有效。据报道,有人试用拟交感胺尤其右旋安菲太明能使水肿减轻。
八、局部水肿
(一)炎症性水肿
这是最常见的局部水肿,尤其急性炎症时,水肿明显。其发生机制的中心环节是微血管壁的通透性增高,后者导致组织间液中胶体渗透压增高和局部血管内胶体渗透压下降,因而有效胶体渗透压低于正常。急性炎症时,不仅毛细血管通透性增高,微静脉壁通透性也增高,关于通透性增高的机制,详见病理解剖学炎症章。
(二)静脉阻塞性水肿
主要原因是静脉管壁受压或腔内阻塞。水肿分布于该静脉的受纳区。但水肿是否发生及其程度,则视阻塞程度、侧支循环是否建立和淋巴回流程度而定。临床上较重要的静脉阻塞性水肿见于:①慢性静脉功能不全:最常见的是髂股血栓静脉炎、长期立位所致的静脉曲张;②上腔静脉阻塞综合片;常见原因是肺癌或转移灶膨胀压迫导致管腔闭塞,表现为颜面、肩及上肢水肿;③下腔静脉阻塞综合征:视阻塞部位而表现不同,肝静脉上方阻塞引起腹水,下段阻塞则水肿限于两下枝。主要原因是肿瘤或腹腔包腹腔包块压迫,或骼静脉栓波及下段下静脉;④其它静脉阻塞:妊娠后期子宫压迫髂静脉,久病卧床引起下肢静脉血栓形成等,都可引起局部水肿。
(三)淋巴水肿
淋巴管系统有很强的适应性,在组织液生成增时能增加回流几十倍,因而淋巴回流不畅或代偿不全可成为局部水肿的重要成因。淋巴干回流受阻引起的水肿称淋巴水肿。原发性淋巴水肿少见,有的与遗传有关,如Milroy氏病时,一个或多个肢体有弥慢性淋巴管扩张致回流不畅,患肢呈管状,粗大膨胀,严重时延伸到外生殖器;继发性淋巴水肿常见,是肿瘤、手术,感染或辐射治疗把淋巴管阻断而引起的。引起淋巴水肿的感染多为反复作发作的淋巴管炎或蜂窝织炎,此时沿淋巴管的行径可见红线。继发于丝虫病的象皮肿,也较常见,多发生于下肢、阴囊、女阴等处,其病变特点是局部表皮的过度角化,直皮及皮下极度增多的纤维组织并有炎性细胞浸润。这些病变使局部皮肤及皮下组织畸形地增厚、肥大、下垂、状似象皮,故名。这些病变的原因是慢性的淋巴水肿和并发的慢性感染。因手术引起的淋巴水肿,以乳腺癌根治术后常见,表现手臂水肿,成因有:①截断了淋巴干道;②切除了腋淋巴结;③术后感染;④腋静脉扭曲;⑤术后放疗损伤;⑥腋静脉血栓静脉炎伴发血管外鞘淋巴管阻塞因此它不是单纯的淋巴水肿。也有静脉阻塞的成因参与。
此外,变态反应性水肿及遗传性血管性水肿也属局部水肿,本章不论述。
第八章 应激
第一节 应激的概念
应激(stress)是指机体在受到各种强烈因素(即应激原)剌激时所出现的以交感神经兴奋和垂体-肾上腺皮质分泌增多为主的一系列神经内分泌反应以及由此而引起的各种机能和代谢的改变。任何躯体的或情绪的剌激,只要达到一定的强度,都可以成为应激原(stressor),例如创伤、烧伤、冻伤、感染、中毒、发热、放射线的作用、出血、缺氧、环境过冷、环境过热、手术、疼痛、体力消耗、饥饿、疲劳、情绪紧张、忧虑、恐惧、盛怒、激动等等。
任何应激原所引起应激,其生理反应和变化都几乎相同,因此,应激的一个重要特征是其非物异的性质(nonspecific nature)。
应激是一种全身性的适应性反应,在生理学和病理学中都有非常重要的意义。应激既可以对人有利,也可以对人有害。
在日常生活中,几乎每一个人都会遇到某些激原的作用。只要这种作用不是过分强烈,作用的时间也不是过分地持久,那么所引起的应激将有利于动员机体身心,以便更好地完成必须完成的任务或者更好地避开可能要发生的危险,也就是说,这种应激将使人们能有效地去应付日常生活中各种各样的困难局面。这种应激,显然对机体是有利的。因而有人称之为良性应激(benign stress)。
如果应激原的作用过于强烈和/或过于持久,那么所引起的应激就属于病理生理学的范畴。许多疾病或病理过程都伴有应激;这些疾病,都有其本身的特异性的变化,又有应激所引起的一系列非特异的变化,因此应激也就是这些疾病的一个组成部分。应激在疾病中,不仅有适应代偿和防御的作用,而且它本身也可以引起病理变化。创伤、烧伤、严重感染性疾病等的发生发展中,都有应激的参与,但这些还不能算是应激性疾病,只有以应激所引起的损害为主要表现的疾病中应激性溃疡等,才可称为应激性疾病。由于应激在上述的情况下可以引起病理变化,故有人称之为劣性应激(malignant stress)。
第二节 应激时的神经内分泌反应
当机体受到强烈剌激时,就会出现以交感神经兴奋、儿茶酚胺分泌增多和下丘脑、垂体-肾上腺皮质分泌增多为主的一系列神经内分泌反应,以适应强烈剌激,提高机体抗病的能力。因此,应激时的神经内分泌反应,是疾病时全身性非特异反应的生理学基础(表8-1)。
表8-1 应激时激素和神经递质的变化
| 激素和神经递质变化 | 激素和递质 |
| 分泌增多 分泌受抑制 |
儿茶酚胺(肾上腺素、去甲肾上腺素、多巴胺) 促肾上腺皮质激素释放因子(CRF) 促肾上腺皮质激素(ACTH) 糖皮质激素(肾上腺) β-内啡肽、生长素、催乳素 胰高血糖素 抗利尿激素 肾素、血管紧张素、醛固酮 组织激素:前列腺素、血栓素、激肽 细胞因子:白细胞介素-1 胰岛素 |
一、交感神经-肾上腺髓质反应
应激时,血浆肾上腺素、去甲肾上腺素和多巴胺的浓度迅速增高。至于这些激素的浓度何时恢复正常,则在不同的应激,情况也各不相同。例如,运动员在比赛结束后一个多小时血浆儿茶酚胺已恢复正常。但大面积烧伤后半个多月,病人尿中儿茶酚胺的排出量仍达正常人的7~8倍。
应激时,交感神经-肾上腺髓质反应既有防御意义又有对机体不利方面。
1.
(1)心率加快、心收缩力加强、外周总阻力增加:有利于提高心脏每搏和每分钟输出量,提高血压。
(2)血液的重分布:交感-肾上腺髓质系统兴奋时,皮肤、腹腔内脏;肾等的血管收缩,脑血管口径无明显变化,冠状血管反而扩张,骨骼肌的血管也扩张(参阅休克章),从而保证了心、脑和骨骼肌的血液供应,这对于调节和维持各器官的功能,保证骨骼肌在应付紧急情况时的加强活动,有很重要的意义。
(3)支气管舒张:有利于改善肺泡通气,向血液提供更多的氧。
(4)促进糖原分解,升高血糖;促进脂肪分解,使血浆中游离脂肪酸增加,从而保证了应激时机体对能量需求的增加。
(5)儿茶酚胺对许多激素的分泌有促进作用(表8-2)。儿茶酚胺分泌增多是引起应激时多种激素变化的重要原因。
表8-2 儿茶酚胺对激素分泌的作用
| 激素 | 作用 | 受体 |
| ACTH | 促进 | β、α(?) |
| 胰高血糖素 | 促进 | β、α(?) |
| 胰岛素 | 促进 | α |
| 生长素 | 促进 | α |
| 甲状腺素 | 促进 | β |
| 甲状旁腺素 | 促进 | β |
| 降钙素 | 促进 | β |
| 肾素 | 促进 | β |
| 促红细胞生成素 | 促进 | β |
| 胃泌素 | 促进 | β |
2.对机体不利方面
⑴外周小血管收缩,微循环灌流量少,导致组织缺血。
⑵儿茶酚胺促使血小板聚集,小血管内的血小板聚集可引起组织缺血。
⑶过多的能量消耗。
⑷增加心肌的耗氧量。
应激一般主要引起交感神经的兴奋,但有时也可引起副交感神经兴奋占优势。例如,突然的情绪剌激,有时可引起人的心率减慢和血压下降就是一个例子。
二、肾上腺糖皮质激素反应
1.应激时糖皮质激素分泌增加 应激时几乎无例外地出现血浆糖皮质激素(glucocorticoid,GC)的浓度升高。反应迅速,升高的幅度大。例如,大面积烧伤休克期病人,血浆皮质醇(hydrocortison或cortisol)含量可高达正常的3~5倍[952~1600nmol/L(34.5~58.0μg/dl)],正常血浆含量为69~276nmol/L(2.5~10μg/dl)。同时,肾上腺皮质细胞的类脂质和维生素C含量减少;肾上腺肥大;外周血液嗜曙红性粒细胞计数减少;尿中17-羟类固醇排出量增加。后二者,再加上上述的血浆皮质醇浓度已下降到近于正常;如术后有并发症,则血浆皮质醇持续升高(图8-1)。大面积烧伤病人,血浆皮质醇维持于高水平的时间可长达2~3个月。死亡病例,在濒死期血浆皮质醇又极度升高。
图8-1① 5例 手术后并发症的病人手术前后血浆皮质醇度的浓度
图8-2② 1例手术后并发肺炎的病 手术前后血浆皮质醇的浓度
(引自Gill GV, et al Brit J Surg62:441,1975)
2.应激时糖皮质激素分泌调节应激时糖皮质激素的分泌加强是通过下丘脑-垂体前叶-肾上腺皮质相互作用而实现的。下丘脑分泌的促肾上腺皮质激素释放因子(corticotropin releasing factor, CRF)通过垂体门脉循环进入垂体前叶,刺激ACTH的释放,后者作用于肾上腺皮质,促进皮质醇的分泌,皮质醇的分泌反过来又抑制CRF和ACTH的释放,即负反馈调节机制。下丘脑受大脑各部的控制,上面主要接受来自边缘系统的纤维,下面主要受脑干网状结构的影响。来自边缘系统杏仁核的纤维调节情绪应缴反应,例如、愤怒、恐惧、忧虑等应激原均通过此通道显著地增加ACTH分泌。而创伤、剧烈湿度变化等应激原则可通过外周感受器传入冲动,引起脑干网状结构的上行激动系统的兴奋,从而引起下丘脑的兴奋,激发ACTH的释放(图8-2)。
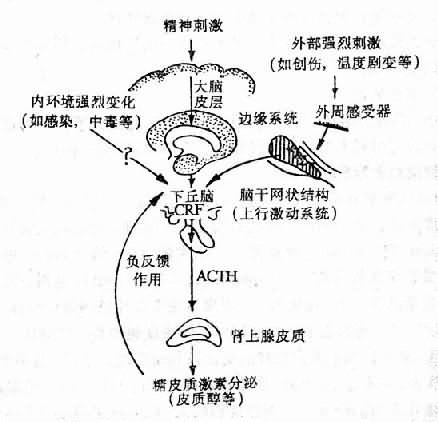
图8-2 应激时糖皮质激素分泌的调节(引自扬钢,1980)
3.应激时糖皮质激素分泌增多的生理意义GC分泌增多是应激最重要的一个反应,对机体的抗有害剌激起着极为重要的作用。动物实验表明,去除肾上腺后,动物可以在适宜条件下生存,但如受到强烈剌激,则容易衰竭、死亡。如给摘除肾上腺的动物注射糖皮质激素,则可使动物恢复抗损害的能力。大量的临床观察也证明,肾上腺皮质功能的过低病人,对应激原的抵抗力明显降低。应激时GC分泌增高,提高机体对剌激的抵抗力的机制目前还不完全清楚,已经知道的有以下四方面:
(1)糖皮质激素有促进蛋白质分解和糖异生作用,从而可以补充肝糖原的储备;GC还能抑制组织对葡萄糖的利用,从而提高血糖水平。
(2)糖皮质激素可提高心血管对儿茶酚胺的敏感性。肾上腺皮质功能不足时,血管平滑肌对去甲肾上腺素变得极不敏感,因而易发生血压下降,循环衰竭。
(3)已经证明,药理浓度的糖皮质激素具有稳定溶酶体膜,防止或减少溶酶本酶外漏的作用。由此可避免或减轻水解酶对细胞及其它方面的损害。但应激时糖皮质激素浓度是否有此作用,尚待有探讨。
(4)抑制化学介质的生成、释放和激活。生理浓度的糖皮质激素对许多化学介质的生成、释放和激活具有抑制作用。,例如,前列腺素(PGs)、白三烯(LTs)、血栓素(Tx)、缓激肽、5-羟色胺、纤溶酶原激活物、胶原酶和淋巴因子等。GC与细胞内GC受体结合后,能诱导一种分子量为40~50kD的蛋白质,称为巨皮质素(macrocortin)或脂调蛋白(lipomodulin)。它具有抑制磷脂酶A2活性的作用,因此可以减少花生四烯酸的释放,从而减少了PGs、LTs和Tx的生成。由于应激时这些化学介质的生成过多,而GC则可以抑制这些介质的产生,因而可以不发生过强的炎症、变态反应等。
三、其他激素反应
1.胰高血糖素应时胰高血糖素分泌增加。胰高血糖素促进糖原异性生和肝糖原分解,是引起应激性高血糖的重要激素。胰高血糖素分泌增加的主要原因可能是交感神经兴奋和儿茶酚胺在血中浓度的升高。
2.生长激素应激时生长激素分泌增多。交感神经过α受体可剌激生长激素的分泌。生长激素的作用是:促进脂肪的分解和动员;又能促进甘油、丙酮酸合成为葡萄糖,抑制组织对葡萄糖的利用,因而具有升高血糖的作用;生长激素还能促进氨基酸合成蛋白质,在这一点上它可以对抗皮质醇促进蛋白质分解的作用,因而对组织有保护作用。
3.胰岛素应激时,血浆胰岛素含量偏低,这是由于交感神经兴奋,血浆中儿茶酚胺升高所致。尽管应激性高血糖和胰高血糖素水平升高都可剌激胰岛素分泌,但应激时胰岛素分泌减少。
4.醛固酮应激时血浆醛固酮水平常升高(图8-3)。这主要是由于交感-肾上腺髓质系统兴奋使肾血管收缩,因而肾素-血管紧张素-醛固酮被激活,此外,ACTH分泌的增多也可刺激醛固酮的分泌。
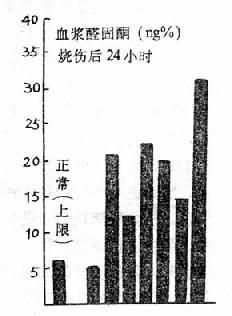
图8-3 烧伤后血浆醛固酮的改变
(每一根线为一个病例)
(引自Bane JW, et al:J Trauma14:605,1974)
5.抗利尿激素情绪紧张、运动、手术、胃肠牵拉、呕吐、缺氧、烧伤等应激原可引起抗利尿激素(ADH)分泌释放增加,使尿量减少。但有些应激原如吸入乙醚或加速度运动不伴有ADH分泌增加。精神剌激在一定条件下,也可因抑制ADH分泌而引起多尿。ADH主要由下丘脑视上核的神经元分泌。剌激边缘系统的某些部位,如杏仁核、隔区和海马等。剌激中脑网状结构,可促进视上核合成和分泌ADH。疼痛、情绪紧张等可能通过这些途径使ADH分泌增加。
6.β-内啡肽许多应激原(手术、分娩、电剌激、注射内毒素、放血、脊髓损伤等)可引起人血浆β-内啡肽明显增多,可达正常的5~10倍。血浆β-内啡肽水平的升高程度与ACTH平行。β-内啡肽在应激中起重要的作用。β-内啡肽和ACTH是同一前体阿片样肽黑素皮质激素原(proopiomelanocortin)的衍生物。β-内啡肽和ACTH都在下丘脑CRF的剌激下分泌入血。β-内啡肽和ACTH都受血浆糖皮质激素的反馈调节。向人输入β-内啡肽可降低血中ACTH和皮质醇的水平,而输入阿片受体拮抗药纳络酮(naloxone)则能使血中ACTH、β-促脂解激素(β-lipotropin,β-内啡肽的前体)和皮质醇的水平升高,提示β-内啡肽能调节ACTH的分泌,并且与ACTH一起经过短反馈或长反馈回路来抑制下丘脑CRF的分泌。β-内啡肽有很强的镇痛作用。应激镇痛(应激时痛阈值升高,称应激镇痛)可部分地为纳铬酮所取消,说明可能与β-内啡肽增多有关。
第三节 应激时的物质代谢变化
应激时物质代谢发生相应变化,总的特点是分解增加,合成减少。表现有以下几方面:
一、高代谢率(超高代谢)
严重应激时,代谢率升高十分显着。一个大面积烧伤病人,对能量需要可高达5,000千卡/天(正常成年人在安静条件下为2,000千卡/天)相当于重力劳动时的代谢率。机体处于分解代谢大于合成代谢状态,造成物质代谢的负平衡,因而患者出现消瘦、衰弱、抵抗力下降等。超高代谢主要与儿茶酚胺分泌量的增加密切相关。
二、糖代谢的变化
应激时,糖代谢变化的主要表现为高血糖。空腹血糖常为6.72~7.84mmol/L(120~140mg/dl),甚至可以超过葡萄糖的肾阈8.96mmol/L(160mg/dl)而出现糖尿。应激时的高血糖合糖尿是由于儿茶酚胺胰高血糖素、生长激素、肾上腺糖皮质激素等促进糖原分解和糖原异生以及胰岛素的相对不足所致。因此,称为应激性高血糖或应激性糖尿。肝糖原和肌糖原在应激的开始阶段有短暂的减少。随后由于糖的异生作用加强而得到补充。组织对葡萄糖的利用减少(但脑组织不受影响)。这些变化与应激的强度相平行,在严重创伤和烧伤时,这些变化可持续数周。因此,称为创伤性糖尿病。
三、脂肪代谢的变化
应激时由于肾上腺素、去甲肾上腺素、胰高血糖素等脂解激素增多,脂肪的动员和分解加强,因而血中游离脂肪酸和酮体有不同程度的增加。同时组织对脂肪酸的利用增加。严重创伤后,机体所消耗的能量有75~95%来自脂肪的氧化。
四、蛋白质代谢的变化
应激时,蛋白质分解加强,尿氮排出量增加,出现负氮平衡。严重应激时,负氮平衡可持续较久。应激病人的蛋白质代谢既有破坏和分解的加强,也有合成的减弱。待至恢复期,才逐渐恢复氮平衡。
上述这些代谢变化的防御意义在于为机体应付“紧急情况”提供足够的能量。但如持续时间长,则病人可因消耗过多而致消瘦和体重减轻。负氮平衡还可使病人发生贫血、创面愈合迟缓和抵抗力降低等不良后果(图8-4)。
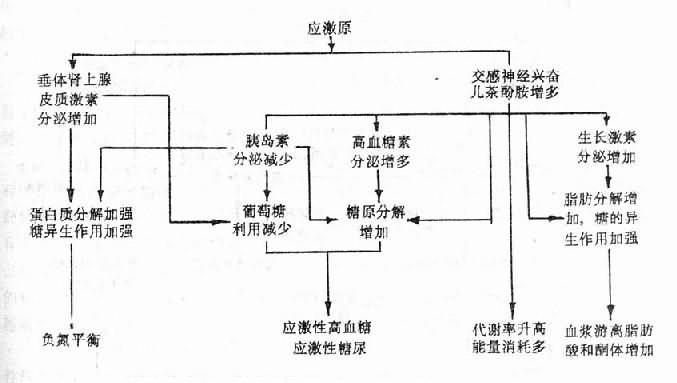
图8-4 应激时糖、脂肪和蛋白质代谢的变化
第四节 应激时机体的机能变化
一、心血管系统的变化
前文已经提到,在应激时,主要因交感-肾上腺髓质系统所引起的心率加快、心收缩力加强、外周总阻力增高以及血液的重分布等变化,有利于提高心输出量、提高血压、保证心、脑和骨骼肌的血液供应(图8-5)。因而有十分重要的防御代偿意义。但同时也有使皮肤、腹腔内脏和肾缺血缺氧、心肌耗氧量增多等不利影响,而且当应激原的作用特别强烈和/或持久时,还可引起休克(参阅第十二章休克)。
此外,在人类应激特别是情绪性应激时,可发生心律失常,这可能与交感神经兴奋时心肌细胞的钙内流增加有关。因为细胞内钙离子浓度升高可使心肌细胞膜电位负值变小,钠离子快通道失活。此时,心肌的去极化只好依赖于钙离子慢通道,其结果是使快反应细胞变成慢反应心肌细胞,不应期相应延长。传导延缓。因此容易产生兴奋的折返而发生心律失常。
应激也可引起心肌坏死,其机制可能是①交感神经兴奋和儿茶酚胺增多使心肌耗氧量增加,使心肌相对缺血;②应激时醛固酮分泌增多,钾的排出增多可引起心肌细胞内缺钾,从而促使心肌细胞坏死;③应激时心肌小血管内可有血小板聚集物出现,从而可以阻塞血管。血小板聚集物的出现与儿茶酚胺的作用有关。
二、消化道的变化
应激时,有消化系统功能障碍者较为常见,但各种应激原所致的消化并不一致。引人注目的是由应激引起消化道溃疡,称为应激性溃疡(stress ulcer)。经内窥镜检查发现,烧伤、严重创伤和败血症病人应激性溃疡的发生率高达80~100%。
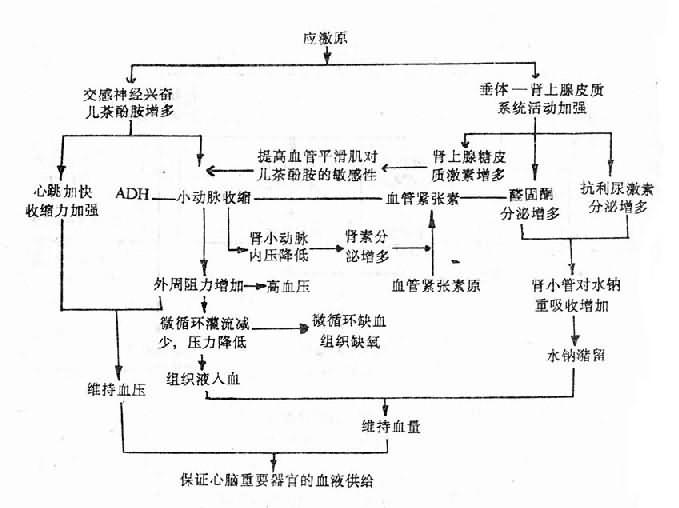
图8-5 应激反应时循环系统的变化
与慢性经过的消化性溃疡(peptic ulcer)不同,应激性溃疡是一种急性溃疡,在病理解剖学上,应激性溃疡主要是胃和/或十二指肠的粘膜缺损,可以严重在严重的应激原作用以后数小时内就出现。粘膜缺损或则表现为多发性糜烂(仅仅到达粘膜肌层的表浅损害)。或则表现为单个的或多发性的溃疡(深达粘膜肌层之下的损害)。溃疡的直径可达20mm。溃汤周围无水肿、炎性细胞浸润或纤维化可见。如果病人存活,应激性溃疡可在数天之内愈合。而且不留疤痕。由于溃汤不侵及肌层。因而在临床上也很少引起疼痛。临床上的主要症状是出血,出血可轻可重,常表现为呕血或黑粪出血严重时可致死。由于溃疡浅表,因而胃或十二脂肠穿孔极为罕见。
应激性溃疡的发病机制尚未完全阐明。总的来说,是由于对胃或十二指肠粘膜的“损害性因素”胜过了,“保护性因素”的结果。
1.粘膜缺血应激时,主要由于交感肾上腺髓质系统的兴奋,使胃和十二指肠粘膜的小血管也发生收缩,粘膜的血液灌流量乃显著减少,于是粘膜发生缺血缺氧。这就使粘膜的“保护性因素”削弱而“损害因素”就得以破坏粘膜而引起溃疡形成,因为:
(1)粘膜缺血使粘膜上皮细胞能量不足,因而粘膜的某些细胞就不能产生足量的碳酸氢盐和粘液。这样就使由粘膜上皮细胞之间的紧密连接和覆盖于粘膜表面的碳酸氢粘液层所组成的胃粘膜屏障遭到破坏,胃腔内的H+就顺着浓度差(胃腔与胃粘膜细胞的[H+]之比为1.6×106:1)进入粘膜;同时,由于粘膜缺血,又不能将侵入粘膜的H+随血液运走,因而H+就在粘膜内积聚。已经证明,H+是主要的“损害性因素”,是形成应激性溃疡的必不可少的原因。如果将腔内的胃酸完全中和,那么在动物实验中就不能造成应激性溃疡的模型。在家兔的实验中,当H+的返流使胃粘膜固有层的pH值降至6.7~6.5时就可以预计会出现溃疡。当溃疡初步形成后,另一“损害性因素”胃蛋白酶也可借其蛋白分解作用分解已经受损的细胞而溃疡扩大。
(2)粘膜缺血使粘膜细胞的再生能力降低,因而使已经发生的缺损不易修复。
2.糖皮质激素分泌增多糖皮质激素使蛋白质的分解大于合成,胃上皮细胞更新减慢再生能力降低。因而胃粘膜对“损害性因素”抵抗力降低,胃粘膜对H+的屏障作用也被削弱,已经发生的缺损也不易修复。
3.胃粘膜合成前列腺素减少胃粘膜上皮细胞不断地合成和释放前列腺素(PGs)。PGs有保护胃粘膜上皮细胞的作用。据报道,应激后胃粘膜PGs含量减少,而且在临床上应用大剂量前列腺素,可以预防某些应激性溃疡的发生。前列腺素保护胃粘膜的机制尚未阐明。据有人研究,前列腺素的这种作用可能与改善细胞内对H+的中和能力有关。当H+进入胃粘膜上皮细胞时,就可以被细胞内的HCO3-所中和。而前列腺素可以加强这种作用,因而具有保护的意义,应激时,一方面由于粘膜缺血而致细胞内HCO3-产生不足,另方面由于胃粘膜上皮细胞前列腺素的合成减少,因而进入细胞内的大量H+就不能被中和而引起细胞的损害。
4.全身性酸中毒某些严重的应激特别是在伴有休克时,往往发生全身性酸中毒。全身性酸中毒也可使胃粘膜上细胞内的HCO3-减少,从而使细胞内中和H+的能力降低而有助于溃疡的发生。
5.β-内啡肽 前文已经提到,应激时血浆β-内啡肽显著增多。最近的一些研究提示,β-内啡肽可能作为一种“损害因子”而引起应激性溃疡。如果事先给予阿片受体拮抗药纳络酮,就可以预防大鼠发生应激性溃疡。
6.胆汁酸和溶血卵磷脂 这是十二指肠内的两种“损害性因素”。胆汁酸来自肝,溶血卵磷脂则是胰腺分泌的酶作用于卵磷脂而形成。生理情况下,通过十二指肠至胃的反流,可有少量胆汁酸和溶血卵磷脂进入胃腔,但由于“保护性因素”占优势,故不能对胃粘膜造成损害。在严重应激特别是伴有休克时,这种十二指肠至胃的反流加强,同时又因缺血等原因使“保护性因素”削弱。因而这两种“损害性因素”既可直接损害胃粘膜。又可因为能使粘膜的通透性增高而导致H+反流的进一步加强,从而使粘膜的损害更严重。有的作者特别提到,胆汁酸和溶血卵磷脂使胃粘膜通透性增高从而促进H+反流的作用,在动物实验中已经得到很好的证实,但在人类则还有待进一步的研究。
三、凝血和纤溶的变化
应激时,有暂时性的血液凝固性升高。外伤后数小时内,病人凝血时间和血凝块溶解时间都缩短。血液凝固性升高和纤溶活性升高的机制,有以下三方面:
1.应激时因儿茶酚胺分泌增加,可使血小板的聚集性增强。
2.应激时血浆中凝血因子Ⅷ、纤维蛋白原和血小板三者均增多,从而使血液凝固性升高。
3.应激时纤溶活性升高是由于纤溶酶原激活物的增多,该激活物存在于血管内皮细胞内。儿茶酚胺等血管活性物质作用于血管内皮细胞,具有剌激纤溶酶原激活物的作用。
应激时凝血和纤溶的变化是严重创伤或感染时易于发生弥散性血管内凝血的因素之一。然而,应激时血液凝固性的增高也不乏有利的一面,因为它可以促进组织损伤时的止血。
四、泌尿机能的变化
应激时,泌尿机能的主要变化是尿少,尿比重升高、水和钠排出减少。这些变化的机制有以下三方面。
1.应激时交感神经兴奋, 肾素-血管紧张素系统增强,肾入球小动脉明显收缩,肾血流量减少,肾小球滤过率减少。
2.应激时醛固酮分泌增多,肾小管钠、水重吸收增加,钠水排出减少,尿钠浓度降低。
3.应激时抗利尿激素分泌增加,从而使肾远曲小管和集合管对水的通透性增高,水的重吸收增加,故尿量少而比重升高。
肾泌尿功能变化的防御意义在于减少水钠的排出,有利于维持循环血量。但肾缺血所致是肾泌尿功能障碍,却可导致内环境的紊乱。
泌尿机能的这些变化,实际上相当于休克早期所伴有的功能性急性肾功能衰竭,如果不及时抢救休克,将发展成为急性肾小管坏死(参阅肾脏病理生理学)。
五、免疫功能的变化
表现为免疫功能的减弱,这是CG分泌增加的结果,与生长激素、盐皮质激素也可能有一定的关系。GC对免疫反应的许多环节都有影响,主要是抑制巨噬细胞对抗原的吞噬和处理,阻碍淋巴细胞DNA合成有丝分裂,破坏淋巴细胞,使外周淋巴细胞数减少,并损伤浆细胞,从而抑制细胞免疫反应和体液免疫反应。此外,GC还能抑制毛细血管壁的通透性升高,抑制胶原纤维和毛细血管的增生,抑制中性粒细胞向炎症灶游出。这些作用就使炎症反应受到抑制。
应当指出,前文所述的一些代谢和机能的紊乱,是在应激原的作用十分强烈和/或持久的情况下发生的,也就是说,这些变化是“劣性应激”的组成部分。
第五节 急性期反应蛋白
在感染、炎症、组织损伤等应激原作用于机体后的短时间(数小时至数日)内,即可出现血清成分的某些变化,称为急性期反应(acute phase reaction),参与急性期反应的物质称为急性期反应物(acute phase reactant)。急性期反应物大多数是蛋白质,称为急性期蛋白(acrte phase protein, AP蛋白)。最早发现的AP蛋白是C-反应蛋白(C-reactive protein);它能与肺炎双球菌的荚膜成分C-多糖体起反应,故起名为C-反应蛋白。
急性期反应时血浆中浓度增加的AP蛋白种类繁多,可分为五类,即参与抑制蛋白酶作用的AP蛋白(如α1抗胰蛋白酶等);参与血凝和纤溶的AP蛋白(如凝血因子Ⅷ,纤维蛋白原、纤溶酶原等);属于补体成分的AP蛋白;参与转运的AP蛋白(如血浆铜蓝蛋白等);其他多种AP蛋白(如C-反应蛋白、纤维连接蛋白、血清淀粉样物质A等)。急性期反应时血浆蛋白浓度也有减少的,称为负性AP蛋白,(如白蛋白,运铁蛋白等)。
现已证明,除了感染以外,创伤、烧伤、手术等许多应激原,均能引起人和许多动物血浆中一些AP蛋白的增多或减少,它是应激的一种重要变化。虽然AP蛋白的变化是非特异性的,但却有广泛的防御意义。
一、AP蛋白的浓度变化和来源
正常血浆中AP蛋白含量一般较低或甚微,有的还不易检出。在炎症、感染、发热、创伤、手术等应激原作用下,有些AP蛋白可增加20~1,000倍。如C-反应蛋白,血清淀粉样物质A等;有些AP蛋白则增加2~5倍,如α1-抗胰蛋白酶等;而有的AP蛋白只增加30~60%,如铜蓝蛋白、C3等。
AP蛋白在血浆中浓度的升高主要是由于合成增强和释放增多。但它们在高水平上保持恒定则主要是合成和分解平衡的结果。有些AP蛋白在急性期反应中合成增加,但消耗也增加,例如,某些补体成分在血中浓度可不增高或增高不多。
AP蛋白来源于何种组织和细胞虽有争论,但灌流实验已证实了肝是AP蛋白的主要来源,肝细胞能合成大多数的AP蛋白。少数AP蛋白来源于巨噬细胞、内皮细胞、成纤维细胞和多形核白细胞等。
二、AP蛋白的生物学功能
1.抑制蛋白酶的作用 创伤、感染等引起的应激时,体内蛋白水解酶增多,过多的蛋白水解酶可引起组织的损害。AP蛋白中有蛋白酶抑制物,例如α1-抗胰蛋白酶、α1-抗糜蛋白酶、C1酯酶抑制因子、α2-抗纤溶酶等。应激时,这些酶的消耗增加,同时合成也增加,以保证蛋白酶抑制物能得到必要的补充。
2.凝血和纤溶纤维蛋白原在凝血酶作用下形成的纤维蛋白在炎症区组织间隙构成网状物或凝块,有利于阻止病原体及其毒性产物的扩散;继而纤溶系统的激活又可在晚些时候溶解这些凝块而使组织间隙恢复原状。然而,凝血和纤溶系统的过度激活却有可能导致DIC而给机体造成严重的后果。
3.清除异物和坏死组织某些AP蛋白具有迅速的非特异性的清除异物和坏死组织的作用。例如C-反应蛋白容易与细菌细胞壁结合,又可激活补体的以经典途经,促进大、小吞噬细胞的功能。这就使得与C-反应蛋白结合的细菌迅速地被清除。
4.清除自由基如铜蓝蛋白能活化超氧化物岐化酶(superoxide dismutase,SOD),故有清除氧自由基的作用。
5.其他如血清淀粉样物质A可能有促使损伤细胞修复的作用;纤维连接蛋白则能促进单核细胞、巨噬细胞和纤维母细胞趋化性,促进单核细胞膜上Fc受体和C3b受体的表达,并活补体旁路,从而促进单核细胞的吞噬功能,等等。
第六节 热休克蛋白
热休克蛋白(heatshock protein,HSP)是指细胞在应激原特别是环境高温诱导下所生成的一组蛋白质。
HSP首先是在果蝇体内发现的。果蝇幼虫唾液腺的多丝染色体(polytene chromosome)比一般染色体粗1~2千倍,故有利于在光学显微镜下进行观察研究。1962年有人发现,将果蝇的培养湿度从25℃提高到30℃(热休克环境温度升高),30分钟后就可在多丝染色体上看到蓬松现象(或称膨突puff),提示这些区带基因的转录加强并可能有某些蛋白质的合成增加。至1974年,后人才从热休克果蝇幼虫的唾液腺等部位分离到了6种新的蛋白质,即HSP。除环境高温以外,其他应激原如缺氧、寒冷、感染、饥饿、创伤、中毒等也能诱导细胞生成HSP。因此,HSP又称应激蛋白(stress protein, SP),但习惯上仍称HSP。
近年研究表明,HSP的生成,不仅见于果蝇,而且是普遍存在于从细菌直至人类的整个生物界(包括植物和动物)的一种现象。例如,1981年有人在实验中证明,将大鼠置于55℃的高温环境,直肠温度迅速升至42~42.5℃,15分钟后使环境温度降至常温,体温也随之于30分钟后降至正常水平。90分钟后处死动物,就可在心、脑、肝、肺等器官的组织内分离出一种分子量为71kD的新的蛋白质,即HSP。
绝大部分生物细胞生成的HSP分子量都在80~110kD、68~74kD和18~30kD之间。不同分子量的HSP,在细胞内的分布也有所不同,例如,在酵母菌中发现的分子量为89kD的HSP是一种可溶性的细胞浆蛋白质,而分子量为68kD、70kD、110kD的HSP却主要分布于核或核仁区域。
HSP在生物界中的一个重要特点是它们在进化过程中的高度保守性。例如。从大肠杆菌、酵母、果蝇和人体分离的分子量为70kD的HSP,如果对它们进行全氨基酸序列分析,就可发现它们具有80%以上的相似性。HSP在进化过程中的高度保守性,说明它们具有普遍存在的重要生理功能。然而在这方面的研究,迄今还很不充分。
一、热休克蛋白的诱导和调节的机制
总的来说,HSP的诱导和调节的机制迄今还不清楚,只有一些推测。
应激原诱导HSP生成的速度很快。将果蝇从25℃移至37℃环境,只要20分钟,就可以检出HSP,因而有人推想高温是通过某种已经存在的调节因子作用于基因并从而使转录加强的。实验证明,用热休克细胞的胞浆提取物可以诱导果蝇幼虫唾液腺细胞核内染色体的蓬松现象,而未经热休克的对照细胞胞浆无此种诱导作用。提示胞浆内存在的某种物质,在应激时可被活化而转位到核内,进而启动基因对HSp mRNA的转录。
上述的染色体蓬松现象,即使是在应激原的持续作用下,一般也都在60分钟以内消失,而HSP则由于降解较慢,故可持续存在6小时,提示HSp mRNA的转录受HSP的负反馈调节。
二、热休克蛋白的功能
HSP可提高细胞的应激能力,特别是耐热能力。预先给生物以非致死性的热剌激,可以加强生物对第二次热剌激的抵抗力,提高生物对致死性热剌激的存活率,这种现象称为热耐受。目前对此现象的分子机制仍不太清楚,但许多研究均发现了HSP的生成量与热耐受呈正相关。
HSP还可调节Na+-K+-ATP酶的活性。某些细胞经热休克丧失的Na+-K+-ATP酶活性可在3℃培养中随着HSP的产生而得到部分恢复。HSP的诱导剂亚砷酸钠亦可使Na+-K+-ATP酶的活性升高。这种现象可被放线菌素D和环已酰亚胺抑制,提示Na+-K+-ATP酶活性升高是一种基因表达的结果,而不是亚砷酸钠直接作用的结果。
有人通过四膜虫属细胞热休克的研究,发现有些HSP具有促进细胞内糖原异生和糖原生成的作用,使细胞内糖原贮量增多,从而提高应能力。
此外,有人还报道,热、乙醇、亚砷酸钠的预处理不仅能使某些细胞产生热耐受,还能使细胞对阿霉素(adriamycin)的耐受性增强,提示HSP可以增强对各种损伤的抵抗力。
至于在人类的应激中,HSP究竟起什么作用,目前还知之甚少。
第七节 应激的生物学意义与防治原则
一、应激的生物学意义
前文已经提到,良性应激或生理性应激,是人们日常生活的重要组成部分,对于人们要想达到生活中的某些目标,是一种促进的、激动的因素。生理性应激时物质代谢和各器官机能的改变,特别是能量提供的增加,心、脑和骨骼肌血液供应的保证等,对于努力完成某种艰巨的任务,对于进行“斗争(fight)”和“脱险(flight)”,都有极为重要的意义。因此,有人把这种反应称为“斗争-脱险反应(fight-flight reaction)”。
许多疾病或病理过程都伴有应激。这时的应激,是由于应激原的作用过于强烈和/或过于持久所引起。应激时的一系列非特异性变化,虽然也有前述的防御和适应的作用,但由于这些变化过剧烈和/或持久,故可导致机能代谢的障碍和组织的损害,严重时甚至可以导致死亡。因而这种应激被称为劣性应激或病理性应激。
二、应激的防治原则
1.避免过于强烈的或过于持久的应激原作用于人体,例如,避免不良情绪和有害的精神剌激,避免过度而持久的精神紧张,避免各种意外的躯体性的严重伤害,等等。
2.及时正确地处理伴有病理性应激的疾病或病理过程如烧伤、创伤、感染、休克等等,以尽量防止或减轻对人体的不利影响。
3.采取一些针对应激本身所造成损害的措施,例如在严重创伤后加强不经胃肠道的营养补充,其目的之一就是弥补应激时因高代谢率和蛋白分解加强所造成的机体的消耗。
4.急性肾上腺皮质功能不全(如肾上腺出血、坏死)或慢性肾上腺皮质功能不全的病人,受到应激原剌激时,不能产生应激;或者由于应激时肾上腺糖皮质激素受体明显减少,病情危急,应及时大量补充肾上腺糖皮质激素。
第九章 弥散性血管内凝血
弥散性血管内凝血(disseminatedor diffuse intravascular coagulation, DIC)是指在某些致病因子作用下凝血因子或血小板被激活,大量可溶性促凝物质(soluble thromboplastin)入血,从而引起一个以凝血功能失常为主要特征的病理过程(或病理综合征)。此时微循环中有纤维蛋白性微血栓或血小板团块形成,同时一系列血浆凝血因子被消耗,血小板减少,并有继发性纤维蛋白溶解(纤溶)过程加强。在临床上,DIC患者主要表现为出血、休克、脏器功能障碍和贫血。
由于DIC的发病机制和临床表现比较复杂,因此长期以来,曾经有过许多不同的命名。近年来,某些学者认为,为了更全面地表达此病理过程的变化特点,建议将DIC称为消耗性血栓-出血性疾病(cosumptive thrombohemorrhagicdisorders)。本章继续使用弥散性血管内凝血的命名。
第一节 弥散性血管内凝血的原因和发病机制
正常机体的血液呈液体状态,在心、血管内流动不止,同时也不发生血凝。这是由于机体存在着凝血、抗凝血和纤维蛋白溶解系统,它们处于动态平衡状态。其中以凝血过程和纤维蛋白溶解过程最为重要,二者保持着极为密切的关系。
DIC的病因众多,引起DIC的发病机制为复杂,但其中以血管内皮细胞的损伤与组织损伤为最重要。
一、血管内皮细胞损伤、激活凝血因子Ⅻ,启动内源性凝血系统
细菌、病毒、螺旋体、高热、抗原抗体复合物、休克时持续的缺血、缺氧和酸中毒、败血症时的细菌内毒素等,在一定条件下皆可使血管内皮细胞发生损伤,使其下面的胶原暴露。胶原、内毒素等均为表面带负电荷的物质,当无活性的凝血因子Ⅻ与这些物质表面发生接触后,其精氨酸残基上的胍基在负电荷影响下分子构型发生改变,它的活性部分——丝氨酸残基暴露,所以因子Ⅻ被激活(此种激活方式称接触激活或固相激活)。另外,也可能在激肽释放酶、纤溶酶或胰蛋白酶等可溶性蛋白水解酶的作用下,因子Ⅻ或Ⅻa通过酶性水解(酶性激活或液相激活)而生成Ⅻf。胶原等激活因子Ⅻ的过程开始时进行得较为缓慢,但因Ⅻ的碎片(Ⅻf),即激肽释放酶原激活物(predallidreinactivator, PKA)可把血浆激肽释放酶原(prekallikrein)激活成激肽释放酶(kallikrein),后者又能反过来使因子Ⅻ进一步活化,从而使内源性凝血系统的反应加速(图9-1)。Ⅻa和Ⅻf还可相继激活纤溶、激肽和补体系统,从而进一步促进DIC发展。
图9-1 血液凝固过程及纤溶系统
以内毒素血症为例,此时,除内毒素可直接激活因子Ⅻ外,内毒素还可引起血管内皮细胞损伤,基底膜暴露后,胶原、胶原与某些糖蛋白的复合物或另一些结缔组织成分也可激活因子Ⅻ。此外,还有某些酸糖脂(acidic glycolipids),硫酸脂(sulfatides),氨基葡聚糖(glycosaminoglycans)或另外一些特殊的因子Ⅻ激活物也可自损伤的内皮细胞释放,因此内源性凝血系统启动。在家兔内毒素引起的DIC中常可有内皮细胞脱落,在循环血中有内皮细胞出现。抗原抗体复合物粘附在肾小球等微血管壁上时,可引起血管内皮细胞损伤。血管炎时也可继发血管内皮细胞损伤,进一步触发DIC。
此外,在内皮细胞受损时,血小板与内皮下结缔组织中的胶原接触后可以产生胶原诱导的促凝活性,此时,因子Ⅺ可不通过Ⅻa而直接被激活,从而推动凝血连锁反应,引起DIC。
表9-1不同的人体组织中凝血因子Ⅲ的含量
| 组织 | 含量 (μ/mg) |
| 肝 | 10 |
| 肌肉 | 20 |
| 脑 | 50 |
| 肺 | 50 |
| 胎盘 | 2,000 |
| 蜕膜 | 2,000 |
二、组织严重破坏使大量组织因子进入血液,启动外源性凝血系统
在外科大手术、严重创伤、产科意外(如胎盘早期剥离、宫内死胎等)、恶性肿瘤或实质性脏器的坏死等情况下均有严重的组织损伤或坏死,所以大量促凝物质入血(表9-1),其中尤以组织凝血活酶(tissuethromboplastin,即凝血因子Ⅲ,或称组织因子)较多。这些促凝物质可通过外源性凝血系统的启动引起凝血。
组织因子是一种脂蛋白复合物,含有大量磷脂。当它进入血浆后。血浆中的钙离子将因子Ⅶ连接于组织因子的磷脂上,形成复合物,后者可使凝血因子X活化为Xa,并与Ca2+、因子V和血小板磷脂相互作用而形成凝血酶原激活物,然后通过与内源性凝血系统后阶段相同的途径,完成凝血的化学反应(图9-1)。
以宫内死胎为例,当胎儿的坏死组织在子宫内滞留超过5周,DIC的发生率可达50%左右,这是因为坏死的胎儿组织释放组织因子,后者大量进入母体循环,启动外源性凝血系统。此外有人证明,当肿瘤组织坏死时,释放出一种蛋白酶,如某些腺癌能分泌一种含有唾液酸的粘蛋白,它可直接激活X因子,从而启动凝血连锁反应。
三、血细胞大量破坏
红细胞大量破坏时常可发生DIC。急性溶血,如大量(>50ml)误型输血、药物引起的免疫性溶血时,抗原-抗体复合物的形成对凝血起主要作用。因为据报道,在蚕豆病中由非免疫因素引起的血管内溶血以及实验性血红蛋白尿等情况下常常不产生DIC。因此,一般认为只有在红细胞大量破坏伴有较强的免疫反应时,DIC才比较容易发生。此外,红细胞大量破坏释出的ADP与DIC的发生有关,因为后者触动了血小板释放反应,使大量血小板第3因子(PF3)入血,促进凝血过程。红细胞膜内大量的磷脂既有直接的促凝作用,又能促进血小板的释放而间接促进凝血过程。
实验研究证明,正常的中性粒细胞和单核细胞内有促凝物质。在内毒素或败血症所引起的DIC时内毒素可使中性粒细胞合成并释放组织因子,同时有大量白细胞在肺血管中停滞,并释放出大量促凝物质(可能就是组织因子),这些物质进入体循环进一步加速了凝血反应,所以肺似乎起了凝血的放大作用。大量促凝物质从崩解的白细胞中释放出来,从肺血管经左心进入主动脉后,肾脏首先受累,因此肾脏微血栓发生率较高,病变程度较重。另外,在病人患急性早幼粒细胞性白血病时,此类白血病细胞浆中含有凝血活酶样物质,当白血病细胞大量坏死或经化疗杀伤时,这些物质就大量释放入血,通过外源性凝血系统的启动而引起DIC。
血小板在DIC的发生发展中起着重要的作用。内毒素、免疫复合物、颗粒物质、凝血酶等都可直接损伤血小板,促进它的聚集。微血管内皮细胞的损伤,内皮下胶原和微纤维的暴露是引起局部血小板粘附、聚集、释放反应的主要原因,这是因为是构成胶原的肽链中,存在着一个与血小板粘附有关的活性部位。血小板表面的糖蛋白Ib(glycoprotein Ib, GPIb)对血小板粘附起重要作用,GPIb通过血浆因子(如Ⅷ相关抗原/vonWillebrand因子,Ⅷ/VWF因子)使血小板与内皮下组织粘连。另外,由于血小板膜上的另一些糖蛋白(GPⅡb,GPⅡa)能结合于纤维蛋白原,后者通过与钙离子的连接,在血小板之间“搭桥”,使血小板聚集。血小板发生粘附、释放和聚集后,除有血小板微集物形成(microaggregateformation,图9-2)堵塞微血管外,还能进一步激活血小板的凝血活性,促进DIC的形成。但是在不同病因所引起的DIC中,血小板所发挥的作用并不一致,它可以起原发的作用,如血栓性血小板减少性紫癜,在发病开始时即可由免疫反应等原因使血小板发生聚集,其中PF3(血小板第3因子)能加速凝血酶原的激活,PF4(血小板第4因子)能中和肝素并使可溶性纤维蛋白多聚体沉淀。β-血栓球蛋白也具有促凝作用,从而加速血液凝固,形成微血栓。但是,一般来说,在DIC发病中,血小板多起继发的作用。在外源性凝血系统被激活所致的DIC中,血小板不起主要作用,在内毒素引起的DIC中,血小板对白细胞的促凝机制还有促进作用。实验证明,人类白细胞与内毒素同时孵育后所产的促凝活性可因加入血小板而增强,这可能是血小板膜上的脂蛋白、白细胞及某些凝血因子相互作用造成的。
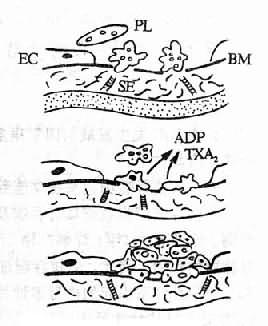
图9-2 血小板微聚物形成机制示意图
PL血小板 EC 内皮细胞SE 内皮下组织 BM 基底膜
四、其它促凝物质进入血液
一定量的羊水、转移的癌细胞或其它异物颗粒进入血液可以通过表面接触使因子Ⅻ活化,从而激活内源性凝血系统。急性胰腺炎时,蛋白酶进入血液能促使凝血酶原变成凝血酶。毒蛇咬伤时,某些蛇毒如蝰蛇的蛇毒含有一种蛋白酶,它可直接水解凝血酶原形成凝血酶。响尾蛇的蛇毒可直接使纤维蛋白原凝固。抗原抗体反应也可以引起DIC,这可能是抗原抗体复合物能激活因子Ⅻ或损伤血小板引起血小板聚集并释放促凝物质(如血小板因子等)所致。补体的激活在DIC的发生发展中也起着重要的作用。有人发现,给正常动物静脉注射内毒素后,出现动脉血压下降,血小板及纤维蛋白原等凝血因子减少;但如事先耗竭动物的补体,然后再注射内毒素,则该动物血压改变不明显,DIC实验室检查的异常变化轻微,存活率比未去除补体的动物高,由此可见补体系统在内毒素引起的的DIC中也起一定的作用。补体系统激活的产物C3a、C5a可引起组织肥大细胞、血液嗜碱性粒细胞的脱颗粒反应,从而释放5-羟色胺、组胺等物质。组胺能使毛细血管、微静脉等部位的血管内皮细胞收缩,内皮细胞之间的裂隙扩大,内皮下的胶原暴露,促使内源性凝血系统激活。此外,补体系统激活后C3b还可通过人单核细胞上的C3b受体而使凝血因子Ⅲ的释放增多。补体系统还能直接或间接地促进血小板释放PF3。
上述各种所致DIC的机制如图9-3所示。它们常常综合或相继起作用。
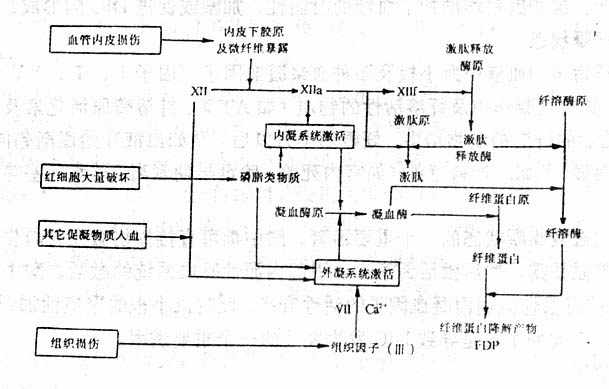
图9-3 DIC的发病机制
第二节 影响弥散性血管内凝血
发生发展的因素
影响DIC发生发展的因素很多,应该引起警惕,尽可能及早采取相应的措施以防止、减轻或排除其作用。
一、单核吞噬细胞系统功能受损
单核吞噬细胞系统具有吞噬及清除循环血液中的凝血酶、其它促凝物质、纤维蛋白、纤维溶酶、纤维蛋白(原)降解产物(fibrin or fibrinogen degradation product, FDP)以及内毒素等物质作用。因此,单核吞噬细胞系统的严重功能障碍会促使DIC的形成。例如在严重的革兰氏阴性细菌引起的内毒素性体克中,单核吞噬细胞系统可因吞噬大量坏死组织、细菌或内毒素而使其功能处于“封闭”状态;同样,在严重的酮症酸中毒时,大量脂质有时也可“封闭”单核吞噬细胞系统,这时机体再与内毒素接触就易于发生DIC。
全身性Shwartzman反应(generalized Shwartzman reaction,GSR)是给家兔间隔24小时静脉内各注射一次小剂量内毒素,在接受第二次注射后家兔就发生休克、出血倾向,甚至因急性肾功能衰竭而死亡。死亡解剖发现各重要脏器的微循环中常有纤维蛋白性微血栓,而且由此产生相应组织的缺血坏死,其中尤以肾、肺、肝等最为明显。如果第一次注射时用具有封闭单核吞噬细胞系统作用的二氧化钍代替内毒素,则第二次注射小剂量内毒素后同样发生DIC。目前一般认为GSR的发生机制之一是由于第一次内毒素注射后单核吞噬细胞系统吞噬了内毒素和纤维蛋白而被“封闭”,因此第二次注射时,单核吞噬细胞系统清除激活的凝血因子的能力降低,并无法使内毒素灭活。内毒素具有损伤血管内皮、激活血小板及凝血因子Ⅻ、促使血小板聚集和收缩血管等作用,故能引起DIC。
二、肝功能严重障碍
肝功能严重障碍时肝脏产生的某些抗凝物质如抗凝血酶Ⅲ(antithrombin Ⅲ,AT-Ⅲ)减少,引起肝功能障碍的某些病因,如肝炎病毒、某些药物、抗原抗体复合物等均可激活凝血因子。肝细胞如有大量坏死,又可释放组织凝血活酶(因子Ⅲ)样物质。此时机体经肝脏处理乳酸的能力降低。这些因素均增加了血液的凝固性,加剧或促进DIC的形成。
三、血液的高凝状态
妊娠后三周开始孕妇血液中血小板及多种血浆凝血因子(因子Ⅰ、Ⅱ、Ⅴ、Ⅷ、Ⅸ、Ⅹ及Ⅻ等)增多,而具有抗凝作用及纤溶活性的物质(如ATⅢ、纤溶酶原活化素及尿中尿激酶等)降低,来自胎盘的纤溶抑制物增多。妊娠四个月以后,孕妇血液开始逐渐趋向高凝状态,到妊娠末期最为明显。因此,产科意外(如宫内死胎、胎盘早期剥离、羊水不栓塞等)时DIC的发生率较高。
酸中毒是引起血液高凝状态的一个重要因素。酸中毒可直接损伤微管内皮细胞,使内皮下的微纤维与胶原暴露,然后激活因子Ⅻ,引起内原凝血系统的激活。酸中毒时,血液pH降低,肝素的抗凝活性减弱而凝血因子的活性升高,此时血小板的聚集性加强,由它释放的促凝因子增加,因此酸中毒是导致DIC发生发展的一个重要诱因。
四、微循环障碍
休克导致的严重微循环障碍,常有血流淤滞,血细胞聚集,血液甚至可呈淤泥状(sludging)。巨大血管瘤时对毛细血管中血流极度缓慢,血流出现涡流,再加上局部内皮细胞损伤与酸中毒,这些因素均有利于DIC的发生。低血容量时,由于肝、肾等脏器处于低灌流状态,无法及时清除某些凝血或纤溶产物,这也是促成DIC发生的因素。
五、其它
不恰当地应用纤溶抑制剂如6-氨基已酸(ε-aminocaproic acid, EACA)、对羧基苄胺(paminomethyl benzoic acid,PAMBA)等药物造成纤溶系统的过度抑制、血液粘度增高时也会促进DIC形成。DIC的发生可能还与病人当时的微血管功能状态有关,例如,有实验证明大剂量长时间地使用α受体兴奋剂会促使DIC形成,但是对其发生机制还未完全阐明。
此外,DIC的发生发展还与促凝物质进入血液的数量、速度和途径有关。促凝物质进入血液少而慢时,如机体代偿功能(如吞噬功能等)健全,可不发生或仅表现为症状不明显的慢性型DIC;促凝物质入血过多过快,超过机体代偿能力时,则可引起急性DIC。此外,DIC的定位与促凝物质入血的途经有重要关系。动物实验证明,股静脉内注入凝血酶所引起的DIC,微血栓的分布以肺为主,主动脉内注入则微血栓主要在肾。
第三节 弥散性血管内凝血的发展
过程(分期)及分型
一、分期
DIC是一个病理过程,根据它的病理生理特点及发展过程,典型者一般可经过三期:
(一)高凝期
由于凝血系统被激活,所以多数患者血中凝血酶含量增多,导致微血栓的形成,此时的表现以血液高凝状态为主。
(二)消耗性低凝期
由于凝血系统被激活和微血栓的形成,凝血因子、血小板因消耗而减少,此时常伴有继发纤溶。所以有出血的表现。
(三)继发性纤溶亢进期
在凝血酶及Ⅻa的作用下,纤溶酶原活化素被激活,从而使大量纤溶酶原变成纤溶酶;此时又有FDP的形成,它们均有很强的纤溶和/或抗凝作用,所以此期出血十分明显。
二、分型
由于引起DIC的原因很多,其发生发展速度也不相同,因此又可将DIC分为以下各型:
(一)按DIC发生快慢分为急性型、亚急性型与慢性型
主要和致病因素的作用方式、强度与持续时间长短有关。当病因作用迅速而强烈时,DIC表现为急性型;相反,作用缓慢而持续时,表现为慢性型或亚急性型。各型的主要特点如下:
1.急性型 DIC可在几小时或1~2天内发生,常见于各种严重的感染,特别是革兰氏阴性菌感染引起的败血症性休克、血型不合的输血、严重创作、移植后急性排异反应等。此时,临床表现明显,常以休克和出血为主,患者的病情迅速恶化,分期不明显,实验室检查结果明显异常。
2.亚急性型 DIC在数天内逐渐形成,常见于恶性肿瘤转移、宫内死胎等患者,表现介于急性型和慢性型之间。
3.慢性型常见于恶性肿瘤、胶原病、慢性溶血性贫血等疾病。此时,由于机体有一定的代偿能力,单核吞噬细胞系统的功能也较健全,所以各种异常表现均轻微而不明显。病程较长,临床诊断较困难,常常以某脏器功能不全的表现为主,有时仅有实验室检查异常,所以出现亚临床型的(subclinical)表现,此类DIC往往在尸解后作组织病理学检查时才被发现。在一定条件下,可转化为急性型。
(二)按DIC代偿情况分为代偿型、失代偿型和过度代偿型
在DIC发生发展过程中,血浆凝血因子与血小板不断消耗,但是骨髓和肝可通过增加血小板和血浆凝血因子的生成而起代偿作用。此时肝脏生成纤维蛋白原的能力可增加5倍,骨髓生成血小板的功能可增国10倍,因此根据凝血物质的消耗与代偿性生成增多之间的对经关系。可将DIC分为以下三型:
1.代偿型凝血因子与血小板的消耗与生成间基本上保持平衡状态。主要见于轻度DIC。此型患者可无明显临床表现或仅有轻度出血和血栓形成的症状。实验室检查无明显异常(如纤维蛋白原无明显减少),易被忽视。但如病情持续加重,则可转化为失代偿型。
2.失代偿型凝血因子和血小板的消耗超过生成。主要见于急性DIC。此型患者出血、休克等表现明显,实验室检查发现血小板和纤维蛋白原等凝血因子均明显减少。
3.过度代偿型机体代偿功能较好,凝血因子和血小板的生成迅速,甚至超过消耗。因此有时出现纤维蛋白原等凝血因子暂时升高的表现。主要见于慢性DIC或DIC恢复期。此型患者出血或栓塞症状可不太明显,但与代偿型相似,在致病因子的性质和强度发生改变时,也可转化为典型的失代偿型。
至于局部型的DIC,主要是指局限于某一脏器的多发性微血栓症,但全身仍有轻度的血管内凝血存在,多见于静脉瘤、主动脉瘤,心脏室壁瘤、人造血管、体外循环、器官移植后的排异反应等,此时常在病变局部有凝血过程的激活。因此严格地说,这是全身性DIC的一种局部表现。
第四节 弥散性血管内凝血时的机能代谢变化与临床表现
DIC时,各种典型病理变化及临床表现主要发生在急性、严重的DIC。形成这些变化的主要基础是凝血酶的生成增加、某些凝血因子的激活、消耗,纤维蛋白性微血栓的形成,以及继发性纤溶的增强。因此其病理与临床表现复杂多样,并随原发疾病的不同而异,但是在各种表现中尤以出血及微血管中微血栓的形成最为突出。
一、出血
虽然DIC病人典型的病理变化是微血栓形成,但是病人最初的临床现表现为出血,引起出血的机制有以下可能:
(一)凝血物质的消耗
在DIC发生发展过程中,各种凝血因子和血小板大量消耗,特别是纤维蛋白原、凝血酶原、因子Ⅴ、Ⅷ、Ⅹ、Ⅷ的血小板普遍减少。因此曾有人将DIC称为消耗性凝血病(consrmptive coagulopathy)。此时。因凝血物质大量减少,因而凝血过程受阻。
(二)纤溶系统的激活
DIC时在凝血系统激活后,常有继发性纤溶系统的激活。这主要是由于在凝血过程中,通过酶性激活(蛋白酶作用造成酶性水解)由Ⅻa形成Ⅻf,Ⅻf使激肽释放酶原转变成激肽释放酶,后者使纤溶酶原变为纤溶酶。一些富含纤溶酶原激活物的器官(如子宫、前列腺、肺等)因血管内凝血而发生变性坏死时,激活物便大量释放入血而激活纤溶系统。血管内皮细胞受损、缺氧、应激等也皆可激活纤溶系统,导致纤溶酶增多。纤溶酶除能使纤维蛋白(原)降解外,还能水解凝血因子Ⅴ、Ⅷ和凝血酶原等,故这些凝血因子进一步减少,从而引起凝血障碍和出血。
(三)纤维蛋白(原)降解产物的形成
凝血过程的激活以及继发性纤溶过程的启动使血中纤溶酶增多,纤维蛋白(原)被降解。纤维蛋白原在纤溶酶作用下先从其分子的Bβ链上裂解出一个小肽,然后又在Aα链上裂解出碎片A、B、C和H,留下的片段即X(分子量240~260KD),后者再在纤溶酶作用下不断裂解先后产生Y(分子量150KD)、D(分子量100KD)及E(分子量50KD)片段。它们统称为纤维蛋白原降解产物(FgDP)。纤维蛋白在纤溶酶作用下形成X'、Y’、D、E’片段,各种二聚体、多聚体及复合物,统称其为纤维蛋白降解产物(FDP)。两类FDP的功能特性基本相似,其中X,Y碎片可与纤维蛋白单体聚合,从而抑制纤维蛋白多聚体生成;Y、E碎片有抗凝血酶作用;D碎片抑制纤维蛋白单体聚合;大部分FDP均抑制血小板的粘附和聚集,因此FDP可通过强烈的抗凝作用引起出血。
临床上一般常用血浆鱼精蛋白副凝试验(plasma protamineparacoagulation test,3P试验)检查FDP存在,其主要原理为纤维蛋白原在凝血酶作用下形成许多纤维蛋白单体,后者在凝因血因子Ⅻ(纤维蛋白稳定因子)作用下形成纤维蛋白。纤维蛋白在纤溶酶作用下分解为X'、Y'、D、E'碎片,这些碎片(主要是X’碎片)可与纤维蛋白单体形成可溶性纤维蛋白单体复合物(soluble fibrinmonomer complex, SFMC)病人血浆中如有SFMC存在,则在体外加入鱼精蛋白后,此种现象称副凝现象。DIC病人血浆中由于有SFMC的存在,3P试验常呈阳性,所以此试验主要是反映SFMC和纤维蛋白降解产物中X'片段的试验。晚期DIC病人血浆中X'片段减少,D、E'明显增多,因此3P试验反而呈阴性。
临床上DIC病人可有轻重不等的多部位出血倾向(图9-4)。病理形成上既可有血管内凝血,也可有出血的表现。实验室检查有凝血时间和凝血酶原时间延长,纤维蛋白原和血小板减少等发现。出血发生在皮肤时,常可见到出血斑或局部坏死,它与周围皮肤分界清楚,边缘不规则,这种现象反映了皮肤下阻塞的终末微动脉的分布,如果较大的血管发生阻塞,则这些病变可发展形成出血性大疱或融合成片,但治疗及时、恰当,也可吸收。在重症病例,出血特别严重时,可以表现为手指或脚趾的坏疽,有时可出现对称性坏死性病变。
出血也可在静脉穿剌部位。血尿常见。此外也可出现牙龈和鼻出血。出血严重而剧烈时可引起死亡,而且用一般止血药物治疗无效。出血常可成为DIC的最初或主要症状,所以有人强调,如病人患有可能引起DIC的原发疾病,病程中出血症状又难以用其它原因解释时,应考虑到DIC的可能。
二、微血管栓塞引起脏器功能障碍
DIC病人尸检或活检时,常发现微血管(特别是毛细血管与微静脉)内有微血栓存在。它可由血小板组成,但典型的是纤维蛋白性微血栓。它们可以在局部形成,也可来自别处,从而阻塞微血管。在某些情况下,患者虽然有典型的DIC临床表现,但病理检查却未能发现阻塞性微血栓,这可能是由于体内凝血系统发动后纤溶系统同时被激活,使微血栓在生前或死后被溶解所致,也可能是纤维蛋白微血栓尚未完全形成,只有在电镜下才能见到。曾有人在研究猴的内毒素性休克与DIC关系时,获得这方面的证据。
微血管中形成的微血栓,可阻塞相应部位的微循环血流,严重时可造成实质脏器的局灶性坏死(图9-4)。严重或持续过久的坏死性病变可成为受累脏器功能衰竭的原因(图9-4)如果微血栓在肾脏形成,则病变可累及入球小动脉或肾小球毛细血管,严重时可出现双侧肾皮质坏死和急性肾功能衰竭,临床上表现为少尿、蛋白尿、血尿等。在肺部,可引起呼吸困难、肺出血,从而导致呼吸衰竭。消化系统的病变可导致恶心、呕吐、腹泻、消化道出血。肝脏受累时可出现黄疸及肝功能衰竭。内分泌腺的病变常见者为肾上腺皮质出血性坏死造成的急性肾上腺皮质功能衰竭,称华-佛氏综合征(Waterhorst-Friderichsen syndrome)。垂体坏死可导致席汉氏综合征(Sheehan's syndrome)。神经系统的病变可导致神志模糊,嗜睡、昏迷、惊厥等非特异症状,这些症状的出现并非是由一个孤立的局部病灶引起,而可能是由蛛网膜下腔出血以及微血管阻塞、脑皮质和脑干的多处出血所致。
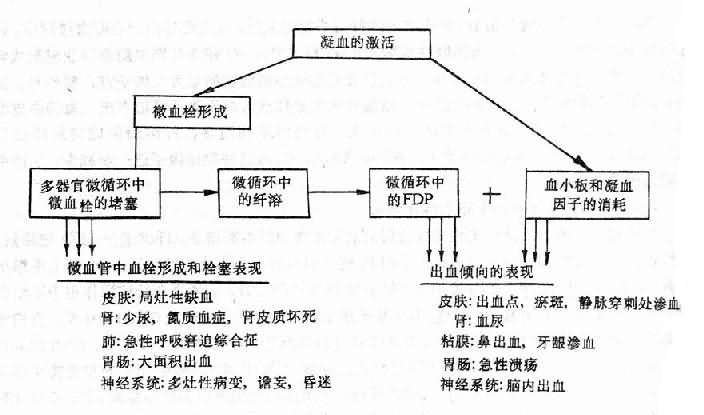
图9-4 DIC的主要临床表现及其机制
DIC时由于凝血及纤溶的轻重程度不一,在不同的病人及病程的不同阶段可有不同的表现,此外,DIC范围大小不一所造成的后果也不同,轻者仅影响个别脏器的部分功能,重者可引起一个或多个脏器的功能衰竭即多器官功能衰竭,甚至造成死亡。
三、循环功能——休克
DIC,特别是急性DIC,常伴有休克。重度及晚期休克又可能促进DIC的形成,二者互为因果,形成恶性循环。
急性DIC常伴发休克,是由于毛细血管和微静脉中有广泛血小板聚集和/或纤维蛋白性微血栓形成,以致回心血量严重不足,再加上心肌损伤,广泛出血所引起的血容量减少等因素,使有效循环血量严重下降,心输出量减少,出现全身微循环障碍。与此同时,中心静脉压也往往降低,若肝和肺内有广泛微血栓阻塞,则又可相应地引起门静脉和肺动脉压升高。前者的临床表现为胃肠道瘀血、水肿,后者为右心排血障碍。此外,在DIC的形成过程中,由于凝血因子Ⅻ被激活,凝血酶增多和继发性纤溶的启动,可使循环血中Ⅻf、凝血酶和纤溶酶增多,它们均能激活补体和激肽系统,使激肽和某些补体成分(如C3a、C5a等)生成增多,激肽能使微动脉和毛细血管前括约肌舒张,从而使外周阻力显著降低;C3a、C5a等则可使肥大细胞和嗜碱性粒细胞脱颗粒,从而通过释放组胺而发挥与激肽类似的作用。这是急性DIC时动脉血压下降的重要原因。FDP的形成,加重了微血管扩张及通透性升高,这是因为FDP的某些部分(如裂解碎片A、B等)能增强组胺和激肽的作用,能使微血管舒张,因而更易产生休克。(图9-5)。
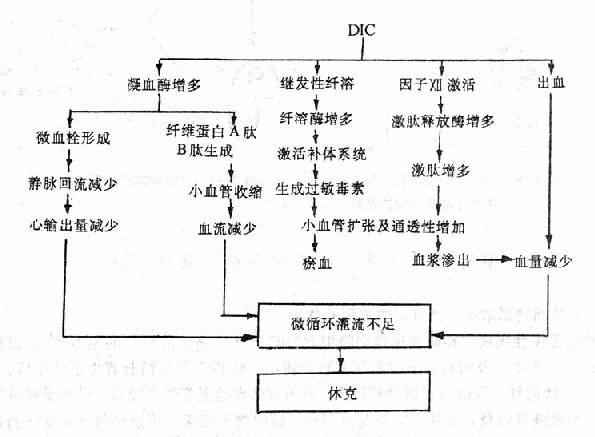
图9-5 DIC产生休克的机理
各种休克发展到一定阶段也往往可以伴发DIC(参阅《休克》)。
四、红细胞机械性损伤引起的微血管病性溶血性贫血
在DIC中有时可伴发一种特殊类型的贫血,即微血管病性溶血性贫血(microangiopathic hemolytic anemia)这种贫血除具备溶性贫血的一般特征外,外周血涂片中发现有某些形态特殊的变形的红细胞如裂体细胞(schistocyte),其外形呈盔甲形、星形、新月形等,统称其为红细胞碎片。这些碎片由于脆性高,故容易发生溶血。
目前认为产生红细胞碎片的原因虽然很多,但DIC是主要因素。其机制是当微血管中有纤维蛋白性微血栓形成时,在早期,纤维蛋白丝在微血管腔内形成细网;当循环中的红细胞流过由纤维蛋白丝构成的网孔时,常会粘着、滞留或挂在纤维蛋白丝上。这样由于血流的不断冲击,引起红细胞破裂(图9-6、图9-7)。在微血流通道发生障碍时,红细胞还可能通过肺组织等的微血管内皮细胞间的裂隙,被“挤压”到血管外组织中去。这种机械损伤同样也可使红细胞扭曲、变形和碎裂。这样就形成了上述各种畸形的红细胞碎片。所以在DIC病人中有时可以有溶血的一系列表现和实验室检查异常,外周血涂片中可出现较多的上述各种红细胞碎片。
图9-6 微血管病性溶血性贫血血片中能见到裂体细胞
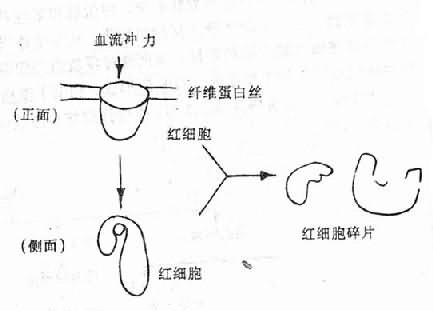
图9-7 红细胞碎片的形成机制
第五节 弥散性血管内凝血的防治原则
DIC的防治要采取综合措施,主要原则如下:
1.以严重感染引起的DIC为例,及时有效地控制原发感染病灶,对DIC的防治起着决定性作用。
2.
研究发现,肝素的抗凝效果与AT-Ⅲ相关。因为AT-Ⅲ是肝素辅助因子,肝素可与AT-Ⅲ的赖氨酸残基形成复合物,从而加速AT-Ⅲ对凝血酶的灭活,此外,AT-Ⅲ对血小板聚集也有一定抑制作用。DIC患者原先存在AT-Ⅲ减少或DIC本身引起的AT-Ⅲ减少均会影响肝素的抗凝效果。因此,有人认为,在应用肝素以前或同时,如能应用AT-Ⅲ制剂,则可提高肝素的抗凝效果。
3.重新建立凝血和纤溶间的动态平衡DIC时由于大量凝血因子及血小板消耗,因此在病情控制或使用肝素治疗后,以及在恢复期可酌情输入新鲜全血、冰冻血浆或纤维蛋白原等,以利凝血、纤溶间恢复新平衡。
第十章 休克
休克(Shock)是平时和战时一种常见的而严重的病理过程,是医学中的一个重要课题。一旦发生如未能及早发现,采取积极的正确的治疗措施,将贻误生命,造成严重后果。为了更有效的预防和治疗,对休克的发生机理及其所引起的病理生理变化应有比较深入的理解。
第一节 休克的概念
休克是指因各种原因(如大出血、创伤、烧伤感染、过敏、心泵衰竭等)引起的急性血液循环障碍,微循环动脉血灌流量急剧减少,从而导致各重要器官机能代谢紊乱和结构损害的复杂的全身性病理过程。
人类对休克的认识,经历了一个由浅入深,从现象到本质的认识过程。很早以前,人们对休克时外部表现作过详细而生动的描述,把机体受到强烈“打击”(这个词原意是“打击、“震荡”)后,面色苍白、四肢厥冷、出冷汗、脉搏快而微弱、表情淡漠或神志不清等综合现象称为休克。随后,人们发现休克是严重的血液循环障碍,认为上述表现是由于血压降低引起的,把血压作为判断休克的标准,并把低血压看作是休克发生发展的主要矛盾,因而采用升压药作为治疗休克的重要手段。但是在医疗实践中发现,休克的早期,往往没有明显的血压降低;使用升压药维持血压,有的不仅不能挽救休克病人,甚至加重休克的发展。近十几年来,通过对组织微循环研究,发现:①休克时有明显的微循环障碍(缺血、淤血、微血栓形成),组织器官的功能和代谢障碍是微循环动脉血灌流不足引起的;②休克时微循环障碍往往发生在血压降低之前,休克早期,由于小动脉收缩,外周阻力增加,血压降低往往不明显,但是微循环已发生明显的缺血;③就大多数休克而言,由于循环血量不足,心输出量减少,加上应激反应,已使小动脉收缩和微循环缺血,不适当地作用升压药,看来血压暂时得以维持在较高水平,但更加重微循环缺血,促使休克进一步发展。因此目前认为微循环血灌流急剧减少,致重要生命器官因缺氧而发生功能和代谢障碍,不论何种原因引起的休克,微循环动脉血灌流急剧减少,致重要生命器官因缺氧而发生功能和代谢障碍,是各型休克发生发展的共同规律。根据这一新的理论,休克的治疗应着重于尽快改善微循环,而不应单纯追求一个“满意”的血压。休克的恢复取决于微循环的改善,而不单纯取决于提高血压,虽然目前对休克本质有了进一步的认识,但还存在许多的争论和没有被认识的领域,目前休克的研究已进入细胞代谢和功能的分子水平,从代谢、功能和结构多方面进行综合研究。近年来的研究发现,休克时细胞损伤还可以由休克的始动因素直接引起,如感染性休克,在有些情况下,它主要不是由于血液灌流不足,而是组织细胞对氧或其它营养物质利用能力降低。相信随着对克本质认识的逐步深入,对休克的防治水平也将不断获得提高。
第二节 休克的原因和分类
引起休克的原因很多,分类方法也不一,比较常用的分类方法是:
一、按休克的原因分类
1.失血后是否发生休克不仅取决于失血的量,还取决于失血的速度。休克往往是在快速、大量(超过总血量的30~35%)失血而又得不到及时补充的情况下发生的。
2.创伤性休克严重创伤特别是在伴有一定量出血时常引起休克,称为创伤性休克(traumaticshock)。
3.烧伤性休克大面积烧伤伴有大量血浆丧失者常导致烧伤性休克(burn shock)。
4.感染性休克严重感染特别是革兰氏阴性细菌感染常可引起感染性休克。在革兰氏阴性细菌引起的休克中,细菌的内毒素起着重要的作用,故亦称内毒素性休克(endotoxin shock)或中毒性休克。感染性休克常伴有败血症,故又称败血症性休克(septic shock)。
5.心源性休克大面积急性心肌梗塞、急性心肌炎、心包填塞等常可导致心源性休克(cardiogenicshock)。
6.过敏性休克给某些有过敏体质的人注射某些药物(如青霉素)、血清制剂或疫苗时可引起过敏性休克(anaphylactic shock)。
7.神经源性休克剧烈疼痛、高位脊髓麻醉或损伤等可引起神经源性休克(neurogenicshock)。
二、按休克发生的始动环节分类
尽管引起休克的原因很多,但休克的始动环节不外乎血容量减少,有效循环血量下降;或心脏泵血功能严重障碍。引起有效循环血量下降和微循环流量减少;或由于大量毛细血管和小静脉扩张,血容量扩大,血容量相对不足,使有效循环血量下降。据此,可将休克作如下的分类。
1.低血容量性休克低血容量性休克(hypovolemic shock)的始动发病环节是血容量减少。快速大量失血、大面积烧伤所致的大量血浆丧失、大量出汗、严重腹泻或呕吐等情况所引起的大量体液丧失都可使血容量急剧减少而导致低血容量性休克。
2.心源性休克心源性休克(cardiogenic shock)的始动发病环节是心输出量的急剧减少,常见于大范围心肌梗塞(梗塞范围超过左心室体积的40%),也可由严重的心肌弥漫性病变如急性心肌炎、严重的心律失常如过度的心动过速、心包填塞等所引起。
3.血管源性休克血管源性休克(vasogenic shock)的始动发病环节是外周血管(主要是微小血管)扩张所致的血管容量扩大。属此者有过敏性休克和神经源性休克等。此时血容量和心泵功能可能正常,但由于广泛的小血管扩张和血管床扩大,大量血液淤积在外周微血管中而使回心血量减少。
三、按休克时血液的动力学的特点分类
1.低排高阻型休克亦称低动力型休克(hypodynamic shock),其血液动力学特点是心脏排血量低,而总外周血管阻力高。由于皮肤血管收缩,血流量减少,使皮肤温度降低,故又称为“冷性休克(coldshock)”。本型休克在临床上最为常见。低血容量性、心源性、创伤性和大多数感染性休克均属本类。
2.高排低阻型休克亦称高动力型休克(hyperdynamic shock),其血液动力学特点是总外周血管阻力低,心脏排血量高。由于皮肤血管扩张,血流量增多,使皮肤温度升高,故亦称“温性休克(warmshock)”。部分感染性休克属本类。
第三节 休克的病理生理变化
一、微循环变化
各种休克虽然由于致休克的动因不同,在各自发生发展过程中各有特点,但微循环障碍(缺血、淤血、播散性血管内凝血)致微循环动脉血灌流不足,重要的生命器官因缺氧而发生功能和代谢障碍,是它们的共同规律。休克时微循环的变化,大致可分为三期,即微循环缺血期、微循环淤血期和微循环凝血期。下面以低血容量性休克为例阐述微循环障碍的发展过程及其发生机理。
低血容量性休克常见于大出血、严重的创伤、烧伤和脱水。其微循环变化发展过程比较典型(图10-1)。
(一)微循环缺血期(缺血性缺氧期)
此期微循环变化的特点是:①微动脉、后微动脉和毛细血管前括约肌收缩,微循环灌流量急剧减少,压力降低;②微静脉和小静脉对儿茶酚胺敏感性较低,收缩较轻;③动静脉吻合支可能有不同程度的开放,血液从微动脉经动静脉吻合支直接流入小静脉。
引起微循环缺血的关键性变化是交感神经——肾上腺髓质系统强烈兴奋。不同类型的休克可以通过不同机制引起交感——肾上腺髓质性休克和心源性休克时,心输出量减少和动脉血压降低可通过窦弓反射使交感——肾上腺髓质系统兴奋;在大多数内毒素性休克时,内毒素可直接剌激交感——肾上腺髓质系统使之发生强烈兴奋。
交感神经兴奋、儿茶酚胺释放增加对心血管系统的总的效应是使外周总阻力增高和心输出量增加。但是不同器官血管的反应却有很大的差别。皮肤、腹腔内脏和肾的血管,由于具有丰富的交感缩血管纤维支配,。而且α受体又占有优势,因而在交感神经兴奋、儿茶酚胺增多时,这些部位的小动脉、小静脉、微动脉和毛细血管前括红肌都发生收缩,其中由于微动脉的交感缩血管纤维分布最密,毛细血管前括约肌对儿茶酚胺的反应性最强,因此它们收缩最为强烈。结果是毛细血管前阻力明显升高,微循环灌流量急剧减少,毛细血管的平均血压明显降低,只有少量血液经直捷通路和少数真毛细血管流入微静脉、小静脉,组织因而发生严重的缺血性缺氧。脑血管的交感缩血管纤维分布最少,α受体密度也低,口径可无明显变化。冠状动脉虽然也有交感神经支配,也有α和β受体,但交感神经兴奋和儿茶酚胺增多却可通过心脏活动加强,代谢水平提高以致扩血管代谢产物特别是腺苷的增多而使冠状动脉扩张。
交感兴奋和血容量的减少还可激活肾素-血管紧张素-醛固酮系统,而血管紧张素Ⅱ有较强的缩血管作用,包括对冠状动脉的收缩作用。
此外,增多的儿茶酚胺还能剌激血小板产生更多的血栓素A2(thromboxane A2,TXA2),而。TXA2也有强烈的缩血管作用。
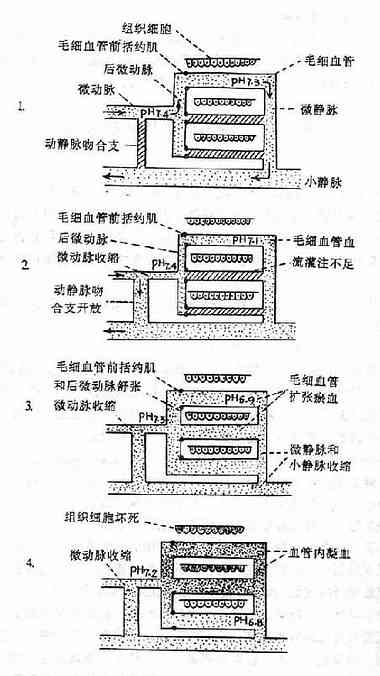
图10-1 微循环障碍的发展过程模式图
1.正常情况
⑴动静脉吻合支是关闭的。
⑵只有20%毛细血管轮流开放,有血液灌流。
⑶毛细血管开放与关闭受毛细血管前括约肌的舒张与收缩的调节。
2.微循环缺血期
⑴交感神经兴奋和肾上腺素、去甲肾上腺素分泌增多,小动脉、微动脉、后微动脉,毛细血管前括约肌收缩。
⑵动静脉吻合支开放,血液由微动脉直接流入小静脉。
⑶毛细血管血液灌流不足,组织缺氧。
3.微循环淤血期
⑴小动脉和微动脉收缩,动静脉吻合支仍处于开放状态,进入毛细血管的血液仍很少。
⑵由于组织缺氧,组织胺、缓激肽、氢离子等舒血管物质增多,后微动脉和毛细血管前括约肌舒张,毛细血管开放,血管容积扩大,进入毛细血管内的血液流动很慢。
⑶由于交感神经兴奋,肾上腺素和去甲肾上腺素分泌增多(可能还有组织胺的作用),使微静脉和小静脉收缩,毛细血管后阻力增加,结果毛细血管扩张淤血。
4.微循环凝血期
⑴由于组织严重缺氧、酸中毒,毛细血管壁受损害和通透性升高,毛细血管内血液浓缩,血流淤滞;另外血凝固性升高,结果在微循环内产生播散性血管内凝血。
⑵由于微血栓形成,更加重组织缺氧和代谢障碍,细胞内溶酶体破裂,组织细胞坏死,引起各器官严重功能障碍。
⑶由于凝血,凝血因子(如凝血酶原、纤维蛋白原等)和血小板大量被消耗,纤维蛋白降解产物增多,又使血液凝固性降低;血管壁又受损害,继而发生广泛性出血。
而TXA2也有强烈的缩血管作用。
还有,溶酶体水解酶-心肌抑制因子系统在休克Ⅰ期微循环缺血的发生中也起一定的作用。休克时,主要由于胰腺血液灌流量减少所引起的缺血、缺氧和酸中毒可使胰腺外分泌细胞的溶酶体破裂而释出组织蛋白酶,后者即可分解组织蛋白而生成心肌抑制因子(myocardial depressant factor, MDF)。小分子肽MDF进入血流后,除了引起心肌收缩力减弱、抑制单核吞噬细胞系统的吞噬功能以外,还能使腹腔内脏的小血管收缩,从而进一步加重这些部位微循环的缺血。
本期的主要临床表现是:皮肤苍白,四肢厥冷,出冷汗,尿量减少;因为外周阻力增加,收缩压可以没有明显降低,而舒张压有所升高,脉压减小,脉搏细速;神志清楚,烦躁不安等。
此期微循环变化具有一定的代偿意义。皮肤和腹腔器官等小动脉收缩,既可增加外周阻力,以维持血压,又可减少这些组织器官的血流量,以保证心脑等重要器官的血液供给;毛细血管前阻力增加,毛细血管流体静压降低,促使组织液进入血管,以增加血浆容量;另外,动静脉吻合支开放,静脉收缩使静脉容量缩小(正常约有70%血液在静脉内),可以加快和增加回心血量,也有利于血压的维持和心脑的血液供给。但是由于大部分组织器官因微循环动脉血灌流不足而发生缺氧,将导致休克进一步发展。如能及早发现,积极抢救,及时补充血量,降低过剧的应激反应,可以很快改善微循环和恢复血压,阻止休克进一步恶化,而转危为安。
这时微循环变化的机理可概括如下(图10-2):
(二)微循环淤血期(淤血性缺氧期)
在休克的循循环缺血期,如未能及早进行抢救,改善微循环,则因组织持续而严重的缺氧,而使局部舒血管物质(如组织胺、激肽、乳酸、腺苷等)增多,后微动脉和毛细血管前括约肌舒张,微循环容量扩大,淤血,发展为休克微循环淤血期。此期微循环变化的特点是:①后微动脉和毛细血管前括约肌舒张(因局部酸中毒,对儿茶酚胺反应性降低),毛细血管大量开放,有的呈不规侧囊形扩张(微血池形成),而使微循环容积扩大;②微静脉和小静脉对局部酸中毒耐受性较大,儿茶酚胺仍能使其收缩(组织胺还能使肝、肺等微静脉和小静脉收缩),毛细血管后阻力增加,而使微循环血流缓慢;③微血管壁通透性升高,血浆渗出,血流淤滞;④由于血液浓缩,血细胞压积增大,红细胞聚集,白细胞嵌塞,血小板粘附和聚集等血液流变学的改变,可使微循环血流变慢甚至停止。⑤由于微循环淤血,压力升高,进入微循环的动脉血更少(此时小动脉和微动脉因交感神经作用仍处于收缩状态)。由于大量血液淤积在微循环内,回心血量减少,使心输出量进一步降低,加重休克的发展。
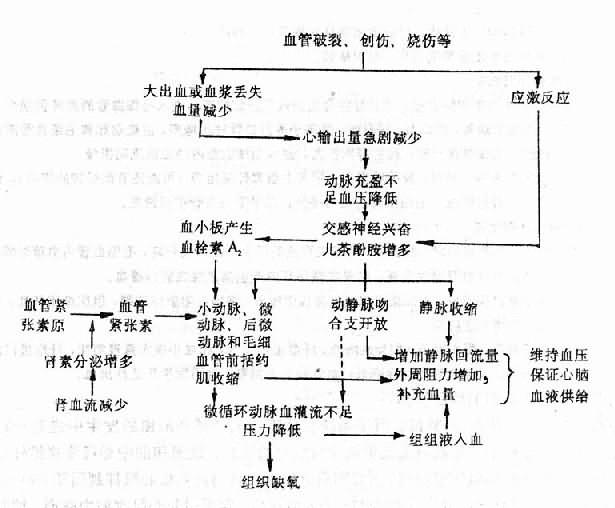
图10-2 缺血性缺氧期微循环变化机理
由于上述微循环变化,虽然微循环内积有大量血液,但动脉血灌流量将更加减少,病人皮肤颜色由苍白而逐渐发绀,特别是口辰和指端。因为静脉回流量和心输出量更加减少,病人静脉萎陷,充盈缓慢;动脉压明显降低,脉压小,脉细速;心脑因血液供给不足,ATP生成减少,而表现为心收缩力减弱(心音低),表情淡漠或神志不清。严重的可发生心、肾、肺功能衰竭。这是休克的危急状态,应立即抢救,补液,解除小血管痉挛,给氧,纠正酸中毒,以疏通微循环和防止播散性血管内凝血。这时微循环变化的机理可概括如下(图10-3):
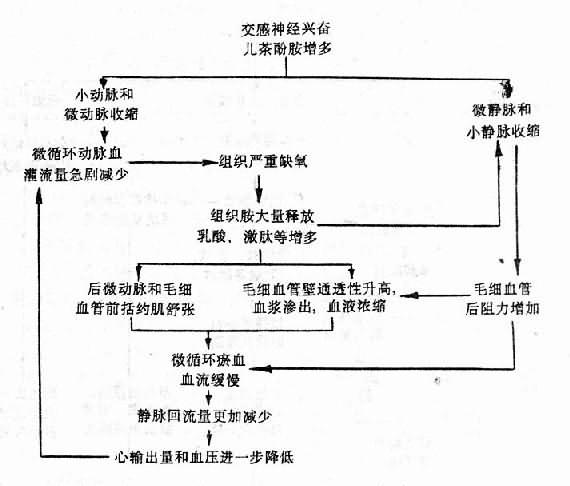
图10-3 淤血性缺氧期微循环变化机理
(三)微循环凝血期(播散性血管内凝血)
从微循环的淤血期发展为微循环凝血期是休克恶化的表现。其特点是:在微循环淤血的基础上,于微循环内(特别是毛细血管静脉端、微静脉、小静脉)有纤维蛋白性血栓形成,并常有局灶性或弥漫性出血;组织细胞因严重缺氧而发生变性坏死。
播散性血管内凝血与休克的联系极为密切。关于播散性血管内凝血引起的病理变化以及它如何引起休克或加重休克的发展,已在《播散性血管内凝血》一章讨论过了,这里再概要地归纳一下休克如何引起播散性血管内凝血。
1.应激反应使血液凝固性升高。致休克的动因(如创伤、烧伤、出血等)和休克本身都是一种强烈的剌激,可引起应激反应,交感神经兴奋和垂体-肾上腺皮质活动加强,使血液内血小板和凝血因子增加,血小板粘附和聚集能力加强,为凝血提供必要的物质基础。
2.凝血因子的释放和激活。有的致休克动因(如创伤、烧伤等)本身就能使凝血因子释放和激活。例如,受损伤的组织可释放出大量的组织凝血活素,起动外源性凝血过程;大面积烧伤使大量红细胞破坏,红细胞膜内的磷脂和红细胞破坏释出的ADP,促进凝血过程。
3.微循环障碍,组织缺氧,局部组织胺、激肽、乳酸等增多。这些物质一方面引起毛细血管扩张淤血,通透性升高,血流缓慢,血液浓缩红细胞粘滞性增加,有利于血栓形成;另一方面损害毛细血管内皮细胞,暴露胶元,激活凝血因子Ⅻ和使血小板粘附与聚集。
4.缺氧使单核吞噬细胞系统功能降低,不能及时清除凝血酶元酶、凝血酶和纤维蛋白。结果在上述因素作用下,而发生播散性血管内凝血(图10-4)。
图10-4 创伤性休克引起播散性血管内凝血的机理
应当指出,在不同类型的休克,播散性血管内凝血形成的早晚可不相同。例如,在烧伤性和创伤性休克时,由于有大量的组织破坏,感染中毒性休克时,由于内毒素对血管内皮的直接损伤,因而都可较早地发生播散性血管内凝血,而在失血性休克等,则播散性血管内凝血发生较晚。
播散性血管内凝血一旦发生,将使微循环障碍更加严重,休克病情进一步恶化,这是因为:①广泛的微血管阻塞进一步加重微循环障碍,使回心血量进一步减少;②凝血物质消耗、继发纤溶的激活等因素引起出血,从而使血容量减少;③可溶性纤维蛋白多聚体和其裂解产物等都能封闭单核吞噬细胞系统,因而使来自肠道的内毒素不能被充分清除。
由于播散性血管内凝血的发生和微循环淤血的不断加重,由于血压降低所致的全身微循环灌流量的严重不足,全身性的缺氧和酸中毒也将愈益严重;严重的酸中毒又可使细胞内的溶酶体膜破裂,释出的溶酶体酶(如蛋白水解酶等)和某些休克动因(如内毒素等)都可使细胞发生严重的乃至不可逆的损害,从而使包括心、脑在内的各重要器官的机能代谢障碍也更加严重(详后),这样就给治疗造成极大的困难,故本期又称休克难治期。
二、血液流变学的变化
血液流变学(hemorheology)是研究血液流动和变形的科学,或者说是研究血液的流变性、凝固性、血液有形成分(主要是红细胞)粘弹性以及心血管的粘弹性和变形的科学。物体在一定外力作用下能流动或变形的特性,称为该物体流变性。一切流体在一定外力作用下,都具有流动性,但流动的难易,则主要取决于流体内部对于流动起阻抗作用的分子之间和颗粒之间的内摩擦力(即流体的粘度)。例如,水的粘度低,容易流动,即流度大;血液的粘度大(红为蒸馏水的4-5倍),不易流动,即流度小。由于流体的流动是以物体的变形为基础,所以流体的粘度是映流体流变性的重要指标。
血液是由水、无机盐、蛋白质、脂类、糖等大小分子所组成的混合液,其中还悬浮着大量具有可塑性的红细胞,所以血液是一种高浓度的悬浊液。因此能够影响血液流变性的因素主要有:血细胞压积(血液粘度随血细胞的压积增加而升高)、血细胞的分散程度(血细胞处于分散状态,血液粘度较低;红细胞或血小板发生聚集,血液粘度升高)、红细胞的可塑性(红细胞可塑性降低,不易变形,血液粘度增加)、血浆内高分子化合物的浓度(血浆粘度大小与其所含蛋白质、脂类、糖等的浓度呈正比)、血管内壁平滑度(血管内皮受损、变形,流经的血液粘度升高)。此外,与血管的长度、口径、血管壁的弹性和张力也有关系。
休克时血液流变学的主要变化是:
1.血细胞比容血细胞比容的改变与休克的原因和发展阶段有关。在低血容量性休克的早期,由于组织间液向血管内转移,导致血液稀释,血细胞比容降低,当休克进入微循环淤血期,由于微血管内流体静压升高和毛细血管通透性增高,液体乃从毛细血管内外渗至组织间隙,因而血液浓缩,血细胞比容升高。血细胞比容越高,血液粘度越大,血流阻力越大,而血流量则越少,血流更加缓慢。
2.红细胞变形能力降低,聚集力加强在正常情况下,红细胞在流经小于其直径的毛细血管时,可折叠、弯曲而发生多种变形以减少其宽度,从而得以顺利通过。现已证明,休克时红细胞的变形能力明显降低,其主要原因是:①休克Ⅱ期时因血液浓缩和组织缺氧所引起的血液渗透压升高和pH降低,可使红细胞膜的流动性和可塑性降低并使红细胞内部的粘度增加;②ATP缺乏(可由缺氧或某些休克动因直接引起)可使红细胞不能维持正常的功能和结构。结果是由于红细胞的变形能力降低而难以通过毛细血管,从而导致血流阻力增高。
红细胞聚集加强,是休克时细胞流变学的重要改变之一。轻者4、5个红细胞聚集在一起,重者20~30个红细胞聚集成长链或团块。引起红细胞聚集的原因是:①血流速度变慢,切变率(shear rate)降低:正常时由于血流速度快和切变率高。可防止红细胞的聚集,并可促使聚集的红细胞解聚。休克时随着血压下降,血液流速减慢和切变率降低,红细胞就易于聚集。②红细胞表面电荷减少:正常红细胞表面因含有唾液酸的羧基,故都带有负电荷。红细胞之间的这种同电荷的排斥力可阻止红细胞互相靠拢和聚集。休克时,尤其是内毒素性休克时,红细胞表面负电荷减少,可能是由于血浆中带正电荷的蛋白质增多,被红细胞吸附所致,从而使红细胞彼此靠拢发生聚集。③血细胞比容增加:已如前述,休克时由于血浆外渗,血液浓缩,故血细胞比容增加,这就可以促进红细胞聚集。④纤维蛋白原浓度增高;纤维蛋白原覆盖于红细胞表面,在红细胞之间可形成有相互聚集作用的“桥力”。休克时由于纤维蛋白原浓度增高,致使“桥力”增大乃至超过负电荷的排斥力。因而就可导致红细胞的聚集。红细胞聚集轻则增加血液粘度和血流阻力,重则可引起红细胞淤滞并阻塞微循环,甚至形成微血栓。
(一)白细胞粘着和嵌塞
正常微循环的血流是红细胞位于中央的轴流,血浆构成边流,虽然也可见到少量白细胞附壁滚动,但不发生附壁粘着现象。休克时可见白细胞附着于小静脉壁,致使血流阻力增高和静脉回流障碍。发生白细胞附壁的原因可能与白细胞和管壁之间吸引力增大,休克时血流变慢和切应力(shear stress)下降等因素有关。休克时,还可见到白细胞嵌塞于血管内皮细胞核的隆起处或毛细血管分支处,这可增加血流阻力和加重微循环障碍,而且嵌塞的白细胞还可释放自由基和溶酶体酶类物质,从而破坏生物膜和引起坏死。休克时白细胞发生嵌塞的原因是:①白细胞的变形能力降低,故不易通过毛细血管而发生嵌塞;②休克时血压下降,脉压差减小,动脉血流量减少,驱动白细胞通过毛细血管的力量减弱,因而易于发生白细胞嵌塞。
(二)血小板粘附和聚集
血小板粘附是指血小板和血小板以外的物质相互粘附的现象,血小板聚集则是血小板之间相互发生反应并形成血小板团(或称血小板聚集物)的过程。粘附一旦开始,聚集过程也随之发生。在血小板聚集开始时,其表面首先失去光滑性,变得粗糙,形成有突剌的球状体(或称聚集型血小板)。在内毒素性、创伤性和烧伤性休克时,血液中这种聚集型血小板的数目增多,而且在微血管中有血小板粘附、聚集和血小板微血栓的形成。这种聚集的血小板不但阻塞微血管,还可释放多种生物活性物质如儿茶酚胺、TXA2、5-羟色胺等,使局部微血管收缩、通透性增高、血管内皮水肿和血流减少。此外,尚可释放促凝血的血小板因子(如PF3等),加速凝血过程,形成DIC。
休克时引起血小板粘附和聚集的主要原因是:①血流减慢,血管内皮完整性破坏,内膜下胶原暴露,为血小板粘附提供了基础;②损伤的内皮组织释放ADP,发生聚集的血小板可释放ADP、TXA2以及血小活化因子(PAF),均可触发并加重血小板的聚集。
(三)血浆粘度增大
休克时,尤其是严重创伤或烧伤休克时,一方面由于机体发生应激,使体内合成纤维蛋白原增多;另一方面,在休克的微循环淤血期,毛细血管内的流体静压增高,微血管周围的肥大细胞又因缺氧而释放组胺并从而使毛细血管通透性增高,液体乃从毛细血管大量外渗至组织间隙,因而血液浓缩,使血浆纤维蛋白原浓度增高,有时纤维蛋白原可高达10g/L(1000mg/dl),故可使血浆粘度增大。这不但影响组织血液流量,并可促进红细胞的聚集。如当纤维蛋白原的浓度增到5~8g/L(500~800mg/dl)时,由于血浆粘度的增高,红细胞就发生聚集,形成缗钱状。
总之,由于发生上述血液流变学的改变,不但会加重微循环障碍和组织的缺血缺氧,还可促进DIC的形成和休克的发展,近年来应用血液稀释治疗休克,其目的在于改善血液流变学,降低血流粘度。这种疗法已取得良好的效果。
三、细胞代谢的变化以及功能、结构的损害
休克时细胞的代谢障碍及其功能、结构的损害,既是组织低灌流、微循环流变学改变和/或各种毒性物质作用的结果,又是引起各重要器官功能衰竭和导致不可逆性休克的原因。
(一)休克时细胞的代谢变化
休克时细胞代谢改变比较复杂。由于休克的类型、发展价段以及组织器官的不同,其代谢改变的特点和程度也都有所不同,但共同的重要改变是:
1.糖酵解加强休克时由于组织的低灌流和细胞供氧减少,使有氧氧化受阻,无氧酵解过程加强,从而使乳酸产生增多,而导致酸中毒。但严重酸中毒又可抑制糖酵解限速酶如磷酸果糖激酶等的活性,使糖酵解从加强转入抑制。
2.脂肪代谢障碍正常情况下,脂肪分解代谢中产生的脂肪酸随血液进入细胞浆后,在脂肪酰辅酶A(脂肪酰CoA)合成酶的作用和ATP的参与下,被活化为水溶性较高的的脂肪酰CoA,后者再经线粒体膜上肉毒碱脂肪酰转移酶的作用而进入线粒体中,通过β-氧化生成乙酰辅酶A,最后进入三羧酸循环被彻底氧化。休克时,由于组织细胞的缺血缺氧和酸中毒,使脂肪酰CoA合成酶和肉毒碱脂肪酰转移的活性降低,因而脂肪酸的活化和转移发生障碍;另方面因线粒体获氧不足和/或某些休克动因(如细菌内毒素)、酸中毒等的直接作用使线粒体呼吸功能被抑制,使转入线粒体内的脂肪酰CoA不能被氧化分解,结果造成脂肪酸和/或脂肪酰CoA在细胞内蓄积,从而加重细胞的损害。
(二)休克时细胞的损害
休克时细胞的损害首先是生物膜(包括细胞膜、线粒体膜和溶酶体膜等)发生损害。
1.细胞膜的损害 最早的改变是细胞膜通透性增高,从而使细胞内的Na+、水含量增加而K+则向细胞外释出,细胞膜内外Na+、K+分布的变化,使细胞膜Na+-K+ATP酶活性增高。因而ATP消耗增加,再加上ATP的供应不足和膜上受体腺苷酸环化酶系统受损,结果使控制细胞代谢过程的第二信使-cAMP含量减少,因此细胞的许多代谢过程发生紊乱,例如休克时肌肉细胞对胰岛素的反应减弱,使胰岛素促进细胞摄取葡萄糖的效应减弱甚至丧失。
休克时引起细胞膜损害的原因是多方面的:
(1)能量代谢障碍休克时因组织细胞的缺血缺氧,一方面ATP生成不足,使细胞膜不能维持正常功能和结构;另一方面脂肪酸氧化受阻,蓄积于细胞内的脂肪酸和脂肪酰CoA与细胞内Na+、K+、Ca+等阳性离子结合形成“皂类”化合物,可直接对膜上脂类起“净化去垢”的破坏作用。
(2)细胞酸中毒休克时细胞发生酸中毒,除与乳酸等蓄积有关外,还可能与下述因素有关:①细胞低灌流,使产生的CO2不易排出;②ATP分解过程中产生H+(MgATP2-→MgADP-+Pi2-+H+);③胞浆Ca2+增多,可促使Ca2+进入线粒体并与其中的磷酸结合,在结合过程中也产生H+(3Ca2++2HPO42-→Ca3(PO4)2+2H+)。酸中毒可直接或间接破坏膜系统的功能和结构。
(3)氧自由基的产生休克时氧自由基产生增多主要是由于①氧代谢途径改变:即休克时由于细胞的缺氧和/或内毒素对线粒体呼吸功能的直接抑制,细胞色素氧化酶系统功能失调,以致进入细胞内的氧经单电子还原而形成的氧自由基增多而经4价还原而形成的水减少;②休克时产生大量乳酸、NADH及由ATP分解产生的次黄嘌吟等物质都可提供电子,使氧发生不全性还原而变成氧自由基。另外,休克时因蛋白水解酶活性增高,可催化黄嘌吟脱氢 酶变为黄嘌呤氧化酶,从而使次黄嘌吟变成黄嘌呤和氧自由基。③感染性炎症,活化补体等可激活中性粒细胞和巨噬细胞,使之释放出氧自由基。
氧自由基可通过膜脂质过氧化反应而破坏生物膜(参阅《缺血与再灌注损伤》)。
此外,溶酶体酶、内毒素等也可破坏细胞膜的功能与结构。
由于细胞膜的完整性在维持细胞的生命活动中起着重要作用。故当膜完整性破坏时,即意味着细胞不可逆性损伤的开始。
2.线粒体损害休克时线粒体最早出现的损害是其呼吸功能和ATP合成受抑制,线粒体ATP酶活性降低。此后发生超微结构的改变,如基质颗粒减少或消失;继之,基质电子密度增加、嵴内腔扩张,随后,嵴明显肿胀,终至破坏。
关于休克时线粒体损害的原因尚不完全清楚。缺氧可减少线粒体合成ATP,但除非在严重缺氧和伴有缺血时,并不引起线粒体膜的明显损害。目前认为,线粒体损害可能与下列因素有关:①内毒素等毒性物质及酸中毒对线粒体各种呼吸酶的直接抑制;②缺血导致线粒体合成ATP的辅助因子(如NAD、CoA和腺苷等)不足和细胞内环境(pH、离子)的改变。③前述的氧自由基对线粒体膜磷脂的过氧化作用等。
线粒体是维持细胞生命活动的“能源供应站”。线粒体损害时,由于氧化磷酸化障碍,产能减少乃至终止,故必然导致细胞损害和死亡。
3.溶酶体破裂溶酶体含有多种水解酶,如组织蛋白酶、多肽酶、磷酸酶等,但在未释放之前都处于无活性状态。一旦释放出来后,它们即转为活性状态而可溶解和消化细胞内、外的各种大分子物质,尤其是蛋白类物质。已证明,休克早期,肝、脾、肠等细胞即出现溶酶体肿大,颗粒丧失和酶释放增加;内毒素休克动物血液和淋巴中水解酶浓度增高,且与休克严重程度呈正相关。给动物注射溶酶体或溶酶体酶,可产生类似休克的各种病理生理改变。
休克时导致溶酶体破裂的主要原因是:①组织的缺血、缺氧、酸中毒以及内毒素对溶酶体膜的直接破坏;②氧自由基对溶酶体膜磷脂的过氧化作用;③血浆补体被激活产生C5a,后者可剌激中性粒细胞释放溶酶体酶。释放的溶酶体酶又可通过多种途径参与休克的发生、发展和细胞的损害,例如:a.释放的组织蛋白酶使蛋白质水解,这不但可以破坏蛋白酶的活性,甚至还可使细胞自溶坏死,而且所产生的多肽类活性物质,还能加重微循环障碍;b.破坏生物膜的完性;c.直接损害血管内皮和血管平滑肌细胞,从而导致血液外渗、出血和血小板的粘附、聚集以及DIC形成;d.激活补体系统产生C5a,后者再进一步促使溶酶体酶的释放。现已证明,休克时使用溶酶体膜稳定药可防止或减轻溶酶体膜的破裂。
总之,休克时生物的损害被认为是细胞发生损害的开始,而细胞的损害又是各脏器功能衰竭的共同机制。
四、器官功能的改变
休克时各器官功能都可发生改变,其中主要是中枢神经系统、心、肾、肺、胃肠及肝脏等重要器官的功能障碍。
(一)中枢神经系统功能的改变
休克早期,如果能通过代偿性调节维持脑的血液供给,除因应激反应而有兴奋性升高外,一般没有明显的脑的功能障碍。休克进一步发展,心输出量减少和血压降低,不能维持脑的血液供给,则发生缺氧。严重的缺氧和酸中毒还能使脑的微循环血管内皮细胞和小血管周围的神经胶质细胞肿胀,致脑微循环狭窄或阻塞,动脉血灌流更加减少。在微循环凝血期,脑循环内可以有血栓形成和出血。大脑皮层对缺氧极为敏感,当缺氧逐渐加重,将由兴奋转为抑制(表情淡漠),甚至发生惊厥和昏迷。皮层下中枢因严重缺氧也可发生抑制,呼吸中枢和心血管运动中枢兴奋性降低(详见《缺氧》)。
(二)心脏功能的改变
除心源性休克伴有原发性心功能障碍外,其它各类型休克也都可引起心功能的改变。一般而言,休克的早期可出现心的代偿性加强,此后心脏的活动即逐渐被抑制,甚至可出现心力衰竭,其主要机制是:
1.冠脉血流量减少和心肌耗氧量增加 由于休克时血压降低以及心率加快所引起的心室舒张期缩短,可使冠脉灌流量减少和心肌供血不足;同进因交感-儿茶酚胺系统兴奋使心率加快、心缩加强,导致心肌耗氧量增加,因而更加重了心脏缺氧。结果心肌因能量不足和酸中毒而使舒缩功能发生障碍,并从而引起心力衰竭,对于原来就有冠状动脉供血不良者,尤其容易出现心力衰竭。
2.酸中毒和高钾血症 酸中毒可通过多种机制影响心脏舒缩功能:①抑制肌膜的Ca2+内流;②H+和Ca2+竞争结合肌钙蛋白;③抑制肌浆网对Ca2+的摄取和释放;④抑制肌球蛋白ATP酶的活性。此外,酸中毒还可通过抑制心肌细胞能量代谢酶的活性、促使生物膜的破坏以及诱发心律失常等多种途径来抑制心肌的舒缩功能,并从而促使心力衷竭的发生。
休克时,组织细胞的破坏可释出大量K+,肾功能的障碍又使K+的排出减少,因而总是伴有高钾血症。高血钾可抑制动作电位复极化2期中Ca2+的内流,从而使心肌兴奋-收缩偶联减弱。
此外,心肌内DIC形成,内毒素对心肌的直接作用等等。都可以促使心肌力衰竭的发生。一旦发生了心力衰竭,将迅速促使休克进一步恶化,并给输液扩容造成一定困难。
3.心肌抑制因子的作用如前文所述,休克时的缺血、缺氧等可使胰腺产生心肌抑制因子(MDF),MDF能使心肌收缩力减弱,从而有助于心力衰竭的发生。
(三)肾功能的改变
肾功能的改变在休克早期就可发生,这时发生的是功能性的急性肾功能衰竭,因为它还不伴有肾小管的坏死。其主要临床表现为少尿(<400ml/d)或无尿(<100ml/d),其发生的主要机制如下:
1.肾小球滤过率减少在休克早期,有效循环血量的减少不仅能直接使肾血流量不足,而且还可通过肾素-血管紧张素系统和交感-儿茶酚胺系统的激活而使肾血管收缩,因而使肾血流量更加减少,结果是肾小球滤过压降低,肾小球滤过率减少。
2.肾小管对钠、水重吸收加强在休克早期,肾小管上皮细胞虽然已经发生缺血,但是因为持续时间不久,故这些细胞仍能保持其正常的重吸收功能,加之此时醛固酮和抗利尿激素分泌增多,所以肾小管对钠水的重吸收加强。肾小球滤过率的减少和肾小管重吸收的增强就可导致少尿或无尿。但此时肾功能的变化是可逆的。一旦休克逆转,血压恢复,肾血流量和肾功能即可恢复正常,尿量也将随之而恢复正常。故尿量变化是临床判断休克预后和疗效的重要指标。
当休克持续时间较长时,可引起急性肾小管坏死,发生器质性的肾功能衰竭。此时即使肾血流量随着休克的好转而恢复,患者的尿量也难以在短期内恢复正常。肾功能的这些改变,将导致严重的内环境紊乱,包括高钾血症、氮质血症和酸中毒等。这样就会使休克进一步恶化,故许多休克患者,尤其是老年患者常死于急性肾功能衰竭。
(四)肺功能的改变
随着休克的发展,肺功能也发生不同程度的改变:在休克早期,由于呼吸中枢兴奋,故呼吸加快加深,通气过度,从而甚至可以导致低碳酸血症和呼吸性碱中毒;继之,由于交感-儿茶酚胺系统兴奋和其他血管活性物质的作用,可使肺血管阻力升高;如果肺低灌流状态持续较久,则可引起肺淤血、水肿、出血、局限性肺不张、微循环血栓形成和栓塞以及肺泡内透明膜形成等重要病理改变,此即所谓休克肺(shock lung)的病理学基础(参阅第十三章)。
上述休克肺的病理变化,有的影响肺的通气功能。有的妨碍气体弥散,有的改变肺泡通气量/血流量的比例,造成死腔样通气和/或功能性分流,从而可以导致呼吸衰竭甚至死亡。休克肺是休克死亡的重要原因之一,约有1/3的休克患者死于休克肺。
(五)肝和胃肠功能的改变
1.肝功能的改变休克时常有肝功障碍,其主要原因有:①低血压和有效循环血量减少可使肝动脉血液灌流量减少,从而引起肝细胞缺血缺氧,严重者可导致肝小叶中央部分肝细胞坏死;②休克时由于腹腔内脏的血管收缩,致使门脉血流量急剧减少。肝约有一半以上血液来自门脉,故门脉血流量减少,也将加重肝细胞的缺血性损害;③肝内微循环障碍和DIC形成,更可引起肝细胞缺血缺氧;④在肠道产生的毒性物质经门脉进入肝,加之肝本身毒性代谢产物的蓄积对肝细胞都有直接损害作用。
肝功能障碍又可通过下列机制加重休克:①肝代谢障碍:肝对糖和乳酸的利用障碍,一方面可促使乳酸蓄积并从而引起酸中毒;另方面又不能为各重要脏器提供充足的葡萄糖。蛋白质和凝血因子合成障碍,可引起低蛋白血症和出血。②肝的生物转化作用(解毒功能)减弱:可增加休克时感染与中毒的危险。
2.胃肠功能的改变 休克早期就有胃肠功能的改变。开始时是因微小管痉挛而发生缺血,继而可转变为淤血,肠壁因而发生水肿甚至坏死。此外,胃肠的缺血缺氧,还可使消化液分泌抑制,胃肠运动减弱。有时可由于胃肠肽和粘蛋白对胃肠粘膜的保护作用减弱,而使胃肠粘膜糜烂或形成应激性溃疡。
由于胃肠上述改变,可通过下列机制促使休克恶化:①肠道粘膜屏障功能减弱或破坏,致使肠道细菌毒素被吸收入血,加之肝的生物转化作用减弱,故易引起机体中毒和感染。②胃微循环淤血,血管内液体外渗,加之胃肠粘膜糜烂坏死和DIC的形成都可导致胃肠道出血,从而使血容量进一步减少。③胃肠道缺血、缺氧,可剌激肥大细胞释放组胺等血管活性物质,因而微循环障碍进一步加剧。
(六)多器官功能衰竭
多器官功能衰竭(mrltiple organ failure MOF)是指心、脑、肺、肾、肝、胃肠、胰腺及血液等器官中,在24小时内有两个或两个以上的器官相继或同时发生功能衰竭。MOF又称多系统功能衰竭或综合器官衰竭。休克的晚期常出现MOF。MOF是致死的重要原因,而且衰竭的器官越多,病死率也就越高。如有三个器官发生功能衰竭时,病死率可高达80%以上。
MOF在临床上有两种表现形式,一是创伤和休克直接引起的速发型,又称单相型,发生迅速,发病后很快出现肝、肾和呼吸功能障碍,在短期内或则死亡,或则恢复;二是创伤、休克后继发感染所致的迟发型,又称双相型。此型患者往往有一个相对稳定的间歇期,多在败血症发生后才相继出现多器官功能衰竭。引起MOF的主要原因是:①重症感染:约有70~80%的MOF是在重症感染的基础上发生的;②休克时组织较长时间的低灌流和交感神经的高反应;③非感染性的严重病变如急性胰腺炎、广泛性组织损伤等。尤其是当机体的免疫功能和单核吞噬细胞系统功能减弱时,或者是治疗不当或延误时,如未及时纠正组织低灌流和酸碱平衡紊乱、过多过快输液、大量输血或过量应用镇静剂、麻醉剂等情况下,更易引起MOF。
MOF的发病机制尚不很清楚,现认为MOF的发生是多因素参与作用的结果,其中休克时组织低灌流所致的组织缺血缺氧、代谢障碍和酸中毒都起着重要作用;在感染或感染中毒性休克时,细菌内毒素在MOF的发生机制中被认为是起着关键的作用。这是因为内毒素不但能直接或间接(如激活补体和凝血系统)损害各器官的功能,而且还可通过激活补体而使中性粒细胞聚集和激活,从而使中粒细胞①释放各种水解酶,包括酸性和碱性蛋白酶,这些酶不但能破坏结构蛋白(如弹性蛋白酶破坏弹性纤维,胶原酶破坏胶原纤维等),而且还可分解血浆蛋白,激活凝血系统并从而导致弥散性血管内凝血;②产生并释放活性氧和脂类代谢产物(如白三烯等),这些物质又可破坏生物膜和/或增高血管通透性,加重微循环障碍。在一般情况下,中性粒细胞向感染和损伤处趋化和聚集,是炎性的正常反应,但在休克或严重感染时,由于机体免疫功能降低等原因,对这种炎性反应失去控制,从而使中性粒细胞释放的上述各种毒性物质得以广泛地破坏各器官细胞的结构和功能。故有人认为MOF是一种非特异性失控的全身性炎性反应。此外,儿茶酚胺-腺苷酸环化酶-cAMP系统异常也可能起着重要的作用,在休克时一方面因为细胞的缺血缺氧,使膜功能异常,腺苷酸环化酶系统的受体受损,对儿茶酚胺的反应减弱;另方面由于组织ATP含量降低,缺乏产生cAMP的底物,结果使细胞内cAMP水平下降,进而影响细胞内的许多代谢过程和功能。
第四节 各型休克的特点
各型休克虽有共同规律,但也各有其特点。前文所述,虽然反映了休克的共同规律,但主要是围绕低血容量性休克的特点来进行分析的。现将其它几种重要的休克类型的特点简述如下。
一、感染性休克
感染性休克(infectious shock)可见于各种微生物引起的败血症(故又称败血症休克,septic shock),特别是革兰氏阴性细菌的感染,由内毒素引起的休克(内毒素性休克,endotoxic shock)这型休克主要有以下特点。
1.感染性休克的发生机理由于细菌的毒素各异,作用不尽相同,感染性休克的发生机理是极为复杂的,不可能是一个模式。感染性休克和内毒素性休克是不一样的,目前研究最多的是内毒素在休克发生中的作用。给狗注射内毒素,可以在几分钟内出现血压急剧降低,未梢血液内血小板和中性粒细胞数减少;随后血压又逐渐升高,血小板中性粒细胞增多;3-4小时后,血压又再降低,血小板和中性粒细胞数又减少,外周血管阻力往往是先降低,而后逐渐升高。关于内毒素如何引起微循环障碍和血液动力学变化,目前尚未完全搞清楚,可能通过以下机制(图10-5)。
图10-5 内毒素性休克的发生机理
(1)内毒素作用于血管内皮细胞、血小板和中性粒细胞,而使大量血小板和中性粒细胞聚集和粘附在微循环内(特别是肝和肺内),血流受阻(血小板和中性粒细胞的聚集和粘附,早期是可逆的,可被血流冲散)。同时,内毒素还可激活补体,使组织胺和5-羟色胺释放,激活激肽系统,产生缓激肽,而使血管扩张,毛细血管开放数目增多(组织胺还使肝、肺微静脉和小静脉收缩),结果大量血液淤积在微循环内,回心血量和心输出量减少,血压降低。
(2)由于心输出量减少,可使交感神经兴奋和儿茶酚胺增多,内毒素还有拟交感作用,可引起小动脉收缩和动静脉吻合支开放,毛细血管内动脉血灌流减少。
(3)内毒素损害血管内皮细胞、激活凝血因子Ⅻ、促使血小板聚集和释放,再加上微循环淤血,通透性升高,血液浓缩,容易产生播散性血管内凝血(详见《播散性血管内凝血》)。播散性血管内凝血在内毒素性休克中较为常见,有的是先发生播散性血管内凝血,而导致休克的发生和发展。
(4)内毒素性休克,除由于微循环动脉血灌流不足,使细胞代谢发生障碍外,内毒素还可直接损害细胞(线粒体肿胀),抑制氧化过程,而引起细胞代谢和功能变化。例如给动物注射内毒素后,在未出现严重的微循环障碍之前,就可发现血浆内溶酶活性升高,心肌抑制因子的产生,心肌收缩性减弱。因此内毒素对细胞的损害在休克发生过程中,也有一定的意义。
2.由于细菌的毒素不同,作用不同,因而各种感染性休克的表现也很不同,有的表现为低动力型(低排高阻型),有的表现为高动力型(高排低阻型)(表10-1)。
表10-1 高动力型休克和低动力型休克比较
| 高动力型休克 | 低动力型休克 | |
| 血 压 | 降 低 | 降 低 |
| 循 环 血 量 | 正 常 | 减 少 |
| 中心静脉 压 | 正常或偏高 | 偏 低 |
| 心输 出 量 | 正常或偏高 | 减 少 |
| 外周血管阻力 | 降 低 | 升 高 |
| 皮肤 颜 色 | 潮红→发绀 | 苍白→发绀 |
| 皮 肤 温 度 | 温暖→湿冷 | 湿 冷 |
| 尿 量 | 减 少 | 少尿或无尿 |
| 动 静 脉氧差 | 缩 小 | 不 定 |
| 发 病机 理 | 以肾上腺素能β受体兴奋为主,动静脉吻合支开放,毛细血管灌流减少。 | 以肾上腺素能α受体兴奋为主,小动脉微动脉收缩,微循环缺血。 |
为什么有这样不同的表现,目前还不清楚,可能与下列因素有关:
⑴细菌种类不同,革兰氏阳性细菌引起的多为高动力型;革兰氏阴性细菌引起的多为低动力型,但也有人报道高动力型休克多数也是由革兰氏阴性细菌引起的。
⑵休克的发展阶段不同,开始阶段和轻型休克,常表现为高动力型;休克进一步发展和重型休克,表现为低动力型。有人把高动力型休克看作是感染性休克发展过程的早期阶段。
⑶休克前的血量和血管反应性不同,休克前已有血量减少,易引起小动脉收缩,表现为低动力型;如休克前汉有血量减少,细菌毒素通过某种机理,使β受体兴奋,动静脉吻合支开放和心收缩力加强,因而外周阻力降低,循环速度加快,心输出量增加,表现为高动力型。因皮肤微循环动静脉吻合支比较丰富,此时血流量增加,所以表现为皮肤潮红温暖。
3.因为有感染存在,在发生休克时,除有休克表现外,还有因感染而引起的其它损害,所以病情更加严重和复杂。
二、心源性休克
凡能严重地影响心脏排血功能,使心输出量急剧降低的原因,都可引起心源性休克(cardiogenicshock)。例如大范围心肌梗塞、弥漫性心肌炎、急性心包填塞、肺动脉栓塞、严重心律失常以及各种严重心脏病晚期。其中主要的是心肌梗塞。这型休克的主要特点是:①由于心泵衰竭,心输出量急剧减少,血压降低;微循环变化的发展过程。基本上和低血容量性休克相同,但常在早期因缺血缺氧死亡;②多数病人由于应激反应和动脉充盈不足,使交感神经兴奋和儿茶酚胺增多,小动脉、微动脉收缩,外周阻力增加,致使心脏后负荷加重;但有少数病人外周阻力是降低的(可能是由于心室容量增加,剌激心室壁压力感受器,反射性地引起心血管运动中枢的抑制);③交感神经兴奋,静脉收缩,回心血量增加,而心脏不能把血液充分输入动脉,因而中心静脉压和心室舒张期末容量和压力升高;④常比较早地出现较为严重的肺淤血和肺水肿,这些变化又进一步加重心脏的负担和缺氧,促使心泵衰竭。
三、过敏性休克
过敏性休克(anaphylactic shock)可见于某些药物(如青霉素、奴夫卡因)和血清制剂(如破伤风抗毒素、白喉类毒素)过敏的人。特应性机体受到过敏原剌激后易产生抗体(IgE)持久地被吸附在细胞膜上(特别是小血管周围的肥大细胞和血液的嗜硷性细胞)。当再次遇到相应的过敏原,细胞膜上的IgE即与过敏原结合,激发细胞释放组织胺和其它血管活性物质(如5-羟色胺、慢反应物质);抗原与抗体在细胞表现结合,还可激活补体系统,并通过被激活的补体激活激肽系统。组织胺、缓激肽、补体C3a、C5a等可使后微动脉和毛细血管前括约肌舒张,大量毛细血管开放,通透性增加,另外组织胺还可选择性地使一些器官的微静脉和小静脉收缩,因而造成微循环淤血,容量扩大,大量血液淤积在微循环内,致静脉回流量和心输出量急剧减少,血压降低。另外,组织胺还能引起支气管平滑肌收缩,造成呼吸困难。这种休克发病非常迅速,可立即注射缩血管药物(如肾上腺素),解除支气管平滑肌收缩,改善通气功能;使小动脉、微动脉收缩,增加外周阻力,提高血压,保证心脑等重要器官的血液供给。
四、神经源性休克
在正常情况下,血管运动中枢不断发放冲动沿传出的交感缩血管纤维到达全身小血管,使其维持着一定的紧张性。当血管运动中枢发生抑制或传出的交感缩血管纤维被阻断时,小血管就将因紧张性的丧失而发生扩张,结果是外周血管阻力降低,大量血液淤积在微循环中,回心血量急剧减少,血压下降,引起神经源性休克(neurogenic shock)。此类休克常发生于深度麻醉或强烈疼痛剌激后(由于血管运动中枢被抑制)或在脊髓高位麻醉或损伤时(因为交感神经传出径路被阻断)。本类休克的病理生理变化和发生机制比较简单,预后也较好,有时不经治疗即可自愈,有的则在应用缩血管药物后迅速好转。有人认为这种情况只能算是低血压状态(hypotensive state),而不能算是休克,因为从休克的概念来看,在这种患者,微循环的灌流并无急剧的减少。
第五节 休克的防治原则
一、及早预防
1.积极防治感染和各种容易引起感染性休克的疾病,例如败血症、细菌性痢疾、肺炎、流行性脑脊髓膜炎、腹膜炎等。
2.做好外伤的现场处理,如及时止血、镇痛、保温等。
3.对失血或失液过多(如呕吐、腹泻、咯血、消化道出血、大量出汗等)的患者,应及时酌情补液或输血。
4.在应用可能引起过敏性休克的药物(如青、链霉素等)或血清制剂(如破伤风、白喉抗毒素)前,务必作皮肤过敏试验,反应阳性者禁用;输血前应严格检查供受者血型是否相符等。
二、积极治疗
对于休克病人,应当分秒必争,尽早抢救。因为如果不然,病情就将不断恶化。治疗开始得愈晚,效果也将愈差。除了采取相应的措施以对抗感染、出血、疼痛等能促进和加重休克的因素外,还应当抓紧下述几个方面的治疗措施。
(一)改善微循环、提高组织灌流量
1.补充血容量各种休克均有有效循环血量不足——或者是由于容量绝对减少(如失血、脱水、血浆丧失)或者是因为血容量相对不足(如血管扩张),故最终都导致组织灌流量的减少。因此,补充血容量是提高心输出量、改善组织灌流的根本措施。
关于补液的量,以往遵循的是“失多少,补多少”的原则。现在看来,根据这个原则决定的补液量,显然是不够的,因为有些休克患者,如感染性和过敏休克患者,可以并无明显的失液,但由于血管容量扩大、微循环淤血、血浆外渗等,有效循环血量也是显著减少的;在失血性、失液性休克患者,除了向体外失液外,到休克Ⅱ期时也有微循环淤血、血浆外渗等变化。因此,补液的量应当大于失液的量,应当遵循,“量需而入”的原则,以达到迅速改善微循环的目的,当然。补液过多也是危险的,因为可能促进休克肺的发生。
为了掌握适当的补液量,应严密观察患者的颈静脉充盈程度、尿量、血压、脉搏等临床指标,作为监护输液的尺度。有条件时,应当动态地监测患者的中心静脉压,最好还能测定肺动脉楔压。中心静脉压和肺动脉楔压低于正常,说明血容量不足,应当继续输液,以使二者保持在正常范围内。如果超过正常,说明补液过多,应当立即停止补液,严密观察病情并采取相应的措施。中心静脉压的测定虽然较为简便,但它只能较好地反映右心的功能。在反映左心功能方面,肺动脉楔压是一个较好的指标。因此,对于心源性休克的患者,应尽可能采用肺动脉楔压作为监护输液的指标。
此外,在补充容量的同时,应考虑纠正血液流变学的改变,例如由于血浆外渗而导致的血液浓缩,白细胞的粘附和阻塞等。故除失血性休克时宜输全血外,对其它休克患者应补充适量的胶体溶液(如血浆及其代用品、右旋糖酐等)及晶体溶液(如生理盐水、任氏液等)。
2.合理应用血管活性药物,调整血管容量在补足血容量的基础上,根据休克的不同类型和不同的发展阶段以及不同的表现,合理选用血管活性药物,对于改善微循环、提高组织灌流量有重要意义。
(1)扩血管药物的应用:a受体阻断药酚妥拉明(phentolamine)、酚苄明(phenoxybenzamine)等适用于低血容量性休克、低动力型感染性休克和高阻力型心源性休克,因为它们能解除小血管和微血管的痉挛,从而改善微循环的灌流和增加回心血量。但扩血管药物不宜用于过敏性休克、神经性休克和高动力型感染性休克,因为在这些休克患者,血管已经扩张。此外,我国学者应用大剂量阿托品、东莨菪碱,山莨菪碱(654-2)等扩血管药物治疗休克,获得了较好的效果,但这些药物的作用机制尚未完全阐明。
应当再次强调,扩血管药物必须在血容量得到充分的先决条件下才能应用,否则,血管的扩张将使血压进一步急剧降低而减少心、脑的血液供应。
(2)缩血管药物的应用:直至本世纪60年代。缩血管药物如去甲肾上腺素(norepinephrine)、甲氧胺(methoxamine)等曾被广泛应用于休克的治疗。但这些药物有进一步减少微循环灌流量的缺点,而且在临床上的效果也不理想,故目前不主张在各型休克患者长期和大量应用。但缩血管药物仍有其适应症:①血压过低而又不能立即补液时,可用缩血管药物来暂时提高血压以维持心、脑的血液供应;②对于过敏性休克和神经源性休克,缩血管药效果良好,应当尽早使用;③对于高动力型感染性休克和低阻力型心源性休克,缩血管药也有疗效。
(3)扩血管药与缩血管药的联合应用;联合应用,可以取长补短,突出某一药物的治疗作用而减轻其副作用,从而有效地改善微循环,提高组织灌流量。例如去甲肾上腺素和α-受体阻断药妥拉唑啉(tolaxoline)联合应用,即可减少去甲肾上腺素的强烈缩血管作用,又可突出其β受体的兴奋作用。
另外,选用能同时兴奋α受体和β受体的药物如多巴胺,既能使皮肤、肌肉等的血管收缩,又能选择性地扩张重要器官(心、脑、肾)的血管,同时还有强心作用,因而不但能提高血压和促进血液的合理分配,且能提高心输出量。故适用于各类休克。
3.防治DIC(参阅第九章)
(二)改善细胞代谢,防治细胞损害
1.自由基清除药目前实验室和临床较常用的自由基清除药有超氧化物歧化酶、亚硒酸钠、谷胱甘肽过氧化物酶等。此外,维生素C.辅酶Q、甘露醇和葡萄糖等都有清除自由基的作用,也可防止或减轻细胞的损害。
2.溶酶体稳定药和钙拮抗药在防止溶酶体酶释放及其破坏作用方面,除了消除破坏溶酶体膜因素(如纠正缺氧和酸中毒、清除自由基等)外,目前常用的是溶酶体膜稳定药,如糖皮质激素、前列腺素(PGI2、PGE1)和组织蛋白酶抑制剂(如parachloromercuribenzoate,PCMB)。此外,由于钙拮抗药能抑制Ca2+的内流和在胞质中的蓄积,从而降低生物膜的磷脂酶活性,故也能保护溶酶体膜。实验证明,山莨菪碱也有抑制Ca2+内流、保护溶酶体膜的作用。
3.纠正酸中毒,提供细胞营养底物和能量酸中毒可加重微循环障碍,促进DIC的形成,抑制心肌收缩和能量代谢,破坏生物膜,并能降低药物效应,故纠正酸中毒是改善心肌代谢、防止细胞损害和提高药物疗效的重要措施。
此外,由于交感-肾上腺髓质系统的兴奋使胰岛素效应被抑制,组织低灌流又引起细胞的缺氧,因而使细胞处于高度“饥饿”状态,故适当补充葡萄糖、胰岛素和能量合剂,对改善细胞营养和代谢,防止细胞损害都有一定的良好作用。
(三)治疗器官功能衰竭
休克时如出现器官功能衰竭,则除了采取一般治疗措施外,尚应针对不同的器官衰竭采取不同的治疗措施,如出现心力竭时,除停止或减慢补液外,尚应强心,利尿,并适当降低前、后负荷;如出现呼吸衰竭时,则应给氧,改善呼吸功能;如发生急性肾功能衰竭时,则可考虑采用利尿,透析等措施。
第十一章 缺血与再灌注损伤
第一节 概 念
由于近年医学的发展进步;使缺血再灌注损伤愈益受到重视。例如断肢再植和器官移植就存在缺血肢体和器官的再灌注问题。又如动脉搭桥术后、溶栓疗法之后,休克治疗之后均可能发生缺血与再灌注损伤。因而研究渐多,已成为一个医学重要问题。上述各种治疗方法十分有效,应用渐广,所以我们要对这些疗法有时可能发生损伤的规律有所认识。
缺血后疏通血管或再造血管使组织得到血液的再灌注,本来是医师们的目的所在,而且事实上在大多数情况下也确能收到良好的治疗效果。但在一定条件下(取决于缺血时间)再灌注反而引起更加严重的后果。这不仅见于临床,而且也为不同种属(兔、大鼠、豚鼠、狗、猪等)的大量动物实验所证明。这是一种反常(paradox)现象,称之为再灌注损伤(reperfusion injury)。这类反常现象目前已知主要有:
钙反常(calciumparadox):这是1966年Zimmerman首先发现的,他在研究心兴奋-收缩偶联时偶然观察到,当以无钙生理溶液灌流离体大鼠心脏很短时间(2分钟)后再以含钙正常溶液灌注时,心脏发生严重的功能和结构变化;心脏电活动消失,机械活动停止;超微结构可见严重的收缩带(肌纤维过度收缩,肌节分不清明暗带,粗细肌丝过度重合)、间盘分离、肌膜破裂;生化学变化包括高能磷酸化合物耗竭,心肌酶、肌红蛋白及其他心肌蛋白以及钾离子的释出,细胞内钙离子大量积聚而形成所谓钙超载(calcirm overload),后者成为细胞致死的原因。钙反常对细胞的损伤程度与无钙灌流的时限有关,随无钙灌流时间的延长而加重。
氧反常(oxygenparadox):这是1973年h 提出的概念,他在以大鼠离体心脏为很我人所证实。其功能结构损伤类似钙反常但不如钙反常那样急剧。肌原纤维呈严重收缩带,线粒体肿胀,嵴断裂,空泡化,肌浆网扩张;生化学变化与钙反常类似。氧反常损程度与缺氧时间、灌流液温度、PH值以及重给氧时的氧分压有关。缺氧时间越长,温度及PH值越高,给氧时的氧分压越高,氧反常性损伤愈重。
缺血/再灌注损伤(ischemia/reperfusion injury):其与氧反常不同之处在于既缺氧又缺营养底物。这是在整体上所发现的现象。在整体动物实验中,结扎冠状动脉造成心肌缺血,梗塞形成,当重新开放血液使心肌得到备注的重新藻流后,在一定条件下心肌损伤反而加重,出现心率失常,出血性坏死,梗塞面积扩大。在临床心脏外科手术后重新恢复血液灌流时有时也出现类似现象,病人在术后死于心肌出血性梗塞。这种在缺血基础上恢复血流后引起的更为剧烈的损伤称为再灌注损伤。其功能结构变化与上述大致相同。
再灌注损伤是否出现及其严重程度,关键在于缺血时间的长短,侧支循环的形成情况以及对氧的需求程度。这是再灌注损伤形成的基础。
第二节 心、脑、肠的缺血/再灌注损伤
一、心肌的缺血/再灌注损伤
心肌缺血/再灌注损伤最为常见。从心功能上看,静止张力(restingtension,指心肌在静息状态下受前负荷作用而被拉长进产生的张力)随缺血时间的延长逐渐升高,发展张力(developed tension,指心肌收缩时产生的主动张力)逐步下降,再灌注时静止张力更加增高,发展张力愈加低下,说明心脏收缩力下降。
从腺苷酸类代谢来看,缺血时心肌ATP、CP含量迅速下降,CP下降尤甚。由于ATP降解,使ADP、AMP含量升高。但由于腺苷酸可进一步降解为核苷类(腺苷adenosine,肌苷,inosine)及硷基(次黄嘌吟等),心肌中这些物质可以增加百倍。这些非磷酸化嘌吟可进入血管,因而ADP、AMP迅速下降。心肌中合成高能磷酸化合物的原材料减少。由于再灌注时血流的冲洗,核苷类物质明显下降。
缺血时肌浆网(sarcoplasmicreticulum, SR)对钙的摄取下降,再灌注时更进一步下降,因而肌浆钙浓度升高,线粒体内钙积聚。
缺血/再灌注损伤时超微结构的变化与单纯心肌缺血时性质基本相同,但前者的程度更为严重(缺血时间相同)。基底膜部分缺失,质膜破坏,损伤迅速扩展到整个细胞使肌原纤维结构破坏(出现严重收缩带或肌丝断裂、溶解),线粒体损伤(极度肿胀、嵴断裂、溶解,空泡形成、基质内致密物增多)。这说明重新恢复血流引起了快速的结构破坏过程,既破坏膜磷脂也破坏蛋白质大分子及肌腺纤维。再灌注损伤与缺血时间的依赖关系,提示在缺血过程中已经奠定了再灌注损伤细胞及生化学的变化基础。
二、脑的缺血/再灌注损伤
脑是一个对缺氧最敏感的器官,它的活动主要依靠葡萄糖有氧氧化提供能量,因此一旦缺血时间较长即可引起严重的不可逆性损伤。脑缺血时生物电发生改变,出现病理性慢波,缺血一定时间后再灌注,慢波持续并加重。
脑缺血后短时间内ATP,CP、葡萄糖、糖原等均减少,乳酸明显增加。环腺苷酸(cAMP)在缺血(结扎砂鼠两侧颈总动脉)30分钟时增加2.2倍,环鸟苷酸(cGMP)则减少53%。恢复血流15分钟后cAMP进一步增加,为缺血前的21倍,cGMP进一步下降21%。这一情况提示缺血及再灌注时过氧化反应增强,因而cAMP上升导致磷脂酶激活,使磷脂降解,游离脂肪酸增多,缺血后血流再灌注时,自由基产生增加,与游离脂肪酸作用而使过氧化脂质生成增多,能引起细胞和组织的损伤。
脑又是一个特别富有磷脂的器官。因此缺血后游离脂肪酸的增加尤为明显。实验证明大鼠断头1分钟后脑内游离脂肪酸含量增加2倍以上,随缺血时间延长持续增加,增加最显著的是花生四烯酸(arachidonicacid)及硬脂酸(18烷酸,stearic acid)。再灌注后游离脂肪酸的增加更为显著,恢复血流90分钟,反而有更多的游离脂肪酸贮留。血流重新恢复时游离脂肪酸的增加,是由于来源于游离脂肪酸的过氧化物进一步损伤膜的同时,由cAMP介导膜磷脂继续降解的结果(cAMP激活磷脂酶)。
缺血时脑的最明显的组织学变化为脑水肿及脑细胞坏死,两者往往又互为因果关系。特别是脑水肿,是各种脑血管意外的常见病理过程。Chan及Fishman等将大脑切片浸于含各种脂肪酸的溶液中,以观察组织肿胀的情况,发现只有在多价不饱和脂肪酸存在下,才能引起水肿,这是因为脂肪酸容易过氧化而引起细胞膜损伤。用砂鼠做实验制备不完全缺血模型,证明缺血及再灌注过程中脑含水量持续增加,缺血时水肿的产生是膜脂降解,游离脂肪酸增加的结果,而过氧化是再灌注后水肿持续加重的原因之一,因为细胞膜脂质过氧化使膜结构破坏。
三、肠缺血/再灌注损伤
肠管缺血时,液体通过毛细血管滤出而形成间质水肿。缺血后再灌注,肠管毛细血管通透性更加升高。
从形态学变化来看,严重肠管缺血所致损伤的特征为粘膜病变(粘膜损伤)。不论人和动物,在出血性休克及局部肠管缺血后出现肠粘膜损伤。粘膜损伤的特点表现为:广泛的上皮与绒毛分离,上皮坏死,固有层破坏,出血及溃疡形成,此必导致广泛的功能,(如吸收)障碍及粘膜屏障的通透性增高,使大分子得以通过。
第三节 缺血/再灌注损伤的机制
关于再灌注损伤的发生机制问题,到目前为止,学说虽多而且有的学说也颇为有力,便尚未得到彻底阐明。兹介绍如下:
一、无复流现象
无复流现象(no-reflow phenomenon)是在犬的实验中发现的。结扎犬的冠状动脉造成局部心肌缺血后,再打开结扎的动脉,使血流重新开放,缺血区并不能得到充分的灌注,故称此现象为无复流或无再灌。这种无复流现象不仅见于心肌,而且也见于脑、肾骼肌缺血后再灌注时。即再灌注损伤实际上是缺血的延续和叠加,缺血细胞并未能得到血液灌注,而是继续缺血,因而损伤加重。所以发生无复流现象,可能与下列因素有关(以心肌为例):
(一)心肌细胞肿胀由于缺血引起细胞膜Na+-K+泵功能障碍,从而使钠、水在细胞内潴留,因而再灌注时缺血区心肌细胞发生肿胀,压迫微血管。
(二)血管内皮细胞肿胀缺血及再灌注时也发生内皮细胞肿胀,内皮细胞向管腔伸出突起造成管腔狭窄,阻碍血液灌流。内皮细胞的肿胀与氧自由基的增多有关,因为氧自由基可以使血管内皮细胞膜受损,水钠乃进入内皮细胞而引起细胞水肿。
(三)心肌细胞的收缩缺血所致的心肌细胞收缩形成严重收缩带,压迫微血管,使缺血区某部分得不到血液重新灌注。心肌细胞的肿胀与收缩带可同时存在。
(四)微血管堵塞Feinburg及其同事曾证明,缺血一定时间后血管内血小板的沉积增加2倍。又证明心肌及肠管缺血后的无复流区内白细胞(主要是中性粒细胞)的聚集明显增加,从组织学上可见白细胞嵌顿、阻塞毛细血管。正常灌注情况下,每2433μm到3261μm长的毛细血管可发现一个白细胞,而在缺血时增加10倍,平均292μm长的毛细血管即有一个白细胞。缺血时红细胞作叠连状聚集,但这不是血管阻塞的主要原因。因为叠连状红细胞的解聚较白细胞与内皮细胞粘着的分离要容易得多。此外也有人解释无复流现象是由于纤维蛋白塞和微血栓形成所致。但有人在再灌前用链激酶进行纤溶并未减轻无复流现象。
微血管的堵塞还与花生四烯酸的代谢产物前列环毒(PGI2)和血栓素A2(TXA2)之间的失衡密切相关。PCI2主要由血管内皮生成,除了有很强的扩血管作用以外,还能抑制血小板的聚集。TXA2主要由血小板生成,不仅是很强的缩血管物质,而且也是一种引起血小板聚集的因子,因此是一个很强的致血栓形成的物质。缺血缺氧时,一方面因为血管内皮细胞受损而致PCI2生成减少,另方面缺氧又可使血小板释放TXA2增多,因而发生强烈的血管收缩和血小板的聚集并进一步释放TXA2,从而促使血栓形成和血管堵塞。动物实验也证明,应用TXA2合成酶抑制药可以使缺血/再灌注以后的冠脉血流改善。
二、钙超载
如前所述,钙反常(无钙灌流后用含钙溶液再灌注)时细胞内钙超载,引起严重的功能及结构障碍。钙反常的原因不甚明了,但主要的损伤是在无钙灌流期出现的细胞膜外板(external lamina)与糖被膜(glycocalyx)表面的分离(两者由Ca2+连结在一起)。细胞膜的这种损伤为再灌注时钙的大量内流提供了条件。在长期缺血缺氧后再给氧或再灌注时也可引起细胞内钙超载。
缺血后再灌注时细胞内钙超载的机制尚无定论,但可能与下列因素有关,如图11-1哺乳类钙代谢模式图所示:
图11-1 哺乳类细胞钙代谢模式图
①电压依赖性钙通道;②质膜钙泵ATP酶;③Na+-Ca2+交换;④线粒体;⑤肌浆网;⑥细胞内蛋白或阴离子结合的钙;⑦膜磷脂的极性头部;⑧结合于质膜糖被的钙
(一)钠的平衡障碍有人证明,再灌注时钙超载是由于钠平衡障碍所致。因为在缺血缺氧时发生了细胞酸中毒,细胞内pH值降低,所以在再灌注时细胞内外形成pH梯度差,由于Na+-H+交换,致细胞内钠增加,然后又依Na+-Ca2+交换机制使细胞外钙大量内流造成细胞钙超载。
(二)细胞膜通透性增高缺血缺氧引起的细胞酸中毒在再灌注时通过细胞内外Na+-H+交换和Na+-Ca2+交换而使细胞内钙增加,而细胞内钙增加可激活磷脂酶,使膜磷脂降解,细胞膜通透性增高,故在灌注时细胞外钙顺着浓度梯度而大量内流,细胞膜通透性增高的更重要的原因可能是再灌注时氧自由基的大量产生。氧自由基可引发细胞膜的脂质过氧化,使膜受损,通透性增高。
(三)线粒体受损有些学者认为原发性损伤在于线粒体。如所周知,缺血时线粒体结构的功能障碍出现最早,表现为线粒体肿胀、嵴断裂。线粒体膜流动性降低,氧化磷酸化功能受损ATP生成障碍。因此,使上述损伤更为严重。
ATP减少使肌膜及肌浆网膜钙泵功能障碍,由于钙泵功能障碍不能排出和摄取细胞浆中过多的钙,致使细胞浆中游离钙浓度增加而造成钙超载。细胞浆中过多的钙最终形成磷酸盐沉积于线粒体,使线粒体结构及功能更加破坏。
细胞钙超载是再灌注损伤的一个重要特征,但目前,仍未能搞清它究竟是再灌注损伤的原因抑制或结果。
三、白细胞的作用
白细胞(主要是中性粒细胞)出现于梗塞的心肌中已为尸检所证实。1984年Mullane及其同事证明,冠状动脉堵塞60分钟时心肌组织就有白细胞出现,5小时后在缺血区有大量的白细胞聚集。根据Engler及其同事的研究,再灌注时白细胞数非但不减少反而增加。以犬心肌缺血为模型,再灌注仅5分钟,心内膜中性粒细胞就增加25%,缺血轻的组织白细胞聚集也少。
组织缺血和再灌注时白细胞浸润增加的机制还不十分清楚。可能是由于组织受损时,细胞膜磷脂降解,花生四烯酸代谢产物增多,其中有些物质具有很强的趋化作用,因而就能吸引大量白细胞进入组织或粘附于血管内皮,而白细胞本身又能释放很多具有趋化作用的炎性介质,如白三烯之一的LTB4,从而使微循环中白细胞进一步增加。
白细胞积聚对组织的损伤作用在于:
(一)嵌顿、堵塞毛细血管有助于形成无复流现象。微动脉及微静脉亦有大量白细胞粘附于内皮细胞,虽不一定堵塞血流,但粘附的白细胞仍可损伤组织并释放趋化因子从而吸引更多的细胞。
(二)白细胞可以增加血管通透性,水肿组织的含水量与白细胞密度呈正相关,说明白细胞可能引发水肿。白细胞增加血管通透性、引发水肿的机制与白细胞释放的某些炎症介质有关。
(三)激活的中性粒细胞释放溶酶体酶,可使组织发生蛋白水解性破坏和液化。
(四)中性粒细胞可通过产生氧自由基而损伤组织。
白细胞在缺血再灌注损伤的作用,可被以下实验结果证明:
1.用除去白细胞的血液进行再灌注,可以防止水肿产生并减轻再灌性损伤。
2.抗炎药减轻组织白细胞浸泣,可缩小梗塞面积。实验证明布洛芬(ibuprofen)对心肌具有保护作用,主要是由于抑制白细胞浸润的作用所致。
3.用补体抑制药降低补体,从而减少白细胞浸润,可能减轻组织损伤。丝氨酸蛋白酶抑制药,可以缩小心肌梗塞,而这种酶恰是一种在心肌缺血时能激活补体的酶。因此这种酶的抑制药,可通过抑制补体的激活而抑制白细胞浸润,从而减轻心肌损伤。
四、高能磷酸化合物的缺乏
心肌正常情况下以有氧代谢形式生成三磷酸腺苷(ATP)供作功需要。心肌缺血时则转为无氧代谢为主,ATP合成减少,以致心舒缩功能障碍。在犬的实验中证明,心肌严重缺血15分钟(结扎冠状动脉左旋支),心肌发生可逆性损伤。此时如果得到血液再灌注,则细胞并不死亡。但有很多报告指出,短时间缺血后,收缩功能长时间不能恢复。究其原因,多认为与ATP水平的低下有关。研究证明缺血15分钟时不仅ATP减少60%。总腺苷酸池也减少50%。ADP也轻度减少(可能转为ATP或AMP),AMP明显升高,但其升高程度小于ATP减少辐度。再灌注20分钟ATP明显回升,但只接近正常的一半,再灌注24小时仍然维持在低水平上,只有在再灌注4天后ATP及总腺苷池才近于恢复,但仍低于非缺血区(图11-2)。
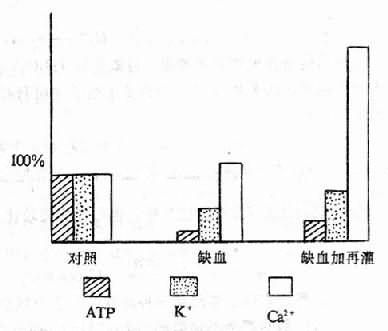
图11-2 心肌缺血/再灌注时ATP、Ca2+、K+的变化
以大鼠离体作功心脏为模型(Neely氏模型),先在20℃低温下给心脏停跳液,再短时间全心缺血(夹住主动脉)后,再灌注生理溶液。结果是给停跳液30分钟再缺血30分钟后,ATP几乎完全丧失,ADP明显减少,AMP明显增加,总腺苷酸量显著降低。再灌注60分钟可使ATP明显回升,但不及正常对照的一半,而总腺苷酸量则明显低于给停跳液后,比正常对照减少50%。上述研究提示缺血及再灌注损伤的心肌有氧代谢性发生严重损伤,影响能量代谢及心肌功能的恢复。
再灌注时高能磷酸化合物之所以恢复慢且总腺苷酸水平明显下降,可能与下列因素有关。
(一)缺血心肌的代谢障碍主要表现为对氧的利用能力受限,有氧代谢严重受损。在缺血进入不可逆阶段再灌注时,氧的利用并不增加,心肌只能利用运至心肌的氧的17%。氧的利用能力受限与缺血及再灌注所致线粒体受损有关。
(二)ATP合成的前身物质(腺苷、肌苷、次黄嘌呤等)在再灌时被冲洗出去,使心肌失去再合成高能磷酸化合物的物质基础。实验证明在再灌注液中补充肌苷或谷氨酸能促进ATP的合成及心功能的恢复。
3.线粒体膜发生氧自由基诱发的脂质过氧化反应使线体受损。线粒体膜富有磷脂,线粒体在缺氧时又是产生自由基的场所,因此极易引起膜脂过氧化使线粒体功能障碍。
五、自由基的作用
自由基(free radical)是具有一个不配对电子的原子和原子团的总称。由氧诱发的自由基称为氧自由基或活性氧,如超氧阴离子(O2)、羟自由基(OH·)及单线态氧(1O2,激发态放出一个光子)等非脂性自由基。H2O2非自身基,但也是一种氧化作用很强的活性氧。氧自由基与多聚不饱和脂肪酸作用后生成的中间代谢产物烷自由基(L·)、烷氧基(LO·)、烷过氧基(LOO·)等属于脂性自由基。氧自由基和脂性自由基的性质极为活泼,易于失去电子(氧化)或夺取电子(还原),特别是其氧化作用强,故具有强烈的引发脂质过氧化的作用。在生理情况下,氧通常是通过细胞色素氧化酶系统接受4个电子还原成水,同时释放能量,但也有1~2%的氧接受一个电子生成O2,或再接受一个电子生成H2O2。但由于细胞内存有超氧化物歧化酶(superoxidedismutase, SOD)和谷胱甘肽过氧化物酶(glrtathione peroxidase,GSH-PX)等抗氧化酶类可以及时清除它们,所以对机体并无有害影响。在病理条件下,由于活性氧产生的过多或抗氧化酶类活性下降,则可引发链式脂质过氧化反应损伤细胞膜系并进而使细胞死亡。
(一)细胞内氧自由基的生成
分子氧在线粒体细胞色素氧化酶系统中接受一个电子而被还原生成O2。
O2e-→O2-
这是其他活性氧产生的基础。过氧化氢(H2O2)及羟自由基(OH·)续发于此。即氧在获得一个电子时还原生成O2-,获得2个电子生成H2O2,获得3个电子生成OH·,获得四个电子生成H2O。
![]()
H2O2既可由O2-自发歧化产生,也可经酶促歧化而生成。
![]()
H2O2本身并非自由基而是一种活性氧,它与氧自由基的产生有密切关系(详下文)。
OH·自由基的产生不仅需要O2-或H2O2,而且要有过渡金属,如铁的螯合物的存在。由铁催化的Fenton型Haber-Weiss反应可迅速形成OH·,而单纯的Haber-W-eiss反应速度很慢,很难由此形成OH·。
O2-+H2O2→O2+OH-+OH·
(Haber-Weiss反应)
Fe(Ⅲ)+O2-→fe(Ⅱ)+O2(2)
Fe(Ⅱ)H2O2→Fe(Ⅲ)+OH-+OH· (3)
也就是说,O2-使铁还原,还原的铁再使H2O2还原生成OH·,OH·是最活跃最强力的氧自由基。缺血与再灌注时氧自由基生成过多,其机制可能是:
1.黄嘌呤氧化酶的形成增多黄嘌呤氧化酶(xanthine ocidase, XO)的前身是黄嘌呤脱氢酶(xanthine dehydrogenase, XD)。这两种酶主要存在于毛细血管内皮细胞内。正常时只有10%以XO的形式存在,90%为XD。缺血时由于ATP减少,膜泵功能失灵,Ca2+进入细胞激活Ca2+依赖性蛋白水解酶,使XD大量转变为XO。缺血时ATP不能用来释放能量,而且还依次降解为ADP、AMP和次黄嘌呤,故在缺血组织内次黄嘌呤大量堆积。再灌注时,大量分子氧随血液进入缺血组织,黄嘌呤氧化酶在催化次黄嘌呤转变为黄嘌呤并进而催化黄嘌吟转变为尿酸的两步反应中,都同时以分子氧为电子接受体,从而产生大量的O2-和H2O2,后者再在金属离子参与下形成OH·。因此,再灌注时组织内O2-、OH·等氧自由基大量增加。
2.中性粒细胞 中性粒细胞在吞噬活动时耗氧量显著增加,所摄取的O2绝大部分经细胞内的NADPH氧化酶和NADH氧化酶的作用而形成氧自由基,并用以杀灭病原微生物。如氧自由基产生过多或机体清除氧自由基的酶系统活性不足或抗氧化剂不够时,中性粒细胞形成的氧自由基就可损害组织。
在再灌注时,由黄嘌呤氧化酶的作用所产生的氧自由基起原发的、主要的作用;这些自由基作用于细胞膜后产生的具有趋化活性的物质如LTB4等可吸引大量中性粒细胞到局部释放氧自由基等物质而进一步损害组织。
3.线粒体 可能是由于缺氧使ATP减少,Ca2+进入线粒体增多而使线粒体功能受损,细胞色素氧化酶系统功能失调,以致进入细胞内的氧,经单电子还原而形成的氧自由基增多而经4价还原而形成的水减少。细胞色素氧化酶的功能失调,也可能是缺氧时细胞内氧分压降低的结果。
4.儿茶酚胺的增加 交感-肾上腺髓质系统是机体在应激时的重要调节系统。在各种应激包括缺氧的条件下,此系统分泌大量的儿茶酚胺,儿茶酚胺一方面具有重要的代偿调节作用,但过多的儿茶酚胺特别是它的氧化产物,往往又成为对机体的有害因素。实验证明,大量的异丙肾上腺素、去甲肾上腺素、肾上腺素均能引起细胞损伤。造成心肌损害的是儿茶酚胺的氧化产物,而非儿茶酚胺本身。儿茶酚胺氧化能产生具有细胞毒性的氧自由基。肾上腺素代谢产生紧上腺素红的过程中有O2-产生。
(二)自由基反应与再灌注损伤
机体在生命过程中所能遇到的自由基种类很多。因此很难概括其生物学反应。自由基参与一个反应系统后能形成新的自由基。因此自由基一旦形成,就成为自由基反应扩展程序的一部分,例如:
R·+XH→RH+H·
R·+CCl4→RCl+Cl3C·
另一个自由基反应则是自由基加入到不饱和键中去。如脂肪酸及芳香族环的不饱和键。
自由基反应既可经自由基中间代谢产物不断向前发展,又可由细胞损伤而终止。自由基反应的扩展可以是无限的,但又可为各种自由基清除剂(free radical scavenger)所终止。
由于自由基有极为活泼的反应性,所以它们能和各种细胞成分(膜磷脂、蛋白、核酸)发生反应。
(1)膜脂:是构成膜脂质双层的重要结构及功能成分,富含不饱和脂肪酸,自由基与不饱和脂肪酸作用引发脂质过氧化(lipid peroxidation)反应。脂质过氧化物的形成使膜受体、膜蛋白酶和离子通道的脂质微环境改变,从而改变它们功能,由于脂质过氧化反应的增强,细胞膜内多价不饱和脂肪酸减少,生物膜不饱和脂肪酸/蛋白质比例失常,膜的液态性、流动性改变,通透性增强。含双键脂肪酸过氧化可生成丙二醛,它的产生与脂质过氧化相平行,因而测定丙二醛含量可代表脂质过氧化物的浓度。丙二醛能使膜成分之间形成交联和聚合(polymerization),使膜的基本特性如变构、离子传递、酶活性等发生改变(图11-3)。
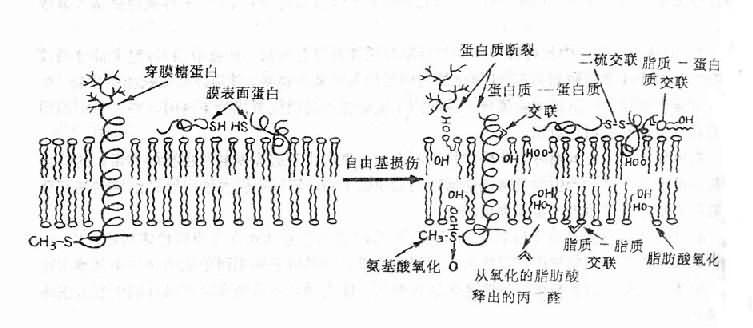
图11-3 自由基对膜的损伤
自由基可通过诱致过氧化而影响脂质,从而产生短链脂酰衍化物和副产物丙二醛。丙二醛反应可介导各种交联反应。自由基也能催化氨基酸氧化、蛋白质-蛋白质交联和蛋白质链的断裂
(2)蛋白质:在自由基的作用下,胞浆及膜蛋白及某些酶可交联成二聚体或更大的聚合物。这种交联既可借助于蛋白质之间的二硫键形成也可由于自由基损伤的氨基酸残基间的反应形成。蛋白质的交联将使其失去活性,结构改变。
(3)核酸:自由基对细胞的毒性作用主要表现为染色体畸变,核酸碱基改变或DNA断裂。80%是OH·的作用。OH·易与脱氧核糖及硷基反应并使其改变。
(三)细胞对自由基损伤的防护
自由基的产生既然是有机体在正常或病理条件下的常见现象,因此在进化过程中也就形成了一系列对抗自由基,防止其损伤的系统。这种生化学防护系统主要有两大类:低分子自由基清除剂及复合酶系统。
1.低分子清除剂 存在于细胞脂质部分的自由基清除剂有维生素E(α-生育酚)和维生素A(β-胡萝卜素);存在于细胞内外水相中的自由基清除剂有半胱氨酸、抗坏血酸和谷胱甘肽等,它们能提供电子使自由基还原,故有重要的防护作用。例如维生素E(生育酚)能还原O2-、单线态氧(1O2)、过氧化脂质自由基等;抗坏血酸具有相同作用而且能协助生育酚维持其具有活性的还原状态。β-胡萝卜素是单线态氧(1O2)的有效清除剂并能抑制脂质过氧化。
胞浆中的还原型谷胱甘肽(GSH)与还原型辅酶Ⅱ(NADPH)在某些酶如过氧化氢酶、谷胱甘肽过氧化物酶等的协同作用下,能还原H2O2、过氧化脂质、二硫化物及某些自由基。
2.酶性清除剂 细胞内有二种酶可以清除H2O2,即过氧化氢酶和过氧化物酶。如所周知H2O2是OH·自由基的前身,上述两酶可使H2O2浓度降低,从而避免高毒性OH·的产生。这两个酶的细胞内分布,尚不清楚。
细胞内具有清除剂作用的另一个重要酶是超氧化物歧化酶(SOD)。它是一种金属蛋白,可以歧化O2-生成H2O2。哺乳类细胞含有两种SOD。其一是位于胞浆中的CuZn 超氧化物歧化酶,另一是位于线粒体中的Mn超氧化物歧化酶。SOD作用的重要意义,在于清除H2O2及OH·的前身O2-,从而保护细胞不受强毒性氧自由基的损伤。
清除剂的浓度及活性下降必将引起自由基所致之细胞损伤。
(四)自由基在缺血再灌注损伤机制中的地位
缺血/再灌注损伤机制中的各种学说,无一不与自由基的作用有关。
1.缺血/再灌注时脂质过氧化增强(自由基引发),组织及血浆中脂质过氧化物显着增高,超威结构严重受损。给予抗氧化剂如:维生素E、硒(谷胱甘肽过氧化物酶辅基所含金属)及SOD能显着减轻缺血/再灌注损伤。
2.细胞膜脂质过氧化改变膜酶、离子通道的脂质微环境,从而使膜通透性增高,细胞外钙离子内流。膜上Na+-K+-ATP酶失活,可使细胞内Na+升高,Na+-Ca2+交换增强。而使细胞内钙超载。
3.线粒体膜富有磷脂,缺血/再灌注时自由基引发的线粒体膜脂质过氧化或细胞内形成脂质过氧化物作用于线粒体膜,使膜的液态及流动性改变,从而导致线粒体功能障碍,高能磷酸化物产生减少,自由基产生增多。细胞丧失能量贮备。依靠能量的质膜及肌浆网膜钙泵,由于能量不足不能将肌浆中过多的Ca2+泵出或吸收入肌浆网,致使肌细胞内Ca2+浓度增加,加上由细胞外来的Ca2+终于造成细胞内Ca2+超载,成为细胞致死原因。
4.自由基引发的脂质过氧化造成细胞成分间的交联(脂质-脂质交联、蛋白-蛋白交联、脂质-蛋白交联、蛋白-胶原交联),使整个细胞丧失功能。
5.缺血/再灌注时,微粒体及质膜上的脂加氧酶(lipoxygenase)及环加氧酶(cyclooxygenase)激活,催化花生四烯酸代谢,在加强自由基产生及脂质过氧化的同时形成具有高度生物活性的物质,如前列腺素、血栓素等。很多实验证明,缺血特别是再灌注时血栓素形成增加,前列环素形成减少,因而造成微循环障碍,出现无复流现象。
总之,自由基即使不是缺血/再灌注损伤的唯一发病学因素,至少也是甚为重要的环节。
第四节 防治缺血/再灌注损伤的展望
迄今为止,对于缺血/再灌注损伤的研究,绝大部分是在实验动物身上进行的,然而这些实验资料却已经为缺血/再灌注损伤的临床防治提供了重要的启示和借鉴,为临床研究奠定了重要的基础。目前,值得临床上参考的防治措施有:
1.尽早恢复血流,尽量减少缺血时间。
2.注意再灌注时的低流、低压、低温。低流低压的意义在于使灌注氧的供应不至突然增加而引起大量氧自由基的形成;低温则是使缺血器官代谢降低,代谢产物聚积减少。
3.缺血组织有氧代谢代下,酵解过程增强,因而补充糖酵解底物如磷酸已糖有保护缺血组织的作用;外源性ATP作用于细胞表面与ATP受体结合,或使细胞膜蛋白磷酸化,有利于细胞膜功能恢复,并可穿过细胞膜进入细胞直接供能;针对缺血时线粒体损伤所致的氧化磷酸化受阻,可以应用氢醌、细胞色素C等进行治疗,以加强NAD-黄素蛋白-细胞色素链的功能,延长缺血组织的可逆性改变期限。实验证明,细胞色素C能增加线粒体的ADP磷酯化;醌类化合物则能加速电子传递或将电子直接传递给氢。
4.清除自由基实验证明,外源性SOD、黄嘌呤氧化酶抑制剂别嘌呤醇(allopurinol)、维生素E、维生素C、过氧化氢酶、二甲基亚砜(dimethylsulfoxide, DMSO)等自由基清除剂对缺血/再灌注损伤的心肌有防护作用。
例如,外源性SOD能显著地降低缺血所致的血管通透性增高。预先用SOD给动物作静脉内注射,然后进行肠管缺血实验,则血管通透性无大变化。
又如,在氧自由基中起主要作用的是继发于O2-的OH·。如预先用OH·清除剂DMSO处理肠管,可以明显地减轻缺血所致的血管通透性增高。
第十二章 心力衰竭及高血压
心力衰竭(rhertfailure)是指由于心脏泵功能障碍,以致心输出量减少,不足以适应全身组织代谢需要的一种病理过程。心力衰竭亦称泵衰竭(prmp failure)。在这一概念中可以包括两种情况:①心肌衰竭(myocardial failure):是指原发性心肌肌原纤维收缩功能障碍所致的心力衰竭,此时泵功能障碍是原发的。如心肌炎时,心肌的变质、渗出或结缔组织增生可使心肌收缩性明显减弱。又如心肌梗死时可因部分心肌坏死而致心肌收缩性减弱等等。②其他原因所致的心力衰竭:如心脏瓣膜病时,由于心肌负荷过重而发生心肌肥大和心脏扩大,继则心肌收缩性相对不足而导致心力衰竭,此时泵功能障碍是继发的,在除去瓣膜障碍时较易逆转。但心肌以外的原因引起的心力衰竭,在晚期往往也伴有心肌损害,故在临床上有时两者不易区分。
应当指出,有人把心脏受压(如心包填塞)等原因引起的心脏泵血减少也包括在心力衰竭或泵衰竭的概念中。但是,由于引起心脏泵功能障碍最常见的原因是心肌收缩性减弱,故本章主要论述因原发性或继发性心肌收缩性减弱而发生的心力衰竭。
心力衰竭时,由于心输出量不能与静脉回流相适应,故血液可在静脉系统中淤积。当心力衰竭呈慢性经过时,往往伴有血容量和组织间液的增多,并出现水肿,临床上称之为充血性心力衰竭(congestive heart failure)。急性心力衰竭多见于原发性心肌病变时,慢性心力衰竭患者如心脏负荷突然加重,也可发生急性心力衰竭。
心功能不全(cardiacinsufficiency)与心力衰竭本质上是相同的,只是在程度上有所区别:心力衰竭一般是指心功能不全的晚期,患者有明显的心力衰竭的临床症状,而心功能不全则指病情从轻到重的全过程,包括没有心力衰竭症状的心功能不全代偿阶段。但是,在实际应用中,这两个概念往往又是通用的。
第一节 心力衰竭的原因和诱因
心力衰竭在临床上十分常见。它可既有心脏本身的疾病引起,也可继发于某些心外疾病如甲状腺功能亢进症,维生素B1缺乏等等。心力衰竭的病因可以概括为下述三类:
一、心脏负荷加重
心脏的负荷可分为前负荷和后负荷两种,前负荷(preload)或容量负荷是指心脏在收缩之前所承受的负荷,相当于心脏舒张末期的容量,前负荷的大小决定了心肌收缩的初长度,后负荷(afterload)或压力负荷是指心腔在收缩时所必须承受的负荷,相当于心腔壁在收缩时的张力,但一般常以主动脉压作为左心室后负荷的指标。心脏负荷过重是心力衰竭的常见原因。例如,在主动脉瓣狭窄,高血压病或肺动脉高压时,心脏后负荷过重,即压力负荷过重,即前负荷(容量负荷)过重、久之也能导致心力衰竭。在动静脉瘘或严重贫血时,心输出量长期增多,回心血量增多,故心脏所受的容量负荷也过重,因而也可引起心力衰竭。在这种情况下,一般都先发生心肌肥大等代偿适应性变化,从而使心功能可长期处于相对正常状态,最后则向代偿不全转化,而出现心力衰竭。
二、心肌代谢障碍
心肌对氧的需求量很大,必须有充分的血液和氧的供给才能保持其正常功能。因此在严重或长期的缺血、,缺氧时可发生心力衰竭。心肌供血不足最常见的原因是冠状动脉粥样硬化。此时,由于冠脉血流量减少,病变部位心肌供血相对或绝对不足,故心肌收缩性可逐渐减弱而导致心力衰竭。冠状动脉粥样硬化所引起的急性心肌梗死,也是心力衰竭的重要原因。在高血压病时,心肌代偿性肥大所致的心肌供血相对不足可能也是引起心力衰竭的因素之一。
此外严重的贫血和维生素B1缺乏,也可分别引起心肌供氧不足和生物氧化过程的障碍,从而也可导致心力衰竭(详后文)。
三、弥漫性心肌病
心肌炎,退行性心肌病等原发性心肌病变时,可因肌原纤维受到损害而使心肌收缩性减弱。如果损害严重或发展迅速,可导致急性心力衰竭(如急性心肌炎时);若损害较轻,或病变呈慢性经过时,则对损害的反应是心肌肥大等代偿适应性变化,因而在相当一段时间内心功能可处于相对正常状态,但在一定条件下,如在某些诱因的作用下,代偿状态可转向代偿不全而发生心力衰竭。
心力衰竭的诱因:促使心力衰竭发生的诱因很多。这些诱因基本上都是使心肌耗氧增加或供氧(供血)减少的因素,如感染(尤其是肺部感染)、体力负荷过重、妊娠、分娩、情绪激动、心率过快或过漫、血压过高或一时性降低、输液过多等都可促使代偿失调而导致心力衰竭。
第二节 心力衰竭的分类
心力衰竭的病因繁多,分类标准不一,常用的有以下几种分类法:
一、根据心脏的受损部位分类
(一)左心衰竭主要是左心室搏出功能障碍,多见于冠状动脉粥样硬化性心脏病(冠心病)高血压病、主动脉瓣狭窄或关闭不全、二尖瓣关闭不全等。机体的病理变化是由心输出量减少以及肺部淤血、水肿所引起。
(二)右心衰竭主要是右心室搏出功能障碍,见于肺心病、三尖瓣或肺动脉瓣的疾病,并常继发于左心衰竭。此时心输出量减少,体循环淤血,静脉压增高,常伴有下肢水肿,严重时可发生全身性水肿。
(三)全心衷竭左、右心都发生衰竭称为全心衰竭,见于:①持久的左心衰竭可使右心负荷长期加重而导致右心衰竭;②心肌炎、心肌病等病变如发生于全心、亦可引起全心衰竭。
二、根据发病的速度分类
(一)急性心力衰竭发病急骤。心输出量急剧减少,机体来不及充分发挥代偿作用。常可伴有心源性休克。常见原因为急性心肌硬死,严重的心肌炎等。
(二)慢性心力衰竭较常见,病人长期处于一种持续的心力衰竭状态,并伴有静脉淤血和水肿。常见原因为心脏瓣膜病、高血压病、肺动脉高压等。
三、根据心力衰竭时心输出量的高低分类
(一)低心输出量性心力衰竭 常见于冠心病、高血压病、心肌病、心脏瓣膜病等。此种病人在基础状态下心输出量就低于正常。
(二)高输出量性心力衰竭 继发于代谢增高或心脏后负荷降低的疾病如甲状腺机能亢进症、贫血、维生素B1缺乏病(脚气病)和动静脉瘘等。在此种情况下,由于循环血量增多或循环速度加快,心室前负荷增加,心输出量代偿性地增高,心脏必须作更多的功。但心肌的能量供给却不足,故容易导致心力衰竭。发生心力衰竭时心输出量比心力衰竭以前有所降低,但可稍高于正常水平。然而,由于组织需氧量增高、外周血管扩张、动静脉短路等原因,这些病人的心输出量虽可比正常水平稍高。但组织的供氧量仍然不足。
第三节 心功能不全发病过程中机体的代偿活动
心肌受损或心脏负荷加重时,体内出现一系列的代偿活动,通过这些代偿活动可使心血管系统的功能维持于相对正常状态。若病因继续作用,则经过相当时间,在一定条件下代偿状态可以向失代偿状态转化而出现力心衰竭。
虽然机能代谢和形态的代偿是密切地相互联系的,但为了理解的方便,可把代偿分为以机能、代谢为主的代偿和以形态结构为主的代偿两个方面。
一、机能和代谢的代偿
正常人在运动或劳动时,才动员心血管系统的代偿活动。而心脏病患者则在基础情况下就需要动员这种代偿活动。能在短时间内被动员起来的代偿活动,主要属于机能和代谢的代偿,有以下几种形式:
(一)通过紧张源性扩张使心输出量增加
正常心脏在回心血量增加,由于心室舒张末期容积及压力增加,心肌初长度增大,心室发生紧张源性扩张,按照Frank-Starling定律,此时心肌收缩力加强,心输出量增加。心功能不全时,由于心泵功能减弱,心输出量减少,故心室舒张末期容积增加,心肌初长度增大;如肌节长度不超过2.2μm,则这种紧张源性扩张也使心肌收缩力有所加强而起到代偿作用。
(二)通过心交感神经和肾上腺髓质释放儿茶酚胺增加使心肌收缩性加强、心率加快
心功能不全病人的交感神经系统活动加强。体力活动增加时血浆中去甲肾上腺素的含量比正常人有较明显的增多;24小时尿中去甲肾上腺素的排出量也显著地高于正常人的排出量,说明心功能不全病人在休息时儿茶酚胺的分泌也是增加的。交感-肾上腺髓质系统的兴奋在维持功能不全的心脏收缩性上起一定作用,另一方面也是心功能不全病人心率加快的基础。
一定程度的心率加快是最容易被迅速动员起来的一种代偿活动。正常人可通过心率加快使心输出量增加数倍。心功能不全时心率加快也是一种重要的代偿形式,借此可使心输出量维持在一定的水平。但心率加快(如超过每分150~160次)时则由于心舒张期缩短,心肌耗氧量过大,故每搏输出量明显减少,甚至因每分输出量减少而失去代偿意义。
(三)心外的代偿
1.心输出量不足时交感-肾上腺系统兴奋,外周小动脉的紧张性增加,有利于动脉血压维持在正常范围内。同时由于肾血流减少,肾素-血管紧张素-醛固酮系统被激活,从而导致体内钠、水潴留,使血容量增加,这对维持动脉血压,也起一定作用。
2.组织利用氧的能力增加心功能不全患者因血流变慢而发生循环性缺氧。与此同时,组织、细胞中线粒体的呼吸酶活性增强,在慢性缺氧时,细胞内线粒体的数量还可增多,因而组织利用氧的能力增强。
3.红细胞增多缺氧又可使血液细胞数和血红蛋白量增多。红细胞增多可提高血液携氧的能力,同时又有助于增加血量,故具有代偿意义。
二、形态结构的代偿-心肌肥大
在心脏负荷过重或心肌受损的初期,首先出现的主要是机能和代谢的代偿,但与此同时也开始出现另一种代偿形式,即心肌形态结构的代偿,表现为心肌肥大。
心肌肥大主要是心肌细胞体积增大的结果。心肌细胞一般不发生增生,但有人报道,当心脏重量超过500g左右时,心肌纤维也可有数量的增多。单位重量肥大心肌的收缩性是降低的,但由于整个心脏的重量增加,所以心脏总的收缩力加强,因此肥大心脏可以在相当长时间内处于功能稳定状态,使每搏和每分输出量维持在适应机体需要的水平,使病人在相当长时间内不致发生心力衰竭。所以心肌肥大是心血管系统疾病时起重要作用的一种代偿形式。
心肌肥大的发生机制至今尚未完全阐明。动物实验表明,在实验性主动脉狭窄时,几秒钟内就可出现快速的适应性反应,表现为心肌收缩加强。与此同时,核酸和蛋白质合成加强。主动脉狭窄后几天内心肌RNA含量可增加30~40%,1g心肌中蛋白质含量每小时可增加1mg,所以心肌总量很快增长。
病理学家早就观察到,心肌肥大的两种形式:向心性肥大和远心性肥大(心脏扩大)。关于其发生机制,Grossman曾提出如下的假设:当心室受到过度的压力负荷时,收缩期室壁压力的增高可引起心肌纤维中肌节的并联性增生,使心肌纤维变粗(在心脏总重量超过一定的监界值时,心肌纤维的数量也可增多),室壁厚度增加,形成向心性肥大。这样,增厚了的心室壁可使收缩期室壁张力保持正常,使心输出量不致降低。当心室受到过度的容量负荷时,舒张期室壁张力的增加可引起心肌纤维中肌节的串联性增生,心肌纤维长度加大,心室腔因而扩大,即发生远心性肥大。这两种肥大在心脏功能的代偿上都起重要作用。
当心血管疾病呈慢性经过时,心肌肥大出现在心力衰竭之前。在相当长的时间内,如数年甚至数十年内,心肌肥大可以代偿心脏的过度负荷或心肌损害,使心功能处于代偿阶段。
若病因历久而未能消除,上述各种休偿仍不足以克服心功能障碍,则心输出量将显著减少而出现心力衰竭的临床症状,此时心脏已从代偿状态发展到失代偿状态。
第四节 心力衰竭的发病机制
心力衷竭的本质是心肌收缩性减弱。为了理解有关的问题,本节将首先简要地复习心肌收缩的分子生物学基础,然后讨论心力衰竭的一般发病机制,以及肥大心肌转向衰竭的机制。
心肌的收缩物质是组成粗、细肌丝的心肌蛋白。粗肌丝的主要成分是肌球蛋白(myosin),其分子量约50万,全长1500A,一端为杆状的尾部,另一端为粗大的头部(S1),二者之间是能弯曲的颈部(S2)。头部又分成两片,是ATP酶的活动中心,它在肌动蛋白和肌球蛋白之间的搭桥和粗细肌丝之间的滑行中起着重要作用。细肌丝的主要成分是肌动蛋白(actin),分子量47.000,分子呈球状,串联而成双链螺旋状的细肌丝纤维。在双链间的沟槽内,杆状的向肌球蛋白(tropmyosin)和肌动蛋白卷曲在一起,每距400A处还有一个肌钙蛋白(troponin)分子。向肌球蛋白和肌钙蛋白是调节蛋白,本身不起收缩作用,但能调节肌动蛋白与肌球蛋白的联结,而使心肌纤维发生收缩和舒张。肌钙蛋白由三个亚单位组成,即向肌球蛋白亚单位(tropotroponin,TnT)、抑制亚单位(inhibrtor troponin,TnI)钙结合亚单位(calciumcombining troponin, TnC)在心肌兴奋-收缩偶联中起重要作用(图12-1、2)。
图12-1 心肌收缩蛋白和调节蛋白
A.肌球蛋白分子结构模式图,说明见正文。
B.肌动蛋白分子呈球形,串联而成双链螺旋状的细肌丝。
向肌球蛋白在两个肌动蛋白链之间。
每隔400A有一肌钙蛋白复合体。
C.示粗、细肌丝在收缩与舒张时的相互关系。
Ca2+在心肌兴奋时的电活动与机械收缩之间起偶联作用。当心肌除极化时,Ca2+从细胞外转移到心肌细胞的胞质中,同时也从肌质网释放入胞质。因此胞质内Ca2+浓度升高(由10-8M升至10-5M)。此时肌钙蛋白的TnC即迅速与Ca2+结合。这种结合相继使TnC和TnI的构型发生变化,其结果是TnI从肌动蛋白移开。这种构型变化又可通过TnT影响向肌球蛋白的位置,使向肌球蛋白旋转到肌动蛋白两条螺旋状链的深沟中,从而使肌动蛋白的受点暴露而与肌球蛋白头部相接触,形成横桥。S1的ATP酶随即作用于ATP而释放能量,肌动球蛋白(actomyosin)乃发生收缩。心肌收缩后,由于Ca2+又重新移到细胞外及进入肌质网,胞质内Ca2+浓度又降至10-8M。此时,肌钙蛋白的TnC失去了Ca2+,TnC和TnI的构型恢复原状,故TnI又与肌动蛋白结合,进而通过TnT使向肌球蛋白从肌动蛋白的深沟中转移出来,而恢复原来的位置。于是肌动蛋白上的受点又被掩盖,肌动球蛋白重新解离为肌动蛋白和肌球蛋白,横桥解除,心肌乃舒张。
图12-2 Ca2+与肌钙蛋白结合引起心肌收缩的示意图
在舒张期(左),肌钙蛋白复合体三成分(I、C、T)使向肌球蛋白
(Tm)位于细肌丝(A)螺旋沟的外侧,从而阻止细肌丝与肌球蛋白
横桥(M)发生作用。当Ca2+与肌钙蛋白C(右)结合引起一系列的
活动使I与细肌丝分开、从而使Tm移进细肌丝的螺旋沟中,细肌丝乃得
以与肌球蛋白横桥相互作用而引起心肌收缩。
下文将着重讨论慢性心力衰竭的发病机制。
从心肌分子结构及兴奋收缩偶联过程的基础出发,目前认为心肌负荷过重和心肌受损等病因引起心肌收缩性减弱的一般机制大致有下述几个方面。
一、心肌能量代谢障碍
(一)能量生成(释放)障碍
心肌主要借各种能源物质包括脂肪酸,葡萄糖等的有氧氧化而获得能量。心肌细胞对氧的需要量很大,摄取能力很强,在正常安静情况下,冠状动静脉血氧含量差可高达14ml%。可见,心肌氧供给不足或有氧氧化过程的障碍,均可使心肌细胞内能量生成不足而导致心肌收缩性减弱。
严重的贫血、冠状动脉硬化等所引起的心肌缺氧,是导致心肌细胞内能量生成不足的常见原因。维生素B1缺乏时,由于焦磷酸硫胺素(丙酮酸脱羧酶的辅酶)生成不足,丙酮酸的氧化发生障碍,故也可引起心肌能量生成不足。肥大的心肌也可因心肌缺氧而导致能量生成不足(详后文)。
(二)能量利用障碍
心肌细胞内氧化磷酸化过程中所产生的ATP,在心肌兴奋-收缩偶联过程中受到肌球蛋白头部ATP酶的作用而水解,为心肌收缩提供能量。实验表明,部分动物的心肌由肥大转向衰竭时,心肌耗氧量和ATP含量并不减少而完成的机械功却显著减少,说明心肌利用ATP中的化学能作机械功的过程有障碍,即心肌的能量利用发生障碍。有人发现,随着心肌负荷过重而发生心肌肥大时,心肌收缩蛋白的结构发生变化,肌球蛋白头部ATP酶的活性降低,ATP水解发生障碍,因此能量利用发生障碍,心肌收缩性乃因而减弱。这种现象也可见于老年人及甲状腺功能低下的心脏。关于心肌收缩蛋白质结构发生变化的机制尚未阐明。
二、兴奋-收缩偶联障碍-Ca[SB]2+[/SB]的运转失常
近年来,在心力衰竭的发病机制中,因Ca2+运转失常引起的心肌兴奋-收缩偶联障碍,受到了很大重视。正常心肌在复极化时,心肌细胞内肌质网的ATP酶(钙泵)被激活,从而使胞质中的Ca2+逆着浓度差被摄取到肌质网中储存;同时,另一部分Ca2+则从胞质中被转运到细胞外。于是心肌细胞胞质Ca2+浓度降低。心肌舒张。心肌除极化时,肌质网向胞质释放Ca2+,同时又有Ca2+从细胞外液进入胞质,因而胞质中Ca2+浓度增高,心肌收缩。心肌兴奋-收缩偶联障碍的发生机制主要有:
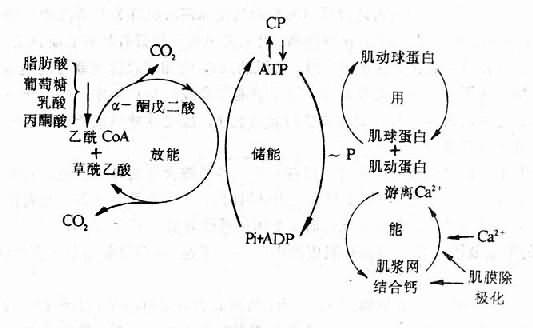
图12-3 心肌能量代谢
(一)肌质网摄取Ca2+减少有人发现,在过度肥大的心肌中,肌质网ATP酶的活性降低,因而在心肌复极化时肌质网摄取和储存Ca2+的量减少,除极化时肌质网向胞质释放的Ca2+也因之减少。由此所引起的心肌细胞除极化时胞质内Ca2+浓度的低下可能是心肌收缩性减弱的重要原因。
另据报道,在肌质网摄取Ca2+减少的同时,线粒体对Ca2+摄取量增多,但线粒体在心肌除极化时向胞质释放Ca2+的速度却非常缓慢。Ca2+在心肌细胞中这种异常的分布也是胞质Ca2+浓度降低的一个原因。此外,还有人认为线粒体内Ca2+的增多可引起氧化磷酸化脱偶联,从而使能量生成不足。
(二)酸中毒和高钾血症 Ca2+的运转也受H+和K+的影响。在心力衰竭时有一定程度的缺氧,故可有细胞外液H+和K+浓度的增高。关于H+如何影响运转的问题也未完全清楚。不同的作者提出过不同的假设:Katz等曾认为H+能在肌钙蛋白上与Ca2+竞争结合位置,因而在H+过多时就能取代Ca2+的位置而使心肌的兴奋-收缩偶联发生障碍。也有人报道,在H+浓度增高时,Ca2+与肌质网的结合比较牢固,除极化时肌质网释放Ca2+减少,故H+增多时,心肌的兴奋-收缩偶联发生障碍。前文已经提到,正常心肌细胞在除极化时有Ca2+从细胞外液进入胞质,而最近有人报道,在细胞外液H+浓度增高时,Ca2+的内流减慢,故心肌兴奋-收缩偶联发生障碍。
细胞外液中的K+和Ca2+在心肌细胞上有互相竞争的作用。当外液中K+浓度升高时,动作电位中Ca2+内流就减少,因而心肌胞质中Ca2+浓度降低,这也是引起心肌兴奋-收缩偶联障碍的一个因素。
(三)心肌内去甲肾上腺素含量减少有人报道,从心力衰竭患者取得的心房活组织中。去甲肾上腺素的含量很低:从有严重心力衰竭而行二尖瓣置换术的患者取得的心室乳头肌活组织中,去甲肾上腺素的含量也很少,在有的病人,含量仅为正常的10%。心力衰竭时心肌去甲肾上腺素含量减少,可能是由于肥大而衰竭的心肌中酪氨酸羟化酶活性降低,因而心肌交感神经纤维中儿茶酚胺的合成减少所致。在正常情况下,去甲肾上腺素与心肌细胞表面的β受体结合后,通过激活腺苷酸环化酶,可使心肌细胞内的ATP转变为cAMP,cCAMP一方面能促使Ca2+内流,另一方面又可通过蛋白激酶的活化而使心肌细胞肌质网的一种蛋白磷酸化,从而使肌质网摄取和释放Ca2+的速度增高。可见去甲肾上腺素有加强心肌兴奋-收缩偶联的作用,而心肌内去甲肾上腺素含量减少时,心肌的兴奋-收缩偶联过程就可能发生障碍。肥大而衰竭的心肌内去甲肾上腺素含量减少,除了可能与合成减少有关外,还可能是由于消耗过多。这是因为心输出量减少时,交感神经的活动加强,故交感神经末梢包括心肌交感神经末梢释放去甲肾上腺素增多。
此外,有些事实还说明心力衰竭时不仅有心肌儿茶酚胺含量的减少,而且心肌细胞肾上腺能受体的功能也发生了改变,例如在严重心力衰竭时,β受体受异丙基肾上腺素作用时细胞内不能产生正常量的cAM,提示可能此时β受体敏感性降低,因而cAMP产生不足。如前所述,cAMP的不足就可使Ca2+内流和肌质网摄取Ca2+不足,从而导致心肌兴奋-收缩偶联障碍。
在这方面,也有一些不同的见解。有人认为,虽然心力衰竭时心肌儿茶酚胺含量减少,但因心力衰竭患者交感神经活动加强,故血液中儿茶酚胺显著增多,而心肌对儿茶酚胺的敏感性也较高,故完全可能弥补心肌中儿茶酚胺的不足。
三、心肌的结构破坏
严重缺血时的心肌坏死,以及急性炎症时的心肌变性,坏死等可导致心肌收缩蛋白大量破坏,从而引起心肌收缩性显着减弱。
关于肥大心肌转向衰竭的机制,前文已经有几处提到。为了加深理解,下文将再就此问题作综合性的概述。
心肌肥大是一种强有力的代偿形式,然而它不是无限度的,如果病因历久而不能被消除,则肥大心肌的功能便不能长期维持正常而终转向心力衰竭。慢性心力衰竭一般都是在心肌代偿性肥大的基础上逐渐发生发展的。
肥大的心肌为何会转向衰竭?这是长期以来为人们进行探讨和研究的问题。目前认为,代偿性心肌肥大是一种不平衡的生长形式。这种在器官、组织、细胞、分子等不同的水平上都有其特征性表现的不平衡生长,是肥大心肌转向功能不全的基础。
(一)器官水平上的特征从整个心脏来看,不平衡生长表现为心脏重量的增长超过了支配心脏的交感神经元轴突的生长,因此心脏内交感神经分布的密集程度显著地低于正常。而且,肥大心肌中儿茶酚胺合成减少而消耗增多,因而心内去甲肾上腺素含量显著减少,这种神经支配和递质含量方面的变化,就会促使心肌兴奋-收缩偶联发生障碍,从而导致心肌收缩性减弱。
(二)组织水平上的特征表现在心肌内微动脉和毛细血管的生长明显地落后于心肌细胞体积的增长,所以单位重量的肥大心肌毛细血管数目减少。对哺乳类动物心肌微循环的活体组织研究表明,安静时,正常动物心肌每1mm3内约有2,300条开放的毛细血管,毛细血管平均间距为16.8μm。当心脏负荷加重或缺氧时,毛细血管前括约肌松弛,原处于贮备状态的约2,100条毛细血管也开放。这样,总的功能性毛细胞管可达4.400条/mm3,毛细血管间距因而减少到5.5μm。因而,由于负荷加重而增高的心肌需氧量,很快通过原运输加快而得到满足。然而在心肌肥大时,则因毛细血管总数相对减少,氧的弥散间距增大,故心肌缺氧。这样的患者在安静的状态下,大部分贮备毛细血管已经开放,故当负荷增加时,功能性毛细血管数不能再有显著的增加,氧的弥散间距也不能明显缩小。因此肥大心肌在负荷增加时常处于缺氧状态,致有氧代谢减弱,能量生成不足,心肌收缩性减弱。
关于肥大心肌是否缺氧的问题,也曾有过不同的见解,因为有人曾经测定过单位重量肥大心肌冠状动、静脉血液氧含量差,结果与正常心肌的情况无明显的差别,指示肥大心肌并不缺氧。
(三)细胞水平上的特征表现为细胞体积和重量的增加大于其表面积的增加,即肥大心肌的表面积与重量之比显著降低。而细胞表面的胞膜(sarcoplasmic membrane)正是Na+-K+、Na+-Ca2+等离子转运所必经的部位。故细胞面积的相对减少可使细胞转运离子的能力减弱,包括Ca2+内流相对不足,从而使心肌细胞的功能降低。近年来电子显微镜的观察还证实、肥大心肌内线粒体数量与心肌细胞体积的比值减小,线粒体膜表面积与心肌纤维重量的比值也明显减少,所以肥大心肌内生物氧化作用相对减弱。这也是肥大心肌能量生成不足的原因之一。
(四)分子水平上的特征 表现为肌球蛋白分子的重节片(头部)和轻节片(尾部)的比值降低,即头部在整个分子中所占的比重减少。而头部正是ATP酶所在的部位,头部比重的减少,就可使ATP酶的活性随之相对降低。此外,ATP酶又受Ca2+的激活,心力衰竭时,由于Ca2+向肌球蛋白横桥部位转运缓慢,故可使ATP酶活性进一步降低。体外实验表明,衰竭心肌中ATP酶的活性约降低20~30%。ATP酶活性的降低使心肌能量利用发生障碍,因而心肌收缩性减弱。
应当强调指出,临床上心力衰竭的发生发展,往往是多种机制共同作用的结果。例如,贫血和维生素B1缺乏主要引起心肌能量生成障碍,但当心肌因负荷加重而代偿性肥大,即发生心肌的不平衡生长时,又可发生心肌能量利用障碍和兴奋-收缩偶联障碍。高血压病慢性心瓣膜病引起心肌肥大时,固然以兴奋-收缩偶联障碍和能量利用障碍为主,然而在高度肥大的心肌中也可能存在着相对的缺血缺氧,因而也可有能量生成障碍。
第五节 心力衰竭时机体的机能和代谢变化
心力衰竭时机体一系列机能代谢变化的根本原因在于心脏泵功能低下,其结果是心输出量减少,动脉系统充盈不足,静脉系统血液淤滞,于是各器官组织血流量不足,发生淤血、水肿和缺氧,并从而引起器官功能障碍和代谢紊乱。
一、心血管系统的变化
(一)心功能的变化
是心力衰竭时最根本的变化,主要表现为心脏泵功能低下,从而可引起一系列血液动力学的变化。通常用于评价心脏泵功能的指标都发生显着的改变:
1.心输出量减少 心力衰竭时每搏及每分心输出量均降低。正常人心输出量(cardiac output, CO)为3.4~5.5L/min,心力衰竭时往往低达2.5L/min以下(指低输出量心力衰竭)。
2.心脏指数降低 心脏指数(cardiac index, CI)是单位体表面积的每分心输出量(CO/m2),正常值为2.5~3.5L/min/m2,心力衰竭时心指数降低,多数在2.5L/min/m2,但在此种情况下,往往由于组织代谢率升高、血液加快等原因,这样的心指数仍嫌相对不足。
3.射血分数降低 射血分数(ejection fraction, EF)是每搏输出量(stroke volume, SV)与心室舒张末期容积(ventricular enddiastolic volume, VEDV)的比值,正常为0.56~0.78。心力衰竭时,由于心肌收缩性减弱,每搏输出量减少。因而心室收缩末期余血较多,心室舒张末期容积也必然增大,故EF降低。
4.心肌最大收缩速度减低 心肌最大收缩速度(Vmax)是指负荷为0时的心肌最大收缩速度,须通过左室压力动态变化所投影的图来计算,测量比较复杂。但它能更准确地反映心肌的收缩性,因为上述CO、CI及EF等指标明显地受负荷状态的影响,不能独立反映心肌的收缩性。
5.心室dp/dt max减少 心室dp/dt max(ventricular dp/dt maximum)表示心室内压力随时间的最大变化率,也即心室内压力上升的最大速度,可反映心肌的收缩性,此值可以在一般多导仪上通过记录心室内压而得出,心肌收缩性减弱时此值减小。
6.心室舒张末期容积增大、压力增高 心力衰竭时心室舒张末期容积(VEDV)增大。垠据Frank-Starling关于长度-张力相关的定律,在一定范围内,心肌初长度的增大可使心肌收缩性加强,表现为每搏输出量增加或搏出功(stroeke work)增大。这个关系可用心功能曲线来表示(图11-4)。在正常情况下,VEDV与心室舒张末期压力(ventricrlarend diastolic pressure, VEDP)大致相当。临床上测定VEDP又比较方便,故常用VEDP来对心肌初长度或VEDV作大致的估计。由此可见,心室的搏出功与VEDP也密切相关:在一定范围内,VEDP的升高伴有心室搏出功的相应增加。但当VEDP达到一定临界水平(例如左心室舒张期末压力LVEDP为3.2kPa(24mmHg),心肌肌节长度为2.2μm)以后,VEDP的进一步升高仅会使搏出功降低。VEDV与VEDP的关系,在正常人和心力衰弱的病人也有所不同。在正常人,由于其心肌顺应性(compliance)正常,故VEDV一定程度的增大不致引起VEDP的急剧升高。在慢性心力衰竭的病人,心肌肥大可使心肌顺应性减低,故VEDV的增大可使VEDP显著增高。LVEDP的正常值在2kPa(15mmHg)以下,左心衰竭时,LVEDP可达2.67kPa(20mmHg)或更高。VEDP愈高,心室肌张力愈大,能量消耗愈多。对于已经衰弱的心肌来说,VEDP的这种增大不能使搏出功增加,反而能使搏出功和每搏输出量减少。
7.肺动脉楔压升高 肺动脉楔压(pulmonary artery wedge pressure, PAWP),也称肺毛细血管楔压(Pulmonary capillarp wedge pressune PCWP)是用漂浮导管通过右心进入肺小动脉末端而测出的。PAWP接近左房压和LVEDP,可以反映左心功能。正常值为0.93kPa(7mmHg)(平均压),左心衰竭时由于LVEDP异常升高,PAWP也明显高于正常。
(二)动脉血压的变化
急性心力衰竭(如何见于急性心肌梗死)时,由于心输出量急剧减少,动脉血压可以下降,甚至可以发生心源性休克。但在慢性心力衰竭时。机体可通过窦弓反射使外周小动脉收缩和心率加快,以及通过血量增多等代偿活动,使动脉血压维持于正常水平。动脉血压的维持正常有利于保证心、脑的血液供应(详后文),故无疑有重要的代偿意义;然而,外周阻力的增高使心脏的后负荷加重,心率加快使心肌的耗氧量增多,血量的增多又使心脏的前负荷加重,这些又是对机体不利的。
(三)器官、组织血流量的改变——血液重分布
心输出量的减少,可使动脉系统充盈不足,同时又通过窦弓反射引起外周小血管收缩,故可使器官组织的血液量减少。由于各脏器的血管对交感神经兴奋的反应不一致,因而发生血液的重分布。由表11-1可见,心力衰竭时,肾脏的血流量减少最显著,其次是皮肤和肝脏等。在重度心力衰竭,肾血流量的减少可使肾小球滤过率减少30~50%。正常人在运动时器官血液量一般都有增加或不减少,而心力衰竭患者在运动时肾、肝的血液量比在安静时更进一步地明显减少。由于交感神经兴奋时脑血管并不收缩而冠状血管反有所舒张,故脑和心脏的血液供应可不减少(指慢性心力衰竭患者动脉血压正常时)。这种血液的重分布具有重要的代偿意义。
表12-1 正常人和心力衰竭患者的心输出量及器官血流量(L/min)
正 常 安 静 运 动 |
心力衰竭 安 静 运动 |
||||
| 心输出量 | 5.4 | 12.5 | 3.9 | 8.0 | |
| 肾血流量 | 1.0 | 1.0 | 0.45 | 0.25 | |
| 肝血流量 | 1.5 | 1.5 | 1.0 | 0.7 | |
| 脑血流量 | 0.8 | 0.8 | 0.8 | 0.8 | |
| 骨骼肌血流量 | 0.9 | 6.5 | 0.8 | 4.4 | |
| 冠脉血流量 | 0.3 | 1.2 | 0.45 | 1.35 | |
| 皮肤脂肪等血流量 | 0.9 | 1.5 | 0.45 | 0.5 | |
(四)淤血和静脉压升高
心力衰竭时,由于钠、水潴留使血量增加,又因有关心腔舒张末期容积增大和压力升高以致静脉回流发生障碍,故血液在静脉系统中发生淤滞,并从而使静脉压升高。使静脉压升高的另一原因是交感神经的兴奋,因为交感神经兴奋时,不仅小动脉发生收缩,而且小静脉也发生收缩。
左心衰竭引起肺淤血和肺静脉压升高,肺泡毛细血管压亦随之升高。严重时可导致水肿。肺淤血和肺水肿可引起呼吸困难、两肺出现湿罗音、咳粉红色泡沫痰甚至咯血等临床症状和体征。右心衰竭引起体循环淤血和静脉压增高。体循环淤血是引起许多器官功能代谢变化的重要原因。此外,淤血和静脉压升高也是引起心性水肿的重要原因之一。
二、肺呼吸功能变化
肺呼吸功能的改变主要是左心衰竭时出现的呼吸困难(dyspnea)。呼吸困难是一种主观的感觉。正常人在剧烈运动时可以发生呼吸困难,即首先是感觉到自已在呼吸,然后是感觉到呼吸费力,伴有“喘不过气”等一系列不适的感觉,左心衰竭较轻时,患者只是在体力活动时发生呼吸困难,称为劳力性呼吸困难;严重时患者在安静情况下也有呼吸困难,甚至不能平卧,必须采取坐位才能减轻呼吸困难。这就是所谓端坐呼吸(orthopnea)。而且,患者还可以发生夜间阵发性呼吸困难(Paroxysmalnocturnal dyspnea)。
左心衰竭时呼吸困难,是由肺淤血、水肿所引起,因为肺淤血、水肿时:①肺的顺应性降低,因而要吸入正常时同样量的空气,就必须增大胸廓运动的幅度,也就是呼吸时作功和耗能增大,患者感到呼吸费力,即出现了呼吸困难。②肺血管感受器受剌激,经迷走神经传入而使呼吸中枢兴奋,因而呼吸运动增强,患者感到呼吸费力。③肺淤血水肿时,支气管静脉内血液含量增多,因而支气管粘膜肿胀,呼吸道阻力增高,患者感到呼吸费力。④肺泡毛细血管与肺泡间气体交换障碍,动脉血氧分压可以降低,从而反射性地引起呼吸中枢兴奋。而且,前文所提到的呼吸困难时呼吸作功和耗能增加,又使全身耗氧量增多,这又可促进缺氧并加重呼吸困难。
左心衰竭严重时出现端坐呼吸,主要是由于平卧位时,下半身静脉血液回流量增多,因而可以进一步加剧肺的淤血水肿。当患者被迫采取端坐位时,肺部淤血水肿和呼吸困难即可有所减轻。
左心衰竭特别是已经生发端坐呼吸的患者,常可发生夜间阵发性呼吸困难,其特征是病人入睡后突然感到气闷而惊醒,并立即坐起喘气和咳嗽。其发生机制是:①端坐呼吸的患者入睡后往往滑向平卧位,因而下半身静脉血液回流增多,而且在白天因重力关系积聚在下垂部位组织间隙中的水肿液也因体位改变而回流入血,故肺部的淤血水肿明显加剧。②入睡时迷走神经中枢紧张性升高,支气管口径变小,通气阻力增大。③熟睡时神经反射的敏感性降低,因而只有当肺淤血发展到比较严重的时候,才能剌激呼吸中枢,引起突然发作的呼吸困难,如果患者在发作时伴有哮鸣音,则称为心性哮喘(cardac asthma),可能与患者有潜在的支气管炎有关。
三、肝脏和消化系统功能的改变
肝脏和消化系统功能的障碍,主要由体循环静脉淤血所引起,当然也与这些器官的动脉血液的灌流不足有关。右心衰竭时肝脏因淤血而肿大,并可伴有压痛和上腹部不适感;长期肝淤血可引起肝脂肪变性,甚至引起黄疸和淤血性肝硬变。胰腺淤血和供血不足可影响其内分泌和外分泌机能,从而可使糖代谢和食物的消化发生障碍。胃肠道的淤血可引起食欲不振、消化和吸收不良以及胃肠道剌激症状如恶心、呕吐、腹泻等等。
四、肾脏功能的改变
左心衰竭和右心衰竭都可使肾血流量减少而导致少尿。尿钠含量低而比重高。除了在严重而持久的右心衰竭以外,肾功能仅有轻度的障碍,可伴有一定程度的氮质血症。
五、水、电解质和酸碱平衡紊乱
心力衰竭时水、电解质平衡紊乱主要表现为钠、水潴留。钠、水潴留的机制在于:①肾小球滤过率减少;前文已经提到,心输出量减少时,各器官中以肾脏血液量的减少最为显着,而右心衰竭引起的肾淤血,也可使肾脏血流量减少。肾血流量的减少即可使肾小球滤过率减少;②肾小管重呼收功能加强:心输出量减少以及通过窦弓反射使肾小血管收缩以致肾血流量减少时,可通过肾素-血管紧张素-醛固酮系统的激活和抗利尿激素的增多,通过肾内血流重分布,通过肾小球滤过分数的升高而使肾小管对钠、水的重吸收加强。上述两方面的因素,特别是肾小管重吸收机能的加强就可引起钠、水潴留。钠、水潴留一方面可引起血量增加,一方面也是导致心性水肿的重要困素之一。
心力衰竭时,体循环静脉淤血和血流速度减慢可引起循环性缺氧,肺淤血水肿则又可引起低氧血症性缺氧。缺氧往往引起代谢性酸中毒,而酸中毒和伴随发生的血钾升高又可进一步使心肌收缩性减弱。
第六节 心力衰竭的防治原则
一、防治原发病(缺)
二、消除诱因
对于心脏负荷已经过重或心肌已经受损的某些患者,体力活动过度紧张、疲劳、心率过快、异位心律,补液过多过快等均能诱发心力衰竭,应尽量消除这此因素。
三、改善心功能
(一)调整前负荷心功能不全的患者,心室充盈压(即前负荷)可高可低(心室充盈压降低可见于急性心力衰竭),应该把充盈压调整到适宜的高度。对于急性心肌梗死的患者而言,肺毛细血管楔压(相当于左室舒张末期压LVEDP)在2-2.4kPa(15~18mmHg)时最为合适。低于此值表示血容量不足,每搏输出量将会减少,而超过此值(前负荷过重)时,不但不能使每搏输出量增加,反而会诱发心力衰竭。因此在给心肌梗死患者或其他心功能不全的病人输液时,应慎重掌握输液的量和速度。
(二)调整后负荷在一定的前负荷下,后负荷的改变会影响心功能。如果后负荷突然增加,每搏输出量便相应的降低,但以后又可由于代偿适应而恢复正常。在后负降低时,则可见每搏输出量升高。这一现象近来受到重视,并已成为使用扩血管药治疗心力衰竭的依据。如应用硝普钠(Na-nitroprusside)、硝酸甘油、苄胺唑啉(rigitine)等扩血管药物时,随着动脉压及外周阻力的降低(后负荷降低),心输出量就相应增加。
(三)加强心肌收缩性 心力衰竭时,由于心肌收缩性减弱,即使VEDP尚未达到临界水平,心室的搏出功就低于正常(心功能曲线向右下移动)。洋地黄类强心剂的作用,就在于加强心肌收缩性,使心脏在承受与治疗前相同的前负荷时,能够作较大的搏出功,即使心功能曲线向左上移动(图12-4)。洋地黄类强心药对慢性心力衰竭的疗效较好,但对急性心肌梗死引起的急性心力衰竭,效果尚不肯定,因为这类药物有引起心律失常和增加心肌耗氧量等不良的副作用。
(四)控制水肿从理论上讲,洋地黄类强心剂是控制水肿的最佳药物,因为它能从根本上改善心脏的泵功能。但实际上单用洋地黄还不能消除水肿,因而,往往还可加用利尿剂,并且要适当限制钠的摄入量。每是钠的摄入量可限制在20~25mmol(20~25mEq)左右,同时要适当补钾,以防大量利尿时引起缺钾。消除水肿的意义,主要在于减少过多的细胞外液,从而使心脏的负荷得以有所减轻。此外,还应当注意,持续过久的不适当的低盐饮食,可引起低钠血症,这对患者很不利的。
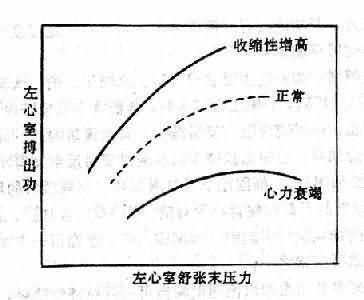
图12-4 心功能曲线示意图
(五)改善组织的供氧给氧(吸氧)是临床以对心力衰竭病人常规治疗措施之一。近年来有人用高压氧治疗多种疾病,其中包括心力衰竭,借以提高血液的携氧能力和改善组织的供氧情况。在严重心力衰竭或急性心肌梗死伴有休克的病人,应用间断的高压氧治疗有一定效果,病死率有所降低,但对这一措施尚需总结经验。
经过上述种种治疗,患者的心输出量有可能满足器官、组织的需要,心力衰竭的临床症状可以消失,但若病因未能去除,则在一定的诱因作用下,可以再次发生心力衰竭,而心力衰竭反复多次发生后,治疗效果可能不够理想。
[附]:高血压
血管内血液对血管壁所产生的侧压力称为血压,分动脉血压、静脉血压和毛细血管血压。高血压(hypertenson)是指以体循环动脉血压升高为特征的疾病或病理过程。由于正常时动脉血压不但受情绪、应激、体位、时间、种族、性别、体力活动等因素的影响,还随着年龄的增长而增高,因此,高血压与正常血压之间的界限划分是比较困难的,应充分考虑这些因素的影响。根据世界卫生组织(WHO)的建议,诊断成人高血压的标准是:静息时收缩压等于或超过21.28kPa(160mmHg)和/或舒张压等于或超过12.46kPa(95mmHg)时,才称为高血压;如果收缩压为18.62~21.28kPa(140~160mmHg),舒张压为11.97~12.46kPa(90~95mmHg)时,谓之临界高血压(borderline hypertension)。
由于血压尤其收缩压受年龄增长的影响比较明显,故有人建议诊断成人高血压也应分为三个不同年龄组(表14-1)。
表12-2 年龄组诊断高血压的标准
| 17~40岁 | 41~60岁 | >60岁 | |
| 正常 | <18.62/11.97kPa(140/90mmHg) | <19.95/11.91kPa(150/90mmHg) | <21.28/11.97kPa(160/90mmHg) |
| 临界高血压 | ≥18.62/11.97kPa(140/90mmHg) | ≥19.95/11.97kPa(150/90mmHg) | ≥21.28/11.97kPa(160/90mmHg) |
| 高血压 | ≥21.28/12.65kPa(160/95mmHg) | ≥21.28/12.65kPa(160/95mmHg) | ≥23.28/12.65kPa(175/95mmHg) |
年龄在15岁以下者如血压超过17.29/10.64kPa(130/80mmHg)时,也应视为高血压。
一、高血压的分类
根据不同的标准可将高血压进行不同的分类。
(一)根据病因分类
最常用,可分为原发性高血压(essential hypertension)和继发性高血压(secondary hypertension)。前者是指病因尚不清楚而以血压高为主要表现的一种独立性疾病,故又称高血压病;因它的发生与多种因素有关,故亦称多原因性高血压,多见于肾脏疾患、神经内分泌障碍、血流机械受阻以及妊娠中毒等时,其发生原因比较简单而清楚,故又称单原因性高因压。
(二)根据收缩压和舒张压升高的情况分类
1.收缩期高血压:即仅出现收缩压升高,而舒张压正常甚至低于正常,多见于老年人大动脉硬化、动脉壁顺应性降低时。
2.舒张期高血压:见于外周血管硬化、阻力较高时。
但大多数情况下舒张压升高往往伴有收缩压的升高。
(三)根据高血压病的发展速度分类
1.缓进型或良性高血压:起病隐匿,病程发展缓慢,开始时多无症状,往往是在体检或因其它病就医时才被发现,此后随着病情的进展,才相继出现有关临床症状和体征。
2.急进型或恶性高血压:少数高血压病起病急骤,发展迅速,血压明显升高,舒张压多在17.3kPa(130mmHg)以上,病情严重,如不及时采取治疗措施,多在一年内死于心、脑、肾等器官功能的严重损害。本病多见于青年人。
此外,根据临床表现及器官受损情况,可将高血压分为三期:一期,即血压达到诊断高血压的水平,尚无器官的损害;二期,已有器官损伤,但其功能还可代偿;三期,即器官的功能受损严重,已失去代偿。
二、高血压的原因和机制
血压是由心输出量和外周血管阻力两个基本因素决定的,心输出量又受心脏舒缩功能、心率、血容量和回心血量等因素的影响,而外周血管阻力主要决定于血管口径和血液粘度,血管口径又受神经、体液和血管本身等各种复杂因素的影响。上述各种因素就其对血压的作用而言,可大致分为两类,一是通过增加外周阻力和/或心输出量,使血压升高;二是通过降低外周阻力和/或心输出量,使血压下降。在正常情况下,两者保持着动态平衡,使血压维持在正常波动范围之内,如上述某种或多种因素导致加压作用大于减压作用时,就会发生高血压。
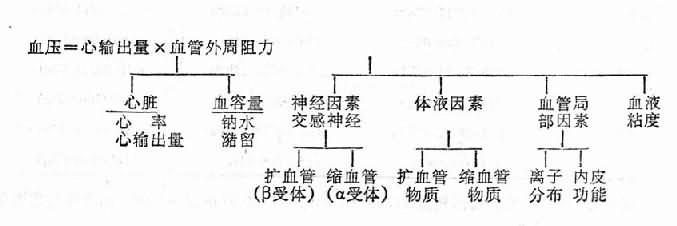
(一)原发性高血压
原发性高血压发生的原因和机制,尚不完全清楚,目前认为是多种因素参与的。
1.精神、神经因素 临床观察和动物实验各方面的资料证明,本病的发生与精神应激和高级神经活动过度紧张有关:①长期从事与精神紧张有关的职业或处于过度精神应激的环境中的人,本病发病率高;②高血压的发生与忧郁、恐惧、悲伤等不良情绪有关;③给犬造成高级神经活动过度紧张状态,可促进高血压的发生和发展;④在本病早期,单纯消除精神应激,常可使血压恢复正常;使自发性高血压大鼠(spontaneous hypertension rat, SHR)脱离应激环境,也可明显推迟高血压的发生。
精神应激引起高血压的机缺可能是由于大脑皮层在各种精神应激长期作用下,通过兴奋下丘脑神经内分泌中枢而使交感神经系统兴奋,儿茶酚胺释放增多。
已有许多事实证明,原发性高血压时交感系统的活动加强;如①SHR大鼠内脏大神经放电率较正常鼠高;②某些本病患者,尤其是青年患者血浆中儿茶酚胺(主要是去甲肾上腺素)含量较高;③临界高血压时中枢和外周交感神经活动性都较血压正常者高;④应用交感神经或α-肾上腺素受体阻滞药可使血压下降。
交感神经引起血压升高的机制是多方面的:①使小动脉收缩,增大外周阻力;使静脉收缩,增加回心血量;②通过兴奋心脏的β受体使心脏收缩加强、加快,从而提高心输出量;③直接或间接激活肾素-血管紧张素系统(renin-angiotensinsystem, RAS),进而收缩血管和通过血管紧张素Ⅱ(angiotensinⅡ,A-Ⅱ)促进醛固酮分泌,增加血容量(图12-5)。
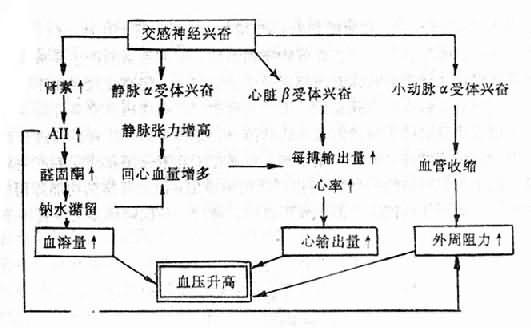
图12-5 交感神经引起血压升高的机制
但须指出,并非原发性高血压患者血中儿茶酚胺的含量都升高;而且,即使升高,其程度(一般升高20~25%)也远不足以导致高血压。说明交感-儿茶酚胺系统的活动增强在原发性高血压的发生中并非起决定性作用。现证明交感神经节后纤维有两类,一类是以神经肽Y(neuropeptide Y, NAY)和去甲肾上腺素为递质的缩血管纤维,另一类是以降钙素基因相关肽(calcitonin gene related peptide, CGRP)和P物质为递质的扩血管纤维。认为这两类纤维的功能失衡即前者功能强于后者才是交感社经参与高血压发生的重要机制。
精神-神经因素虽与高血压发生有关,但并非唯一重要因素,因为远非所有长期处于精神应激环境中的人都发生高血压病;反之,发生高血压病者也不一定有精神应激史。另外,由于本类因素引起的血压升高多为一时性的,目前多数人认为精神-神经因素在原发性高血压发生的始动机制中所起的作用较在维持机制中所起的作用为大。
2.肾素-血管紧张素系统(RAS)原发性高血压时血浆肾素活性升高者占20%,正常者60%,降低者10~20%。过去认为RAS只有在肾素升高组高血压的发生中可能起一定作用,而在其他两组不起作用。但晚近证明并不能排除RAS在其他两组中的作用,因为:①应用RAS抑制药,尤其是血管紧张素转换酶抑制药(angiotensin converting enzyme inhibitor ACEI)治疗高血压病时,不但对高肾素型有效,且对其它型都有效。目前已证明,除肾脏产生肾素影响血浆RAS水平外,心肌、血管平滑肌等组织也存在完整的局部RAS。血浆平滑肌的RAS也具较强的收缩血管加压作用。高血坟时局部RAS活性升高并不影响血浆肾素的水平,但对高血压的发生却起着一定作用;②因高血压病患者血管对加压物质(包括血管紧张素)反应性增高,故即使肾素活性正常甚至低于正常时,对本病患者仍可能起着加压作用。目前认为本系统在高血压病尤其对高血压的维持起着较重要的作用。
③钠、钾、钙与高血压临床与实验材料证明,食盐与高血压的发生有密切关系:例如①食盐摄入量与本病发病率呈正相关,摄取食盐多的地区(如日本本士和我国北京地区)发病率较食盐较少地区(如阿拉斯加的爱斯基摩人和我国广州地区)高;而牙买加某岛上居民食盐摄入量每人<2g/d,就无高血压发生。②给SHR大鼠喂低盐饮食,仅发生轻度高血压;但给予高盐饮食则迅速出现重度高血压。③限制食盐的摄入量或服用利尿药增加排钠量,对某原发性高血压患者有较好的降压作用。
关于食盐引起高血压的机制,尚不完全清楚,可能是多方面的:如①钠潴留导致细胞外液增加,从而可加大心输出量,而且血管壁的钠水溜留可使管腔狭窄,从而使外周阻力增大。②高盐负荷可促使下丘脑产生利钠因子(natriuretic factor, NF),又称下丘脑抑制因子(hypothalamic inhibitor factor, HI)具有所谓利钠激素样的特性,为一种低分子量(500)的非肽类物质。它可抑制细胞膜Na+-K+-ATP酶的活性,故又称Na+-K+-ATP酶抑制因子或钠泵抑制因子。NF也可被看成是一种内源性洋地黄样物质。这样,NF一方面可降低肾小管上皮细胞对钠的重吸收,引起利钠效应;另方面使血管平滑肌细胞等细胞内的钠潴留起来,进而加强Na+-Ca2+交换,致使细胞内Ca2+增加,血管收缩(图12-6)。③增高中枢和外周交感神经的活性;④高食盐可使扩血管物质如激肽、前列腺素的产生释放减少,使缩血管物质如血管紧张素产生增多并加强其与相应受体的亲和力。但高食盐引起高血压是有条件的。高食盐能否引起高血压,关键在于肾脏能否将摄入过多的钠排出。绝大多数人的肾脏通过排钠利尿机制(食盐过多,血Na+浓度增高,肾血容量增加,可通过①肾素分泌和醛固酮产生减少,和②肾内血压增高等机制加强对钠的排泄),可把过多的钠排出;但当肾排钠功能障碍时,由于不能把过多的钠排出,即可导致钠潴留而出现高血压。肾排钠功能障碍多与遗传因素有关。
流行病学调查证明,钾的摄入量与血压呈负相关;给高血压患者补充钾盐可使血压下降,并能提高限钠饮食疗法的降压效果。钾的降压机制可能是:①抑制血浆肾素活性,减少血管紧张素Ⅱ受体数目和降低其与血管素Ⅱ的亲和力;②抑制肾小管对钠的重吸收,促进钠排出;③减弱交感神经的敏感性。此外,高能尚能激活血管平滑肌细胞膜Na+-K+泵,防止钠、钙在细胞内蓄积([Na]i、[Ca]i)。
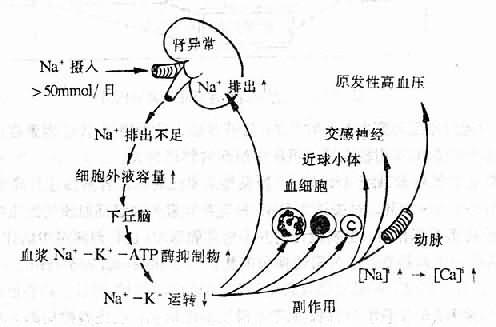
图12-6 高食盐和利钠因子引起高血压机制示意图
据调查,某些原发性高血压患者饮食钙摄入量较正常人低,其血清Ca+和肾素水平也低,对这些患者长期补充钙可使血压降低,并能对抗高盐所致的升压效应。实验也证明,减少饮食中的钙,可使自发高血压大鼠(SHR)血压明显升高;相反,如增加钙摄入量,可延缓SHR幼鼠血压的升高,并使SHR成年鼠血压降低。钙的降压机制可能是:①稳定细胞膜结构,控制膜离子通透性,使Ca+不致大量进入细胞内;②钙与钙调蛋白结合,激活细胞膜钙泵,增加钙的外流,使细胞内钙浓度降低。防止血管平滑肌细胞内Ca2+的积聚就可防止血管的收缩,从而防止血压的升高。
4.遗传因素已有充分证据说明高血压病与遗传基因有关:①有家族性:双亲都有高血压病史的子女发病率为50%,即两个子女一个发病;双亲之一有高血压史的子女,发病率为25%;双亲没有高血压病史的子女,发病率为5%。②自幼收领的养子和亲生子虽然生活环境完全相同,但后者的高血压病发病率与双亲的发病率更为接近。③对食盐敏感和不敏感的两种大鼠,虽然在同样高食的饲养条件下,前者发生高血压而后者则否。
遗传基因主要决定高血压病发生的易感性,而非高血压的本身。由于血压受多种因素控制,故遗传的“易感性”也是多基因决定的。其表现是多方面的。例如肾脏排钠的先天性缺陷,细胞膜的先天性功能异常和血管平滑肌对加压物质的敏感性高等,今分述如下:
(1)肾先天性排钠缺陷:在同样大量摄取食盐的条件下,对盐敏感鼠发生高血压,而对盐不敏感鼠则不发生高血压。如把对盐敏感鼠的两个肾脏移植给对盐不敏感鼠,又把对盐不敏感鼠的两个肾移植给对盐敏感鼠,则对盐不敏感鼠发生高血压而对盐敏感鼠不发生高血压。这说明对敏感鼠所以发生高血压,是由于肾排钠障碍,而肾排钠障碍是由遗传因素决定的。人类高血压病易感者也可能存在类似的遗传性肾排钠缺陷。
(2)细胞膜先天性功能异常:主要表现在膜的钠、钙离子运转异常。细胞对钠的运转有四个系统,即①Ca+-K+泵:为主动的运转系统;②被动性运转,决定于细胞内外K+、Na+离子梯度;③Na+-K+同向运转(co-transport)和④Na+-Li+逆向运转(counter-transpot)(图12-7)。
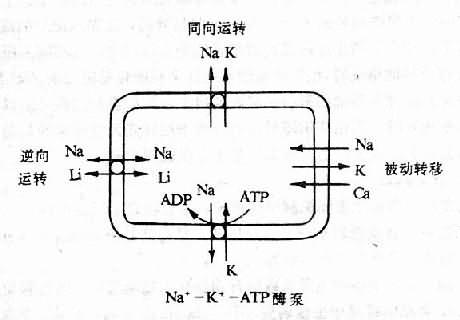
图12-7 细胞内钠运转的四个系统
上述的③和④两个运转系统,虽不需ATP水解供能,但需在特异的载体参与下使相应的离子进行跨膜的易化扩散。这两个系统都是协同运转系统,即一种离子与载体结合后又可提高载体与另一种高子的亲和力,进而使两种离子偶联协同扩散。除了Na+-K+泵的主动运转外,Na+-K+同向运转也是细胞用以防止细胞内Na+过度负荷的重要机制。Na+-Li+逆向运转与肾脏近曲小管对钠、水的重吸收呈正相关,即当Na+-Li+逆向运转加强时,近曲小管对钠、水的重吸收也加强。据报道,高血压病患者和SHR大鼠上述四种钠运转系统都有可能发生改变,主要是Na+-K+泵运转被抑制;同时,因膜通透性增高,故Na+被动转入细胞增加;此外还有Na+-Li+逆向运转加强和Na+-K+同向运转减弱。其结果是细胞内钠蓄积。研究结果也证明SHR大鼠和部分原发笥高血压患者细胞内的钠含量高于正常。细胞内钠的蓄积。可以导致:①血管壁钠水负荷增加,管壁增厚,血管腔狭窄,从而引起外周阻力增高;②Na+-Ca2+交换加强,使血管平滑肌细胞内Ca2+增加(可能还有其它机制),从而促使血管收缩。尤其是在管壁增厚的情况下,轻度的血管收缩就可产生较大的阻力。此外,由于机体的钠水潴留,又可促使利钠因子的分泌和释放,通过抑制Na+-K+泵,进一步加重钠在细胞内蓄积。
在双亲有高压病史而血压正常的子女中,约有一半有细胞膜对钠运转的异常。
原发性高血压患者细胞内Ca2+增多,除继发于细胞内钠升高外,还与膜对钙的运转系统异常有关,主要表现在:①SHR大鼠和原发性高血压者红细胞和血管平滑肌细胞膜对Ca2+的通透性增加,故Ca2+内流加强:②质膜对Ca2+的结合力决定于细胞膜表面多种Ca2+结合蛋白的量及其与Ca+的亲和力。SHR大鼠和原发性高血压患者可能由于结合蛋白量减少和/或亲和力降低,从而导致膜结合钙减少而胞浆中的游离钙增加;③ATP依赖性钙泵运转障碍:现证明SHR大鼠的血管平滑肌肌膜、心肌肌浆网以及红细胞等都有ATP依赖性钙泵的运转障碍,从而不能把胞浆中的Ca2+分别转运到细胞外和肌浆网中,结果使Ca2+在胞浆中增加。血管平滑肌细胞内游离钙的增多,被认为是原发性高血压发生机制的最后共同途径。现认为膜对钠、钙离子运转的障碍是遗传因素决定的膜功能异常的表现,而高血压病可能是一种细胞膜病。
此外,实验还证明SHR大鼠出生后血压升高之前,其血管壁的收缩成分较正常鼠多,对加压物质的收缩反应也远较正常鼠强。双亲有高血压病史的血压正常子女也存在有类似的现象。同时这类子女血中的利钠因子(NF)也明显升高,这提示血管的高反应性和NF的升高都与遗传有关。总之,高血压病遗传的易感性是多因素的,它在高血压病发生中的作用是和环境因素的作用相辅相成的,也即环境因素往往是在遗传易感性的基础上发挥致病作用的,而遗传易感性又是通过对环境因素的反应表现出来。例如食盐过多之所以能引起钠水潴留和血压升高,常是在肾脏先天性排钠障碍和膜对离子运转先天性异常的基础上发生的,而患者对高血压病的遗传易感性又是通过食盐过量才得以表现出来。
(二)继发性高血压
继发性高血压发生的原因和机制比较清楚,主要决定于原发疾病。
1.肾性高血压 肾疾患时出现的高血压谓之肾性高血压(renal hypertension),为继发性高血压中最常见者,发病的主要机制为:
(1)RAS激活:见于各种原因所致的肾动脉狭窄或阻塞时,例如肾动脉粥样硬化(多见于男性老年人)、肾动脉纤维增生性病变(fibroplsatic disease,多见于中青年)和肾动脉先天性发育不良(见于儿童)等。由于肾脏缺血促进肾素分泌,激活了RAS并进而引起高血压。其主要依据是:①手术缩窄犬一侧肾动脉后,与血压升高的同时,手术侧肾静脉血中肾素明显高于对侧,且近球小体中含肾素颗粒也明显增加;解除狭窄后,随着肾素含量的下降血压下降血压也降低;②用血管肾张素转换酶抑制剂可使血压下降;③大部分本类高血压患者血浆中的肾素活性增高。RAS在活性增高的升压机制主要是使血管收缩,使外周阻力增加。
(2)肾排水、排钠能力减弱或丧失:多见于急性或慢性肾实质广泛性病变时(如急性或慢性肾炎、肾盂肾炎、多囊肾等),由于大量肾单位丧失了排水、排钠能力,而剩余肾单位又不能充分代偿,结果导致钠水潴留、血容量增加和心输出量增大,产生高血压。此时血管外周阻力可正常甚至低于正常。采取利尿措施以减少血容量可有效地降低血压。
(3)肾减压物质生成减少:肾不但分泌加压物质,肾髓质间质细胞还分泌多种减压物质,如前列腺素E2(PGE2)和具有抗高血压作用脂质等物质。这些物质都具有排、扩血管和降低交感神经活性的作用,和RAS既互相对抗又维持着平衡。
现证明这些特质的消长与高血压的发生有密切关系,例如:①肾髓质乳头移植,可防止肾实质性高血压的发展,但移植肾皮质则无此作用:②在钠负荷的情况下,切除肾髓质可很快发生高血压,但不切除髓质则不易发生。所以当肾髓质受到破坏或其间质细胞产生减压物质减少(或被抑制)时,由于RAS与减压物质失去平衡,即可引起血压升高。
上述三种机制,在肾性高血压发病中的作用因肾疾患的种类、部位和程度不同而异。例如:肾血管疾患时以第一种机制为主。肾实质性病变尤其是伴有肾功能不全者以第二种机制为主;肾髓质破坏时则有第三种机制参与;但在慢性肾疾患时,由于病变性质和部位的复杂,三种机制常同时参与作用。
2.内分泌性高血压由内分泌紊乱引起的高血压谓之内分泌性高血压(endocrinic hypertension),主要见于:
(1)嗜铬细胞瘤:嗜铬细胞瘤(pheochromocytoma)多发生于肾上腺髓质。由于嗜铬瘤细胞大量分泌和释放去甲肾腺素和肾上腺素,使小血管收缩和心输出量增加,故可导致血压升高。这种血压升高多为阵发性的,即当缺氧、麻醉、肌肉活动、性活动或肾上腺部位受剌激时,就可激发瘤细胞释放这些物质而血压突然升高,并多伴有心悸、出汗、烦躁、头痛、胸前区痛和血糖升高等临床表现;当血中此类物质含量降低时,血压也随之下降。但也有少数患者血压呈持续性升高。应用α-肾上腺体拮抗剂可使血压恢复正常,用β肾上腺素受体拮抗剂可有效地控制心输出量增加和其它临床表现。
(2)原发性醛固酮增多症:原发性醛固酮增多症(primary aldosteronism),多见于肾上腺皮质球状带肿瘤或双侧肾上腺皮质增生时。血压升高主要是由于醛固酮分泌过多导致血容量和心输出量的增加。血容量增加可抑制肾近球小体细胞的肾素分泌,故血浆肾素低于正常,此和继发性醛固酮增多症有些不同(后者肾素活性升高)。另外,由于醛固酮促进肾曲小管的Na+-K+交换使排钾增加,故常导致低钾血症。
(3)皮质醇增多症;皮质醇增多症(hypercortionlism)是由于肾上腺皮质分泌过量的糖皮质激素(主要是皮质醇)所致。该症如由肾上腺皮质肿瘤所引起,称为Cushing综合征;本症约有80%伴有高血压。血压升高的主要机制是:糖皮质激素可①促进的潴留和增加血浆容量;②剌激肾素的合成,激活RAS;③加强血管对加压物质(如去甲肾上腺素)的加压反应。如果伴有盐皮质激素(脱氧皮质酮、醛固酮)增加时,则更使钠水潴留加重。
(4)肾上腺某些酶的先天性缺陷:常见的是11-β羟化酶和17-α羟化酶的缺乏。当前一种酶缺乏时,皮质醇生成减少,反馈性地促进ACTH分泌增加,使脱氧皮质酮(DOC)生成增多,结果是钠水潴留、血压升高;同时因17-羟孕烯醇酮增多,使雄性激素的生成也增多,故女性患者常出理男性化。当后一种酶缺乏时,因17-羟孕烯醇酮的减少,不但反馈性地促进ACTH分泌增加,从而引起高血压,并且雄性激素生成也减少,故男性患者出现女性化(图12-8)。
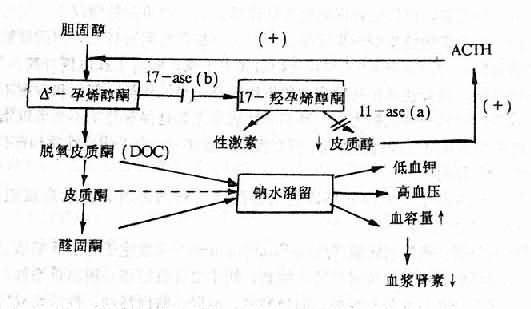
图12-8 11-β羟化酶(11-ase)和17-α 羟化酶(17-ase)缺乏时发生高血压机理示意图,
11-ase(a)示11-β羟化酸缺乏影响部位;
17-ase(b)示17-α羟化酶缺乏影响部位;(+)反馈性兴奋
3.妊娠高血压妊娠期发生和发展起来的高血压谓之妊娠高血压(pregenancy-induced hypertension)。由于正常妊娠期的血压较未妊娠时低,故判断妊娠高血压的血压水平也应较一般的诊断标准低。在妊娠4~6个月时舒张压超过10.7kPa(80mmHg)或妊娠7~9个月时超过11.3kPa(85mmHg)时,或者高于妊娠早期4.00/2.00kPa(30/15mmHg)时,即可视为高血压。包括①先兆子痫高血压(又称妊娠中毒性高血压);②慢性高血压,多来源于原发性或肾性高血压;和③子痫前合并慢性高血压。
先兆子痫高血压(preeclamptic hypertension)是妊娠期特发的高血压,多发生在妊娠的后期,伴有蛋白尿和/或水肿。主要是由于胎盘组织供血绝对减少(因血液循环障碍)和/或相对减少(因胎盘组织增大)从而导致胎盘缺血的结果。此时,①缺血的胎盘可产生较多的肾素和血管紧张素类物质(患者子宫和胎盘中含量较正常妊娠者高);②胎盘组织产生的抗RAS的减压物质减少。正常妊娠的胎盘所产生的减压物质(如前列腺素)能和RAS保持对抗性的动态平衡,从而抵消RAS的加压效应。先兆子痫高血压时,因胎盘减压物质产生减少,故可使两者失去平衡;③可产生和释放组织因子(凝血因子Ⅲ)而引起DIC,当肾小球微血管中出现DIC时,可导致小球滤过率严重减少而发生水潴留。
4.主动脉狭窄引起的高血压高血压可由于主动脉狭窄所引起。例如可见于先天性主动脉狭窄(coarctation of aorta)。此时,心脏收缩代偿性加强,而射出的血液又不能顺利通过狭窄部,致使大量血液蓄积在容量有限的狭窄部近心端的主动脉及其分支中,因而使狭窄部近心端的上肢以上的动脉血压升高,而下肢血压不高;尤其当运动时,由于心输出量的增加,可使其收缩压突然升高,此外,曾有人提出主动脉缩窄时,起源于缩窄部远心端的肾动脉可能因血灌流不足而引起肾缺血,从而促使肾素的分泌增多,但尚无有力的根据。
5.收缩期高血压 凡收缩压≥21.3kPa(≥160mmHg)而舒张压<12.7kPa(<95mmHg)者,则可定为收缩期高血压。单纯收缩期高血压在老年人最常见,尤其超过55岁的妇女。这是由于动脉硬化(主要是粥样硬化)使大动脉顺应性降低所致;主要表现是脉压增宽,左室射血速率和心脏指数降低,总血管外周阻力增高。少数收缩期高血压继发于主动脉瓣关闭不全、严重贫血和甲状腺机能亢进时,主要表现是左室射血速率和心脏指数增高,但总血管外周阻力正常。
总之,①继发性高血压发生的原因和机制比较简单,最先多是由于某个参与调压机制的环节(如肾、肾上腺或垂体)发生障碍的结果;②由于各种调压机制之间常是相互联系着,在高血压的发生中,一个加压机制的激活,常可导致另一个加压机制的激活(如交感神经兴奋→肾素分泌↑→血管紧张素↑)或者通过负反馈作用,一个加压机制的激活又对另一个加压机制起着抑制作用(如盐皮质激素↑→血容量↑→肾素↓);这样,继发性高血压一旦发生后,其发展过程又表现出一定的复杂性;③一般认为,不管引起继发性高血压的始动机制如何,肾对钠水排泄能力的降低,是维持各种继发性高血压的重要机制,故利尿,排钠对不同种类的继发性高血压都有明显的降压效果。
三、高血压时血液动力学的改变及其对机体的影响
(一)高血压时血液动力学改变
高血压时血液动力学的改变比较复杂,不但决定于高血压的发生原因和机制,还决定于高血压的发展速度、程度和发展阶段。
1.心输出量(CO)的改变高血压时心输出量可以升高,也可降低。
(1)心输出量增高:多见于原发性高血压早期和临床性高血压以及嗜铬细胞瘤、原发性醛固酮增多症和肾性高血压等继发性高血压。心输出量增高的机制各有不同。原发性醛固酮增多症和某些肾性高血压(如急性肾炎等引起的高血压)时主要是由于血容量增加;嗜铬细胞瘤时,是因为肾上腺髓质激素对心脏的正性肌力和正性变时作用;原发性高血压时心输出量增高的机制尚不清楚,可能与交感神经兴奋、肾上腺髓质激素分泌增多和/或血管对加压物质的敏感性增高,使外周容量血管收缩,促使回心血量增加有关。
(2)心输量降低:常见于严重高血压和高血压心脏病时。CO降低的原因,或者是因为心脏射血阻抗过大,或者是因为肥大心肌舒缩功能和/或顺应性降低,或者是由于冠脉供血不足使心肌收缩性减弱,可能是上述诸因素综合作用的结果。一旦CO降低,即可加重外周血管的代偿性收缩,外周血管阻力进一步增大,从而促进高血压的发展,并可进一步影响重要器官的血液供应。
2.血管阻力的改变 除因甲状腺机能亢进引起的血压升高外,其他各种高血压时外周血管阻力都呈不同程度的升高。血管阻力升高是外周血管功能和/或结构改变的结果。血管功能性改变主要是血管平滑肌对各种神经、体液加压因素作用的敏感性增高,多发生于血压升高之前,故为引起高血压的原因。待高血压发生后,由于血管的持续痉挛和高血压的长期作用,可发生血管结构改变,主要是小动脉和微动脉(arteriole)壁增厚和纤维化,从而进一步使外周阻力增高,使高血压进一步发展并减少各重要器官的血液供应。此外,较大血管的管壁在长期高压机械力的冲击之下,可发生内膜损伤、平滑肌细胞增生和胆固醇的沉积,从而促进较大血管粥样硬化病变的发生和发展。
外周血管阻力和心输出量的改变在高血压不同发展阶段也是不同的。例如原发性高血压的早期阶段心输出量增加,此时的外周血管阻力基本正常或稍高;随着高血压的发展,外周血管阻力逐渐增高,心输出量则逐渐降低甚至低于正常(图12-9)。
图12-9 原发性高血压时血液动力学的改变
3.细胞外液的改变某些高血压时细胞外液容量可以增多,例如在钠负荷过高、盐皮质激素过多、原发性醛固酮增多症以及肾实质病变所致的高血压,部分原发性高血压尤其是伴有心力衰竭时,细胞外液容量都可增加。血容量增加可通过加大心输出量而使血压升高。故此时利尿即可降压。
(二)高血压对机体的影响
高血压对机体的影响取决于血压升高和血液动力学改变的速度、程度、持续时间以及原发性疾病的情况等各种因素。其影响主要表现在心、脑、肾等重要器官的功能和结构的改变以及眼底血管的变化上。
1.对心脏的影响高血压对心脏的影响表现在两方面。一是适应代偿性改变,如心脏高功能状态和心肌的肥大;二是代偿失调性改变和损害,如冠状动脉粥样硬化、心力衰竭和心律失常等。开始时多为适应代偿性改变,随着高血压的发展逐渐转为代偿失调和损害性变化。
(1)心脏的高功能状态:为高血压时心脏最早发生的一种适应代偿性变化。主要表现是前负荷增加、心肌收缩力加强和心脏输出量增大,前负荷增加或者是由于血浆容量的增加,或者是因交感-儿茶酚胺系统的兴奋,使外周容量血管收缩,迫使回心血量增多。心脏收缩力加强主要是因为交感神经兴奋和肾上腺髓质激素的作用。心脏前负荷增中和肾上腺髓质激素的作用。心脏前负荷增神以兴奋和肾上腺髓质激素的作用。心脏前负荷增加和收缩力加强可使心输出量增大,从而使心脏能在较高的射血阻力下,保证各重要器官的血液供应。
(2)心肌肥大:是心脏长期对压力负荷过度发生的一种慢性适应代偿性改变。主要表现是向心性肥大,此时虽然单位重量心肌的收缩性在压力负荷过的情况下有所降低,但由于心脏肥大。总的心功能是提高了(参见本章心力衰竭节)。
早期的代偿性心肌肥大和心功能适应性改变一般是可逆的,即当血压恢复正常时,这些改变可恢复正常。
(3)心力衰竭:是高血压时常见的严重合并症。发生心力衰竭的主要原因是:①压力负荷过度、心肌耗氧量增多和冠脉供血减少(因小动脉硬化和冠状动脉粥样硬化使管腔狭窄和阻塞)导致心肌缺血缺氧和能量利用障碍;②心肌向心性肥大引起心脏舒张充盈障碍(参见本章心力衰竭节)。高血压所致的心力衰竭,多为慢性充血性心力衰竭,发生率随年龄的增长和血压升高程度而升高。约有50%伴发心力衰竭的患者在5年内死亡。
2.对大脑的影响大脑是最易受高血压影响的靶器官。对大脑的影响是通过高血压对脑血管损害(包括功能和结构)和压力本身的作用引起的。
(1)脑血管自身调节障碍(高血压脑病,hypertensive encephalopathy):是因脑血管在血压持续性升高时,发生自身调节失控而导致的一种可逆性脑血管综合征。主要临床表现是剧烈头痛、呕吐、抽搐、意识模糊、视力障碍等。
正常情况下,当血压升高时,脑血管即自动收缩;反之,血压下降时,脑血管又自动舒张。大脑通过脑血管的这种自身调节机制才能使脑血流量不受血压波动的影响,经常保持相对稳定状态。正常人脑血管的自身调节范围是8.00~16.00kPa(60~120mmHg)。高血压患者,由于对高血压产生了慢性适应,其调节范围可变为14.7~24.0kPa(110~180mmHg)。血压波动在上述范围内,通过脑血管的自身调节,可使脑血流量维持稳定;但若血压突然升高且超过此调节上限时,脑血管的自身调节机制就会失效,因而不再继续收缩发生被动扩张,结果脑血流量突然增加,毛细血管的压力急剧升高,体液外渗,引起水肿,甚至发生斑点出血等病理变化,从而引起高血压脑病。
(2)脑小血管阻塞:脑微动脉(内径50~200μm)在长期痉挛和高血压的机械性冲击的影响下,可发生纤维性坏死、管腔阻塞,其支配的脑组织乃因血供被阻断而发生梗塞,出现直径为0.5~15mm大小的小灶性空腔病变,即腔隙性脑梗塞(lacunar infarction)。好发于脑深部神经核如壳核、尾状核以及内囊后段等处。临床的表现取决于病变数目和部位,有的出现相应的临床症状,有的则无。因病灶较小,血管造影也往往难以发现。
(3)小动脉破裂:所引起的高血压性脑出血(hypertensivve hemorrhage),为高血压常见的致命脑并发症。脑小动脉和微动脉在高压长期作用下,发生机械性扩张,造成动脉瘤或动脉壁纤维性坏死,在此基础上,当血压突然升高时(如体力活动、精神激动或用力排便等),即可引起这小血管的破裂而出血。内囊附近出现的好发部位。发病前多无预兆,发病后常伴有剧烈头痛、呕吐和意识丧失等。
此外,有部分人,由于Willis环中等大小的动脉壁先天性内膜缺乏,在高血压的作用下,也易形成动脉瘤和破裂出血。
(4)动脉粥样硬化与脑血栓形成:主要发生于较大的脑血管,虽非高血压所引起,但高血压可促进本病变的发生和发展。由于动脉粥样硬化,致使管腔狭窄,可引起脑缺血。如在这些病变的基础上形成脑血栓堵塞血管,则出现支配区脑组织坏死,患者突然出现失语、偏瘫、半身感觉缺失、同侧偏盲等。
高血压合并症以脑合并症为最多,约占高血压总合并症的50~70%。
3.对肾的影响高血压与肾的相互关系,一是高血压引起肾脏病变,而肾病变又加重高血压;二是肾病变引起高血压,高血压又促进肾病变。
(1)高血压引起肾病变:见于非肾性高血压,尤其原发性高血压。持续性高血压可引起肾小动脉和微动脉的硬化、纤维组织增生,促进肾大血管的粥样硬化与血栓形成,从而使肾缺血、肾单位萎缩和纤维化。轻者可致肾功能降低,出现多尿,夜尿和等渗尿等;重者可导致肾功能衰竭。
肾病变发生后,可反过来加重高血压。这是由于:①肾缺血激活RAS;②当大量肾单位被破坏时,使肾小球滤过率降低,导致钠水潴留。
(2)肾病变引起高血压:见于肾性高血压。高血压发生后,又可通过肾小血管的功能和结构改变,加重肾缺血,促进肾病变和肾功衰竭。
因高血压而发生肾功衰竭者约占高血压合并症的5%。
4.对视网膜血管的影响高血压时视网膜血管出现不同程度的改变和损害如血管痉挛、硬化、渗出和出血等,有时还发生视神经乳头水肿。
视网膜血管痉挛是对血压升高的自身调节反应;渗出是小血管壁通透性增高和血管内压增高所致;出血则是小血管在高血压作用下管壁破坏的结果。关于高血压时发生视神经乳头水肿的原因尚不完全清楚,可能是因为高血压时脑血管自身调节失效引起脑水肿,水肿液沿视神经流出并蓄积于视神经乳头所致。
上述渗出、出血和视神经乳头水肿多在舒张压明显升高[>16.7kPa(>125mmHg)]或收缩压急剧增高的情况下出现,一旦出现这些改变则预示病情严重,故检查眼底血管的变化是评定高血压严重程度的重要参考指标。
四、高血压的防治原则
(一)、预防
原发性高血压是常见多发病,特别应当重视人群一级综合预防。要消除和控制与本病发生有关的危险因素,如调整人群的饮食、生活习惯、改善生活和工作劳动环境等等。对临界性高血压或有家族史的子女则应采取个体二级预防措施,如严密随访观察、控制饮食质量,避免精神应激、加强体育锻炼,必要时给予适当的调压药等等。对继发性高血压的预防,关键在于防治原发病。
(二)、治疗
已如前述,尽管引起高血压的原因很多,但其发病的基本环节是外周血管阻力升高和/或心输出量与血容量的增加。因此,治疗高血压的基本原则是降低外血管阻力和/或心输出量与血容量。
1.降低外周血管阻力降低外周血管为治疗高血压的基本措施。最常用的是交感神经阻断药、血管扩张以及抗RAS的药物等。它们可以通过不同环节降低外周血管阻力,以达到降压目的。
2.减少血浆容量 用利尿药减少血浆容量,对容量依赖性高血压有较好的降压效果。
3.联合措施 为了提高降压效果和减少药物副作用,不但应把药物和非药物性(如控制食盐摄入量、加强体育锻炼、心理精神疗法等)降压措施结合起来,而且必要时还应把各种降压物如利尿和不同的降低血管阻力的药物联合应用。
最后尚需指出:在高血压持续时间较久以后,尤其是在老年患者,机体的各种重要器官对高血压状态已经发生了适应,即器官的基本灌流量都是较高的血压水平的基础上维持的。在这种情况下,如果采用的措施使血压降得过急过剧,那么即便血压水平还高于正常人,但对高血压患者来说,已难以维持各重要器官的基本灌流量,以致可以发生缺血缺氧,甚至引起器官功能衰竭,故应特别加以注意。
第十三章 呼吸衰竭与成人呼吸窘迫综合征
呼吸的主要功能是不断地给机体提供氧气和从机体排出多余的二氧化碳
■[此处缺少一些内容]■
);根据主要的发病机制不同,也可将呼吸衰竭分为通气性和换气性两大类;根据原发病变部位不同又可分为中枢性和外周性;根据病程经过不同还可将呼吸衰竭分为急性和慢性。
1kPa=7.5006mmHg,下同。
第一节 呼吸衰竭的病因
很多疾病都能直接或间接影响肺的呼吸功能而导致呼吸衰竭。重要的病因如下:
一、神经系统疾病
(一)中枢或周围神经的器质病变 如脑或脊髓外伤、脑肿瘤、脑血管意外、脑部感染、脑水肿、脊髓灰质炎、多发性神经炎等。
(二)呼吸中枢抑制如镇静药、安眼药或麻醉药过量等。
二、骨骼、肌肉和胸膜疾病
(一)胸廓骨骼病变 如脊柱后侧凸、多发性肋骨骨折等。
(二)呼吸肌活动障碍如重症肌无力症、有机磷中毒、多发性肌炎、肌营养不良、低钾血症以及腹压增大或过度肥胖膈肌活动受限等。
(三)胸膜病变如胸膜纤维化、胸腔大量积液、张力性气胸等。
三、肺和气道的疾病
(一)气道病变 如异物、肿瘤、炎症使中央气道狭窄或阻塞。更为多见的是细支气管炎、支气管哮喘、慢性支气管炎、慢性阻塞性肺气肿等引起的外周气道阻塞。
■[此处缺少一些内容]■
第二节 呼吸衰竭的发病机制
外呼吸包括通气和换气两个基本环节。各种病因不外乎通过引起肺泡通气不足、弥散障碍、肺泡通气与血流比例失调、肺内短路增加等机制,使通气和/或换气过程发生严重障碍而导致呼吸衰竭。不同的病因常通过相似的机制引起呼吸衰竭,因而使不同病因引起的呼吸衰竭具有共性;但由于病变的部位、性质以及机体反应性不同,故其发病又往往存在特殊性。
一、肺泡通气不足
肺泡通气量即有效通气量,正常成人静息时约为4L/min。除死腔通气量增加可直接减少肺泡通气量外,凡能减弱呼吸的动力或增加胸壁与肺的弹性阻力或非弹性阻力的任何原因,都可引起肺泡通气不足而导致呼吸衰竭。
(一)限制性通气不足
肺泡扩张受限制所引起的肺泡通气不足称为限制性通气不足(restrictivehypoventilation),其发生机制如下:
1.呼吸肌活动障碍当脑部病变或药物使呼吸中枢受损或抑制,或神经肌肉疾患累及呼吸肌时,均可因吸呼肌收缩减弱或膈肌活动受限,以致肺泡不能正常扩张而发生通气不足。呼吸肌的病变往往需同时累及肋间肌和膈肌时才会引起明显的血气变化。
2.胸壁和肺的顺应性降低呼吸肌收缩使胸廓与肺扩张时,需克服组织的弹性阻力,肺的弹性回缩力使肺趋向萎陷(collapse)。胸廓的弹性在静息呼气末时是使胸廓扩大的力量,但当吸气至肺活量的75%以上时,胸廓对吸气也构成弹性阻力。故弹性阻力的大小直接影响肺与胸廓在吸气时是否易于扩张。肺与胸廓扩张的难易程度通常以顺应性(Compliance)表示,它是弹性阻力的倒数。
胸廓顺应性降低见于胸廓骨骼病变或某些胸膜病变时。肺顺应性除直接与肺容量有关(肺容量小,肺顺应性也低)外,主要取决于其弹性回缩力。肺泡的弹性回缩力是由肺组织本身的弹性结构和肺泡表面张力所决定。肺淤血、水肿、纤维化等均可降低肺的顺应性,增加吸气时的弹性阻力。肺泡表面张力有使肺泡回缩的作用。生理情况下,由肺泡Ⅱ型上皮细胞产生的表面活性物质覆盖于肺泡、肺泡管和呼吸性细支气管液层表面,它能降低肺泡表面张力。降低肺泡回缩力,提高肺顺应性,维持肺泡膨胀的稳定性。它与维持肺泡的干燥也有关。Ⅱ型肺泡上皮受损(如循环灌流不足、氧中毒、脂肪栓塞)或发育不全(婴儿呼吸窘迫综合征)以致表面活性物质的合成与分泌不足,或者表面活性物质被大量破坏或消耗(如急性胰腺炎、肺水肿、过度通气)时,均可使肺泡表面活性物质减少,肺泡表面张力增加而降低肺顺应性,从而使肺泡不易扩张而发生限制性通气不足。
限制性通气不足时,由于病变往往并不均匀对称,故不仅有肺泡通气不足,而且往往伴有肺泡通气与血流比例失调,故换气功能也遭到损害(详后文)。
(二)阻塞性通气不足
气道狭窄或阻塞引起的肺泡通气不足称为阻塞性通气不足(obstructivehypoventilation)。气道阻力是通气过程中主要的非弹性阻力,正常约为0.1~0.3kPa(1~3cmH2O)/L·sec-1呼气时略高于吸气时。其中80%以上发生于直径大于2mm的支气管与气管,直径小于2mm的外周小气道的阻力仅占总阻力的20%以下。
影响气道阻力的因素有气道内径、长度和形态、气流速度和形式(层流、湍流)、气体的密度和粘度,其中最主要的是气道内径。气道内外压力的改变,管壁痉挛、肿胀或纤维化,管腔被粘液、渗出物、异物或肿瘤等阻塞,肺组织弹性降低以致对气道管壁的牵引力减弱等,均可使气道内径变窄或不规则而增加气流阻力,引起阻塞性通气不足。气道阻塞有中央性外周性两类:
1.中央气道阻塞指声门至气管隆凸间的气道阻塞。急性阻塞较慢性的多见,可立即威胁生命。气道阻塞有的是固定不变的,有的是可变的。固定阻塞见于疤痕形成。可变阻塞若位于胸外(如声带麻痹、炎症等),则吸气时气流经病灶引起的压力下降,可使气道内压明显小于大气压,故可使气道狭窄加重;呼气时则因气道内压力大于大气压而可使阻塞减轻,故此类患者吸气更为困难,表现出明显的吸气性呼吸困难。可变阻塞如位于中央气道的胸内部分,则由于吸气时气道内压大于胸内压,故可使阻塞减轻,用力呼气时则可因胸内压大于气道内压而加重阻塞(图13-1)。
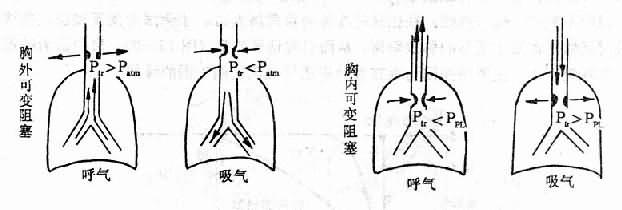
图13-1 不同类型中央气道阻塞呼气与吸气时气道阻力变化
Ptr气管内压;Patm大气压;Ppl胸膜腔压
2.外周气道阻塞 内径小于2mm的细支气管无软骨支撑,管壁薄,与管周的肺泡结构又紧密相连,因此随着吸气与呼气,由于跨壁压的改变,其内径也发生动力学变化。吸气时胸内压降低,而且随着肺泡的扩张,细支气管受到周围弹性组织的牵拉,故其口径可变大,管道伸长。呼气时则相反,小气道缩短变窄。慢性阻塞性肺疾患主要侵犯这些小气道,不仅可使管壁增厚或平滑肌紧张性升高和管壁顺应性降低,而且管腔还可因分泌物潴留而发生狭窄阻塞;此外,由于肺泡壁损伤,对细支气管周围的弹性牵引力也大大减弱。因此,管腔也变得狭窄而不规则,气道阻力大大增加。尤其是在用力呼气时,由于胸内压增高,而小气道内压力却因肺泡弹性回缩力减弱而降低,当气流通过狭窄部位时,气道内压降低更加明显,甚至低于胸内压,因而小气道被压而易于闭合阻塞,故患者常发生呼气性呼吸困难。
外周气道阻塞时除有肺泡通气不足外,还因为阻塞的部位与程度几乎都是不均匀的,所以往往同时有肺泡通气与血流比例失调而引起换气功能障碍。
二、弥散障碍
肺泡与血流经肺泡-毛细血管膜(下简称肺泡膜)进行气体交换的过程是一个物理性弥散过程。单位时间内气体的弥散量取决于肺泡膜两侧的气体分压差、肺泡的面积与厚度和气体的弥散常数。弥散常数又与气体的分子量和溶解度相关。此外,气体总弥散量还决定于血液与肺泡接触的时间。肺的病变引起弥散障碍(diffusion impairment)可发生于下列情况:
(一)肺泡膜面积减少 正常成人肺泡总面积约为80m2,静息呼吸时参与换气的肺泡表面积约仅35~40m2,运动时增加。由于储备量大,因此只有当肺泡膜面积极度减少时,才会引起换气功能障碍,肺泡膜面积减少可见于肺实变、肺不胀、肺叶切除等时。
(二)肺泡膜厚度增加肺泡膜的薄部为气体交换的部位,它是由肺泡上皮、毛细血管内皮及二者共有的基底膜所构成,其厚度小于1μm。虽然气体从肺泡腔到达红细胞内还需经过肺泡表面的液体层、管内血浆层和红细胞膜,但总厚度也不到5μm。故正常气体交换是很快的。当肺水肿、肺泡透明膜形成、肺纤维化、肺泡毛细血管扩张或稀血症导致血浆层变厚等时,都可因肺泡膜通透性降低或弥散距离增宽而影响气体弥散。
(三)血液与肺泡接触时间过短 正常静息时,血液流经肺泡毛细血管的时间约为0.75sec,由于肺泡膜很薄,与血液的接触面又广,故只需0.25sec血红蛋白即可完全氧合。当血液流经肺泡毛细血管的时间过短时,气体弥散量将下降。上述肺泡膜面积减少和厚度增加的病人,虽然肺毛细血管血液中氧分压上升较慢,但一般在静息时肺内气体交换仍可达到平衡,因而不致产生低氧血症,往往只是在体力负荷增大时,才会因为血流加快,血液和肺泡接触时间缩短而发生明显的弥散障碍,从而引起低氧血症(图13-2)。目前认为肺泡膜病变时发生呼吸衰竭,主要还是因为存在着肺泡通气血流比例失调的缘故。
三、肺泡通气与血流比例失调
有效的换气不仅取决于肺泡膜面积与厚度、肺泡总通气量与血流量,还要求肺泡的通气与血流配合协调。肺部疾病时肺的总通气量与总血流量有时可以正常,但通气与血流的分布不均匀以及比例的严重失调(ventilation-perfusion imbalance)却可使患者不能进行有效的换气。这是肺部疾病引起呼吸衰竭最常见的机制。
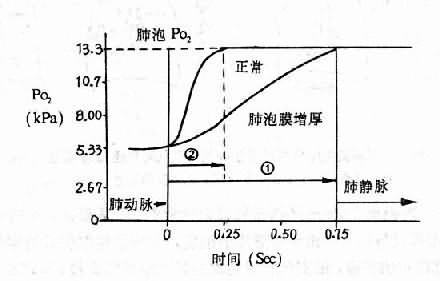
图13-2 正常人与肺泡膜增厚时肺循环中血红蛋白氧合的时间过程
①休息时红细胞经过肺毛细血管的时间;
②运动时红细胞经过肺毛细血管的时间
正常人在静息状态下,肺泡每分通气量(VA)每分约为4L,肺血流量(Q)约为每分5L,二者的比率(VA/Q)约为0.8左右。但是即使在健康人,肺的各部分通气与血流的分布也都不是均匀的。直位时,肺泡的通气量和血流量都是自上而下递增的,而血流量的上下差别更大,其结果是各部肺泡的V/Q比率自上而下递减。在正常青年人VA/Q比率的变动范围自上而下约为0.6~3;随着年龄增大,变动范围扩大。尽管如此,PaO2和PaCO2最终仍可维持在正常范围。肺部疾病时,若肺泡通气不足与血流量减少发生于同一部位(如肺叶切除、大叶性肺炎灰色肝变期),其功能可由其余的健肺以适当的比例加强通气与血流来代偿,因而对换气功能影响可以不大。但大多数呼吸系统疾病时肺泡通气和血流量的改变多不相平行配合,使部分肺泡V/Q比率降低或增高,而且VA/Q比率的变动范围也扩大,因而使肺泡通气血流比例严重失调,不能保证有效的换气而导致呼吸衰竭。肺泡通气血流比例失调有两种基本形式:
(一)部分肺泡V/Q比率降低支气管哮喘、慢性支气管炎、阻塞性肺气肿等引起的气道阻塞或狭窄性病变,以及肺与胸廓顺应性降低在肺的各个部分所造成的影响,往往都不是均匀一致,而是轻重不一的。因此都可导致肺泡通气分布的严重不均。如肺泡通气明显降低而血流无相应减少甚至还增多,即VA/Q比率降低,则流经这部分肺泡的静脉血未经充分氧合便掺入动脉血内。这种情况类似肺动-静脉短路,故称功能性分流增加。正常成人由于肺内通气分布不均形成的功能性分流约仅占肺血流量的3%。慢性阻塞性肺疾患严重时,功能性分流明显增加,可相当于肺血流量的30~50%,因此可以严重地影响换气功能而导致呼吸衰竭(图13-3(3))。
(二)部分肺泡V/Q比率增高某些肺部疾患,如肺动脉压降低、肺动脉栓塞、肺血管受压扭曲和肺壁毛细血管床减少等时,VA/Q比率增高。患部肺泡血流少而通气多,吸入的空气没有或很少参与气体交换,因而与气道的情况类似,即犹如增加了肺泡死腔量,故这种情况又称死腔样通气。此时肺脏总的有效通气量必然减少,因而也会引起血气异常。正常人的生理死腔量(VD)约占潮气量(VT)的30%,上述疾病时可使死腔气量明显增多,VD/QT可高达60~70%(图13-3(4))。
总之,在通气分布不均或血流分布不均以及通气和血流配合不当时,有的肺泡通气与血流比率明显降低,可低至0.01;有的肺泡通气血流比率明显增高,可高达10以上,也有的仍接近0.83,各部分肺泡的V/Q比率的变动范围明显扩大,严重偏离正常的范围。这样就可引起换气障碍并从而导致呼吸衰竭。
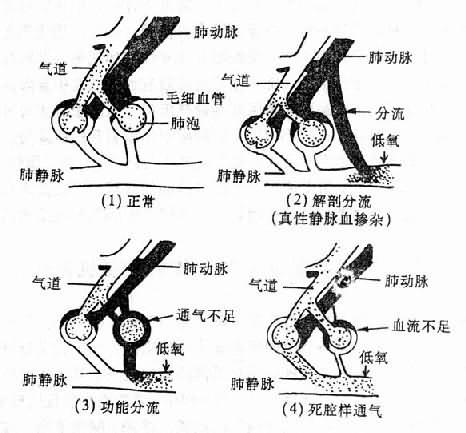
图13-3 肺泡通气与血流比例失调模式图
四、肺循环短路增加
生理情况下肺内有一部分静脉血经支气管静脉和极少数的肺内动-静脉交通支直接流入肺静脉,称“短路”(shunt)或右-左分流。此外,心内最小静脉的静脉血也直接流入左心。这些都属解剖分流,其血流量约占心输出量的2~3%。因为静脉血未经动脉化即掺入动脉血中,故又称静脉血掺杂。支气管扩张(伴支气管血管扩张)、先天性肺动脉瘘、肺内动静脉短路开放等病变,可增加解剖分流,使静脉血掺杂显着增多而引起血液气体异常(图13-3(2))。有人将肺内病变所引起的肺泡完全不通气但仍有血流者也视为短路,见于肺实变、肺不张等。
呼吸衰竭的发病机制中,单纯的通气不足、单纯的弥散障碍、单纯的肺内短路增加或单纯的肺泡通气与血流比例失调是较少的,这些因素往往同时存在或相继发生作用。现以成人呼吸窘迫综合征(adult respiratory distress syndrome, ARDS)为例概要说明之。
成人呼吸窘迫综合征是在原无心肺疾患的病人因急性弥漫性肺泡-毛细血管膜损伤,以致外呼吸功能严重障碍而发生的急性呼吸衰竭,常见于休克、创伤、败血症、过量输液、体外循环术和氧中毒等时。其早期病变主要为肺严重水肿、出血、透明膜形成、肺不胀、微血栓形成、肺血管内皮细胞及肺泡Ⅰ型上皮细胞肿胀变性与坏死等。此时呼吸衰竭的发生可有下述多种机制参与:①由于肺水肿、肺不胀等使肺顺应性降低而引起限制性通气障碍,也可因支气管痉挛和气道内液体增加而导致阻塞性通气障碍,但因患者多有呼吸加速,故肺泡总通气量可无明显减少,但其分布则不均匀。②肺泡膜增厚引起弥散障碍。③肺小动脉内微血栓或脂肪栓塞,使部分肺泡血流不足,形成死腔样通气。另一方面,因肺顺应性降低、肺不胀、肺泡内充满水肿液或气道受阻等原因,使部分肺泡通气减少而血流未相应减少,故可造成大量肺内短路或功能性分流增加,这些因素均导致肺泡的通气与血流比例失调。
第三节 呼吸衰竭时机体的主要机能代谢变化
外呼吸功能障碍引起的直接效应是血液气体的变化,即PaO2降低或同时伴有PaCO2增高或降低。呼吸衰竭时机体各系统的机能变化的最重要的原因就是低氧血症,高碳酸血症和酸碱平衡紊乱。低氧血症和高碳酸血症对机体的影响取决于其发生的急缓、程度、持续的时间以及机体原有的机能代谢状况等。在发病过程中,尤其是在病程迁延的慢性呼吸衰竭病人,常出现一系列代偿适应反应,可改善组织的供氧,并调节酸碱平衡,或改变组织器官的机能代谢以适应新的环境。严重时,呼吸系统以外的器官也可发生功能紊乱,甚至成为死亡的直接原因。
一、气体代谢变化
呼吸衰竭时必定有血液气体的变化,有时因原发疾病的掩盖,呼吸衰竭本身引起的临床表现可不明显,但根据血液气体的变化可判断是否发生了呼吸衰竭。由于病变部位、性质与程度以及机体反应性和代偿功能的不同,呼吸衰竭时的血液气体变化也可不全相同,大致有下列四种类型:
(一)PaO[XB]2[/XB]下降PaCO[XB]2[/XB]上升,二者成一定比例关系
各种原因只要引起总肺泡通气量不足,就会使肺泡气氧分压(PAo2)下降和肺泡气二者氧化碳分压(PAco2))增高,流经肺泡毛细血管的血液不能充分动脉化,因而必然导致(Pao2)降低和PAco2增高。PAco2取决于每分肺泡通气量(VA)与体内每分钟产生的二氧化碳量(Vco2),可以下式表示之,PAco2=0.86×Vco2/VA。如Vco2不变,只要通气减少,PAco2必增,而肺泡毛细血管末端血液的二氧化碳分压几乎和PAco2相等,故Paco2增高是全肺通气不足的特征。根据肺泡气体公式(Pao2=Pio2-PAco2/R,如吸入气氧分压(Pio2)为150mmHg,当通气减少一半时,PAco2即由正常的5.33kPa(40mmHg)增至10.7kPa(80mmHg)。在R(即RQ,呼吸商)为0.8时,PAo2就由15.3kPa(100mmHg)降至6.67kPa(50mmHg)。这种变化反映在动脉血液气体分压也有相似的变化,即Pao2下降和Paco2上升,二者呈一定比例地加重(图13-4A)。通常见于呼吸中枢抑制与中央气道狭窄阻塞等所引起的通气不足时。
(二)PaO[XB]2[/XB]下降而Paco[XB]2[/XB]变动不大
这种血液气体的变化可见于下列情况:
1.肺泡通气血流比例失调肺功能分流增加时,流经此处的静脉血不能充分动脉化,因此氧分压降低二氧化碳分压增高。由于Paco2升高剌激中枢化学感受器以及PaO2降低剌激主动脉体,颈动脉体化学感受器,故呼吸快速,每分通气量增加,代偿性过度通气的肺泡的PAo2增高而PAco2降低,血液流经这些肺泡,其氧分压也会有所增高,二氧化碳分压也必然降低。混合的肺静脉血最终往往是氧分压低于正常,而二氧化碳分压正常或略有降低。死腔样通气增多时,病变区域的血液固然可以充分氧合,但浪费了通气,其余肺泡就会相对通气不足,因而也出现功能分流增加,其血液气体变化也是PaO2降低而PaCO2变动较小(图13-4C)。
图13-4 呼吸衰竭时不同类型的血气变化A. PaO2↓PaCO2↑,二者变动值呈一定比例B. PaO2↓PaCO2↑,二者变动值不呈一定比例C.PaO2↓PaCO2,不变E. D用氧后D.PaO2↓PaCO2明显降低 F. B用氧后
部分肺泡通气血流比例失调时,往往只引起低氧血症而无高碳酸血症,主要是由血液二氧化碳解离曲线和氧离曲线的特性所决定。在PaCO2相当于5.00-8.00kPa(40-60mmHg)时,血液二氧化碳分压改变与二氧化碳的含量改变几乎呈直线关系(图13-5),在代偿性过度通气的肺泡,只要PAco2降低,血液中二氧化碳就可得到更多的排除,因此可以代偿那些通气不足的肺泡所造成的二氧化碳潴留。而氧离曲线的特点则与此不同。当氧分压为13.3kPa(100mmHg)时,血氧饱和度已达95~98%,过度通气的肺泡即使提高了氧分压,流经的血液氧饱和度和氧含量的增加也是极微少的,因此不能代偿通气不足的肺泡所造成的低氧血症(图13-6)。
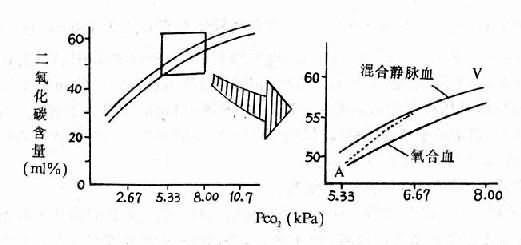
图13-5 血液二氧化碳解离曲线图中kPa相当于mmHg的数值kPa mmHg
| 2.67 | 20 |
| 5.33 | 40 |
| 8.00 | 60 |
| 10.7 | 80 |
| 13.3 | 100 |
| 16.0 | 120 |
| 18.67 | 140 |
肺顺应性降低引起的限制性通气障碍的患者,呼吸常加快,总通气量可增加。同时因其病变常不是均匀一致的,还有肺泡通气血流比例失调,故体内二氧化碳可化偿性地排出增多,其血液气体变化亦常为Pao2下降,而Paco2不变或略低。
功能性分流增加和死腔样通气增多引起的缺氧,都可用吸入高浓度氧治疗得到缓解。肺内短路也可引起类似功能分流的血气变化,唯吸入高浓度氧不能改变缺氧。
2.弥散障碍肺泡膜面积减少、厚度增加或通透性降低时,除因同时存在的肺泡通气血流比例失调可引起低氧血症外,严重时本身也可因氧从肺泡弥散到血液的过程受阻而使PaO2下降。但因二氧化碳的弥散能力很强(约比氧大20倍)其排出受影响较小,故PaCO2多正常,甚至因为代偿性通气过度而有所下降。这类病人吸入高浓度氧也可解除低氧血压。
弥散障碍与肺泡通气血流比例失调都使换气不能有效地进行,因此Pao2低于PAo2,PAo2和Pao2的差值增大是它们的共同特征。
(三)PaO[XB]2[/XB]下降,Paco[XB]2[/XB]升高,二者变化不成一定比例关系
这种血液气体变化可见于下列情况:
1.胸廓顺应性降低引起的限制性通气不足 由于胸廓的病变常不对称均匀,故此时呼吸衰竭的发病机制,不仅是肺泡通气不足,还有肺泡通气血流比例失调。此时,胸廓的的病变又限制了通气反应,使之不能充分加强,二氧化碳虽可代偿性排出一部分,但又排出不足,故血液气体变化为Pao2降低,PaCO2上升,但二者的变化不是一致的比例关系,其中以Pao2下降程度较为严重(图13-4B)。
图13-6 肺泡通气与肺泡PO2、肺泡Pco2、
动脉血氧饱和度和动脉血pH的关系
2.慢性阻塞性肺疾患由于此时外周小气道阻塞等病变也不均匀一致,故肺泡通气血流比例明显失调。这是导致血液气体异常的重要机制。血液气体变化除可表现为Pao2降低Paco2不变外,在有些患者可能因中枢神经系统反应性不同而使增强通气的反应有所减弱,故不仅有Pao2降低,而且早期就有Paco2升高,只是Paco2变动程度小于Pao2变动的程度,二者不呈固定的比例关系。这些慢性呼吸衰竭患者因肺泡通气血流比例失调而CO2增强呼吸的作用又减弱,故Paco2升高,直到每分钟产生的CO2全由通气良好的肺泡排出,则又可使二氧化碳的生成和排出在高Paco2水平上达到新的平衡,因此Paco2可较长期维持于较高水平,患者尚能继续生存。
(四)Pao[XB]2[/XB]下降,Paco[XB]2[/XB]也明显下降
在肺泡通气血流比例失调引起呼吸衰竭的患者中,有些在Pao2下降的同时Paco2也明显降低(图13-4D)。这种情况可见于成人呼吸窘迫综合征和肺广泛纤维性变等。血液气体的这种变化,可能是由于此时肺间质中感受器受到强烈刺激而反射性引起通气极度加强所致,故即使纠正缺氧,通气过度仍可持续存在。
二、酸碱平衡及电解质紊乱
呼吸衰竭时,不仅因外呼吸障碍可引起酸碱平衡紊乱,而且还可因并发肾功能障碍、感染、休克以及某些治疗措施不当等因素而出现不同类型的酸碱平衡紊乱。因此患者的表现可能是多样的。由于外呼吸功能严重障碍本身引起的有呼吸性酸中毒、呼吸性碱中毒、代谢性酸中毒或呼吸性酸中毒合并代谢性酸中毒。
(一)呼吸性酸中毒 Ⅱ型呼吸衰竭时,大量二氧化碳潴留,可造成原发性血浆碳酸过多。发病急骤者,往往代偿不全而出现失代偿性呼吸性酸中毒,如发病较缓慢,则可出现代偿性呼吸性酸中毒。此时血液电解质主要有如下的变化。①血清钾浓度增高:急性呼吸性酸中毒时,主要是由于细胞内外离子分布改变,细胞内钾外移而引起血清钾浓度增高;慢性呼吸性酸中毒时,则是由于肾小管上皮细胞泌氢和重吸收碳酸氢钠增多而排钾减少,故也可导致血清钾浓度增高。②血清氯浓度降低,碳酸氢根增多;当血液中二氧化碳潴留时,在碳酸酐酶及缓冲系统作用下,红细胞中生成碳酸氢根增多,因而进入血浆的碳酸氢根也增多,同时发生氯转移,血浆中氯离子进入红细胞增多,因此血清氯离子减少而碳酸氢根增加。另一方面,由于肾小管泌氢增加,碳酸氢钠重吸收和再生增多,而较多氯离子则以氯化钠和氯化铵的形式随尿排出,因而也可引起血清氯离子减少和碳酸氢根增多。
(二)代谢性酸中毒或呼吸性酸中毒合并代谢性酸中毒 由于严重缺氧,无氧代谢加强,酸性代谢产物增多,可引起代谢性酸中毒,或呼吸性酸中毒合并代谢性酸中毒。如患者合并肾功能不全或感染、休克等、则因肾脏排酸保碱功能障碍或体内固定酸产生增多,将更加重代谢性酸中毒。此时血清钾浓度增高可更明显。
(三)呼吸性碱中毒 Paco2明显下降的患者,可因原发性碳酸过低而发生呼吸性碱中毒,由于发病急骤,故多为失代偿性呼吸性碱中毒。此时因细胞外钾离子进入细胞内,可发生血清钾浓度降低。由于二氧化碳排出过多,血浆中碳酸氢根移入红细胞增多,氯离子则转移至红细胞外,加之肾排出氯也减少,故血清氯浓度增高。血浆碳酸氢根则因移入红细胞以及肾小管重吸收和再生碳酸氢钠减少而浓度降低。
此外,某些呼吸衰竭患者可以发生代谢性碱中毒,多属医源性,发生于治疗过程中或治疗后。如使用人工呼吸机,过快排出大量二氧化碳,而原来代偿性增加的碳酸氢根又不能迅速排出,因此发生代谢性碱中毒;由于钾摄入不足、应用排钾利尿剂和肾上腺皮质激素等可导致低钾症性碱中毒等。
三、呼吸系统变化
呼吸衰竭病人的呼吸功能变化,很多是由原发疾病引起的。如阻塞性通气障碍时,由于气流受阻,呼吸可减慢。随着阻塞部位的不同,以及阻塞是可变还是固定,呼气与吸气困难的程度亦可不一。在肺顺应性降低的疾病,则因牵张感受器或肺-毛细血管旁感受器(juxtapulmonary-capillary receptor,J感受器)兴奋而反射地引起呼吸浅快。中枢性呼吸衰竭可出现呼吸浅慢,或出现潮式呼吸、间歇呼吸、抽泣样呼吸、吸气样呼吸、下颌呼吸等呼吸节律紊乱。
外呼吸功能障碍造成的低氧或高碳酸血症可进一步影响呼吸功能。Pao2降低作用于颈动脉体与主动脉体化学感受器(其中主要是颈动脉体化学感受器),反射性增加通气,但此反应要Pao2低于8.0kPa(60mmHg)时才明显。二氧化碳潴留主要作用于中枢化学感受器,使呼吸中枢兴奋,从而引起呼吸加深加快,增加肺泡通气量。但Pao2低于4.0kPa(30mmHg)时或Paco2超过12.0kPa(90mmHg)时,将损害或抑制呼吸中枢。
在慢性Ⅱ型呼吸衰竭病人,随着低氧血症和高碳酸血症发展的逐渐严重,其呼吸调节也将发生变化:此类病人的中枢化学感受器常被抑制而对二氧化碳的敏感性降低,此时引起通气的冲动大部来自缺氧对外周化学感受器的剌激。如果给予高浓度的氧吸入,则虽可缓解低氧血症,但却因此解除了缺氧反射性兴奋呼吸中枢的作用,故易招致呼吸进一步的抑制,使通气更减弱而二氧化碳的潴留更加重。此外,吸入高浓度氧使血氧饱和度回升后,由于霍尔登(Haldane)效应使二氧化碳解离曲线向右下移动(图13-5),也可引起Paco2进一步增高。
四、中枢神经系统变化
呼吸衰竭时,由于低氧血症与高碳酸血症的作用,中枢神经系统的功能可发生明显变化,轻度时可使兴奋性升高,严重时将发生一系列中枢神经系统的功能障碍,直接威胁生命。低氧血症和高碳酸血症的作用很难截然分开。
中枢神经对缺氧很敏感,故最易受损。Pao2为8.0kPa(60mmHg)时可出现智力和视力轻度减退。如Pao2迅速降至5.33~6.66kPa(40~50mmHg)以下时,就会引起一系列神经精神症状,如头痛、不安、定向与记忆障碍、精神错乱、嗜睡,以致惊厥和昏迷,Pao2低于2.67kPa(20mmHg)时,只需几分钟就可造成神经细胞的不可逆性损害。
二氧化碳潴留发生迅速而严重时,也能引起严重的中枢神经系统功能障碍,称为二氧化碳麻醉。一般认为,当Paco2超过10.7kPa(80mmHg)时,可引起头痛,头晕、烦躁不安、言语不清、扑翼样震颤、精神错乱、嗜睡、昏迷、抽搐等。其可能的作用机制如下:
(一)二氧化碳直接作用于脑血管,使之扩张。一般认为Paco2升高1.33kPa(10mmHg),脑血流量约可增50%;由此可以影响脑循环,并引起毛细血管通透性增高,其结果是脑血管充血、间质水肿、颅内压升高和视神经乳头水肿。严重时还可导致脑疝形成。
(二)正常脑脊液的缓冲作用较血液为弱,其pH也较低(7.33~7.40),而Pco2却比动脉血的高,血液中的碳酸氢离子及氢离子又不易进出脑脊液,故后者的酸碱调节需时较长。Ⅱ型呼吸衰竭病人的脑脊液中二氧化碳也增多,但因脑脊液缓冲能力差,故氢离子浓度增高的程度大于血液,继而又可加重细胞内酸中毒,使神经细胞的功能发生障碍,细胞膜结构受损,通透性增高。这些变化一方面改变神经细胞内外离子分布,另一方面使溶酶体膜稳定性降低,释出的各种水解酶,能促使蛋白分解与细胞死亡。细胞内外离子分布的改变和细胞内蛋白分解又可使细胞内渗透压升高,促使脑细胞肿胀,颅内压升高。
目前国内已普遍地将呼吸衰竭时由于中枢神经功能障碍而出现一系列神经精神症状的病理过程称为肺性脑病。
五、循环系统变化
一定程度的缺氧可反射性兴奋心血管运动中枢,从而使心率加快,心输出量增加,皮肤及腹腔内脏血管收缩,因而发生血液重分布和血压轻度升高。此外,缺氧时也可间接地因通气加强,胸腔负压增大,回心血量增加而影响循环功能。这种变化在急性呼吸衰竭时较为明显,且有代偿意义。严重低氧血症时,因循环中枢与心血管受损,可发生低血压,心收缩力降低,心律失常等后果。缺氧尤其是肺泡气氧分压降低可使肺小动脉收缩,这是呼吸衰竭时引起肺动脉高压与右心衰竭的主要原因。
一定程度的二氧化碳潴留可与缺氧协同作用,反射性地引起循环功能的代偿性变化。二氧化碳可反射性地引起外周血管收缩,但其直接作用,则除肺动脉外多为引起血管扩张,故二氧化碳潴留的最终效应通常为:①皮肤血管扩张,因而肢体皮肤温暖红润,常伴大量出汗;②睑结膜血管扩张充血;③脑血管扩张,脑血流量增加;④广泛的外周血管扩张,可引起低血压;⑤肾与肺小动脉收缩。
呼吸衰竭可伴发心力衰竭,尤其是右心衰竭,其发生原因为肺动脉高压和心肌受损。目前认为,不论是急性或慢性呼吸衰竭,肺血管功能性改变在肺动脉高压的发病中都具有极重要意义。缺氧(主要是肺泡气氧分压降低)可引起肺血管收缩,若合并二氧化碳潴留,血液氢离子浓度增高,就更可增加肺血管对缺氧的敏感性,使肺血管收缩进一步加重,从而大大增加肺循环的阻力。如原发肺部疾病引起肺小动脉壁增厚、管腔狭窄或纤维化、肺毛细血管网受压破坏与减少、毛细血管内皮细胞肿胀或微血栓阻塞等变化,则亦可增加肺循环阻力而导致肺动脉高压。有的慢性呼吸衰竭患者血液中的红细胞增多,因而血液粘滞性增高,而后者又可因合并酸中毒而加重,这也是肺动脉高压发病的一个因素。某些患者可因血量增多,或因呼吸深快以致胸腔负压增大,或因体循环外周血管扩张,阻力降低,以致静脉回流增加而加重右心负荷。呼吸衰竭引起右心衰竭的另一发病因素是心肌受损。缺氧,高碳酸血症、酸中毒和电解质代谢紊乱均可损害心肌。长期持续缺氧还可引起心肌变性、坏死、纤维化等病变。心肌受损加上负荷过重,就可导致右心衰竭。
六、肾功能变化
呼吸衰竭时肾功能也可遭到损害,轻者尿中出现蛋白、红细胞、白细胞及管型等。严重时可发生急性肾功能衰竭,出现少尿、氮质血症和代谢性酸中毒等变化。此时肾脏结构往往无明显变化,故常为功能性肾功能衰竭。只要外呼吸功能好转,肾功能就可较快恢复。肾功能衰竭的基本发病机制在于缺氧与高碳酸血症反射性引起肾血管收缩,从而使肾血流量严重减少。若患者并发心力衰竭、播散性血管内凝血或休克,则肾脏的血液循环障碍将更严重而肾功能障碍也将加重。
七、胃肠道变化
严重缺氧可使胃壁血管收缩,因而能降低胃粘膜的屏障作用。二氧化碳潴留可增强胃壁细胞碳酸酐酶活性,使胃酸分泌增多,而且有的患者还可合并播散性血管内凝血、休克等,故呼吸衰竭时可出现胃肠道粘膜糜烂、坏死、出血与溃疡形成等变化。
第四节 呼吸衰竭的防治原则
一、防治原发病
针对引起呼吸衰竭的原发疾病进行预防,或在发病后及时进行积极处理。
二、防止与去除诱因的作用
对于可能引起呼吸衰竭的疾病,还必须同时防止诱因的作用。例如对于创伤、休克患者,要避免吸入高浓度氧、输给久存血库的血液或输液过量等,以免诱发成人呼吸窘迫综合征。有呼吸系统疾病的患者必须作手术时,应先检查病人的肺功能储备力。对肺功能已有损害或慢性呼吸衰竭的病人更应积极防止及去除各种诱因的作用,以免诱发急性呼吸衰竭。
三、畅通气道和改善通气
常用的方法有:①清除气道内容物或分泌物;②解除支气管痉挛;③用抗炎治疗减轻气道的肿胀与分泌;④必要时作气管插管或气管切开术;⑤给以呼吸中枢兴奋剂;⑥掌握适应症,正确使用机械辅助通气。
四、改善缺氧
呼吸衰竭时必定有严重缺氧,因此纠正缺氧,提高Pao2水平对每个患者都是必要的。其目的在于短期内争取使Pao2升至6.67~8.0kPa(50~60mmHg),动脉血氧饱和度升至85%左右。
Ⅰ型呼吸衰竭有缺氧而无二氧化碳潴留,可吸入较高浓度的氧(一般不超过50%)。慢性Ⅱ型呼吸衰竭时,由于呼吸中枢反应性的变化,一般认为给氧原则上以持续低浓度低流量为宜。应使Pao2达到安全水平8.0~9.33kPa(60~70mmHg),以求能供给组织以必要的氧而不致引起二氧化碳麻醉,然后根据病人情况调整并逐渐提高吸入氧的浓度及流量。如在给氧时出现二氧化碳分压进行性上升,则须助以人工通气以促进二氧化碳的排出。
五、密切观察监护,综合治疗
注意纠正酸碱平衡紊乱与水电解质紊乱;维持心、脑、肾等重要器官的功能;防治常见的严重并发症。
[附]:成人呼吸窘迫综合征
成人呼吸窘迫综合征(adultrespiratory distress syndrome, ARDS)是指在某些疾病过程中(例如创伤、烧伤、感染等)。特别是在休克初期复苏后,突然出现以进行性缺氧和呼吸困难为特征的急性呼吸衰竭综合征。此时即使吸入高浓度氧,也难于纠正低氧血症。Ashbaugh于1967年首先报导了这种综合征。由于其临床过程与新生儿呼吸窘迫综合征类似,故命名为成人呼吸窘迫综合症。
一、其临床主要表现及诊断依据为:
1.败血症、休克、创伤或误吸等患者突然发生进行性呼吸困难,呼吸频率在20次/分以上。
2.顽固性低氧血症 Pao2<8kPa(<60mmHg),吸纯氧15分钟后Pao2仍低于46.7kPa(350mmHg)[正常人吸纯氧可使Pao2高达73.3kPa(550mmHg)]、动脉血二氧化碳分压也降低,Pao2<4.7kPa(<35mmHg)。晚期Paco2也可高于正常。
3.胸廓肺顺应性降低 低于50ml/98Pa,正常约为100ml/98Pa。
4.胸部X光检查 可见肺纹理增加,发展为斑片状阴影,甚至弥漫性呈毛玻璃样。
5.必须排除由慢性肺部疾病及左心疾病引起肺水肿,肺动脉楔压小于Pao2为1.76kPa(18cmH2O)。
成人呼吸窘迫综合症发病率高,病死率也很高,大多数病人在发病后二周内死亡。
二、成人呼吸窘迫综合征的原因
ARDS是由许多原因引起的急性肺泡-毛细血管膜损伤。有些原因可直接损伤肺:如①吸入烟雾、毒气、胃内容物及溺水;②服用过量海洛因或水杨酸盐;③细菌、病毒及真菌等所致肺部感染;④脂肪、羊水及血栓等引起肺栓塞;以及⑤肺挫伤、放射线损伤与氧中毒等。有些全身性病理过程可引起肺损伤,如败血症、休克、弥散性血管内凝血、过敏反应、创伤及烧伤等。烧伤面积超过40%就可能导致Pao2明显降低。有些治疗措施也可能引起ARDS,如血液透析、体外循环、用尼龙丝去除白细胞等。根据病因的不同及病变特点,ARDS曾有20多个名称,如创伤后湿肺、败血症肺、休克肺、输血后肺、微血管漏出综合征,充血性肺不张、透明膜病、出现性肺综合征、僵肺综合症、进行性肺实变等。 二、成人呼吸窘迫综合征的病理变化各种原因所致急性肺泡-毛细血管膜损害伤的病理变化均类似,可分为急性阶段与慢性阶段病变:
(一)急性阶段病变
主要为广泛肺泡血管内皮和肺泡上皮损伤所致肺水肿,首先是肺间质水肿,后出现肺泡水肿,肺重可达正常值之三倍。肺泡腔内液体蛋白质含量高,甚而是血性液体,并有血细胞、巨噬细胞、细胞碎片、无定形物质、纤维蛋白条和表面活性物质的残存物,偶而可见细胞碎片和蛋白等物质在纤维蛋白网眼中形成透明膜。
(二)慢性阶段病变
发病数天后进入慢性阶段,病变以细胞增生为主,两周后即可出现纤维化。Ⅱ型上皮细胞增生取代了变性坏死的Ⅰ型肺泡上皮细胞,加上各种细胞的浸润使肺泡间隔增厚,肺泡腔与肺泡管内富含蛋白质的的液体机化而形成纤维化。
三、成人呼吸窘迫综合征的发病机制
ARDS的病变主要是肺泡-毛细血管膜损伤引起肺水肿和继后的细胞增生和纤维化。细胞增生反应和纤维化的机制和炎症修复过程相同。故此处着重讨论急性肺损伤的机制。ARDS病人均有肺动脉高压,故有人强调肺微血管内高压为肺水肿的原因。但病人肺动脉楔压往往并不高,说明毛细血管压并不一定高。水肿液蛋白质含量丰富,动物实验也证明,类似ARDS的动物模型肺淋巴液流量增大,肺淋巴液中蛋白质浓度与血浆蛋白质浓度之比值大于0.7;此时,静脉注入的高分子右旋糖酐(分子量500,000)可透入肺泡液,均说明ARDS时的肺水肿主要是渗透性肺水肿,由于肺泡-毛细血管膜损伤使其通透性增高所致。ARDS中肺泡毛细血管膜通透性增高的机制并未完全阐明。有些原始病因能直接损伤肺泡毛细血管膜使其通透性增高,如吸入胃酸、毒气,烟熏、放射性损伤及细胞毒素作用等。大量实验表明,更主要的是继发性损伤,即通过白细胞和血小板在肺内聚集引起肺泡-血管膜损伤使其通透性增高。
(一)中性粒细胞在ARDS发病中的作用
ARDS病人外周血液中中性粒细胞数减少。肺活检可见肺内有中性粒细胞聚集和浸润。支气管肺泡洗出液中中性粒细胞可增加20~100倍。现在一般认为中性粒细胞在肺中聚集、激活、释放氧自由基、蛋白酶和脂质代谢产物,从而导致肺微血管膜及肺泡上皮的损伤,是ARDS肺水肿的主要发病机制。
1.中性粒细胞在肺血管中聚集 正常人在直立体位时约有10~20%的中性粒细胞聚集在肺毛细血管床中,这是一种物理性扣留。由于一个肺泡有近千节(segment)毛细血管,每节毛细血管长1~30μm(平均8μm),直径1~15μm(平均5μm),一个血细胞从肺动脉流入肺静脉要经过100个以上的毛细血管节,中性粒细胞直径大于毛细血管口径,其形状与红细胞相比更接近球形,故变形能力较差,变形速度较慢,所以中性粒细胞易被扣留在肺毛细血管床中。由于肺毛细血管床容量较大,白细胞的扣留对肺血管的阻力和肺动脉压的影响不大。物理性扣留的中性粒细胞一般不进入肺泡腔,故正常人支气管肺泡洗出液中的细胞90~95%是巨噬细胞。
ARDS时中性粒细胞在肺血管中的聚集是化学性粘附(adhesion),是由于趋化因子作用的结果。实验证明,中性粒细胞经趋化因子激活后与血管内皮细胞之间的亲和力显著增强。肺泡毛细血管总面积达60m2,可粘附大量中性粒细胞,以至使外周血液中白细胞数减少。
趋化因子种类很多,主要有补体激活产物C5a、纤维蛋白降解产物(FDP),花生四烯酸代谢产物如白三烯B4(LTB4)、羟花生四烯酸(HETE)和血栓素A2(AXA2),血小板活化因子(PAF),以及其他具趋化作用的蛋白质、多肽和脂质。其中研究较多的是补体的作用。在临床确诊ARDS前8小时血浆C5a往往已升高。激活的补体本身并不直接损伤肺血管内皮细胞,它是通过激活中性粒细胞起损伤作用。给出羊灌注激活的补体可导致肺水肿,如先用氮芥使粒细胞减少后再灌注激活之补体,则肺水肿的发生明显较经。激活的中性粒细胞可浸润入肺间质和进入肺泡腔。ARDS病人支气管肺泡洗出液中存在补体碎片及大量中性粒细胞。近年来发现中性粒细胞表面有一组糖蛋白,后者与粒细胞的的粘附和吞噬功能有关,它们是巨噬细胞分子-1(macrophage-1,Mac-1)淋巴细胞功能相关抗原-1(lymphocyte function associatedantegen-1,LFA-1),其中Mac-1与粘附功能关系最密切。正常中性粒细胞表面仅有少量Mac-1表达,在病理情况下,趋化因子的作用使中性粒细胞表面Mac-1表达量增加,促进中性粒细胞与血管内皮细胞间的粘附。
2.中性粒细胞对肺泡-毛细血管膜的损伤动物实验中静脉注入内毒素、空气等可复制急性肺微血管损伤的模型,用佛波豆蔻醚乙酸盐(phorbol myristate acetate,PMA)激活的中性粒细胞灌注离体肺也可使肺毛细血管通透性增高。如先用羟基脲、氮芥等使动物中性粒细胞数减少,则内毒素、空气栓子等对肺微血管的损伤明显减轻。细胞培养中发现,中性粒细胞秘须紧密粘附于内皮细胞才能使单层肺动脉内皮的通透性增高。以上说明减轻。细胞培养中发现,中性粒细胞必须紧密粘附于内皮细胞才能使单层肺动脉内皮的通透性增高。以上说明,中性粒细胞的粘附与激活在ARDS发病中具有重要作用,很可能是中性粒细胞激活时释放的氧自由基、蛋白酶、脂质代谢产物和肽类物质等造成肺泡-毛细血管膜的损伤。
(1)氧自由基的作用:中性粒细胞被激活时,耗氧量急剧上升,比静息时增加数倍至数十倍。此时细胞膜上的NADPH氧化酶(NADPhoxidase)被激活,将还原型辅酶Ⅱ(NADPH)转变为氧化型(NADP),氧分子则获得电子形成超氧阴离子O2-,由O2-又可生成H2O2和OH·。将正常中性粒细胞和其激活剂PMA一起灌注离体肺可引起肺水肿,水肿液蛋白质含量高;如将遗传性慢性肉芽肿病患者的中性粒细胞与PMA灌注则不引起肺水肿,因为慢性肉芽肿病患者的中性粒细胞缺乏NADPH氧化酶、氧自由基的生成少。另外,静脉内注射氧自由基清除剂,如超氧化物歧化酶(SOD)、过氧化氢酶、过氧化物酶、二甲本硫脲等,可减轻实验动物的急性肺损伤。可见,中性粒细胞激活时通过释放氧自由基引起肺损伤。
氧自由基损伤肺微血管内皮及肺泡上皮细胞的作用可能有以下几个方面:①作用于细胞膜和细胞器膜,使其脂质过氧化,从而损害细胞膜和细胞膜的结构和功能;②作用于酶,使之失活;③作用于α1—蛋白酶抑制物,使之失活,从而增强溶酶体释放的蛋白酶对组织的破坏作用;④作用于血浆成份,可形成一种很强的趋化物,引起更多的中性粒细胞在肺内聚集与激活,产生更多的氧自由基,由此形成阳性反馈,加重肺的损伤。
(2)蛋白酶的作用:中性粒细胞中溶酶体含有多种中性蛋白酶和酸性蛋白酶。当中性粒细胞被激活或破坏时,释出的这些酶可引起周围蛋白质的分解和组织结构的破坏,使肺泡-毛细血管膜的通透性增高。其中研究较多的是中性粒细胞弹性蛋白酶。例如实验中发现,ARDS病人支气管肺泡洗出液中弹性蛋白酶活性很高,给动物注射内毒素或油酸复制ARDS模型时,其血浆及肺泡洗出液中弹性蛋白酶含量也增多;给动物注射中性粒细胞弹性蛋白酶可引起肺血管内皮及肺泡上皮的通透性增高;组织培养中加入弹性蛋白酶导致内皮细胞分散等,说明中性粒细胞弹性蛋白酶与ARDS中的肺损伤有关。弹性蛋白酶可降解弹性蛋白、胶原蛋白、纤维连接蛋白(fibronectin,FN)等。纤维连接蛋白在内皮细胞之间和内皮细胞与基底膜之间起“锚连”作用。纤维连接蛋白受损,则血管通透性增高。
肝脏和肺泡巨噬细胞能合成α1-蛋白酶抑制物(α1-protease inhibitor,α1-PI),后者能抑制弹性蛋白酶。虽然ARDS病人血浆α1-PI可正常,支气管肺泡洗出液中α1-PI活性却降低,可能由于中性粒细胞产生的自由基使其氧化灭活所致。蛋白酶与蛋白酶抑制物间的失衡更加重了蛋白酶对组织的损伤,使肺泡-毛细血管膜通透性增高。
3.脂类代谢产物的作用内毒素等许多致病因素激活中性粒细胞、巨噬细胞、肥大细胞、内皮细胞等细胞膜上的磷脂酶A2,使膜磷脂裂解为花生四烯酸,后者通过环加氧酶途生成前列腺素,通过脂加氧酶途径生成白三烯。一般认为白三烯,TXA2、和PGF2α既可收缩肺小动脉引起肺动脉高压,也能增加肺微血管的通透性;而PGI2和PGE1则有扩张血管、降低血压,和使血管通透性降低的作用。急性肺损伤的动物及病人肺泡洗出液及血液中TXA2、PGF2α和LTs均增多。有实验证明PGI2和PGE1对急性肺损伤有一定的治疗作用。白细胞、巨噬细胞、肥大细胞和内皮细胞等激活后还可释放血小板活化因子(PAF)。PAF可促使血小板聚集和TXA2合成,从而导致微血管通透性增高。
4.蛋白类物质的作用巨噬细胞等激活后尚可释放蛋白类物质,其中较重要的有肿瘤坏死因子(TNF)及白细胞介素ⅠIL-1)。人及动物在内毒素血症时血浆中TNF和IL-1增多。TNF能使肺血管通透性增高,并促使中性粒细胞在肺中聚集;IL-1剌激T淋巴细胞产生白细胞介素2(IL-2),后者也可使肺血管通透性增高。
总之,现在一般认为,中性粒细胞巨噬细胞在肺内聚集、激活释出大量氧自由基和蛋白酶及脂类代谢产物和蛋白类,引起肺泡-毛细血管膜的损伤和通透性增高,导致肺水肿,这是ARDS的主要发病机制。虽然有报导白细胞减少的病人患败血症时也可发生急性肺损伤,用药物使动物中性粒细胞减少对注油酸引起肺损伤并无明显影响,但这些事实并不足以否定中性粒细胞的致病作用,因为外周血液中的中性粒细胞数不一定能反映肺循环中的粒细胞数。而且究竟需要多少中性粒细胞激活就足以引起急性肺损伤尚不了解,也可能正常聚集在肺血管中的中性粒细胞只有一部分被激活就足以损伤肺而导致ARDS。
(二)凝血系统在ARDS发病中的作用
ARDS病人肺活检及死后尸解发现,肺小动脉血栓可发生于肺充血、水肿、出血及透明膜的形成之前。ARDS病人合并有弥散性血管内凝血者,其低氧血症和肺顺应性降工远较未合并DIC者为重,中性粒细胞激活和肺组织损伤所释放的促凝物质,肺血管内皮损伤和血液停滞,可导致血小板聚集和血管内凝血形成微血栓。肺内广泛微血栓形成可能引起:①肺循环阻力增加使肺动脉压升高,未堵塞的肺血管则血液量增大和毛细血管压升高,导致压力性肺水肿;②血栓损伤血管壁和血小板释放的血管活性物质以纤维蛋白降解产物,可使血管通透性增高致渗透性肺水肿;③血小板的消耗、纤维蛋白降解产物的抗凝作用、和血管壁的损伤可引起肺内出血;④血小板释放的5-HT等介质使支气管收缩,影响肺通气。近年特别引人注目的是纤维蛋白降解产物(FDP)的作用,发现严重创伤烧伤或感染病人中,已合并ARDS者血中FDP水平比未发生ARDS者高得多,而且ARDS病情与FDP浓度有一定平行关系。将纤维蛋白碎片D(FD)注入家兔血管,可引起进行性外周血液血小板减少、肺间质内白细胞浸润,肺血管通透性增高和肺功能不全;如注入血浆白蛋白、纤维蛋白及纤维蛋白碎片E,则不出现以上病变。很可能小板上有FD特异的膜受体,后者与FD结合可激活血小板,引起血小板聚集和释放反应。另外,FD也是趋化物,能促使中性粒细胞在肺内聚集、粘附和激活,由此加重肺的损害。
正常肺毛细血管内皮的通透性比肺泡上皮高10倍。ARDS中肺毛细血管通透性的变化早于肺泡上皮,故先发生肺间质水肿,后出现肺泡水肿。肺泡上皮的损伤使Ⅱ型上皮细胞生成的表面活性物质减少,可导致肺不张,形成功能性分流。
在全身性病理过程如败血症、休克等,中性粒细胞粘附于血管内皮以及血管内凝血引起的组织损伤,不仅发生于肺内,也可发生于肝、肾、肠、心、内分泌器官等处,故不能把ARDS看成仅仅是肺的损伤。但肺的血流量最大,毛细血管床面积也最大,故肺受累最重,使病人主要表现为急性呼吸衰竭.
四、成人呼吸窘迫综合征时肺呼吸功能变化
ARDS所致外呼吸功能障碍以肺泡通气-血流比例失调为主,加上弥散功能障碍,表现为低氧血症性呼吸衰竭。极严重病例有总的肺泡通气量减少时可出现高碳酸血症性呼吸衰竭。
(一)肺泡通气-血流比例失调
由于Ⅱ型肺泡上皮细胞受损致表面活性物质的生成减少,肺泡水肿使表面活性物质被稀释和破坏,和肺泡过度通气引起的表面活性物质消耗,以致肺泡表面张力升高,肺顺应性降低,导致肺不张,由此形成功能性分流和真性分流。中性粒细胞等释出的白三烯等介质使支气管收缩,和水肿液堵塞小气道,气可造成肺通气障碍而形成功能性分流。ARDS病人分流量可达肺血流量的30%。肺血管内微血栓形成、血管活性物质引起不均匀的肺血管收缩、以及肺间质水肿对血管的压迫,不仅可增加肺血管阻力使肺动脉压升高。尚可增加死腔样通气。因此,肺泡通气-血流比例失调是病人发生呼吸衰竭最主要的原因。
(二)弥散功能障碍
肺间质和肺泡水肿、透明膜的形成和慢性阶段细胞的增生及肺纤维化,均可增加弥散膜的厚度,导致弥散功能障碍。
(三)肺泡通气量减少
ARDS时肺部病变的分布是不均的。肺顺应性降低引起的限制性通气障碍和小气道阻塞引起的阻塞性通气障碍,造成部分肺泡通气量减少,未受累或病变较轻的肺泡反而代偿性通气增强,排出过多的二氧化碳,故病人Paco2反而降低。当肺泡-毛细血管膜损伤更广泛更严重时,全肺总的肺泡通气量将减少,CO2将潴留而发生高碳酸血症,此时Pao2将进一步下降。
肺通气障碍、Pao2降低对血管化学感受器的剌激、肺充血和肺水肿对J感觉器的剌激,导致病人呼吸窘迫。[J感受器(juxtapulmonary capillaryreceptor)位于肺泡毛细血管旁,能感受毛细血管压力剌激,肺充血、肺水肿受剌激反射性地引起呼吸加快。
五、成人呼吸窘迫综合征的防治原则
(一)治疗原发疾病
消除ARDS的原因,如抗感染、抗休克治疗等。
(二)吸氧与呼气末正压呼吸
ARDS病人发生低氧血症的主要机制是肺内功能性分流,所以吸氧疗法对提高其Pao2的作用较小。吸呼高浓度氧可提高Pao2,但吸入氧浓度在60%以上2~3天就可能引起氧中毒,反而加重ARDS。呼气末正压(positiveend expiratory pressure, PEEP)呼吸使呼气末时气道及肺泡压大于大气压,可将原来萎陷的气道和肺泡张开,恢复其气体交换功能,从而减少肺内分流,提高Pao2用PEEP呼吸尚可降低吸入气的氧浓度。但呼气末压力过高会压迫肺血管和心脏,使心输出量减少,导致循环性缺氧。
(三)维持液体平衡,控制肺水肿
如适当限制入水量、利尿等。
(四)用药物减轻肺泡-毛细血管膜的损伤和降低膜通透性
针对肺泡-毛细血管膜损伤的机制,采用相应的药物进行发病学治疗,有的已取得一定疗效,有的还处于实验阶段。曾试用的药物有:抗氧化剂(如超氧化物歧化酶)、蛋白酶抑制剂、磷脂酶A2抑制剂(如阿的平)、TXA2合成酶抑制剂(如咪唑)、脂加氧酶抑制剂(如U-60,257)、肝素和右旋糖酐、C5a及TNF的抗体、钙通道阻滞剂(如戊脉安)、其他扩血管药(如腺苷)、PGE和PGI2、以及肾上腺皮质激素等。
第十四章 肾功能不全
肾脏是人体的重要排泄器官,具有排泄体内代谢产物、药物、毒物和解毒产物,以及调节体内水、电解质、酸碱平衡的功能。此外,肾脏还能分泌肾素、前列腺素、促红细胞生成素、1,25-二羟维生素D3(1,25-(OH)2-D3)等,借以调节机体的重要生理功能。因此肾脏又是一个多功能器官,它在维持人体内环境的稳定性中起着重要的作用。当各种病因引起肾功能严重障碍时,人体内环境就会发生紊乱,其主要表现为代谢产物在体内蓄积,水、电解质和酸碱平衡紊乱,并伴有尿量和尿质的改变以及肾脏内分泌功能障碍引起一系列病理生理变化,这就是肾功能不全(renal insufficiency)。
引起肾功能不全的原因可概括如下:
1.肾脏疾病 如急性、慢性肾小球肾炎,肾盂肾炎,肾结核,化学毒物和生物性毒物引起的急性肾小管变性、坏死,肾脏肿瘤和先天性肾脏疾病等。
2.肾外疾病如全身性血液循环障碍(休克、心力衰竭、高血压病),全身代谢障碍(如糖尿病)以及尿路疾患(尿路结石、肿瘤压迫)等。
第一节 肾功能不全的基本发病环节
各种病因引起肾功能不全的基本环节是肾小球滤过功能或/和肾小管重吸收、排泄、分泌功能障碍,以及肾脏的内分泌功能障碍。
一、肾小球滤过功能障碍
肾脏滤过功能以肾小球滤过率(glomerularfiltration rate ,GRF)来衡量,正常约为125ml/min。GFR受肾血流量、肾小球有效滤过压及肾小球滤过膜的面积和通透性等因素的影响。肾功能不全可能是下列有关肾小球滤过功能的因素发生改变的结果。
(一)肾血流量减少
正常成人两肾共重仅300g左右,但其血灌流量却高达心输出量的20~30%,即两侧肾脏的血液灌流量约为1200ml/min。其中约94%的血液流经肾皮质,6%左右的血液流经肾髓质。实验证明,当全身平均动脉压波动在10.7~24kPa(80~180mmHg)时,通过肾脏的自身调节,肾脏血液灌流量仍可维持相对恒定。但当平均动脉压低于8.0kPa(60mmHg)(例如在休克时)时,肾脏血液灌流量即明显减少,并有肾小动脉的收缩,因而可使GFR减少,并可使肾小管因缺血缺氧而发生变性、坏死,从而加重肾功能不全的发展。此外,肾脏内血流分布的异常,也可能是造成肾功能不全的重要原因。其主要表现为肾皮质血流量明显减少。而髓质血流量并不减少,甚至可以增多,这可见于休克、心力等竭等。
(二)肾小球有效滤过压降低
肾小球有效滤过压=肾小球毛细血管血压-(肾小球囊内压+血浆胶体渗透压)。在大量失血、脱水等原因引休克时,由于全身平均动脉压急剧下降,而肾小球毛细血管血压也随之下降,故肾小球有效滤过压降低,GFR减少。此外,肾小球入球及出球小动脉的舒缩状态,也会影响肾小球有效滤过压及滤过率。当入球小动脉舒张或出球小动脉收缩时,可提高肾小球毛细血管血压,故GFR增多;反之,当入球小动脉收缩或出球小动脉舒张时,则会降低肾小球毛细血管血压而使GRR减少。
肾小球囊内压一般比较恒定,然而在尿路梗阻,管型阻塞肾小管以及肾间质水肿压迫肾小管时,则会引起囊内压升高,肾小球有效滤过压降低,原尿形成减少。
血浆胶体渗透压与血浆白蛋白含量有关。血浆胶体渗透压的变化对肾小球有效滤过压的影响并不明显。因为血浆胶体渗透压下降后,组织间液的形成增多,可使有效循环血量减少,进而通过肾素-血管紧张素系统使肾脏入球小动脉收缩而降低肾小球毛细血管血压。可见在血浆胶体渗透压下降时,肾小球有效滤过压不会发生明显改变。在大量输入生理盐水,引起循环血量增多和血浆胶体渗透压下降时,则会造成肾小球有效滤过压及GFR增高,出现利尿效应。
(三)肾小球滤过面积减少
单个肾小球虽然很小,但成人两肾约有200万个肾单位,肾小球毛细血管总面积估计约为1.6m2,接近人体总体表面积,因而能适应每天约180L的肾小球滤过量。在病理条件下,肾小球的大量破坏,可引起肾小球滤过面积和CFR的减少,但肾脏具有较大的代偿储备功能。切除一侧肾脏使肾小球滤过面积减少50%后,健侧肾脏往往可以代偿其功能。在大鼠实验中,切除两肾的3/4后,动物仍能维持泌尿功能。但在慢性肾炎引起肾小球大量破坏后,因肾小球滤过面积极度减少,故可使GFR明显减少而导致肾功能衰竭。
(四)肾小球滤过膜的通透性改变
肾小球滤过膜具有三层结构,由内到外为:内皮细胞,基底膜和肾小球囊的脏层上皮细胞(足细胞)。内皮细胞间有小孔,大小约为500~1,000A。小的溶质和水容易通过这种小孔。基底膜为连续无孔的致密结构、厚度为3200~3400A,表面覆有胶状物,胶状物的成分似以粘多糖为主,带负电荷。肾小球囊的脏层上皮细胞(足细胞)具有相互交叉的足突,足突之间有细长的缝隙,宽约100~400A,长200~900A,上覆有一层薄膜,此薄膜富含粘多糖并带负电荷。过去认为肾小球滤过膜小孔的大小是决定其通透性的因素,而小孔只允许相当于或小于白蛋白分子量大小(约68,000)的分子滤过,因而滤过的蛋白质主要为白蛋白以及其他低分子量蛋白如溶菌酶(分子量14,000)、β2-微球蛋白(分子量11,800)及胰岛素等。这些滤过的蛋白质绝大部分又都在近曲小管被重吸收。当炎症、缺氧等因素使肾小球滤过膜通透性增高时,滤过的蛋白质就增多并可出现蛋白尿。但是近年发现,某一物质能否经肾小球滤过,不仅取决于该物质的分子量,而且还和物质所带的电荷有关。因为肾小球滤过膜表面覆盖一层带负电荷的粘多糖,所以带负电荷的分子如白蛋白因受静电排斥作用,正常时滤过极少。只有在病理情况下,滤过膜表面粘多糖减少或消失时,才会出现蛋白尿。抗原-抗体复合物沉积于基底膜时,可引起基底膜中分子聚合物结构的改变,从而使其通透性增高。这也是肾炎时出现蛋白尿的原因之一。肾小球滤过襄上皮细胞的间隙变宽时,也会增加肾小球滤过膜的通透性。肾小球滤过膜的通透性增高是引起蛋白尿的重要原因。
二、肾小管功能障碍
肾小管具有重吸收、分泌和排泄的功能。在肾缺血、缺氧、感染及毒物作用下,可以发生肾小管上皮细胞变性甚至坏死,从而导致泌尿功能障碍。此外,在醛固酮、抗利尿激素(antidiuretic hormone, ADH)利钠激素及甲状旁腺激素作用下,也会发生肾小管的功能改变。
由于肾小管各段的结构和功能不同,故各段受损时所出现的功能障碍亦各异。
(一)近曲小管功能障碍
肾小球滤液中60~70%的钠以等渗形式由近曲小管主动重吸收;同时,近曲小管上皮细胞内分泌H+以利碳酸氢钠的重吸收。此外,葡萄糖、氨基酸、磷酸盐、尿酸、蛋白质、钾盐等经肾小球滤过后,绝大部分也由近曲小管重吸收。因此近曲小管重吸收功能障碍时,可引起肾小管性酸中毒(renal tubular acidosis)。此外,近曲小管具有排泄功能,能排泄对氨马尿酸、酚红、青霉素以及某些用于泌尿系造影的碘剂等。近曲小管排泄功能障碍时,上述物质随尿排出也就减少。
(二)髓袢功能障碍
髓袢重吸收的钠约占肾小球液中钠含量的10~20%。当原尿流经髓袢升支粗段及其相邻部分的远曲小管(称为远曲小管稀释段)时,Na+被肾小管上皮细胞主动重吸收,而Cl-则属于继发性的主动重吸收。由于此处肾小管上皮细胞对水的通透性低,因此原尿逐渐转变为低渗,而髓质则呈高渗状态,且越到髓质深部,高渗程度越高。此高渗状态是造成尿液浓缩的重要生理条件。高渗状态的形成,还与尿素由内髓集合髓质间质有关。髓袢功能障碍可致钠、水平衡失调。慢性肾盂肾炎使肾髓质高渗状态破坏时,可出现多尿、低渗尿和等渗尿。
(三)远曲小管和集合管*功能障碍
这两部分重吸收的钠占肾小球滤液中钠含量的8~10%。远曲小管在醛固酮的作用下,具有重吸收Na+和分泌H+、K+和NH3的功能。远曲小管功能障碍可导致钠、钾代谢障碍和酸碱平衡紊乱。远曲小管和集合管在髓质高渗区受ADH的调节而完成肾脏对尿浓缩与稀释功能。集合管的功能障碍可引起肾性尿崩症。
三、肾脏内分泌功能障碍
肾脏具有分泌肾素、前列腺素、促红细胞生成素和形成1,25-(OH)2-D3等内分泌功能。
肾素由近球细胞分泌。全身平均动脉压降低、脱水、肾动脉狭窄、低钠血症、交感神经紧张性增高等,可分别通过对入球小动脉壁牵张感受器、致密斑钠受体,以及直接对近球细胞的作用而引起肾素释放增多。肾素进入血液循环后,即使由肝细胞生成的血管紧张素原(angiotensinogen,一种α2-球蛋白)分解成为血管紧张素Ⅰ(angiotensin Ⅰ);后者在转化酶的作用下形成血管紧张素Ⅱ(angiotensin Ⅱ);血管紧张素Ⅱ在血管紧张素酶A的作用下,分解而形成血管紧张素Ⅲ(angiotensin Ⅲ)。血管紧张Ⅱ、Ⅲ均具有明显的生理效应,主要具有收缩血管(血管紧张素Ⅱ>血管紧张素Ⅲ)和促进肾上腺皮质分泌醛固酮(血管紧张素Ⅲ>血管紧张素Ⅱ)的作用。血管紧张素Ⅱ、Ⅲ除可被靶细胞摄取外,主要为血浆中血管紧张分解酶所灭活。在休克、脱水等肾前性因素作用时,肾素-血管紧张素-醛固酮系统活性即可增加,从而可提高平均动脉压,促进钠水潴留,因而具有代偿意义。但如血管紧张素形成过多,作用延续过久,则也可因肾脏血管的过度收缩而使肾小球血液灌流量和GFR显著减少,从而造成肾脏泌尿功能严重障碍。肾脏疾病如肾小球肾炎、肾小动脉硬化症等,均可使肾素-血管紧张素系统活性增强,从而引起肾性高血压,醛固酮分泌过多,则是造成体内钠、水潴留的重要发病因素。
(* 集合管不属于肾小管,也不包括在肾单位内,但在功能上与远曲小管密切联系,在尿生成过程中,特别是在尿浓缩过程中起重要作用。因此,为了理解方便,此处把远曲小管和集合管的功能障碍一并论述。)
肾髓质间质细胞可形成前列腺素E2(prostaglandin E2,PGE2)、A2(porstaglandinA2,PGA2)、和F2α(porstaglandinF2α,PGF2α),其中PGE2、PGA2具有扩张肾血管和促进排钠、排水的作用。因此有人认为肾内PGE2、PGA2形成不足可能是引起肾性高血压的发病因素之一。它们对体内钠、水潴留可能也起一定的作用。
促红细胞生成素(erythropoietin)是由肾脏皮质形成的一种激素,具有促进骨髓造血干细胞分化成原始红细胞,加速幼红细胞增殖分化,促进血红蛋白合成等作用。因此当肾脏疾病使这种激素形成减少时,就可引起贫血,这是慢性肾炎引起贫血的重要原因之一。但在两侧肾脏切除后依赖透析疗法生存的病人,其血红蛋白浓度仍可保持在10%左右。因此有人认为,肾脏以外的组织也可产生促红细胞生成素。
维生素D3本身并无生物学活性,它在体内首先必须在肝细胞微粒体中经25-羟化酶系统在C-25中羟化而生成25-(OH)-D3,然后25-(OH)-D3在肾脏近曲小管上皮细胞线粒体中,经1-羟化酶系进一步羟化生成1,25-(OH)2-D3,才具有生物学活性。1,25-(OH)2-D3进入血液循环后,就能作用于远隔靶组织而显示其生理功能,例如促进肠道对钙、磷的吸收,促进成骨作用等等。因此可以把肾脏形成1,25-(OH)-D3看成是肾脏的内分泌功能。在慢性肾功能衰竭时,由于肾脏生成1,25-(OH)-D3减少,故肠道对钙的吸收减少,因而可发生低钙血症。这种低钙血症用维生素D治疗并无效果。故肾脏生成1,25-(OH)2-D3减少是慢性肾功能衰竭患者具有抗维生素D作用的重要原因。
第二节 急性肾功能衰竭
急性肾功能衰竭(acute renal failure)是各种原因引起肾脏泌尿功能急剧降低,以致机体内环境出现严重紊乱的临床综合症。临床上主要表现为氮质血症、高钾血症和代谢性酸中毒,并常伴有少尿或无尿。
一、急性肾功能衰竭的原因与分类
根据发病原因可将急性肾功能衰竭分为肾前性、肾性和肾后性三大类。
(一)肾前性急性肾功能衰竭
肾前性急性肾功能衰竭是由于肾脏血液灌流量急剧减少所致,常见于休克的早期。此时,有效循环血量减少和血压降低除直接导致肾血流量减少外,还可通过交感-肾上腺髓质系统和肾素-血管紧张素系统使肾脏小动脉强烈收缩,从而进一步降低肾脏血液灌流量和有效滤过压。故GFR显着减少。同时,继发性醛固酮和ADH分泌增多,又可增强远曲小管和集合管对钠、水的重吸收,因而尿量显着减少,尿钠含量低于20mmol(mEq)/L,尿比重较高。GFR的急剧减少,还可引起高钾血症和酸碱平衡紊乱。
由于肾前性急性肾功能衰竭时尚无肾实质的器质性损害,故当血容量、血压及心输出量因及时的治疗而恢复正常时,肾脏泌尿功能也随即恢复正常。因此,一般认为这是一种功能性急性肾功能衰竭,但若肾缺血持续过久就会引起肾脏器质性损害,从而导致肾性急性肾功能衰竭。
(二)肾性急性肾功能衰竭
肾脏器质性病变所引起的急性肾功能衰竭称为肾性急性肾功能衰竭。例如,急性肾小球肾炎和狼疮性肾炎(见于全身性红斑狼疮)时,由于炎性或免疫性损害,可使大量肾小球的功能发生障碍,故可引起急性肾功能衰竭。双侧肾动脉栓塞亦可引起急性肾功能衰竭。此外,急性肾盂肾炎、子痫、结节性多动脉炎等也都能引起急性肾功能衰竭。
但是,临床上较为常见的是肾缺血及肾毒物引起的急性肾小管坏死所致的急性肾功能衰竭。急性肾小管坏死的原因有以下两类。
1.肾缺血多见于各种原因引起的休克病例而又未得到及时有效的抢救时。此时,严重和持续性的血压下降和肾小动脉的强烈收缩,可使肾脏血液灌流量显著而持续地减少。因此,肾小管可发生缺血性损害,甚至发生坏死。在已经出现肾小管器质性病变后,即使纠正血容量并使血压恢复正常,也不能使肾脏泌尿功能迅速恢复。患者尿中含有蛋白质,红、白细胞及各种管型。尿钠浓度一般可升高到40~70mmol(40~70mEq)/L或更高,说明肾小管已因受损而致保钠功能减退。
2.肾毒物重金属(汞、砷、锑、铅),抗菌素(二甲氧苯青霉素、新霉素、多粘菌素、庆大霉素、先锋霉素等),磺胺类,某些有机化合物(四氯化碳、氯仿、甲醇、酚、甲苯等),杀虫药,毒蕈。某些血管和肾脏造影剂、蛇毒、肌红蛋白等经肾脏排泄时,均可直接损害肾小管,甚至引起肾小管上皮细胞坏死。此时若并发肾脏血液灌流量不足,则更会加剧肾小管的损害。
在许多病理条件下,肾缺血与肾毒物经常同时或相继发生作用。例如在肾毒物作用时,肾内可出现局部血管痉挛而致肾缺血;反之,肾缺血也常伴有毒性代谢产物的堆积。一般认为肾缺血时再加上肾毒物的作用,最易引起急性肾功能衰竭。
急性肾小管坏死所致的急性肾功能衰竭。在临床上根据有无少尿可分为少尿型和非少尿型两大类。少尿型较为常见,患者空然出现少尿(成人24小时的尿量少于400ml)甚至无尿(24小时的尿量少于100ml)。非少尿型患者尿量并不减少,甚至可以增多,但氮质血症逐日加重,此型约占20%。
(三)肾后性急性肾功能衰竭
从肾盏到尿道口任何部位的尿路梗阻,都有可能引起肾后性急性肾功能衰竭。膀胱以上的梗阻,多由结石引起。然而由于肾脏的代偿储备功能强大,因此只有当结石使两侧尿路同时梗阻或一侧肾已丧失功能而另一侧尿路又被阻塞时才会引起肾后性急性肾功能衰竭。膀胱及尿道的梗阻可由膀胱功能障碍(如脊髓痨、糖尿病假性脊髓痨等引起的慢性尿潴留)或前列腺肥大、前列腺癌等引起。
在肾后性急性肾功能衰竭的早期并无肾实质的器质性损害。及时解除梗阻。可使肾脏泌尿功能迅速恢复。因此对这类病人,应及早明确诊断,并给予适当的处理。
二、急性肾功能衰竭的发病机制
急性肾功能衰竭的发病机制目前是指急性肾小管坏死引起的肾功能衰竭的发病机制。在探讨急性肾功能衰竭的发病机制时,除临床观察、尸体解剖或活体组织检查外,还常采用各种方法造成急性肾功能衰竭的动物模型,以供分析研究。例如可给动物注射升汞、硝酸氧铀(uranyl nitrate)、铅化合物等造成中毒性急性肾功能衰竭,或者用缩窄肾动脉,肾动脉内持续注入去甲肾上腺素、造成失血性休克等方法以引起缺血性肾功能衰竭,也可以肌肉内注入甘油造成肌红蛋白症并从而导致急性肾功能衰竭等。在上述动物模型中,由于引起肾脏损害的因素较为复杂,故任何一种实验模型都不能全面说明急性肾功能衰竭的发病机制而只能从某一个方面提示有关因素的作用。因此应当指出,下文所述急性肾功能衰竭的发病机制,是从不同动物实验模型中所得结果的综述,而这些实验资料也未必能充分阐明临床所见的急性肾功能衰竭的发病机制。
(一)原尿回漏入间质
应用显微穿刺法将14C-菊粉直接注入因缺血或肾动脉内注射硝酸氧铀而受损的大鼠一侧肾脏的肾小管腔后,可在对侧肾脏生成的尿液内发现有大量放射性菊粉排出。这证实受损肾脏的肾小管上皮细胞有较高的通透性,因而菊粉得以通过回漏而进入全身血液循环,并被对侧肾脏排出。所以有人认为,持续性肾缺血或肾毒物引起肾小管上皮坏死并进而导致急性肾功能衰竭时,肾小管管腔内原尿向肾间质的回漏,一方面可直接使尿量减少,另一方面又可通过形成肾间质水肿而压迫肾小管和阻碍原尿通过,其结果是肾小球囊内压增高,GFR进一步减少。但最近有人在缺血性及中毒性肾功能衰竭的实验中发现,在肾小管上皮细胞出现坏死以前已有尿生成减少。临床上给某些急性肾功能衰竭的患者施行肾包囊切除术以减轻肾间质水肿,也并不能改善肾脏泌尿功能。因此现在认为,肾小管坏死引起的原尿回漏,不是急性肾功能衰竭少尿的原发机制,但能使少尿加重。
(二)肾小管阻塞
异型输血、挤压伤等引起急性肾功能衰竭时,在病理组织切片中可发现有坏死脱落的上皮细胞碎片、肌红蛋白、血红蛋白等所形成的管型阻塞肾小管。在急性肾功能衰竭的动物实验中,也有人看到肾小管内有各种管型存在。因此,肾小管阻塞可能是引起急性肾功能衰竭时少尿的发病机制之一。肾小管阻塞后,可提高肾小管阻塞上段的管腔内压,从而使肾小球囊内压增高,GFR减少。但是在肾缺血及肾毒物引起急性肾功能衰竭的实验中,用显微穿剌术测定梗阻近侧的肾小管内压时,大部分实验资料表明管内压并不升高,甚至反而降低。有人认为这是通过管-球反馈调节机制(tubuloglo-merular feedbackmechanism)使肾小球动脉收缩,GFR减少所致。因此很难肯定肾小管阻塞引起急性肾功能衰竭时少尿的原发机制。实验还证明,当GFR恢复正常,原尿形成充足时,管型即可被冲走而不易形成肾小管阻塞。这更说明肾小管被管型阻塞是GFR减少的结果。但是在已有肾小管阻塞之后,则会促进肾功能衰竭的恶化。
(三)肾小球滤过功能障碍
在急性肾功能衰竭的发病机制中,肾小球滤过功能障碍日益受到重视。引起肾小球滤过功能障碍的主要因素有肾脏血液的灌流量减少,肾小球有效滤过压降低及肾小球滤过膜的通透性改变等,今分述如下。
1.肾脏血液灌流量减少 分别采用惰性气体洗出术(inert gas washout)、染料稀释法、同位素标记微球体灌注法及电磁流量计测定肾脏血液灌流量时,发现急性肾功能衰竭患者以及肾缺血或肾毒物引起急性肾功能衰竭的实物动物,均有肾脏血液灌流量的减少,而且一般约减少45~60%之多,其中以肾皮质血流量的减少最为明显,即出现了肾脏血流的异常分配,从而使肾脏泌尿功能发生严重障碍。但是应用双肼苯哒嗪、乙酰胆碱;前列腺素等治疗急性肾功能衰竭时,在肾脏血流量及肾皮质血流量增加的情况下,并不能提高GFR。因此,肾脏血液灌流量的减少可能不是急性肾功能衰竭的主要发病机制。
2.肾小球有效滤过压降低实验资料指出,肾缺血及肾毒物引起急性肾功能衰竭时,有时可见血液中儿茶酚胺及血管紧张素Ⅱ的含量增多,因而可能导致肾小球入球小动脉收缩,使肾小球有效滤过压和滤过率降低,其中,血管紧张素Ⅱ增多尤受重视。最近实验资料表明,急性肾功能衰竭时,主要是肾脏皮质外层血管紧张素的含量增多。近来还发现,肾脏组织内的肾素,在肾内也可形成血管紧张素Ⅱ,并从而引起入球小动脉收缩,使肾小球的血液灌流量减少,有效滤过压和滤过率降低。
急性肾功能衰竭时出现肾内肾素-血管紧张素系统活性增高,可能是通过管-球反馈调节机制所致。实验证明,肾缺血和肾毒物引起肾小管功能障碍后,近曲小管对钠的重吸收功能降低,远曲小管内的钠的浓度增高,从而使致密斑受到钠负荷的剌激而引起肾素分泌增多,肾内血管紧张素Ⅱ的形成也因而增多,其结果是入球小动脉收缩,肾小球有效滤过压及滤过率降低。
在急性肾功能衰竭的发病机制中,肾内肾素-血管紧张素系统的作用,并未完全肯定。例如,实验资料表明,慢性盐负荷肾内肾素耗竭时并不能防止某些实验性急性肾功能衰竭的发生,当肾组织中含有高浓度肾素时(进行性肾性血管性高血压),通常并不伴发急性肾功能衰竭。因此,这一问题尚待进一步探讨。
3.肾小球滤过膜通透性的改变实验证明,给狗的一侧肾动脉内持续滴注高浓度去甲肾上腺素造成急性肾功能衰竭时,用扫描电镜可观察到肾小球囊脏层上皮细胞出现明显的形态学改变——上皮细胞相互融合,正常的滤过缝隙消失。此时如给实验狗输入盐水使肾血流量增加,并不能增加尿量,因而认为肾小球的上述形态学改变,可能是造成肾小球滤过功能障碍的原因。但在其他动物模型中,肾小球一般并不出现这种形态学改变。也有人在缺血和中毒性肾功能衰竭的动物模型中,发现有肾小球毛细血管内皮细胞和肾小球囊上皮细胞的肿胀,认为这些变化能减少肾小球的血液灌流量,并改变滤过膜的通透性。但是一般认为这些改变只出现在急性肾功能衰竭的最初阶段,因而不是引起急性肾功能衰竭时GFR减少的主要机制。
综上所述,可见急性肾小管坏死所致肾功能衰竭的发病机制是复杂的。其中肾小球滤过功能障碍可能起着较重要的作用。引起肾小球滤过功能障碍的机制,除肾小球病变和肾血流量减少直接引起肾小球血液灌流量减少外,还可因肾小管受损,通过管-球反馈调节机制,引起肾内肾素-血管紧张素系统活性增高。从而导致入球小动脉收缩,使肾小球有效滤过压和滤过率持续降低。肾小管上皮细胞坏死引起的原尿回漏以及肾小管被管型阻塞,在急性肾功能衰竭的发病过程中,也可能起一定作用。图14-1可作为了解急性肾功能衰竭少尿发病机制的参考。应当注意,在少尿或无尿发生的同时,肾小球和/或肾小管的严重障碍也必然引起机体的环境紊乱,如氮质血症、水电解质和酸碱平衡紊乱等等。
当病变逐渐减轻,并开始出现肾小管上皮细胞的再生修复,GFR又逐渐恢复至正常时,急性肾功能衰竭患者即由少尿进入多尿期。多尿是由于再生的肾小管上皮细胞对水和电解质的重吸收功能尚不完善所致。待肾小管功能基本恢复正常后,肾脏泌尿功能和机体内环境才逐渐恢复正常。
在严重烧伤、创伤、大量失血及手术后,有些急性肾功能衰竭患者为非少尿型,其发病机制和体内机能代谢变化详见后文。
图14-1 急性肾小管坏死引起的肾功能衰竭时少尿发病机制示意图
还应当指出近年对急性肾功能衰竭病人的研究表明,病理组织学检查证明只有一部分病人的肾小管有真正的坏死,而不少病人的肾小管只有轻微的病变,甚至看不到病变。然而用电子显微镜观察时,则可见无明显坏死的肾小管上皮细胞的细胞器有损害,而且细胞生物化学方面的变化更为明显和广泛,因而有人认为,在这样的病人,急性肾功能衰竭的致病因素可能是肾内小血管的收缩和肾小管上皮细胞的重要功能的损害。
三、急性肾功能衰竭发病过程中各期的机能代谢变化
急性肾功能衰竭少尿型的发病过程一般可分为少尿期、多尿期和恢复期三个阶段。
(一)少尿期
此期尿量显着减少,并有体内代谢产物的蓄积,水、电解质和酸碱平衡紊乱。它是病程中最危险的阶段。
1.少尿或无尿及尿成分的变化如前所述,急性肾功能衰竭出现少尿或无尿的机制与GFR减少,原尿由坏死的肾小管漏回间质以及肾小管阻塞等因素有关。当原尿通过受损的肾小管时,由于肾小管上皮重吸收水和钠的功能障碍,故尿比重低,尿渗透压低于350mOsm/L,尿钠含量高于40mmol(40mEq)/L,常达80~100mmol(80~100mEq/L)。由于肾小球滤过功能障碍和肾小管上皮坏死脱落,尿中含有蛋白,红、白细胞和各种管型。这些改变与功能性急性肾功能衰竭时的尿液变化有明显差别,见表14-1。
表14-1 功能性急性肾功能衰竭与急性肾小管坏死少尿期尿液变化的比较
| 功能性肾功能衰竭 | 急性肾小管坏死 | |
| 尿比重 | >1.020 | <1.015 |
| 尿渗透压(mOsm/L) | >700 | <250 |
| 尿钠含量(mmol(mEq)/L) | >40:1 | >40 |
| 尿/血肌酐比值 | >40:1 | <10:1 |
| 尿蛋白含量 | 阴性至微量 | + |
| 尿沉渣镜检 | 基本正常 | 透明、颗粒、细胞管型,红细胞、白细胞和变性坏死上皮细胞 |
2.水中毒由于肾脏排尿量严重减少,体内分解代谢加强以致内生水增多,以及输入葡萄糖溶液过多等原因,可引起体内水潴留。当水潴留超过钠潴留时,可引起稀释性低钠血症,水分可向细胞内转移而引起细胞水肿。严重患者可并发肺水肿、脑水肿和心功能不全。因此对急性肾功能衰竭患者,应严密观察和记录出入水量,严重控制补液速度和补液量。
3.高钾血症这是急性肾功能衰竭患者最危险的变化。引起高钾血症的原因是①尿量的显著减少,使尿钾排出减少,②组织损伤、细胞分解代谢增强、缺氧、酸中毒等因素均可促使钾从细胞内血细胞外转移,③摄入含钾食物或大量输入含高浓度钾的库血,等等。高钾血症可引起心脏兴奋性降低,诱发心律失常,甚至导致心跳骤停而危及病人生命。
4.代谢性酸中毒主要是由于肾脏排酸保碱功能障碍所致,具有进行性、不易纠正的特点。酸中毒可抑制心血管系统和中枢神经系统,并能促进高钾血症的发生。
5.氮质血症由于体内蛋白质代谢产物不能由肾脏充分排出,而且蛋白质分解代谢往往增强,故血中尿素、肌酐等非蛋白含氮物质的含量可大幅度的增高,称为氮质血症(azotemia)。一般在少尿期开始后几天,就有血中非蛋白氮的明显增多。感染、中毒、组织严重创伤等都会使血中非蛋白氮水平进一步升高,有关尿素等非蛋白含氮物质对机体的影响可参阅本章第四节。
少尿期可持续几天到几周,平均为7~12天。少尿期持续愈久,预后愈差。患者如能安全度过少尿期,而且体内已有肾小管上皮细胞再生时,即可进入多尿期。
(二)多尿期
当急性肾功能衰竭患者尿量逐渐增多至每日1,200ml以上时,即进入多尿期,说明病情趋向好转。此期尿量可达每日3,000ml以上。产生多尿的机制为:①肾小球滤过功能逐渐恢复正常;②间质水肿消退,肾小管内的管型被冲走,阻塞解除;③肾小管上皮虽已开始再生修复,但其功能尚不完善,故重吸收钠、水的功能仍然低下,原尿不能被充分浓缩;④少尿期中潴留在血中的尿素等代谢产物开始经肾小球大量滤出,从而增高原尿的渗透压,引起渗透性利尿。
多尿期中患者尿量虽已增多,但在早期由于GFR仍较正常为低,溶质排出仍然不足,肾小管上皮细胞的功能也不完善,因此氮质血症、高钾血症和酸中毒等并不能很快改善,只有经过一定时间后,血钾和非蛋白氮才逐渐下降至正常水平,肾脏排酸保硷的功能才恢复正常。多尿期间,患者每天可排出大量水和电解质,若不及时补充,则可发生脱水、低钾血症和低钠血症。对此,应给予充分的注意。
多尿期历时约1~2周后病程进入恢复期。
(三)恢复期
此期患者尿量和血中非蛋白氮含量都基本恢复正常。水、电解质和酸碱平衡紊乱及其所引起的症状也完全消失。但是,肾小管功能需要经过数月才能完全恢复正常;因而在恢复期的早期,尿的浓缩和尿素等物质的消除等功能仍可以不完全正常。少数病例(多见于缺血性损害病例)由于肾小管上皮和基底膜的破坏严重和修复不全,可出现肾组织纤维化而转变为慢性肾功能不全。
非少尿型急性肾功能衰竭患者,肾内病变可能较轻。虽然也有GFR减少和肾小管的损害,便以肾小管浓缩功能的障碍较为明显,因此虽有血浆非蛋白氮的增高,但尿量并不减少,尿比重(<1.020)尿钠含量也较低,预后较好。由于非少尿型的尿量排出较多,故一般很少出现高钾血症。
四、急性肾功能衰竭的防治原则
(一)由于许多药物及毒性物质能损害肾小管,因此应合理用药,以避免毒性物质对肾脏的损害作用。
(二)积极抢救危重病人,预防休克的发生,如已发生休克伴有功能性急性肾功能衰竭时,应及时采用抗休克措施,迅速恢复有效循环血量,使肾血流量和GFR恢复正常,以利肾功能的恢复。如通过尿液分析,发现患者已发生急性肾小管坏死所致的急性肾功能衰竭时,应按急性肾功能衰竭的治疗原则进行处理。
(三)由于急性肾功能衰竭的发病机制尚未完全清楚,因此常采用下述综合治疗措施。
1.适当输入液体,以维持体内水电解质平衡。在少尿期应严重控制液体输入量,以防水中毒的发生。在多尿期,除注意补液外,还应注意补钠、补钾、以防脱水、低钠血症和低钾血症的发生。
2.处理高钾血症。高钾血症是少尿期威胁生命的变化,应进行紧急处理,治疗原则是:
①促进细胞外钾进入细胞内,如静脉内滴注葡萄糖和胰岛素,使细胞内糖原合成增多,从而促使细胞外液中的钾进入细胞内;②静脉内注入葡萄糖酸钙,对抗高钾血症对心脏的毒性作用;③应用钠型阳离子交换树脂如聚苯乙烯磺酸钠口服或灌肠,使钠和钾在肠内进行交换,钾即可随树脂排出体外;④严重高钾血症时,应用透析疗法(详后文)。
3.控制酸中毒。
4.控制氮质血症。如①滴注葡萄糖以减轻蛋白质的分解代谢;②静脉内缓慢滴注必需氨基酸,以促进蛋白质的合成,降低尿素氮上升的速度,并加速肾小管上皮的再生;③采用透析疗法以排除非蛋白氮等(详后文)。
5.积极抗感染。此时应选用合适的药物和剂量,以免加重肾中毒。
6.透析疗法包括血液透析(人工肾)和腹膜透析。其原理是通过透析作用,使半透膜两侧溶液中的小分子物质如尿素、葡萄糖、电解质、H+等进行交换,以矫正水、电解质、酸碱平衡紊乱和降低尿素氮。透析效应取决于半透膜的孔径大小,以及膜两侧溶质的浓度差。腹膜透析是利用腹膜作为半透膜,人工肾则利用一种赛璐玢(cellophane)或铜玢(cupraphane)的透明薄膜作为半透膜。透析液的配置很重要。为了降低血浆中K+、H+和非蛋白氮等物质的浓度,透析液中的钾浓度应比正常血钾浓度为低(如2mmol(2mEq)/L),而且不应含有非蛋白氮类物质,人工肾的透析效果最好,但设备及条件要求较高,不易推广。因此临床上常将透析液注入腹腔内,利用腹膜进行透析。留置1~2小时再将透析液放出。透析疗法已广泛应用于急性、慢性肾功能衰竭,取得了较好的疗效,但也不应因此而忽视其他治疗措施。
第三节 慢性肾功能衰竭
各种慢性肾脏疾病均可引起肾实质的破坏和肾功能障碍。由于肾脏具有强大的储备代偿功能,因此在肾实质尚未受到广泛而严重的损害时,肾脏尚能维持内环境的稳定。当疾病进一步恶化以致有功能的肾单位残存不多时,就会发生内环境紊乱,主要表现为代谢产物及毒性物质在体内潴留,以及水、电解质和酸碱平衡紊乱,并伴有一系列临床症状。这就是慢性肾功能衰竭(chronic renal failure)。
一、慢性肾功能衰竭的病因和发病机制
引起慢性肾功能衰竭的疾病,以慢性肾小球肾炎为最常见,约占50~60%。肾小动脉硬化症、慢性肾盂肾炎以及全身性红斑狼疮等也是较为常见的原因。其他如肾结核、糖尿病性肾小球硬化症、多囊肾、肾脏发育不全,以及结石、肿瘤、前列腺肥大等引起的尿道梗阻也可导致慢性肾功能衰竭。在发生慢性肾功能衰竭之前,由于各种慢性肾脏疾病可分别引起以肾小球或肾小管损害为主的病变,故在临床上可出现不同的症状和体征。但是在各种慢性肾脏疾病的晚期,由于大量肾单位的破坏和功能的丧失却可出现相同的后果,即残存肾单位过少所致的肾功能衰竭。因此慢性肾功能衰竭是各种慢性肾脏疾病最后的共同结局。由于肾脏有强大的储备代偿功能,故慢性肾功能衰竭的发展过程可以随着肾脏受损的逐步加重而分为下列四个时期。
第一期——肾脏储备功能降低期在较轻度或中度肾脏受损时,未受损的肾单位尚能代偿已受损的肾单位的功能。故在一般情况下肾脏泌尿功能基本正常。机体内环境尚能维持在稳定状态,内生性肌酐清除率仍在正常值的30%以上,血液生化指标无明显改变,也无临床症状。但在应激剌激作用下,如钠、水负荷突然增大或发生感染等时,可出现内环境紊乱。
第二期——肾脏功能不全期由于肾脏进一步受损,肾脏储备功能明显降低,故肾脏已不能维持机体内环境的稳定。内生性肌酐清除率下降至正常值的25~30%。有中度氮质血症和贫血,肾脏浓缩功能减退,常有夜尿和多尿,一般临床症状很轻,但在感染、手术及脱水等情况下,肾功能即明显恶化,临床症状加重。
第三期——肾功能衰竭期 肾脏内生性肌酐清除率下降至正常值的20~25%,有较重的氮质血症,血液非蛋白氮多在60mg%以上。一般有酸中毒、高磷血症、低钙血症,也可出现轻度高钾血症。肾脏浓缩及稀释功能均有障碍,易发生低钠血症和水中毒,贫血严重。有头痛,恶心,呕吐和全身乏力等症状。临床称为氮质血症期或尿毒症前期。
第四期——尿毒症期为慢性肾功能衰竭的晚期。内生性肌酐清除率下降至正常值的20%以下。血液非蛋白氮在80~100mg%或更高。毒性物质在体内的积聚明显增多,有明显的水、电解质和酸碱平衡紊乱及多种器管功能衰竭。临床不有一系列尿毒症症状即自体中毒的症状出现。图14-2表示内生性肌酐清除率(基本上代表GFR)和临床表现的关系。由此可见,肾功能衰竭的临床表现和GFR的减少有密切关系。
图14-2 慢性肾功能衰竭的临床表现与肾功能的关系
有关慢性肾功能衰竭的发病机制,一般采用完整的肾单位学说(intactnephron hypothesis)来解释。此学说认为:虽然引起慢性肾损害的原始病因各不相同,但是最终都会造成病变肾单位的功能丧失,肾只能只能由未受损的残存肾单位来承担。丧失肾功能的肾单位越多,残存的完整肾单位就越少;最后,当残存的肾单位少到不能维持正常的泌尿功能时,内环境就开始发生紊乱,亦即慢性肾功能衰竭开始发生发展。
Bricker在七十年代提出的矫枉失衡假说(trade-off hypothesis)可以认为是对完整肾单位学说的一个补充。根据动物实验和临床研究的结果,Bricker指出,当肾单位和GFR进行性减少以致某一溶质(例如某一电解质)的滤过减少时,作为一种适应性反应,血液中一种相应的抑制物(例如某一激素)就会抑制残存肾单位肾小管对该溶质的重吸收,从而使该溶质随尿排出不致减少而在血浆中的水平也不致升高。可见,这种适应性反应有稳定内环境的作用。随着肾单位和GFR的进一步减少,该溶质的滤过也进一步减少。此时,尽管血浆中抑制物仍起抑制作用,但因残存肾单位过少,故不能维持该溶质的排出,结果是溶质在血浆中的浓度升高,即内环境发生紊乱。该溶质浓度的升高又可使血浆中的抑制物也随之增多,而此时抑制物的增多,非但不能促进溶质的排泄而有助于机体内环境恒定性的维持,反而可以作用于其他器管而起不良影响,从而使内环境的紊乱进一步加剧。
下文在论述慢性肾功能衰竭者钙、磷代谢障碍时,将对矫枉失衡假说作具体的解释。
二、慢性肾功能衰竭时机体的机能及代谢变化
(一)氮质血症
当血液中非蛋白氮(NPN)浓度水平超过正常时称为氮质血症。正常人血中NPN为25~30mg%;其中尿素氮为10~15mg%,尿酸为3~5mg%,肌酐为0.9~1.8mg%。慢性肾功能衰竭时,由于GFR减少,上述NPN浓度均有不同程度升高。
1.血浆尿素氮(blood urea nitrogen, BUN)浓度的变化尿素是由肝脏合成的蛋白质分解代谢的产物,主要由肾脏排泄。慢性肾功能衰竭患者,BUN的浓度与GFR的变化有密切关系。如图14-3所示。
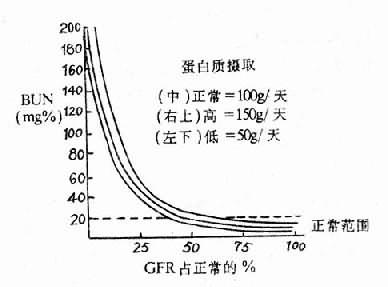
图14-3 BUN和GFR的关系
在肾功能衰竭的早期,当GFR减少到正常值的40%以前,BUN浓度虽有缓慢地升高,但仍在正常范围内。当GFR进一步减少时,BUN浓度就明显上升,当GFR减少到正常值的20%以下时,血中BUN可高达100mg%以上。由此可见,BUN浓度的变化并不是反映肾功能改变的敏感指标;而且BUN值还与外源性(与蛋白质摄入量有关)及内源性(与感染、肾上腺皮质激素的应用、胃肠道出血等有关)尿素负荷的大小有关,因此根据BUN值判断肾功能变化时,应考虑这些尿素负荷的影响。
2.血浆肌酐浓度的变化血浆肌酐浓度与蛋白质的摄入量无关,而主要与肌肉中磷酸肌酸自身分解产生的肌酐量及肾脏排泄肌酐的功能有关,因此血浆肌酐浓度的改变更能反映GFR的变化。但在GFR变化的早期,血中肌酐浓度的改变与BUN一样,也并不明显。因此,在临床上必须同时测定血浆和尿液的肌酐含量,以计算肌酐清除率(肌酐清除率=UV/P,U=尿中肌酐浓度,V=每分钟尿量,P=血浆肌酐浓度)。肌酐清除率与GFR的变化具有平行关系。但在严重肾功能衰竭并伴有食欲丧失和恶病质时,由于肌肉组织分解代谢明显增强,内生性肌酐形成过多,故血清肌酐浓度可迅速增高,此时肌酐清除率降低,并不能确切地反映GFR的变化。
3.血浆尿酸氮浓度的变化慢性肾功能竭衰时,血清尿酸氮浓度虽有一定程度的升高但较尿素、肌酐为轻,这主要与肾脏远曲小管分泌尿酸增多和肠道尿酸分解增强有关。
慢性肾功能衰竭患者NPN的增高还包括有中分子量肽类、氨基酸、胍类等蛋白质分解产物的增多,这些物体对机体具有毒性作用。
(二)电解质及酸碱平衡紊乱
1.对多尿的患者,特别是在伴在呕吐、腹泻时,如不及时补充足够的水分,则因肾脏浓缩功能减退,尿量不能相应的减少,故容易发生严重脱水从而使酸中毒、高钾血症、高磷血症、氮质血症加重,病情恶化。反之,当静脉输血过多时,又易发生水潴留,甚至引起肺水肿和脑水肿。当慢性肾功能衰竭引起GFR过度减少时,则会出现少尿和水肿。
(1)夜尿:正常成人每日尿量约为1,500ml,白天尿量约占总尿量的2/3,夜间尿量只占1/3。慢性肾功能衰竭患者,早期即有夜间排尿增多的症状,夜间尿量和白天尿量相近,甚至超过白天尿量,这种情况称为夜尿。其形成机制尚不清楚。
(2)多尿:多尿是慢性肾功能衰竭较常见的泌尿功能变化,但尿量很少超过每天3,000ml。其形成机制可能为:①大量肾单位被破坏后,残存肾单位血流量增多,其肾小球滤过率增大,原尿形成增多。由于原尿流速较快和溶质含量较多,因而产生了渗透性利尿效应;②慢性肾盂肾炎导致慢性肾功能衰竭时,常有肾小管上皮细胞对ADH的反应减弱;③慢性肾盂肾炎,慢性肾小球肾炎等患者髓袢主动重吸收Cl-的功能减弱时,髓质间质不能形成高渗环境,因而尿的浓缩功能降低。但是当肾单位大量破坏,肾血流量极度减少时,则可发现少尿。
(3)等渗尿:慢性肾功能衰竭时,由于肾脏浓缩和稀释功能障碍,尿溶质接近于血清浓度,尿比重固定在1.008~1.012,称为等渗尿。由于各种尿溶质的分子量不一致,故同样是1克分子浓度和不同溶质,对尿比重所起的影响却各不相同。因此最好用冰点降低法测定尿的渗透压来反映尿中溶质的浓度。等渗尿的渗透压为250~400mOsm/L。
2.钠代谢障碍慢性肾功能衰竭时,由于大量肾单位被破坏,因此残存肾单位维持钠平衡的功能大为降低,如图14-4所示。
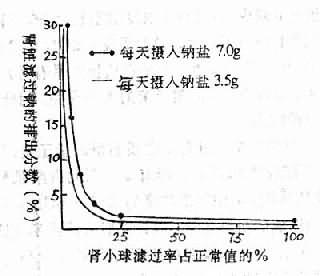
图14-4 慢性肾功能衰竭时肾脏滤过钠的排出分数与肾小球滤过率关系
无论在高盐或低盐饮食条件下,随着GFR的下降,肾脏滤过钠的排出分数(fractional exeretion of filtered sodium)均上升;即当GFR由100%下降到5%时,滤过钠的排出分数也相应的由0.5上升到32%以上,因此在低盐饮食时易引起低钠血症,并可导致细胞外液和血浆容量减少,使GFR进一步下降,从而加重内环境的紊乱。如患者同时因食欲缺乏、恶心呕吐而使钠的摄入减少,则更会促进低钠血症的发生;因此对慢性肾功能衰竭患者,应适当补充钠盐。对失盐性肾炎所致的慢性肾功能衰竭患者,更应注意补充钠盐。关于慢性肾功能衰竭患者失钠的机制,尚有争论。多数学者认为主要是因渗透性利尿引起失钠。因为流经少数残存肾单位的原尿中溶质(主要为尿素)浓度较高,原尿流速也快,钠、水重吸收因而减少,于是钠盐排出过多。
慢性肾功能衰竭虽易发生失钠,但补充钠盐却不宜过多。因为残存肾单位的残存肾单位的滤过率已很低,补充钠盐过多后,易造成钠水潴留,使细胞外液及血浆容量扩大,从而进一步使血压升高,加重心脏负荷,并可能导致心力衰竭。当患者已有心力衰竭时,更不宜过多的补充钠盐。
3.钾代谢障碍慢性肾功能衰竭患者,虽有GFR减少,但只要尿量不减少,血钾可长期维持在正常水平。多尿及反复使用失钾性利尿剂引起的尿钾排出过多,以及厌食、呕吐、腹泻所致的钾摄入不足和丧失过多等还可导致低钾血症。
慢性肾功能衰竭患者一般不易出现高钾血症。但在晚期尿量过少(每天尿量低于600~900ml),以致钾排出过少时,就可发生高钾血症。引起高钾血症的其他因素有:①严格控制钠盐的摄入,使尿钠排出过低,因而尿钾排出减少;②长期使用保钾利尿剂;③代谢性酸中毒;④溶血及感染等。
高钾血症和低钾血症均可影响神经肌肉的应激性,严重时可引起致命的心律失常。
4.代谢性酸中毒慢性肾功能衰竭患者发生代谢性酸中毒的机制如下。
(1)肾小管排NH4+减少:慢性肾功能衰竭时,由于肾小管上皮细胞产NH3减少,肾小管排NH4+降低,可致H+排出障碍而发生代谢性酸中毒。
(2)肾小管重吸收重碳酸盐减少:慢性肾功能衰竭时继发性甲状旁腺激素(parathyroid hormone, PTH)分泌增多(详见后文)可抑制近曲小管上皮细胞碳酸酐酶的活性,使近曲小管对重碳酸盐的重吸收降低,因而造成重碳酸盐的丧失。
(3)肾小球滤过率明显下降:当GFR降低到20ml/min时,体内酸性代谢产物如碳酸、硫酸、磷酸、有机酸等从肾小球滤过减少而致潴留体内。
酸中毒时氢离子对神经肌肉系统具有抑制作用。此时患者虽可有明显低钙血症(详见后文),但因血液pH降低可提高钙的离解度,血浆[Ca2+]水平可以不低,因此临床上不出现抽搐。但在快速纠正酸中毒后,钙的离解度随即降低而使血浆[Ca2+]下降,患者因而可以发生手足搐搦。酸中毒能使细胞内K+外移而促进高钾血症的发生,酸中毒又能促使骨盐溶解,引起骨骼脱钙。
5.镁代谢障碍体内镁代谢平衡主要受肠道对镁的吸收和肾脏排镁的影响。慢性肾功能衰竭伴有少尿时,可因尿镁排出障碍而引起高镁血症。若同时用硫酸镁以降低血压或导泻,更易造成血镁升高。但一般血镁升高的程度并不严重,高镁血症对神经肌肉具有抑制作用。
6.钙、磷代谢障碍慢性肾功能衰竭时,钙磷代谢障碍主要表现为血磷升高,血钙降低及骨质营养不良。
(1)血磷升高:在肾功能衰竭早期(GFR>30ml/min),因GFR减少而引起的肾脏排磷减少,可引起磷酸盐潴留和血磷暂时性升高。血磷升高可使血钙降低,而血钙降低又可剌激甲状旁腺,引起续发性PTH分泌增多。按照Bricker所提出的矫枉失衡假说,PTH就是针对血磷滤过减少而在血液中增多的抑制物。PTH能抑制近曲小管对磷酸盐的重吸收,故可使尿磷排出增多,从而使血降低到正常水平。因此慢性肾功能衰竭患者可以在很长一段时间内不发生血磷过高。由此可见,抑制物PTH的增多是一种适应性反应,具有稳定内环境的作用。在慢性肾功能衰竭的晚期,GFR和血磷的滤过都进一步显著减少。此时,由于残存肾单位太少,继发性PTH分泌增多已不能维持磷的充分排出,故血磷水平显著升高。PTH的增多又可加强溶骨活性,使骨磷释放增多,从而形成恶性循环,使血磷水平不断上升。
(2)血钙降低:慢性肾功能衰竭出现血钙降低的原因是:①血液中钙、磷浓度之间有一定关系,当血磷浓度升高时,血钙浓度就会降低:②肾实质破坏后,25-(OH)-D3羟化为1,25-(OH)2-D3的功能发生障碍,肠道对钙的吸收因而减少;③血磷过高时,肠道分泌磷酸根增多,故可在肠内与食物中的钙结合而形成不易溶解的磷酸钙,从而妨碍钙的吸收;④尿毒症时,血液中潴留的某些毒性物质可使胃肠道粘膜受损,钙的吸收因而减少。
(3)肾性骨质营养不良:肾性骨质营养不良(renal osteodystrophy)是慢性肾功能衰竭,尤其是尿毒症的严重并发症。其中包括有骨囊性纤维化,骨软化症和骨质疏松等病变,其发病机制与慢性肾功能衰竭时出现的高磷血症、低钙血症、PTH分泌增多、1,25-(OH)2-D3形成减少、胶原蛋白代谢障碍以及酸中毒等有关,其相互关系如图14-5所示。
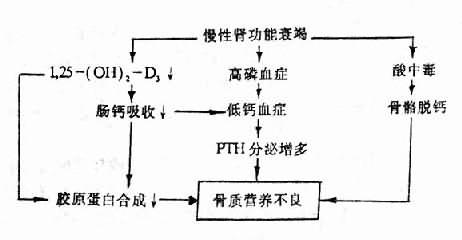
图14-5 肾性骨质营养不良的发病机制示意图
临床上如采取一定措施降低血磷和控制低钙血症,则可减轻继发性PTH分泌增多和骨质营养不良。
(三)肾性高血压
高血压是慢性肾功能衰竭患者的常见症状之一,故称为肾性高血压,其发病机制与下列因素有关。
1.钠、水潴留慢性肾功能衰竭时,由于肾脏排钠、排水功能降低,钠水可在体内潴留而引起血容量增高和心输出量增多,从而可导致血压升高,这种高血压称为钠依赖性高血压(sodium-dependent hypertension)。对这种病人限制钠盐的摄入,并就用利尿剂以加强尿钠的排出,可以收到较好的降压效果。
2.肾素-血管紧张素系统的活性增高慢性肾小球肾炎、肾小动脉硬化症、肾硬化症等疾病引起的慢性肾功能衰竭,常伴有肾素-血管紧张素系统的活性增高,血液中血管紧张素Ⅱ形成增多。血管紧张素Ⅱ可直接引起小动脉收缩,又能促使醛固酮分泌,导致钠水潴留,并可兴奋交感-肾上腺髓质系统,引起儿茶酚胺释放和分泌增多,故可导致血压上升,这种高血压称为肾素依赖性高血压(renin-dependent hypertension)。对此类患者限制钠盐摄入和应用利尿剂,不能收到良好的降压效果。只有采用药物疗法等减轻肾素-血管紧张素系统的活性,消除血管紧张素Ⅱ对血管的作用,才有明显的降压作用。
3.肾脏形成血管舒张物质减少正常肾髓质能生成前列腺素A2(PGA2)和E2(PGE2)等血管舒张物质。此类物质能舒张肾皮质血管,增加肾皮质血流量和抑制肾素的分泌,从而具有抗高血压的作用。此外,这类物质还具有排钠排水的效应。因此有人认为肾实质破坏引起这类物质形成减少,也可促进高血压的发生;但此问题尚待进一步研究。肾性高血压的形成机制,概括如图14-6。
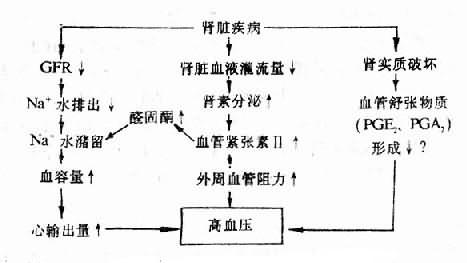
图14-6 肾性高血压发病机制的示意图
(四)贫血
慢性肾脏疾病经常伴有贫血。贫血原发病机制可能与下列因素的作用有关:①肾脏组织严重受损后,肾脏形成促红细胞生成素减少;②血液中潴留的毒性物质对骨髓造血功能具有抑制作用,如甲基胍对红细胞的生成具有抑制作用;③慢性肾功能障碍可引起肠道对铁的吸收减少,并可因胃肠道出血而致铁丧失增多;④毒性物质的蓄积可引起溶血及出血,从而造成红细胞的破坏与丢失。
(五)出血倾向
慢性肾功能衰竭的患者常有出血倾向,其主要临床表现为皮下淤斑和粘膜出血,如鼻出血和胃肠道出血等。一般认为血小板数量减少不是造成出血的主要原因,而血小板的功能障碍,才是其主要病因,血小板功能障碍表现为:①血小板第3因子(磷脂,是Ⅸ、Ⅹ、凝血酶原活化场所)的释放受到抑制,因而凝血酶原激活物生成减少;②血小板的粘着和聚集功能减弱,因而出血时间延长。上述血小板的功能改变可能是毒性物质在体内蓄积所引起,例如尿素、胍类、酚类化合物等都可能有改变血小板功能的作用。
(六)免疫功能障碍
慢性肾功能衰竭晚期常并发免疫功能障碍,而且以细胞免疫异常为主。如尿毒症病人血中淋巴细胞减少,T淋巴细胞的绝对数降低,迟发型皮肤变态反应减弱,同种异体移植的皮肤和肾脏存活时间延长等。由于中性粒细胞趋化性降低,尿毒症患者对细菌感染的敏感性有所增高。体液免疫变化不大,大多数尿毒症病人的抗体生成未见明显异常,血清补体水平也属正常。慢性肾功能衰竭时出现细胞免疫功能异常,可能与毒性物质对淋巴细胞的分化和成熟有抑制作用,或者对淋巴细胞有毒性作用等因素有关。
第四节 尿毒症
急性和慢性肾功能衰竭均可导致终末代谢产物和内源性毒性物质在体内潴留,水、电解质、酸碱平衡紊乱以及内分泌功能失调,从而引起一系列自体中毒症状,称为尿毒症(uremia)。
一、尿毒症的主要临床表现及其发病机制
在尿毒症期,除上述水、电解质、酸碱平衡紊乱、贫血、出血倾向、高血压等进一步加重外,还可出现各器官系统功能障碍以及物质代谢障碍所引起的临床表现,兹分述如下。
(一)神经系统症状
神经系统的症状是尿毒症的主要症状。在尿毒症早期,患者往往有头昏、头痛、乏力、理解力及记忆力减退等症状。随着病情的加重可出现烦躁不安、肌肉颤动、抽搐;最后可发展到表情淡漠、嗜睡和昏迷。这些症状的发生与下列因素有关:①某些毒性物质的蓄积可能引起神经细胞变性;②电解质和酸碱平衡紊乱;③肾性高血压所致的脑血管痉挛,缺氧和毛细血管通透性增高,可引起脑神经细胞变性和脑水肿。
(二)消化系统症状
尿毒症患者消化系统的最早症状是食欲不振或消化不良;病情加重时可出现厌食,恶心、呕吐或腹泻。这些症状的发生可能与肠道内细菌的尿素酶将尿素分解为氨,氨剌激胃肠道粘膜引起炎症和多发性表浅性小溃疡等有关。患者常并发胃肠道出血。此外恶心、呕吐也与中枢神经系统的功能障碍有关。
(三)心血管系统症状
慢性肾功能衰竭者由于肾性高血压、酸中毒、高钾血症、钠水潴留、贫血及毒性物质等的作用,可发生心力衰竭,心律失常和心肌受损等。由于尿素(可能还有尿酸)的剌激作用,还可发生无菌性心包炎,患者有心前区疼痛;体检时闻及心包摩擦音。严重时心包腔中有纤维素及血性渗出物出现。
(四)呼吸系统症状
酸中毒时患者呼吸慢而深,严重时可见到酸中毒的特殊性Kussmaul呼吸。患者呼出的气休有尿味,这是由于细菌分解睡液中的尿素形成氨的缘故。严重患者可出现肺水肿,纤维素性胸膜炎或肺钙化等病变,肺水肿与心力衰竭、低蛋白血症、钠水潴留等因素的作用有关。纤维素性胸膜炎是尿素剌激引起的炎症;肺钙化是磷酸钙在肺组织内沉积所致。
(五)皮肤症状
皮肤瘙痒是尿毒症患者常见的症状,可能是毒性产物对皮肤感受器的剌激引起的;有人则认为与继发性甲状旁腺功能亢进有关,因为切除甲状旁腺后,能立即解除这一痛苦的症状。此外,患者皮肤干燥、脱屑并呈黄褐色。皮肤颜色的改变,以前认为是尿色素增多之故,但用吸收分光光度计检查,证明皮肤色素主要为黑色素。在皮肤暴露部位,轻微挫伤即可引起皮肤淤斑。由于汗液中含有较高浓度的尿素,因此在汗腺开口处有尿素的白色结晶,称为尿素霜。
(六)物质代谢障碍
1.糖耐量降低尿毒症患者对糖的耐量降低,其葡萄糖耐量曲线与轻度糖尿病患者相似,但这种变化对外源性胰岛素不敏感。造成糖耐量降低的机制可能为:①胰岛素分泌减少;②尿毒症时由于生长激素的分泌基础水平增高,故拮抗胰岛素的作用加强;③胰岛素与靶细胞受体结合障碍,使胰岛素的作用有所减弱;④有关肝糖原合成酶的活性降低而致肝糖原合成障碍。目前认为引起上述变化的主要原因可能是尿素、肌酐和中分子量毒物等的毒性作用。
2.负氮平衡负氮平衡可造成病人消瘦、恶病质和低白蛋白血症。低白蛋白血症是引起肾性水肿的重要原因之一。引起负氮平衡的因素有:①病人摄入蛋白质受限制或因厌食、恶心和呕吐而致蛋白质摄入减少;②某些物质如甲基胍可使组织蛋白分解代谢加强;③合并感染时可导致蛋白分解增强;④因出血而致蛋白丢失;⑤随尿丢失一定量的蛋白质等。
尿毒症时大量尿素可由血液渗入肠腔。肠腔细菌可将尿素分解而释放出氨,氨被血液运送到肝脏后,可再合成尿素,也可合成非必需氨基酸,后者对机体是有利的。因此有人认为,尿毒症病人蛋白质的摄入量可低于正常人,甚至低于每天20g即可维持氮平衡,但必须给予营养价值较高的蛋白质,即含必需氨基酸丰富的营养物质。近年来有人认为。为了维持尿毒症病人的氮平衡,蛋白质摄入量应与正常人没有明显差异;而且认为,单纯为了追求血液尿素氮的降低而过分限制蛋白质的摄入量,可使自身蛋白质消耗过多,因而对病人有害而无益。
3.高脂血症尿毒症病人主要由于肝脏合成甘油三酯所需的脂蛋白(前β-脂蛋白)增多,故甘油三酯的生成增加;同时还可能因脂蛋白脂肪酶(lipoprotein lipase)活性降低而引起甘油三酯的清除率降低,故易形成高甘油三酯血症。此种改变可能与甲基胍的蓄积有关。
二、尿毒症毒性物质的作用
在肾功能衰竭时,体内许多蛋白质最终代谢产物不能由肾脏排出而蓄积于体内,因而可引起一系列中毒症状,故这类物质称为尿毒性的毒性物质。尿毒症的毒性物质的作用机制,迄今尚未阐明。有资料表明,胍类尤其是甲基胍,以及未知结构的中分子量物质可能是尿毒症时的主要毒性物质,而尿素、肌酐、酚类等?
■[此处缺少一些内容]■
2.胍基琥珀酸的作用尿毒症时胍基琥珀酸可通过以下两个途径生成:
(1)在正常情况下,精氨酸和甘氨酸可在甘氨酸精氨酸脒基移换酶的作用下,生成胍乙酸和鸟氨酸;胍乙酸又可转变为肌酐。尿毒症时上述酶的活性降低,且因肌酐在体内蓄积,故使上述反应不能进行。此时组织中的精氨酸易于和门冬氨酸在门冬氨酸精氨酸脒基移换酶的作用下,生成胍基琥珀酸。
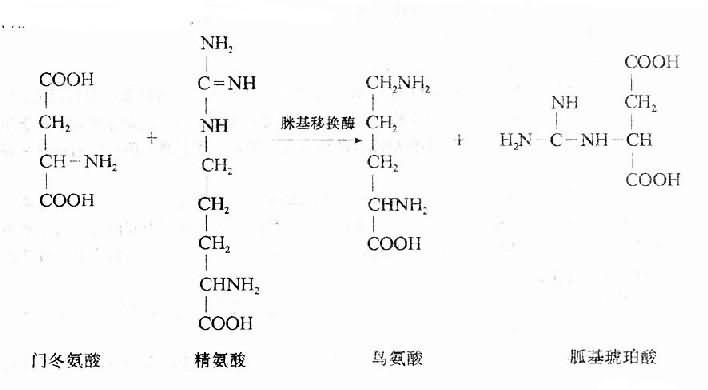
(2)在体内瓜氨酸和门冬氨酸可以生成精氨酸代琥珀酸。正常情况下,精氨酸代琥珀酸裂合酶活性高,故精氨酸代琥珀酸在B键处断裂,而生成延胡索酸和精氨酸。尿毒症时,有人认为血中尿素浓度增高可能引起此酶的活性降低,因而精氨酸代琥珀酸在A键处被裂解而生成鸟氨酸和胍基琥珀酸。
正常人血浆中胍基琥珀酸的浓度约为0.03mg%,而在尿毒症患者可高达8.3mg%,增高200多倍。胍基琥珀酸可抑制血小板第3因子的活性,又能促进溶血,因而可能与尿毒症的出血倾向和贫血有关。
(二)未知中分子量物质的作用
腹膜透析与人工肾透析相比,尽管前一方法清除低分子量毒性物质的能力远远低于后一方法,但两者的临床效果相差不大。因此有人推测,除低分子量物质外,可能还有借腹膜透析能有效清除的其他物质与尿毒症的发生有关。以后的研究证实,腹膜比人工肾用的赛璐玢膜更易于让某些中分子量物质透过。因此提出中分子量物质学说以解释尿毒症的中毒症状。中分子量物质的分子量在300~1,500之间,其化学结构不明,推测为多肽类物质。有人发现,从尿毒症患者透析液中提出的中分子量物质,在体外对成纤维细胞的增生、白细胞吞噬作用、淋巴细胞的增生以及细胞对葡萄糖的利用等具有抑制作用。这可能与尿毒症病人糖耐量降低和免疫抑制等变化有关。
(三)其他毒性物质的作用
1.尿素的作用一般认为尿毒症病人血浆尿素浓度的增高不会引起明显的毒性的反应;但是尿素可抑制单胺氧化酶,黄嘌呤氧化酶,以及ADP对血小板第3因子的激活作用。并能使胍基琥珀酸产生增多,从而导致血小板功能异常和出血。此外,尿素增高还会引起糖耐量降低。
2.肌酐的作用在尿毒症期,体内蓄积的肌酐可能并无明显的毒性作用;但在体外将肌酐加到血液中,使其浓度相当于重症尿毒症病人血中肌酐浓度时,却可引起溶血。给正常狗注入肌酐,可降低红细胞的存活时间。此外,肌酐还可引起动物倦睡和糖耐量降低,故不能认为肌酐是完全无毒的物质。
3.尿酸的作用血浆尿酸浓度很高的病人,并发心包炎也多;因此认为尿酸在心包炎的发病机制中可能起一定作用。
4.酚和酚酸的作用肠道细菌可将芳香族氨基酸转变成酚和酚酸。在正常人,这些物质被吸收后,可经肝脏解毒而由肠道和肾脏排出。肾功能衰竭时,由于肝脏解毒功能降低和肾脏排泄功能减弱,故血浆中酚类含量可以增高。酚类能促进溶血。酚酸如羟苯乙酸在体外可抑制血小板的聚集;因此酚酯可能是导致尿毒症时出血倾向的原因之一。
三、尿毒症的防治原则
(一)积极防治原发疾病以防止肾实质的继续破坏。
(二)慢性肾功能衰竭患者的肾功能主要依靠残存的完整肾单位来维持。任何加重肾脏负荷的因素,均可加重肾功能衰竭;因此应积极消除诱发肾功能恶化的有害因素,例如控制感染,减轻高血压等。此外,还应矫正水、电解质紊乱,纠正酸中毒等以维持内环境的稳定。
(三)肾功能衰竭患者出现尿毒症时,应采取抢救措施以维持内环境的稳定。常用的措施有腹膜透析、血液透析(人工肾)等。必要和可能时也可进行同种肾移值以取代患病的肾脏。
第十五章 肝脏病理生理学
第一节 肝功能不全
肝脏的主要功能是参与物质代谢、生物转化(解毒与灭活)、凝血物质的生成和消除、胆汁的生成与排泄。肝脏有丰富的单核吞噬细胞,在特异和非特异免疫中具有重要的作用。
当肝脏受到某些致病因素的损害,可以引起肝脏形态结构的破坏(变性、坏死、肝硬化)和肝功能的异常。但由于肝脏具有巨大的贮备能力和再生能力,比较轻度的损害,通过肝脏的代偿功能,一般不会发生明显的功能异常。如果损害比较严重而且广泛(一次或长期反复损害),引起明显的物质代谢障碍、解毒功能降低、胆汁的形成和排泄障碍及出血倾向等肝功能异常改变,称为肝功能不全(hepatic insufficiency)。严重肝功能损害,不能消除血液中有毒的代谢产物,或物质代谢平衡失调,引起中枢神经系统功能紊乱(肝性脑病),称为肝功能衰竭(hepatic failure)。
引起肝功能不全的原因很多,可概括为以下几类:
1.感染寄生虫(血吸虫、华枝睾吸虫、阿米巴)、钩端螺旋体、细菌、病毒均可造成肝脏损害;其中尤以病毒最常见(如病毒性肝炎)。
2.化学药品中毒 如四氯化碳、氯仿、磷、锑、砷剂等,往往可破坏肝细胞的酶系统,引起代谢障碍,或使氧化磷酸化过程受到抑制,ATP生成减少,导致肝细胞变性坏死;有些药物,如氯丙嗪、对氨柳酸、异菸肼、某些碘胺药物和抗菌素(如四环素),即使治疗剂量就可以引起少数人的肝脏损害,这可能与过敏有关。
3.免疫功能异常肝病可以引起免疫反应异常,免疫反应异常又是引起肝脏损害的重要原因之一。例如乙型肝炎病毒引起的体液免疫和细胞免疫都能损害肝细胞;乙型肝炎病毒的表面抗原(HBsAg)、核心抗原(HBcAg)、e抗原(HBeAg)等能结合到肝细胞表面,改变肝细胞膜的抗原性,引起自身免疫。又如原发性胆汁性肝硬化,病人血内有多种抗体(抗小胆管抗体、抗线粒体抗体、抗平滑肌抗体、抗核抗体等),也可能是一种自身免疫性疾病。
4.营养不足缺乏胆硷、甲硫氨酸时,可以引起肝脂肪性变。这是因为肝内脂肪的运输须先转变为磷脂(主要为卵磷脂),而胆硷是卵磷脂的必需组成部分。甲硫氨酸供给合成胆硷的甲基。当这些物质缺乏时,脂肪从肝中移除受阻,造成肝的脂肪性变。
5.胆道阻塞胆道阻塞(如结石、肿瘤、蛔虫等)使胆汁淤积,如时间过长,可因滞留的胆汁对肝细胞的损害作用和肝内扩张的胆管对血窦压迫造成肝缺血,而引起肝细胞变性和坏死。
6.血液循环障碍 如慢性心力衰竭时,引起肝淤血和缺氧。
7.肿瘤 如肝癌对肝组织的破坏。
8.遗传缺陷有些肝病是由于遗传缺陷而引起的遗传性疾病。例如由于肝脏不能合成铜蓝蛋白,使铜代谢发生障碍,而引起肝豆状核变性;肝细胞内缺少1-磷酸葡萄糖半乳糖尿苷酸转移酶,1-磷酸半乳糖不能转变为1-磷酸葡萄糖而发生蓄积,损害肝细胞,引起肝硬化。
一、物质代谢的改变
肝功能不全时,代谢的变化是多方面的,包括蛋白质、脂质、糖、维生素等。而且能反映在血液内血浆蛋白、胆固醇和血糖含量的变化。
(一)蛋白质代谢变化
主要表现为血浆蛋白的含量改变。
血浆蛋白主要有白蛋白、球蛋白、纤维蛋白原,以及微量的酶及酶原(如凝血酶原)等。正常人血浆蛋白总量为6-7.5克%,其中白蛋白3.8-4.8克%,球蛋白(α1、α2、β、γ)2-3克%,纤维蛋白原0.2-0.4克%,白蛋白/球蛋白的比值为1.5-2.5。
1.血浆白蛋白减少血浆白蛋白由肝细胞合成,肝细胞损害时,血浆白蛋白降低。肝脏每天合成白蛋白约12-18克,半衰期约为13.5天,因此急性肝炎在短期内,血浆白蛋白改变不明显。肝细胞受到极其严重的损害(急性或慢性),如急性或亚急性肝坏死、慢性肝炎、肝硬化等,由于白蛋白合成减少,血浆白蛋白才明显减少。血浆白蛋白减少(低于2.0克%),血浆胶体渗透压降低,是产生腹水或全身性水肿的重要原因之一。
2.纤维蛋白原和凝血酶原等凝血物质减少纤维蛋白原、凝血酶原及凝血因子Ⅴ、Ⅶ、Ⅷ、Ⅸ、Ⅹ,均在肝细胞内合成。肝细胞严重损害,凝血因子(Ⅰ、Ⅱ、Ⅴ、Ⅶ、Ⅷ、Ⅸ、Ⅹ)生成减少,血液凝固性降低,是肝病患者出血倾向的重要原因。
3.球蛋白增多,主要是γ-球蛋白增多。γ球蛋白是由浆细胞产生的。肝脏疾患时,由于抗原的剌激,γ-球蛋白产生增多。β-球蛋白是由肝细胞、浆细胞、淋巴细胞合成的,其主要成分是β-脂蛋白。肝脏疾患时,β-球蛋白常常也是增多,特别是在胆汁淤滞时,如阻塞性黄疸患者,血中β-球蛋白明显升高,这可能与脂类代谢障碍有一定关系。
肝脏疾患时,由于白蛋白合成减少,球蛋白增多。因此,虽然血浆总蛋白可以没有明显改变,但是白蛋白/球蛋白的比值降低,可以小于1.5-1,甚至倒置(即球蛋白多于白蛋白)。
(二)血浆胆固醇含量变化
人体内胆固醇有两个来源:一是来自动物性食物,二是在体内合成。肝脏、小肠粘膜、皮肤合成胆固醇的能力很强。血浆胆固醇大部分来自肝脏,一部分来自食物,肝外组织合成的胆固醇一般很少进入血液。肝细胞分泌卵磷脂胆固醇脂酰转移酶,在血浆中将卵磷脂分子中β位置上的不饱和脂酰基转移至游离胆固醇的分子上,生成胆固醇脂(图15-1),肝脏本身也能将游离胆固醇转变为胆固醇脂。因此,血浆中胆固醇有两种存在形式,一是游离胆固醇(占20-40%),二是胆固醇脂(占60-80%)。正常血浆胆固醇总量为150-250毫克%。胆固醇一部分由肝桩经胆道系统直接排入肠内,绝大部分(约占80%)在肝内先转变为胆酸和脱氧胆酸,以胆盐的形式经胆道系统排入肠内,。肝功能不全时,胆固醇的形成、酯化、排泄发生障碍,引起血浆胆固醇含量的变化。
图15-1 胆固醇的酯化
LCAT 卵磷脂胆固醇脂酰转移酶
1.单纯胆道阻塞,胆固醇排出受阻,血浆胆固醇总量明显增高,而胆固醇酯占胆固醇总量的百分比正常。
2.肝细胞受损害,胆固醇酯生成减少,血浆胆固醇酯含量减少,在胆固醇总量中所占的百分比降低,血浆胆固醇总量降低或在正常范围内。
3.肝细胞受损害同时伴有胆道阻塞(如黄疸型肝炎伴有小胆管阻塞),血浆胆固醇总量可以增高,但胆固醇酯在胆固醇总量中的百分比降低。
(三)血糖的变化
肝脏在糖代谢中具有合成、贮藏及分解糖原的作用,使肝糖原与血糖之间保持动态平衡,维持血糖浓度在一定水平。正常血糖含量为80-120毫克%。一般地说,轻度肝脏损害往往很少出现糖平衡紊乱。当肝细胞发生弥漫性的严重损害时,由于肝糖原合成障碍及贮存减少,表现为空腹时血糖降低。当血糖低于60-70毫克%时,就会出现低血糖症,此时病人感到软弱、疲乏、头晕。脑的能量来源主要靠葡萄糖的氧化,而脑糖原的贮存量极少,主要依靠血液供给葡萄糖。当血糖急剧降低至40毫克%时,由于脑的能量供应不足,发生低血糖性昏迷。低血糖性昏迷常见于急性坏死、肝硬化及肝癌的晚期。由于肝细胞损害,不能及时地把摄入的葡萄糖合成肝糖原,食多量糖后,可发生持续时间较长的血糖升高。
二、血清酶的改变
肝脏是物质代谢最活跃的器官,酶的含量极为丰富。肝细胞受损或肝功能障碍时,也可反映到血清中某些酶的改变,有的升高,有的降低。临床上常利用血清中某些酶的变动来衡量肝脏功能,了解肝细胞的损害程度或胆道系统的阻塞情况。
(一)有些血清酶升高
1.在肝细胞内合成并在肝细胞内参与代谢的酶,例如转氨酶,(谷—丙转氨酶、谷—草转氨酶)、乳酸脱氢酶,由于肝细胞受损害(变性、坏死、细胞膜通透性升高)而释放入血,使这些酶在血清中升高。在肝细胞中谷—丙转氨酶活力比较高,因此当肝细胞损害时,血清谷—丙轻氨酶升高比较明显。正常值;金氏单位<100,穆氏单位<40。测定血清谷—丙转氨酶有助于判断病情的变化。
2.从胆道排出的酶,因排泄障碍或生成增多,而在血清内增多。例如碱性磷酸酶、γ谷氨酰转肽酶。
碱性磷酸酶(AKP)的作用是在碱性环境中水解有机磷酸脂类化合物,并促进磷酸钙在骨骼中沉积。正常人血清AKP主要来自肝脏,正常成人为3-13单位(金氏法),在正常情况下可经胆道排出。当胆道阻塞、胆内胆汁淤积时,该酶从胆道排出受阻,而随胆汁逆流入血,与此同时,肝内AKP的合成也增加,故血清AKP的活性明显升高。而在肝炎或肝硬化等肝细胞病变时,此酶活性变化不大,据此可以为区别阻塞性和肝细胞性黄疸指标之一。此外,当肝脏中有原发性肝癌或肝内占位性病变(如肝脓肿)时,也可见血清AKP增高,尤以转移性肝癌病人,增高更显著。
γ-谷氨酰转肽酶(γ-GT)对于体内氨基酸和蛋白质的吸收、分泌和合成都是必需的。主要存在于肾小管及肝毛细胆管处,血清中γ-GT主要来自肝脏和由胆道排出。它能将谷胱甘肽中的γ-谷氨酰基团转移到其它氨基酸或多肽上。
病毒性肝炎或慢性活动性肝炎时,此酶可轻度升高,而在阻塞性黄疸、原发性肝癌或转移性肝癌时明显升高。无黄疸而γ-GT明显升高,注意排除肝癌。
(二)有些血清酶降低
在肝细胞内合成并不断释放入血的酶,例如血清胆碱脂酶(或称假性胆碱脂酶),因肝细胞受损害,合成减少,血清胆硷脂酶降低。正常值:比色法为30-80单位。
血清内酶活性的变化,取决于组织内酶释放的多少、组织内酶产生的改变和酶排泄的异常三个因素。这些改变缺乏特异常性,不同的疾病均可引起同一酶活性的变化,但如果把各种不同的酶组合成酶谱,用以分析不同疾病时酶谱的谱型,则能弥补单项酶活性测定之不足。在由谷丙转氨酶(GPT)、碱性磷酸酶(AKP)、乳酸脱氢酶(LDH)、磷酸已糖异构酶(PHI)和γ谷氨酶转肽酶(γ-GT)组成的酶谱中,若GPT、PHI显著高,其余各酶活性正常或轻度升高,则提示肝细胞受损,称为“肝细胞损伤型酶谱”、若以γ-GT和AKP活性升高为主,则称为“梗阻型酶谱”、如GPT正常或轻度升高,其余和酶显著升高,则称为“肝癌型酶谱”。临床上所作的酶谱测定,利用不同的谱型对肝胆疾病作早期诊断和鉴别诊断有一定帮助。
三、生物转化和排泄功能的变化
(一)解毒功能降低
肝脏是人体重要的解毒器官。机体代谢过程中产生的有毒物质,例如蛋白质代谢产生的氨,在肝内变成无毒的尿素,从大肠吸收的有毒物质(如氨、胺类、吲哚、酚类等)以及直接来自体外的毒物,随血液进入肝脏后,在肝细胞中经生物转化作用,变成无毒或毒性较小随尿或胆汁排出体外。这些变化称为解毒功能。
肝脏的解毒功能有氧化、还原、结合、水解、脱氨等方式,其中主要是氧化和结合解毒。
1.氧化解毒氧化解毒是最常见的解毒方式。许多有毒物质在肝内经氧化后,即被破坏而失去毒性。例如,在肠内经腐败作用所产生的胺类,可由肝组织内活性很强的单胺氧化酶及二胺氧化酶的作用,先被氧化成醛及氨。醛再被氧化成酶,最后变成二氧化碳及水;氨在肝内合成尿素。
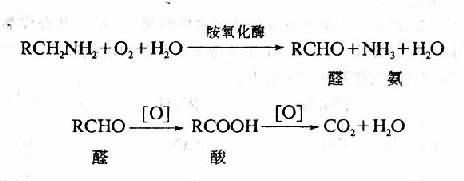
2.结合解毒结合解毒是体内最重要的解毒方式。许多有毒物质常不能在体内被氧化或还原,或虽经氧化或还原仍有毒性。这类物质的解毒方式是在肝细胞的内质网中与葡萄糖醛、硫酸盐、甘氨酸等结合,生成无毒、毒性较小而易于溶解的化合物,然后从体内排出。由于肝脏能合成葡萄糖醛酸,因此与葡萄糖醛酸结合的解毒方式最常见。例如食物残渣在大肠内腐败后,常产生许多有毒的酚类化合物,这些有毒物质被吸收后,在肝内与葡萄糖醛酸结合解毒。也能与硫酸盐结合解毒。
又如色胺酸在大肠内腐败生成有毒性的吲哚,被吸收后先在肝内氧化成为吲哚(吲哚酚),然后再与硫酸盐(或葡萄糖醛酸)结合成无毒的尿蓝母,随尿排出。
当肝功能不全时,肝解毒功能降低,引起机体中毒。
(二)对激素的灭能作用低
正常有些激素是肝脏内破坏的(称为肝脏对激素的灭能作用),例如雌激素、抗利尿激素、醛固酮等。
雌激素在体内降解主要是在肝内进行,雌激素在羟化酶作用下,生成雌三醇,孕酮被还原为孕二醇。雌三醇和孕二醇在肝内与葡萄糖醛酸或硫酸盐结合,随胆汁和尿排出。动物实验及人体研究证明,肝脏受损害后,对激素的灭能作用减退,使体内及尿内的雌激素含量增加。有些肝病,(如门脉性肝硬化)患者,血与尿中的雌激素都增加,并出现蛛蜘痣(皮肤上以小动脉为中心及其向周围放射状毛细血管组成的一种小血管扩张现象)、肝掌(手掌充血发红)。蜘蛛痣及肝掌的出现,与肝脏的灭能作用减退,体内雌激素增多有关。此外,雌激素破坏减少,男子出现乳房发育,睾丸萎缩;女子可出现月经失调。
肝脏对抗利尿激素及醛固酮也具灭能作用。实验证明,肝浸出物有破坏抗利尿激素的作用。肝脏损害时,对抗利尿激素的灭能作用减弱,引起体内抗利尿激素增多。实验证明,将醛固酮和肝脏切片放在一起,置于保温箱内,醛固酮潴留钠的作用即可消失。当肝脏受损害时,醛固酮在肝内破坏减少,在体内增多。因此,在肝功能不全时,尤其是肝硬化患者,体内抗利尿激素及醛固酮增多是引起水肿及腹水的原因之一。
(三)排泄功能降低
肝脏有一定的排泄功能,如胆色素、胆盐、胆固醛、碱性磷酸酶以及Ca++、Fe+++等,可随胆汁排出。解毒作用后的产物除一部分由血液运到肾脏随尿排出外,也有一部分从胆汁排出;As+++、Hg++及某些药物和色素在某种情况下进入机体后,也是胆道排出。肝脏对一些内源性或外源性有毒物质的排泄,必须经过肝细胞的摄取、生物转化、输送及排出等一系列过程。肝脏排泄功能降低时,由肝道排泄的药物或毒物在体内蓄积,导致机体中毒。
临床上常用酚四溴酞钠(BSP)清除试验,来判断肝脏的排泄功能。BSP是一种无毒性、在血液内不变化的染料,注入血液后,大部分与白蛋白及α1-球蛋白结合。在健康人体内约80%由肝细胞摄取,15-20%由骨骼肌摄取,仅2%由肾脏排出。BSP在肝细胞内与谷胱甘肽等结合的形式排入胆管。试验时,由静脉注入BSP5mg/公斤体重,正常注射后一小时,血内已不能查出这种染料,或只有很微量,如果注射后30分钟,血内还滞留有注入量的10-40%,表示有轻度肝功能减退;滞留50-80%,表示中等度肝功能减退;滞留90%以上,表示有严重的肝功能不全。
四、肝性脑病(肝性昏迷)
肝性脑病(hepatic encephalopathy)是指继发于严重肝脏疾患的中枢神经系统机能障碍所呈现的精神、神经综合病症。它包括从轻度的精神、神经症状、到陷入深度昏迷的整个过程。早期有性格改变(欣快或沉默少言,烦躁或淡漠);进一步发展,可发生精神错乱,行动异常,定向障碍(什么时候、地点、是谁分辨不清),两手有扑翼样震颤(让患者平举两上肢,两手呈扑翼样抖动);严重时发展为嗜睡,昏迷。
肝性脑病常见于急性或亚急性肝坏死(重型病毒性肝炎、中毒)肝硬化和肝癌的晚期,以及一部分门体分流手术后的病人,上述情况造成的肝功能严重损害和门体分流是导致肝昏迷的重要原因。肝功能受到严重损害,不能消除血液中有毒的代谢产物;由于门腔静脉分流术或自然形成的侧枝循环,使门静脉中的有毒物质不经过肝脏这个起屏障作用的重要脏器,而进入体循环,从而引起中枢神经系统代谢紊乱。
急性型肝性脑病,起病急骤,迅速出现躁动、谵妄以至昏迷,大多数短期内死亡。多见于重型病毒性肝炎及中毒性肝炎引起的广泛而急剧的肝细胞破坏。
慢性型肝性脑病,起病较缓,往往有明显的诱因(如上消化道出血),常在慢性肝疾患(如肝硬化)或门腔静脉分流术后的基础上发生。
(一)肝性脑病的发生机理
关于肝性脑病的发病机理至今尚未完全阐明。肝性脑病时脑的形态变化,在急性型除少数可见脑水肿外,大多无特殊的病理形态变化;而在慢性型,特别是有反复发作史的患者,通常可见明显的星形细胞肥大和增生;在少数的慢性型特殊病例,可见脑神经元变性和髓鞘脱失现象。由于星形细胞的生理意义还不完全清楚,因此在目前,还很难以脑的形态变化来解释肝性脑病的发生机理,脑的形态变化和功能变化之间的关系,也还有待进一步的研究。目前认为,肝性脑病时中枢神经系统的机能障碍主要是代谢性的或功能性的,是多种发病因素综合作用的结果。一般认为与下列因素有关。
1.血氨增多(氨中毒学说theory of ammonia intoxication)正常人血氨浓度低于100微克%,80-90%的肝性脑病的病人,有血氨升高,有的增高到正常人的2-3倍以上(200-500微克%),而且有时还可看到血氨增高与神经精神症状严重程度相平行。给动物注入氨化铵可引起中枢神经系统机能障碍。因此血氨增多,可能是肝性脑病发生的一个重要因素。
正常血氨的主要来源、①组织代谢过程中形成的氨,包括氨基酸脱氨基过程中产生的氨以及肾小管上皮细胞内的谷氨酰胺经谷氨酰胺酶水解产生的NH3。由肾小管上皮细胞产生的NH3,除了扩散到肾小管与H+结合形成NH4+,起着排NH4+保碱的作用外,也有部分氨弥散入血。②肠道内形成的氨。未被吸收的氨基酸以及经肠壁渗入肠腔的尿素,在大肠内经细菌产生的氨基酸氧化酶和尿素酶的作用,产生氨,由肠道吸收入血。正常对氨的处理,绝大部分在肝脏通过鸟氨酸循环形成尿素,再从肾脏排出和经肠壁渗入肠腔(图15-2),部分氨与谷氨酸合成谷氨酰胺。
(二)血氨增多的原因
1)尿素合成障碍:肝功能不全时,由于代谢障碍,ATP供给不足以及肝内酶系统受损害,导致鸟氨酸循环障碍,尿素合成能力降低,由组织代谢过程中形成的氨及肠道吸收的氨在肝内合成尿素减少,血氨增多(图15-2)。
2)门体侧枝循环形成:肝硬化时,由于门静脉高压,门腔静脉侧支循环形成,由肠道吸收门静脉血的氨,经侧支循环绕过肝脏,直接流入体循环,血氨增多(图15-2)。
3)产氨增多:门脉高压时,可因胃肠道粘膜于血水肿或胆汁分泌减少,而使消化吸收功能减弱,胃肠运动迟缓,肠内蛋白质及其含氮的分解产物,受细胞作用(腐败),产氨增多,物别在进食高蛋白膳食或上消化道出血时(每100毫升血液约含15-20克蛋白质,还有尿素),将更加重血氨的升高。
图15-2 正常人和肝性脑病患者的氨及胺类代谢示意图
(2)血氨增多对中枢神经系统的损害正常时血氨含量很少,脑组织内少量的氨与α-酮戊二酸在谷氨酸脱氢酶的作用下形成谷氨酸。谷氨酸在谷氨酰合成酶及ATP、Mg++的参与下,再与氨结合形成谷氨酰胺(图15-3)。谷氨酰胺是运送氨的一种形式,在肝及肾内经谷氨酰胺酶的作用,再分解成谷氨酸和氨。氨在肝内经鸟氨酸循环形成尿素,在肾内形成的NH4+,由肾排出。
血氨增多时,血氨通过血脑屏障进入脑组织的氨增多,氨损伤中枢神经系统机能的机理,至今尚未完全阐明。可能通过下列几个环节干扰神经细胞代谢(图15-3):
1)氨与脑细胞中的α-酮戊二酸结合,形成谷氨酸,消耗了大量的α-酮戊二酸。有人认为在一般组织内,α-酮戊二酸用去后,可以很快从血液中得到补充,而脑组织因α-酮戊二酸很难通过血脑屏障,α-酮戊二酸消耗后,不易从血液中得到补充。α-酮戊二酸是三羧酸循环的中间反应物,当α-酮戊二酸减少后,三羧酸循环不能正常进行,ATP生成减少,能量供应不足,不能维持大脑的正常活动,从而产生机能紊乱,以至发生昏迷。
2)在谷氨酸形成过程中,大量消耗了还原型辅酶Ⅰ(NADH),妨碍了呼吸链中递氢过程,ATP生成减少。
3)谷氨酸在谷氨酰胺合成酶及ATP参与下,再与氨结合,形成谷氨酰胺,这样又大量消耗ATP。
4)高浓度氨对丙酮酸和a-酮戊二酸的脱氢酶系有抑制作用(这可能与维生素B1不能在肝脏有效地转变成焦磷酸硫胺素有关),影响三羧酸循环,而使ATP生成减少。
图15-3 氨干扰脑组织代谢的可能环节①α-酮戊二酸减少,影响三羧酸循环②NADH减少,使呼吸链生成ATP减少③谷氨酰胺合成时,消耗ATP④丙酮酸和α-酮戊二酸脱氢酶系受抑制⑤γ-氨基丁酸含量改变⑥乙酰胆硷减少
5)γ-氨基丁酸生成减少。γ-氨基丁酸是一种中枢抑制性神经递质,是由谷氨酸经谷氨酸脱羧酶作用脱羧而形成的。当脑中氨增多时,谷氨酸被消耗,因而γ-氨基丁酸形成减少。目前认为肝性脑病时的躁动、精神错乱、抽搐等神经精神症状与γ-氨基丁酸减少有关。故对兴奋型肝性脑病患者,有人主要给予γ-氨基丁酸治疗,以制止抽搐和躁动。但也有报导,肝性脑病时γ-氨基丁酸含量增多(可能通过对转氨酶的抑制,使γ-氨基丁酸不能转变为琥珀酸半醛),加重中枢的抑制和昏迷。
6)乙酰胆硷含量减少。高浓度氨抑制丙酮酸的氧化脱羧过程,使乙酰辅酶A生成减少,从而影响乙酰胆硷的合成。乙酰胆硷是中枢兴奋性神经递质,它的减少可导致脑功能抑制。
综上所述,脑内氨浓度的升高主要引起:①能量代谢受干扰,ATP消耗增加和因三羧酸循环受影响而使ATP的生成减少,造成脑内ATP供应不足;②通过对谷氨酸代谢和丙酮酸代谢的干扰,改变了脑内某些神经递质的浓度和相互平衡关系。
2.假性神经递质的形成(假递质学说,theory of false neurotransmitter)由于肝性脑病的病人,不是所有患者都有血氮增多,或经治疗后血氨虽已下降,但病人的精神神经症状并未得到改善,于是有人对氨中毒的观点提出了怀疑,并认为肝性脑病的发生可能与中枢神经系统正常的神经递质被假性神经递质所取代有关。
在我们的食物蛋白中,包含一些芳香族氨基酸,如苯丙氨酸及酪氨酸,经肠内细菌脱羧酶的作用,形成苯乙胺及酪胺。这些胺类物质从肠道吸收,经门静脉到达肝脏,经肝脏单胺氧化酶的作用氧化分解,而被清除(图15-4,15-5)。
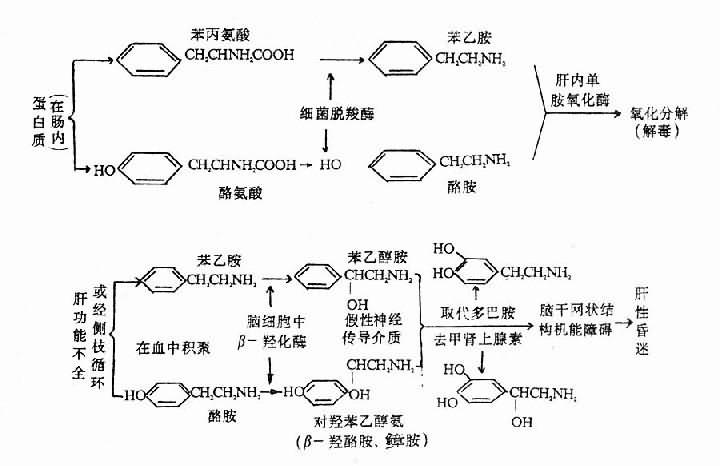
图15-4 假性神经递质的形成
肝功能不全时,由于肝脏解毒功能降低,或经侧枝循环,使血液中的苯乙胺及酷胺积聚,随体循环进入脑组织,在脑细胞内经非特异性的β-羟化酶的作用,在侧链β位置上被羟化,形成苯乙醇胺和对羟苯乙醇胺(鱆胺)。
当脑组织中的苯乙醇胺及对羟苯乙醇胺增多,特别是在脑干网状结构中,由于苯乙醇胺和对羟苯乙醇胺在结构上与脑干网状结构以及黑质、纹状体的正常递质去甲肾上腺素和多巴胺很相似,于是发生竟争,而被儿苯酚胺能神经元所摄取、贮存,并作为神经递质释放出来。脑中苯丙氨酸还具有抑制酪氨酸羟化酶的作用,使多巴胺和去甲肾上腺素生成减少,由于苯乙醇胺和对羟苯乙醇胺的作用远不如正常递质强(如羟苯乙醇胺的作用不到去甲肾上腺素的1/50),因而不能产生正常的效应,故称为假性神经递质(falseneurotransmitter),使脑组织中这类神经细胞功能失常。脑干网状结构中,此类儿苯酚胺能神经元的上行神经纤维属于非特异性投射系统,经丘脑后再向大脑处于觉醒状态有关,故当其功能失常时,可导致昏迷。而大脑基底核内有关神经元的功能失常,造成锥体外系失调,可能与扑翼样震颤的发生有关。
在一些肝性脑病的患者,采用左旋多巴治疗(去甲肾上腺素及多巴胺不易通过血脑屏障),由于增加了中枢神经系统内儿茶酚胺的合成与贮存,在恢复神志上常有明显效果,但只有短时清醒的效应。
3.胰岛素、血浆氨基酸失衡学说(theory of insulin, plasma amino acid inbalance)正常血浆及脑内各种氨基酸的含量有适当的比例。近年来证实,肝脏受损害时,许多氨基酸的含量有变化,其中主要有芳香族氨基酸(苯丙氨酸、酪氨酸、色氨酸)增多,支链氨基酸(缬氨酸、亮氨酸、异亮氨酸)减少。
肝功能不全时,苯丙氨酸、酪氨酸和色氨酸在肝内分解代谢发生障碍,致血液和脑组织内苯丙氨酸、酪氨酸和色氨酸含量增多。苯丙氨酸和酪氨酸则在脑组织内形成假性神经递质(图15-5)。色氨酸则在脑组织内经色氨酸羟化酶和5-羟色氨酸脱羟酶的作用,生成过多的5-羟色胺。5-羟色胺是中枢神经系统中的一个抑制性递质,是去甲肾上腺素的拮抗物。脑中的5-羟色胺大量集中在脑干的中缝核(此神经核与睡眠有关),脑内5-羟色胺增高可引起睡眠,故认为它可能是引起肝性昏迷的一个重要原因。
芳香族氨基酸为什么进入脑增多呢?目前认为芳香族氨基酸与支链氨基酸都是在正常pH下不电离的氨基酸,它们通过血脑屏障是由同一载体转运的,因此它们之间有竞争作用。肝功能不全病人,血中支链氨基酸浓度降低,其竞争力量减弱,芳香族氨基酸争得载体而入脑的机会就增多。
酪氨酸在脑内的浓度升高,使脑内对羟苯乙醇胺生成增多。苯丙氨酸的升高,使脑内苯乙醇胺生成增加,而且高浓度的苯丙氨酸对酪氨酸羟化酶有抑制作用,阻碍多巴和多巴胺以及去甲肾上腺素的生成,而有利于对羟苯乙醇胺的生成(图15-5)。
严重肝损害病人血中支链氨基酸为什么会降低呢?原因是血中胰岛素浓度升高。正常胰岛素在肝脏失活,一次循环,门静脉中的胰岛素就可被肝脏清除40-50%。以此维持血中胰岛素于一定水平。支链氨基酸的分解代谢主要在骨骼肌和肾脏等组织中进行,而肝脏处理支链氨基酸的能力极为有限。当肝功能受损时,血中岛素因清除减少而增多。胰岛素不仅有降低血糖的作用,还能增加肌肉对支链氨基酸的摄取和分解,使血中支链氨基酸浓度降低。
肝功能不全,一方面由于芳香族氨基酸分解代谢障碍,使血浆芳香族氨基酸含量增多;另一方面血中胰岛素升高,使血液支链氨基酸含量减少,结果芳香族氨基酸进入脑组织增多,在脑组织内生成假性神经递质和过多的5-羟色胺,儿茶酚胺生成减少和儿茶酚胺能神经元功能降低,而引起中枢神经系统功能障碍,这就是肝性脑病的胰岛素、血浆氨基酸失衡学说的主要论点。
血浆氨基酸谱(plasma aminogram),除上述六种氨基酸有变化外,还有其他氨基酸的变化,如甲硫氨酸、门冬氨酸、谷氨酸含量增多等。
血浆氨基酸的变动不仅表现为氨基酸绝对值的变化,而且表现为氨基酸的克分子比率(molar ratio)的变化。正常人,(缬+亮+异亮)/(苯丙+酷氨酸)的平均克分子比率为3.0-3.5,而在有肝性脑病患者,其比率明显降低,可降到1.0以下(表15-1)。若用中性氧基酸混合液将此比率矫正到3-3.5,则中枢神经系统功能可得到改善,神志恢复。无脑病的慢性肝病患者也有上述克分子比率的变化,因此,某些研究者试图纠正血浆氨基酸比例来治疗肝性脑病,例如输注特制的氨基酸葡萄糖混合液,其中支链氨基酸含量高而芳香族氨基酸含量低,使之有利于纠正已经失衡了的血浆氨基酸谱,以改善中枢神经系统的功能。动物实验证明,给肝昏迷动物输注特制的氨基酸平衡液,使失衡的比值恢复正常,动物也随之苏醒。
图15-5 假递质生成过程
→:减弱 ↑:升高 ↓减低 (—):抑制
表15-1 肝性脑病病人的血浆氨基酸含量的变化
| 对 照 组 | 肝硬化组 | 肝昏迷组 | |
| 总色氨酸(微克分子%) | 4.7±0.5 | 4.4±0.3 | 2.8±0.3 |
| 游离色氨酸(微克分子%) | 0.43±0.03 | 0.54±0.05 | 1.94±0.23 |
| 缬氨酸(微克分子%) | 22.6±2.3 | 13.5±1.5 | 13.4±1.3 |
| 异亮氨酸(微克分子%) | 7.2±0.5 | 3.7±0.3 | 4.3±0.3 |
| 亮氨酸(微克分子%) | 12.4±1.3 | 9.4±0.5 | 7.7±0.5 |
| 苯丙氨酸(微克分子%) | 6.1±0.4 | 6.7±0.3 | 9.5±0.9 |
| 酪氨酸(微克分子%) | 6.1±0.3 | 10.1±1.3 | 19.6±0.9 |
| 蛋氨酸(微克分子%) | 5.1±0.2 | 8.5±0.5 | 9.1±0.5 |
| 游离脂肪酸(毫克当量/升) | 0.63±0.06 | 0.83±0.06 | 1.04±0.05 |
| 白蛋白(毫克%) | 4.1±0.1 | 3.7±0.1 | 2.3±0.3 |
4.短链脂肪酸中毒学说(theory of short chain fatty caid intoxication)肝病患者的血及脑脊液中,某些短链脂肪酸(指4-10碳原子的低级脂肪酸)含量较高;在肝性脑病患者的血液中,其浓度特别高。将大量短链脂肪酸给动物注射可引起昏迷。因此有人认为短链脂肪酸增多与肝病的发生有关。正常从肠道吸收的短链脂肪酸在肝内进行氧化分解;肝功能严重损害及门体分流存在时,可大量出现于血循环,并进入脑组织中。脑电图变化的研究表明,短链脂肪酸主要作用于脑干网状结构。关于短链脂肪酸对脑的损害机制,目前还不清楚。主要材料来自离体实验。①在体外,高浓度的短链脂肪酸对氧化磷酸有解偶联作用,使ATP生成减少;②直接与神经细胞膜或突触部位的某些成分结合,从而影响神经的电生理效应;③在突触处与正常神经递质(如多巴胺等)结合,从而干扰正常脑功能。
5.其它除上述因素较重要外,下面一些因素在肝性脑病的发生中,可能也起一定的作用。
(1)严重的肝功能不全时,由于血糖降低(特别是空腹时),脑组织能量供应不足,可促使肝性脑病的发生。
(2)电解质代谢紊乱及酸碱平衡失调。
1)低血钾:肝病病人常因饮食减少,腹泻,放腹水,特别是长期使用利尿剂(如双氢克尿噻等),使钾摄入不足,丢失过多。低血钾可以引起中枢神经系统兴奋性降低,严重的可以发生昏迷。
2)碱中毒:肝功能不全时,可能由于血氨增多,氨剌激呼吸中枢,使呼吸中枢兴奋,换气过度,而出现呼吸性碱中毒。低血钾时伴有代谢性碱中毒。血液的pH正常时,只有4%的NH3呈游离状态,96%是成离子状态的NH4+,NH4+容易通过细胞膜,进入脑细胞。当血液的pH值增高时,则下面的反应
![]()
朝着NH3的方向进行。因此,随着血液pH值的增高,游离的NH3增多,大量的NH3进入脑细胞,促使肝性脑病的发生。
3)代谢性酸中毒:肝功能不全时,促进丙酮酸氧化脱羧的脱氢酶的辅酶,如焦磷酸硫胺素(在肝内合成)合成减少,于是丙酮酸氧化脱羟过程障碍,三羟酸循环不能正常进行,丙酮酸堆积,乳酸生成增多,同时乳酸在肝内合成糖元的功能也降低。由于血液中丙酮酸和乳酸增多,引起代谢性酸中毒。酸中毒时,脑组织氧化过程受抑制,能量供应不足,可以发生昏迷。
综上所述,肝性脑病主要是由脑性毒物在体内增多,作用于中枢神经系统的结果,而在肝病时,脑对一些毒物的敏感性增加,更促进了脑病的发生。慢性肝功能不全的患者,镇静剂、感染、电解质紊乱、缺氧等因素,易引起昏迷。脑的敏感性升高,可能与脑长期受各种毒物剌激或肝脏提供的某些物质(如白蛋白)减少有关。
(三)肝性脑病发生的诱因
1.出血肝硬化时上消化道出血(如食道静脉破裂)、外伤、手术、产后大出血等,往往促使肝性脑病的发生。可能是由于循环血量减少,肝脏血液供应不足,发生缺氧,肝细胞进一步受损害,引起肝功能衰竭。脑组织也因缺血、缺氧,使脑细胞的代谢和机能障碍。如果为消化道大出血,血液在肠腔内分解生成氨增多,使血氨增高。
2.感染肝脏病患者如果合并肺炎、胆囊炎、胃肠道感染时,可能由于细菌及其毒素侵入肝脏,加重肝细胞的变性坏死及肝功能减退,往往可以促使肝性脑病的发生。
3.腹腔穿剌放液肝脏病患者有腹水,如果放液量过多或过速,由于腹腔内压突然降低,门静脉系统淤血,流入肝脏的血液减少,引起肝脏缺氧,肝细胞进一步受损害;或因放液丢失电解质过多,都可促使肝性脑病的发生。
4.药物损害麻醉及镇静药(特别是巴比妥类及吗啡)使用过多、饮酒等、都可以增加肝脏的负担,加重肝脏的损害,促使肝性脑病的发生。
5.进食蛋白质过量及口服铵盐
临床上常根据上述肝性脑病发生的机理及诱发因素,采取预防和治疗措施:
(1)避免或纠正各种诱发肝性脑病的因素。
(2)降低血液中氨及胺类的措施:以糖类食物为主,禁用高蛋白饮食,减少氨及胺类的来源;采用清洁灌肠或口服硫酸镁导泻,使肠腔内容物迅速排出;口腔或灌肠给予不易吸收的广谱抗菌素如新霉素等,抑制肠内细菌的繁殖,使氨及胺类产生减少;采用谷氨酸钠或谷氨酸钾静脉注入,降低血氨;采用左旋多巴治疗,增加中枢神经系统内儿茶酚胺的合成和贮存,恢复神经的正常功能。目前已有人研究人工肝脏辅助装置,即将病人的血液流经一种透析膜或吸附剂灌流装置,以清除血液中某些有害物质。
(3)低血糖时,补充葡萄糖。
(4)及时纠正电解质及酸碱平衡失调。
第二节 黄 疸
一、黄疸的概念
黄疸(jaundice)是由于胆色素代谢障碍,血浆中胆红素含量增高,使皮肤、巩膜、粘膜等被染成黄色的一种病理变化和临床表现。正常血清总胆红素含量在1.0毫克%以下,当超过2毫克%时,临床上出现黄疸。若血胆红素的浓度已超过正常范围,而临床上未表现出黄疸,称为隐性黄疸。
黄疸是肝功能不全的一种重要的病理变化,但并非所有的黄疸都是肝功能障碍引起的,例如红细胞破坏对多引起的溶血性黄疸,肝外胆管阻塞引起的阻塞性黄疸,为了叙述方便,合并一起讨论。
二、胆色素的正常代谢
首先复习一下胆色素的正常代谢过程(图15-6)。
图15-6 正常胆色素代谢途径
UDP:尿嘧啶核苷二磷酸UDPGA:尿嘧啶核苷二磷酸-葡萄糖醛酸基转移酶
(一)胆红素的来源
80-85%的胆红素来自衰老的红细胞崩解。约15%左右是由在造血过程中尚未成熟的红细胞在骨髓中被破坏(骨髓内无效性红细胞生成)而形成的。少量来自含血红素蛋白(hemoprotein),如肌红蛋白、过氧化物酶、细胞色素等的破坏分解。有人把这种不是由衰老红细胞分解而产生的胆红素称为“旁路性胆红素”(shuntbilirubin)。
(二)未结合胆红素的形成
肝、脾、骨髓等单核吞噬细胞系统将衰老的和异常的红细胞吞噬,分解血红蛋白,生成和释放游离胆红素,这种胆红素是非结合性的(未与葡萄糖醛酸等结合)、脂溶性的,在水中溶解度很小,在血液中与血浆白蛋白(少量与α1-球蛋白)结合。由于其结合很稳定,并且难溶于水,因此不能由肾脏排出。胆红素定性试验呈间接阳性反应。故称这种胆红素为未结合胆红素,也称间应胆红素。
(三)结合胆红素的形成
肝细胞对胆红素的处理,包括三个过程。
“摄取”:未结合胆红素随血流至肝脏,很快就被肝细胞摄取,与肝细胞载体蛋白-Y蛋白和Z蛋白结合(这两种载体蛋白,以Y蛋白为主,能够特异地结合包括胆红素在内的有机阴离子)被动送至滑面内质网。
“结合”:Y蛋白—胆红素和Z蛋白—胆红素在滑面内质网内,未结合胆红素通过微粒体的UDP-葡萄糖醛酸基转移酶(UDPGA)的作用,与葡萄糖醛酸结合,转变为结合胆红素。结合胆红素主要的是胆红素双葡萄糖醛酸酯,另外有一部分结合胆红素为胆红素硫酸酯。这种胆红素的特点是水溶性大,能从肾脏排出,胆红素定性试验呈直接阳性反应。故称这种胆红素为结合胆红素,也称直应胆红素。
“分泌”:结合胆红素在肝细胞浆内,与胆汁酸盐一起,经胆汁分泌器(高尔基复合体在细胞分泌过程中有重要作用),被分泌入毛细胆管,随胆汁排出。由于毛细胆管内胆红素浓度很高,故胆红素由肝细胞内分泌入毛细胆管是一个较复杂的耗能过程。
(四)胆红素在肠内的转化和肝肠循环
结合胆红素经胆道随胆汁排入肠内,被细胞还原为尿(粪)胆素元。绝大部分尿(粪)胆素元随粪便排出,小部分(约1/10)被肠粘膜吸收经门静脉到达肝窦。到达肝窦的尿(粪)胆素元,大部分通过肝脏又重新随胆汁由胆道排出(肝肠循环),仅有小部分经体循环,通过肾脏排出。
在胆红素代谢过程中,任何一个环节发生了障碍,都将引起胆红素在血浆内含量升高,产生高胆红素血症(hyperbilirubinaemia)。
三、胆色素代谢障碍的基本环节及各型黄疸胆色素代谢的特点
(一)未结合胆红素生成过多
这主要是由于红细胞本身的内有缺陷(如某些酶的缺乏或血红蛋白异常)或红细胞受外源性溶血因素的损害(如疟疾、免疫性溶血、蛇毒、苯胺等),造成大量红细胞破坏,产生大量的未结合胆红素,若超过了肝细胞的处理能力,则使血液中未结合胆红素增多,而出现黄疸。在一些贫血的病人,由于骨髓红细胞系统增生,骨髓内无效性红细胞生成增多,这种红细胞多在“原位”破坏,而未能进入血循环,或是进入血循环后红细胞生存的时间很短(数小时),而使未结合胆红素增多。
由于红细胞破坏过多,使未结合胆红素增多而引起的黄疸,称为溶血性黄疸(hemolyticjaundice)。其胆色素代谢特点是(表15-2):
表15-2 各型黄疸的胆色素代谢变化特点
| 黄疸 类 型 | 胆红 素 | 尿(粪)胆素元 | |||||
| 血 清 | 尿 | 尿 | 粪 | ||||
| 未结合 | 结 合 | ||||||
| 高未结胆合红素黄疸 | 肝前性 | 胆红素生成过多性—溶血性黄疸 | ↑↑ | -~↑ | - | ↑ | ↑↑ |
| 肝性 | 胆红素摄取障碍性—吉伯特氏病 | ↑ | -~↑ | - | -~↓ | -~↓ | |
| 胆红素结合障碍性—克里格勒—纳亚二氏综合征 | ↑↑↑ | ↓ | - | ↓ | ↓ | ||
| 胆红素分泌障碍性一杜宾-约翰森综合征,罗特综合征 | - | ↑ | ↑ | -~↓ | -~↓ | ||
| 高结合胆红素黄疸 | |||||||
| 胆汁分泌障碍性一肝内胆汁淤积 | -~↑ | ↑↑↑ | ↑↑ | ↓ | ↓ | ||
| 胆红素摄取、结合和胆汁分泌障碍混合性一肝细胞性黄疸 | ↑↑ | ↑↑ | ↑ | ↑~↓ | ↓ | ||
| 肝后性 | 胆道阻塞性一阻塞性黄疸 | -~↑ | ↑↑↑ | ↑↑ | ↓~○ | ↓~○ | |
-正常 增加↑ 减少↓ 没有○
1.血清未结合胆红素增多由于肝脏对未结合胆红素的处理有很大的储备力,一般血清总胆红素含量不超过3-5毫克%。血清胆红素定性试验呈间接阳性反应。
2.粪内尿(粪)胆素元增多这是由于肝脏加强制造结合胆红素,排入肠道的胆红素增多所致。
3.尿内尿(粪)胆素元增多,胆红素阴性。
(二)肝细胞对胆红素摄取障碍,
肝细胞摄取未结合胆红素障碍,可见于下列原因:
1.
2.新生儿肝脏的发育尚未完善,肝细胞内载体蛋白少,因而肝细胞摄取胆红素的能力不足。
3.吉尔伯特(Gibert)氏病是一种先天性、非溶血性黄疸,它是由于肝细胞窦侧微绒毛对胆红素的摄取障碍所致。临床检验发现,这种病人的肝脏对未结合胆红素的清除能力只有正常人的1/3,其血清胆红素一般不超过3毫克%(在血清胆红素高于5毫克%和重型病例中,还发现肝组织内UDP-葡萄糖醛酸基转移酶活性降低)。
肝细胞摄取障碍的胆色素代谢特点是(表15-2):血中未结合胆红素增高,血清胆红素定性试验呈间接阳性反应;尿内无胆红素;粪和尿排出的尿(粪)胆素元偏低。
(三)肝细胞内胆红素结合障碍
肝细胞内胆红素结合障碍可见于下列原因:
1.肝细胞受损害(如病毒性肝炎或药物中毒),使肝内葡萄糖醛酸生成减少或UDP-葡萄糖醛酸基转移酶受抑制。
2.新生儿肝内UDP-葡萄糖醛酸基转移酶的生成不足(要在出生后10个月左右才渐趋完善)。而且母乳汁内的孕二醇,对UDP-葡萄糖醛酸基转移酶有抑制作用。
3.克里格勒—纳亚(Crigler-Najiar)二氏综合征:这是一种伴有核黄疸的新生儿非溶血性、家族性黄疸。用同位素标记胆红素所作的试验证明,肝脏不能使胆红素与葡萄糖醛酸结合。这是由于肝脏缺少UDP-葡萄糖醛酸基转移酶所致。这种黄疸危害性大,大多数患儿死于核黄疸(nuclearjaundice),或称胆红素脑病。因为未结合胆红素毒性比较大,高浓度的未结合胆红素有抑制氧化磷酸化作用。另外未结合胆红素是脂溶性的,和脂质多的组织亲和力大;再加上新生儿或婴幼儿血脑屏障发育还不完善,未结合胆红素容易透入脑组织,沉积在神经细胞内,特别是在大脑基底核、丘脑、海马被胆红素所深染(故称核黄疸),引起中枢神经系统功能障碍,表现为精神不振、嗜睡、肌肉张力降低或增强,甚至发生角弓反张、肌肉痉挛和强直。
肌细胞内胆红素结合障碍,胆色素的代谢特点是(表15-2)。
(1)血清未结合胆红素增高(Grigler-Najiar二氏综合征Ⅰ型,UDP-葡萄糖醛酸基转移酶完全缺乏,血清未结合胆红素可高达25-45mg%),血清胆红素定性试验呈间接阳性反应。
(2)尿内无胆红素。
(3)由于结合胆红素生成减少,因此,尿(粪)胆素元从粪和尿排出明显减少。
(四)肝细胞对胆红素分泌障碍
肝细胞内结合胆红素是与胆固醇、胆汁酸盐、卵磷脂、水及电解质组成肝胆汁,通过高尔基复合体和微绒毛,分泌到毛细胆管的。“单纯的”或选择性胆红素分泌障碍是很少的。杜宾—约翰森(Dubin-Johnson)综合征和罗特(Rotor)综合征,是两种很相似的慢性特发性黄疸,可发生在同一家族中。其胆色素代谢特点是:血清内结合胆红素增多,呈直接阳性反应;尿中胆红素阳性。同时肝细胞对酚四溴酞钠(BSP)的排泄也有障碍,但胆汁酸盐分泌和胆流正常,没有胆汁淤积。目前认为可能是由于肝细胞对胆红素和带阴性离子异性染料的分泌有先天性缺陷,胆红素不能定向地向毛细胆管分泌而返流入血窦,使血清内结合胆红素增多。
(五)胆汁的分泌和排泄障碍
这里包括两种情况,一是肝内胆汁淤积(肝内胆汁淤积性黄疸)。二是胆汁由胆管排入肠道受阻,引起的阻塞性黄疸。胆道阻塞,毛细胆管内胆汁流不出去,以致压力升高,胆汁浓缩或胆栓形成,也反过来阻碍胆汁的分泌.
1.肝内胆汁淤积(intrahepatic cholestasis)由于各种原因使肝细胞排泌胆汁障碍,以致肝内胆汁淤滞和血中胆汁成分增多,而无胆道阻塞者,称为肝内胆汁淤积。可见于原发性胆汁性肝硬变、病毒性肝炎、某些药物的损害(如酒精、氯丙嗪、睾丸酮、妊酮等)及个别孕妇。其病变特点:光镜下,可见有肝细胞肿胀,内有胆色素沉积;严重的,有少数肝细胞变性坏死,毛细胆管内胆汁浓缩和有胆栓形成。电镜下,毛细胆管扩张,微绒毛减少,变平、甚至消失,滑面内质网呈不规则之增生,线粒体的嵴卷曲,毛细胆管周围溶酶体增多等。
肝内胆汁淤积的发病机制,目前还不完全清楚,可能与下列因素有关:
(1)自身免疫机制 原发性胆汁性肝硬化被认为与自身免疫有密切关系,早期见小胆管周围有淋巴细胞浸润和小胆管上皮细胞增生和坏死;以后发展成小胆管周围纤维组织增生和小叶间胆管萎缩,数目减少。病人血中抗线粒体抗体、抗小胆管抗体检出率高,免疫球蛋白浓度高。
(2)滑面内质网功能障碍胆固醇先在肝细胞滑面内质网中羟化,再在线粒体中进行侧链氧化,生成游离型初级胆汁酸(primary bile acibs),其中主要是三羟基的胆酸(cholic acid)和二羟基的鹅脱氧胆酸(lithochocholic acid)很少。游离型胆汁酸再与牛磺酸或甘氨酸结合,形成结合型初级胆汁酸(如牛磺胆酸或甘氨胆酸、牛磺鹅脱氧胆酸或甘氨鹅脱氧胆),与胆红素、胆固醇形成亲水性强的乳粒,分泌入毛细胆管,经胆管排入肠道。胆汁酸盐在肠内受细菌作用,水解脱羟,成为次级胆汁酸(secondary bile acids),例如牛磺胆酸变为脱氧胆酸(deoxycholicacid),甘氨鹅脱氧胆酸变为石胆酸,然后被重吸收入肝,再经过羟化生成胆酸和鹅脱氧胆酸(图15-7)。
肝细胞主动分泌胆汁酸盐,使毛细胆管内渗透压升高,使水和电解质进入毛细胆管,因此胆汁酸盐是决定毛细胆管胆汁流量的重要因素,这部分胆汁称为“胆盐依赖性胆流”(bile saltdependent bile flow, BSDF)。滑面内质网质损,羟化胆固醇功能降低,此时胆固醇直接经侧链氧化而生成大量的石胆酸;从肠道重吸收入肝的石胆酸也增多,而在肝内又不能再羟化,因而在细胞内石胆酸过多。胆汁酸为两亲性化合物,一端为极性基团(-0H等),另一端为非极性基团(CH等),胆酸和鹅脱氧胆酸含羟基较多,极性较高,与胆红素和胆固醇形成的乳粒亲水性强,较易通过毛细胆管微绒毛分泌入毛细胆管,而石胆酸的羟基少,极性低,与胆红素和胆固醇形成的乳粒亲水性差,在毛细胆管微绒毛内发生淤积,渗透活性降低,从而使胆盐依赖性胆流减少,引起肝内胆汁淤积。
(3)毛细胆管内胆流受到抑制胆盐分泌可以带动水分流入毛细血管,这是推动毛细胆管内胆管内胆汁流动的一个因素。但是,在毛细胆管内胆汁流动率中还有40-70%是由另外的因素推动的,叫做“不依赖于胆盐的胆流(bile salt independent bile flow, BSIF)。BSIF在毛细胆管的胆汁流中似有更重要作用。实验证明,氨丙嗪、雌激素、特别是炔雌醇可抑制BSIF。凡能抑制细胞质膜Na+-K+-ATP酶的物质(如毒毛旋花苷),可抑制BSIF。氯丙嗪和炔雌醇(避孕药)在人可引起肝内胆汁淤积,可能是通过抑制Na+-K+-ATP酶而起作用的。
图15-7 胆汁酸的生成和肝肠循环
正常石胆酸生成很少,结合石胆酸约占结合型胆汁酸总量的1-2%,滑面内质网功能障碍时,石胆酸生成增多。
(4)微丝的功能降低毛细胆管微绒毛中有丰富的微丝,其中含有肌动蛋白样的收缩蛋白。微丝在胆汁分泌中的重要性可用下述实验说明;用细胞松弛素(选择性地降低微丝的功能)灌流大鼠肝脏1小时,胆汁流量减少50%,灌流2小时,胆流完全停止,微丝的作用大概是通过它的收缩、松弛,使微绒毛变形,毛细胆管扩张或缩小,从而推动胆汁流动。
各种原因引起的肝内胆汁淤积,几乎无例外地都有毛细胆管微绒毛的形态变化,如肿胀、变形、数目减少等。
由于肝细胞胆汁分泌障碍,发生肝内胆汁淤积,胆汁则从肝细胞弥散入血,造成肝内胆汁淤积性黄疸(图15-8),血液中胆汁酸盐增多。其胆色素代谢变化:血清结合胆红素明显增多,胆红素定性试验直接阳性反应;粪和尿内尿(粪)胆素元减少;尿内胆红素阳性。除胆红素代谢变化外,绝大多数病人血清硷性磷酸酶显著升高,还有其它肝功能损害的变化。
2.胆汁由胆管排入肠道受阻—阻塞性黄疸
常见于胆道结合、蛔虫、肿瘤或胆管炎,使胆道狭窄或阻塞。胆汁排入肠道受阻,阻塞上部的胆管内则有大量的胆汁淤积,胆管扩张,压力升高,胆汁通过破裂的小胆管和毛细胆管而流入组织间隙和血窦,引起血内胆红素增多(胆汁酸盐也进入血循环),产生黄疸(图15-6,15-8)。
阻塞性黄疸(obstructive jaundice)的胆色素代谢特点和肝内胆汁淤积性黄疸很难加以区别(表15-2),都表现为血清结合胆红素明显增多,尿内胆红素阳性,尿和粪内尿(粪)胆素元减少,如胆道完全阻塞,尿(粪)胆素元可以没有,但是阻塞上部胆道有感染,结合胆红素可被细菌还原为尿(粪)胆素元,吸收入血由肾脏排出。由于对这两种黄疸治疗不同,必须认真加以区别,肝内胆汁淤积性黄疸,除胆色素代谢障碍外,往往有其它肝功能的损害。另外,肝脏穿剌取活组织检查和胆道造影,对确定诊断有较大的帮助。
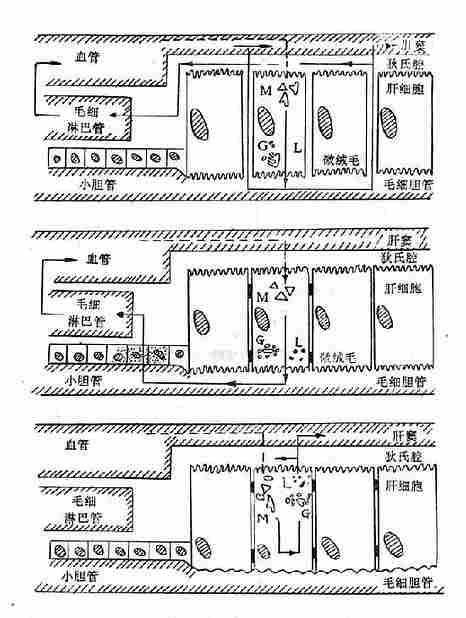
图15-8 结合型胆红素返流入血机理
……未结合胆红素
——结合胆红素
M:滑面内质网,L:溶酶体,G:Golgi(高尔基)氏器
1.肝细胞之间的桥粒体受破坏,毛细胆管内胆汁流入狄氏腔和血窦。
2.小胆管破裂,胆汁流入组织间隙,再吸收入血。
3.肝胆汁分泌方向发生改变,由肝细胞分泌入狄氏腔和血窦。
肝内胆汁淤积和阻塞性黄疸时,不仅血清结合胆红素增加,胆汁酸盐在血液中也大量增加。胆汁酸盐可剌激皮肤感觉神经,引起搔痒;通过对迷走神经作用而引起心动徐缓,血压降低。胆汁酸盐也有抑制脑的呼吸作用,表现为疲倦,精神不振。
胆汁酸盐排入肠道减少,可引起脂肪消化吸收不良,维生素K等脂溶性维生素吸收减少。正常凝血酶原和凝血因子Ⅶ、Ⅸ、Ⅹ的生成需要有维生素K参与,如果维生素K缺乏,上述凝血因子的谷氨酸残基不能羟化成γ-羧基谷氨酸残基,就不能Ca++结合参与凝血过程,而使血液凝固性降低。
(六)肝细胞对胆红素的摄取、结合和胆汁分泌综合性障碍
上面我们从胆色素代谢障碍的各个环节,讨论了黄疸发生的机理。但在疾病过程中,黄疸的发生,往往不是某单一环节障碍的结果,常涉及到多个环节,例如肝细胞性黄疸和新生儿生理性黄疸等。
1.肝细胞性黄疸肝细胞性黄疸(hepatocellular jaundice)是由于感染或中毒,使肝细胞受损害(肝窦面肝细胞膜微突减少,内质网减少,变得稀疏和空泡形成,线粒体肿胀,基质混浊,内嵴呈灶性消失,毛细胆管微绒毛减少,变平、不规则),肝细胞对未结合胆红素的摄取、结合胆红素的形成和肝胆汁的分泌发生了一系列障碍而引起的黄疸。其胆色素代谢变化比较复杂,主要变化是(表15-2):
(1)由于肝细胞对未结合胆红素摄取障碍和结合胆红素生成减少,血清未结合胆红素增多。
(2)由于肝细胞胆汁分泌功能受损,肝胆汁分泌障碍,肝内胆汁淤积,或由于肝内小胆管炎,引起机械性阻塞,而使胆汁从肝细胞反流入血;分泌到毛细胆管的胆汁,还可通过变性坏死的肝细胞或肝细胞之间的间隙反流入血,而使血清结合胆红素增多。
(3)血清未结合胆红素和结合胆红素增多,胆红素定性试验阳性双相反应。
(4)由于排入肠道的胆汁减少,粪和尿内尿(粪)胆素元一般是减少的。
(5)尿内胆红素阳性。
2.新生儿生理性黄疸 新生儿生理性黄疸(icterus neonatorum)可见于50-60%出生后第一周的新生儿,这种黄疸的发生有以下一些原因:
(1)生下后,血液内原来过多的红细胞被破坏,未结合胆红素生成过多。
(2)肝细胞内载体蛋白—Y蛋白少,肝细胞摄取未结合胆红素的能力不足。
(3)肝细胞内胆红素葡萄糖醛酸基转移酶生成不足,结合胆红素生成少。
(4)肝细胞胆汁分泌器发育不完善,对肝胆汁分泌的潜力不大。
这种黄疸以血清未结合胆红素增多为主,如没有先天性胆红素代谢缺陷,可以逐渐消退。
3.药物性黄疸
药物可干扰胆红素代谢各个环节,例如:
退热镇痛药非那西汀,可引起溶血,使胆红素生成过多。
磺胺类、水杨酸类药,可竞争性地与白蛋白结合,使血中未结合胆红素转运障碍。
驱虫药黄棉马酸,能争夺Z蛋白,胆道造影剂能争夺Y蛋白,使肝细胞对胆红素的摄取及在肝细胞内的运输受影响。
新生霉素、氯霉素、利福平可抑制葡萄糖醛酸基转移酶,使结合胆红素形成受影响。
氯丙嗪甲基睾丸酮等作用于微胆管的排泌装置,造成肝内胆汁淤滞。
药物性黄疸只要及时发现,及时停药,一般予后良好。
第三节 肝性肾功能不全(肝肾综合征)
肝性肾功能不全(hepatic renal insufficiency)是指在肝硬化晚期和暴发性肝炎等严重实质性肝病时发生的肾功能不全。“肝肾综合征”(hepatorenal syndrome)这一术语,过去用得过于广泛,造成概念上的混乱,1979年一个国际会议建议:肝肾综合征仅用于肝硬化和暴发性肝炎等实质性肝病时发生的进行性肾功能不全。用肝性肾功能不全似乎比肝肾综合征更确切一些。
肝性肾功能不全的临床特点是:①主要见于晚期肝硬化,也可见于暴发性肝功能衰竭,有明显的肝病症状、体征和肝功能异常,常同时伴有腹水、黄疸、肝昏迷;②无原发性肾脏病存在的证据,突然或逐渐出现急性肾功能衰竭的临床表现,如尿少、氮质血症和尿毒症等;③可发生在大量利尿、大量放腹水或消化道大出血后;④多有稀释性低血钠症和低血钾症;⑤化验多无或仅有轻度蛋白尿,尿沉渣变化轻微,尿钠肌酐/血浆肌酐的比值常大于20,而急性肾小管坏死引起的急性肾功能衰竭,尿肌酐/血浆肌酐比值小于10;⑥在疾病的末期,出现深度昏迷,血压下降,严重少尿和氮质血症,尿钠浓度很低,最后主要由于肝功能衰竭而死亡。予后主要取决于肝脏功能的改善。
肝性肾功能不全的发生机理:
肝性肾功能不全的病理生理改变,主要是肾血流量减少与肾小球滤过率降低。
尸检结果表明,大多数肝性肾功能不全的肾脏,在解剖学、组织学方面是正常的。近年来对本病的大量研究证明,本病是一种肾脏缺血所致的功能性改变。将7例本病死者的肾移植给别人,结果6例成功。另方面,有报告用正常人的肝移植于本病患者后,其衰竭的肾脏也恢复了正常功能。这都说明本病的肾功能变化是可逆的。但也有报告,急性暴发性肝炎可引起急性肾小管坏死。
肝性肾功能不全,肾缺血的原因有以下两个方面:
1.有效血浆容量下降严重肝病时,由于大量腹水(使血浆大量漏出)、大量利尿(丧失大量体液)、上消化道出血及低蛋白血症等,可使血容量减少,心输出量降低,致流经肾脏的血液减少,造成肾缺血。这是所谓的低动力型肝肾综合征(此型较少见)。多数肝硬化腹水肝肾综合征患者属于高动力型,其心输出量及血浆容量并不减少,甚或增加,但由于肝内外存在着大量的侧枝循环和毛细血管扩张,使大量血流分布在侧枝循环和淤积在毛细血管内,而使肾血流量减少和肾小球滤过率降低,发生少尿和氮质血症。后期由于腹水增多,使血容量降低,同时下腔静脉和心肝受压,心输出量降低,肾缺血进一步加重。
2.肾小血管收缩和肾内血液重新分布患者肾血管造影,可看到叶间动脉和弓形动脉呈串珠状或扭曲状,但患者死后,肾动脉造影发现上述变化消失,说明生前有强烈的肾血管收缩。肾血管收缩的原因可能是:①有效血浆容量减少,通过交感—肾上腺素系统及肾素—血管紧张素系统的作用,使肾血管收缩;②从肠道吸收的内毒素不能在肝内被清除而进入血流。内毒素的拟交感神经的作用和使肾素—血管紧张素活性加强,而引起肾血管收缩,肾缺血;③产生于胃肠道内的假性交感神经递质—胺类物质,取代了外周交感神经末梢的正常神经递质,使血流重新分布,而引起肾血流量减少。
用133氙测定肾血流量及其在肾内的分布,发现肾皮质外层血流量低于正常,而皮质内层和髓质血流量不变,说明血液在肾内重新分布,结果是血液流经皮质肾小球减少,而导致肾小球滤过率降低。
综上所述,严重的肝功能不全,通过各种机制,使肾血流减少,是引起肝性肾功能不全的主要原因(图15-9)。早期肾功能的变化是功能性的,可逆的。但是严重缺血或持续时间过久,也可使肾小管上皮细胞变性,甚至坏死。胆汁对肾小球上细胞有一定的毒性作用,给肾缺血的大白鼠静脉滴注胆汁酸,可以加重缺血对肾小管上皮细胞的损害。暴发性肝炎时,不仅肾血流量减少,而且有胆汁逆流入血(肝细胞性黄疸),在肾小管内有许多胆色素管型形成,肾小管上皮细胞常有的明显的变性,甚至坏死。
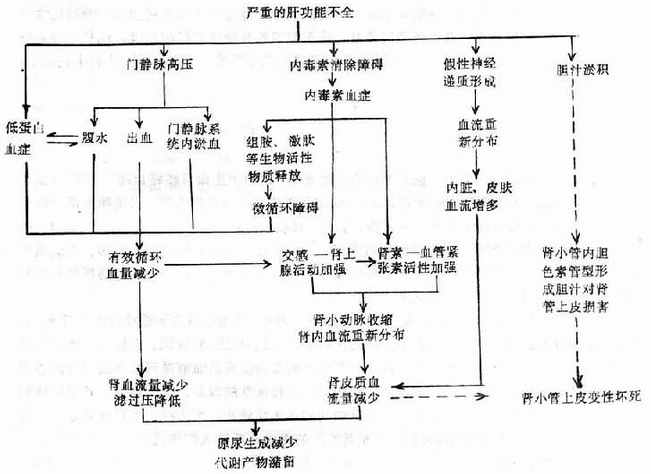
图15-9 肝性肾功能不全的发生机理
虚线:表示可能有肾小管上皮变性坏死
肝功能不全患者,一旦发生肝性肾功能不全,将促使和加重肝性脑病的发生和发展。因为:①氮质血症,有更多的尿素透入肠腔,氨生成增多;②芳香族氨基酸代谢产物,如对羟苯乙醇胺经肾排出减少,而在体内潴留;③代谢性酸中毒,血钾升高,血钠降低,都可加重中枢神经系统功能障碍。
第十六章 死亡与复苏
第一节 死亡(缺)
第二节 复苏
病员呼吸心跳骤然停止时所采取的一切急救措施称为复苏术。鉴于复苏的最终目的是恢复病人神志和工作能力,不少学者主张把心肺复苏改为心肺脑复苏。
复苏(Reuscitation)又称生命支持(Life support),全过程可分三个阶段,①初级复苏或基础生命支持;②高级复苏或进一步生命支持,以心脏为重点;③后期复苏或持续生命支持,以脑为重点。
一、复苏的条件
上述三大死亡原因中只有重要器官没有明显器质性损伤,同时代谢的物质基础尚未耗竭的急死患者有复苏的可能。各种原因引起的心跳、呼吸骤停是临床上最常见的复苏对象,心钟之内也丧失功能,也有因呼吸先停止后,由于缺氧,心脏也在2-4分钟内相继停跳。下面简单介绍心跳呼吸停止的原因及原理。
(一)心跳骤停:
心跳骤停是指心脏突然丧失舒缩功能,不能射出足够的血液以供给脑的需要。此时心脏可以是完全停止活动,也可能是处于心室纤维性颤动状态。临床上以后者为多见。
1.心跳骤停的临床上表现:病员突然惊厥、昏迷;呼吸呈喘息状或呼吸停止;瞳孔扩大;眼球偏斜;皮肤粘膜呈死灰色;心跳及动脉搏动消失等,但其中最主要的是两大症状;即突然意识丧失及颈动脉或股动脉搏动消失。凡具备此二症状者,即可诊断心跳骤停。其他指征均不确切,仅供参考。
2.心跳骤停的原因有两大类:
(1)原发性心脏疾患:缺血性心脏病是心跳骤停的最常见原因,例如冠心病。此外也可见于心肌炎、心瓣膜病及先天性心脏病等。
(2)心外疾患:包括各种急性窒息、各型休克、药物中毒、电解质紊乱、麻醉及手术意外等情况。
3.心跳骤停的原理较复杂,常为多种因素共同作用引起的。主要有以下几点:
(1)迷走神经过度兴奋:例如机械性剌激气管、肺门、心脏、肠系膜等内脏器官时反射性引起迷走神经兴奋,从而抑制了窦房结及其它室上的起搏点,以致引起心跳骤停。如胸部或腹部手术时可见。
(2)严重缺氧:例如大出血、麻醉意外等。此时除缺氧引起迷走神经过度兴奋外,心肌处于无氧状态,局部发生酸中毒及钾离子释放,结果导致心自律性传导性受抑制,最后发生停搏。
(3)CO2贮留和酸中毒:例如窒息时。CO2和酸性产物的贮留可兴奋迷走神经,此外它们也可抑制心肌特殊组织的传导性,导致异位节律。并抑制心肌的氧化磷酸化过程,从而直接减弱心肌收缩力,终导致心跳停止。
(4)电解质紊乱:心肌细胞功能与细胞膜内外离子浓度变化密切相关,例如不论高血钾还是低血钾,严重时均可导致心跳停止或心室纤颤。参阅第五章。
(5)其它:例如电击时,电流通过心脏引起心室颤动或心肌变性坏死、断裂,从而心跳骤停。又如心跳中枢衰竭时也可致心跳骤停。
(二)呼吸停止:
有些急死首先发生呼吸性停止,比如药物过量、颅内压增高、传染、中毒、窒息、溺水、电击等情况下引起的呼吸停止常常是死亡的直接原因。
此时呼吸停止的主要原理是脑干损伤和呼吸中枢局部血流供应不足;而中毒、传染本身又可使呼吸中枢功能高度抑制。此外,由于各种原因使呼吸肌麻痹(例如脊髓前角灰白质炎等),气道梗阻、或肺膨胀严重障碍时(例如胸部挤压伤),也可发生呼吸停止。
呼吸停止后的主要危险是低氧血症,当动脉血氧张力低于5.433kPa(40mmHg)时,可出现明显的紫绀;当低于2.67kPa(20mmHg)时,脑摄氧停止,可立即引起死亡。
二、复苏的原则及方法
不论急死的情况是否相同,复苏的原则基本上是一致的,所采取措施的基本目的是使脑实质不受损伤,保护其功能,恢复自动心跳和呼吸,避免和克服合并症。复苏大致可分为三个方面,下面简述其原则和原理。
(一)保护脑功能
迅速以含氧血供应脑以保护脑功能,所采取的措施是进行有效的心脏挤压和人工呼吸。
1.脑缺氧的严重性 机体全身缺氧后,各组织器官对缺氧的耐受力有很大差异,最敏感的是中枢神经系统,其它组织耐受缺氧的次序是:心肌10-20分,骨骼肌2-4小时,骨和结缔组织可耐缺氧10小时以上也不出现明显损害。即使是中枢神经系统,其各部分的耐受缺氧能力也很不相同,最敏感的是大脑皮层,一般心脏停跳3-4分后就有可能引起严重的损害。其余各部分脑组织耐受缺氧的次序为:中脑(瞳孔及睑反射中枢)5-10分钟、小脑10-15分钟、延髓(呼吸、血管运动中枢)20-30分钟、脊髓约45分。
为什么脑组织对缺氧最敏感呢?因为脑虽小(仅占体重的2%),但其需氧量最大;正常人安静状态下,脑血流量约为心输出量的15%,而脑之耗氧量却占全身总摄氧量的20%,灰质耗氧量又比白质多5倍。脑需氧量这样大,但脑本身的无氧代谢能力及能量贮备能力极差,它所需要的能量几乎全部来自葡萄糖的有氧代谢。心脏停跳后,脑动脉床内所含的氧仅够供脑消耗几秒钟,脑贮备的葡萄糖也只能维持几分钟的消耗。一般脑供血量至少达到正常量的14%才能保护脑不受损害。所以一旦心跳停止后,最敏感的症状是意识丧失,而抢救的主要目的也是迅速将含氧及葡萄糖的血不断输送给脑——这就是人工呼吸、心脏按压的作用。此外,及时给头部及全身降温,使体温降至31-33℃左右可使脑组织代谢率降低约50%左右,这对保护脑功能、防止脑并发症具有很大意义。
2.心脏按压的作用 抢救时脑是否能受保护而不受到缺氧损害,取决于心脏按压能否维持足够的脑血流量。正常时尽管心输出量及血压有变化,但通过脑血管口径变化的调节,脑血流量几乎是恒定的。脑血管口径的改变主要受脑部小动脉平滑肌处pH变化来调节,而与全身酸碱度变化的关系不大。在心脏停跳后,由于缺氧或心输出量下降,可发生颅内进行性酸中毒,从而引起脑血管口径的自家调节功能丧失,表现为脑血管麻痹。这实际是一种脑血流供应的有意义的代偿反应。此时脑血管口径扩大,脑血流量直接与全身血压有关。只要有足够高的血压,甚至可出现脑组织“奢侈灌注”(luxury perfusion)现象(即脑耗氧下降,而血流供应却相对增加)。
有效的心脏按压可以使血压维持在8kPa(60mmHg)以上,从而保证全身重要器官的血流量。同时,可使脑血流量维持在正常脑血流量的30%(约为最低需要量的两倍)。所以在自主心跳恢复前,应坚持心脏按压。据报导,在心脏按压持续两小时内,仍有恢复意识的可能。自主心跳恢复后,意识恢复较慢者,不一定是由于脑缺氧损伤所引起,也可能是较重的颅内酸中毒的抑制作用所致。
3.人工呼吸作用 呼吸停止后,机体内之贮存氧仅1升左右,足够维持生命活动约4分钟。此时肺泡内之残留气体仍不断通过弥散与血中气体进行交换。但由于CO2从组织向肺泡之弥散量较O2由肺泡向血液内的弥散量约小10倍,结果肺泡内气体总量逐渐减少而形成负压,使空气不断自外界流入,此过程叫做“列通气气流”。靠此种方式供氧约可维持30秒;如果呼吸停止前肺泡内充满氧,则此过程可维持10分钟左右。如以后仍得不到氧,则由于缺氧可导致体内代谢性酸中毒。
CO2分压增高在开始时并不严重,只有当体内缓冲系统代偿能力耗尽后,动脉血内CO2分压才以0.4-0.8kPa(3-6mmHg)/分的速度逐步上升。要达到CO2分压12kPa(90mmHg)以上(一般动脉血CO2分压小于9.33kPa(70mmHg)者,不考虑呼吸性酸中毒),约需15-40分以后,所以一般说来呼吸停止后的早期很少引起呼吸性酸中毒。
呼吸停止后,为维持有效的气体交换,首先要清洁呼吸道使之通畅,并立即采取压胸法或口对口呼吸,其中尤以口对口呼吸最为简便有效。一般一个操作者的呼气一次吹气1250毫升左右,其中含氧18%,含CO22%,足够预防病员严重缺氧和高碳酸血症的发生。但如病员仍处于低心排出量状态,则尽管维持有效通气量,组织内却仍然缺氧,尤其在合并肺功能异常时,更是如此,此时应优先选用人工呼吸器正压呼吸给氧,效果更好。
4.心脏按压及人呼吸的效果判定脑活动的存在是复苏效果的最主要指标。由于脑血流量下降,此时病员很少有清醒的意识,而常常陷于昏迷。但下面五个体征中,只要存在一项就表示已有足够的含氧血供给脑:
(1)瞳孔瞳孔逐渐由大缩小是予后良好的灵敏指征。瞳孔逐渐增大但未完全扩大时并不说明抢救无效,此时应注意其它生命指征存在与否。扩大的瞳孔不缩小,则表示可能复苏无效或有脑损伤。迅速扩大至最大的瞳孔反应肯定是不良的指征。
(2)角膜反射是重要的指标。它的出现说明可迅速复苏。但因频繁剌激角膜易损害眼睛,所以仅在其它指征均不能确定时,才使用它。
(3)挣扎它的出现说明复苏是有效的,但挣扎可引起复苏操作困难,增加耗氧,并加重酸中毒及高血钾,从而影响自主心跳的恢复及造成脑损伤,值得警惕。
(4)肌张力 出现良好的肌张力,下颌紧闭等。
(5)呼吸形式因延脑对缺氧耐受性较大,而且在呼吸停止后脑缺氧及颅内酸中毒都是有力的呼吸剌激剂,所以经有效复苏后常可出现正常呼吸。不规则或喘息式呼吸的出现,常说明仍有脑缺氧存在。
(二)恢复自主的心跳呼吸
第一阶段保护脑机能的同时也是为恢复自主呼吸和心跳作准备。因复苏过程拖得愈久,成功率愈低,所以必须在心脏按压,人工呼吸的同时采取更有力的恢复自主心跳、呼吸的措施。
1.复苏时循环、呼吸及体液成分的变化
(1)循环功能的变化:
心脏按压时,由于房室瓣口的肌肉松弛,瓣膜关闭不灵,所以心输出量常常是低的,很少能超过正常心输出量的50%,结果引起组织缺氧。缺氧及动脉血CO2分压增高都可引起全身外周血管扩张、麻痹,从而使外周血管阻力下降(约为正常的一半),舒张压下降,组织灌流更不足。由于心输出量低及外周血管扩张的影响,尽管脑血管呈现扩张状态但脑组织却得不到优先的血液供应,所以应脑血流仍是低的,中枢机能处于高度抑制状态。此时适当给予缩血管药物,使增加外周血管收缩反应而提高血压,以保证心、脑血流的供应是很重要的。
复苏时的低氧血、高血钾、酸中毒都严重的影响心肌代谢及心律改变(见后),从而造成自主心跳恢复困难,所以在心脏按压的同时要积极纠正酸中毒,并给予钙剂以拮抗高血钾的毒性作用。
(2)呼吸功能的变化:
心脏停跳前,病人就可能存在各种各样的心肺功能异常,例如心衰、各型休克等;抢救当时又可并发肺水肿、呕吐物吸入,低心排出量所致的肺灌注不足、气胸等情况,将更进一步引起肺功能下降。
无论是上述何种原因,最终都不外乎通过五种基本病理变化而影响肺气体交换,从而产生低氧血症。这五种基本病变即:低通气量、无效死腔增加、肺泡弥散不足,通气量与血灌注分布不协调、动—静脉流增加。
有效的人工呼吸或减少低通气量及无效死腔的有害作用。纯氧吸入可消除因低通气量、弥散不足所引起的低氧血症,也可减少因通气—血灌注不协调而引起的血氧不饱和现象。此外纯氧吸入还可减少肺动—静脉分流所引起的恶果,因为在正常时,生理性肺动—静脉分流不超过心排出量的5%,而此时可高达心输出量的50%,结果引起动脉血—肺泡气氧分压差增大(>2.67kPa(20mmHg)),使动脉血氧分压降到10.67kPa(80mmHg)以下,因此更加重了低氧血症,因此用纯氧,可部分纠正动脉血氧不饱和现象(如果肺动—静脉分流原为心输出量的50%,现可纠正为20%)。综上所述,即使并无严重心肺合并症时,在自主心跳恢复后,仍应继续给氧12-24小时。
心脏复苏时高通气量的人工呼吸常预防动脉血CO2分压过度增高(一般均不会超过8kPa(60mmHg))。有时反应可以出现呼吸性碱中毒,这对脑并无损害。有时由于颅内酸中毒剌激呼吸中枢,可出现高通气量(呼吸加强),此时静脉注射NaHCO3纠正颅内酸中毒无效。因为NaHCO3很难通过血脑屏障而进入脑内。
(2)体液成分的变化:
①血钾:由于组织缺氧或损伤常释放钾;挣扎时肌肉活动增加,分解代谢增强也释放钾;此外,肾上腺素分泌增加引起肝糖元分解也释放钾,所以高血钾是复苏时的重要生化改变之一。一般测定动脉血钾含量较能正确反映整体情况。高血钾可使心传导系统抑制,心肌应激性下降,并可干扰心脏去极化过程,常常是长期复苏后心跳不恢复的重要原因。高血钾的毒害作用又因细胞外低钠、低钙而加重。此时如肾功正常,可采用脱水利尿疗法。
②血钠:可正常或下降。因为缺氧,细胞膜正常离子交换的功能失调,钠可进入细胞内。
③细胞外液容量:由于酸中毒时钠、水均能被吸收到细胞外胶元组织中去;同时因细胞内渗透压增加,细胞外液又流入细胞内,所以细胞外液减少。此外,由于微循环淤滞,液体外渗到组织间隙内,更引起循环血量下降。但此时病员一般均有水贮留现象,在密切观察中心静脉压(正常为5-10cmH2O)、尿量等情况下,应将补液量限制在1500-2000ml左右,以便减少脑水肿及肺水肿等并发症。
④酸碱平衡障碍:代谢性酸中毒是抢救时影响复苏效果的重要问题之一。当动脉血氧分压降至2.67-4kPa(20-30mmHg)时,细胞代谢将转为以无氧酵解为主,此时产生ATP的效率仅为氧代谢的1/20;同时将有大量酸性中间代谢产物积聚。
心跳暂停瞬间,不一定伴有酸中毒。即使因窒息而造成乳酸增高,一旦自主心跳恢复后,酸中毒可很快消失。但心跳暂停超过几分钟,则不可避免地产生代谢性酸中毒。如继续合并低心排出量,则酸中毒更加重,必须给以治疗。
在心脏停跳初期呼吸性酸中毒并不严重。有时因低心排出量和代谢性酸中毒或脑损伤等反而引起高通气量反应,使动脉血CO2分压下降,以致发生呼吸性碱中毒。
当长时间复苏合并肺功能显著低下时,则可表现为混合性酸中毒。此时应先纠正呼吸性酸中毒,否则单纯使用碱性药物很难纠正代谢性酸中毒。
酸中毒可以抑制氧化磷酸化过程,使能量产生下降,同时表现出心肌利用葡萄糖能力低下,从而导致心肌收缩无力,心肌驰缓,搏出量下降,心动徐缓,甚至窦性停博。此外,在酸中毒时,由于CO2分压增高及乳酸的抑制作用,使心肌和周围血管对儿茶酚胺的反应性降低,心室颤动的阈值降低,因而除颤不易成功。这些酸碱平衡的紊乱,即使在复苏成功又无合并症的病员也可延续三日之久,所以心脏复苏时必须及时给以碱性药物,才能使抢救具有好的效果。
2.常用的抢救措施心室纤维性颤动时,在改善心肌缺氧的情况下,用电击或肾上腺素等药物注射除颤,常可恢复有自主心跳。当心跳完全停止后2分内,因心肌应激性增高,所以以拳叩打心区,有时可恢复心跳,当无效时可试用人工起搏器,帮助恢复自主节律。
呼吸中枢对缺氧耐受性大于脑皮层,所以一旦心脏复跳后,自主呼吸均能随之恢复。如迟迟不恢复常说明缺氧或脑水肿影响了呼吸中枢功能,此时应考虑气管插管、正压给氧及用脱水剂解除脑水肿。此外适当地使用呼吸兴奋剂常有助于呼吸的早期恢复。
输血和补液可在心脏按压的同时进行,以保证心脏充盈。必要时可采用股动脉逆血流输血或注入50%葡萄糖液,通过剌激心、血管的感受器,有时可以反射性地使停博的心脏复跳,并且可以使脑及冠状动脉迅速得到大量血液而纠正缺氧。
常用的药物疗法:
(1)肾上腺素老三联针(肾上腺素、异丙肾上腺、去甲肾上腺素)已废用,新三联针(肾上腺素、利多卡因、阿托品)亦不提倡,目前主张首选肾上腺素,以近心端静脉素以近心端静脉注射为首选途径,剂量为每5分钟0.5~1.0mg。研究证明在持续心肺复苏时,发现肾上腺素能增加主动脉舒张压而不增加右心房舒张压,从而增加心肌血灌流量,增加脑血流和减少颅外血流。人们发现当主动脉舒张压大于5.33kPa(40mmHg)时心律紊乱易恢复,生存率高,其作用机制是通过α-肾上腺素能作用或缩血管特性,而不是它的β-肾腺素能作用。
(2)NaHCO3停跳超过10min,有高血钾症及酸中毒存在者,应补充NaHCO3纠正酸中毒,首剂为1mmol/kg,以后每10min给初量的一半。但对无有效通气者不宜使用。也可用THAM。它对呼吸性酸中毒和细胞内酸中毒也有效。
(3)利多卡因 特别是在无电除颤的条件下,为除颤首选药。负荷量为1mg/kg,以后每8~10min给0.5mg/kg,总剂量不大于3.0mg/kg,维持量为2mg/min。
(4)溴苄胺 可替代利多卡因。
目前主张禁用钙剂。
3.自主心跳恢复的指征
1.颈部或股动脉搏动的存在,是最可靠的指征。检查时,应持续按脉5分钟,搏动持续不消失者才可说明复苏有效。
2.意识恢复。但在长期心脏停跳后,此指征常不明显(多半仍处于昏迷状态)。
3.出现心电图波形。
4.瞳孔缩小、胸膜反射存在、呼吸改善等。
(三)防治并发症
当自主心跳、呼吸恢复后,继续维持循环、呼吸功能;防治脑水肿;纠正酸中毒;注意肾脏功能;防治感染等仍是重要的问题,否则可使心跳、呼吸再度停止。
1.维持有效的循环低心输出量和低血压是复苏后经常发生的并发症,由于心肌损伤,心律不齐使心脏泵血的作用发生严重紊乱;如同时存在循环血量不足,则心脏复跳后循环功能更低下。常表现为心排出量、心力衰竭、心律不齐等循环功能障碍,若不及时纠正,必然会影响整个机体组织的血液灌注和供氧量,导致脑水肿,肾功能衰竭等,终可致心跳、呼吸再度停止。
现着重说明低心排出量问题:
(1)表现:低血压、少尿、四肢发凉且可呈青紫色、意识障碍(嗜睡或无意识)、代谢性酸中毒等均是低心排出量的症状。一般无心肌器质性改变时,经抢救后予后良好。未纠正的低血压状态可在几小时或数日内引起死亡。
(2)原因:
①心源性休克:常发生于心肌梗死后,急性左室衰竭、心脏填塞等也可引起。如不及时抢救,死亡率高达90%。
②严重的心节律不整:实验证明在室上性心动过速或房颤时,冠状动脉和脑动脉的血流量较正常心律时减少35~40%。在室性心动过速时可减少60%。心律紊乱可引起低血压;多源性室性期外收缩、室性动过速、房室传导阻滞及心搏过缓常是心跳骤停的先兆,应及时给予防治。
③血容量不足:由于大失血或微循环衰竭,使大量血液淤滞在毛细血管内,以致有效循环血量下降,造成回心血量不足,心排出量下降。
④中枢神经系统(尤其是心血管运动中枢)调节障碍:由于脑缺氧、脑水肿、或心血管中枢直接受累引起心搏出量下降。
(3)后果:
①组织灌注不足:由于心肌收缩不全及严重心律不齐可引起心排出量大大下降,在外周血管代偿不足的情况下就发生血压下降,微循环血液淤滞,此时常不可避免地要发生代谢性酸中毒。
②低氧血症:由于低心排出量造成肺泡通气量-血灌注的比例异常,终可导致功能下降;同时由于肺动静脉分流增加,使大量肺动脉血混入肺静脉内,以致动脉血氧分压下降。
低氧血症,心血管功能低下与组织灌流不足三者构成恶性循环,最后可导致重要器官的功能衰竭。
2.维持有效的呼吸除原有肺疾患外,复苏过程中常出现呼吸功能不全,在心跳恢复后几小时内也常因肺损伤、胸壁损伤、脑损伤或心衰等原因使呼吸不能立即恢复正常。此时应持续给氧,尤其在急性心肌梗死后或合并低心排出量的更属必要。在有条件的情况下还应做胸透的检查。一般在肺损伤中,较常见的是肺水肿:
(1)肺水肿时表现:患者常突然出现严重的呼吸困难、不能平卧、胸前紧迫感、烦躁不安、紫绀、咳大量粉红色泡沫样痰、肺部满布湿罗音。动脉血氧张力通常是低的。尽管无效死腔增加,但动脉血CO2常常正常或下降,只是在严重肺水肿时CO2分压才增高,此时常预示病情的恶化。
(2)肺水肿的原因:当心跳骤停时,由于心房压力突然升高,可以引起肺水肿,此外,引起肺水肿最常见的原因是急性左心衰竭。其它如心肌梗死、二尖瓣或主动脉瓣疾患,以及输液过度、肺毛细血管床减少的疾患等都可促使肺水肿的发生。这些情况下造成肺水肿的直接因素是肺血流量增加,引起肺毛细血管的过度扩张,最后导致肺毛细血管静力压过高,当大于血浆胶体渗透压(3.33kPa(25mmHg))时,液体渗透到肺间质就可引起肺水肿。如果此时因缺氧性损害肺毛细血管通透性增加,则更易发生肺水肿。此外,昏迷或脑损伤时,丘脑下部缺氧,可以引起肺静脉痉挛,造成毛细血管床压力增高;同时,由于交感神经兴奋,外周血管收缩,大量血液涌向肺循环,也易引起肺水肿。总之,心跳骤停后的肺水肿,其原理并不完全清楚。可能系多种因素所致。详见成人呼吸窘迫综合征。
(3)肺水肿的后果:肺水肿分间质性和肺泡性两型,后者常自前者发展而来,临床上很难分清两个阶段。二者引起的后果是一致的。
①严重的低氧血症:由于肺间质内液体增多,使肺泡、毛细血管壁增厚,不但引起气体弥散困难;还可造成肺泡通气-血灌流的紊乱,从而增加了无效死腔,以致大量未经氧合的血液流回左心。同时肺动脉-静脉分流增加,也更进一步加重了动脉血氧不饱和状态,结果动脉血氧分压下降,肺泡气-动脉血氧分压差也增大。严重的低氧血症还可导致心律紊乱。
②呼吸困难和/或窒息:由于肺泡及气管内液体增多,以及气管粘膜极度充血、水肿,同时还可合并支气管痉挛,造成呼吸阻力增加,以致发生通气困难。严重时可由于窒息而引起死亡。
③增加右心负荷:肺血管周围包围着液体,因而增加了肺血管的抵抗力,所以右心室的负荷增加,有时可引起右心室衰竭。
3.神经系统功能的恢复在及时而有效的复苏后,意识常很快恢复。心脏复跳后1-2小时意识仍不恢复者,通常有两个原因:①脑损伤;②脑血流供应不足,此时多不可避免地合并脑水肿,可在几小时内或2-3日达到高峰,于5日后脑水肿逐渐减退和消失。
(1)急性脑损伤的表现:缺氧轻时可出现不安、定向力丧失、意识朦胧等。一般脑循环完全停止5-10秒钟就可发生晕厥;停止10-15秒钟以上可发生昏迷及抽搐、肢体强直或驰缓性瘫痪、瞳孔散大等。个别还可出现反应性高热。现按其临床经过分为三期:
①昏迷期:皮层受缺氧影响发生广泛性抑制,若抑制涉及皮层下和脑干时,病员可呈深度昏迷(各种深、浅反射消失,肌张力减退且可不对称,呼吸异常等)。脑缺氧轻时,短暂昏迷后即清醒,严重时昏迷可持续数日、数周后逐渐进入去皮层综合征期。
②去皮层综合征期:此时大脑皮层仍处于抑制状态。皮层下及脑干部分因受伤较轻,抑制逐渐解除,瞳孔、角膜、咽或咳嗽等反射可逐渐出现,肌张力增高,各种腱反射活跃或亢进,并可出现病理反射,但无意识存在。大部分病例经进一步抢救可好转,个别病例可长期维持此状态。
③恢复期:此时皮层抑制基本解除,意识活动相继出现,智力逐渐恢复正常。个别病例可残留中枢局部损害的功能障碍如失明、失语、失听、瘫痪等症状。但经刻苦锻炼后仍可能痊愈。
(2)脑水肿:在脑水肿开始前就可因脑内血管扩张充血而出现脑体积增大,颅内压增高的症状,但脑水肿系指脑实质细胞肿胀、或/和组织间液含量增加的情况。
①表现:由于颅内压增高,引起脑血管张力增加及脑膜紧张,常可出现头痛。当脑水肿剌激了延脑的呕吐中枢时,可引起有不伴恶心的喷射状呕吐。此外,由于脑细胞缺氧、缺血,可出现皮层机制抑制的障碍,可出现中枢神经系统代偿性的反应,表现为血压升高,心率减慢,呼吸深而慢等症状。此时眼底检查常出现视网膜及视乳头水肿;外观可出现球结膜水肿、两眼外突等现象。
②病因及发病原理:心跳骤停后的完全缺氧或复苏后的脑“无再流”现象都易并发脑水肿。此外,急性颅脑损伤、毒物中毒时也可伴有脑水肿。脑水肿的发病原理远未搞清。但如及时预防脑水肿及纠正酸中毒,可进一步减少脑组织损伤,避免复苏后的脑后遗症。近年来对脑水肿的发病原理有以下几点看法:
a 脑细胞的肿胀:灰质水肿主要发生在神经胶质细胞,而损伤的神经元并不肿胀。正常时神经元与周围的星形细胞毛细血管三者构成一个功能单位,即血脑屏障。星形细胞在其中起着“代谢桥”的作用,主动传递葡萄糖物质给神经元。神经胶质细胞对缺氧耐受性很强,其耗氧量仅为神经元的1/50。当缺氧或/和细胞内酸中毒抑制神经元活动时,星形细胞的代谢反而增强,使原供应神经元的葡萄糖转而合成类糖元物质。大量类糖元之积聚增加了星形细胞的渗透压,因而星形细胞吸水、肿胀甚至破裂。此过程开始得很早,在缺氧13分钟后,可出现星形细胞肿胀,肿胀的神经胶质细胞通过压迫,阻断血循环可“绞死”损伤的神经元,从而加重了缺氧损伤的不可逆性。从开始的脑血管麻痹发展到脑水肿压迫引起的脑血流下降约需数小时,类糖元的逐步积累正好解释脑水肿的缓慢发生过程。此外,由于钠泵破坏,Na+转移到细胞内,也使脑细胞的含水量增加,尤其是胶质细胞膜对水和电解质渗透性较高,所以更易发生细胞内水肿。
b 细胞外液容量增多:主要发生在白质。当脑血管麻痹时,脑血管丧失了舒缩反应时的缓冲作用,使毛细血管的静压力增加;此外,缺氧时毛细血管内皮受损害;CO的积聚也可使脑毛细血管的通透性增加,导致大量液体漏到细胞外,造成细胞间液含量增加。
③后果:
a 脑循环障碍:星形细胞肿胀及细胞外液量的增加均可压迫脑内的小血管,使管腔变狭窄。更重要的是完全缺氧十几分钟后,脑毛细血管内皮因缺氧而变性、肿胀、血流不通畅,并可有微血栓形成,这些因素都严重地引起脑循环的障碍,使呼吸循环功能进一步受抑制,结果脑细胞缺氧更加恶化,形成恶性循环(见图16-1)。严重时患者可出现去大脑强直现象及两侧病理反射阳性,瞳孔扩大,对光反应迟钝等,此时常说明脑水肿,氧已波及到脑干功能。
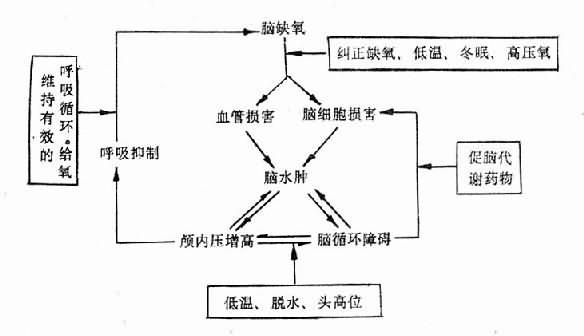
图16-1 脑水肿时的恶性循环
脑循环的恢复过程较慢,常常在循环重建30-40分钟后,脑组织才逐步获得较充分的血流灌注,缺氧缓解。
b 脑疝:颅内压急剧增高可导致突然脑疝形成,使呼吸骤停,同时出现一侧或两侧的瞳孔继续扩大、严重威胁生命。
c 脑细胞坏死:由于脑循环不足及脑组织水肿造成的血氧弥散障碍。最终引起局部酸性代谢产物堆积:它们可损害氧化酶系统的功能,并使线粒体、溶酶体膜通透性增加,释出蛋白酶、核酸酶等,使细胞发生自溶,产生不可逆的坏死病灶,表现为神经系统的各种定位症状。
④防治原则:高压氧气治疗,可改善脑组织缺氧状态,并减少脑血流之需要量,从而对防治脑水肿有良好作用。低温可以降低脑代谢率,并可降低颅内压(体温每下降1℃可使颅内压下降约5.5%),是极其有效的防治脑水肿的方法。此外,脱水疗法减少脑组织水份、减轻脑组织病变、降低颅内压等也有很显著的效果。
4.保护肾功能心跳恢复后,就应及时考虑保护肾脏。首先,复苏后的低血压(低于8kPa(60mmHg))可使肾血流急剧下降,引起肾皮质缺血、缺氧,从而导致肾功能衰竭。所以减少复苏后其它系统的合并症,对保护肾脏有很大意义。
反之,只有具有良好功能的肾脏,才能很好的利尿脱水,减轻脑水肿,改善体内酸中毒及高血钾等症状,从而巩固复苏的效果。