一、代偿性反应
动脉血氧分压一般要降至8kPa(60mmHg)以下,才会使组织缺氧,才引起机体的代偿反应,包括增强呼吸血液循环,增加血液运送氧和组织利用氧的功能等。
(一)呼吸系统
PaO2降低(低于8kPa)可刺激颈动脉体和主动脉体化学感受器。反射性地引起呼吸加深加快,从而使肺泡通气量增加,肺泡气氧分压升高,PaO2也随之升高。吸入10%氧时,通气量可增加50%;吸入5%氧可使通气量增加3倍。胸廓呼吸运动的增强使胸内负压增大,还可促进静脉回流,增加心输出量和肺血流量,有利于氧的摄取和运输。但过度通气使PaO2降低,减低了CO2对延髓的中枢化学感受器的刺激,可限制肺通气的增强。
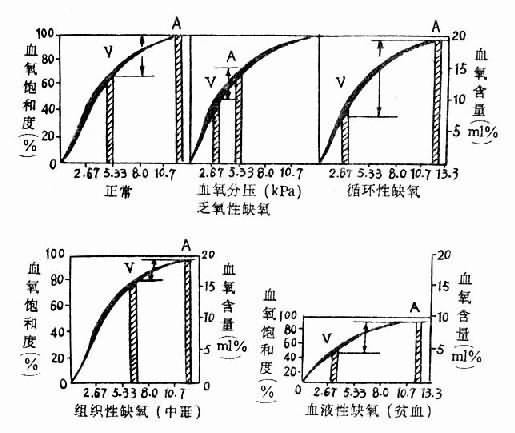
图3-2 各型缺氧的血氧变化特点
A动脉V静脉图中kPa相当于mmHg的数值
| kPa | mmHg |
| 2.67 | 20 |
| 5.33 | 40 |
| 8.0 | 60 |
| 10.7 | 80 |
低张性缺氧所引起的肺通气变化与缺氧持续的时间有关。如人达到400m高原后,肺通气量立即增加,但仅比在海平面高65%。数日后,肺通气量可高达在海平面的5~7倍。但久居高原,肺通气量逐渐回降,至仅比海平面者高15%左右。在急性缺氧早期肺通气增加较少,可能因过度通气形成的低碳酸血症和呼吸性碱中毒对呼吸中枢的抑制作用,使肺通气的增加受限。2~3日后,通过肾脏代偿性地排出HCO3-,脑脊液内的HCO3-也逐渐通过血脑屏障进入血液,使脑组织中pH逐渐恢复正常,此时方能充分显示缺氧兴奋缺氧的作用。久居高原肺气量回降,可能与外周化学感受器对缺氧的敏感性降低有关。据观察,世居高原者之颈动脉体的平均体积比世居海平面者大6.7倍,患慢性阻塞性肺病的病人的颈动脉比正常人大一倍以上。电镜观察表明,在慢性低张性缺氧的早期,颈动脉体增大,其中I型细胞增多,因I型细胞中嗜锇体含儿茶酚胺类神经介质,其增多可能具代偿意义。但在缺氧晚期,在增大的颈动脉体中嗜锇体的中心(core)缩小、晕轮(halo)加宽,有时整个嗜锇体为空泡所取代。这可能是颈动脉化学感受器敏感性降低的原因。长期缺氧使肺通气反应减弱,这也是一种慢性适应性反应。因为肺通气每增加1L,呼吸肌耗氧增加0.5ml,可能加剧机体氧的供求矛盾,故长期呼吸运动增强显然是对机体不利的。
肺通气量增加是对急性低张性缺氧最重要的代偿性反应。此反应的强弱存在显著的个体差异,代偿良好者肺通气量增加较多,PaO2比代偿不良者高。PaCO2也较低。
血液性缺氧和组织性缺氧因PaO2不低,故呼吸一般不增强;循环性缺氧如累及肺循环,如心力衰竭引起肺淤血、水肿时,可使呼吸加快。
(二)循环系统
低张性缺氧引起的代偿性心血管反应,主要表现为心输出量增加、血流分布改变、肺血管收缩与毛细血管增生。
1、心输出量增加 有报道进入高原(6100m)30天的人,其心输出量比平原居民高2~3倍。在高原久住后,心输出量逐渐减少。心输出量增加可提高全身组织的供氧量,故对急性缺氧有一定的代偿意义。心输出量增加主要是由于:
(1)心率加快:过去认为心率加快是颈动脉体和主动脉体化学感受器刺激反射性地引起。但有人实验,在控制呼吸不变的情况下,缺氧刺激血管化学感受器却使心率变慢。因此缺氧时心率加快很可能是通气增加所致肺膨胀对肺牵张感受器的刺激,反射性地通过交感神经引起的。
然而呼吸运动过深反而通过反射使心率减慢,外周血管扩张和血压下降。
(2)心收缩性增强:缺氧作为一种应激原,可引起交感神经兴奋,作用于心脏β—肾上腺素能受体,使心收缩性增强。
(3)静脉回流量增加:胸廓呼吸运动及心脏活动增强,可导致静脉回流量增加和心输出量增多。
2、血流分布改变器官血流量取决于血液灌注的压力(即动、静脉压差)和器官血流的阻力。后者主要取决于开放的血管数量与内径大小。缺氧时,一方面交感神经兴奋引起的血管收缩;另一方面局部组织因缺氧产生的乳酸、腺苷等代谢产物则使血管扩张。这两种作用的平衡关系决定器官的血管是收缩或扩张,以及血流量是减少或增多。急性缺氧时,皮肤、腹腔内脏交感神经兴奋,缩血管作用占优势,故血管收缩;而心、脑血管因以局部组织代谢的产物的扩血管作用为主,故血管扩张,血流增加。这种血流分布的改变显然对于保证生命重要器官缺氧的供应是有利的。
心肌活动消耗的能量主要来自有氧代谢。心脏重量约占体重之0.4~0.5%,静息时冠脉流量约占心输出量之4~5%,其动—静脉血氧含量差约为12ml%,表明心肌耗氧量大,由单位容积血液摄取的氧量多。心肌缺氧时,进一步提高对单位容积血液中氧的摄取率很有限,主要依靠扩张冠状血管以增加心肌的供氧。冠脉扩张由局部代谢产物(腺苷、H+、K+、PGI2等)与冠脉平滑肌中β—肾上腺能受体占优势所致,其中腺苷的作用最为重要。当心肌细胞缺氧时,由ATP、ADP生成的AMP增多,AMP在5—核苷酸酶的作用下,脱去磷酸,形成腺苷。腺苷易透过细胞膜进入组织液,作用于冠状血管,使之扩张。通常组织液中的腺苷大部分进入细胞,重新磷酸化生成AMP,一部分被腺苷脱氨酶灭活。缺氧时,腺苷脱氨酶活性可能降低,这也是局部腺苷增多的一个原因。
3、肺血管收缩肺血管直接对缺氧的反应与体血管相反。肺泡缺氧及混合静脉血的氧分压降低都引起肺小动脉收缩,从而使缺氧的肺泡的血流量减少。如果是由肺泡通气量减少引起的肺泡缺氧,则肺血管的收缩反应有利于维持肺泡通气与血流的适当比例,使流经这部分肺泡的血液仍能获得较充分的氧,从而可维持较高的PaO2。此外,正常情况下由于重力作用,通过肺尖部的肺泡通气量与血流量的比值过大,肺泡气中氧不能充分地被血液运走。当缺氧引起较广泛的肺血管收缩,导致肺动脉压升高时,肺上部的血流增加,肺上部的肺泡通气能得到更充分的利用。
缺氧引起肺血管收缩的机制较复杂,尚未完全阐明,研究结果也有矛盾。当前具倾向性的观点:①交感神经作用:缺氧所致交感神经兴奋可作用于肺血管的α受体引起血管收缩反应。②体液因素作用:缺氧可促使肺组织内肥大细胞、肺泡巨噬细胞、血管内皮细胞等释放组胺、前列腺素和白三烯等血管活性物质,其中有的能收缩肺血管,如白三烯(leukotriene,LTs)、血栓素A2(thromboxane A2、TXA2)、前列腺素F2a(prostaglandin F2a,PGF2a)等,有的扩张血管,如前列环素(prostacyclin,PGI2)、前列腺素E1(prostaglandin E1 PGE1)等。在肺血管收缩反应中,缩血管物质生成与释放增加,起介导作用;扩血管物质的生成与释放也可增加,起调节作用。两者力量对比决定肺血管收缩反应的强度。组胺作用于H1受体使肺血管收缩,作用于H2受体则使之扩张。在缺氧性肺血管收缩反应中,组胺释放增多,主要作用于H2受体以限制肺血管的收缩。③缺氧直接对血管平滑肌作用:缺氧使平滑肌细胞膜对Na+、Ca2+的通透性增高,促使Na+、Ca2+的通透性增高,促使Na+、Ca2+内流,导致肌细胞兴奋性与收缩性增高。这一观点还有待进一步证实。看来缺氧性肺血管收缩反应是多因素综合作用的结果。
4、毛细血管增生长期慢性缺氧可促使毛细血管增生。尤其是脑、心脏和骨骼肌的毛细血管增生更显著。毛细血管的密度增加可缩短血氧弥散至细胞的距离,增加对细胞的供氧量。
(三)血液系统
缺氧可使骨髓造血增强及氧合血红蛋白解离曲线右移,从而增加氧的运输和释放。
1、红细胞增多 移居到3600m高原的男性居民红细胞计数通常约为6×1012/L(6×106/mm3),Hb为210g/L(21g/dl)左右。慢性缺氧所致红细胞增多主要是骨髓造血增强所致。当低氧血流经肾脏近球小体时,能刺激近球细胞,使其中颗粒增多,生成并释放促红细胞生成素(erythropoietin),促红细胞生成素能促使红细胞系单向干细胞分化为原红细胞,并促进其分化、增殖和成熟,加速Hb的合成和使骨髓内的网织红细胞和红细胞释放入血液。当血浆中促红细胞生成素增高到一定水平时,可因红细胞增多使缺氧缓解,肾脏促红细胞生成素的产生因而减少,通过这种反馈机制控制着血浆促红细胞生成素的含量。红细胞增多可增加血液的氧容量和氧含量,从而增加组织的供氧量。
2、氧合血红蛋白解离曲线右移缺氧时,红细胞内2,3—DPG增加,导致氧离曲线右移,即血红蛋白与氧的亲和力降低,易于将结合的氧释出供组织利用。但是,如果PaO2低于8kPa,则氧离曲线的右移将使血液通过肺泡时结合的氧量减少,使之失去代偿意义。
2,3—DPG是红细胞内糖酵解过程的中间产物。缺氧时红细胞中生成的2,3—DPG增多是因为:①低张性缺氧者氧合血红蛋白(HbO2)减少,脱氧血红蛋白(Hb)增多,前者中央孔穴小,不能结合2,3—DPG;后者中央孔穴较大,可结合2,3—DPG。故当脱氧血红蛋白增多,红细胞内游离的2,3—DPG减少,使2,3—DPG对二磷酸甘油酶变位酶(diphosphoglycerate mutase, DPGM)及磷酸果糖激酶的抑制作用减弱,从而使糖酵解增强及2,3—DPG的生成增多;②低张性缺氧时出现的代偿性肺过度通气所致呼吸性碱中毒,以及由于脱氧血红蛋白稍偏碱性,致使pH增高,pH增高能激活磷酸果糖激酶使糖酵解增强,2,3—DPG合成增加,另一方面,pH增高还能抑制2,3—DPG磷酸酶(2,3—DPg phosphatase, 2,3—DPG)的活性,使2,3—DPG的分解减少(图3-3,图3-4)。
2,3—DPG增多使氧离曲线右移,是因为:①2,3—DPG与脱氧血红蛋白结合,可稳定后者的空间构型,使之不易与氧结合;②2,3—DPG是一种不能透出红细胞的有机酸,增多时能降低红细胞内pH,而pH下降通过Bohr效应可使血红蛋白与氧的亲和力降低。(Bohr效应系指H+和Pco2对Hb与O2亲和力的影响,当H+浓度或Pco2增高时,Hb与O2的亲和力降低,氧离曲线右移)。
P50为反映Hb与O2的亲和力的指标,指的是血红蛋白氧饱和度为50%时的氧分压,正常为3.47~3.6kPa(26~27mmHg)。红细胞内2,3—DPg 浓度每增高1μm/gHb,P50将升高约0.1kPa。
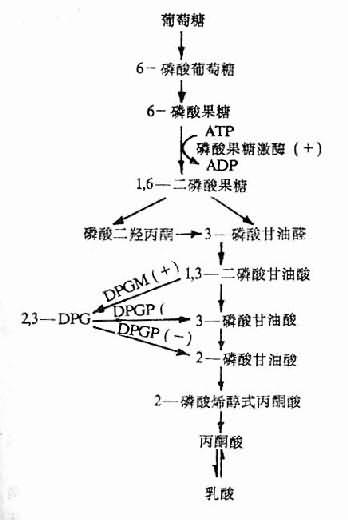
图3-3,2,3—DPG的生成与分解
DPGM二磷酸甘油酸变位酶;
DPGP二磷酸甘油酸磷酸酶;
(+)pH增高时促进反应;
(-)PH增高时抑制反应
图3-4 2,3-DPG结合于HHb分子的中央空穴示意图
(四)组织细胞的适应
在供氧不足的情况下,组织细胞可通过增强利用氧的能力和增强无氧酵解过以获取维持生命活动所必须的能量。
1、组织细胞利用氧的能力增强 慢性缺氧时,细胞内线粒体的数目和膜的表面积均增加,呼吸链中的酶如琥珀酸脱氢酶、细胞色素氧化酶可增加,使细胞的内呼吸功能增强。如胎儿在母体内处于相对缺氧的环境,其细胞线粒体的呼吸功能为成年动物的3倍,至出生后10~14天,线粒体呼吸功能才降至成年动物水平。
2、无氧酵解增强发 严重缺氧时,ATP生成减少,ATP/ADP比值下降,以致磷酸果糖激酶活性增强,该酶是控制糖酵解过程最主要的限速酶,其活性增强可促使糖酵解过程加强,在一定的程度上可补偿能量的不足。
3、肌红蛋白增加 慢性缺氧可使肌肉中肌红细胞蛋白含量增多。肌红蛋白和氧的亲和力较大,当氧分压为1.33kPa(10mmHg)时,血红蛋白的氧饱和度约为10%,而肌红蛋白的氧饱和度可达70%,当氧分压进一步降低时,肌红蛋白可释出大量的氧供细胞利用。肌红蛋白的增加可能具有储存氧的作用。
肺通气及心脏活动的增强可在缺氧时立即发生。但这些代偿功能活动本身消耗能量和氧,红细胞的增生和组织利用氧能力的增强需较长的时间,但为较经济的代偿方式。急性缺氧时以呼吸系统和循环系统的代偿反应为主;慢性缺氧者,如世居高原的居民,主要靠增加组织利用氧和血液运送氧的能力以适应慢性缺氧。其肺通气量、心率及输出量并不多于居住海平面者。
二、缺氧时机体的机能代谢障碍
严重缺氧,如低张性缺氧者PaO2低于4kPa(30mmHg)时,组织细胞可发生严重的缺氧性损伤,器官可发生功能障碍甚而功能衰竭。
(一)缺氧性细胞损伤
缺氧性细损伤(hypoxic cell damage)主要为细胞膜、线粒体溶酶体的变化。
1、细胞膜的变化 在细胞内ATP含量减少以前,细胞膜电位已开始下降。其原因为细胞膜对离子的通透性增高,导致离子顺浓度差透过细胞膜。
(1)钠离子内流:Na+内流使细胞内Na+浓度增加,可激活Na+-K+泵以泵出Na+,从而消耗ATP。ATP消耗量增多可促使线粒体氧化磷酸化过程增强,严重缺氧时,线粒体呼吸功能降低使ATP生成减少,以至Na+-K+泵不能充分运转,进一步使细胞内Na+增多。细胞内Na+的增多促使水进入细胞,导致细胞水肿。血管内皮细胞肿胀可堵塞微血管,加重微循环缺氧。
(2)钾离子外流:K+外流使细胞内缺K+。而K+为蛋白质包括酶等合成代谢所必需。细胞内缺钾将导致合成代谢障碍,酶的生成减少,将进一步影响ATP的生成和离子泵的功能。
(3)钙离子的内流:细胞外Ca2+浓度比胞浆中游离Ca2+高1000倍以上。细胞内Ca2+逆浓度外流和肌浆网、线粒体逆浓度差摄Ca2+均为耗能过程。当严重缺氧时使细胞膜对Ca2+的对通透性增高量Ca2+内流将增加;ATP减少将影响Ca2+的外流和摄取,使胞浆Ca2+浓度增高。Ca2+增多可抑制线粒体的呼吸功能;可激活磷脂酶,使膜磷脂分解,引起溶酶体的损伤及其水解酶释出;还可激活一种蛋白酶,使黄嘌呤脱氢酶(D型)转变为黄嘌呤氧化酶(O型)。由此增加自由基的形成,加重细胞的损伤。
2、线粒体的变化 细胞内的氧约有80-90%在线粒体内用于氧化磷酸化生成ATP,仅10~20%在线粒体外用于生物合成、降解及生物转化(解毒)作用等。轻度缺氧或缺氧早期线粒体呼吸功能是增强的。严重缺氧首先影响线粒体外氧的作用,使神经介质的生成和生物转化过程等降低,当线粒体部位氧分压降到监界点0.1kPa(<1mmHg)时,可降低线粒休的呼吸功能,使ATP生成减少。呼吸功能降低主要因脱氢酶活性下降,严重时线粒体可出现肿胀、嵴崩解、外膜破裂和基质外溢等病变。
3、溶酶体的变化缺氧时因糖酵解增强,乳酸生成增多,和脂肪氧化不全使其中间代谢产物酮体增多。导致酸中毒。pH降低可引起磷脂酶活性增高,使溶酶体膜磷脂被分解,膜通透性增高,结果使溶酶体肿胀、破裂,和大量溶酶体酶的释出,进而导致细胞本身及其周围组织的溶解、坏死。
(二)中枢神经系统的机能障碍
脑重仅为体重为2%左右,而脑血流量约占心输出量之15%,脑耗氧量约为总耗氧量的23%,所以脑对缺氧十分敏感。脑灰质比白质的耗氧量多5倍,对缺氧的耐受性更差。急性缺氧可引起头痛、情绪激动、思维力、记忆力、判断力降低或丧失以及运动不协调等。慢性缺氧者则有易疲劳、思睡、注意力不集中及精神抑郁等症状。严重缺氧可导致烦躁不安、惊厥、昏迷甚而死亡。正常人脑静脉血氧分压约为4.53kPa(34mmHg),当降至3.73kPa(28mmHg)以下可出现神经错乱等;降至2.53kPa(19mmHg)以下时可出现意识丧失;低达1.6kPa(12mmHg)时将危及生命。缺氧引起脑组织的形态学变化主要是脑细胞变性、坏死、脑细胞肿胀及脑水肿。
缺氧引起中枢神经系统机能障碍的机制较复杂。神经细胞膜电位的降低、神经介质的合成减少、ATP的生成不足、酸中毒、细胞内游离Ca2+增多、溶酶体酶的释放以及细胞水肿等,均可导致神经系统的功能障碍,甚而神经细胞结构的破坏、当PaO2低于6.67kPa(50mmHg)时,可使脑血管扩张。缺氧与酸中毒还使脑微血管通透性增高,从而导致脑水肿(图3-5)。脑血管扩张、脑细胞及脑间质水肿可使颅内压升高,由此引起头痛、呕吐等症状。
图3-5 缺氧时脑水肿发生机理
(三)外呼吸功能障碍
急性低张性缺氧,如快速登上4000m以上的高原时,可在1-4天内发生肺水肿,表现为呼吸困难、咳嗽、咳出血性泡沫痰、肺部有湿性罗音、皮肤粘膜发绀等。因高原肺水肿的动物模型难以复制成功,故其发病机制至今尚不清楚。因为肺水肿与肺动脉高压呈正相关,故有人强调肺毛细血管压力增高的作用。可能缺氧所致外周血管收缩使回心血量增加。和肺血量增多;加上缺氧性肺血管收缩反应使肺血流阻力增加,导致肺动脉高压。由于肺血管收缩强度不一,致使肺血流分布不均,在肺血管收缩较轻或不收缩的部位肺泡毛细血管血流增加,毛细血管压力增高,从而引起压力性肺水肿。也有人强调肺微血管通透性增高的作用。因为患者支气管肺泡洗出液中蛋白质含量较高,并有大量肺泡巨噬细胞,可测得补体C3a、LTB4、TXB2等;尸检可见肺泡水肿、炎性细胞浸润及透明膜形成。但高原性肺水肿不同于其它原因引起的成人呼吸窘迫综合征,前者经休息、氧疗或下山后短期内即可痊愈;而成人呼吸窘迫综合征经治疗往往要数月后才能痊愈。肺内血压高和流速对微血管的切应力(流动的血液作用于血管壁的力与管壁平等方向的分力)可能是导致微血管内皮损伤和血管通透性增高的一个因素。肺水肿影响肺的换气功能,可使PaO2进一步下降。
PaO2过低可直接抑制呼吸中枢,使呼吸抑制,肺通气量减少,导致中枢性呼吸衰竭。
(四)循环功能障碍
严重的全身性缺氧时,心脏可受累,如高原性心脏病、肺原性心脏病、贫血性心脏病等,甚而发生心力衰竭。今以高原性心脏病为例说明缺氧引起循环障碍的机制。
1、肺动脉高压 肺泡缺氧所致肺血管收缩反应可增加肺循环阻力,可导致严重的肺动脉高压。慢性缺氧使肺小动脉长期处于收缩状态,可引起肺血管中膜平滑肌肥大,血管硬化,形成稳定的肺动脉高压。肺动脉高压增加右室射血的阻力。另外,缺氧所致红细胞增多,使血液粘度增高,也可增加肺循环阻力。肺动脉高压可导致右心室肥大,甚至心力衰竭。
2、心肌的收缩与舒张功能降低心肌缺氧可降低心肌的舒缩功能,甚而使心肌发生变性、坏死。(参阅第十四章心血管系统病理生理学)
3、心律失常 严重缺氧可引起窦性心动过缓、期前收缩、甚至发生心室纤颤致死。心动过缓可能为严重的PaO2降低对颈动脉体化学感受器的刺激,反射性地兴奋迷走神经所致。此外,持久缺氧也往往显示副交感优势使心率变慢。期前收缩与室颤的发生与心肌细胞内K+减少、Na+增加使静息膜电位降低、心肌兴奋性增高、和传导性降低有关。缺氧部位的心肌静息膜电位降低,使其与相邻较完好的心肌之间形成电位差,从而产生“损伤电流”,可成为异位激动的起源,严重的心肌受损可导致完全的传导阻滞。
4、静脉回流减少 脑严重缺氧时,呼吸中枢的抑制使胸廓运动减弱,可导致静脉回流减少,全身性极严重而持久的缺氧使体内产生大量乳酸、腺苷等代谢产物,后者可直接扩张外周血管,使外周血管床扩大,大量血液淤积在外周,回心血量减少,使心输出量减少,而引起循环循衰竭。
除以上所述神经系统、呼吸与循环系统机能障碍外,肝、肾、消化道、内分泌等各系统的功能均可因严重缺氧而受损害。