心力衷竭的本质是心肌收缩性减弱。为了理解有关的问题,本节将首先简要地复习心肌收缩的分子生物学基础,然后讨论心力衰竭的一般发病机制,以及肥大心肌转向衰竭的机制。
心肌的收缩物质是组成粗、细肌丝的心肌蛋白。粗肌丝的主要成分是肌球蛋白(myosin),其分子量约50万,全长1500A,一端为杆状的尾部,另一端为粗大的头部(S1),二者之间是能弯曲的颈部(S2)。头部又分成两片,是ATP酶的活动中心,它在肌动蛋白和肌球蛋白之间的搭桥和粗细肌丝之间的滑行中起着重要作用。细肌丝的主要成分是肌动蛋白(actin),分子量47.000,分子呈球状,串联而成双链螺旋状的细肌丝纤维。在双链间的沟槽内,杆状的向肌球蛋白(tropmyosin)和肌动蛋白卷曲在一起,每距400A处还有一个肌钙蛋白(troponin)分子。向肌球蛋白和肌钙蛋白是调节蛋白,本身不起收缩作用,但能调节肌动蛋白与肌球蛋白的联结,而使心肌纤维发生收缩和舒张。肌钙蛋白由三个亚单位组成,即向肌球蛋白亚单位(tropotroponin,TnT)、抑制亚单位(inhibrtor troponin,TnI)钙结合亚单位(calciumcombining troponin, TnC)在心肌兴奋-收缩偶联中起重要作用(图12-1、2)。
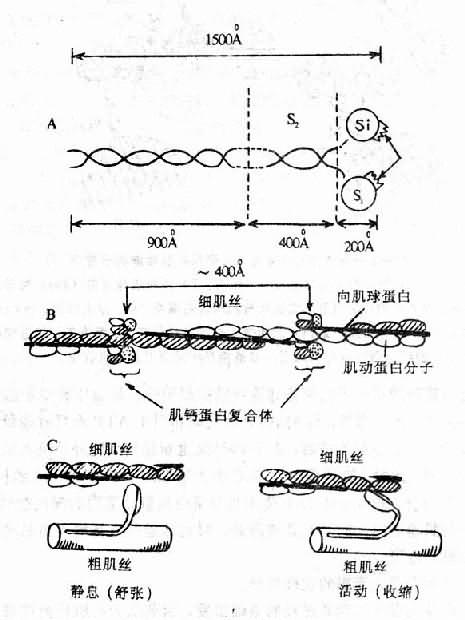
图12-1 心肌收缩蛋白和调节蛋白
A.肌球蛋白分子结构模式图,说明见正文。
B.肌动蛋白分子呈球形,串联而成双链螺旋状的细肌丝。
向肌球蛋白在两个肌动蛋白链之间。
每隔400A有一肌钙蛋白复合体。
C.示粗、细肌丝在收缩与舒张时的相互关系。
Ca2+在心肌兴奋时的电活动与机械收缩之间起偶联作用。当心肌除极化时,Ca2+从细胞外转移到心肌细胞的胞质中,同时也从肌质网释放入胞质。因此胞质内Ca2+浓度升高(由10-8M升至10-5M)。此时肌钙蛋白的TnC即迅速与Ca2+结合。这种结合相继使TnC和TnI的构型发生变化,其结果是TnI从肌动蛋白移开。这种构型变化又可通过TnT影响向肌球蛋白的位置,使向肌球蛋白旋转到肌动蛋白两条螺旋状链的深沟中,从而使肌动蛋白的受点暴露而与肌球蛋白头部相接触,形成横桥。S1的ATP酶随即作用于ATP而释放能量,肌动球蛋白(actomyosin)乃发生收缩。心肌收缩后,由于Ca2+又重新移到细胞外及进入肌质网,胞质内Ca2+浓度又降至10-8M。此时,肌钙蛋白的TnC失去了Ca2+,TnC和TnI的构型恢复原状,故TnI又与肌动蛋白结合,进而通过TnT使向肌球蛋白从肌动蛋白的深沟中转移出来,而恢复原来的位置。于是肌动蛋白上的受点又被掩盖,肌动球蛋白重新解离为肌动蛋白和肌球蛋白,横桥解除,心肌乃舒张。
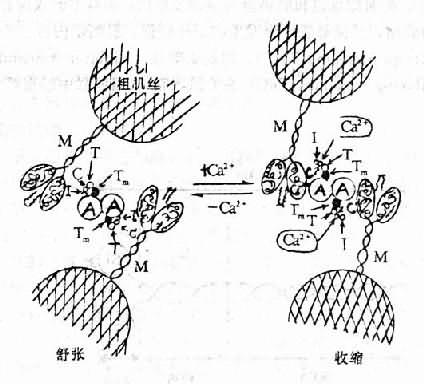
图12-2 Ca2+与肌钙蛋白结合引起心肌收缩的示意图
在舒张期(左),肌钙蛋白复合体三成分(I、C、T)使向肌球蛋白
(Tm)位于细肌丝(A)螺旋沟的外侧,从而阻止细肌丝与肌球蛋白
横桥(M)发生作用。当Ca2+与肌钙蛋白C(右)结合引起一系列的
活动使I与细肌丝分开、从而使Tm移进细肌丝的螺旋沟中,细肌丝乃得
以与肌球蛋白横桥相互作用而引起心肌收缩。
下文将着重讨论慢性心力衰竭的发病机制。
从心肌分子结构及兴奋收缩偶联过程的基础出发,目前认为心肌负荷过重和心肌受损等病因引起心肌收缩性减弱的一般机制大致有下述几个方面。