第一章 药理学总论
绪言
一、药理学的性质与任务
药理学(pharmacology)是研究药物的学科之一,是一门为临床合理用药防治疾病提供基本理论的医学基础学科。药理学研究药物与机体(包括病原体)相互作用的规律及其原理。药物(drug)是指用以防治及诊断疾病的物质,在理论上说,凡能影响机体器官生理功能及(或)细胞代谢活动的化学物质都属于药物范畴,也包括避孕药及保健药。药理学一方面研究在药物影响下机体细胞功能如何发生变化,另一方面研究药物本身在体内的过程,即机体如何对药物进行处理,前者称为药物效应动力学(pharmacodynamics),简称药效学;后者称为药物代谢动力学(pharmacokinetics),简称药动学。可见药理学研究的主要对象是机体,属于广义的生理科学范畴。它与主要研究药物本身的药学科学,如生药学、药物化学、药剂学、制药学等学科有明显的区别。药理学是以生理学、生化学、病理学等为基础,为指导临床各科合理用药提供理论基础的桥梁学科。药理学的学科任务是要为阐明药物作用机制、改善药物质量、提高药物疗效、开发新药、发现药物新用途并为探索细胞生理生化及病理过程提供实验资料。药理学的方法是实验性的,即在严格控制的条件下观察药物对机体或其组成部分的作用规律并分析其客观作用原理。近年来逐渐发展而设立的临床药理学是以临床病人为研究和服务对象的应用科学,其任务是将药理学基本理论转化为临床用药技术,即将药理效应转化为实际疗效,是基础药理学的后继部分。学习药理学的主要目的是要理解药物有什么作用、作用机制及如何充分发挥其临床疗效,要理论联系实际了解药物在发挥疗效过程中的因果关系。
二、药物与药理学的发展史
远古时代人们为了生存从生活经验中得知某些天然物质可以治疗疾病与伤痛,这是药物的源始。这些实践经验有不少流传至今,例如饮酒止痛、大黄导泻、楝实祛虫、柳皮退热等。以后在宗教迷信与邪恶斗争及封建君王寻求享乐与长寿中药物也有所发展。但更多的是将民间医药实践经验的累积和流传集成本草,这在我国及埃及、希腊、印度等均有记载,例如在公元一世纪前后我国的《神农本草经》及埃及的《埃伯斯医药籍》(Ebers’Papyrus)等。明朝李时珍的《本草纲目》(1596)在药物发展史上有巨大贡献,是我国传统医学的经典著作,全书共52卷,约190万字,收载药物1892种,插图1160帧,药方11000余条,是现今研究中药的必读书籍,在国际上有七种文字译本流传。在西欧文艺复兴时期(十四世纪开始)后,人们的思维开始摆脱宗教束缚,认为事各有因,只要客观观察都可以认识。瑞士医生Paracelsus(1493-1541)批判了古希腊医生Galen恶液质唯心学说,结束了医学史上1500余年的黑暗时代。后来英国解剖学家W.Harvey (1578-1657)发现了血液循环,开创了实验药理学新纪元。意大利生理学家F.Fontana (1720-1805)通过动物实验对千余种药物进行了毒性测试,得出了天然药物都有其活性成分,选择作用于机体某个部位而引起典型反应的客观结论。这一结论以后为德国化学家F.W.Serturner(1783-1841)首先从罂粟中分离提纯吗啡所证实。18世纪后期英国工业革命开始,不仅促进了工业生产也带动了自然科学的发展。其中有机化学的发展为药理学提供了物质基础,从植物药中不断提纯其活性成分,得到纯度较高的药物,如依米丁、奎宁、士的宁、可卡因等。以后还开始了人工合成新药,如德国微生物学家P.Ehrlich从近千种有机砷化合物中筛选出治疗梅毒有效的新胂凡纳明(914)。药理学作为独立的学科应从德国R.Buchheim(1820-1879)算起,他建立了第一个药理实验室,写出第一本药理教科书,也是世界上第一位药理学教授。其学生O.Schmiedeberg(1838-1921)继续发展了实验药理学,开始研究药物的作用部位,被称为器官药理学。受体原是英国生理学家J.N.Langley(1852-1925)提出的药物作用学说,现已被证实是许多特异性药物作用的关键机制此后药理学得到飞跃发展,第二次世界大战结束后出现了许多前所未有的药理新领域及新药,如抗生素、抗癌药、抗精神病药、抗高血压药、抗组胺药、抗肾上腺素药等。近年来药动学的发展使临床用药从单凭经验发展为科学计算,并促进了生物药学(biopharmaceutics)的发展。药效学方面逐渐向微观世界深入,阐明了许多药物作用的分子机制也促进了分子生物学本身的发展。展望今后,药理学将针对疾病的根本原因,发展病因特异性药物治疗,那时将能进一步收到药到病除的效果。
三、新药开发与研究
人们生活水平提高要求更多更好的新药,药物科学的发展为新药开发提供了理论基础和技术条件,市场经济竞争也促进了新药快速发展。美国食品与药物管理局(FDA)近十年来每年批准上市的新药都在20种以上。我国近年来引进新药品种很多,但需要加快创新。新药开发是一个非常严格而复杂的过程,各药虽然不尽相同,药理研究却是必不可少的关键步骤。临床有效的药物都具有相应的药理效应,但具有肯定药理效应的药物却不一定都是临床有效的药物。例如抗高血压药都能降低血压,但降压药并不都是抗高血压药,更不一定是能减少并发症、延长寿命的好药。因此新药开发研究必需有一个逐步选择与淘汰的过程。为了确保药物对病人的疗效和安全,新药开发不仅需要可靠的科学实验结果,各国政府还对新药生产上市的审批与管理制定了法规,对人民健康及工商业经济权益予以法律保障。
新药来源包括天然产物、半合成及全合成化学物质。过去选药主要方法是依靠实践经验,现在可以根据有效药物的植物分类学找寻近亲品种进行筛选或从有效药物化学结构与药理活性关系推断,定向合成系列产品,然后进行药理筛选。近年来对于机体内在抗病物质(蛋白成分)利用DNA基因重组技术,即将DNA的特异基因区段分离并植入能够迅速生长的细菌或酵母细胞,以获得大量所需蛋白药物。此外,还可对现有药物进行化学结构改造(半合成)或改变剂型,也可获得疗效更好,毒性更小或应用更方便的药物。
新药研究过程大致可分三步,即临床前研究、临床研究和售后调研。临床前研究包括用动物进行的系统药理研究及急慢性毒性观察。对于具有选择性药理效应的药物,在进行临床试验前还需要测定该药物在动物体内的吸收、分布及消除消除过程。临床前研究是要弄清新药的作用谱及可能发生的毒性反应。在经过药物管理部门的初步审批后才能进行临床试验。目的在于保证用药安全。
临床研究首先在10~30例正常成年志愿者观察新药耐受性,找出安全剂量。再选择有特异指征的病人按随机分组、设立已知有效药物及空白安慰剂双重对照(对急重病人不得采用有损病人健康的空白对照),并尽量采用双盲法(病人及医护人员均不能分辨治疗药品或对照药品)观察,然后进行治疗结果统计分析,客观地判断疗效。与其同时还需进行血药浓度监测计算药动学数据(详第三章)。受试病例数一般不应少于300例,先在一个医院以后可扩大至三个以上医疗单位进行多中心合作研究。对那些需要长期用药的新药,应有50~100例病人累积用药半年至一年的观察记录。由此制定适应证、禁忌证、剂量疗程及说明可能发生的不良反应后,再经过药政部门的审批才能生产上市。
售后调研(postmarketing surveillance)是指新药问市后进行的社会性考查与评价,在广泛的推广应用中重点了解长期使用后出现的不良反应和远期疗效(包括无效病例)。药物只能依靠广大用药者(医生及病人)才能作出正确的历史性评价。
第二章 药物效应动力学
第一节 药物的基本作用
一、药物作用与药理效应
药物作用(drug action)是指药物与机体细胞间的初始作用,是动因,是分子反应机制,有其特异性(specificity)。药理效应(pharmacological effect)是药物作用的结果,是机体反应的表现,对不同脏器有其选择性(selectivity)。因此,药理效应实际上是机体器官原有功能水平的改变,功能的提高称为兴奋(excitation)、亢进(augmentation),功能的降低称为抑制(inhibition)、麻痹(paralysis)。过度兴奋转入衰竭(failure),是另外一种性质的抑制。近年来生命科学的迅速发展,能引起细胞形态与功能发生质变的药物受到注意,例如某些物质可以引起细胞癌变,基因疗法能使机体引出遗传缺陷时或原来没有的特殊功能。药物作用特异性强的药物不一定引起选择性高的药理效应,二者不一定平行。例如阿托品特异性阻断M-胆碱受体,但药理效应选择性并不高,对心脏、血管、平滑肌、腺体及中枢神经功能都有影响,而且有的兴奋、有的抑制。作用特异性强及(或)效应选择性高的药物应用时针对性较好。反之,效应广泛的药物副反应较多。但广谱药物在多种病因或诊断未明时也有其方便之处,例如广谱抗生素、广谱抗心律失常药等。
药理效应与治疗效果,后者简称疗效(therapeutic effect)并非同义词,例如具有扩张冠脉效应的药物不一定都是抗冠心病药,抗冠心病药也不一定都会取得缓解心绞痛临床疗效,有时还会产生不良反应(adverse reaction),这就是药物效应的两重性:药物既能治病也能致病。
二、治疗效果
1.对因治疗(etiological treatment)用药目的在于消除原发致病因子,彻底治愈疾病称为对因治疗,或称治本,例如抗生素消除体内致病菌。
2.对症治疗(symptomatic treatment)用药目的在于改善症状称为对症治疗,或称治标。对症治疗未能根除病因,但在诊断未明或病因未明暂时无法根治的疾病却是必不可少的。在某些重危急症如休克、惊厥、心力衰竭、高热、剧痛时,对症治疗可能比对因治疗更为迫切。
三、不良反应
凡不符合用药目的并为病人带来不适或痛苦的反应统称为药物不良反应。多数不良反应是药物固有效应的延伸,在一般情况下是可以以预知的,但不一定是可以避免的。少数较严重的不良反应是较难恢复的,称为药原性疾病(drug induced disease),例如庆大霉素引起神经性耳聋,肼屈嗪引起红斑性狼疮等。
1.副反应(side reaction)由于药理效应选择性低,涉及多个效应器官,当某一效应用作治疗目的时,其他效应就成为副反应(通常也称副作用)。例如阿托品用于解除胃肠痉挛时,将会引起口干、心悸、便秘等副反应。副反应是在常用剂量下发生的,一般不太严重,但是难以避免的。
2.毒性反应(toxic reaction)毒性反应是指在剂量过大或蓄积过多时发生的危害性反应,一般比较严重,但是可以预知也是应该避免发生的不良反应。急性毒性多损害循环、呼吸及神经系统功能,慢性毒性多损害肝、肾、骨髓、内分泌等功能。致癌(carcinogenesis)、致畸胎(teratogenesis)、致突变(mutagenesis)三致反应也属于慢性毒性范畴。企图增加剂量或延长疗程以达到治疗目的是有限度的,过量用药是十分危险的。
3.后遗效应(residual effect)后遗效应是指停药后血药浓度已降至阈浓度以下时残存的药理效应。例如长期应用肾上腺皮质激素停药后肾上腺皮质功能低下数月内难以恢复。
4.停药反应(withdrawal reaction)突然停药后原有疾病的加剧,又称回跃反应(rebound reaction),例如长期服用可乐定降血压,停药次日血压将激烈回升。
5.变态反应(allergic reaction)变态反应是一类免疫反应。非肽类药物作为半抗原与机体蛋白结合为抗原后,经过接触10天左右敏感化过程而发生的反应,也称过敏反应(hypersensitive reaction)。常见于过敏体质病人。临床表现各药不同,各人也不同。反应性质与药物原有效应无关,用药理拮抗药解救无效。反应严重度差异很大,与剂量也无关,从轻微的皮疹、发热至造系统抑制,肝肾功能损害、休克等。可能只有一种症状,也可能多种症状同时出现。停药后反应逐渐消失,再用时可能再发。致敏物质可能是药物本身,可能是其代谢物,也可能是药剂中杂质。临床用药前常做皮肤过敏试验,但仍有少数假阳性或假阴性反应。可见这是一类非常复杂的药物反应。
6.特异质反应(idiosyncrasy)少数特异体质病人对某些药物反应特别敏感,反应性质也可能与常人不同,但与药物固有药理作用基本一致,反应严重度与剂量成比例,药理拮抗药救治可能有效。这种反应是免疫反应,故不需预先敏化过程。现在知道这是一类药理遗传异常所致的反应,例如对骨骼肌松弛药司可林特异质反应是由于先天性血浆胆碱酯酶缺乏。
第二节 药物剂量与效应关系
药理效应与剂量在一定范围内成比例,这就是剂量-效应关系(dose-effect rela-tion-ship)。由于药理效应与血药浓度的关系较为密切,故在药理学研究中更常用浓度-效应关系(concentration-effect relationship)。用效应强弱为纵座标、药物浓度为横座标作图得直方双曲线(rectangular hyperbola)。如将药物浓度改用对数值作图则呈典型的对称S型曲线,这就是通常所讲的量效曲线(图2-1)。药理效应强弱有的是连续增减的量变,称为量反应(graded response),例如血压的升降、平滑肌舒缩等,用具体数量或最大反应的百分率表示。有些药理效应只能用全或无,阳性或阴性表示称为质反应(all-or-noneresponse或quantalresponse),如死亡与生存、抽搐与不抽搐等,必需用多个动物或多个实验标本以阳性率表示。用累加阳性率对数剂量(或浓度)作图也呈典型对称S型量效曲线(图2-2)。
从上述两种量效曲线可以看出下列几个特定位点:最小有效浓度(minimumeffective concentration),即刚能引起效应的阈浓度(thresholdconcentration)。如果横座标用剂量表示,将“浓度”改为“剂量”即可,下同。半数有效量(medianeffective dose)是能引起50%阳性反应(质反应)或50%最大效应(量反应)的浓度或剂量,分别用半数有效浓度(EC50)及半数有效剂量(ED50)表示。如果效应指标为中毒或死亡则可改用半数中毒浓度(TC50)、半数中毒剂量(TD50)或半数致死浓度(LC50)、半数致死剂量(LD50)表示。继续增加浓度或剂量而效应量不再继续上升时,这在量反应中称为最大效能(maximumefficacy),反映药物的内在活性。在质反应中阳性反应率达100%,再增加药量也不过如此。如果反应指标是死亡则此时的剂量称为最小致死量(minimumlethal dose)。药物效应强度(potency)是指能引起等效反应(一般采用50%效应量)的相对浓度或剂量,反映药物与受体的亲和力,其值越小则强度越大。药物的最大效能与效应强度含意完全不同,二者并不平行。例如利尿药以每日排钠量为效应指标进行比较氢氯噻嗪的效应强度大于呋塞米,而后者的最大效能大于前者(图2-3)。药物的最大效能值有较大实际意义,不区分最大效能与效应强度只讲某药较另药强若干倍是易被误解的。量效曲线中段斜率(slope)较陡的提示药效较激烈,较平坦的提示药效较温和。但在质反应曲线,斜率较陡的曲线还提示实验个体差异较小。曲线上的每个具体数据常用标准差(standarddeviation)表示个体差异(individualvariation)。
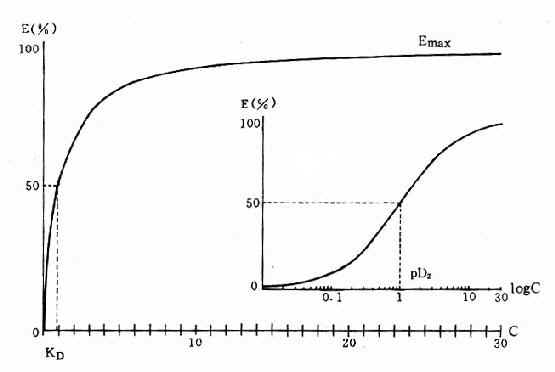
图2-1 药物作用的量效关系曲线
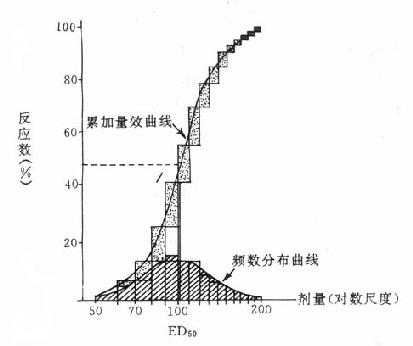
图2-2 质反应的频数分布曲线和累加量效曲线
频数分布曲线:100个人的有限剂量分布情况
(常态分布);累加量效曲线:频数分布曲线中每个长方形的累加曲线
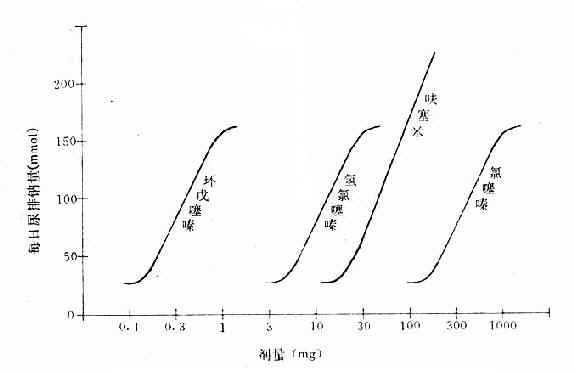
图2-3 各种利尿药的作用强度及最大效能比较
TD50/ED50或TC50/EC50的比值称为治疗指数(therapeutic index),是药物的安全性指标。治疗指数为4的药物相对较治疗指数为2的药物安全。由于TD与ED两条量曲线的首尾可能重叠,即ED95可能大于TD5,就是说在没能获得充分疗效的剂量时可能已有少数病人中毒,因此不能认为治疗指数为4的药物是安全的。还由于该指标所指的药物效应及毒性反应性质不明确,这一安全指标并不可靠。在动物实验常用LD50/ED50作为治疗指数,性质相似。较好的药物安全性指标是ED95~TD5之间的距离,称为安全范围(margin ofsafety),其值越大越安全。药物的安全性与药物剂量(或浓度)有关,因此如果将ED与TD两条量效曲线同时画出并加以比较则比较具体(图2-4)。
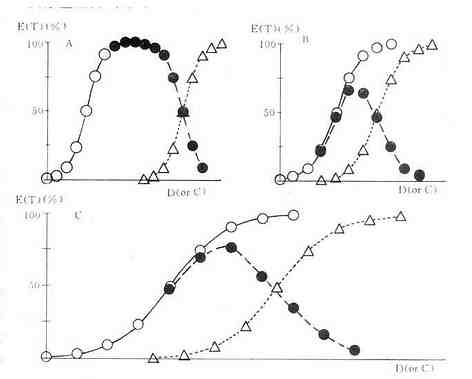
图2-4 药物的安全性指标:治疗指数及安全范围
A药的治疗指数比B药大 A药与C药的治疗指数相等,但A药的安全范围较大C药的治疗指数比B药大,而安全范围无区别
○有效量的量效关系 △中毒量的量效关系
●有效百分数减中毒百分数
关于药物剂量各国药典都制定了常用剂量范围,非药典药药厂在说明书上也有介绍。药典对于剧毒类药品还规定了极量(包括单剂量、一日量及疗程量),超限用药造成不良后果医生应负法律责任。
第三节 药物作用机制
药物效应多种多样,是不同药物分子与机体不同靶细胞间相互作用的结果。药物作用的性质首先取决于药物的化学结构,包括基本骨架、活性基团、侧链长短及立体构形等因素。这些构效关系(structure-activityrelationship)是药物化学研究的主要问题,但它有助于加强医生对药物作用的理解。药理效应是机体细胞原有功能水平的改变,从药理学角度来说,药物作用机制(mechanismof action)要从细胞功能方面去探索。
1.理化反应抗酸药中和胃酸以治疗溃疡病,甘露醇在肾小管内提升渗透压而利尿等是分别通过简单的化学反应及物理作用而产生的药理效应。
2.参与或干扰细胞代谢补充生命代谢物质以治疗相应缺乏症的药例很多,如铁盐补血、胰岛素治糖尿病等。有些药物化学结构与正常代谢物非常相似,掺入代谢过程却往往不能引起正常代谢的生理效果,实际上导致抑制或阻断代谢的后果,称为伪品掺入(counterfeitincorporation)也称抗代谢药(antimetabolite)。例如5-氟尿嘧啶结构与尿嘧啶相似,掺入癌细胞DNA及RNA中干扰蛋白合成而发挥抗癌作用。
3.影响生理物质转运很多无机离子、代谢物、神经递质、激素在体内主动转运需要载体参与。干扰这一环节可以产生明显药理效应。例如利尿药抑制肾小管Na+-K+、Na+-H+交换而发挥排钠利尿作用。
4.对酶的影响酶的品种很多,在体内分布极广,参与所有细胞生命活动,而且极易受各种因素的影响,是药物作用的一类主要对象。多数药物能抑制酶的活性,如新斯的明竞争性抑制胆碱酯酶,奥美拉唑不可逆性抑制胃粘膜H+-K+ATP酶(抑制胃酸分泌)。尿激酶激活血浆溶纤酶原,苯巴比妥诱导肝微粒体酶,解磷定能使遭受有机磷酸酯抑制的胆碱酯酶复活,而有些药本身就是酶,如胃蛋白酶。
5.作用于细胞膜的离子通道细胞膜上无机离子通道控制Na+、Ca2+、K+、Cl-等离子跨膜转运,药物可以直接对其作用,而影响细胞功能。
6.影响核酸代谢核酸(DNA及RNA)是控制蛋白质合成及细胞分裂的生命物质。许多抗癌药是通过干扰癌细胞DNA或RNA代谢过程而发挥疗效的。许多抗生素(包括喹诺酮类)也是作用于细菌核酸代谢而发挥抑菌或杀菌效应的,这将在有关章节详述。
7.影响免疫机制除免疫血清及疫苗外,免疫增强药(如左旋咪唑)及免疫抑制药(如环孢霉素)通过影响免疫机制发挥疗效。某些免疫成份也可直接入药。第50章将有专题介绍。
8.非特异性作用一些药
■[此处缺少一些内容]■
第四节 药物与受体
受体(receptor)是细胞在进化过程中形成的细胞蛋白组分,能识别周围环境中某种微量化学物质,首先与之结合,并通过中介的信息转导与放大系统,触发随后的生理反应或药理效应。自从Langley 提出受体学说100年后,受体已被证实为客观存在的实体,类型繁多,作用机制多已被阐明,现在受体已不再是一个空泛笼统的概念。受体分子在细胞中含量极微,1mg 组织一般只含10fmol左右。能与受体特异性结合的物质称为配体(ligand)。受体仅是一个“感觉器”,对相应配体有极高的识别能力。受体-配体是生命活动中的一种偶合,受体都有其内源性配体,如神经递质、激素、自身活性物(autocoid)等。能激活受体的配体称为激动药(agonist),能阻断其活性的配体称为拮抗药(antagonist)。根据受体与配体结合的高度特异性,受体被分为若干亚型,如肾上腺素受体又分为α1、α2、β1和β2等亚型,其分布及功能都有区别。受体与配体有高度亲和力,多数配体在1pmol~1nmol/L的浓度时即可引起细胞的药理效应。反应之所以如此灵敏主要是靠后续的信息转导系统,如细胞内第二信使(secondmessenger)的放大、分化及整合功能。酶、载体、离子通道及核酸也可与药物直接作用,但这些物质本身具有效应力,故严格地说不应被认为是受体。某些细胞蛋白组分可与配体结合,但没有触发效应的能力,称为结合体(acceptor)。
一、受体动力学
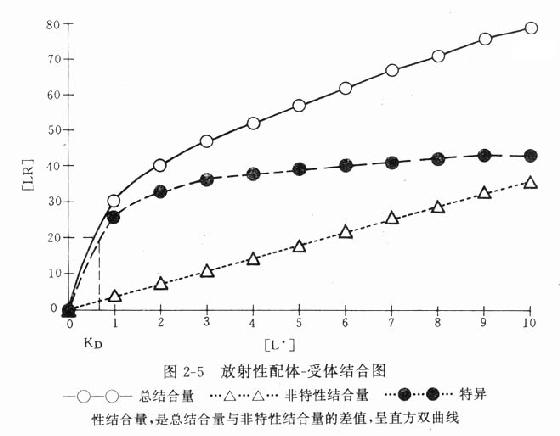
受体动力学一般用放射性同位素标记的配体(L)与受体(R)做结合试验研究。取一定量组织,磨成细胞匀浆,分组加入不同浓度的放射性同位素标记的配体(药物),温孵待反应达平衡后,迅速过滤或离心分出细胞,用缓冲液洗去尚未结合的放射性配体,测定标本的放射强度,这是药物与细胞结合的总量,此后用过量冷配体(未用同位素标记的配体)洗脱特异性与受体结合的放射性配体再测放射强度,这是药物非特性结合量。将总结合量减去非特性结合量就可以获得L-R结合(B)曲线。如果L只与单一R可逆性结合,以B为纵座标,[L]为横座标,L-R结合曲线为直方双曲线(图2-5)。如将横座标改用log[L]([]表示摩尔浓度)则呈典型的S形量效曲线。
按质量作用定律
![]()
(E代表效应)
反应达到平衡时
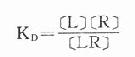
(KD是解离常数)
因为[RT]=[R]+[LR](RT为受体总量),代入上式并经推导得
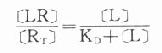
由于只有LR才发挥效应,故效应的相对强弱与LR相对结合量成比例,即
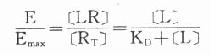
按此公式以E为纵座标,log[L]为横座标作图,结果与实验数据图形完全一致。
当[L]=0时,效应为0,
当[L]>>KD时,[LR]/[RT]=100%,达最大效能,即[LR]max=[RT]。
当[LR]/[RT]=50%时,即EC50时,KD=[L]。
KD表示L与R的亲和力(affinity),单位为摩尔。各药(L)与R亲和力不同,KD越大时亲和力越小,二者成反比。令pD2=-logKD则其值不必用摩尔单位、数值变小且与亲和力成正比,在半对数座标上也较易理解,故pD2较为常用。
药物与受体结合产生效应不仅要有亲和力,还要有内在活性(intrinsicactivity),后者用α表示,0≤α≤100%。故上述公式应加入这一参数:E/Emax=α[LR]/[RT]。两药亲和力相等时其效应强度取决于内在活性强弱,当内在活性相等时则取决于亲和力大小(图2-6)。
将上述受体动力学基本公式([LR]/[RT]=[L]/KD+[L])加以推导改变可将S形量效曲线改变为直线关系,使计算方便很多也准确很多:
1.双倒数图 将上述基本公式两侧取倒数后加以推导得1/[LR]=KD/[L][RT]+1/[RT]。以1/[LR]为纵座标、1/[L]为横座标作图得直线(图2-7),斜率为KD[RT],即KD/Emax,与纵座标交点为1/[RT],即1/Emax,与横座标交点为-1/KD。
2.Scatchard图 推导得公式[LR]/[L]=[RT]/KD-[LR]/KD以[LR]/[L],为纵座标,[LR]为横座标作图也呈直线(图2-8),斜率为-1/[KD],与纵座标交点为[RT]/KD,与横座标交点为[RT]。
这些直线关系图解在受体研究中有重要用途,也可加深对受体动力学的理解
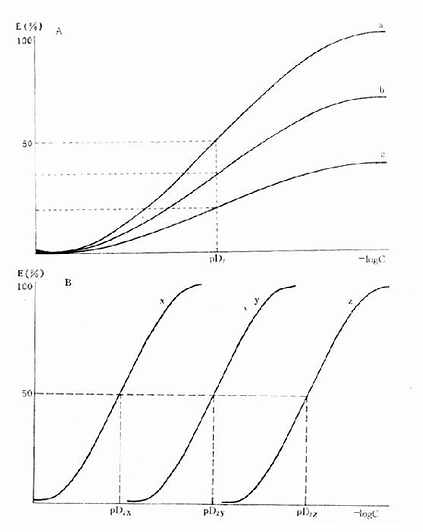
图2-6 药物与受体的亲和力及其内在活性对量效曲线的影响
A图 a,b,c三药与受体的亲和力(pD2)相等,但内在活性(Emax)不等
B图 a,b,c 三药与受体的亲和力(pD2)不等,但内在活性(Emax)相等
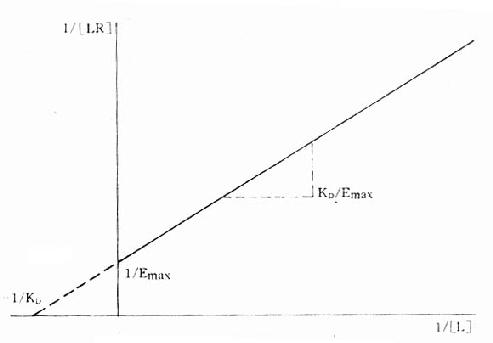
图2-7 受体结合量效关系的双倒数作图
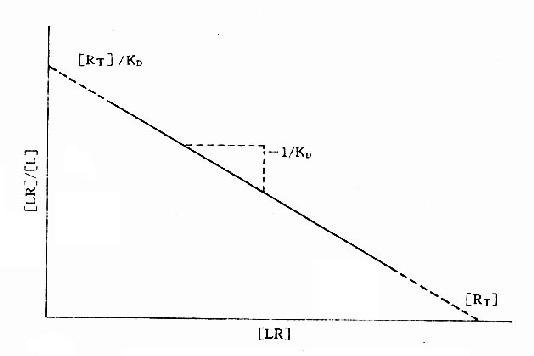
图2-8 受体结合量效关系的Scatchard作图
一些活性高的药物与相应受体结合的量效曲线 (B-log[L]曲线)并不一定与结合后产生效应的量效曲线(E-log[L]曲线)相重合。因为这类药物只需与一部分受体结合就能发挥最大效应(Emax),剩余下未结合的受体为储备受体(spare receptor)。这对理解拮抗药作用机制有重要意义,因为这类拮抗药必须在完全占领储备受体后才能发挥其拮抗效应。
受体激动药(L)对相应受体有较强的亲和力,也有较强的内在活性,α达100%。受体拮抗药(I)虽然也有较强的亲和力,但缺乏内在活性,α=0,本身不能引起效应,却占据一定量受体,拮抗激动药的作用。竞争性拮抗药(competitiveantagonist)能与激动药互相竞争与受体结合,这种结合是可逆性的。在实验中如果L与I同时存在则[RT]=[R]+[LR]+[IR],代入上述基本公式并加推导得
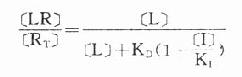
可见L和I同时存在时,如L这一因素固定不变,药理效应大小取决于[i]/K1(K1是I的解离常数)。[i]越高及(或)K1越小时效应越弱,即拮抗效果越强。当[L]>>[i]时,[LR]/[RT]→100%,这就是竞争性拮抗药使量效曲线平行右移(Emax不变)的理论解释(图2-9)。
在有一定量的竞争性拮抗药[i]存在时,增加[L]至[L’]仍可使药理效应维持在原来单用[L]时的水平。据此,
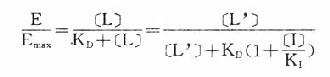
将之推导得
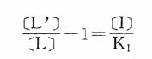
[L’]/[L]是剂量比 (dose ratio),即将[L]增加[L’]/[L]倍就能克服[i]的拮抗作用。该比值也取决于[i]/K1而与[L]绝对值或KD无关。将此公式两侧取log,并以log([L’]/[L]-1)为纵座标、以-log[i]为横座标作图,呈直线,斜率为1,与横座标交点为-logK1,即pA2此即Schild 图(图2-10)。按Schild定义,拮抗参数pAx是指剂量比为X时竞争性拮抗药浓度的负对数值。常用pA2,即[L’]/[L]=2时的数值,则pA2=-log[i]=-logK1,些参数反映拮抗药的拮抗强度,其值越大表示拮抗作用越强。
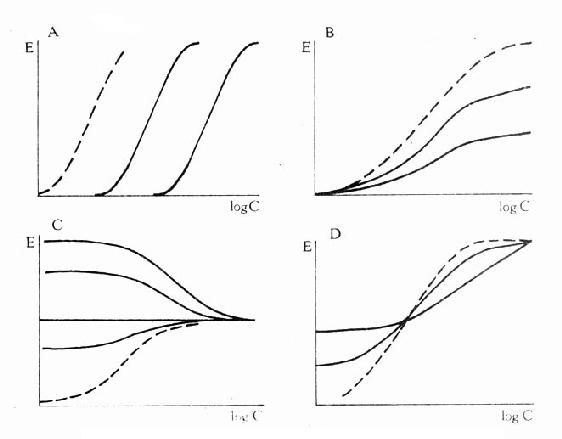
图2-9 竞争性拮抗药(A图)、非竞争性拮抗药(B图)及部分
激动药(D图)对激动药(虚线)量效的影响及激动药(C图)
对部分激动药(虚线)量效曲线的影响
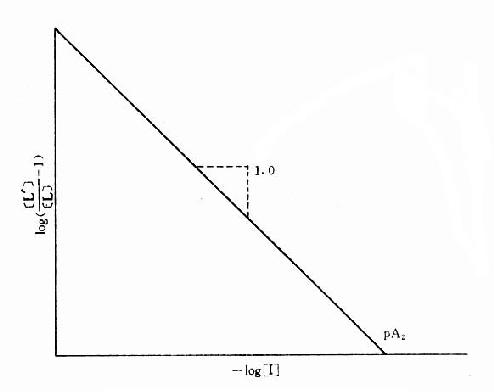
图2-10 竞争性拮抗作用的Schild作图
非竞争性拮抗药(noncompetitiveantagonist)与R结合非常牢固,分解很慢或是不可逆转,使能与L结合的R数量减少。另一类非竞争性拮抗药可阻断受体后某一中介反应环节而使受体-效应功能容量减少。二者共同特点是使量效曲线高度(Emax)下降。但L与剩余的R结合动力学不变,即KD不变。在双倒数图中更易看出这一关系(图2-11)。
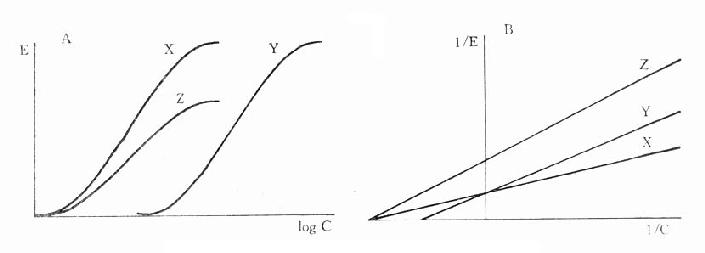
图2-11 竞争性拮抗作用与非竞争性拮抗作用比较
A图 量效曲线 B图 双倒数曲线
X 单用激动药 Y 竞争性拮抗药对激动药的拮抗作用
Z 非竞争性拮抗药对激动药的拮抗作用
还有一类药物称为部分激动药(partialagonist)和R结合的亲和力不小,但内在活性有限,α<100%,量效曲线高度(Emax)较低。与激动药同时存在时,当其浓度尚未达到Emax时,其效应与激动药协同,超过此限时则因与激动药竞争R而呈拮抗关系,此时激动药必需增大浓度方可达到其最大效能。可见部分激动药具有激动药与拮抗药两重特性。(图2-9C、D)
目前放射性配体-受体结合技术已普遍用于受体研究,但必需和药理效应实验结合进行才有意义。
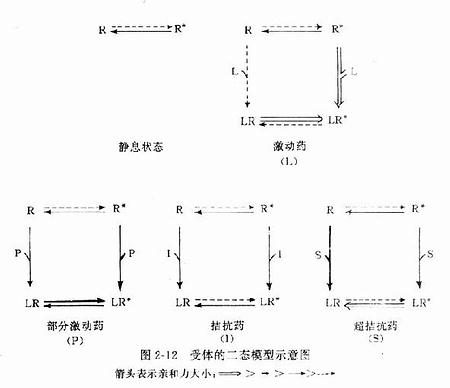
为什么化学结构类似的药物作用于同一受体有的是激动药,有的是拮抗药,还有的是部分拮抗药?还可用二态模型(two-statemodel) 学说解释。按此学说,受体蛋白有两种可以互变的构型状态:静息状态(R)与活动状态(R*)(图2-12)。静息时平衡趋向R。活动药只与R*有较大亲和力,L-R*结合后充分发挥药理效应。部分激动药(P)与R及R*都能结合但对R*的亲和力大于对R的亲和力,故只有部分受体被激活而发挥较小的药理效应。拮抗药对R及R*亲和力相等,且能牢固结合,但保持静息状态时两种受体状态平衡,拮抗药不能激活受体但能阻断激动药作用。个别药物(如苯二氮![]() 类)对R亲和力大于R*,结合后引起与激动药相反的效应,称为超拮抗药(superantagonist)。这一学说容易理解,但有待进一步实验证实。
类)对R亲和力大于R*,结合后引起与激动药相反的效应,称为超拮抗药(superantagonist)。这一学说容易理解,但有待进一步实验证实。
二、受体类型
根据受体蛋白结构、信息传导过程、效应性质、受体位置等特点,受体大致可分为下列4类:
1. 含离子通道的受体又称直接配体门控通道型受体,它们存在于快速反应细胞的膜上,由单一肽链反复4次穿透细胞膜形成1个亚单位,并由4~5个亚单位组成穿透细胞膜的离子通道,受体激动时离子通道开放使细胞膜去极化或超极化,引起兴奋或抑制效应。最早发现的N型乙酰胆碱受体就是由α×2、β、γ、δ5个亚单位组成的钠离子通道,在α亚单位上各有一个乙酰胆碱结合点(图2-13A)与乙酰胆碱结合后,钠离子通道开放,胞外钠离子内流、细胞膜去极化、肌肉收缩。这一过程在若干毫秒内完成(钠离子通道开放时间仅1ms)。脑中γ氨基丁酸(GABA)受体情况类似,其他如甘氨酸、谷氨酸、天门冬氨酸受体都属于这一类型。
2.G-蛋白偶联受体这一类受体最多,数十种神经递质及激素的受体需要G-蛋白介导其细胞作用,例如肾上腺素、多巴胺、5-羟色胺、M-乙酰胆碱、阿片类、嘌呤类、前列腺素及一些多肽激素等的受体,这些受体结构非常相似,都为单一肽链形成7个α-螺旋来回穿透细胞膜,N-端在细胞外,C-端在细胞内,这两段肽链氨基酸组成在各种受体差异很大,与其识别配体及转导信息各不相同有关。胞内部分有G-蛋白结合区(图2-13B)。G-蛋白(G-protein)是鸟苷酸结合调节蛋白的简称,存在于细胞膜内侧,由三个亚单位组成。主要有两类,其一为兴奋性G-蛋白(GS),霍乱弧菌毒素能使之活化,激活腺苷酸环化酶(AC);另一为抑制性G-蛋白(Gi),抑制AC,百日咳杆菌素抑制之。G-蛋白还介导心钠素及NO对鸟苷酸环化酶(GC)的激活作用。此外G-蛋白对磷脂酶C、磷脂酶A2、Ca2+、K+离子通道等有重要调节作用。一个受体可激活多个G-蛋白,一个G-蛋白可以转导多个信息给效应机制,调节许多细胞功能。
3.具有酪氨酸激酶活性的受体这一类细胞膜上的受体由三个部分组成(图2-13C),细胞外有一段与配体结合区,中段穿透细胞膜,胞内区段有酪氨酸激酶活性,能促其本身酪氨酸残基的自我磷酸化而增强此酶活性,再对细胞内其他底物作用,促进其酪氨酸磷酸化,激活胞内蛋白激酶,增加DNA及RNA合成,加速蛋白合成,从而产生细胞生长分化等效应。胰岛素、胰岛素样生长因子、上皮生长因子、血小板生长因子及某些淋巴因子(lymphokines)的受体属于这一类型。
4.细胞内受体甾体激素受体存在于细胞浆内,与相应甾体结合后分出一个磷酸化蛋白,暴露与DNA结合区段,进入细胞核能识别特异DNA碱基区段并与之结合促进其转录及以后的某种活性蛋白增生(图2-13D)。甲状腺素受体存在于细胞核内,功能大致相同。这两种受体触发的细胞效应很慢。需若干小时。
A.直接配体门控通道型
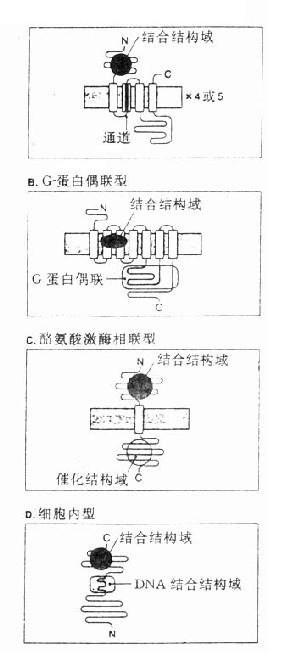
图2-13 受体类型示意图
三、第二信使
受体在识别相应配体并与之结合后需要细胞内第二信使(secondmessenger) 将获得信息增强、分化、整合并传递给效应机制才能发挥其特定的生理功能或药理效应。最早发现的第二信使是环磷腺苷(cAMP),现在知道还有许多其他物质参与细胞内信息转导。这是一个非常复杂的系统,简示如下(图2-14),很多问题尚有待进一步阐明。
1.G-蛋白G蛋白是一类存在于细胞膜内侧的调节蛋白,都是由三个不同亚单位α、β、γ组成的三聚体。静息状态时与GDP结合。相应受体激活后GDP-α、β、γ复合物在Mg2+参与下,结合的GDP与胞浆中GTP交换,GTP-α与β、γ分离并与相应的效应机制结合,同时配体与受体分离。α亚单位内在的GTP酶活性促使GTP水解为GDP,激活效应机制,从而恢复原来静息状态(图2-15)。GS激活腺苷酸环化酶(AC),使cAMP增加。Gi抑制AC,使cAMP减少,G-蛋白还激活磷脂酶C(PLC),调节Ca2+、K+等离子通道。对鸟苷酸环化酶也有激活作用,作用非常广泛,介导多种效应。近来发现G-蛋白还介导激活磷脂酶A2(PLA2)而产生花生四烯酸(AA),后者是各种前列腺素及白三烯的前体。
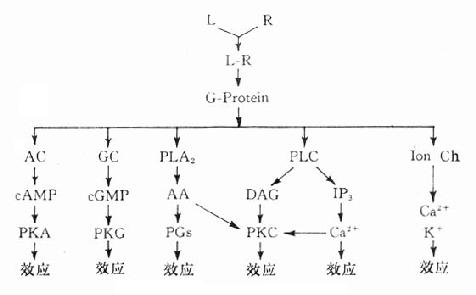
图2-14 第二信使系统示意图
2. 环磷腺苷(cAMP)cAMP是ATP经AC作用的产物。β受体、D1受体、H2受体等激动药通过GS作用使AC活化,ATP水解而使细胞内cAMP增加。α受体、D2受体、MACh受体、阿片受体等激动药通过Gi作用抑制AC,细胞内cAMP减少。cAMP受磷酸二酯酶(phosphodiesterase,PDE)水解为5’AMP后灭活。茶碱抑制PDE而使胞内cAMP增多。cAMP能激活蛋白激酶a (PKA)而使胞内许多蛋白酶磷酸化(ATP提供磷酸基)而活化,例如磷酸化酶、脂酶、糖原合成酶等活化而产生能量。钙离子通道磷酸化后激活,钙离子内流而使神经、心肌、平滑肌等兴奋。
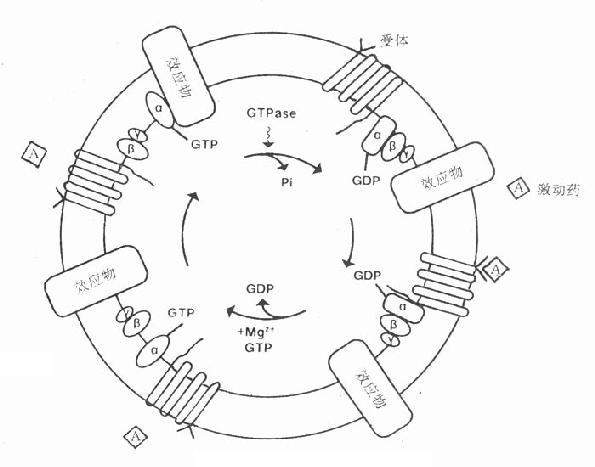
图2-15 G-蛋白作用示意图
3.环磷鸟苷(cGMP)cGMP是GTP经鸟苷酸环化酶(GC)作用的产物,也受PDE灭活。cGMP作用与cAMP相反,使心脏抑制、血管舒张、肠腺分泌等。CGMP可以独立作用而不受cGMP制约。cGMP可激活蛋白酶G而引起各种效应。
4.肌醇磷脂(phosphatidylinositol)细胞膜肌醇磷脂的水解是另一类重要的受体信息转导系统。α、H1、5-HT2、M1、M3等受体激动药与其受体结合后通过G-蛋白介导激活磷脂酶C(PLC)PLC使4,5-二磷酸肌醇磷脂(PIP2)水解为二酰甘油(DAG)及1,4,5-三磷酸肌醇(IP3)。DAG在细胞膜上激活蛋白激酶C(PKC),使许多靶蛋白磷酸化而产生效应,如腺体分泌,血小板聚集,中性粒细胞活化及细胞生长、代谢、分化等效应。IP3能促进细胞内钙池释放Ca2+,也有重要的生理意义。
5.钙离子细胞内Ca2+浓度在1μmol/l以下,不到血浆Ca2+的0.1%,对细胞功能有着重要的调节作用,如肌肉收缩、腺体分泌、白细胞及血小板活化等。细胞内Ca2+可从细胞外经细胞膜上的钙离子通道流入,也可从细胞内肌浆网等钙池释放,两种途径互相促进。前者受膜电位、受体、G-蛋白,蛋白激酶A(PKA)等调控,后者受IP3作用而释放。细胞内Ca2+激活蛋白激酶C(PKC),与DAG有协同作用,共同促进其他信息传递蛋白及效应蛋白活化。很多药物通过对细胞内Ca2+影响而发挥其药理效应,故对细胞内Ca2+调控及其作用机制近年来受到极大的重视。
四、受体的调节
受体虽是遗传获得的固有蛋白,但并不是固定不变的,而经常代谢转换处于动态平衡状态,其数量,亲和力及效应力经常受到各种生理及药理因素的影响。连续用药后药效递减是常见的现象,一般称为耐受性(tolerance)、不应性(refractoriness)、快速耐受性(tachyphylaxis)等。由于受体原因而产生的耐受性称为受体脱敏(receptordesensitization)。N2-ACh受体在受激动药连续作用后若干秒内发生脱敏现象,这是由于受体蛋白构象改变,钠离子通道不再开放所致。β-Adr受体脱敏时不能激活AC是因为受体与G-蛋白亲和力降低,或由于cAMP上升后引起PDE负反馈增加所致。具有酪氨酸激酶活性的受体可被细胞内吞(endocytosis)而数目减少,这一现象称为受体数目的向下调节(down regulation)。受体与不可逆拮抗药结合后其后果等于失去一部分受体,如银环蛇咬伤中毒时,N2-ACh受对激动药脱敏。与此相反,在连续应用拮抗药后受体会向上调节(up regulation),反应敏化。例如长期应用β-Adr受体拮抗药后,由于受体向上调节,突然停药时会出现反跳反应。
第三章 药物代谢动力学
药物代谢动力学,简称为药动学,研究药物体内过程及体内药物浓度随时间变化的规律。药物在体内虽然不一定集中分布于靶器官,但在分布达到平衡后药理效应强弱与药物血浆浓度成比例。医生可以利用药动学规律科学地计算药物剂量以达到所需的血药浓度并掌握药效的强弱久暂。这样可以比单凭经验处方取得较好的临床疗效。
第一节 药物体内过程
一、吸收
药物的吸收(absorption)是指药物自体外或给药部位经过细胞组成的屏蔽膜进入血液循环的过程。多数药物按简单扩散(simplediffusion)物理机制进入体内。扩散速度除取决于膜的性质,面积及膜两侧的浓度梯度外,还与药物的性质有关。分子量小的(200D以下),脂溶性大的(油水分布系数大的),极性小的(不易离子化的)药物较易通过。药物多是弱酸性或弱碱性有机化合物,其离子化程度受其pKa(酸性药物解离常数的负对数值)及其所在溶液的pH而定,这是影响药物跨膜被动转运,吸收分布排泄的一个可变因素。按Handerson-Hasselbalch公式:

由此可见不论弱酸性或弱碱性药物的pKa都是该药在溶液中50%离子化时的pH值,各药有其固定的pKa值。当Pka与pH的差值以数学值增减时,药物的离子型与非离子型浓度比值以指数值相应变化。非离子型药物可以自由穿透,而离子型药物就被限制在膜的一侧,这种现象称为离子障(iontrapping)。例如弱酸性药物在胃液中非离子型多,在胃中即可被吸收。弱碱性药物在酸性胃液中离子型多,主要在小肠吸收。碱性较强的药物如胍乙啶(pKa=11.4)及酸性较强的药物如色甘酸钠(pKa=2.0)在胃肠道基本都已离子化,由于离子障原因,吸收均较难。pKa小于4的弱碱性药物如安定(pKa=3.3)及pKa大于7.5的弱酸性药物如异戊巴比妥(pKa=7.9)在胃肠道pH范围内基本都是非离子型,吸收都快而完全。
少数与正常代谢物相似的药物,如5-氟尿嘧啶、甲基多巴等的吸收是靠细胞中的载体主动转运(activetransport)而吸收的,这一主动转运机制对药物在体内分布及肾排泄关系比较密切。易化扩散(facilitateddiffusion)是靠载体顺浓度梯度跨膜转运方式,如葡萄糖的吸收,吸收速度较快。固体药物不能吸收,片剂、胶囊剂在胃肠道必须先崩解(disintegration)、溶解(dissolution)后才可能被吸收。
1.胃肠道给药口服(per os)给药是最常用的给药途径。小肠内pH接近中性,粘膜吸收面广,缓慢蠕动增加药物与粘膜接触机会,是主要吸收部位。药物吸收后通过门静脉进入肝脏。有些药物首次通过肝脏就发生转化,减少
■[此处缺少一些内容]■
达200m2),与血液只隔肺泡上皮及毛细管内皮各一层,而且血流量大,药物只要能到达肺泡,吸收极其迅速,气体及挥发性药物(如全身麻醉药)可直接进入肺泡。药物溶液需要经喷雾器分散为微粒,气雾剂(aerosol)可将药液雾化为直径达5μm左右微粒,可以达到肺泡而迅速吸收,如在雾化器及口鼻罩间加用一个气室则效果更好。2~5μm直径以下的微粒可重被呼出,10μm直径微粒可在小支气管沉积。后者可用于异丙肾上腺素治疗支气管哮喘。较大雾粒的喷雾剂(nebula)只能用于鼻咽部的局部治疗,如抗菌、消炎、祛痰、通鼻塞等。
4.经皮(transdermal)给药除汗腺外,皮肤不透水,但脂溶性药物可以缓慢通透。许多杀虫药可以经皮吸收中毒。利用这一原理可以经皮给药以达到局部或全身药效,近年来有许多促皮吸收剂加氮酮(azone),可与药物制成贴皮剂,如硝苯地平贴皮剂以达到持久的全身疗效,对于容易经皮吸收的硝酸甘油也可制成缓释贴皮剂预防心绞痛发作,每日只贴一次。
二、分布
药物进入循环后首先与血浆蛋白结合(plasmaprotein binding)。酸性药物多与清蛋白结合,碱性药物多与α1酸性糖蛋白结合,还有少数药物与球蛋白结合。这种结合和药物与受体蛋白结合情况相似:
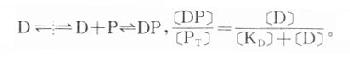
可见药物的血浆蛋白结合量([DP])受药物浓度([D]),血浆蛋白(P)的质和量及解离常数(KD)影响,各药不同而且结合率(血中与蛋白结合的药物与总药量的比值)随剂量增大而减少。药理学书籍收载药物的血浆蛋白结合率是在常用剂量范围内对正常人测定的数值。药物与血浆蛋白的结合是可逆性的,结合后药理活性暂时消失,结合物分子变大不能通过毛细管壁暂时“储存”于血液中。上述反应式中纵向虚线代表毛细管壁,在吸收过程中游离药物穿透毛细管壁进血液后与血浆蛋白结合,反应平衡向右移,有利于吸收。在消除过程中(如肝摄取及肾小管分泌),血中游离药物被除去,反应平衡左移,有利于消除。药物与血浆蛋白结合特异性低,而血浆蛋白结合点有限,两个药物可能竞争与同一蛋白结合而发生置换现象。如某药结合率达99%,当被另药置换而下降1%时,则游离型(具有药理活性)药物浓度在理论上将增加100%,可能导致中毒。但一般药物在被置换过程中,游离型药物会加速被消除,血浆中游离型药物浓度难以持续增高。药物也可能与内源性代谢物竞争与血浆蛋白结合,例如磺胺药置换胆红素与血浆蛋白结合,在新生儿可能导致核黄疸症。血浆蛋白过少(如肝硬化)或变质(如尿毒症)时药物血浆蛋白结合率下降,也容易发生毒性反应。
吸收的药物通过循环迅速向全身组织输送,首先向血流量大的器官分布(distribution),然后向血流量小的组织转移,这种现象称为再分布(redistribution),如硫喷妥先在血流量大的脑中发挥麻醉效应,然后向脂肪等组织转移,效应很快消失。经过一段时间后血药浓度趋向“稳定”,分布达到“平衡”,但各组织中药物并不均等,血浆药物浓度与组织内浓度也不相等。这是由于药物与组织蛋白亲和力不同所致。因此这种“平衡”称为假平衡(pseudoequilibrium),这时血浆药物浓度高低可以反映靶器官药物结合量多少。药物在靶器官浓度决定药物效应强弱,故测定血浆药物浓度可以估算药物效应强度。某些药物可以分布至脂肪、骨质等无生理活性组织形成储库,或结合于毛发指(趾)甲组织。药物的pKa及体液pH是决定药物分布的另一因素,细胞内液pH(约为7.0)略低于细胞外液(约7.4),弱碱性药物在细胞内浓度略高,弱酸性药物在细胞外液浓度略高,根据这一原理,弱酸性药物苯巴比妥中毒时用碳酸氢钠碱化血液及尿液可使脑细胞中药物向血浆转移并加速自尿排泄,是重要救治措施之一。
血脑屏障(blood-brainbarrier)脑是血流量较大的器官,但药物在脑组织浓度一般较低,这是由于血脑屏障所致。在组织学上血脑屏障是由血-脑、血-脑脊液及脑脊液-脑三种屏障的总称,实际上能阻碍药物穿透的主要是前二者。脑毛细血管内皮细胞间紧密联接,基底膜外还有一层星状细胞包围,药物较难穿透。脑脊液不含蛋白质,即使少量未与血浆蛋白结合的脂溶性药物可以穿透进入脑脊液,其后药物进入静脉的速度较快,故脑脊液中药物浓度总是低于血浆浓度,这是大脑自我保护机制。治疗脑病可以选用极性低的脂溶性药物,例如磺胺药中的磺胺嘧啶。为了减少中枢神经不良反应,对于生物碱可将之季铵化以增加其极性,例如将阿托品季铵化变为甲基阿托品后不能通过血脑屏障,即不致发生中枢兴奋反应。
胎盘屏障(placentabarrier)是胎盘绒毛与子宫血窦间的屏障,由于母亲与胎儿间交换营养成分与代谢废物的需要,其通透性与一般毛细管无显著差别,只是到达胎盘的母体血流量少,进入胎儿循环慢一些罢了。例如母亲注射磺胺嘧啶2小时后才能与胎儿达到平衡。利用这一原理可以在预期胎儿娩出前短时内注射镇静镇痛药,新生儿不致遭受影响。应该注意的是几乎所有药物都能穿透胎盘屏障进入胚胎循环,在妊娠期间应禁用对胎儿发育有影响的药物。
三、生物转化
药物,作为外来活性物质(xenobiotic),机体首先要将之灭活,同时还要促其自体内消除。能大量吸收进入体内的药物多是极性低的脂溶性药物,在排泄过程中易被再吸收,不易消除。体内药物主要在肝脏生物转化(biotransformation)而失去药理活性,并转化为极性高的水溶性代谢物而利于排出体外。生物转化与排泄统称为消除(elimination)。
生物转化分两步进行,第一步为氧化、还原或水解,第二步为结合。第一步反应使多数药物灭活,但少数例外反而活化,故生物转化不能称为解毒过程。第二步与体内物质结合后总是使药物活性降低或灭活并使极性增加。各药在体内转化过程不同,有的只经一步转化,有的完全不变自肾排出,有的经多步转化生成多个代谢产物。
肝脏微粒体的细胞色素P-450酶系统是促进药物生物转化的主要酶系统,故又简称肝药酶,现已分离出70余种。此酶系统的基本作用是从辅酶Ⅱ及细胞色素b5获得两个H+,另外接受一个氧分子,其中一个氧原子使药物羟化,另一个氧原子与两个H+结合成水(RH+NADPH+O2+2H+→ROH+NADP++H2O),没有相应的还原产物,故又名单加氧酶,能对数百种药物起反应(图3-1)。此酶系统活性有限,在药物间容易发生竞争性抑制。它又不稳定,个体差异大,且易受药物的诱导或抑制。例如苯巴比妥能促进光面肌浆网增生,其中P-450酶系统活性增加,加速药物生物转化,这是其自身耐受性及与其他药物交叉耐受性的原因。西米替丁抑制P-450酶系统活性,可使其他药物效应敏化。该酶系统在缺氧条件下可对偶氮及芳香硝基化合物产生还原反应,生成胺基(图3-2)。微粒体内还存在水解酶及葡萄糖醛酸转移酶。
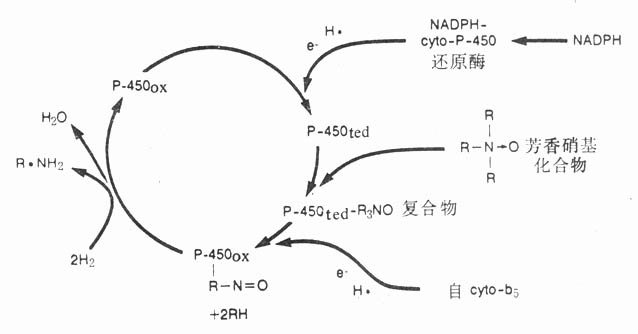
图3-1细胞色素P-450酶系统对药物氧化过程示意图
图3-2 细胞色素P-450酶系统对药物还原过程示意图
生物转化的第二步反应是结合。多数经过氧化反应的药物再经肝微粒体的葡萄糖醛酸转移酶作用与葡萄糖醛酸结合。有些药物还能和乙酰基、甘氨酸、硫酸等结合。这些结合反应都需要供体参加,例如二磷酸尿嘧啶是葡萄糖醛酸的供体。药物在体内转化过程,举例说明见表3-1。
表3-1药物生物转化类型举例
| 转化类型 | 转化反应通式 | 酶系 | 药物举例 |
| 1.氧化 脂肪族羟化 芳香族羟化 N去烷基 O去烷基 硫氧化 去硫 去卤 环氧化 醇类氧化 醛类氧化 胺类氧化 嘌呤氧化 2.还原 硝基还原 偶氮还原 醛类还原 酮类还原 3.水解 酰胺键水解 酯键水解 4.结合 葡萄糖醛酸 结合 乙酰化 |
R→ROH Ar→ArOH CH3 | R1―N―R2→R1―NH―R2 R―O―CH3→ROH O ‖ R1―S―R2→R1―S―R2 S O ‖ ‖ R1―P―R2→R1―P―R2 X OH | | R1―CH―R2→R1―CH―R2+HX O / / R1―CH=CH―R2→R1―CH―CH―R2 R―CH2OH→RCHO RCHO→RCOOH RCH2NH2→RCHO+NH2 Ar(N)→Ar(O) ArNO2→ArNH2 Ar1―N=N―Ar2→Ar1NH2+Ar2NH2 RCHO→RCH2OH O OH ‖ | R1―C―R2→R1―CH―R2 R1―CONH―R2→R1COOH+R2NH2 R1COOR2→R1COOH+R2OH 载体:UDP-葡萄糖醛酸 载体:乙酰辅酶A |
微粒体酶 微粒体酶 微粒体酶 微粒体酶 微粒体酶 微粒体酶 微粒体酶 微粒体酶 非微粒体酶 非微粒体酶 非微粒体酶 非微粒体酶 微粒体酶 微粒体酶 非微粒体酶 非微粒体酶 微粒体酶 非微粒体酶 微粒体酶 非微粒体酶 |
司可巴比妥 苯妥英 地西泮 可待因 氯丙嗪 对硫磷 氟烷 苯并芘(致癌物) 乙醇 乙醛 肾上腺素,组胺 茶碱 氯硝西泮 百浪多息 水合氯醛 纳洛酮 利多卡因,普鲁卡因胺 乙酰胆碱,普鲁卡因 氯霉素,吗啡 异烟肼 |
四、排 泄
药物在体内最后的过程是排泄(excretion),肾脏是主要排泄器官。游离的药物能通过肾小球过滤进入肾小管。随着原尿水分的回收,药物浓度上升。当超过血浆浓度时,那些极性低、脂溶性大的药物反向血浆扩散(再吸收),排泄较少也较慢。只有那些经过生物转化的极性高、水溶性代谢物不被再吸收而顺利排出。有些药物在近曲小管由载体主动转运入肾小管,排泄较快。在该处有两个主动分泌通道,一是弱酸类通道,另一是弱碱类通道,分别由两类载体转运,同类药物间可能有竞争性抑制。例如丙磺舒抑制青霉素主动分泌,使后者排泄减慢,药效延长并增强。碱化尿液使酸性药物在尿中离子化,酸化尿液使碱性药物在尿中离子化,利用离子障原理阻止药物再吸收,加速其排泄,这是药物中毒常用的解毒方法(图3-3)。
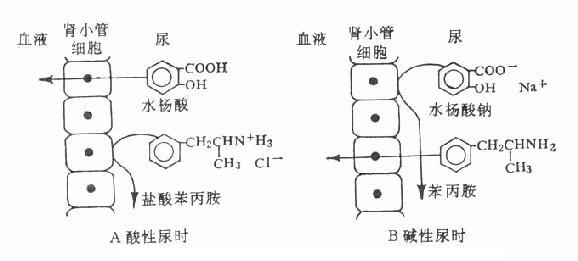
图3-3尿液酿碱度对弱酸性(水杨酸)及弱碱性
(苯丙胺)药物在肾小管内再吸收的影响
药物可自胆汁排泄,原理与肾排泄相似,但不是药物排泄的主要途径。药物自胆排泄有酸性、碱性及中性三个主动排泄通道。有些药物在肝细胞与葡萄糖醛酸等结合后排入胆中,随胆汁到达小肠后被水解,游离药物被重吸收,称为肝肠循环(hepato-enteralcirculation)。在胆道引流病人,药物的血浆半衰期将显著缩短,如氯霉素、洋地黄等。乳汁pH略低于血浆,碱性药物可以自乳汁排泄,哺乳婴儿可能受累。胃液酸度更高,某些生物碱(如吗啡等)注射给药也可向胃液扩散,洗胃是中毒治疗和诊断的措施。药物也可自唾液及汗液排泄。粪中药物多数是口服未被吸收的药物。
肺脏是某些挥发性药物的主要排泄途径,检测呼出气中的乙醇量是诊断酒后驾车的快速简便的方法。
第二节 体内药量变化的时间过程
体内药量随时间而变化的过程是药动学研究的中心问题。药量与效应的关系(量效关系)已在药效学章详述。加入时间因素就引出时量关系(time-concentrationrelationship)与时效关系(time-responserelationship)。大多数情况下由于量效关系基本固定,在达到“平衡”后两条曲线平行一致。整体动物一次血管外给药的时量(效)曲线见图3-4。按一室模型理解,曲线升段主要是吸收过程(此时消除过程已经开始)。曲线在峰值浓度(peakconcentration, Cmax)时吸收速度与消除速度相等。从给药时至峰值浓度的时间称为达峰时间(peak time, Tpeak),曲线降段主要是药物消除过程。血药浓度下降一半的时间称为消除半衰期(elimination half-lifetime)。血药浓度超过有效浓度(低于中毒浓度)的时间称为有效期(effective peroid)。曲线下面积(area under the curve, AUC)与吸收入体循环的药量成比例,反映进入体循环药物的相对量。AUC是血药浓度(C)随时间(t)变化的积分值:
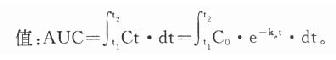
当t1为0,t2为∞时,AUC=Co/Ke,单位是g.h.L-1。
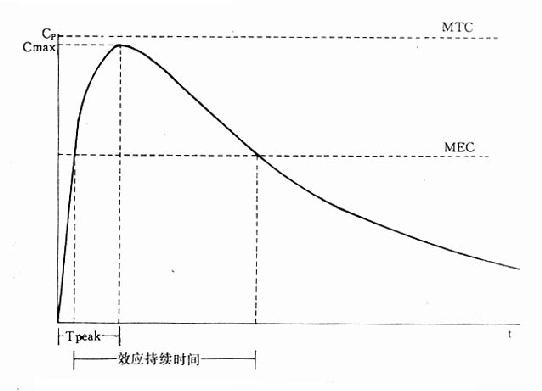
图3-4 ,典型时量曲线图
MTC最小中毒浓度 MEC最小有效浓度
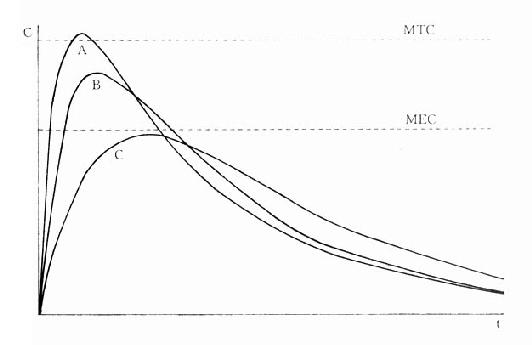
图3-5 某药剂量相等的三种制剂的生物利用度比较
F(AUC)相等,但Tpeak及 Cmax不等
生物利用度(bioavailability)是指经过肝脏首关消除过程后能被吸收进入体循环的药

剂口服后测得的量效曲线,其AUC相等(表示F值相等),但Tpeak及Cmax不等,吸收快的Cmax可能已超过最低中毒浓度,吸收慢的Cmax可能还在有效浓度以下。生物利用度是药物制剂质量的一个重要指标。
第三节 药物消除动力学
从生理学看,体液被分为血浆、细胞间液及细胞内液几个部分。为了说明药动学基本概念及规律现假定机体为一个整体,体液存在于单一空间,药物分布瞬时达到平衡(一室模型)。问题虽然被简单化,但所得理论公式不失为临床应用提供了基本规律。按此假设条件,药物在体内随时间变化可用下列基本通式表达:dC/dt=kCn。C为血药浓度,常用血浆药物浓度。k为常数,t为时间。由于C为单位血浆容积中的药量(A),故C也可用A代替:dA/dt=kCn,式中n=0时为零级动力学(zero-order kinetics),n=1时为一级动力学(first-order kinetics),药物吸收时C(或A)为正值,消除时C(或A)为负值。在临床应用中药物消除动力学公式比较常用,故以此为例如以推导和说明。
一、零级消除动力学
当n=0时,-dC/dt=KC=K(为了和一级动力学中消除速率常数区别,用K代k),将上式积分得:
Ct=C- Kt,C为初始血药浓度,Ct为t时的血药浓度,以C为纵座标、t为横座标作图呈直线(图3-6),斜率为K,当Ct/C=1/2时,即体内血浆浓度下降一半(或体内药量减少一半)时,t为药物消除半衰期(half-life time, t1/2)。
按公式1/2C=C-Kt1/2
![]()
可见按零级动力学消除的药物血浆半衰期随C下降而缩短,不是固定数值。零级动力学公式与酶学中的Michaelis-Menten公式相似: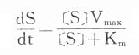 ,式中S为酶的底物,Vmax为最大催化速度,Km为米氏常数。当[S]>>Km时,Km可略去不计,ds/dt=Vmax,即酶以其最大速度催化。零级动力学公式与此一致,说明当体内药物过多时,机体只能以最大能力将体内药物消除。消除速度与C高低无关,因此是恒速消除。例如饮酒过量时,一般常人只能以每小时10ml乙醇恒速消除。当血药浓度下降至最大消除能力以下时,则按一级动力学消除。
,式中S为酶的底物,Vmax为最大催化速度,Km为米氏常数。当[S]>>Km时,Km可略去不计,ds/dt=Vmax,即酶以其最大速度催化。零级动力学公式与此一致,说明当体内药物过多时,机体只能以最大能力将体内药物消除。消除速度与C高低无关,因此是恒速消除。例如饮酒过量时,一般常人只能以每小时10ml乙醇恒速消除。当血药浓度下降至最大消除能力以下时,则按一级动力学消除。
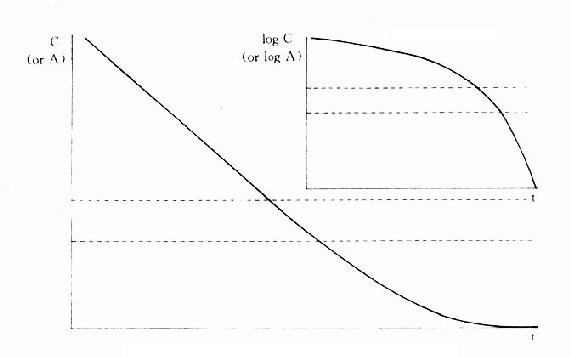
图3-6 药物在体内消除过程的时量曲线
体内药物过多,超过机体最大消除能力(虚线)时为零级动力学恒速消除
体内药物降至虚线以下时为一级动力学恒比消除。插图纵坐标为对数标尺
二、一级消除动力学
当n=1时,-dC/dt=keC1=keC,式中k用ke表示消除速率常数 (elimination rate constant)。将上式积分得

可见按一级动力学消除的药物半衰期与C高低无关,是恒定值。体内药物按瞬时血药浓度(或体内药量)以恒定的百分比消除,单位时间内实际消除的药量随时间递减。消除速率常数(ke)的单位是h-1,它不表示单位时间内消除的实际药量,而是体内药物瞬时消除的百分率。例如ke=0.5h-1不是说每小时消除50%(如果t1/2=1小时则表示每小时消除50%)。按t1/2=0.693/ke计算t1/2=1.39h,即需1.39h后才消除50%。再按 计算,1小时后体内尚存60.7%。绝大多数药物都按一级动力学消除。这些药物在体内经过t时后尚存

当n=5时,At≈3%A,即经过5个t1/2后体内药物已基本消除干净。与此相似,如果每隔一个t1/2给药一次(A),则体内药量(或血药浓度)逐渐累积,经过5个t1/2后,消除速度与给药速度相等,达到稳态(steady state):
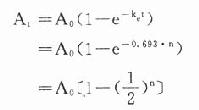
当n=5时,At≈97%A。这一时间,即5个t1/2不因给药剂量多少而改变。具体数值见表3-2。
药物自体内消除的一个重要指标是血浆清除率(plasmaclearance,Cl),是肝肾等的药物消除率的总和,即单位时间内多少容积血浆中的药物被消除干净,单位用L·h-1(也有人用ml·min-1,和肌酐消除率一致)或按体重计算 L·kg-1·h-1。按定义,CL=RE/Cp,RE是消除速率(rate of elimination),即单位时间内被机体消除的药量,Cp为当时的血浆药物浓度。由于RE非固定值也不易检测,故常用表观分布容积(apparent volume ofdistribution, Vd)计算。 Vd是指静脉注射一定量(A)药物待分布平衡后,按测得的血浆浓度计算该药应占有的血浆容积。事实上静注药物后未待分布平衡已有部分药物自尿排泄及(或)在肝转化而消除,故必需多次检测Cp,作时量曲线图,将稳定下降的消除段向O时延升至和Y轴交点以求得理论上静注药量A在体内分布平衡时的血浆浓度C,以此算出Vd=A/C(图3-7)。按RE=keA,Cp=A/Vd,故Cl=keVd。在一级动力学的药物中,Vd及Cl是两个独立的药动学指标,各有其固定的数值,互不影响,也不因剂量大小而改变其数值。Vd是表观数值,不是实际的体液间隔大小。除少数不能透出血管的大分子药物外,多数药物的Vd值均大于血浆容积。与组织亲和力大的脂溶性药物其Vd可能比实际体重的容积还大。Cl也不是药物的实际排泄量。它反映肝和(或)肾功能,在肝和(肾)功能不足时Cl值会下降,因为Cl是肝肾等消除能力的总和。肝清除率虽然难测,但有重要的理论意义。肝清除率小的药物,首关消除少,其口服生物利用度大,但易受肝功能,血浆蛋白结合力及肝药酶诱导或抑制药的影响。肝清除率大的药物,首关消除多,其口服生物利用度小。有些药物的肝清除率很高,接近肝血流量,称为灌流限制性清除,其肝清除率受肝血流量影响较大。药物以原形自肾消除的百分率比较容易测定。自肾排泄多的药物易受肾功能影响,自肾排泄少的药物易受肝功能影响。医生可以据此在肝或肾功能不足病人适当调整剂量。在零级动力学的药物中,RE以恒速消除,不随Cp下降而改变,故Cl 值不固定,与Cp成反比。
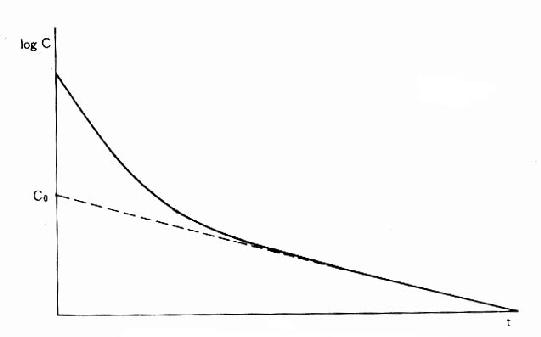
图3-7 表观分布容积计算法
C是静注药量A在0时理论上的血药浓度
Cl值实际上常用静脉或肌肉注射药物A后测定Cp,绘出时量曲线,算出AUC再按CL=A/AUC取得。因为AUC=C/ke,代入得
CL=keVd=CVd/AUC=A/AUC。
三、连续恒速给药
临床治疗常需连续给药以维持有效血药浓度。在一级动力学药物中,开始恒速给药时药物吸收快于药物消除,体内药物蓄积。按 计算约需5个t1/2达到血药稳态浓度(Css)(图3-8),此时给药速度(RA)与消除速度(RE)相等。
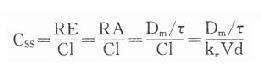 (τ为给药间隔时间)可见Css随给药速度(RA=Dm/τ)快慢而升降,到达Css时间不因给药速度加快而提前,它取决于药物的ke或t1/2。据此,可以用药物的keVd或Cl计算给药速度以达到所需的有效药物浓度。静脉恒速滴注时血药浓度可以平稳地到达Css。分次给药虽然平均血药浓度上升与静脉滴注相同,但实际上血药浓度上下波动(图3-8)。分药间隔时间越长波动越大,其峰值浓度
(τ为给药间隔时间)可见Css随给药速度(RA=Dm/τ)快慢而升降,到达Css时间不因给药速度加快而提前,它取决于药物的ke或t1/2。据此,可以用药物的keVd或Cl计算给药速度以达到所需的有效药物浓度。静脉恒速滴注时血药浓度可以平稳地到达Css。分次给药虽然平均血药浓度上升与静脉滴注相同,但实际上血药浓度上下波动(图3-8)。分药间隔时间越长波动越大,其峰值浓度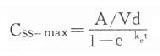 ,谷值浓度 Css-min=Css- maxe 。如果实际Css过高或过低,可以按已达到的Css与需要达到的Css比值调整给药速度,即Css(已达到的)/Css(需要的)=RA(现用的)/RA(将调整的)
,谷值浓度 Css-min=Css- maxe 。如果实际Css过高或过低,可以按已达到的Css与需要达到的Css比值调整给药速度,即Css(已达到的)/Css(需要的)=RA(现用的)/RA(将调整的)
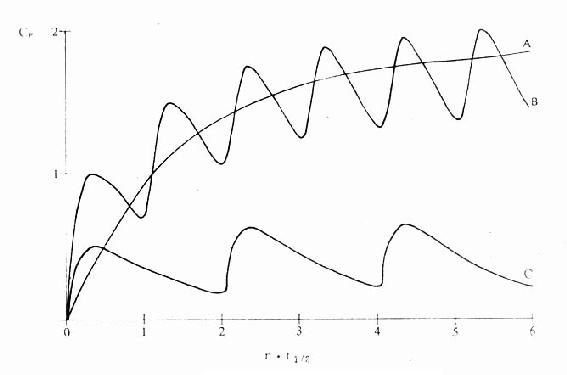
图3-8 连续恒速给药时的时量曲线
约经5个半衰期血药浓度达到稳态。给药间隔越短,
血药浓度波动越小。给药剂量越大,血药浓度越高
A.静脉滴注,Dm/t1/2B.肌肉注射,Dm/t1/2C.肌肉注射,1/2Dm/2t1/2。Dm维持剂量
但从调整剂量时开始需再经过5个t1/2方能达到需要的Css。
在病情危重需要立即达到有效血药浓度时,可于开始给药时采用负荷剂量(loading dose,D1),因为
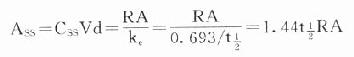
Ass就是负荷剂量。可将第一个t1/2内静脉滴注量的1.44倍在静脉滴注开始时推注入静脉即可立即达到并维持Css。在分次恒速给药达到Css时,体内Ass是维持剂量(maintenance dose, Dm)与体内上一剂量残留药物的和,即

当给药间隔时间τ=t1/2时,

即每隔一个t1/2给药一次时采用首剂加倍剂量的D1可使血药浓度迅速达到Css。
理想的给药方案应该是使CSS- max略小于最小中毒血浆浓度(MTC)而CSS-min略大于最小有效血浆浓度(MEC),即血药浓度波动于MTC与MEC之间治疗窗,这一Dm可按下列公式计算:
Dm=(MTC - MEC)Vd
负荷剂量计算法与上同,即D1=ASS=1.44t1/2 RA=1.44t1/2 Dm/τ,τ为给药间隔时间。τ可按一级消除动力学公式推算得
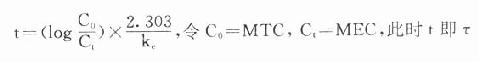
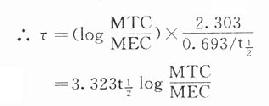
因此可以根据药物的MTC及MEC利用这些公式计算出D1,Dm及τ。注意此时τ≠t1/2,D1≠2Dm(图3-9)。
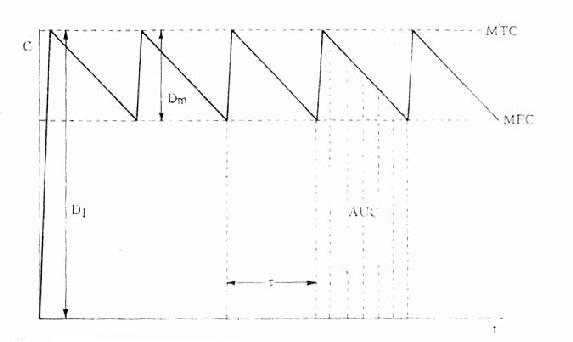
图3-9 负荷剂量、维持剂量、给药间隔与血药浓度关系
Dm维持剂量所形成的C D1负荷剂量所形成的C
在零级动力学药物中,体内药量超过机体最大消除能力。如果连续恒速给药,RA>RE,体内药量蓄积,血药浓度将无限增高。停药后消除时间也较长,超过5个t1/2。因为t1/2=0.5C0/K,达到C0越高t1/2越长。
临床用药可根据药动学参数如Vd、Cl、ke、t1/2及AUC等按以上各公式计算剂量及设计给药方案以达到并维持有效血药浓度。除了少数t1/2特长或特短的药物,或零级动力学药物外,一般可采用每一个半衰期给于半个有效量(half dose athalf life interval)并将首次剂量加倍是有效、安全、快速的给药方法。
有些药在体内转化为活性产物则需注意此活性产物的药动学,如果活性产物的消除是药物消除的限速步骤的话,则应按该产物的药动学参数计算剂量及设计给药方案。
四、一级药动学指标间的相互关系
1.F=A/D×100%口服剂量(D)由于不能100%吸收及存在首关消除效应,能进入体循环的药量(A)只占D的一部分,这就是生物利用度(F)。药动学计算时应采用绝对生物利用度,相对生物利用度作为评比药物制剂质量的指标。生物利用度还包括吸收速度问题,达峰时间(Tpeak)是一个参考指标。
2.A=C·Vd或C=A/Vd体内药量(A)与血药浓度(C)比值固定,在许多药动学公式中,A与C可以通用,如At=也可用Ct=。
3.Cp=[D]+[DP] 血浆中药物有游离型(D)与血浆蛋白结合型(DP),定量测定时需将血浆蛋白沉淀除去,故通常所说的血浆药物浓度(Cp)是指[D]与[DP]的总和。只有透析法或超离心法才可能将二者分离以计算药物的血浆蛋白结合率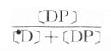 ×100% 。
×100% 。
4.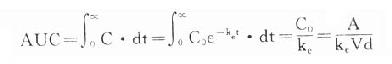 曲线下面积(AUC)是一个可用实验方法测定的药动学指标。它反映进入体循环药量的多少。时量曲线某一时间区段下的AUC反映该时间内的体内药量。AUC是独立于房室模型的药动学参数,常用于估算血浆清除率(Cl)。
曲线下面积(AUC)是一个可用实验方法测定的药动学指标。它反映进入体循环药量的多少。时量曲线某一时间区段下的AUC反映该时间内的体内药量。AUC是独立于房室模型的药动学参数,常用于估算血浆清除率(Cl)。
5.ke=0.693/t1/2=RE/A=CL/Vd消除速率常数是药物瞬时消除的百分率而不是单位时间药物消除速率(RE),是决定t1/2的参数,但其本身又取决于Cl及Vd,故不是独立的药动学指标。
6.Vd=A/C0=A/AUC ke 表现分布容积(Vd)是独立的药动学指标,不是实际的体液容积,取决于药物在体液的分布。Vd大的药物与组织蛋白结合多,主要分布于细胞内液及组织间液。Vd小的药物与血浆蛋白结合多,较集中于血浆。Vd不因A多少而变化。
7.CL=keVd=RE/Cp=A/AUC 血浆清除率(Cl)是肝肾等清除率的总和,也不是实际的药物消除速率(RE),是另一个独立于A的重要药动学指标,但受肝肾功能的影响。
8.t1/2=0.693/ke=0.693Vd/CL 血浆药物消除半衰期(t1/2)是一个非常实用的药动学指标,虽然独立于A,但受Cl及Vd双重制约,Cl大时t1/2短,Vd大时t1/2长。例如庆大霉素Cl小(60ml·min-1),Vd也小(0.25L·kg-1),其t1/2不长(2~3h)。氯喹Cl大(700ml·min-1),Vd也大(185L·kg-1),其t1/2并不短(8天)。药物在吸收及分布过程中也有半衰期,分别用t1/2a及t1/2α表示。
9.稳态时RA=RE=CSS·Cl=CSS·Vd·ke
故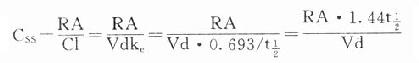 CSS是恒速连续给药达到稳态时平均血药浓度,应该和预期的有效浓度相等。必要时可以按达到的CSS与预期的CSS比值调整剂量或给药速度(RA)。
CSS是恒速连续给药达到稳态时平均血药浓度,应该和预期的有效浓度相等。必要时可以按达到的CSS与预期的CSS比值调整剂量或给药速度(RA)。
10. 分次定时定量给药时,CSS上下波动。当每t1/2给药一次时,其峰值(CSS- max)与谷值(CSS- min)的比值为2,缩短给药间隔可以减少CSS波动。
分次定时定量给药时,CSS上下波动。当每t1/2给药一次时,其峰值(CSS- max)与谷值(CSS- min)的比值为2,缩短给药间隔可以减少CSS波动。
11.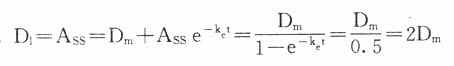 每t1/2给药一次时,首次给予加倍剂量,即负荷剂量(D1)可以立即达到CSS。
每t1/2给药一次时,首次给予加倍剂量,即负荷剂量(D1)可以立即达到CSS。
五、房室模型
以上所述各种药动学公式都是将机体视为一个整体空间,假设药物在其中转运迅速,瞬时达到分布平衡的条件下推导而得的。实际上机体绝非如此简单,不仅有血浆、细胞外液及细胞内液等间隔,而且各组织细胞间存在着无数的区间。静脉注射药物的时量(对数标尺)关系并非直线,而是一条由无数区段组成的连续弧线。粗略地看可见早期一段快速下降,后来才逐渐稳定缓慢下降。这是因为药物进入血液循环后快速向组织分布,首先进入血注量大的肺、肾、心、脑等器官,然后再向其他组织分布,最后达到平衡(假平衡)。因此设想机体由几个互相连通的房室(compartment)组成。这个房室不是解剖学上分隔体液的房室,而是按药物分布速度以数学方法划分的药动学概念。多数药物按二房室模型转运(少数单房室或多房室),中央室大致包括血浆及那些血流量多的器官,周边室包括机体其余部分,界限并不明确。时量曲线因此也只能大致分为分布相及消除相两个指数衰减区段(图3-10)。其药动学规律与单房室不同,如C=Ae-αt +Be-βt,α及β分别为分布相(A)及消除相(B)的消除速率常数。而且在分布相中Vd逐渐增大,ke(α)逐渐减少,t1/2逐渐延长,因此药动学计算需要特殊处理。即使在消除相,血药浓度稳定线性下降,各组织浓度及其下降速度也不尽相等,故称假平衡。可见问题非常复杂。
图3-10 二房室模型时量曲线
A.分布相(实线)及分布曲线(虚线)
B.消除相(实线)与消除曲线(虚线)
正由于问题过于复杂,临床应用诸多不便,实际运算也存在诸多困难。房室模型并非药物固有的药动学指标,机体也无此解剖学间隔,即使运用电子计算机拟合也不一定获得明确的划分。用同一药物试验,在某些人呈二室模型,另些人可能呈一室或三室模型。同一药物静脉注射时呈二室模型而口服则呈单一房室模型。在分布相时药物实际上已开始消除,到达消除相时可能已有相当分量的药物已被消除。如果用血管外给药(口服、肌注等)分布相常被吸收相掩盖。这些时相的划分仅靠血药浓度的测定。如果早期(此时血药浓度变化较快)取样间隔过疏,很难据此准确划分时相,因此,越来越多的临床家及研究者逐渐放弃房室模型而转向采用适用于所有药物的无房室方法(noncompartmentalmedtod)来解决实际问题,对此,有待今后深入学习。
从另一方面看,时量曲线在达到假平衡后已呈单一指数衰减的直线,此时房室划分已无需要,可以按β值计算t1/2及其他实用的药动学指标。
AUC是与房室无关的药动学指标,可用实验方法测定。AUC(0-∞)=C/ke,或AUC(T-∞)=CT/β,T是消除相开始的时间。再用AUC算出Vd及Cl:
Vd=A/AUCβ,CL=A/AUC
第四章 影响药物效应的因素及合理用药原则
同样剂量的某一药物在不同病人不一定都能达到相等的血药浓度,相等的血药浓度也不一定都能达到等同的药效。差异可能很大,甚至出现质的差异,即一般病人不会出现的异常危害性反应。这种随人而异的药物反应称为个体差异(individual variation)。产生个体差异的原因可以存在于药物产生效应的任何一个环节,包括药物剂型、药动学、药效学及临床病理等许多因素。如果不了解这些因素,不结合病人具体情况,不考虑如何加以调整,就难以达到最大疗效和最小反应的治疗目的。
第一节 药物方面的因素
一、药物剂型
同一药物可有不同剂型适用于不同给药途径。不同给药途径药物的吸收速度不同,一般规律是静脉注射>(快于)吸入>肌肉注射>皮下注射>口服>经肛>贴皮。不同药剂所含的药量虽然相等,即药剂当量(pharmaceutical equivalance) 相同,药效强度不尽相等。因此需要用生物当量(bioequivalance),即药物不同制剂能达到相同血药浓度的剂量比值,作为比较标准。不同药物剂型,其中药物剂量不同,应用时亦应注意区分选择。硝酸甘油静脉注射5~10μg,舌下含锭0.2~0.4mg,口服2.5~5mg,贴皮10mg,剂量相差更大。近年来生物药学随着药动学的发展,为临床用药提供了许多新的剂型。缓释制剂(slow release preparation)利用无药理活性的基质或包衣阻止药物迅速溶出以达到比较稳定而持久的疗效。口服缓释片剂或胶囊每日一次可维持有效血药浓度一天。肠外给药除一般油溶长效注射剂外还有控释制剂(controlled release preparation)可以控制药物按零级动力学恒速释放,恒速吸收。例如硝酸甘油贴皮剂每日贴一次。匹鲁卡品眼片置结膜囊内每周一次。子宫内避孕剂每年放置一次。不仅保证长期疗效,也大大方便了病人。
二、联合用药及药物相互作用
临床常联合应用两种或两种以上药物,除达到多种治疗目的外都是利用药物间的协同作用(synergism)以增加疗效或利用拮抗作用(antagonism)以减少不良反应。不恰当的联合用药往往由于药物间相互作用(interaction)而使疗效降低或出现意外的毒性反应。固定剂量比例的复方制剂虽然应用方便,但针对性不强,较难解决个体差异问题。
1.配伍禁忌(incompatibility)
药物在体外配伍直接发生物理性的或化学性的相互作用而影响药物疗效或毒性反应称为配伍禁忌。在静脉滴注时尤应注意配伍禁忌。
2.影响药动学的相互作用
⑴吸收:空腹服药吸收较快,饭后服药吸收较平稳。促进胃排空的药如甲氧氯普胺能加速药物吸收,抑制胃排空药如各种具有抗M胆碱作用药物能延缓药物吸收。对于吸收缓慢的灰黄霉素加快胃排空反而减少其吸收,而在胃中易被破坏的左旋多巴减慢胃排空反而使吸收减少。食物对药物吸收总的来说影响不大,因此基本上没有特异性禁忌。药物间相互作用影响吸收却不少见,如四环素Fe2+,Ca2+等因络合互相影响吸收。
⑵血浆蛋白结合:对于那些与血浆蛋白结合率高的、分布容积小的、安全范围窄的及消除半衰期较长的药物易受其他药物置换与血浆蛋白结合而致作用加强,如香豆素类抗凝药及口服降血糖药易受阿司匹林等解热止痛药置换而分别产生出血及低血糖反应。
⑶肝脏生物转化:肝药酶诱导药如苯巴比妥、利福平、苯妥英及香烟、酒等能增加在肝转化药物的消除而使药效减弱。肝药酶抑制药如异烟肼、氯霉素、西米替丁等能减慢在肝转化药物的消除而使药效加强。
⑷肾排泄:利用离子障原理,碱化尿液可加速酸性药物自肾排泄,减慢碱性药物自肾排泄。反之,酸化尿液可加速碱性药物排泄,减慢酸性药物排泄已如前述(第三章第一节,四)。水杨酸盐竞争性抑制甲氨蝶呤自肾小管排泄而增加后者的毒性反应。
3.影响药效学的相互作用
⑴生理性拮抗或协同:服用催眠镇静药后饮酒或喝浓茶或咖啡会加重或减轻中枢抑制作用,影响疗效。抗凝血药华法林和抗血小板药阿司匹林合用可能导致出血反应。
⑵受体水平的协同与拮抗:许多抗组胺药,酚噻嗪类,三环抗抑郁药类都有抗M胆碱作用,如与阿托品合用可能引起精神错乱,记忆紊乱等不良反应,β-受体阻断药与肾上腺素合用可能导致高血压危象等,都是非常危险的反应。
⑶干扰神经递质的转运:三环类抗抑郁药抑制儿茶酚胺再摄取,可增加肾上腺素及其拟似药如酪胺等的升压反应,而抑制可乐定及甲基多巴的中枢降压作用。由于药物相互作用而影响药物效应的实例不胜枚举,已有多本专著出版,在国外还有电脑检索系统,在此仅举例说明相互作用机制,目的在于引起警惕。
第二节 机体方面的因素
一、年龄
1.小儿特别是新生儿与早产儿,各种生理功能,包括自身调节功能尚未充分发育,与成年人有巨大差别,对药物的反应一般比较敏感。新药批准上市不需要小儿临床治疗资料,缺少小儿的药动学数据,这是主要困难。新生儿体液占体重比例较大,水盐转换率较快;血浆蛋白总量较少,药物血浆蛋白结合率较低;肝肾功能尚未充分发育,药物清除率低,在半岁以内与成人相差很多;小儿的体力与智力都处于迅速发育阶段,易受药物影响等都应引起用药注意,予以充分考虑。例如新生儿肝脏葡萄糖醛酸结合能力尚未发育,应用氯霉素或吗啡将分别导致灰婴综合征及呼吸抑制。新生儿肾功能只有成人的20%,庆大霉素的血浆半衰期长达18h,为成人(2h)的9倍。中枢兴奋药安非他明在小儿科却用于治疗学龄儿童多动症,作用性质也有所改变,儿童服用同化激素影响长骨发育,服用四环素可使牙齿变灰褐色。
2.老人老人实际年龄与其生理年龄并不一致,即老人生理功能衰退的迟早快慢各人不同,因此没有按老人年龄计算用药剂量的公式,也没有绝对的年龄划分界线,在医学方面一般以65岁以上为老人。老人对药物的吸收变化不大。老人血浆蛋白量较低,体水较少、脂肪较多、故药物血浆蛋白结合率偏低,水溶性药物分布容积较小而脂溶性药物分布容积较大。肝肾功能随年龄增长而自然衰退,故药物清除率逐年下降,各种药物血浆半衰期都有程度不同的延长,例如在肝灭活的地西泮可自常人的20~24h延长4倍。又如自肾排泄的氨基甙类抗生素可延长2倍以上。以药效学方面,老人对许多药物反应特别敏感,例如中枢神经药物易致精神错乱,心血管药易致血压下降及心律失常,非甾体抗炎药易致胃肠出血,抗M胆碱药易致尿潴留、大便秘结及青光眼发作等。
二、性别
除大白鼠外,一般动物对药物反应的性别差异不大,男人对醋氨酚及阿司匹林的清除率分别高于妇女40%及60%。妇女月经期不宜服用峻泻药和抗凝药以免盆腔充血月经增多。50年代末期在西欧因孕妇服用反应停(沙利度胺,催眠镇静药)而生产了一万余例海豹畸形婴儿的悲惨结果引起了对孕妇用药的警惕。对于已知的致畸药物如锂盐、酒精、华法林、苯妥英及性激素等在妊娠第一期胎儿器官发育期内应严格禁用。此后,在妊娠晚期及授乳期间还应考虑药物通过胎盘及乳汁对胎儿及婴儿发育的影响,因为胎盘及乳腺对药物都没有屏障作用。孕妇本身对药反应也有其特殊情况需要注意,例如抗癫痫药物产前宜适当增量,产前还应禁用阿司匹林及影响子宫肌肉收缩的药物。
三、遗传异常
先天性遗传异常(geneticpolymorphism) 对药物效应的影响近年来日益受到重视,至少已有一百余种与药物效应有关遗传异常基因被发现。过去所谓的特异体质(idiosyncrasy)药物反应多数已从遗传异常表型获得解释,现在已形成一个独立的药理学分支──遗传药理学(geneticpharmacology) 。遗传异常主要表现在对药物体内转化的异常,可分为快代谢型(extensivemetabolizer, EM) 及慢代谢型(poormetabolizer, PM) 。前者使药物快速灭活,后者使药物灭活较缓慢,因此影响药物血浆浓度及效应强弱久暂。又如6-磷酸葡萄糖脱氢酶(G6PD)缺乏者对伯氨喹,磺胺药、砜类等药物易发生溶血反应。这两种遗传异常的人在我国都不鲜见,这些遗传异常只有在受到药物激发时方出现异常,故不是遗传性疾病。
四、病理情况
疾病的严重度固然与药物疗效有关,同时存在的其他疾病也会影响药物的疗效。肝肾功能不足时分别影响在肝转化及自肾排泄药物的清除率,可以适当延长给药间隔及(或)减少剂量加以解决。神经功能抑制时,如巴比妥类中毒时能耐受较大剂量中枢兴奋药而不致惊厥,惊厥时却能耐受较大剂量苯巴比妥。此外要注意患者有无潜在性疾病影响药物疗效,例如氯丙嗪诱发癫痫,非甾体抗炎药激活溃疡病,氢氯噻嗪加重糖尿病,抗M胆碱药诱发青光眼等。在抗菌治疗时白细胞缺乏、未引流的脓疡、糖尿病等都会影响疗效。
五、心理因素
患者的精神状态与药物疗效关系密切,安慰剂(placebo)是不具药理活性的剂型(如含乳糖或淀粉的片剂或含盐水的注射剂),对于头痛、心绞痛、手术后痛、感冒咳嗽、神经官能症等能获得30%~50%的疗效就是通过心理因素取得的。安慰剂对心理因素控制的自主神经系统功能影响较大,如血压、心率、胃分泌、呕吐、性功能等。它在病人信心不足时还会引起不良反应。安慰剂在新药临床研究时双盲对照中极其重要,可用以排除假阳性疗效或假阳性不良反应。安慰剂对任何病人都可能取得阳性效果,因此医生不可能单用安慰剂作出真病或假病(心理病)的鉴别诊断。医生的任何医疗活动,包括一言一行等服务态度都可能发挥安慰剂作用,要充分利用这一效应。但医生不应利用安慰剂去敷衍或欺骗病人,因为这样会延误疾病的诊治并可能破坏病人对医生的信心。对于情绪不佳的病人尤应多加注意,氯丙嗪,利血平,肾上腺皮质激素及一些中枢抑制性药物在抑郁病人可能引发悲观厌世倾向,用药时应慎重。
六、机体对药物反应的变化
■[此处缺少一些内容]■
(addiction)。由于习惯及成瘾性都有主观需要连续用药,故统称依赖性(dependence) 。药物滥用(drugabuse) 是指无病情根据的大量长期的自我用药,是造成依赖性的原因。麻醉药品的滥用不仅对用药者危害极大,对社会危害也大。吗啡,可卡因,印度大麻及其同类药都属于麻醉药品。苯丙胺类、巴比妥类,苯二氮![]() 类等亦被列入国际管制的成瘾性精神药物。
类等亦被列入国际管制的成瘾性精神药物。
4.耐药性 病原体及肿瘤细胞等对化学治疗药物敏感性降低称为耐药性(drugresistance),也称抗药性。有些细菌还可对某些抗生素产生依药性(dependence)。在抗癌化学治疗中也有类似的抗药性问题。
第三节 合理用药原则
怎样才算合理用药现尚缺一具体标准,对某一疾病也没有统一的治疗方案。由于药物的有限性,即品种有限及疗效有限,和疾病的无限性,即疾病种类无限及严重度无限,因此不能简单以疾病是否治愈作为判断用药是否合理的标准。从理论上说合理用药是要求充分发挥药物的疗效而避免或减少可能发生的不良反应。当然这也不够具体,因此只能提几条原则供临床用药参考。
1.明确诊断选药不仅要针对适应证还要排除禁忌证。
2.而且容易发生相互作用。
3.了解并掌握各种影响药效的因素用药必须个体化,不能单纯公式化。
4.祛邪扶正并举在采用对因治疗的同时要采用对症支持疗法。这在细菌感染及癌肿化学治疗中尤其不应忽视。
5.对病人始终负责开出处方仅是治疗的开始,必需严密观察病情反应,及时调整剂量或更换治疗药物。要认真分析每一病例的成功及失败的关键因素,总结经验教训,不断提高医疗质量,使用药技术更趋合理化。
第五章 传出神经系统药理概论
传出神经系统包括植物神经系统和运动神经系统。植物神经系统(vegetativenervous system)也称自主神经系统(autonomicnervous system),主要支配心肌、平滑肌和腺体等效应器;运动神经系统则支配骨骼肌。
自主神经系统排除传出神经外,尚包括内脏传入感觉神经,然对后者的生理和药理研究不多。国外文献沿用自主神经药理一词,实际上主要指传出而不包括传入神经药理。此外,自主神经系统不应包括运动神经,但运动神经系统的递质和受体与植物神经系统同属一个体系,而传出神经系统药理一词自可将这两类都概括进来。因此,我国沿用传出神经系统药理一词较为合理。
植物神经自中枢神经系统发出后,都要经过神经节中的突触,更换神经元,然后才达到效应器(effector)。因此,植物神经有节前纤维和节后纤维之分(图5-1,5-2)。
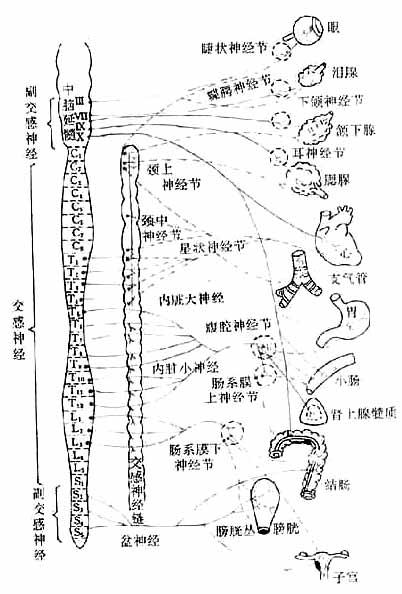
图5-1 植物神经系统分布示意图
神经纤维:蓝色:胆碱能神经 实线:节前纤维
红色:去甲肾上腺素能神经 虚线:节后纤维
运动神经自中枢发出后,中途不更换神经元,直接达到骨骼肌,因此无节前和节后纤维之分。
一、传出神经系统的递质及受体
当神经冲动达到神经末梢时,在突触部位从末梢释放出化学传递物,称为递质(transmitter)。通过递质作用于次一级神经元或效应器的受体(receptor),发生效应,从而完成神经冲动的传递过程。作用于传出神经系统的药物主要是在突触部位影响递质或受体而发挥作用。
(一)传出神经系统的递质
1.递质学说的发展 1921年Loewi通过动物实验证明递质的存在。实验是用两个离体蛙心进行,当刺激甲蛙心的迷走交感神经干以引起迷走神经兴奋时,甲蛙心受到抑制,这时将甲蛙心的灌注液注入乙蛙心,则乙蛙心也表现出抑制。这就说明甲蛙心迷走神经兴奋时,必定释出一种抑制性物质,才能使乙蛙心也受到抑制。后来证明这种物质就是乙酰胆碱。此后相继发现神经节中的节前纤维末梢和运动神经末梢兴奋时,都能释放乙酰胆碱。本世纪四十年代,通过von Euler的工作证明交感神经节后纤维的神经递质是去甲肾上腺素。至此,传出神经系统的化学传递学说才臻完善。
2.传出神经突触的超微结构突触中神经末梢与效应器细胞或次一级神经元间有一定的间隙,称突触间隙。传出神经末梢邻近间隙的细胞膜称为突触前膜;效应器或次一级神经元邻近间隙的细胞膜称为突触后膜。在运动神经与骨骼肌的接头(也称终板),这个间隙约为15~20nm;终板的突触后膜有许多皱褶,其中聚集着胆碱酯酶,能迅速水解已释放的乙酰胆碱。在神经末梢内靠近突触前膜处,聚集着很多直径为20~50nm的囊泡(vesicle),囊泡内含有大量递质乙酰胆碱。
交感神经末梢分成许多细微的神经纤维,分布于平滑肌细胞之间。这些细微神经纤维都有稀疏串珠状的膨胀部分,称为膨体(varicosity)。膨体中含有线粒体和囊泡等亚细胞结构,一个膨体内囊泡的数目约在1000个左右。囊泡内含有高浓度的去甲肾上腺素。
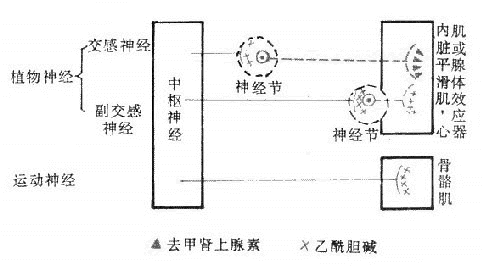
图5-2 传出神经分类模式图
蓝色:胆碱能神经 实线:节前纤维
红色:去甲肾上腺素能神经 虚线:节后纤维
去甲肾上腺素能神经内的囊泡有大小之分,大囊泡在神经节细胞内形成,以每小时数mm的速度沿轴突向末梢运行;小囊泡主要在神经末梢形成。运行到末梢的还有合成去甲肾上腺素所必需的酶,如酪氨酸羟化酶、多巴脱羧酶和多巴胺β-羟化酶等,后者存在于囊泡内;前二者存在于胞质液中。
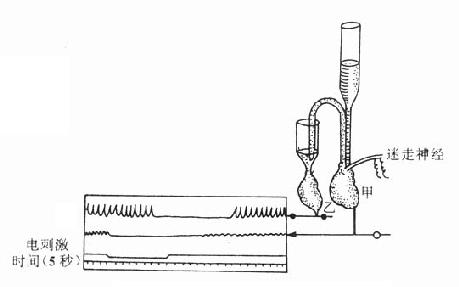
图5-3 证明迷走神经兴奋时释放递质的双蛙心实验
3.递质的生物合成与贮存 去甲肾上腺素的生物合成在去甲肾上腺素能神经细胞体内和轴突中即开始进行,不过在此含量较少,愈到神经末梢,含量愈多,末梢内的含量约为细胞体内的3~300倍。酪氨酸从血液进入神经元后,在酪氨酸羟化酶催化下生成多巴(dopa)再经多巴脱羧酶的催化,脱羧后生成多巴胺(dopamine),后者进入囊泡中,经多巴胺β-羟化酶的催化,转变为去甲肾上腺素。酪氨酸羟化酶的活性较低,反应速度慢,底物要求专一,当胞浆中多巴胺或游离的去甲肾上腺素浓度增高时,对该酶有反馈性抑制作用,反之,当胞浆中多巴胺或去甲肾上腺素浓度降低时,对该酶的抑制作用减弱,催化反应则加速,故这一步骤是去甲肾上腺素生物合成过程的限速因素,是调节去甲肾上腺素生物合成的重要环节。去甲肾上腺素形成后,与ATP的嗜铬颗粒蛋白结合,贮存于囊泡中(图5-4),并可避免被胞质液中的单胺氧化酶(MAO)所破坏。
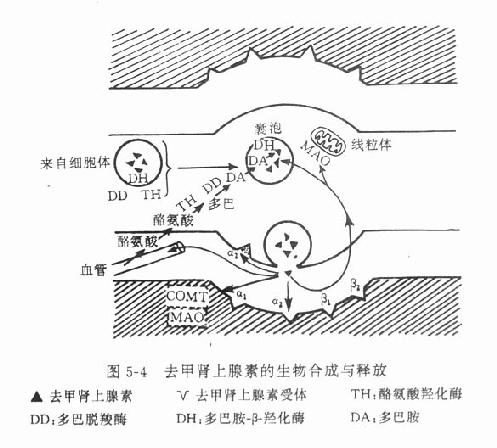
乙酰胆碱主要在胆碱能神经末梢形成,与其合成有关的酶和辅酶有胆碱乙酰化酶(cholineacetylase)和乙酰辅酶A(acetyl coenzyme A)。前者在细胞体内形成并沿轴突转运至末梢,后者则在末梢线粒体内合成,须先与草酰乙酸缩合成枸橼酸盐,才能穿过线粒体膜进入胞质液中,然后在枸橼酸裂酶的催化下再形成乙酰辅酶A。胆碱乙酰化酶和乙酰辅酶A在胞质液内促进胆碱形成乙酰胆碱。乙酰胆碱形成后,即进入囊泡并与ATP和囊泡蛋白共同贮存于囊泡中(图5-5)。
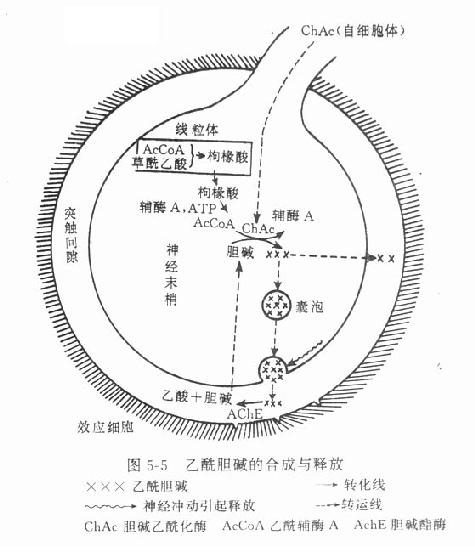
4.递质的释放 现认为当神经冲动到达末梢时,产生除极化,引起Ca2+内流促使靠近突触前膜的一些囊泡的囊泡膜与突触前膜融合,形成裂孔,通过裂孔将囊泡内的递质、ATP和蛋白质等排出至突触间隙,这称为胞裂外排(exocytosis)。每一囊泡约含有1000~50000分子乙酰胆碱或约10000分子去甲肾上腺素。骨骼肌或平滑肌细胞有自发性小终板电位(miniatuse endplate potential)或接头电位,其电位幅度有分极现象;因此提出递质的量子化释放(quantalrelease)概念。每一量子相当于一个囊泡的释放量。由于这种电位幅度很小(0.3~3.0mv),故不会引起动作电位和效应。冲动到达时,可有上百个囊泡同时外排,才引起动作电位和效应。近知嗜铬细胞胞浆中的会合素(synexin),在有Ca2+时,能融合嗜铬颗粒(相当于神经末梢的囊泡)膜与细胞膜。
5.递质作用的消失 乙酰胆碱作用的消失主要是被神经突触部位的胆碱酯酶水解,一般在释放后一至数毫秒之内即被此酶水解而失效。去甲肾上腺素主要靠突触前膜将其摄取入神经末梢内而使作用消失;这种摄取称为摄取1(uptake1)。摄取1是一种主动的转运机制,也称胺泵(amine pump),能逆浓度梯度而摄取内及外源性去甲肾上腺素。其摄取量为释放量的75%~95%,摄取入神经末梢的去甲肾上腺素尚可进一步被摄取入囊泡,贮存起来以供下次的释放。部分未进入囊泡的去甲肾上腺素可被胞质液中线粒体膜上的单胺氧化酶(mono-amineoxidase,MAO)破坏。非神经组织如心肌、平滑肌等也能摄取去甲肾上腺素,称为摄取2。此种摄取之后,即被细胞内的儿茶酚氧位甲基转移酶(catechol-O-methyltransferase,COMT)和MAO所破坏;因此摄取1可称为摄取-贮存型,摄取2可称为摄取-代谢型。此外,尚有小部分去甲肾上腺素释放后从突触间隙扩散到血液中,最后被肝、肾等的COMT和MAO所破坏。
(二)传出神经系统的受体
1.受体的命名 受体的命名常根据能与之选择性地相结合的递质或药物而定。能与乙酰胆碱结合的受体,称为胆碱受体(cholinoceptor)。由于在早期的研究中,发现位于副产感神经节后纤维所支配的效应器细胞膜的胆碱受体对以毒蕈碱为代表的拟胆碱药较为敏感,故这部分受体称为毒蕈碱(muscarine)型胆碱受体(M胆碱受体)。位于神经节细胞膜和骨骼肌细胞膜的胆碱受体对烟碱比较敏感,故这些部位的受体称为烟碱(nicotine)型胆碱受体(N胆碱受体),也可将前者称为N1受体,后者称为N2受体。近年发现M胆碱受体也有M1受体、M2受体和M3受体之分。M1受体主要分布于神经节细胞和腺体细胞,哌仑西平能选择性地阻断之;M2受体似主要分布于心脏;M3受体似主要分布于平滑肌和腺体细胞。哌仑西平对M2和M3受体的亲和力较低,阿托品对三种M受体都能阻断。
能与去甲肾上腺素或肾上腺素结合的受体称为肾上腺素受体(adrenoceptor)。肾上腺素受体又可分为α肾上腺素受体(α受体)和β肾上腺素受体(β受体)。β受体又分为β1受体和β2受体两种亚型,例如心脏的β受体主要为β1受体,支气管和血管平滑肌的β受体主要为β2受体。
70年代发现外源去甲肾上腺素可抑制去甲肾上腺素能神经内[3H]标记去甲肾上腺素的释放(图5-6)。因此认为突触前膜也有α受体,激动时可使递质释放减少,这是一种递质释放的自身调节机制。当时将突触后膜α受体命名为α1受体,突触前膜受体命名为α2受体。后来在许多血管组织突触后膜都发现有不受α1受体阻断药哌唑嗪阻断的α2受体。血管平滑肌突触后膜的α1和α2受体的共存似为普遍现象,可见以突触前、后膜的解剖部位进行α受体分类是不妥的。而以特异的阻断剂和激动剂来区分受体亚型更为合适。
![NA对电刺激所致[3H]NA释放的影响](/lilunshuji/yaolixue/yaolixue058.jpg)
图5-6 NA对电刺激所致[3H]NA释放的影响
猫脾脏灌流,每次刺激共200脉冲,30Hz,
括号内数字为实验次数,竖线为SE(自Bevan,1978)
2.受体按偶联和结构的分类根据第二章对受体的叙述,肾上腺素受体和M胆碱受体属于G-蛋白偶联受体,N胆碱受体属于配体门控通道型受体。近年由于单克隆抗体和DNA重组等技术的应用,许多受体的一级结构得以阐明。现已知G-蛋白偶联受体一级结构的特点是都有7个跨膜区段,以β2受体为例,自图5-7可见,每个跨膜区段有20余个氨基酸残基组成的亲脂性螺旋结构。其伸出细胞膜外的N端较短,伸入细胞内的C端较长。处于β受体第3跨膜区细胞膜内深1.1nm的门冬氨酸似为与配体的主要结合点。
N胆碱受体是由两个单体形成的二聚体。每个单体由5个亚基组成,包括两个α亚基(分子量为40000),一个β亚基(50000),一个γ基(57000)和一个δ亚基(64000)。每个亚基都有4个跨膜区段。5个亚基围绕成环状而形成离子通道(图5-8)。
二、传出神经按递质的分类
一般都根据所释放递质的不同,将传出神经分为胆碱能神经和去甲肾上腺素能神经两大类。
(一)胆碱能神经(cholinergicnerve)能合成Ach,兴奋时从末梢释放Ach。
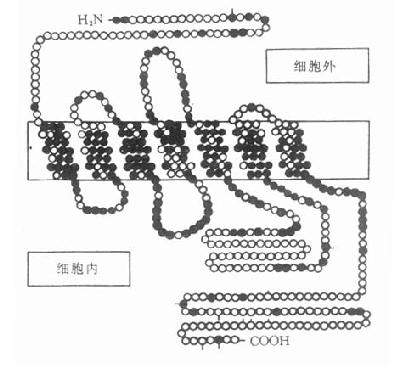
图5-7 人体β1-肾上腺受体
含477氨基酸,肽链跨膜7次,N端在细胞外,C端在
细胞内,胞内带短线的是丝氨酸,能被磷酸化。黑实心
圆是人体β1、β2受体所共有的氨基酸,在跨膜区较多。
(自TINS 11,321,1988)
1.全部交感神经和副交感神经的节前纤维;
2.运动神经;
3.全部副交感神经的节后纤维;
4.极少数交感神经节后纤维,如支配汗腺的分泌神经和骨骼肌的血管舒张神经。
(二)去甲肾上腺素能神经(noradrenergic nerve)能合成NA,兴奋时能释放NA。
几乎全部交感神经节后纤维都属此类。
传统认为一类神经元只释放一种递质,现知情况远较复杂。实际上许多神经元都贮有二或三种递质可供释放,例如颌下腺胆碱能神经元除有能促分泌的乙酰胆碱外,还随同释放血管活性肠肽(VIP),它能扩张血管,阿托品能抑制分泌,却不能拮抗血管扩张。大脑皮质神经元同此。肠壁神经元能共同释放VIP、强啡肽和甘丙肽。许多去甲肾上腺素能神经元也共同释放NA、ATP和神经肽Y。上述现象现称共同传递(cotransmission)。
三、传出神经系统效应产生的生化过程
神经递质或激动药受体结合后,触发一系列瀑布式的生化过程,通过一级一级地放大,最终导致效应,这一过程称为受体-效应偶联(receptor-effect coupling)。现介绍有关传出神经系统的两种受体反应偶联如下:
(一)受体与离子通道的偶联
神经递质或激动药与受体间相互作用可使受体操纵性离子通道(receptor-operatedclannel)开放,从而产生效应。例如β受体激动使钙离子通道开放,Ca2+进入细胞而产生各种生理效应。
有些受体含有离子通道,如上述N胆碱受体,与配体结合就能直接使通道开放。故称这种受体为门控通道型受体。
(二)受体与酶的偶联
传出神经系统的G蛋白偶联受体是通过腺苷酸环化酶(adenyl cyclase,AC)或磷脂酶C(phospholipase C, PLC)而产生效应的(图5-9)。
四、传出神经系统的生理功能
传出神经系统药物种类繁多,但它们药理作用的共性不外是影响传出神经系统的功能,或是拟似药,或是拮抗药。因此,如果熟悉两大类传出神经即去甲肾上腺素能神经和胆碱能神经的生理功能,再结合各药的特性,自易掌握每个药物的药理作用。
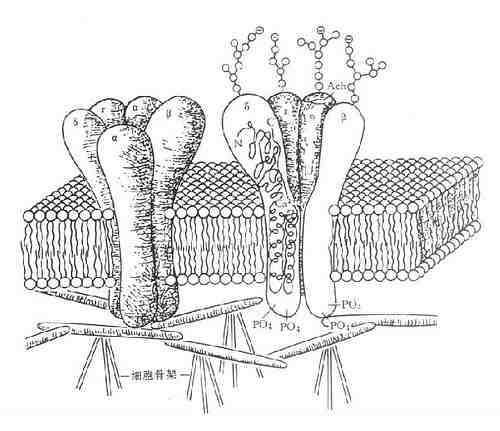
图5-8 N2烟碱受体
5个亚基各含约450个氨基酸,此5个肽链形成一个跨膜的环,在细胞内固定于细胞骨架上,每一肽链跨膜4次,N端和C端都位于胞外部(如δ亚单位剖面所示)。肽链在胞外被糖基化。在胞内被磷酸化,导致受体脱敏,2个α单位各有1个Ach结合位点,二者都结合1分子Ach后,钠通道开放,细胞除极兴奋。(引自F.Hucho)
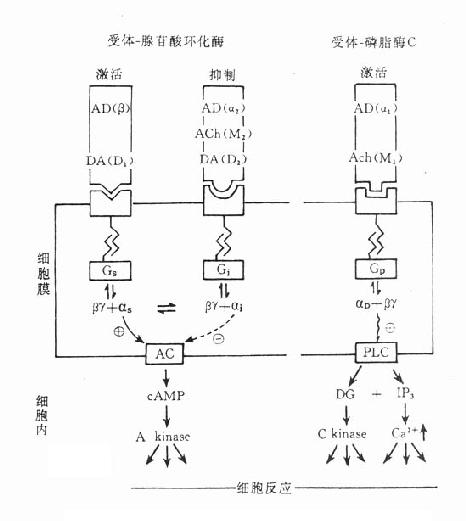
图5-9 受体-腺苷酸环化酶偶联和受体-磷脂酶偶联示意图
多数器官都接受上述两大类传出神经的双重支配。去甲肾上腺素能神经兴奋时(相当于递质去甲肾上腺素的作用),可见心脏兴奋、皮肤粘膜和内脏血管收缩、血压升高、支气管和胃肠道平滑肌抑制、瞳孔扩大等。这些功能变化,有利于机体适应环境的急聚变化。胆碱能神经兴奋时(相当于递质乙酰胆碱作用),节前与节后纤维的功能有所不同,当节后纤维兴奋时,基本上表现与上述相反的作用,有利于机体进行休整和积蓄能量。当节前纤维兴奋时,可引起神经节兴奋和肾上腺髓质分泌的增加,细节见表5-1。
表5-1 传出神经的受体-效应表*
| 效应器 | 肾上腺素能神经兴奋 | 胆碱能神经兴奋 | |||||
| 效 应 | 受 体 | 效应 | 受体 | ||||
| 心脏 | 心肌 窦房结 传导系统 |
收缩力加强⑧ 心率加快 传导加快 |
β1① | 收缩力减弱 心率减慢 传导减慢 |
M M |
||
| 平 滑 肌 |
血 管 |
皮肤、粘膜 腹腔内脏② 骨骼肌 冠状动脉 |
收缩 收缩 舒张 收缩 舒张 舒张 |
α α β2 α β2 β2 |
舒张④ 舒张(交感神经) |
||
| 支气管,气管 胃肠壁⑤ 膀胱逼尿肌 胃肠和膀胱括约肌 胆囊与胆道 |
舒张 舒张 舒张 收缩 舒张 |
β2 α、β2 β2 α β2 |
收缩 收缩 收缩 舒张 收缩 |
||||
| 子宫 | 收缩⑥ 抑制 |
α β2 |
不定 | ||||
| 眼 | 虹膜 睫状肌 |
瞳孔扩大肌收缩(扩瞳) 舒张(远视) |
α β2 |
瞳孔括约肌收缩(缩瞳) 收缩(近视) |
|||
| 腺 体 |
汗腺 唾液腺 胃肠道及呼吸道腺体 |
手心脚心分泌 分泌K+及H2O 分泌淀粉酶 |
α′ α β2 |
全身分泌(交感神经) 分泌K+及H2O 分泌 |
|||
| 代谢 | 肝脏糖代谢 骨骼肌糖代谢 脂肪代谢 |
肝糖原分解及异生 肌糖原分解 脂肪分解 |
α、β2 β2 α、β2⑦ |
||||
| 植物神经节 肾上腺髓质 |
兴奋 分泌(交感神经节前纤维) |
N1 | |||||
| 骨骼肌 | 收缩 | β2 | 收缩(运动神经) | N2 | |||
①心肌也具α和β2受体;人的心肌β1受体与β2受体之比约为80:20。
②肾脏和肠系膜血管尚有多巴胺受体,激动时血管舒张。
③虽也有α受体,但在整体动物,由于自身调节机制而表现为舒张。
④无生理意义。
⑤消化道神经丛副交感神经节前纤维末梢可能有α受体,激动时抑制乙酰胆碱的释放,产生抑制效应。
⑥雌激素占优势时,可致收缩。
⑦不同动物,受体亚型各异。
⑧黑体字表示占优势。
*许多组织都有不同型的受体共存,本表所列是在各组织中占多数的主要受体。
人体内的生理调节是在对立统一规律下进行的。在同一器官上,胆碱能神经和去甲肾上腺素能神经的作用大多是互相对抗的,但在中枢神经系统的调节下,它们的功能既是对立的,又是统一的。
近年来,在受体水平的研究中,也发现胆碱能神经和去甲肾上腺素能神经的功能并非截然分割,而是互相调节和互相制约的。例如有此去甲肾上腺素能神经和胆碱能神经突触前膜可能兼具抑制性的α受体和M受体,即受其本身所释放递质的反馈性调节,也受其生理拮抗性神经元所释放的递质的控制。
五、传出神经系统药物的基本作用
(一)直接作用于受体
许多传出神经系统药物能直接与胆碱受体或肾上腺素受体结合。结合后,如果产生与递质相似的作用,就称激动药。如果结合后不产生或较少产生拟似递质的作用,相反,却能妨碍递质与受体的结合,从而阻断了冲动的传递,产生与递质相反的作用,就称为阻断药(blocker);对激动药而言,可称拮抗药。
这类药物品种很多,也较常用。由于胆碱受体分为M和N两型,肾上腺素受体也有α和β两型。因此,选择性地作用于不同型受体的激动药和阻断药也具有相应的分类。
(二)影响递质
1.影响递质的生物合成直接影响递质生物合成的药物较少,且无临床应用价值,仅作药理学研究的工具药。
2.影响递质的转化如乙酰胆碱的灭活主要是被胆碱酯酶水解。因此,抗胆碱酯酶药就能妨碍乙酰胆碱的水解,提高其浓度,产生效应。
表5-2常用传出神经系统药物的分类
| 拟 似 药 | 拮 抗 药 |
| 1.M,N受体激动药(氨甲酰胆碱) 2.M受体激动药(毛果云香碱) 3.N受体激动药(烟碱) (二)抗胆碱酯酶药(新斯的明) (三)肾上腺素受体激动药 1.α受体激动药 (1)α1,α2受体激动药(去甲肾上腺素) (2)α1受体激动药(去氧肾上腺素) (3)α2受体激动药(可乐定) 2.α、β受体激动药(肾上腺素) 3.β受体激动药 (1)β1,β2受体激动药(异丙肾上腺素) (2)β1受体激动药(多巴酚丁胺) (3)β2受体激动药(沙丁胺醇) |
(一)胆碱受体阻断药 1.M受体阻断药 (1)非选择性M受体阻断药(阿托品) (2)M1受体阻断药(哌仑西平) 2.N受体阻断药 (1)N1受体阻断药(六甲双铵) (2)N2受体阻断药(琥珀胆碱) (二)胆碱酯酶复活药(碘解磷定) (三)肾上腺素受体阻断药 1.α受体阻断药 (1)α1,α2受体阻断药 ①短效类(酚妥拉明) ②长效类(酚苄明) (2)α1受体阻断药(哌唑嗪) (3)α2受体阻断药(育亨宾) 2.β受体阻断药 1A类(普萘洛尔) 1B类(吲哚洛尔) 2A类(阿替洛尔) 2B类(艾司洛尔,esmolol,短效) 3类(拉贝洛尔) |
去甲肾上腺素作用的消失与乙酰胆碱不同,它主要靠突触前膜的摄取,因此现有的MAO抑制药或COMT抑制药并不能成为理想的外周拟肾上腺素药。
3.影响递质的转运和贮存药物可通过促进递质的释放而发挥递质样作用。例如麻黄碱促进去甲肾上腺素的释放、氨甲酰胆碱促进乙酰胆碱的释放而发挥作用,虽然它们同时尚有直接与受体结合的作用。
药物也可通过影响递质在神经末梢的贮存而发挥作用。例如利血平抑制神经末梢囊泡对去甲肾上腺素的摄取,使囊泡内去甲肾上腺素逐渐减少以至耗竭,从而表现为拮抗去甲肾上腺素能神经的作用。
六、传出神经系统药物的分类
传出神经系统药物可按其作用性质(激动受体或阻断受体)和对不同类型受体的选择性进行分类如表5-2。也可将影响胆碱酯酶的药单列一类。
(上海医科大学 杨藻宸)
第六章 胆碱受体激动药
胆碱受体激动药(cholinoceptor agonists)与胆碱受体结合,激动受体,产生与递质乙酰胆碱相似的作用。按其对不同胆碱受体亚型的选择性,可分为:①M,N胆碱受体激动药;②M胆碱受体激动药;③N胆碱受体激动药。
第一节 M,N胆碱受体激动药
本类药物既用于节后胆碱能神经支配的效应器内的M胆碱受体,也作用于神经节和骨骼肌的N胆碱受体。
乙酰胆碱
乙酰胆碱(acetylcholine, Ach)是胆碱能神经递质,化学性质不稳定,遇水易分解。由于作用十分广泛,且在体内为胆碱酯酶迅速破坏,故除作为药理学研究的工具药外,无临床实用价值。但如了解其生理、药理作用,将便于学习和掌握胆碱受体激动药和胆碱受体阻断药的药理。
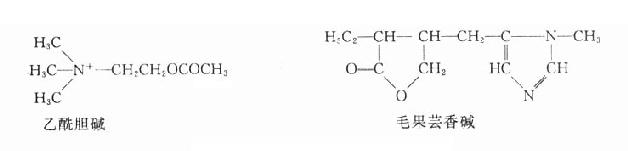
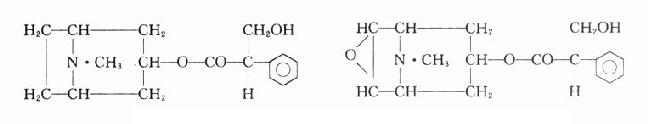
乙酰胆碱和毛果芸香碱的化学结构
【药理作用】
1.M样作用 静脉注射小剂量Ach即能激动M胆碱受体,产生与兴奋胆碱能神经节后纤维相似的作用,引起心率减慢、血管舒张、血压下降,支气管和胃肠道平滑肌兴奋,瞳孔括约肌和睫状肌收缩以及腺体分泌增加等。舒张血管可能是激动血管内皮细胞的M受体使内皮细胞释放依赖性舒张因子(endothelium-dependentrelaxing factor,EDRF,现多认为EDRF即是一氧化氮(NO))所致。
2.N样作用 剂量稍大时,Ach也能激动N胆碱受体,产生与兴奋全部植物神经节和运动神经相似的作用。还能兴奋肾上腺髓质的嗜铬组织(此组织在胚胎发育中与交感神经节的来源相同,受交感神经节前纤维支配),使之释放肾上腺素。许多器官是由胆碱能和去甲肾上腺素能神经双重支配的,通常是其中一种占优势。例如,在胃肠道、膀胱平滑肌和腺体是以胆碱能神经占优势,而心肌收缩和小血管方面则以去甲肾上腺素能神经占优势。故在大剂量Ach作用下,全部神经节(具N1胆碱受体)兴奋的结果是胃肠道、膀胱等器官的平滑肌兴奋,腺体分泌增加,心肌收缩力加强,小血管收缩,血压升高。Ach还激动运动神经终板上的N2胆碱受体,表现为骨骼肌兴奋。过大剂量的Ach很易使神经节从兴奋转入抑制。
氨甲酰胆碱
氨甲酰胆碱(carbamylcholine,carbachol)的化学结构和作用都与Ach相似,能直接激动M和N受体,也可能促进胆碱能神经末梢释放Ach而发挥间接作用。可用于手术后腹气胀和尿潴留。副作用较多,加以阿托品对它的解毒效果差,故目前主要局部滴眼用于治疗青光眼,引起缩瞳以降低眼内压。
第二节 M胆碱受体激动药
毛果芸香碱
又名匹鲁卡品(pilocarpine)是从毛果芸香属(pilocarpus属)植物中提出的生物碱,基水溶液稳定,也能人工合成。
【药理作用】 能选择性地激动M胆碱受体,产生M样作用。对眼和腺体的作用最明显。
1.眼 滴眼后能引起缩瞳、降低眼内压和调节痉挛等作用:
(1)缩瞳虹膜内有两种平滑肌,一种是瞳孔括约肌,受动眼神经的副交感神经纤维(胆碱能神经)支配,兴奋时瞳孔括约肌收缩,瞳孔缩小;另一种是瞳孔开大肌,受去甲肾上腺素能神经支配,兴奋时瞳孔开大肌向外周收缩,瞳孔扩大。用毛果芸香碱后,可激动瞳孔活约肌的M胆碱受体,表现为瞳孔缩小。
(2)降低眼内压房水是从睫状体上皮细胞分泌及血管渗出而产生,经瞳孔流入前房,到达前房角间隙,主要经小梁网(滤帘)流入巩膜静脉窦,最后进入血流(图6-1,6-2)。毛果芸香碱可通过缩瞳作用使虹膜向中心拉紧,虹膜根部变薄,从而使处在虹膜周围部分的前房角间隙扩大,房水易于通过小梁网及巩膜静脉窦而进入循环,结果使眼内压下降。
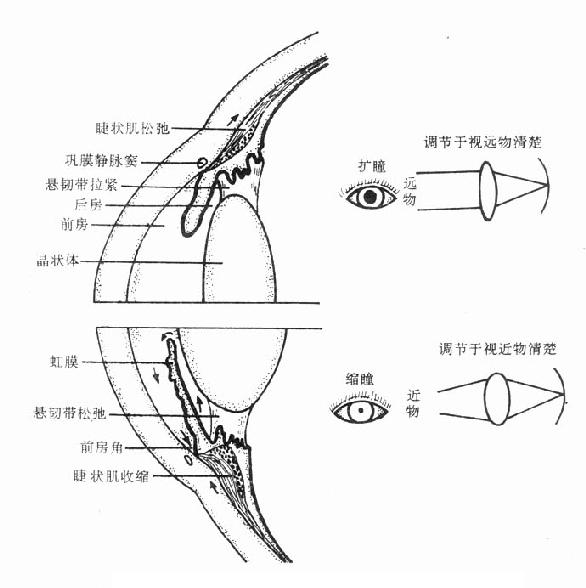
图6-1 M胆碱受体激动药和阻断药对眼的作用
上:胆碱受体阻断药的作用 下:胆碱受体激动药的
作用,箭头表示房水流通及睫状肌收缩或松弛的方向
(3)调节痉挛使晶状体聚焦,适合于视近物的过程,称为调节。眼睛的调节主要取决于晶状体的曲度变化。晶状体囊富有弹性,使晶状体有略呈球形的倾向,但由于睫状小带(悬韧带)向外缘的牵拉,通常使晶状体维持于比较扁平的状态。睫状小带又受睫状肌控制,睫状肌由环状和辐射状两种平滑肌纤维组成,其中以胆碱能神经(动眼神经)支配的环状肌纤维为主。动眼神经兴奋时或用拟胆碱药如毛果芸香碱时使环状肌向瞳孔中心方向收缩,结果使睫状小带放松,晶状体变凸,屈光度增加,只适合于视近物,而看远物则难以使其清晰地成像于视网膜上;故看近物清楚,看远物模糊。拟胆碱药的这种作用称为调节痉挛。
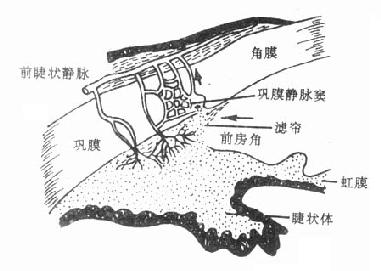
图6-2 房水出路(箭头示房水回流方向)
睫状肌的辐射状肌纤维受去甲肾上腺素能神经支配,但这在眼睛调节中不占重要地位,故拟肾上腺素药一般不影响调节。
2.腺体 吸收后能激动腺体的M胆碱受体,汗腺和唾液腺分泌增加最明显。
【临床应用】其吸收作用除用作抗胆碱药阿托品等中毒的抢救外,其它应用价值不大。临床上主要局部用于治疗青光眼。滴眼后易透过角膜进入眼房,作用迅速,10分钟后出现作用,半小时达高峰。与毒扁豆碱比较,毛果芸香碱作用温和而短暂,故用药间隔时间宜短,水溶液比较稳定。吸收后的不良反应主要由其M胆碱作用所致,可用阿托品对抗。
1,青光眼眼内压增高是青光眼的主要特征,可引起头痛、视力减退等症状,严重时可致失明。闭角型青光眼(急性或慢性充血性青光眼)患者前房角狭窄,眼内压增高。毛果芸香碱能使眼内压迅速降低,从而缓解或消除青光眼症状。
毛果芸香碱也适用于开角青光眼(慢性单纯性青光眼)的治疗。这种青光眼无前房角狭窄和闭塞情况,而是由于小梁网本身及巩膜静脉窦发生变性或硬化,阻碍了房水循环,引起眼内压升高。毛果芸香碱可能的通过扩张巩膜静脉窦周围的小血管以及收缩睫状肌后,小梁网结构发生改变而使眼内压下降。
常用1%~2%溶液滴眼,用后30~40分钟缩瞳作用达高峰,降低眼内压作用可维持4~8小时,调节痉挛作用在2小时左右消失。滴眼时应压迫内眦,避免药液流入鼻腔,因吸收而产生副作用。
2.虹膜炎 与扩瞳药交替应用,可防止虹膜与晶状体粘连。
氨甲酰甲胆碱
氨甲酰胆碱(carbamylmethylcholine)又名乌拉胆碱(urecholine)为M受体激动药,对胃肠道及膀胱平滑肌作用的选择性最高。用于手术后腹气胀和尿潴留。
第三节 N胆碱受体激动药
烟碱(nicotine)是N胆碱受体激动药的代表,它是烟叶(tobacco)的重要成分。作用很复杂,既作用于N1受体,也作用于N2受体,此外,尚可作用于中枢神经系统,而且具有小剂量激动,大剂量阻断N受体的双相作用。因此无临床治疗应用价值,但为烟草制品所含毒物之一,在吸烟的毒理中具有重要意义。
胆碱受体激动药 制剂及用法
氯化氨甲酰胆碱(carbamylcholine chloride) 皮下注射,0.25~0.5mg/次,0.5%~1.5%溶液滴眼。
硝酸毛果芸香碱(pilocarpine nitrate)滴眼液或眼膏,1%~2%,滴眼次数按需要决定,晚上或需要时涂眼膏。
第七章 抗胆碱酯酶药和胆碱酯酶复活药
第一节 胆碱酯酶
胆碱酯酶(cholinesterase)是一类糖蛋白,以多种同功酶形式存在于体内。一般可分为真性胆碱酯酶和假性胆碱脂酶。真性胆碱酯酶也称乙酰胆碱酯酶(acetylcholinesterase),主要存在于胆碱能神经末梢突触间隙,特别是运动神经终板突触后膜的皱折中聚集较多;也存在于胆碱能神经元内和红细胞中。此酶对于生理浓度的Ach作用最强,特异性也较高。一个酶分子可水解3×105分子Ach,一般常简称为胆碱酯酶。假性胆碱酯酶广泛存在于神经胶质细胞、血浆、肝、肾、肠中。对Ach的特异性较低,假性胆碱酯酶可水解其他胆碱酯类,如琥珀胆碱。
胆碱酯酶蛋白分子表面的活性中心有两个能与乙酰胆碱结合的部位,即带负电荷的阴离子部位和酯解部位。酯解部位含有一个由丝氨酸的羟基构成的酸性作用点和一个由组氨酸咪唑环构成的碱性作用点,两者通过氢键结合,增强了丝氨酸羟基的亲核活性,使之易于与乙酰胆碱结合(图7-1)。
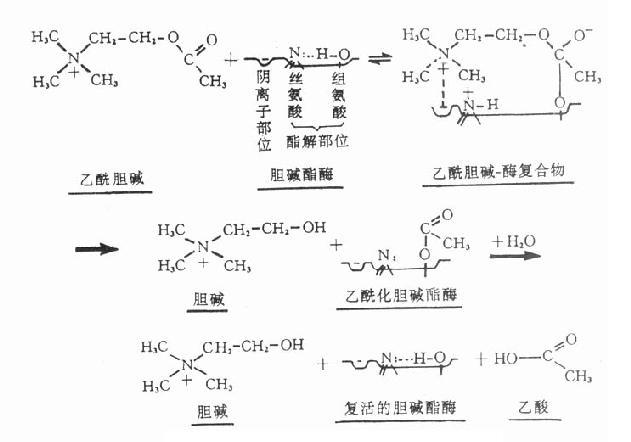
图7-1 胆碱酯酶水解乙酰胆碱碱过程的示意图
胆碱酯酶水解乙酰胆碱的过程可分为三个步骤:①乙酰胆碱分子结构中带正电荷的季铵阳离子头,以静电引力与胆碱酯酶的阴离子部位相结合;同时乙酰胆碱分子中的羰基碳与胆碱酯酶酯解部位的丝氨酸的羟基以共价键形式结合,形成乙酰胆碱和胆碱酯酶的复合物。②乙酰胆碱与胆碱酯酶复合物裂解成胆碱和乙酰化胆碱酯酶。③乙酰化胆碱酯酶迅速水解,分离出乙酸,酶的活性恢复。
第二节 抗胆碱酯酶药
抗胆碱酯酶药和乙酰胆碱一样,也能与胆碱酯酶结合,但结合较牢固,水解较慢,使酶失去活性,胆碱能神经末梢释放的乙酰胆碱便大量堆积,表现M及N样作用。
抗胆碱酯酶药可分为两类:一类是易逆性抗胆碱酯酶药,如新斯的明等;另一类为持久性抗胆碱酯酶药,如有机磷酸酯类。
一、易逆性抗胆碱酯酶药
新斯的明
新斯的明(neostigmine,prostigmine)是人工合成品。
化学结构中具有季铵基因,故口服吸收少而不规则。一般口服剂量为皮下注射量的10倍以上。不易透过血脑屏障,无明显的中枢作用。溶液滴眼时,不易透过角膜进入前房,故对眼的作用也较弱。
【作用机制】新斯的明能可逆地抑制胆碱酯酶,表现乙酰胆碱的M和N样作用。其结构中的季铵阳离头以静电引力与胆碱酯酶的阴离子部位结合,同时其分子中的羰基碳与酶的酯解部位丝氨酸羟基形成共价键结合,生成胆碱酯酶和新斯的明复合物。由复合物进而裂解成的二甲胺基甲酰化胆碱酯酶的水解速度较乙酰化胆碱酯酶的水解速度为慢,故酶被抑制的时间较长,但较有机磷酸酯类为短,故属易逆性类。二甲胺基甲酰化胆碱酯酶水解后,形成二甲胺基甲酸和复活的胆碱酯酶,酶的活性才得以恢复(图7-2)。
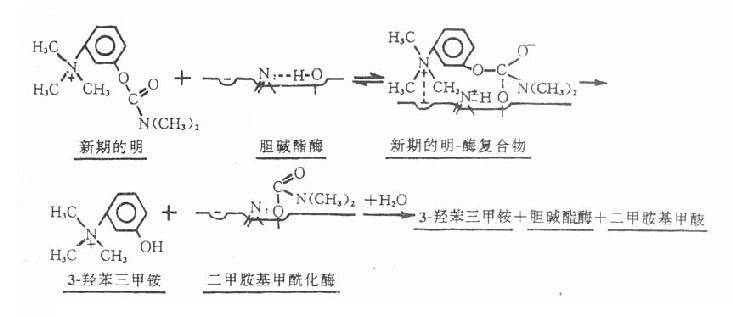
图7-2 新斯的明抑制胆碱酯酶过程的示意图
【药理作用】新斯的明对心血管、腺体、眼和支气管平滑肌作用较弱,对胃肠道和膀胱平滑肌有较强的兴奋作用;而对骨骼肌的兴奋作用最强,因为它除通过抑制胆碱酯酶而发挥作用外,还能直接激动骨骼肌运动终板上的N2胆碱受体以及促进运动神经末梢释放乙酰胆碱。
【临床应用】
1.重症肌无力其主要特征是肌肉经过短暂重复的活动后,出现肌无力症状。这是一种自身免疫性疾病。多数患者血清中有抗胆碱受体的抗体,其终板电位的胆碱受体数量减少70%~90%。皮下或肌内注射新斯的明后,约经15分钟左右即可使症状减轻,约维持2~4小时左右。除严重和紧急情况外,一般采用口服给药,因需经常给药,故要掌握好剂量,以免因过量转入抑制,引起“胆碱能危象”使肌无力症状加重。如疗效不够满意时,可并用糖皮质激素制剂或硫唑嘌呤等免疫抑制药。
2.腹气胀和尿潴留新斯的明能兴奋胃肠道平滑肌及膀胱逼尿肌,促进排气和排尿,适用于手术后腹气胀和尿潴留。
3.阵发性室上性心动过速在压迫眼球或颈动脉窦等兴奋迷走神经措施无效时,可用新斯的明,通过拟胆碱作用使心室频率减慢。
4.可用于非去极化型骨骼肌松弛药如筒箭毒碱过量时的解毒(见第九章)。
【不良反应】副作用较小,过量可产生恶心、呕吐、腹痛、肌肉颤动等,其M样作可用阿托品对抗。禁用于机械性肠梗阻、尿路梗塞和支气管哮喘患者。
其他易逆性抗胆碱酯酶药
吡啶斯的明(pyridostigmine)作用较新斯的明稍弱。主要用于治疗重症肌无力,因肌力改善作用维持较久,故适于晚上用药。也可用于手术后腹气胀和尿潴留。过量中毒的危险较少。禁忌证同新斯的明。
安贝氯铵(ambenonium,酶抑宁mytelase)的抗胆碱酯酶作用和兴奋骨骼肌作用都较新斯的明强,作用持续时间也较长,可口服给药。主要用于重症肌无力,不良反应和应用时注意事项与新斯的明相似。
毒扁豆碱(physostigmine)又名依色林(eserine),是从非洲出产的毒扁豆(Phsostigmavenenosum)种子中提出的生物碱,现已能人工合成。水溶液不稳定,滴眼剂应以pH4~5的缓冲液配制,否则易氧化成红色,疗效减弱,刺激性增大,应保存在棕色瓶内。为叔胺类化合物。易通过粘膜吸收。口服及注射都易吸收,也易于透过血脑屏障。具有与新斯的明相似的可逆性抑制胆碱酯酶的作用,吸收后在外周可出现拟胆碱作用。对中枢神经系统,小剂量兴奋,大剂量抑制,中毒时可引起呼吸麻痹。现主要局部用于治疗青光眼,能缩小瞳孔,降低眼内压,收缩睫状肌而引起调节痉挛等。常用0.005%溶液滴眼,作用较毛果芸香碱强而持久,但刺激性较大。又由于收缩睫状肌的作用较强,可引起头痛。滴眼后5分钟即出现缩瞳,眼内压下降作用可维持1~2天,调节痉挛现象消失较快。滴眼时应压迫内眦,避免药液流入鼻腔后吸收,引起中毒。
加兰他敏(galanthamine)也是可逆性抗胆碱酯酶药,体外抗胆碱酯酶效价约为毒扁豆碱的1/10。可用于重症肌无力,但疗效较差,也用于脊髓前角灰白质炎(小儿麻痹症)后遗症的治疗。
二、难逆性抗胆碱酯酶药一有机磷酸酯类
有机磷酸脂类(organophosphate)与胆碱酯酶结合后,时间稍久,胆碱酯酶即难以恢复,故称难逆性抗胆碱酯酶药,毒性很强。主要用作农业杀虫剂,有的可用作环境卫生杀虫剂,如敌百虫(dipterex)、乐果(rogor)、马拉硫磷(malathion)、敌敌畏(DDVP)和内吸磷(E1059)等。只有少数可医用作为滴眼剂如异氟磷(isoflurophate)等发挥缩瞳作用。
【化学】 有机磷酸酯类的基本化学结构见图7-3,R和R′多是烷基,如CH3、C2H5、C3H7等;O一般是氧或硫;X是烷氧基、烷硫基或卤素等。
【毒性作用机制】有机磷酸酯类的作用机制与可逆性抗胆碱酯酶相似,只是与胆碱酯酶的结合更为牢固。结合点也在胆碱酯酶的酯解部位丝氨酸的羟基。此羟基的氧原子具有亲核性,而有机磷酸酯类分子中的磷原子是亲电子性的,因此磷、氧二原子间易于形成共价键结合,生成难以水解的磷酰化胆碱酯酶(见图7-3)。结果使胆碱酯酶失去水解乙酰胆碱的能力,造成乙酰胆碱在体内大量积聚,引起一系列中毒症状。若不及时抢救,酶在几分钟或几小时内就“老化”。“老化”过程可能是磷酰化胆碱酯酶的磷酰化基团上的一个烷氧基断裂,生成更稳定的单烷氧基磷酰化胆碱酯酶。此时即使用胆碱酯酶复活药,也不能恢复酶的活性,必须等特新生的胆碱酯酶出现,才有水解乙酰胆碱的能力,此一恢复过程需15~30天。因此一旦中毒,必须迅速抢救,而且要持续进行。
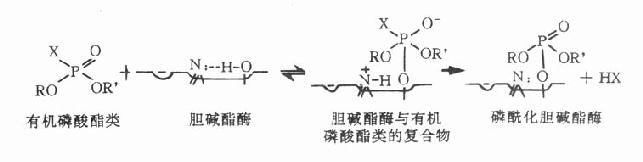
图7-3 有机磷酸酯类抗胆碱酯酶作用示意图
【体内过程及中毒途径】有机磷酸酯类在胃肠道、呼吸道、皮肤和粘膜都可吸收。经胃肠道吸收中毒的多由误食农药而引起。许多有机磷酸酯类容易挥发,因此也易吸入中毒。皮肤沾染了一定量的有机磷酸酯类时,也可引起全身性中毒。吸收后可分布全身,以肝浓度最高,大部分经肾排泄,一般不易蓄积。
【急性毒性】本类药物阻扰乙酰胆碱的消除,而乙酰胆碱的作用又极其广泛,故有机磷酸酯类的中毒症状表现多样化。轻者以M样症状为主,中度者可同时有M样症状和N样症状,严重中毒者除外周M样和N样症状外,还出现中枢神经系统症状。
1.M样症状
(1)眼瞳孔缩小,严重中毒者几乎全部出现,但中毒早期可能并不出现。因此,缩瞳不宜作为早期诊断的依据。此外,可出现视力模糊或因睫状肌痉挛而感觉眼痛者。
(2)腺体分泌增多,引起流涎和出汗。重者可口吐白沫,大汗淋漓。
(3)呼吸系统支气管平滑肌收缩和腺体分泌增加,引起呼吸困难甚至肺水肿。
(4)胃肠道由于胃肠道平滑肌的兴奋和有机磷酸酯类对胃肠道粘膜的刺激作用,可引起恶心、呕吐、腹痛和腹泻等。
(5)泌尿系统严重病倒可由于膀胱逼尿肌收缩而引起小便失禁。
(6)心血管系统 M样作用可引起心率减慢和血压下降,但由于同时有N样作用,故有时也可引起血压升高。
2.N样症状 交感和副交感神经节的N1受体和骨骼肌运动终板的N2受体都被激动。其神经节兴奋症状在胃肠道、腺体、眼等方面,是胆碱能神经占优势;因此结果和M样作用一致。在心血管,则去甲肾上腺素能神经占优势,故常表现为心收缩力加强、血压上升。N2受体激动则表现为肌束颤动,常先自小肌肉如眼睑、颜面和舌肌开始,逐渐发展至全身;严重者可因呼吸肌麻痹而死亡。
3.中枢症状有机磷酸类可使脑内乙酰胆碱含量升高,从而影响神经冲动在中枢突触的传递。表现为先兴奋、不安、谵语以及全身肌肉抽搐;进而由过度兴奋转入抑制,出现昏迷,并因血管运动中枢抑制而血压下降及呼吸中枢麻痹而呼吸停止。
【慢性毒性】多发生在生产农药的工人或长期接触农药的人员中。突出表现为血中胆碱酯酶活性显著而持久地下降,但与临床症状并不平行。主要症状有神经衰弱征候群和腹胀、多汗、偶有肌束颤动及瞳孔缩小。在慢性中毒的基础上,一次稍大剂量的吸收,也可能引起急性毒性发作。
第三节 胆碱酯酶复活药
胆碱酯酶复活药(cholinedterasereactivators)是一类能使已被有机磷酸酯类抑制的胆碱酯酶恢复活性的药物,其出现使有机磷酸酯类中毒的治疗获得了新的发展,它不但使单用阿托品所不能控制的严重中毒病倒得到解救,而且显着地缩短了一般中毒的病程。常用的有碘解磷定和氯磷定,二者均为肟类(oxime)化合物。
碘解磷定
碘解磷定(pyraloximemethoiodide)简称派姆(PAM),为最早应用的胆碱酯酶复活药。水溶性较低,水溶液不稳定,久置可释放出碘。
【药理作用】碘解磷定进入有机磷酸酯类中毒者体内,其带正电荷的季铵氮即与被磷酰化的胆碱酯酶的阴离子部位以静电引力相结合,结合后使其肟基趋向磷酰化胆碱酯酶的磷原子,进而与磷酰基形成共价键结合,生成磷酰化胆碱酯酶和碘解磷定复合物,后者进一步裂解成为磷酰化碘解磷定。同时使胆碱酯酶游离出来,恢复其水解乙酰胆碱的活性(见图7-4)。
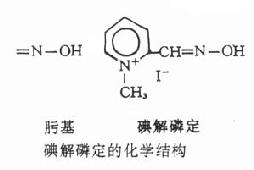
图7-4 碘解磷定复活胆碱酯酶过程示意图
磷酰化胆碱酯酶水解率很低(用┄ →表示),与PAM作用后产生复合物,
后者进一步裂解成胆碱酯酶和磷酰化解磷定,胆碱酯酶因而复活
此外,碘解磷定也能与体内游离的有机磷酸酯类直接接合,成为无毒的磷酰化碘解磷定,由尿排出,从而阻止游离的有机磷酸脂类继续抑制胆碱酯酶。
【解毒疗效】碘解磷定使酶复活的效果因不同有机磷酸酯类而异,例如对内吸磷、马拉硫磷和对硫磷中毒的疗效较好,对敌百虫、敌敌畏中毒的疗效稍差,而对乐果中毒则无效。因乐果中毒时所形成的磷酰化胆碱酯酶比较稳定,几乎是不可逆的,加以乐果乳剂含有苯,可能同时有苯中毒。
碘解磷定类恢复酶活性作用对骨骼肌最为明显,能迅速制止肌束颤动;对植物神经系统功能的恢复较差。对中枢神经系统的中毒症状(如昏迷)似也有疗效。
由于碘解磷定不能直接对抗体内积聚的乙酰胆碱的作用,故应与阿托品合用,以便及时控制症状。动物实验证明;单用碘解磷定仅能提高对氧磷(为内吸磷在体内的活性体)对小鼠致死量至2~4倍;单用阿托品时,也只提高2倍;两者合用时可提高128倍。
【体内过程】静脉注射后在肝、肾、脾、心等器官的念量较多,肺、骨骼肌和血中次之。从临床对中枢症状有效来看,似有少量进入脑内的可能。本药由肾脏排泄,部分在肝代谢。静脉注射碘解磷定后,30分钟内即有原形药物由尿中排出,6小时内约排出80%,故治疗中毒时需足量和反复给药。
【不良反应】一般治疗量时,毒性不大,但如静脉注射过快和剂量超过2g时,可产生轻度乏力、视力模糊、眩晕,有时出现恶心、呕吐和心动过速等。偶有咽痛和其它碘反应。剂量过大,碘解磷定本身也可抑制胆碱酯酶,加重有机磷酸酯类的中毒程度。
氯磷定
氯磷定(pyraloximemethylchloride, PAM-CI)的药理作用和用途与碘解磷定相似,但水溶性高,溶液较稳定,可肌内注射或静脉给药。特别适用于农村基层使用和初步急救。氯磷定经肾排泄也较快,生物t1/2约1.5小时。副作用较碘解磷定小,偶见轻度头痛、头晕、恶心、呕吐等。
由于氯磷定给药方便,不良反应较小,现已逐渐取代了碘解磷定。
第四节 有机磷酸酯类中毒的防治
1.预防按照预防为主的方针,如能严格执行农药的管理制度,加强生产及使用农药的劳动保护措施,有机磷酸酯类农药中毒是可以预防的。
2.急性中毒的治疗
(1)迅速消除毒物以免继续吸收发现中毒时,应立即将患者移出有毒场所。对经皮肤侵入的中毒者,应清洗皮肤,最好用温水和肥皂彻底清洗。经口中毒时,应首先抽出胃液和毒物,并立即以微温的2%碳酸氢钠溶液或1%食盐水反复洗胃,直至洗出液不再有有机磷农药的特殊气味为止。然后再给硫酸镁导泻。敌百虫口服中毒时不能用碱性溶液洗胃,因它在碱性溶液中可转化成敌敌畏而增加毒性。眼部染毒,可用2%碳酸氢钠溶液或0.9%盐水冲洗数分钟。
(2)积极使用解毒药须及早、足量、反复地注射阿托品,能迅速解除有机磷酸酯类中毒时的M样症状,缓解呼吸道和胃肠道平滑肌的兴奋性;也能解除一部分中枢神经系统中毒症状,使昏迷病人苏醒。此外,大剂量阿托品还具阻断神经节作用,从而对抗有机磷酸酯类的兴奋神经节的作用。但阿托品对N2受体无效,因此不能制止骨骼肌震颤,对中毒晚期的呼吸肌麻痹也无效,也无复活胆碱酯酶作用,疗效不易巩固。因此须与胆碱酯酶复活药合用,对中度和重度中毒病例,更须如此。但在两药合用的患者,当胆碱酯酶复活后,机体可恢复对阿托品的敏感性,易发生阿托品中毒。因此,两药合用时,应适当减少阿托品的剂量。
3,慢性中毒的治疗对慢性中毒,目前尚无特殊治疗方法,使用阿托品和解磷定类药物,疗效并不理想。对生产工人,或经常接触者,当血中胆碱酯酶活性下降至50%以下时,应暂时脱离与有机磷酸酯类的接触,以免中毒。
胆碱酯酶药和胆碱酯酶复活药 制剂及用法
水杨酸毒扁豆碱(physostigmine salicylate) 滴眼液或眼膏, 0.25%,每4小时一次,或按需要决定滴眼次数,溶液变红色后不可用。
溴化新斯的明(neostigmine bromide)口服,15mg/次,3次/日或按需要而定。极量:30mg/次,100mg/日。
甲基硫酸新斯的明(neostigmine methylsulfate) 皮下或肌内注射,0.25~0.5mg/次,极量,1mg/次。
溴吡斯的明(phyridostigmine bromide)口服,60mg/次,3次/日,极量,120mg/次,360mg/日。
氢溴酸加兰他敏(galanthamine hydrobromide)肌内注射,2.5~10mg/次,1次/日。
碘解磷定(pyraloxime methoiodide)0.5~1g/次,缓慢静脉注射。
氯磷定(pyraloxime methylchloride)0.25~0.75g/次,肌内注射。0.5~0.75g/次,加于等渗盐水500ml中,静脉滴注。
第八章 胆碱受体阻断药(Ⅰ)M胆碱受体阻断药
第一节 阿托品和阿托品类生物碱
胆碱受体阻断药(cholinoceptorblocking drugs)能与胆碱受体结合而不产生或极少产生拟胆碱作用,却能妨碍乙酰胆碱或胆碱受体激动药与胆碱受体的结合,从而拮抗拟胆碱作用。按其对M和N受体选择性的不同,可分为M1,M2,M3胆碱受体阻断药和N1,N2胆碱受体阻断药。按用途的不同,可分为平滑肌解痉药,神经节阻断药,骨骼肌松弛药和中枢性抗胆碱药。本章先讨论以阿托品为代表的M胆碱受体阻断药。
【来源与化学】 从植物中提取的M胆碱受体阻断药有阿托品、东莨菪碱和山莨菪碱等,其植物来源如表8-1。天然存在于植物的是左旋莨菪碱,在提取过程中,得到比较稳定的消旋莨菪碱,即阿托品。东莨菪碱是左旋品。左旋体较其右旋体作用强许多倍。
表8-1 阿托品类生物碱及其来源
| 植物名称 | 主要生物碱 |
| 颠茄(Atropa belladonna) | 莨菪碱 |
| 曼陀罗(Datura stramonium) | 莨菪碱 |
| 洋金花(Datura sp) | 东莨菪碱 |
| 莨菪(Hyoscyamus niger) | 莨菪碱 |
| 唐古特莨菪(Scopolia tangutica) | 山莨菪碱 |
阿托品及东莨菪碱的化学结构
阿托品
【药理作用】阿托品(atropine)的作用机制为竞争性拮抗乙酰胆碱或胆碱受体激动药对M胆碱受体的激动作用。阿托品与M胆碱受体结合,因内在活性很小,一般不产生激动作用,却能阻断乙酰胆碱或胆碱受体激动药与受体结合,结果拮抗了它们的作用。阿托品对M受体有相当高的选择性,但很大剂量或中毒剂量也有阻断神经节N1受体的作用。阿托品对各种M受体亚型的选择性较低,对M1,M2,M3受体都有阻断作用。
阿托品的作用非常广泛,各器官对阿托品敏感性不同。随剂量的增加可依次出现下列现象:腺体分泌减少,瞳孔扩大和调节麻痹,膀胱和胃肠道平滑肌的兴奋性下降,心率加快,中毒剂量则出现中枢作用。现依次叙述如下:
1.腺体 阿托品因阻断M胆碱受体而抑制腺体分泌;唾液腺和汗腺最敏感,在用0.5mg阿托品时,就显著受抑制,引起口干和皮肤干燥,同时泪腺和呼吸道分泌也大为减少。较大剂量可减少胃液分泌,但对胃酸浓度影响较小,因胃酸分泌还受体液因素如胃泌素的调节,同时又因胃中HCO3-的分泌也受到抑制。
2.眼 阿托品阻断M胆碱受体,因而使瞳孔括约肌和睫状肌松弛,出现扩瞳、眼内压升高和调节麻痹(图6-1),导致畏光。这些作用在局部滴眼和全身给药时,都可出现,需要注意。
(1)扩瞳阿托品松弛瞳孔括约肌,故使去甲肾上腺素能神经支配的瞳孔扩大肌的功能占优势,从而扩瞳。
(2)眼内压升高由于瞳孔扩大,使虹膜退向四周边缘,因而前房角间隙变窄,阻碍房水回流入巩膜静脉窦,造成眼内压升高。因此阿托品禁用于青光眼或有眼内压升高倾向者。
(3)调节麻痹阿托品能使睫状肌松弛而退向外缘,从而使悬韧带拉紧,使晶状体变为扁平,其折光度减低,只适于看远物,而不能将近物清晰地成像于视网膜上,故看近物模糊不清,这一作用称为调节麻痹。
3.平滑肌阿托品能松弛许多内脏平滑肌,对过度活动或痉挛的内脏平滑肌,松弛作用较显著。它可抑制胃肠道平滑肌的强烈痉挛,降低蠕动的幅度和频率,缓解胃肠绞痛;对膀胱逼尿肌也有解痉作用;但对胆管、输尿管和支气管的解痉作用较弱。胃肠道括约肌的反应主要取决于括约肌的机能状态。例如胃幽门括约肌痉挛时,阿托品具有松弛作用,但作用不显著和不恒定。阿托品对子宫平滑肌影响较小。
4.心脏
(1)心率 治疗剂量阿托品(0.5mg)在一部分病人可使心率轻度短暂地减慢,一般每分钟减少4~8次,这可能是阿托品阻断突触前膜M1受体,从而减少突触中Ach对递质释放的抑制作用所致。较大剂量(1~2mg)则阻断窦房结起搏点的M2受体,解除迷走神经对心脏的抑制作用,使心率加速,加速程度取决于迷走神经张力;在迷走神经张力高的青壮年,心率加速作用显著,如肌内注射2mg阿托品,心率可增加35~40次/分。
(2)房室传导阿托品能拮抗迷走神经过度兴奋所致的传导阻滞和心律失常。但在心肌梗塞时,要慎用阿托品,由于其加速心率,加重心肌缺血缺氧,可能会激发室颤。
5.血管与血压治疗量阿托品对血管与血压无显著影响,这可能因许多血管缺少胆碱能神经支配。大量阿托品有解除小血管痉挛的作用,尤其以皮肤血管扩张为显著,可产生潮红温热。扩血管作用的机制未明,但与抗M胆碱作用无关,可能机体对阿托品所引起的体温升高的代偿性散热反应,也可能是阿托品的直接扩张血管的作用。
6.中枢神经系统 较大剂量1~2mg可较度兴奋延髓和大脑,2~5mg时兴奋加强,出现焦虑不安、多言、谵安;中毒剂量(如10mg以上)常致幻觉、定向障碍、运动失调和惊厥等,也可由兴奋转入抑制,出现昏迷及呼吸麻痹。
【体内过程】口服吸收迅速,1小时后血药浓度即达峰值,生物处用度为50%,t1/2为4小时,作用可维持约3~4小时。吸收后很快离开血液而分布于全身组织,可透过血脑屏障,也能通过胎盘进入胎儿循环。阿托品从其它粘膜也可吸收。肌内注射后12小时内有85%~88%经尿排出,其中原形阿托品约占1/3,其余为水解和与葡萄糖醛酸结合的代谢物,在粪及其它分泌物包括乳汁中仅发现少量阿托品。
【临床应用】阿托品在临床上有广泛的用途。
1.解除平滑肌痉挛适用于各种内脏胶痛,如胃肠绞痛及膀胱刺激症状如尿频、尿急等,疗效较好。对胆绞痛及肾绞痛的疗效较差。在治疗这两种绞痛时,常和吗啡类镇痛药合用。用阿托品治疗遗尿症,是利用其松弛膀胱逼尿肌的作用。一般用硫酸阿托品0.5~1mg皮下注射,对于轻症可口服其0.3mg的片剂,也可口服更温和的含莨菪碱颠茄酊或复方颠茄片(见制剂及用法项)。
2.制止腺体分泌用于全身麻醉前给药,以减少呼吸道分泌,防止分泌物阻塞呼吸道及吸入性肺炎的发生,也可用于严重的盗汗和流涎症。
3.眼科
(1)虹膜睫状体炎 0.5%~1%阿托品溶液滴眼,松弛虹膜括约肌和睫状肌,使之充分休息,有利于炎症的消退;同时还可预防虹膜与晶体的粘连。
(2)检查眼底如需扩瞳,可用阿托品溶液滴眼,但因其扩瞳作用可维持1~2周,调节麻痹也可维持2~3天,视力恢复较慢,目前常以作用较短的后马托品溶液取代之。
(3)验光配眼镜滴用阿托品类可使睫状肌的调节功能充分麻痹,晶状体固定,以便正确地检验出晶状体的屈光度,但阿托品作用持续时间过长,现已少用。只有儿童验光时,仍用之。因儿童的睫状肌调节机能较强,须阿托品发挥充分的调节麻痹作用。
4.缓慢型心律失常临床上常用阿托品治疗迷走神经过度兴奋所致窦房阻滞、房室阻滞等缓慢型心律失常,还可用于治疗继发于窦房结功能低下而出现的室性异位节律。
5.抗休克对暴发型流行性脑脊髓膜炎、中毒性菌痢、中毒性肺炎等所致的感染性休克,可用大剂量阿托品治疗,可能解除血管痉挛,舒张外周血管,改善微循环。对于休克伴有心率过速或高烧者,不用阿托品。
6.解救有机磷酸酯类中毒(见第七章)和有些毒蕈类的中毒。
【不良反应及中毒】阿托品的作用广泛,当利用某一作用时,其它作用便成为副作用,这应预先告诉病人,以免惊慌。不同剂量的阿托品的不良反应表现大致如下:
0.5mg:轻微心率减慢,略有口干及乏汗;1mg:口干,心率加速,瞳孔轻度扩大;2mg:心悸,显著口干,瞳孔扩大,有时出现视近物模糊;5mg:上述症状加重,语言不清,烦躁不安,皮肤干燥发热,小便困难,肠蠕动减少;10mg以上:上述症状更重,脉速而弱,中枢兴奋现象严重,呼吸加快加深,出现谵安、幻觉、惊厥等。严重中毒时,可由中枢兴奋转入抑制,产生昏迷和呼吸麻痹等。
阿托品的最低致死量在成人约为80~130mg ,儿童约为10mg。
误服中毒量的颠茄果、曼陀罗果、洋金花或莨菪根茎等,也可逐次出现上述症状。中毒的解救除洗胃排出胃内药物等措施外,可注射新斯的明、毒扁豆碱或毛果芸香碱等。当解救有机磷酸酯类的中毒而用阿托品过量时,当然不能用新斯的明、毒扁豆碱等抗胆碱酯酶药。中枢症状明显时,可用安定或短效巴比妥类,但不可过量,以避免与阿托品类药的中枢抑制作用产生协同作用。
【禁忌证】青光眼及前列腺肥大者禁用,后者因其可能加重排尿困难。老年人慎用。
山莨菪碱
山莨菪碱(anisodamine)是我国从茄科植物唐古特莨菪中提出的生物碱。
山莨菪碱对抗乙酰胆碱所致的平滑肌痉挛和抑制心血管的作用,与阿托品相似而稍弱,同时也能解除血管痉挛,改善微循环。但它的抑制唾液分泌和扩瞳作用则仅为阿托品的1/20~1/10。还因不易穿透血脑屏障,中枢兴奋作用很少。和阿托品相比,其毒性较低,解痉作用的选择性相对较高,副作用与阿托品相似。适用于感染性休克、内脏平滑肌绞痛。青光眼禁用。
东莨菪碱
东莨菪碱(scopolamine)对中枢神经的抑制作用较强,小剂量主要表现为镇静,较大剂量时,则致催眠作用。个别人可能出现不安、激动等类似阿托品的兴奋症状。主要用于麻醉前给药。此外还有抗晕动病和抗震颤麻痹的作用和用途。防晕作用可能和抑制前庭神经内耳功能或大脑皮层及抑制胃肠道蠕动有关,可与苯海拉明合用以增加效果。预防性给药效果好,如已发生呕吐再用药则疗效差。也用于妊娠呕吐及放射病呕吐。对震颤麻痹症有缓解流涎、震颤和肌肉强直的效果,可能与其拮抗中枢神经的乙酰胆碱作用有关。
我国医药工作者阐明了中药麻醉处方的主要药物洋金花,而洋金花的主要成分就是东莨菪碱。故也用东莨菪碱来代替洋金花作为中药麻醉剂。
本品的外周作用和阿托品相似,仅在作用强度上略有不同。扩瞳、调节麻痹和抑制腺体分泌较阿托品强,而对心血管作用较弱。由于能升高眼内压,故青光眼患者忌用。
第二节 阿托品的合成代用品
由于阿托品用于眼科作用太持久,用于内科,副作用又较多,针对这些缺点,通过改变其化学结构,合成了不少代用品,主要有两类,即扩瞳药和解痉药。
近年尚出现一类以哌仑西平(pirenzepine)为代表的M1受体阻断药,选择性地抑制胃酸分泌,用于消化性溃疡病,成为一种新类型。
一、合成扩瞳药
后马托品和托吡卡胺
后马托品(homatropine)的扩瞳作用与调节麻痹作用都比阿托品明显短暂,调节麻痹作用约在用药后24~36小时消退,适用于一般眼科检查。其调节麻痹作用高峰出现较快,但不如阿托品完全,特别是对于儿童。托吡卡胺(tropicamide)的特点是起效快而持续时间最短。
表8-2 几种扩瞳药滴眼作用的比较
| 药 物 | 浓度(%) | 扩瞳作用 | 麻痹调节作用 | ||
| 高峰(分) | 消退(日) | 高峰(小时) | 消退(日) | ||
| 硫酸阿托品 | 1.0 | 30~40 | 7~10 | 1~3 | 7~12 |
| 氢溴酸后马托品 | 1.0 | 40~60 | 1~2 | 0.5~1 | 1~2 |
| 托吡卡胺 | 0.5~1.0 | 20~40 | 1/4 | 1/2 | <1/4 |
二、合成解痉药
研究其合成代用品的目的在于寻找选择性较高的药物,但迄今尚未找到显着优于阿托品者。现有代用品的作用性质、副作用、禁忌证与阿托品相似,仅有量的差别。
(一)季铵类解痉药
常用的为丙胺太林(普鲁本辛,propanthelinebromide)。它有不列特点:如口服给药吸收较差,不易透过血脑屏障,很少发生中枢作用;注射给药时对胃肠道平滑肌的解痉作用较强,并有不同程度的神经节阻断作用。中毒量可致神经肌肉传递阻断,引起呼吸麻痹。
除上述特点外,它对胃肠道M胆碱受体的选择性较高,治疗剂量时抑制胃肠道平滑肌的作用较强和持久,并能不同程度地减少胃液分泌。一次用量(15mg)维持作用6小时。可用于胃十二指肠溃疡、胃肠痉挛和妊娠呕吐等。
(二)叔胺类解痉药
胃复康(benactyzinehydrochloride)含叔胺基因,口服较易吸收,解痉作用较明显,也有抑制胃液分泌作用。此外尚有安定作用。适用于兼有焦虑症的溃疡病、胃酸过多、肠蠕动亢进或膀胱刺激症状的患者。
胆碱受体阻断药(Ⅰ)M胆碱受体阻断药 制剂及用法
硫酸阿托品(atropinesulfate )口服:0.3~0.6mg/ 次,3次/日。皮下、肌内或静脉注射0.5mg/次。滴眼液:0.5%,1%。眼膏:1%。极量:口服,1mg/次,3mg/日。
颠茄酊(tincturebelladonna)由颠茄叶制成的酊剂,主要成分是莨菪碱,所含生物碱作为莨菪碱计算应为0.33%。口服,0.3~1.0ml/次,极量:口服:1.5ml/次,4.5ml/日。一般用前加水配成含6%酊剂的药水,10ml/次,3次/日。
颠茄片(tab.belladonna)每片含颠茄浸膏10mg(颠茄浸膏的主要成分是莨菪碱,所含生物碱按莨菪碱计算应为1%)。口服1~2片/次,3次/日。
氢溴酸山莨菪碱(anisodaminehydrobromide)口服,5~10mg/次,3次/日。静脉注射或肌内注射,5~10mg/次,1~2次/日。
氢溴酸东莨菪碱(scopolaminehydrobromide)口服,0.2~0.3mg/次,3次/日。皮下或肌内注射,0.2~0.5mg/次。极量:口服,0.6mg/次,2mg/日;注射0.5mg/次,1.5mg/日
氢溴酸后马托品(homatropinehydrobromide)滴眼液:1%~2%,滴眼,按需要而定滴数。
溴化丙胺太林(propanthelinebromide)口服,15mg/次,3次/日。
胃复康(benactyzinehydrochloride)口服,1mg/次,3次/日。
第九章 胆碱受体阻断药(Ⅱ)-N胆碱受体阻断药
第一节 N1胆碱受体阻断药-神经节阻断药
【药理作用】 N1胆碱受体阻断药(N1-cholinoceptor blocking drugs)能选择性地与神经节细胞的N1胆碱受体结合,竞争性地阻ACh与受体结合,使ACh 不能引起节细胞的除极化,从而阻断了神经冲动在神经节中的传递,故也称神经节阻断药(ganglion blocking drugs)。
神经节阻断药对交感神经节和副交感神经节都有阻断作用,它对效应器的具体效应则视两类神经对该器官的支配以何者占优势而定。例如交感神经对血管的支配占优势,用神经节阻断药后,则使血管,特别是小动脉扩张,总外周阻力下降,加上静脉扩张,回心血量和心输出量减少,结果常使血压显著下降。又如在胃肠道、眼、膀胱等平滑肌和腺体则以副交感神经占优势,因此,用药后常出现便秘、扩瞳、口干和尿潴留等。
【临床应用】神经节阻断药在过去曾用于治疗高血压,但由于其作用过于广泛,副作用多,且其降压作用过强过快,故现已少用于治疗高血压,主要用于麻醉辅助药以发挥控制性降压作用。曾经应用的神经节阻断药有季铵类的六甲又铵(hexamethonium,C6)以及非季铵类的美加明(mecamylamine)和咪噻吩(trimethaphan)等。
第二节 N2胆碱受体阻断药—骨骼肌松弛药
N2胆碱受体阻断药(N2-cholinoceptorblocking drugs)也称骨骼肌松弛药(skeletalmuscular relaxants),简称肌松药,阻断神经肌肉接头的N2胆碱受体妨碍神经冲动的传递,使骨骼肌松弛,便于在较浅的麻醉下进行外科手术。根据其作用方式的特点,可分为除极化型和非除极化型两类。
一、除极化型肌松药
这类药物与运动终板膜上的N2胆碱受体相结合,产生与乙酰胆碱相似但较持久的除极化作用,使终板不能对乙酰胆碱起反应(处于不应状态),骨骼肌因而松弛。除极化型肌松药(depolarizingmuscular relaxants)的特点是:①常先出现短时的肌束颤动。这是由于不同部位的骨骼肌在药物作用下除极化出现的时间先后不同所致。②连续用药可产生快速耐受性。③抗胆碱酯酶药不仅不能拮抗这类药的肌松作用,却反能加强之。④在临床用量时,并无神经节阻断作用。⑤肌松作用可有2个时相。第1相是持久除极阻断,此时终板四周钠道关闭而不可激活,故致肌松,大量或反复给药后进入第2相,是N2受体脱敏所致,周围钠通道关闭而激活,呈可兴奋状。
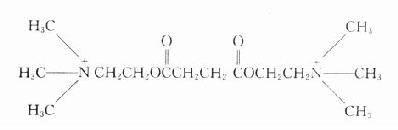
琥珀胆碱
琥珀胆碱(succinylcholine)又称司可林(scoline),由琥珀酸和两分子胆碱组成。
【药理作用】静脉注射10~30mg琥珀胆碱后,患者先出现短时间肌束颤动。一分钟内即转为松弛,通常从颈部肌肉开始,逐渐波及肩胛、腹部和四肢。约在2分钟时肌松作用最明显,在5分钟内作用消失。为了达到较长时间的肌松作用,可采用持续静脉滴注法。肌松作用出现时,四肢和颈部肌肉所受影响最大,而、舌、咽、喉部肌肉次之,呼吸肌无力现象不明显,肺通气量仅降低25%。
【体内过程】琥珀胆碱在血液中被血浆假性胆碱酯酶迅速水解,1分钟内血浆中总量的90%已被水解,其余部分在肝中被水解。首先水解成琥珀单胆碱,肌松作用大为减弱;然后又缓慢水解成为琥珀酸和胆碱,肌松作用消失。仅有不到2%琥珀胆碱以原形从肾排泄。新斯的明抑制血浆假性胆碱酯酶而加强和延长琥珀胆碱的作用。
【临床应用】静脉注射作用快而短暂,对喉肌的麻痹力强,故适用于气管内插管、气管镜、食道镜等短时的操作。静脉滴注适用于较长时手术。
因可引起强烈的窒息感,故对清醒的患者禁用。一般继硫喷妥钠静脉注射后给药,成人短时外科手术,通常用1%~5%氯化琥珀胆碱溶液静脉注射,剂量为0.5~1.0mg/kg,为了达到长时间肌松作用,可用5%葡萄糖液配制后静脉滴注,对此药反应的个体差异较大,须按反应情况控制滴速,以达到满意的肌松程度。
【不良反应及应用注意】过量致呼吸肌麻痹,用时必须备有人工呼吸机。肌束颤动的危害是损伤肌梭,引起肌肉酸痛甚至有形态的改变。一般3~5天自愈。
使眼外骨骼肌短暂地收缩,能升高眼内压,青光眼和白内障晶体摘除术患者禁用。
遗传性血浆胆碱酯酶活性降低的特异质病人和有机磷酸酯类中毒者对琥珀胆碱高度敏感,易中毒,应提高警惕。国外报道3000人中有一人具有上述遗传性缺陷,我国人可能少些,但仍应注意。特异质反应尚可表现为恶性高热。
此外,琥珀胆碱能使肌肉持久性除极化而释出钾离子,使血钾升高,故在烧伤,广泛性软组织损伤、偏瘫和脑血管意外的患者(一般血钾已较高),禁用琥珀胆碱,以免产生高血钾症性心跳骤停。
有些氨基甙类抗生素和多肽类抗生素在大剂量时,也有肌肉松弛作用,与琥珀胆碱合用时,易致呼吸麻痹,应注意。
严重肝功能不全、营养不良和电解质紊乱者慎用。
二、非除极化型肌松药
又称竞争型肌松药(competitivemuscular relaxants),此类药物与运动神经终板膜上的N2胆碱受体结合,能竞争性地阻断ACh的除极化作用,使骨骼肌松弛。
竞争型肌松药的作用特点是:①在同类阻断药之间有相加作用。②吸入性全麻药(特别如乙醚等)和氨基甙类抗生素(如链霉素)能加强和延长此类药物的肌松作用。③与抗胆碱酯酶药之间有拮抗作用,故过量时可用适量的新斯的明解毒。④兼有程度不等的神经节阻断作用,可使血压下降。
筒箭毒碱
筒箭毒碱(d-tubocurarine)是从南美洲生产的植物浸膏箭毒(curarc)中提出的生物碱,右旋体具药理活性。口服难吸收,静脉注射后3~4分钟即产生肌松作用,头颈部小肌肉首先受累;然后波及四肢、躯干和颈部的其它肌肉;继而因肋间肌松弛,出现腹式呼吸;如剂量过大,进而累及膈肌,病人可因呼吸肌麻痹而死亡。由于筒箭毒碱的作用维持时间较短,如及时进行人工呼吸,可挽救生命。同时可用新斯的明解救。
因筒箭毒碱具有神经节阻断和促进组胺释放等作用,故可使血压短时下降、心跳减慢、支气管痉挛和唾液分泌过多。禁用于重症肌无力、支气管哮喘和严重休克患者。10岁以下儿童对此药高敏反应较多,故不宜用于儿童。由于本药来源有限,并有一定缺点,故已少用。国产的氯甲左箭毒(dimethyl-L-curine dimethochloride)可供代用。
加拉碘铵
加拉碘铵(三碘季铵酚gallaminetricthiodide,flexedil)是含有三个季铵基团的人工合成的非除极化型肌松药。
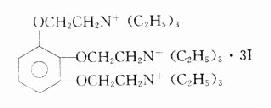
肌松作用和筒箭毒碱相似,但无阻断神经节和释放组胺的作用,却有较强的阿托品样作用,能明显解除迷走神经的张力,使心率加快,血压轻度升高,心输出量增加。静脉给药大部分经肾排出。重症肌无力、心动过速、高血压及肾功能不全者忌用。
泮库溴铵类
近年研制出几种更较安全的新的非除极化型肌松药,它们是:
泮库溴铵(pancuronium),肌松作用较筒箭毒碱强5~10倍。静脉注射量为0.04~0.1mg/kg,在体内约20%在肝代谢,余者经肾与胆汁排泄。
维库溴铵(vecuronium),肌松作用强,静脉注射0.08mg/kg经3~5分钟,血药浓度即达峰值,维持效应20~35分钟,介于琥珀胆碱与筒箭毒碱之间。在体内部分经胆汁排泄,部分被酯解(非胆碱酯酶所催化)。
阿曲库铵(atracutium),静脉注射0.4mg/kg也迅速见效,维持20~35分钟,也属中等强度肌松药。在体内很少被排泄,大部分被酯解。
以上三药都不阻断神经节N1受体,较少促组胺释放。不良反应也较少。适于在各类手术、气管插管术、破伤风及惊厥时作肌松药用。
胆碱受体阻断药(Ⅱ)-N胆碱受体阻断药 制剂及用法
氯化琥珀胆碱(succinylcholinechloride)用量见上文。极量:0.25g/次。
氯化筒箭毒碱(tubocurarinechloride)6~9mg/次,重复时用量减半。
加拉碘铵(三碘季铵酚gallaminetriethiodide,flexedil)1mg/kg/次,重复时用量减半。
第十章 肾上腺素受体激动药
第一节 化学、构效关系及分类
一、化学
肾上腺素受体激动药(adrenoceptoragonists)与肾上腺素受体结合,激动受体,产生肾上腺素样的作用。它们都是胺类,而作用又与兴奋交感神经的效应相似,故又称拟交感胺类(sympathomimeticamine),其基本化学结构是β-苯乙胺。
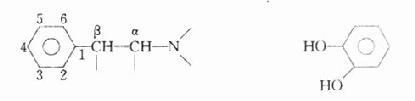
β-苯乙胺 儿茶酚
表10-1 肾上腺素受体激动药的化学结构和受体选择性
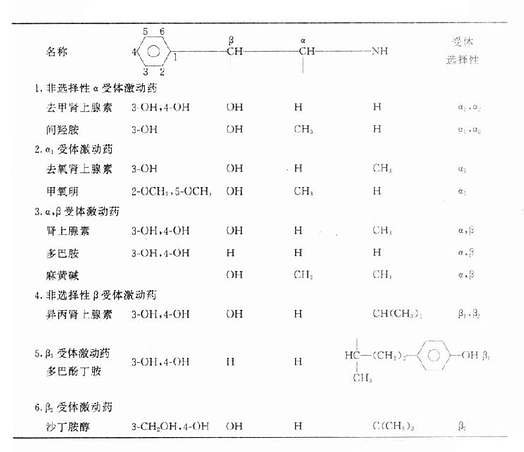
二、构效关系
1.肾上腺素、去甲肾上腺素、异丙肾上腺素和多巴胺等在苯环3、4位C上都有羟基形成儿茶酚,故称儿茶酚胺类(catecholamines)。它们的外周作用强而中枢作用弱,作用时间短。如果去掉一个羟基,其外周作用将减弱,而作用时间延长,特别是去掉3位羟基,如将两个羟基都去掉,则外周作用减弱,中枢作用加强,如麻黄碱。
2,烷胺侧链α碳原子上的氢如被甲基取代,可阻碍MAO的氧化,作用时间延长。易被摄取1所摄入在神经元内存在时间长,从而发挥促进递质释放的作用,如间羟胺和麻黄碱。
3.氨基上氢原子如被取代,则药物对α、β受体选择性将发生变化。取代基团从甲基到叔丁基,对α受体的作用逐渐减弱,β受体作用却逐渐加强。
三、分类
按其对不同肾上腺素受体的选择性而分为三大类:①α受体激动药(α-adrenoceptoragonists)。②α,β受体激动药(α,β-adrenoceptoragonists)。③β受体激动药(β-adrenoceptoragonists)。
第二节 α受体激动药
一、α1,α2受体激动药
去甲肾上腺素
【来源及化学】 去甲肾上腺素(noradrenaline ,NA;norepinephrine,NE)是去甲肾上腺素能神经末梢释放的主要递质,也可由肾上腺髓质少量分泌。药用的是人工合成品,化学性质不稳定,见光易失效,在中性尤其在碱性溶液中迅速氧化变为粉红色乃至棕色失效。在酸性溶液中较稳定。
【体内过程】
1.皮下注射时,因血管剧烈收缩,吸收很少,且易发生局部组织坏死,一般采用静脉滴注法给药。
2.分布 静脉内注射[3H]-去甲肾上腺素后,很快自血中消失,较多分布于受去甲肾上腺素能神经支配的心脏等脏器以及肾上腺髓质中。外源性去甲肾上腺素很少到达脑组织,可能是不易透过血脑屏障之故。
3.摄取(uptake)内源性和外源性去甲肾上腺素都可被去甲肾上腺素能神经末梢和非神经细胞所摄取(分别称为摄取1和摄取2,见第五章)。被摄取的去甲肾上腺素大多又经囊泡摄取贮存起来。被摄取入非神经细胞内者,大多被代谢而失活。
4.代谢 灭活去甲肾上腺素的酶,COMT和MAO广泛存在于许多组织内,特别是肝、肾、肠和血管壁细胞中。去甲肾上腺素能神经元内主要含MAO,也曾发现有COMT,但含量极少。肝是外源性去甲肾上腺素的主要代谢器官。大部分注入的去甲肾上腺素首先在COMT催化下,代谢为活性很低的间甲去甲肾上腺素,其中一部分再经MAO的作用脱胺形成3-甲氧-4羟扁桃酸(vanillyl mandelic acid,VMA)。从去甲肾上腺素能神经释放的去甲肾上腺素则主要先被摄取入神经末梢,部分经MAO脱胺,然后在非神经细胞内经COMT转甲基最后仍形成VMA。部分去甲肾上腺素或其间甲化合物尚可与硫酸或葡萄糖醛酸结合(图10-1)。
由于去甲肾上腺素进入体内后迅速被摄取和代谢,故作用短暂。
5.排泄 正常人24小时尿中儿茶酚胺的代谢产物以VMA为主,约占儿茶酚胺代谢产物总量的90%,静脉注射或滴注去甲肾上腺素后96小时尿中代谢物的比率为:VMA32%,原形去甲肾上腺素4%~16%,与葡萄糖醛酸或硫酸结合的去甲肾上腺素8%,结合的间甲去甲肾上腺素18%。
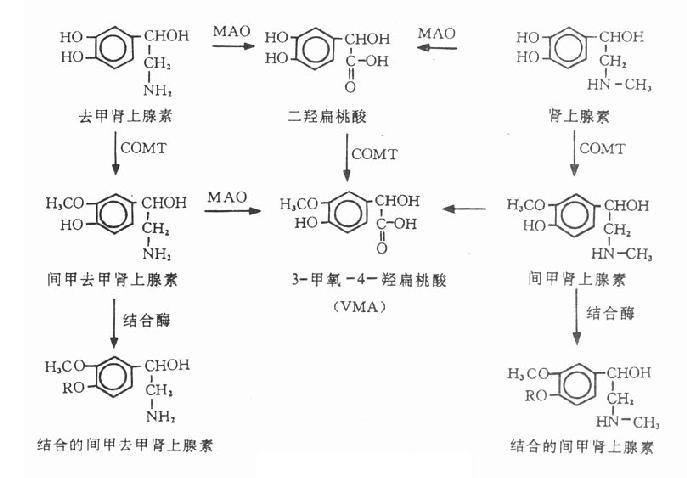
图10-1 儿茶酚胺的代谢
R:硫酸根或葡萄糖醛酸根
【药理作用】非选择性激动α1和α2受体,与肾上腺素比较在某些器官其α作用比肾上腺素略弱,对心脏β1受体作用较弱,对β2受体几无作用。
1.血管 激动血管的α1受体,使血管收缩,主要是使小动脉和小静脉收缩。皮肤粘膜血管收缩最明显,其次是对肾脏血管的收缩作用。此外脑、肝、肠系膜甚至骨骼肌的血管也都呈收缩反应。冠状血管舒张,这主要由于心脏兴奋,心肌的代谢产物(如腺苷)增加,从而舒张血管所致,同时因血压升高,提高了冠状血管的灌注压力,故冠脉流量增加。在一定情况下,也可激动血管壁的去甲肾上腺素能神经突触前α2受体,抑制递质的释放(见第五章)。
2,心脏 作用较肾上腺素为弱,激动心脏的β1受体,使心肌收缩性加强,心率加快,传导加速,心搏出量增加。在整体情况下,心率可由于血压升高而反射性减慢。过大剂量,心脏自动节律性增加,也会出现心律失常,但较肾上腺素少见。
表10-2 拟肾上腺素药基本作用的比较
| 分类 | 对不同肾上腺素受体作用的比较 | 作用方式 | ||||
| α受体 β1受体 β2受体 | 直接作用于受体 释放递质 | |||||
| 去甲肾上腺素 | α | +++ | ++ | ± | + | |
| 间羟胺 | α | ++ | + | + | + | + |
| 去氧肾上腺素 | α | ++ | ± | ± | + | ± |
| 甲氧明 | α | ++ | - | - | + | - |
| 肾上腺素 | α,β | ++++ | +++ | +++ | + | |
| 多巴胺 | α,β | + | ++ | ± | + | + |
| 麻黄碱 | α,β | ++ | ++ | ++ | + | + |
| 异丙肾上腺素 | β | - | +++ | +++ | + | |
| 多巴酚丁胺 | β | + | ++ | + | + | ± |
3.血压小剂量滴注时由于心脏兴奋,收缩压升高,此时血管收缩作用尚不十分剧烈,故舒张压升高不多而脉压加大(图10-2)。较大剂量时,因血管强烈收缩使外周阻力明显增高,故收缩压升高的同时舒张压也明显升高,脉压变小。
![]()
图10-2 去甲肾上腺素、肾上腺素、异丙肾上腺素及多巴胺作用比较
(静脉滴注,除多巴胺500μg/分外,其余均10μg/分)
4.其他其他作用都不显著,对机体代谢的影响较弱,只有在大剂量时才出现血糖升高。对中枢作用也较肾上腺素为弱。
【临床应用】
1.休克目前去甲肾上腺素类血管收缩药在休克治疗中已不占主要地位,仅限于某些休克类型如早期经原性休克及药物中毒引起的低血压等,用去甲肾上腺素静脉滴注,使收缩压维持在12kPa左右,以保证心、脑等重要器官的血液供应。休克的关键是微循环血液灌注不足和有效血容量下降,故其治疗关键应是改善微循环和补充血容量。去甲肾上腺素的应用仅是暂时措施,如长时间或大剂量应用反而加重微循环障碍。现也主张去甲肾上腺素与α受体阻断剂酚妥拉明合用以拮抗其缩血管作用,保留其β效应。
2.上消化道出血 取本品1~3mg ,适当稀释后口服,在食道或胃内因局部作用收缩粘膜血管,产生止血效果。
【不良反应】
1.局部组织缺血环死静脉滴注时间过长、浓度过高或药液漏出血管,可引起局部缺血坏死,如发现外漏或注射部位皮肤苍白,应更换注射部位,进行热敷,并用普鲁卡因或α受体阻断药如酚妥拉明作局部浸润注射,以扩张血管。
2.急性肾功能衰退滴注时间过长或剂量过大,可使肾脏血管剧烈收缩,产生少尿、无尿和肾实质损伤,故用药期间尿量至少保持在每小时25ml以上。
【禁忌证】高血压、动脉硬化症及器质性心脏病人禁用。
间羟胺
间羟胺(metaraminol)又名阿拉明(aramine),性质较稳定,主要作用于α受体,对β1受体作用较弱。间羟胺可被肾上腺素能神经末梢摄取、进入囊泡,通过置换作用促使囊泡中的去甲肾上腺素释放,间接地发挥作用。本品不易被单胺氧化酶破坏,故作用较持久。短时间内连续应用,可因囊泡内去甲肾上腺素减少,使效应逐渐减弱,产生快速耐受性。
间羟胺收缩血管,升高血压作用较去甲肾上腺素弱而持久,略增心肌收缩性,使休克病人的心输出量增加。对心率的影响不明显,有时血压升高反射地使心率减慢,很少引起心律失常,对肾脏血管的收缩作用也较弱,但仍能显著减少肾脏血流量。由于间羟胺升压作用可靠,维持时间较长,比去甲肾上腺素较少引起心悸和少尿等不良反应,还可肌内注射,故临床上作为去甲肾上腺素的代用品,用于各种休克早期,手术后或脊椎麻醉后的休克。
二、α1受体激动药
脱氧肾上腺素和甲氧明
脱氧肾上腺素(苯肾上腺素,phenylephrine;新福林,neosynephrine)和甲氧明(methoxamine)都是人工合成品。主要激动α1受体,作用与去甲肾上腺素相似而较弱,少具或不具β型作用,在产生与去甲肾上腺素相似的收缩血管升高血压的作用时,使肾血流的减少比去甲肾腺素更为明显。作用维持时间较久,除可静脉滴注外也可肌内注射。用于抗休克,也可用于防治脊椎麻醉或全身麻醉的低血压。
甲氧明与脱氧肾上腺素均能收缩血管,升高血压,通过迷走神经反射地使心率减慢,故也可用于阵发性室上性心动过速。去氧肾上腺素还能兴奋瞳孔扩大肌,一般不引起眼内压升高(老年人前房角狭窄者可能引起眼内压升高)。用其1%~2.5%溶液滴眼,在眼底检查时作为快速短效的扩瞳药。
α2受体激动药可乐定将在第二十章介绍。
第三节 α,β受体激动药
肾上腺素
【来源】肾上腺素(adrenaline,epinephrine,AD)是肾上腺髓质的主要激素,其生物合成主要是在髓质铬细胞中首先形成去甲肾上腺素(见第五章),然后进一步经苯乙胺-N-甲基转移酶(phenylethanolamine N-methyl transferase,PNMT)的作用,使去甲肾上腺素甲基化形成肾上腺素。
药用肾上腺素可从家畜肾上腺提取,或人工合成。理化性质与NA相似。
【体内过程】口服后在碱性肠液及肠粘膜和肝内破坏,吸收很少,不能达到有效血药浓度。皮下注射因能收缩血管,故吸收缓慢。肌内注射的吸收远较皮下注射为快。肾上腺素在体内的摄取与代谢途径与去甲肾上腺素相似(图10-2)。肌内注射作用维持约10~30分钟,皮下注射作用维持1小时左右。
【药理作用】肾上腺素能激动α和β两类受体,产生较强的α型和β型作用。
1. 心脏 作用于心肌、传导系统和窦房结的β1受体,加强心肌收缩性,加速传导,加速心率,提高心肌的兴奋性。对离体心肌的β作用特征是加速收缩性发展的速率(正性缩率作用,positive klinotropic effect)。由于心肌收缩性增加,心率加快,故心输出量增加。肾上腺素又能舒张冠状血管,改善心肌的血液供应,且作用迅速,是一个强效的心脏兴奋药。其不利的一面是提高心肌代谢,使心肌氧耗量增加,加上心肌兴奋性提高,如剂量大或静脉注射快,可引起心律失常,出现期前收缩,甚至引起心室纤颤。
2.血管肾上腺素主要作用于小动脉及毛细血管前括约肌,因为这些小血管壁的肾上腺素受体密度高;而静脉和大动脉的肾上腺素受体密度低,故作用较弱。此外,体内各部位血管的肾上腺素受体的种类和密度各不相同,所以肾上腺素对各部位血管的效应也不一致,以皮肤粘膜血管收缩为最强烈;内脏血管,尤其是肾血管,也显著收缩;对脑和肺血管收缩作用十分微弱,有时由于血压升高而被动地舒张;骨骼肌血管的β2受体占优势,故呈舒张作用;也能舒张冠状血管,机制见去甲肾上腺素。
3.血压 在皮下注射治疗量(0.5~1mg)或低浓度静脉滴注(每分钟滴入10μg)时,由于心脏兴奋,心输出量增加,故收缩压升高(图10-2);由于骨骼肌血管舒张作用对血压的影响,抵消或超过了皮肤粘膜血管收缩作用的影响,故舒张压不变或下降;此时身体各部位血液重新分配,使更适合于紧急状态下机体能量供应的需要。较大剂量静脉注射时,收缩压和舒张压均升高。此外,肾上腺素尚能作用于邻肾小球细胞(juxtaglomerular cells)的β1受体,促进肾素的分泌。
4.支气管 能激动支气管平滑肌的β2受体,发挥强大舒张作用。并能抑制肥大细胞释放过敏性物质如组胺等,还可使支气管粘膜血管收缩,降低毛细血管的通透性,有利于消除支气管粘膜水肿。
5. 代谢 能提高机体代谢,治疗量下,可使耗氧量升高20%~30%,在人体,由于α受体和β2受体的激动都可能致肝糖原分解,而肾上腺素兼具α、β作用,故其升高血糖作用较去甲肾上腺素显著。此外,肾上腺素尚具降低外周组织对葡萄糖摄取的作用。肾上腺素还能激活甘油三酯酶加速脂肪分解,使血液中游离脂肪酸升高。
【临床应用】
1. 心脏骤停用于溺水、麻醉和手术过程中的意外,药物中毒、传染病和心脏传导阻滞等所致的心脏骤停。对电击所致的心脏骤停也可用肾上腺素配合心脏除颤器或利多卡因等除颤,一般用心室内注射,同时必须进行有效的人工呼吸和心脏挤压等。
2. 过敏反应对于急性的、严重的过敏反应(变态反应),除糖皮质激素制剂外,肾上腺素也是一个重要药物。本品也适用于过敏性休克。
3.支气管哮喘控制支气管哮喘的急性发作,皮下或肌内注射能于数分钟内奏效。
4. 与局麻药配伍及局部止血肾上腺素加入局麻药注射液中,可延缓局麻药的吸收,减少吸收中毒的可能性,同时又可延长局麻药的麻醉时间。一般局麻药中肾上腺素的浓度为1:250000,一次用量不要超过0.3mg。
当鼻粘膜和齿龈出血可将浸有0.1%盐酸肾上腺素的纱布或棉花球填塞出血处。
【不良反应和禁忌证】主要不良反应为心悸、烦躁、头痛和血压升高等,血压剧升有发生脑溢血的危险,故老年人慎用。也能引起心律失常,甚至心室纤颤,故应严格掌握剂量。禁用于高血压,器质性心脏病。糖尿病和甲状腺功能亢进症等。
多巴胺
多巴胺(dopamine)是去甲肾上腺素生物合成的前体,药用的是人工合成品。
【体内过程】口服易在肠和肝中破坏而失效。一般用静脉滴注给药,在体内迅速经MAO和COMT的催化而代谢失效,故作用时间短暂。因多巴胺不易透过血脑屏障,故外源性多巴胺难于引起中枢作用。
【药理作用】
1.心脏 主要激动心脏β1受体,也具释放去甲肾上腺素作用,能使收缩性加强,心输出量增加。一般剂量对心率影响不明显,大剂量可加快心率。与异丙肾上腺素比较,多巴胺增加心输出量的作用较弱,对心率影响较少,并发心律失常者也较少。
2,血管和血压 能作用于血管的α受体和多巴胺受体,而对β2受体的影响十分微弱。多巴胺能增加收缩压和脉压,而对舒张压无作用或稍增加(图10-2),这可能是心输出量增加,而肾和肠系膜动脉阻力下降,其它血管阻力微升使总外周阻力变化不大的结果。多巴胺的血管舒张作用不能为β受体阻断药、阿托品以及抗组胺药所拮抗,故认为是选择性地作用于血管的多巴胺受体(D1受体)之故。大剂量给药则主要表现为血管收缩,引起外周阻力增加,血压上升。这一效应可被α受体阻断药所对抗,说明这一作用是激动α(α1受体)受体的结果。
3.肾脏多巴胺能舒张肾血管,使肾血流量增加,肾小球的滤过率也增加。有排钠利尿作用,可能是多巴胺直接对肾小管多巴胺受体的作用。用大剂量时,也可使肾血管明显收缩。
【临床应用】用于抗休克,对于伴有心收缩性减弱及尿量减少而血容量已补足的休克患者疗效较好。此外,本品尚可与利尿药合并应用于急性肾功能衰竭。也可用于急性心功能不全。
【不良反应】一般较轻,偶见恶心、呕吐。如剂量过大或滴注太快可出现心动过速、心律失常和肾血管收缩引致肾功能下降等,一旦发生,应减慢滴注速度或停药。
麻黄碱
麻黄碱(ephedrine)是从中药麻黄中提取的生物碱。两千年前的《神农本草经》即有麻黄能:“止咳逆上气”的记载,麻黄碱现已人工合成,药用其左旋体或消旋体。
【药理作用】麻黄碱作用主肾上腺素的相似,能激动α、β两种受体。与肾上腺素比较,麻黄碱具有下列特别:①性质稳定,口服有效;②拟肾上腺素作用弱而持久;③中枢兴奋作用较显着;④易产生快速耐受性。
1.心血管兴奋心脏,使心收缩加强、心输出量增加。在整体情况下由于血压升高,反射性减慢心率,这一作用抵消了它直接加速心率的作用,故心率变化不大。麻黄碱的升高血压作用出现缓慢,但维持时间较长(3~6小时)。一般内脏血流量减少,但冠脉、脑血管和骨骼肌血流量增加。
2.支气管平滑肌 松弛作用较肾上腺素弱,起效慢但持久。
3.中枢神经系统具有较肾上腺素为显著的中枢兴奋作用,较大剂量可兴奋大脑和皮层下中枢,引起精神兴奋、不安和失眠等。
4.快速耐受性麻黄碱、间羟胺等当短期内反复给药,作用可逐渐减弱,称为快速耐受性(tachyphylaxis),也称脱敏(desensitization)。停药数小时后,可以恢复。每日用药如不超过三次则快速耐受性一般不明显。
【作用机制】麻黄碱兼具有直接和间接作用,它的直接作用在不同组织可表现为激动α1,α2,β1和β2受体,加上其释放NA而发挥的间接作用,故其药理作用比较复杂。
至于麻黄碱的快速耐受性产生的机制,一般认为有受体逐渐饱和与递质逐渐耗损两种因素。最近通过放射性配体结合实验方法证明离体豚鼠肺组织在连续给予麻黄碱后,其与β受体的亲和力显著下降。
【体内过程】口服易吸收,可通过血脑屏障进入脑脊液。小部分在体内经脱胺氧化而被代谢,大部分以原形自尿排出。代谢和排泄都缓慢,故作用较肾上腺素持久。
【临床应用】
1.支气管哮喘用于预防发作和轻症的治疗,对于重症急性发作效果较差。
2.鼻粘膜充血引起鼻塞 常用0.5%~1%溶液滴鼻可消除粘膜肿胀。
3.防治某些低血压状态如用于防治硬膜外和蛛网膜下麻醉所引起的低血压。
4.缓解荨麻疹和血管性神经水肿的皮肤粘膜症状。
【不良反应与禁忌证】有时出现中枢兴奋所致的不安,失眼等,晚间服用宜加镇静催眠药以防止失眠。禁忌证同肾上腺素。
第四节 β受体激动药
一、β1,β2受体激动药
异丙肾上腺素
异丙肾上腺素(isoprenaline)是人工合成品,化学结构是去甲肾上腺素氨基上的一个氢原子被异丙基所取代。是经典的β1,β2受体激动剂。
【体内过程】口服易在肠粘膜与硫酸结合而失效,气雾剂吸入给药,吸收较快。舌下含药因能舒张局部血管,少量可从粘膜下的舌下静脉丛迅速吸收。吸收后主要在肝及其它组织中被COMT所代谢。异丙肾上腺素较少被MAO代谢,也较少被去甲肾上腺素能神经所摄取,因此其作用维持时间较肾上腺素略长。
【药理作用】对β受体有很强的激动作用,对β1和β2受体选择性很低。
1.对心脏的作用 具典型的β1受体激动作用,表现为正性肌力和正性缩率作用,缩短收缩期和舒张期。与肾上腺素比较,异丙肾上腺素加快心率、加速传导的作用较强,对窦房结有显著兴奋作用,也能引起心律失常,但较少产生心室颤动。
2.对血管和血压的影响对血管有舒张作用,主要是使骨骼肌血管舒张(激动β2受体),对肾血管和肠系膜血管舒张作用较弱,对冠状血管也有舒张作用。当静脉滴注每分钟2~10μg,由于心脏兴奋和外周血管舒张,使收缩压升高而舒张压略下降(图10-2),此时冠脉流量增加;但如静脉注射给药,则可引起舒张压明显下降,降低了冠状血管的灌注压,冠脉有效血管流量不增加。
3. 缓解支气管平滑肌痉挛 激动β2受体,舒张支气管平滑肌比肾上腺素略强,也具有抑制组胺等过敏性物质释放的作用。但对支气管粘膜的血管无收缩作用,故消除粘膜水肿的作用不如肾上腺素。久用可产生耐受性。
4. 其他 能增加组织的耗氧量.与肾上腺素比较,其升高血中游离脂肪酸作用相似,而升高血糖作用较弱。不易透过血脑屏障,中枢兴奋作用微弱。
【临床应用】
1.支气管哮喘舌下或喷雾给药,用于控制支气管哮喘急性发作,疗效快而强。
2.房室传导阻滞治疗Ⅱ、Ⅲ度房室传导阻滞,采用舌下含药,或静脉滴注给药。
3.心脏骤停适用于心室自身节律缓慢,高度房室传导阻滞或窦房结功能衰竭而并发的心脏骤停,常与去甲肾上腺素或间羟胺合用作心室内注射。
【不良反应】常见的是心悸、头晕。用药过程中应注意控制心率。在支气管哮喘病人,已具缺氧状态,加以用气雾剂剂量不易掌握,如剂量过大,可致心肌耗氧量增加,易引起心律失常,甚至产生危险的心动过速及心室颤动。禁用于冠心病、心肌炎和甲状腺功能亢进症等。
二、β1受体激动药
多巴酚丁胺
临床应用的多巴酚丁胺(dobutamine)是含有右旋多巴酚丁胺和左旋多巴酚丁胺的消旋体。前者阻断α1受体,后者激动α1受体。两者都激动β受体,但前者激动β受体作用为后者的10倍,消旋多巴酚丁胺的作用是两者的综合表现。由于其对β1受体激动作用强于β2受体,故此药属于β1受体激动药。与异丙肾上腺素比较,本品的正性肌力作用比正性频率作用显着。这可能由于外周阻力变化不大和心脏α1受体激动时正性肌力作用的参与。而外周阻力的稳定又可能是因为α1受体介导的血管收缩作用与β2受体介导的血管舒张作用相抵消所致。
静脉滴注短期治疗心脏手术后或心肌梗塞并发心力衰竭,可增加心输出量。连续应用可产生快速耐受性。梗阻型肥厚性心肌病者禁用。
β2受体激动药主要用于哮喘的治疗,见第三十章。
肾上腺素受体激动药 制剂及用法
重酒石酸对去甲肾上腺素(noradrenaline bitartrate )2mg相当于去甲肾上腺素1mg,一般以本品2mg加于5%葡萄糖液500ml中,静脉滴注,每分钟滴入0.004~0.008mg。
重酒石酸间羟胺(metaraminol bitartrate)19mg相当于间羟胺10mg,肌内注射,间羟胺10mg/次;或10~20mg以葡萄糖液100亳升稀释后静脉滴注。极量静脉滴注100mg/次(每分钟0.2~0.4mg)。
盐酸脱氧肾上腺素(phenylephrine hydrochloride)肌内注射,2~5mg/次;或10mg以葡萄糖液100ml稀释后静脉滴注。极量肌内注射10mg/次,静脉滴注每分钟0.18mg.
盐酸甲氧明(methoxamine hydrochloride) 肌内注射,10~20mg/次,或缓慢静脉注射,5~10mg/次;或20mg/次,用葡萄糖液稀释,缓慢静脉滴注。极量肌内注射20mg/次,60mg/日;静脉注射10mg/次。
盐酸肾上腺素(adrenaline hydrochloride)皮下或肌内注射0.25~0.5mg/次。必要时可心室内注射,0.25~0.5mg/次,用生理盐水稀释10倍。极量皮下注射1mg/次。
盐酸多巴胺(dopamine hydrochloride)20mg加入5%葡萄糖液200~500ml内,静脉滴注,75~100μg/分钟。极量静脉滴注20μg/分/公斤。
盐酸麻黄碱(ephedrine hydrochloride)口服,25mg/次,3次/日。皮下或肌内注射,15~30mg/次。极量0.06g/次,0.15g/日,口服、皮下或肌内注射。
硫酸异丙肾上腺素(isoprenaline sulfate)静脉滴注,以0.1~0.2mg加于5%葡萄糖液100~200ml中,每分钟滴入0.5~2ml,或按需要而定。
盐酸异丙肾上腺素(isoprenaline hydrochloride)气雾剂:0.25%气雾剂喷雾吸入,0.1~0.4mg/次。舌下含,10mg/次,3次/日。极量喷雾吸入,0.4mg/次,2.4mg/日。舌下含,20mg/次,60mg/日。
第十一章 肾上腺素受体阻断药
肾上腺素受体阻断药(adrenoceptor blocking drugs)能阻断肾上腺素受体从而拮抗去甲肾上腺素能神经递质或肾上腺素受体激动药的作用。对于整体动物,它们的作用强度取决于机体的去甲肾上腺素能神经张力。这类药物按对α和β肾上腺素受体选择性的不同,分为α肾上腺素受体阻断药(简称α受体阻断药)和β肾上腺素受体阻断药(β受体阻断药)二大类。
第一节 α肾上腺素受体阻断药
α受体阻断药能选择性地与α肾上腺素受体结合,其本身不激动或较少激动肾上腺素受体,却能妨碍去甲肾上腺素能神经递质及肾上腺素受体激动药与α受体结合,从而产生抗肾上腺素作用。它们能将肾上腺素的升压作用翻转为降压,这个现象称为“肾上腺素作用的翻转”(adrenalinereversal)。这可解释为α受体阻断药选择性地阻断了与血管收缩有关的α受体,留下与血管舒张有关的β受体;所以能激动α受体和β受体的肾上腺素的血管收缩作用被取消,而血管舒张作用得以充分地表现出来。对于主要作用于血管α受体的去甲肾上腺素,它们只能取消或减弱其升压效应而无“翻转作用”。对于主要作用于β受体的异丙肾上腺素的降压作用则无影响(图11-1)。
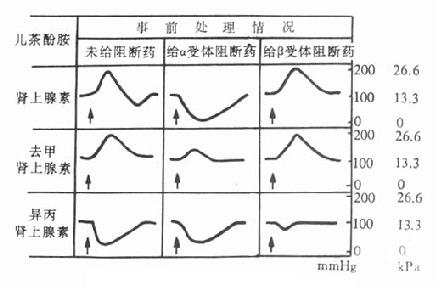
图11-1 在给肾上腺素受体阻断药前后,儿茶酚胺对狗血压的作用
一、α1,α2肾上腺素受体阻断药
早期的α受体阻断药多属此类,其中,酚妥拉明和妥拉唑啉对α1受体和α2受体的选择性很低,酚苄明对α1受体仅具中等程度的选择性。从作用时间看,又有长效与短效之分。
(一)短效类
本类药物与α受体结合较松,易于解离。是竞争性α受体阻断药。它们可使激动药的量效曲线平行右移,增加激动药的剂量仍可达到最大效应(图11-2)。
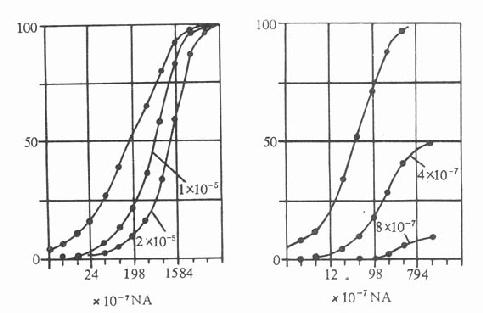
图11-2 短效(左,妥拉唑淋)和长效(右,二苄明)
α受体阻断药对NA收缩猫脾脏条的影响(自Bickerson)
横坐标:NA深度;从坐标,最大收缩的%;图内为阻断药浓度
酚妥拉明
【体内过程】酚妥拉明(phentolamine)又名瑞支亭(regitine),生物利用度低,口服效果仅为注射给药的20%。口服后30分钟血药浓度达峰值,作用维持约3~6小时;肌内注射作用维持30~45分钟。大多以无活性的代谢物从尿中排泄。
【药理作用】选择性地阻断α受体,拮抗肾上腺素的α型作用,但作用较弱。
1.血管静脉注射能使血管舒张,血压下降,肺动脉压和外周血管阻力降低。其机制主要是对血管的直接舒张作用,大剂量也出现阻断α受体的作用。
2.心脏对心脏有兴奋作用,使心收缩力加强,心率加快,输出量增加。这种兴奋作用部分由血管舒张,血压下降,反射地引起;部分是阻断神经末梢突触前膜α2受体,从而促进去甲肾上腺素释放的结果。偶可致心律失常。
3.其他有拟胆碱作用,使胃肠平滑肌兴奋。用组胺样作用,使胃酸分泌增加,皮肤潮红等。
【临床应用】
1.用于外周血管痉挛性疾病如肢端动脉痉挛性病等。
2.在静脉滴注去甲肾上腺素发生外漏,可用本品5mg溶于10~20ml生理盐水中,作皮下浸润注射。也用于肾上腺素等拟交感胺过量所致高血压。
3.用于肾上腺嗜铬细胞瘤的诊断和此病骤发高血压危象以及手术前的准备,能使嗜铬细胞瘤所致的高血压下降。作诊断试验时,曾有致死的报告,故应特别慎重。
4.用于抗休克,能使心搏出量增加,血管舒张,外周阻力降低,从而改善休克状态时的内脏血液灌注,解除微循环障碍。并能降低肺循环阻力,防止肺水肿的发生,但给药前必需补足血容量。有人主张合用去甲肾上腺素,目的是对抗去甲肾上腺素的α型收缩血管的作用,保留其β型加强心肌收缩力的作用。
5.有报告用酚妥拉明等血管扩张药治疗其他药物无效的急性心肌梗塞及充血性心脏病所致的心力衰竭,在心力衰竭时,因心输出量不足,交感张力增加,外周阻力增高,肺充血和肺动脉压力升高,易产生肺水肿。应用酚妥拉明扩张血管,降低外周阻力,使心脏后负荷明显降低,左室舒张末期压与肺动脉压下降,心搏出量增加,心力衰竭得以减轻。
【不良反应】常见的反应有低血压,胃肠道平滑肌兴奋所致的腹痛、腹泻、呕吐和诱发溃疡病(可能与其胆碱受体激动作用有关。)静脉给药有时可引起严重的心率加速,心律失常和心绞痛,因此须缓慢注射或滴注。胃炎,胃、十二指肠溃疡病,冠心病患者慎用。。
妥拉唑啉
妥拉唑淋(toalzoline,苄唑淋)对α受体阻断作用与酚妥拉明相似,但较弱,而组胺样作用和拟胆碱作用较强。口服和注射都易吸收,大部分以原形从肾小管排泄。口服吸收较慢,排泄较快,效果远不及注射给药。主要用于血管痉挛性疾病的治疗,局部浸润注射用以处理去甲肾上腺素静脉滴注时药液外漏。不良反应与酚妥拉明相同,但发生率较高。
(二)长效类
以酚苄明和二苄明为代表的长效α受体阻断药与α受体形成牢固的共价键。在离体实验时,即使加入高浓度的儿茶酚胺,也难与之竞争,达不到最大效应,故可压低激动剂的量效曲线,所以也称非竞争性α受体阻断药。
酚苄明
酚苄明(phenoxybenzamine)又史苯苄胺(dibenzyline),是人工合成品。
【体内过程】口服有20%~30%吸收。因刺激性强,不作肌内或皮下注射仅作静脉注射。静脉注射1小时后可达最大效应。本品的指溶性高,大剂量用药可积蓄于脂肪组织中,然后缓慢释放。12小时排泄50%,24小时排泄80%,一周后尚有少量存留在体内。
【药理作用】酚苄明进入体内后,其分子中的氯乙胺基须环化形成乙撑亚胺基,后者才能与α受体牢固结合,加以排泄缓慢,故阻断α受体作用起效慢,但作用强大而持久。一次用药,作用可维持3~4天。能舒张血管降低外周阻力。对于静卧的正常人,缓慢静脉注射一般剂量(1mg/kg),收缩压改变很少而舒张压下降。但当伴有代偿性交感性血管收缩如血容量减少或直立时,就会引起显著的血压下降。由于血压下降所引起反射作用,加上阻断突触前α2受体作用和对摄取1,摄取2的抑制作用,可使心率加速。
【临床应用】用于外周血管痉挛性疾病,也可用于休克和嗜铬细胞瘤的治疗。
【不良反应】常见的有体位性低血压,心悸和鼻塞;口服可致恶心,呕吐及思睡,疲乏等。静脉注射或用于休克时必须缓慢,充分补液和密切监护。
二、α肾上腺素受体阻断药
哌唑嗪(prazosin)选择性地阻断α1受体而对α2受体的阻断极少,因此不促进去甲肾上腺素的释放,加快心率的副作用较轻,口吸取有效。近年合成不少哌唑嗪的衍生物,成为一类新型降压药,将在第二十五章中叙述。
三、α2肾上腺素受体阻断药
育亨宾(yohimbine)能选择性地阻断α2受体,主要用于作科研的工具药。
第二节 β肾上腺素受体阻断药
β受体阻断药能与去甲肾上腺素能神经递质或肾上腺素受体激动药竞争β受体从而拮抗其β型拟肾上腺素的作用。它们与激动剂呈典型的竞争性拮抗。目前文献常以pA2表示受体阻断药的作用强度。能使激动药在提高一倍浓度后,仍产生原浓度作用强度时所需阻断药摩尔浓度的负对数值就是阻断药的pA2,pA2愈大,其阻断作用愈大。
在整体动物,β受体阻断药的作用也依赖于机体去甲肾上腺素能神经张力。例如,它对正常人休息时心脏的作用较弱,但当心脏交感神经张力增高时(如运动或病理情况),则对心脏的抑制作用明显。
【化学】从化学结构上看,β受体阻断药与β受体激动药异丙肾上腺素有相似之处,它们都有下述基本结构:一端为带异丙基的仲胺,另端的芳香环可以是一个或两个苯环也可以是一杂环。前者似与β受体的亲和力有关;后者可能决定其结合后发挥激动作用还是拮抗作用。再者,左旋体的作用为右旋体50~100倍,说明构效关系中的立体特异性。
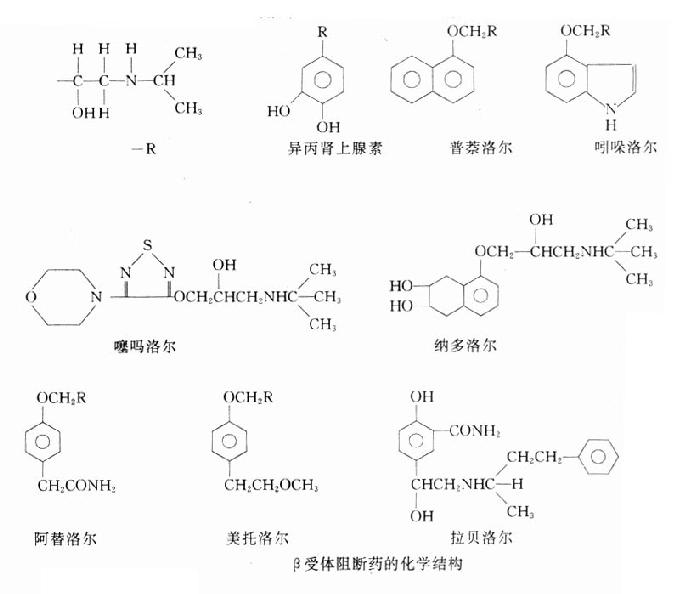
β受体阻断药的化学结构
【药理作用】
1.β受体阻断作用
(1)心血管系统对心脏的作用是这一类药物的重要作用。主要由于阻断心脏β1受体,可使心率减慢,心收缩力减弱,心输出量减少,心肌耗氧量下降,血压稍降低。β受体阻断药还能延缓心房和房室结的传导,延长ECG(心电图)的P-R间期(房室传导时间)。由于非选择性β受体阻断药如普萘洛尔对血管β2受体也有阻断作用,加上心脏功能受到抑制,反射地兴奋交感神经引起血管收缩和外周阻力增加,肝、肾和骨骼肌等血流量减少;在犬和人(包括冠心病人)都发现普萘洛尔能使冠状血管血流量降低。
(2)支气管平滑肌 支气管的β2受体激动时使支气管平滑肌松弛,β受体阻断药则使之收缩而增加呼吸道阻力。但这种作用较弱,对正常人影响较少,只有在支气管哮喘的患者,有时可诱发或加重哮喘的急性发作。选择性β1受体阻断药此作用较弱。
(3)代谢 一般认为人类脂肪的分解主要与β2受体激动有关,而肝糖原的分解与α和β2受体有关。因此β受体阻断药可抑制交感神经兴奋所引起的脂肪分解,当β受体阻断药与α受体阻断药合用时则可拮抗肾上腺素的升高血糖的作用。普萘洛尔并不影响正常人的血糠水平也不影响胰岛素的降低血糖作用,但能延缓用胰岛素后血糖水平的恢复。
■[此处缺少一些内容]■
4.其他普萘洛尔有抗血小板聚集作用。β受体阻断药尚有降低眼内压作用,这可能由减少房水的形成所致。
【临床应用】
1.心律失常对多种原因引起的过速型心律失常有效,如窦性心动过速,全身麻醉药或拟肾上腺素药引起的心律失常等(见第二十二章)。
2.心绞痛和心肌梗塞对心绞痛有良好的疗效。对心肌梗塞,两年以上的长期应用可降低复发和猝死率,用量比抗心律失常的剂量要大(见第二十四章)。
3.高血压能使高血压病人的血压下降,伴有心律减慢(见第二十六章)。
4.其他用于甲状腺功能亢进及甲状腺中毒危象,对控制激动不安,心动过速和心律失常等症状有效,并能降低基础代谢率。也用于嗜铬细胞瘤和肥厚性心肌病,普萘洛尔并试用于偏头痛、肌震颤、肝硬化的止消化道出血等。噻吗洛尔常局部用药治疗青光眼,降低眼内压。
【不良反应】一般的不良反应如恶心、呕吐、轻度腹泻等,停药后迅速消失。偶见过敏反应如皮疹、血小板减少等。严重不良反应为急性心力衰竭,有时可突然出现,可能与个体差异有关。此外,由于β2受体的阻断,可增加呼吸道阻力,诱发支气管哮喘。普萘洛尔等无内在拟交感活性(ISA)的β受体阻断药长期用后突然停药,可使原来病症加剧。
禁用于心功能不全、窦性心动过缓、重度房室传导阻滞和支气管哮喘等患者,慎用于心肌梗塞患者。即使是β1受体选择性阻断药,仍应慎用于支气管哮喘患者。主要由肝脏消除的β受体阻断药,当肝功能不良时应慎用。
【分类】根据对β1受体的选择性和无ISA两种重要特性,β受体阻断药可分为下列五类:
1A类 无内在活性的β1、β2受体阻断药,如普萘洛尔。
1B类 有内在活性的β1、β2受体阻断药,如吲哚洛尔。
2A类 无内在活性的β1受体阻断药,如阿替洛尔。
2B类 有内在活性的β1受体阻断药,如醋丁洛尔。
3类 α、β受体阻断药,如拉贝洛尔。
1A类 无内在活性的β1、β2受体阻断药也称非选择性β受体阻断药,是较早应用而目前仍广泛应用的一类β受体阻断药。
普萘各尔
普萘洛尔(propranolol,心得安)是等量的左旋和右旋异构体混合得到的消旋品,仅左旋体有阻断β受体的活性。
【体内过程】口服吸收率大于90%,主要在肝脏代谢,首关效应60%~70%,生物利用度仅为30%。口服后血浆高峰时间为1~3小时,t1/2为2~5小时。老年人肝功能减退,t1/2可延长。当长期给药或大剂量时,肝的消除功能被饱和,其生物利用度可提高。血浆蛋白结合率大于90%。易于通过血脑屏障和胎盘,也可分泌于乳汁中。其代谢产物90%以上从肾排泄。不同个体口服相同剂量的普萘洛尔,血浆高峰浓度相差可达20倍之多,这可能由于肝消除功能不同所致;因此临床用药需从小剂量开始,逐渐增加到适当剂量。
表11-1 β受体阻断药比较表(摘自新药与临床 1986;5:143)
| 药物名称 | 普萘洛尔 | 噻吗洛儿 | 吲哚洛尔 | 阿替洛尔 | 美托洛尔 | 拉贝洛尔 | 纳多洛尔 |
| 分布系数(辛醇/水) | 5.93 | 0.3 | 0.12 | <0.02 | 0.18 | 11.5 | 0.066 |
| 阻断β受体的作用强度① | 1 | 100 | 15 | 0.5 | 1 | 0.25 | 2~4 |
| β1受体选择性 | - | - | - | + | + | -② | - |
| 内在拟交感活性 | - | - | ++ | - | - | ± | - |
| t1/2(小时)(静注) | 2.5 | 3.1 | 3.2 | 3.4~4.5 | |||
| (口服) | 2~5 | 2~5 | 2~5 | 6~9 | 3~4 | 5.5 | 14~24 |
| 首过效应(%) | 60~70 | 25~30 | 10~13 | 0~10 | 50~60 | 60 | |
| 口服生物利用度%) | 30 | 30~75 | 87~95 | 50~60 | 40~50 | 33 | 30~40 |
| 口服血浆浓度高峰时间(小时) | 1~3 | 2~3 | 1.5~2 | 2~4 | 0.5~1.5 | 1~2 | 2~4 |
| 血浆蛋白结合率%) | 80~95 | 10~80 | 40~60 | 3~40 | 12 | 50 | 20~30 |
| 口服血浆高峰浓度个体差异 | 20倍 | 4倍 | 较低 | 5倍 | |||
| 主要消除器官 | 肝 | 肝 | 肝、肾 | 肾 | 肝 | 肝 | 肾 |
注:①犬对标准剂量异丙肾上腺素心率过速的拮抗作用强度
②兼有α受体阻断作用
【药理作用和临床应用】普萘洛尔具较强的β受体阻断作用,对β1和β2受体的选择性很低,没有内在拟交感活性。用药后使心率减慢,心收缩力和输出量减低,冠脉血流量下降,心肌耗氧量明显减少,对高血压病人可使血压下降,支气管阻力也有一定程度的增高。可用于治疗心律失常、心绞痛、高血压、甲状腺功能亢进等。其他见上文。
噻吗洛尔
噻吗洛尔(timolol,噻吗心安)是已知作用最强的β受体阻断药。我国现常用其滴眼剂,降低眼内压治疗青光眼。作用机制主要在于减少房水的生成。本品0.1%~0.5%疗效与毛果芸香碱1%~4%相近可较优,每日滴眼二次即可,且无缩瞳和调节痉挛等不良反应。
1B类 有内在活性的β1、β2受体阻断药
吲哚洛尔
吲哚洛尔(pindolo,心得静)的内在活性较强,主要表现在激动β2受体方面。激动血管平滑肌β2受体而致的舒张血管作用有利于血压的治疗。对于心肌所含少量β2受体(人类心室肌β1与β2受体比率为74:26,心房为86:14)的激动又可减少心肌抑制作用。
2A类 无内在活性的β1受体阻断药
阿替洛尔和美托洛尔
阿替洛尔(atenolol,氨酰心安)和美托洛尔(美多心安,metoprolol)对β1受体有选择性阻断作用,对β2受体作用较弱,故增加呼吸道阻力作用较轻,不过对哮喘病人仍需慎用。临床试验证明,阿替洛尔每日75~600mg降压效果比心得安每日60~480mg为佳。阿替洛尔的t1/2和作用维持时间均较普奈洛尔和美托洛尔长,临床应用时每日口服一次即可,普萘洛尔和美托洛尔则需每日2~3次。
3类 α、β受体阻断药
拉贝洛尔
拉贝洛尔(labetolol,柳胺苄心定)的特点是兼具β和α受体阻断作用。根据麻醉犬实验,其β1受体阻断作用为普奈洛尔的1/4,对β2受体作用为普奈洛尔的1/11~1/17。其对β受体阻断作用为对α受体阻断作用的4~16倍。
多用于中度和重度高血压病,也有用于绞痛的报告。主要通过阻断β受体抑制心脏和阻断α受体舒张血管,从而发挥较好的疗效。此外尚有增加肾血流量的作用。
肾上腺素受体阻断药 制剂及用法
甲磺酸酚妥拉明(phentolamine methanesulfonate)肌内或静脉注射,5mg/次。
盐酸妥拉唑淋(tolazoline hydrochloride)口服,25mg/次,3次/日。肌内注射,25mg/次。
盐酸酚苄明(phenoxybenzamine hydrochloride)口服,10~20mg/次,2次/日。抗休克,0.5~1mg/kg,加入5%葡萄糖液200~500ml中静脉滴注,最快不得少于2小时内滴完。
盐酸普萘洛尔(propranolol hydrochloride)抗心绞痛及抗高血压,口服,10mg/次,3次/日,每4~5日增加10mg,直至每日80~100mg,或至症状明显减轻或消失。抗心律失常,口服,10~20mg/次,3次/日
■[此处缺少一些内容]■
[此处缺少一些内容]
第十四章 镇静催眠药
能缓和激动,消除躁动,恢复安静情绪的药物称镇静药(sedatives)。能促进和维持近似生理睡眠的药物称催眠药(hypnotics)。但同一药物,在较小剂量时起镇静作用,在较大剂量时则起催眠作用。可见,
■[此处缺少一些内容]■
的衍生物。临床常用的有20余种。虽然它们结构相似,但不同衍生物之间,抗焦虑、镇静催眠、抗惊厥、肌肉松弛和安定作用则各有侧重。本节只讨论主要用于镇静催眠的衍生物,包括地西泮(diazepam,安定)、氟西泮(flurazepam,氟安定)、氯氮![]() (chlordiazepoxide)、奥沙西泮(oxazepam)和三唑仑(triazolam)。
(chlordiazepoxide)、奥沙西泮(oxazepam)和三唑仑(triazolam)。
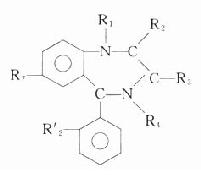
表14-1 苯二氮![]() 类的结构、半衰期和剂量比较表
类的结构、半衰期和剂量比较表
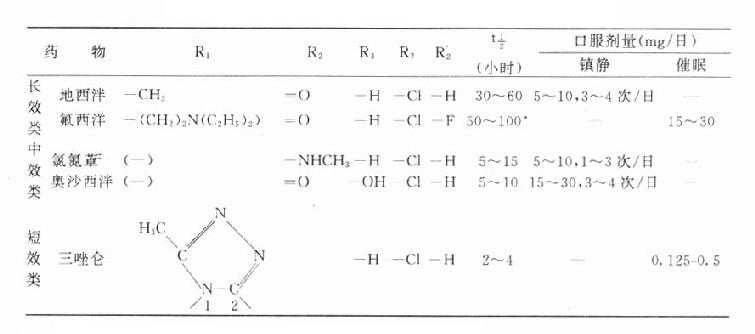
*包括活性代谢产物的半衰期;氯氮 R4为→O,其他药R4无取代
【药理作用和临床应用】
1.抗焦虑作用 苯二氮![]() 类小于镇静剂量时即有良好的抗焦虑作用,显著改善紧张、忧虑、激动和失眠等症状。这可能是选择性作用于边缘系统的结果。主要用于焦虑症。对持续性焦虑状态则宜选用长效类药物。对间断性严重焦虑患者则宜选用中、短效类药物。临床常用地西泮和氯氮
类小于镇静剂量时即有良好的抗焦虑作用,显著改善紧张、忧虑、激动和失眠等症状。这可能是选择性作用于边缘系统的结果。主要用于焦虑症。对持续性焦虑状态则宜选用长效类药物。对间断性严重焦虑患者则宜选用中、短效类药物。临床常用地西泮和氯氮
■[此处缺少一些内容]■
类药物都有抗惊厥作用,其中地西泮和三唑仑的作用尤为明显,临床用于辅助治疗破伤风、子痫、小儿高热惊厥和药物中毒性惊厥。地西泮是目前用作癫痫持续状态的首选药。对于其他类型的癫痫发作则以硝西泮和氯硝西泮的疗效为较好(详第十五章)。 4.中枢性肌肉松弛作用动物实验证明,本类药物对去大脑僵直有明显肌肉松弛作用。对人类大脑损伤所致肌肉僵直也有缓解作用。但有报道认为其疗效并不比其他中枢抑制药为佳。 【作用机制】放射配体结合试验证明,脑内有地西泮的高亲和力的特异结合位点苯二氮![]() 受体。其分布以皮质为最密,其次为边缘系统和中脑,再次为脑干和脊髓。这种分布状况与中枢抑制性递质γ-氨基丁酸(GABA)的GABAA受体的分布基本一致。电生理实验证明,苯二氮
受体。其分布以皮质为最密,其次为边缘系统和中脑,再次为脑干和脊髓。这种分布状况与中枢抑制性递质γ-氨基丁酸(GABA)的GABAA受体的分布基本一致。电生理实验证明,苯二氮![]() 类能增强GABA能神经传递功能和突触抑制效应;还有增强GABA与GABAA受体相结合的作用。GABAA受体是氯离子通道的门控受体,由两个α和两个β亚单位(α2β2)构成Cl-通道(图14-1)。β亚单位上有GABA受点,当GABA与之结合时,Cl-通道开放,Cl-内流,使? ■[此处缺少一些内容]■
类能增强GABA能神经传递功能和突触抑制效应;还有增强GABA与GABAA受体相结合的作用。GABAA受体是氯离子通道的门控受体,由两个α和两个β亚单位(α2β2)构成Cl-通道(图14-1)。β亚单位上有GABA受点,当GABA与之结合时,Cl-通道开放,Cl-内流,使? ■[此处缺少一些内容]■
口服吸收较慢,肌肉给药吸收也缓慢,且不规则。欲快速显效时,应静脉注射。 苯二氮![]() 类血浆蛋白结合率较高。其中地西泮的血浆蛋白结合率高达99%。由于脂溶性很高,使之能迅速向组织中分布并在脂肪组织中蓄积。静脉注射时首先分布至脑和其他血流丰富的组织和器官。脑脊液中浓度约与血清游离药物浓度相等。随后进行再分布而蓄积于脂肪和肌组织中。其分布容积很大,老年患者更大。 此类药物主要在肝药酶作用下进行生物转化。但多数药物的代谢产物(尤其是其N-去甲基代谢物—去甲地西泮)具有与母体药物相似的活性,而其半衰期则比母体药物更长。例如氟西泮的血浆t1/2仅2~3小时,而其主要活性代谢产物N-去烷基烷氟西泮的t1/2却在50小时以上。连续应用长效类药物时,应注意药物及其活性代谢物在体内蓄积。苯二氮
类血浆蛋白结合率较高。其中地西泮的血浆蛋白结合率高达99%。由于脂溶性很高,使之能迅速向组织中分布并在脂肪组织中蓄积。静脉注射时首先分布至脑和其他血流丰富的组织和器官。脑脊液中浓度约与血清游离药物浓度相等。随后进行再分布而蓄积于脂肪和肌组织中。其分布容积很大,老年患者更大。 此类药物主要在肝药酶作用下进行生物转化。但多数药物的代谢产物(尤其是其N-去甲基代谢物—去甲地西泮)具有与母体药物相似的活性,而其半衰期则比母体药物更长。例如氟西泮的血浆t1/2仅2~3小时,而其主要活性代谢产物N-去烷基烷氟西泮的t1/2却在50小时以上。连续应用长效类药物时,应注意药物及其活性代谢物在体内蓄积。苯二氮![]() 类及其代谢物最终均与葡萄糖醛酸结合而失活,经肾排出。结构中含羟基者可直接与葡萄糖醛酸结合而失活,这一途径较少受其他因素影响。结构上7位上有硝基者(如硝西泮)在生物化转化时,硝基还原为氨基,进一步乙酰化为乙酰氨基,这两种代谢物均无生物活性,且此代谢途径也较少受其他因素影响。但本类药物在体内的氧化代谢过程则易受肝功能、老年和同时饮酒的抑制,使t1/2延长。图14-2示苯二氮
类及其代谢物最终均与葡萄糖醛酸结合而失活,经肾排出。结构中含羟基者可直接与葡萄糖醛酸结合而失活,这一途径较少受其他因素影响。结构上7位上有硝基者(如硝西泮)在生物化转化时,硝基还原为氨基,进一步乙酰化为乙酰氨基,这两种代谢物均无生物活性,且此代谢途径也较少受其他因素影响。但本类药物在体内的氧化代谢过程则易受肝功能、老年和同时饮酒的抑制,使t1/2延长。图14-2示苯二氮![]() 类几种药物的生物转化过程。
类几种药物的生物转化过程。
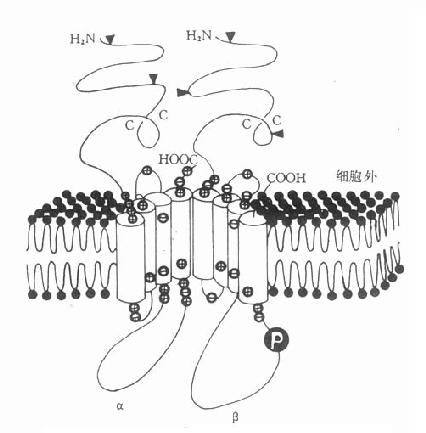
图14-1GABAA受体在细胞膜上的模式图
▲加糖基的位点;cAMP依赖性磷酸化部位;
细胞外亲水序列中两个C构成β-结构环(自BornardE.A.et al.Trends in pharmacol Sci 1987;10:502) 【不良反应】治疗量连续用药可出现头昏、嗜睡、乏力等反应,长效类尤易发生。大剂量偶致共济失调。过量急性中毒可致昏迷和呼吸抑制,但安全范围大,发生严重后果者少。静脉注射对心血管有抑制作用,治疗量口服则无此作用。同时应用其他中枢抑制药、吗啡和乙醇等可显著增强毒性。因可透过胎盘屏障和随乳汁分泌,孕妇和哺乳妇女忌用。本类药物虽无明显药酶诱导作用,但长期用药仍可产生一定耐受性,需增加剂量。久服可发生依赖性和成瘾,停药时出现反跳和戒断症状(失眠、焦虑、激动、震颤等)。与巴比妥类相比,本类药物的戒断症状发生较迟、较轻。 图14-2苯二氮
图14-2苯二氮![]() 类的代谢(*=活性代谢物)
类的代谢(*=活性代谢物)
第二节 巴比妥类
巴比妥类(barbiturates)为巴比妥酸在C5位上进行取代而得的一组中枢抑制药。取代基长而有分支(如异戊巴比妥)或双键(如司可巴比妥),则作用强而短;以苯环取代(如苯巴比妥)则有较强的抗惊厥作用;C2位的O被S取代(如硫喷妥),则脂溶性增高,静脉注射立即生效,但维持时间很短。
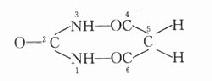
表14-2 巴比妥类作用与用途比较表
| 亚类 | 药物 | 显效时间 (小时) |
作用维持时间 (小时) |
主要用途 |
| 长效 | 苯巴比妥 巴比妥 |
0.5~1 0.5~1 |
6~8 6~8 |
抗惊厥 镇静催眠 |
| 中效 | 戊巴比妥 异戊巴比妥 |
0.25~0.5 0.25~0.5 |
3~6 3~6 |
抗惊厥 镇静催眠 |
| 短效 | 司可巴比妥 | 0.25 | 2~3 | 抗惊厥镇静催眠 |
| 超短效 | 硫喷妥 | iv立即 | 0.25 | 静脉麻醉 |
【中枢抑制作用】巴比妥类是普遍性中枢抑制药。随剂量由小到大,相继出现镇静、催眠、抗惊厥和麻醉作用。10倍催眠量时则可抑制呼吸,甚至致死。
巴比妥类在非麻醉剂量时主要抑制多突触反应,减弱易化,增强抑制。此作用主要见于GABA能神经传递的突触。它增强GABA介导的Cl-内流,减弱谷氨酸介导的除极。但与苯二氮![]() 类不同,巴比妥类是通过延长氯通道开放时间而增加Cl-内流,引起超极化。较高浓度时,则抑制Ca2+依赖性动作电位,抑制Ca2+依赖性递质释放,并且呈现拟GABA作用,即在无GABA时也能直接增加Cl-内流。
类不同,巴比妥类是通过延长氯通道开放时间而增加Cl-内流,引起超极化。较高浓度时,则抑制Ca2+依赖性动作电位,抑制Ca2+依赖性递质释放,并且呈现拟GABA作用,即在无GABA时也能直接增加Cl-内流。
此类药物需用至镇静剂量时才显示抗焦虑作用。由于本类药物的安全性远不及苯二氮![]() 类,且较易发生依赖性,因此,目前已很少用于镇静和催眠。其中只有苯巴比妥和戊巴比妥仍用于控制癫痫持续状态;硫喷妥偶而用于小手术或内窥镜检查时作静脉麻醉。
类,且较易发生依赖性,因此,目前已很少用于镇静和催眠。其中只有苯巴比妥和戊巴比妥仍用于控制癫痫持续状态;硫喷妥偶而用于小手术或内窥镜检查时作静脉麻醉。
【不良反应】催眠剂量的巴比妥类可致眩晕和困倦,精细运动不协调。偶可致剥脱性皮炎等严重过敏反应。中等量即可轻度抑制呼吸中枢,严重肺功能不全和颅脑损伤致呼吸抑制者禁用。其药酶诱导作用可加速其他药物的代谢,影响药效。
巴比妥类连续久服可引起习惯性。突然停药易发生“反跳”现象。此时,快动眼睡眠时间延长,梦魇增多,迫使病人继续用药,终至成瘾。成瘾后停药,戒断症状明显,表现为激动、失眠、焦虑,甚至惊厥。
第三节 其他镇静催眠药
水合氯醛(chloralhydrate)口服易吸收,用于催眠,约15分钟起效,维持6~8小时。此药不缩短快动眼睡眠的时间,停药时也无代偿性快动眼睡眠时间延长。对胃有刺激性,须稀释后口服。久服也可引起耐受性
■[此处缺少一些内容]■
苯巴比妥(phenobarbital,luminal,鲁米那):镇静:15~30mg/次,催眠:60~100mg/次,睡前服。抗癫痫:大发作从小剂量开始,15~30mg/次,3次/日,最大剂量60mg/次,3次/日。
苯巴比妥钠(phenobarbital,sodium)抗惊厥:0.1~0.2g/次,肌内注射。癫痫持续状态:0.1~0.2g/次,缓慢静脉注射。
异戊巴比妥(amobarbital)催眠:0.1~0.2g/次,睡前服。
司可巴比妥(secobarbital,seconal)催眠:0.1~0.2g/次,睡前服。麻醉前给药:0.2~0.3g/次。
硫喷妥钠(thiopentalsodium)临用前配成1.25%~2.5%溶液,缓慢静脉注射,至病人入睡为止。极量:1g/次。
水合氯醛(chloralhydrate)10%溶液,催眠:5~10ml/次,睡前服。抗惊厥:10~20ml/次。
甲丙氨酯(meprobamate,眠尔通,安宁)镇静、抗焦虑:0.2~0.4g/次,3次/日。催眠:0.4~0.8g/次。睡前服。
甲喹酮(methaqualone)催眠:0.1~0.2g/次,睡前服。
格鲁米特(glutethimide)催眠:0.1~0.2g/次,睡前服。
第十五章 抗癫痫药和抗惊厥药
第一节 抗癫痫药
癫痫是一类慢性、反复性、突然发作性大脑机能失调,其特征为脑神经元突发性异常高频率放电并向周围扩散。由于异常放电神经元所在部位(病灶)和扩散范围不同,临床就表现为不同的运动、感觉、意识和植物神经功能紊乱的症状。由此可将癫痫分为以下几型(见表15-1)
表15-1 抗癫的分型
| 部分性发作(局部开始的发作)] 单纯部分性发作(相当于局灶性癫痫) 1.运动性发作 2.感觉性发作 3.植物神经性发作 4.精神性发作 复杂部分性发作(相当于颞叶癫痫) 1.意识障碍起病者(精神运动性发作) 2.继发于单纯部分性发作的意识障碍者 全身性发作(非局部开始的发作) 1.大发作(强直-阵挛发作) |
2.小发作(失神发作) 3.不典型小发作 4.强直发作 5.阵挛发作 6.无张力发作 未能分型的发作 1.新生儿错乱性发作 2.婴儿痉挛 3.偏侧性发作 4.良性中央区癫痫 5.间脑癫痫 |
部分性发作 大脑局部异常放电且只扩散至局部者,只表现大脑局部功能紊乱的症状。其中单纯部分性发作不超过1分钟,意识多不受影响。复杂性部分发作相当于颞叶癫痫,除有部分发作的表现外,还有意识障碍和自动症,包括精神运动性发作。
全身性发作 异常放电涉及全脑,导致突然意识丧失。其中大发作先出现全身肌肉强直性痉挛,约20秒钟后转入阵挛,持续数分钟。大发作连续发生,患者持续昏迷,则称为癫痫持续状态,为危重急症。
小发作又称失神发作,以突然神志丧失为主要表现,伴有脑电图特征性3Hz棘-慢波发放,持续5~20秒,无运动紊乱。小发作对药物的反应与其他型癫痫明显不同。
抗癫痫药的作用机制,从电生理学观点看,有两种方式:抑制病灶神经元过度放电,或作用于病灶周围正常神经组织,以遏制异常放电的扩散。上述效应的基础可能与增强脑内GABA介导的抑制作用有关,如苯二氮![]() 类和苯巴比妥(见第十四章);也可能与干扰Na+、Ca2+、K+等阳离子通道有关,如苯妥英钠。
类和苯巴比妥(见第十四章);也可能与干扰Na+、Ca2+、K+等阳离子通道有关,如苯妥英钠。
一、苯妥英钠
苯妥英钠(phenytoinsodium)为二苯乙内酰脲的钠盐。作为最常用的抗癫痫药已有半个多世纪的历史。
【药理作用机制】苯妥英钠对各种组织的可兴奋膜,包括神经元和心肌细胞膜,有稳定作用,降低其兴奋性。这与其治疗浓度(10μmol/L以下)时即阻滞Na+通道,减少Na+内流有关。苯妥英钠的这一作用具有明显的使用-依赖性(use-dependence)。因此,对高频异常放电的神经元的Na+通道阻滞作用明显,抑制其高频反复放电,而对正常的低频放电并无明显影响。苯妥英钠还抑制神经元的快灭活型(T型)Ca2+通道,抑制Ca2+内流。此作用也呈使用信赖性。较大浓度时,苯妥英钠能抑制K+外流,延长动作电位时程和不应期。
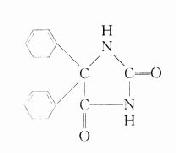
近又知,高浓度苯妥英钠能抑制神经末梢对GABA的摄取,诱导GABA受体增生,由此间接增强GABA的作用,使Cl-内流增加而出现超极化,也可抑制异常高频放电的发生和扩散。
【临床应用】
1.抗癫痫苯妥英钠是治疗大发作和部分性发作的首选药。但对小发作(失神发作)无效,有时甚至使病情恶化。
2.治疗中枢疼痛综合征中枢性疼痛综合征包括三叉神经痛和舌咽神经痛等,其神经元放电与癫痫有相似的发作机制。感觉通路神经元在轻微刺激下即产生强烈放电,引起剧烈疼痛。苯妥英钠能使疼痛减轻,发作次数减少。
3.抗性律失常 见第二十二章
【体内过程】苯妥英钠口服吸收慢而不规则,达峰浓度时间可早于3小时,也可迟于12小时。不同制剂的生物利用度显著不同,且有明显的个体差异。由于本品呈强碱性(pH=10.4),刺激性大,故不宜肌内注射。癫痫持续状态时可作静脉注射。血浆蛋白结合率约90%。60~70%在肝内质网中代谢为无活性的对羟基苯基衍生物,以原形由尿排出者不足5%。消除速率与血浆浓度有密切关系。低于10μg/ml时,按一级动力学消除,血浆t1/2约6~24小时;高于此浓度时,则按零级动力学消除,血浆t1/2可延长至20~60小时,且血药浓度与剂量不成比例地迅速升高,容易出现毒性反应。由于常用量时血浆浓度有较大个体差异,又受诸多因素影响,最好在临床药物监控下给药。
【不良反应】除对胃肠道有刺激外,苯妥英钠的其他不良反应都与血药浓度大致平行。一般血药浓度10μg/ml时可有效地控制大发作,而20μg/ml左右则可出现毒性反应。
轻症反应包括眩晕、共济失调、头痛和眼球震颤等。血药浓度大于40μg/ml可致精神错乱;50μg/ml时以上出现严重昏睡以至昏迷。
长期用药可致牙龈增生,虽无痛苦,但影响美观。发生率约20%,多见于青少年,为胶元代谢改变引起结缔组织增生的结果。注意口腔卫生,经常按摩牙龈,可防止或减轻。一般停药3~6个月后可恢复。
久服可致叶酸吸收及代谢障碍,抑制二氢叶酸还原酶,有时可发生巨幼细胞性贫血。补充甲酰四氢叶酸治疗有效。
过敏反应如皮疹亦较常见。还见粒细胞缺乏、血小板减少、再生障碍性贫血。偶见肝脏损害。应定期作血常规和肝功能检查。
妊娠早期用药,偶致畸胎,如腭裂等。静脉注射过快时,可致心律失常、心脏抑制和血压下降,宜在心电图监护下进行。
【药物相互作用】苯妥英钠为肝药酶诱导剂,能加速多种药物,如皮质类固醇和避孕药等的代谢而降低药效。卡马西平能降低苯妥英钠的血浓度,苯妥英钠也能降低卡马西平的血浓度。苯巴比妥能通过药酶诱导作用,加速苯妥英钠的代谢,又可通过竞争性抑制减弱其灭活,因此影响不确定。但苯妥英钠能提高苯巴比妥的血浓度。
二、卡马西平
卡马西平(carbamazepine)又称酰胺咪嗪。
【药理作用和临床应用】卡马西平的作用机制与苯妥英钠相似。治疗浓度时能阻滞Na+通道,抑制癫痫灶及其周围神经元放电。对复杂部分发作(如精神运动性发作)有良好疗效,至少2/3病例的发作可得到控制和改善。对大发作和部分性发作也为首选药之一。对癫痫并发的精神症状,以及锂盐无效的躁狂、抑郁症也有效。
卡马西平对中枢性痛症(三叉神经痛和舌咽神经痛)有效,其疗效优于苯妥英钠。
【体内过程】口服吸收良好,约2~6小时达血药峰浓度。血浆蛋白结合率为80%。在肝中代谢为有活性的环氧化物。血浆半衰期在用药之初平均为35小时。因本品为药酶诱导剂,连续用药3~4周后,半衰期可缩短50%。
【不良反应】用药早期可出现多种不良反应,如头昏、眩晕、恶心、呕吐和共济失调等,亦可有皮疹和心血管反应。但一般并不严重,不须中断治疗,一周左右逐渐消退。
少见而严重而反应,包括骨髓抑制(再生障碍性贫血、粒细胞减少和血小板减少)、肝损害和心血管虚脱。
三、苯巴比妥和朴米酮
【药理作用】苯巴比妥的中枢抑制作用第十四章已作详细介绍。近发现,苯巴比妥的作用与苯妥英钠相似,也抑制Na+内流和K+外流,但需较高浓度。对异常神经元有抑制作用,抑制其异常放电和冲动扩散。
扑米酮(primidone,扑痫酮)在体内代谢成苯巴比妥和苯乙基丙二酰胺。曾认为此二代谢产物是其抗癫痫作用的基础。但有报道认为扑米酮本身的抗癫痫机制更像苯妥英钠,具有独立的抗癫痫作用。
【临床应用】苯巴比妥对除失神小发作以外的各型癫痫,包括癫痫持续状态,都有效。但因其中枢抑制作用明显,都不作为首选药,仅癫痫持续状态时常用以静脉注射。但临床更倾向于用戊巴比妥钠静脉注射以控制癫痫持续状态。
扑米酮对部分性发作和大发作的疗效优于苯巴比妥;但对复杂部分发作的疗效不及卡巴西平和苯妥英钠。
【不良反应】常见的不良反应为镇静、嗜睡、眩晕和共济失调等。偶可发生巨幼细胞性贫血、白细胞减少和血小板减少。
四、乙琥胺
乙琥胺(ethosuximide)只对失神小发作有效。其疗效不及氯硝西泮,但副作用较少。至今仍是治疗小发作的常用药。对其他型癫痫无效。
常见副作用有嗜睡、眩晕、呃逆、食欲不振和恶心、呕吐等。偶见嗜酸性白细胞增多症和粒细胞缺乏症。严重者可发生再生障碍性贫血。
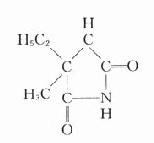
五、丙戊酸钠
丙戊酸钠(sodiumvalproate)对各种类型的癫痫发作都有一定疗效。对失神小发作的疗效优于乙琥胺,但因丙戊酸钠有肝毒性,临床仍愿选用乙琥胺。对全身性肌强直-阵挛性发作有效,但不及苯妥英钠和卡马西平。对非典型小发作的疗效不及氯硝西泮。对复杂部分性发作的疗效近似卡马西平。对其他药物未能控制的顽固性癫痫有时可能奏效。
丙戊酸钠抗癫痫作用与抑制电压敏感性Na+通道有关,也有认为它能抑制GABA代谢酶。使脑内GABA积聚。
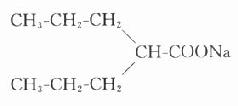
丙戊酸钠口服吸收良好,生物利用度有80%以上。它能显著提高苯妥英钠、苯巴比妥、氯硝西泮和乙琥胺的血药总浓度和游离浓度。而苯妥英钠、苯巴比妥、扑米酮和卡马西平则能降低丙戊酸钠的血药浓度和抗癫痫作用。
丙戊酸钠的不良反应较轻。但近偶见有肝损害,表现为谷草转氨酶升高,少数有肝炎发生,个别肝功能衰竭而死。儿童耐受性较好。对胎儿有致畸作用,常见脊椎裂。
六、苯二氮类
苯二氮![]() 类用于癫痫治疗者有地西泮、氯硝西泮(clonazepam)、硝西泮(nitrazepam)和氯巴占(clobazam)。
类用于癫痫治疗者有地西泮、氯硝西泮(clonazepam)、硝西泮(nitrazepam)和氯巴占(clobazam)。
表15-2 抗癫痫药总结表
| 药物 | 作用 | 用途 | 主要不良反应 | |
| 苯妥英钠 | 阻滞使用-依赖性Na+通道和T型Ca2+通道,增强GABA能抑制效应。 | 除失神小发作以外的所有各型癫痫,尤其用于大发作和部分性发作。中枢性疼痛综合征。心律失常。 | 胃肠道反应,牙龈增生,粒细胞缺乏,再障,致畸。 | |
| 卡马西平 | 与苯妥英钠相似。 | 同上。对中枢性疼痛综合征的疗效优于苯妥英钠。 | 头昏,共济失调,剥脱性皮炎,再障,多动。 | |
| 苯巴比妥 | 与苯妥英钠相似。 | 除失神小发作以外的所有各型癫痫。 | 中枢抑制,眩晕,共济失调,造血障碍。 | |
| 扑米酮 | ||||
| 乙琥胺 | 机制未明 | 小发作常用药。对其他类型发作无效。 | 眩晕,嗜睡,胃肠道反应,粒细胞缺乏,再障。 | |
| 丙戊酸钠 | 阻滞Na+通道,抑制GABA代谢酶 | 各型癫痫 | 胃肠道反应,肝脏损害,共济失调,致畸。 | |
| 苯二氮 |
地西泮 | 增强GABA能抑制作用,使神经元超极化 | 癫痫持续状态首选药。 | 静脉注射偶可致呼吸抑制。 |
| 硝西泮 | 肌阵挛性癫痫,不典型小发作,婴儿痉挛。 | 嗜睡,头昏,共济失调。 | ||
| 氯硝西泮氯巴占 | 各型癫痫,尤其用于不典型小发作,失神小发作,肌阵挛发作。 | 啫睡,共济失调,白细胞减少,行为障碍。 | ||
地西泮是控制癫痫持续状态的首选药之一。静脉注射见效快,安全性较大。但偶可引起呼吸抑制,宜缓慢注射(1mg/min)。
硝西泮对肌阵挛性癫痫、不典型小发作和婴儿痉挛有较好疗效。
氯硝西泮和氯巴占对各型癫痫都有效,尤以对失神小发作、肌阵挛发作和不典型小发作为佳。
苯二氮![]() 类的副作用是中枢抑制作用明显,甚至发生共济失调。久用可产生耐受性,骤然停药时发生症状反跳和戒断症状,原有发作加剧。
类的副作用是中枢抑制作用明显,甚至发生共济失调。久用可产生耐受性,骤然停药时发生症状反跳和戒断症状,原有发作加剧。
第二节 抗惊厥药
惊厥是各种原因引起的中枢神经过度兴奋的一种症状,表现为全身骨骼肌不自主的强烈收缩。常见于小儿高热、破伤风、癫痫大发作、子痫和中枢兴奋药中毒等。
常用抗惊厥药有巴比妥类、水合氯醛和地西泮等,已于镇静催眠药章中讨论。本节只介绍硫酸镁。
硫酸镁
【药理作用和临床应用】 神经化学传递和骨骼肌收缩均需Ca2+参与。Mg2+与Ca2+由于化学性质相似,可以特异地竞争Ca2+受点,拮抗Ca2+的作用,抑制神经化学传递和骨骼肌收缩,从而使肌肉松弛。与此同时,也作用于中枢神经系统,引起感觉和意识消失。对于各种原因所致的惊厥,尤其是子痫,有良好的抗惊厥作用。过量时,引起呼吸抑制、血压骤降以至死亡。静脉缓慢注射氯化钙,可立即消除Mg2+的作用。口服不易吸收,仅有致泻作用(见第三十一章)。
抗癫痫药和抗惊厥药 制剂及用法
苯妥英钠(phenytoin sodium) 0.3~0.6g/次,分2~3次,或于晚上一次顿服。极量:0.3g/次,0.6g/日。癫痫持续状态:若患者未用过苯妥英钠,可用0.25~0.5g,加5%葡萄糖20~40ml,在6~10分钟内缓慢静脉注射。
卡马西平(carbamazepine,酰胺咪嗪)开始剂量:100mg,2次/日,以后逐渐增至600~900mg/日或8~10mg/kg/日,分次服用。用于抗癫痫时,剂量可偏大。用于三叉神经痛等症时,剂量一般宜小,每日1.2g,常不能耐受。
扑米酮(primidone,扑痫酮)开始0.06g,3次/日;渐增至0.25g,3次/日。每日总量不超过1.5g。
乙琥胺(ethosuximide)儿童15~35mg/kg/日;成人0.5g,2~3次/日。
丙戊酸钠(sodium valproate)儿童15~60mg/kg/日;成人0.6~1.8g/日,分3次服。
氯硝西泮(clonazepam,氯硝安定)起始儿童0.01~0.03mg/kg/日;成人不超过1.5mg/日,分3次服。以后儿童每3天加0.25~0.5mg /日;成人加0.5~1mg/日。最大耐受量儿童0.2mg/kg/日;成人20mg/日。
硝西泮(nitrazepam,硝基安定)用于婴儿痉挛和不典型小发作,0.5~1.0mg/kg/日。
地西泮(diazepam,安定)用于癫痫持续状态,5~10mg静脉注射,间隔10~15分钟一次,最大量可至30mg.注射速度以下超过5mg/min为宜。必要时在2~4小时内重复上述方案。亦可静脉滴入,至发作仃止。
硫酸镁(magnesium sulfate)1.25~2.5g/次,肌内注射或静脉滴注。静脉滴注时以5%葡萄糖注射液将硫酸镁稀释成1%浓度进行滴注,直至惊厥停止。使用时宜备有氯化钙或葡萄糖酸钙注射液,以备万一过量时作静脉注射对抗之。
第十六章 抗帕金森病药
帕金森病(Parkinsondisease)又称震颤麻痹。临床主要症状为进行性运动徐缓、肌强直及震颤,此外尚有知觉、识别及记忆障碍等症状。
现认为帕金森病是因纹状体内缺乏多巴胺所致,主要病变在黑质-纹状体多巴胺能神经通路。黑质中多巴胺能神经元(图16-1)发出上行纤维到达纹状体(尾核及壳核),其末梢与尾-壳核神经元形成突触,以多巴胺为递质,对脊髓前角运动神经元起抑制作用。同时尾核中也有胆碱能神经元,与尾-壳核神经元所形成的突触以乙酰胆碱为递质,对脊髓前角运动神经元起兴奋作用。正常时两种递质处于平衡状态,共同调节运动机能。
图16-1 黑质-纹状体多巴胺能神经通路
帕金森病患者因黑质有病变,多巴胺合成减少,使纹状体内多巴胺含量降低,造成黑质-纹状体通路多巴胺能神经功能减弱,而胆碱能神经功能相对占优势,因而产生帕金森病的张力增高症状。
上述理论不仅能说明以往使用胆碱受体阻断药治疗的理由,而且也提示补充脑内多巴胺是治疗帕金森病的新途径。
老年性血管硬化、脑炎后遗症及长期服用抗精神病药等均可引起类似帕金森病的症状,称为帕金森综合征,其药物治疗与帕金森病相似。
抗帕金森病药分为拟多巴胺药和胆碱受体阻断药两类。
第一节 拟多巴胺类药
左旋多巴
左旋多巴(levodopa)又称L-多巴(L-dopa),为酪氨酸的羟化物,在体内是左旋酪氨酸合成儿茶酚胺的中间产物。
【体内过程】口服左旋多巴后,通过芳香族氨基酸的主动转运系统从小肠迅速吸收,约0.5~2小时,血药浓度达峰值,血浆t1/2为1~3小时。其吸收速率受多种因素影响,如胃排空延缓(同服胆碱受体阻断药)、胃液酸度高或小肠中有其他氨基酸与之竞争主动转运系统(如高蛋白饮食)等,均可降低其生物利用度。吸收后,首次通过肝时大部分即被脱羧,转变成多巴胺。也有相当部分在肠、心、肾中被脱羧生成多巴胺。而多巴胺又不易透过血脑屏障,因此进入中枢神经系统的左旋多巴不到用量的1%。在外周组织中形成大量多巴胺是造成不良反应的原因。若同时服用外周脱羧酶抑制剂(卡比多巴)可减少不良反应。小部分左旋多巴转变为黑色素(melannin);另有一部分左旋多巴经儿茶酚氧位甲基转移酶(COMT)而甲基化,转变为3-甲氧基多巴;以上代谢物均由肾迅速排泄。这种代谢过程消耗较多的COMT,而COMT反应中甲基主要来自食物中的蛋氨酸,故长期服用左旋多巴可导致蛋氨酸缺乏。
【药理作用及应用】
1.抗帕金森病左旋多巴在脑内转变为多巴胺,补充纹状体中多巴胺的不足,因而具有抗帕金森病的疗效。研究表明,曾用过大量左旋多巴治疗的患者,死后纹状体中多巴胺浓度比未用药治疗者高5~8倍;而且脑内多巴胺浓度与左旋多巴的疗效相一致。说明患者黑质-纹状体通路中残存的多巴胺能神经元仍有储存多巴胺的能力。其纹状体多巴脱羧酶仍有足够的酶活性可使左旋多巴转变为多巴胺。
用左旋多巴治疗后,约75%的患者获得较好疗效。治疗初期疗效更显。左旋多巴的作用特点是:①对轻症及较年轻患者疗效较好,而重症及年老衰弱患者疗效差;②对肌肉僵直及运动困难疗效较好,而对肌肉震颤症状疗效差,如长期用药及较大剂量对后者仍可见效;③作用较慢,常需用药2~3周才出现客观体征的改善,1~6个月以上才获得最大疗效,但作用持久,且随用药时间延长而递增。
左旋多巴对其他原因引起的帕金森综合征也有效。但对吩噻嗪类等抗精神病药所引起的无效,因这些药有阻断中枢多巴胺受体的作用。
2.治疗肝昏迷肝昏迷发病学说中的伪递质学说认为,正常机体蛋白质代谢产物苯乙胺和酪胺都在肝内被氧化解毒。肝功能障碍时,血中苯乙胺和酪胺升高,在神经细胞内经β-羟化酶分别生成伪递质,苯乙醇胺和羟苯乙醇胺(鱆胺),它们取代了正常递质——去甲肾上腺素,妨碍神经功能。用左旋多巴能在脑内转变去甲肾上腺素。使正常神经活动得以恢复,患者可由昏迷转为苏醒。因不能改善肝功能,作用只是暂时性的。
【不良反应】左旋多巴的不良反应较多,因其在体内转变为多巴胺所致
1.胃肠道反应 治疗初期约80%患者出现恶心、呕吐、食欲碱退等。用量过大或加量过快更易引起,继续用药可以消失。偶见溃疡出血或穿孔。
2.心血管反应 治疗初期,约30%患者出现轻度体位性低血压,原因未明。少数患者头晕,继续用药可减轻。多巴胺对β受体有激动作用,可引起心动过速或心律失常。
3.不自主异常运动为长期用药所引起的不随意运动,多见于面部肌群,如张口、咬牙、伸舌、皱眉、头颈部扭动等。也可累及肢体或躯体肌群,偶见喘息样呼吸或过度呼吸。另外还可出现“开-关现象”(on –off phenomenon),患者突然多动不安(开),而后又出现全身性或肌强直性运动不能(关),严重妨碍病人的正常活动。疗程延长,发生率也相应增加。此时宜适当减少左旋多巴的用量。
4.精神障碍出现失眠、焦虑、恶梦、狂躁、幻觉、妄想、抑郁等。需减量或停药。此反应可能与多巴胺作用于大脑边缘叶有关。
【药物相互作用】
1.维生素B6是多巴脱羧酶的辅基,可增强左旋多巴的外周副作用。
2.抗精神病药能引起帕金森综合征,又能阻断中枢多巴胺受体,所以能对抗左旋多巴的作用。
卡比多巴
α-甲基多巴肼(α-methyldopahydrazine)有两种异构体,其左旋体称卡比多巴(carbidopa)是较强的L-芳香氨基酸脱羧酶抑制剂,由于不易通过血脑屏障,故与左旋多巴合用时,仅能抑制外周多巴脱羧酶的活性,从而减少多巴胺在外周组织的生成,同时提高脑内多巴胺的浓度。这样,既能提高左旋多巴的疗效,又能减轻其外周的副作用,所以是左旋多巴的重要辅助药。卡比多巴单独应用基本无药理作用。将卡比多巴与左旋多巴按1:10的剂量合用,可使左旋多巴的有效剂量减少75%,苄丝肼(benserazide)与卡比多巴有同样的效应,它与左旋多巴按1:4制成的复方制剂称美多巴(madopar),应用于临床。
金刚烷胺
金刚烷胺(amantadine)原是抗病毒药(见第四十三章),后发现其也有抗帕金森病的作用,疗效不及左旋多巴,但优于胆碱受体阻断药。见效快而持效短,用药数天即可获最大疗效,但连用6~8周后疗效逐渐减弱。与左旋多巴合用有协同作用。其抗帕金森病的机制可能在于促使纹状体中残存的完整多巴胺能神经元释放多巴胺;并能抑制多巴胺的再摄取;且有直接激动多巴胺受体的作用及较弱的抗胆碱作用。长期用药后,常见下肢皮肤出现网状青斑,可能是由儿茶酚胺释放引起外周血管收缩所致。偶致惊厥,故癫痫患者禁用。每日剂量超过300mg,可致失眠、精神不安及运动失调等。
溴隐亭
溴隐亭(bromocriptine)是一种半合成的麦角生物碱。口服大剂量对黑质-纹状体通路的多巴胺受体有较强的激动作用,其疗效与左旋多巴相似。小剂量激动结节漏斗部的多巴胺受体,因此可减少催乳素和生长激素的释放。用于回乳、治疗催乳素分泌过多症和肢端肥大症等。
第二节 胆碱受体阻断药
胆碱受体阻断药可阻断中枢胆碱受体,减弱纹状体中乙酰胆碱的作用。本类药物曾是沿用已久的抗帕金森病药,但自使用左旋多巴以来,它们已退居次要地位,其疗效不如左旋多巴。现适用于①轻症患者;②不能耐受左旋多巴或禁用左旋多巴的患者;③与左旋多巴合用,可使50%患者症状得到进一步改善;④治疗抗精神病药引起的帕金森综合征有效。传统胆碱受体阻断药阿托品、东莨菪碱抗帕金森病有效,但因外周抗胆碱作用引起的副作用大,因此合成中枢性胆碱受体阻断药以供应用,常用者为苯海索。
苯海索(trihexyphenidyl)又称安坦(artane),其外周抗胆碱作用为阿托品的1/10~1/2。抗震颤疗效好,但改善僵直及动作迟缓较差,对某些继发性症状如过度流涎有改善作用。不良反应似阿托品,对心脏的影响比阿托品弱,故应用较安全。但仍有口干、散瞳、尿潴留、便秘等副作用。窄角型青光眼、前列腺肥大者慎用。
卡马特灵(kemadrin)又称开马君,其药理作用及应用与苯海索相似。
抗帕金森病药 制剂及用法
左旋多巴(L-dopa)抗帕金森病:开始口服0.1~0.25g/次,2~4次/日。以后每隔2~4天递增0.25~0.75g,通常有效量为2~5g/日。最大日用量不超过8g。如与卡比多巴合用,左旋多巴600mg/日,最多不超过2g/日。治疗肝昏迷:先0.3~0.4g/日,加入5%葡萄糖溶液500ml中静滴,清醒后减量至0.2g/日。
卡比多巴(carbidopa)开始口服卡比多巴10mg/次,左旋多巴100mg/次,一日4次,以后递增至每日量卡比多巴200mg,左旋多巴达2g为限。
溴隐亭(bromocriptine)开始1.25mg/次,2次/日,以后每日递增2.5mg。
金刚烷脘(amantadine)0.1g/次,早晚各服一次。
盐酸苯海索(trihexyphenidyl hydrochloride)开始1~2mg/次,3次/日;以后递增,每日不超20mg。
卡马特灵(kemadrine)开始2.5~5mg/次,3次/日,以后可递增至15~30mg/日。
第十七章 抗精神失常药
精神失常(psychiatric disorders)是由多种原因引起的精神活动障碍的一类疾病。治疗这类疾病的药物统称为抗精神失常药。根据临床用途,分为三类:即抗精神病药(antipsychotic drugs)、抗躁狂抑郁症药(antimanic and antidepressive drugs)及抗焦虑药(antianxiety drugs)
第一节 抗精神病药
抗精神病药主要用于治疗精神分裂症及其他精神失常的躁狂症状。
根据化学结构可将常用抗精神病药分为吩噻嗪类(phenothiazines)、硫杂蒽类(thioxanthenes)、丁酰苯类(butyrophenones)、及其他药物。
一、吩噻嗪类
吩噻嗪是由硫、氮原子联结两个苯环(称为吩噻嗪母核)的一类化合物。根据其侧链基团不同,分为二甲胺类、哌嗪类及哌啶类(表17-1)。
表17-1常用吩噻嗪类药的化学结构
以上三类中,以哌嗪类抗精神病作用最强,其次是二甲胺类,哌啶类最弱。目前国内临床常用的有氯丙嗪、氟奋乃静及三氟拉嗪等,以氯丙嗪应用最广。
氯丙嗪
氯丙嗪(chlorpromazine)又称冬眠灵(wintermin)。
【体内过程】口服或注射均易吸收,但吸收速度受剂型、胃内食物的影响,如同时服用胆碱受体阻断药,可显着延缓其吸收。口服氯丙嗪2~4小时血浆药物浓度达峰值,肌注吸收迅速,但因刺激性强应深部注射,其生物利用度比口服大3~4倍,这与口服具有首过效应有关。吸收后,约90%与血浆蛋白结合。氯丙嗪具有高亲脂性,易透过血脑屏障,脑组织中分布较广,以下丘脑、基底神经节、丘脑和海马等部位浓度最高,脑内浓度可达血浆浓度的10倍。氯丙嗪主要经肝微粒体酶代谢成去甲氯丙嗪、氯吩噻嗪、甲氧基化或羟化产物及葡萄糖醛酸结合物。氯丙嗪及其代谢物主要经肾排泄。老年患者对氯丙嗪的代谢与消除速率减慢。不同个体口服相同剂量氯丙嗪后,血浆药物浓度相差可达10倍以上,因此,临床用药应个体化。氯丙嗪排泄缓慢,停药后2~6周,甚至6个月,尿中仍可检出,这可能是氯丙嗪脂溶性高,蓄积于脂肪组织的结果。
【药理作用及临床应用】氯丙嗪主要对DA受体有阻断作用,另外也能阻断α受体和M受体等。因此,其药理作用广泛而复杂。
DA受体存在于外周神经系统和中枢神经系统。至少有D1和D2二种亚型。D1受体与GS蛋白相偶联,激动时可经GS蛋白激活腺苷酸环化酶,使cAMP增加。在外周引起血管扩张,心肌收缩增强。但在中枢神经系统的功能尚不清楚。D2受体在中枢神经系统见于脑内DA能神经通路。脑内DA通路有多条,其中主要的是黑质-纹状体通路、中脑-边缘叶通路和中脑-皮质通路。前者与锥体外系的运动功能有关,后两条通路与精神、情绪及行为活动有关。此外还有结节-漏斗通路,与调控下丘脑某些激素的分泌有关。D2受体与Gi蛋白相偶联,激动时抑制腺苷酸环化酶,另还能开放钾通道。氯丙嗪对脑内DA受体缺乏特异的选择性,因而作用多样。
1.中枢神经系统
(1)抗精神病作用 正常人一次口服氯丙嗪100mg后,出现安定、镇静、感情淡漠和对周围事物少起反应,在安静环境中易诱导入睡。精神病患者用药后,在不引起过分镇静的情况下,可迅速控制兴奋躁动。继续用药,可使幻觉、妄想、躁狂及精神运动性兴奋逐渐消失,理智恢复,情绪安定,生活自理。氯丙嗪抗幻觉及抗妄想作用一般需连续用药6周至6个月才充分显效,且无耐受性。但连续用药后,安定及镇静作用则逐渐减弱,出现耐受性。
临床上主要应用氯丙嗪治疗各型精神分裂症,对急性患者疗效较好,但无根治作用,必须长期服用以维持疗效,减少复发。此外,也可用于治疗躁狂症及其他精神病伴有的兴奋、紧张及妄想等症状。
氯丙嗪抗精神病的作用机制未定。早年发现吩噻嗪类可提高动物脑内DA的主要代谢产物高香草酸(HVA)的生成率。这意味着DA的更新增加,因而推测这是由于吩噻嗪类阻断了脑内DA受体,代偿性地加速了突触前膜中DA的合成与代谢的结果。还发现吩噻嗪类增加DA的更新率与其作用强度相平行。后又发现,吩噻嗪类可抑制脑内腺苷酸环化酶活性。对该酶的抑制程度又与它们的临床疗效相一致。此外,以放射配体结合分析法又发现吩噻嗪类可与3H-氟哌啶醇及3H-DA竞争脑内特异性结合部位(DA受),而竞争力的强弱与吩噻嗪类抗精神病作用强度相平行。目前认为精神分裂症的临床症状是由于脑内DA功能过强所致,且脑内D2受体密度已特异性地增高。吩噻嗪类是D2受体的强大拮抗剂。因此认为吩噻嗪类抗精神病的作用是通过阻断中脑-边缘叶及中脑-皮质通路中的D2受体而发生的。
(2)镇吐作用氯丙嗪有强大镇吐作用,可对抗去水吗啡的催吐作用,大剂量则直接抑制呕吐中枢。去水吗啡对延脑第四脑室底部极后
■[此处缺少一些内容]■
系的影响 氯丙嗪阻断黑质-纹状体通路的D2受体,导致胆碱能神经功能占优势。因而在长期大量应用时可出现锥体外系反应。
2.植物神经系统氯丙嗪具有明显的α受体阻断作用,可翻转肾上腺素的升压效应,同时还能抑制血管运动中枢,并有直接舒张血管平滑肌的作用,因而扩张血管、降低血压。但反复用药降压作用减弱,故不适于高血压病的治疗。氯丙嗪尚可阻断M胆碱受体,但作用弱,无治疗意义。
3.内分泌系统 结节-漏斗处DA通路的主要功能是调控下丘脑某些激素的分泌。氯丙嗪可阻断该通路的D2受体,减少下丘脑释放催乳素抑制因子,因而使催乳素分泌增加,引起乳房肿大及泌乳。乳腺癌患者禁用氯丙嗪。此外能抑制促性腺释放激素的分泌,使卵泡刺激素和黄体生成素释放减少,引起排卵延迟;以及抑制促皮质激素和生长激素的分泌。后一作用可试用于治疗巨人症。
【不良反应】氯丙嗪安全范围大,但长期大量应用,不良反应较多。
1.一般不良反应有嗜睡、无力、视力模糊、鼻塞、心动过速、口干、便秘等中枢神经及植物神经系统的副作用。长期应用可致乳房肿大、闭经及生长减慢等。氯丙嗪局部刺激性较强,不应作皮上注射。静脉注射可引起血栓性静脉炎,应以生理盐水或葡萄糖溶液稀释后缓慢注射。静注或肌注后,可出现体位性低血压,应嘱患者卧床1~2小时后方可缓慢起立。
2.锥体外系反应是长期大量应用氯丙嗪治疗精神分裂症时最常见的副作用,其发生率与药物剂量、疗程和个体因素有关。其表现为:①帕金森综合征,出现肌张力增高、面容呆板(面具脸)、动作迟缓、肌肉震颤、流涎等;②急性肌张力障碍多出现于用药后1~5天,由于舌、面、颈及背部肌肉痉挛,患者出现强迫性张口、伸舌、斜颈、呼吸运动障碍及吞咽困难;③静坐不能(akathisia)患者出现坐立不安,反复徘徊。以上三种症状可用胆碱受体阻断药安坦缓解之。此外还可引起一种少见的锥体外系反应症状,迟发性运动障碍(tardivedyskinesia)或迟发性多动症,表现为不自主、有节律的刻板运动,出现口-舌-颊三联症,如吸吮、舐舌、咀嚼等。若早期发现及时停药可以恢复,但也有停药后仍难恢复。应用胆碱受体阻断药反可使之加重。造成迟发性运动障碍的原因可能与氯丙嗪长期阻断突触后DA受体,使DA受体数目增加,即向上调节有关。
3.过敏反应常见皮疹、光敏性皮炎。少数患者出现肝细胞内微胆管阻塞性黄疸。也有少数患者出现急性粒细胞缺乏,应立即停药,并用抗生素预防感染。
【急性中毒】一次吞服超大剂量(1~2克)氯丙嗪后,可发生急性中毒,出现昏睡、血压下降达休克水平,并出现心动过速、心电图异常(P-R间期或Q-T间期延长,T波低平或倒置),应立即进行对症治疗。
【禁忌证】氯丙嗪能降低惊厥阈,诱发癫痫,有癫痫史者禁用。昏迷患者(特别是应用中枢抑制药后)禁用。伴有心血管疾病的老年患者慎用,冠心病患者易致猝死应加注意。严重肝功能损害者禁用。
其他吩噻嗪类药物
奋乃静(perphenazine)、氟奋乃静(fluphenazine)及三氟拉嗪(trifluoperazine)是吩噻嗪类中的哌嗪衍生物,其共同特点是抗精神病作用强,锥体外系副作用也很显着,而镇静作用弱。其中以氟奋乃静和三氟拉嗪疗效较好,最为常用,而奋乃静疗效较差。硫利达嗪(thioridazine,甲硫达嗪)是吩噻嗪类的哌啶衍生物,疗效不及氯丙嗪,但锥体外系反应少见,而镇静作用强。各药特点见表17-2。
表17-2吩噻嗪类抗精神病药作用比较
| 药物 | 抗精神病剂量(mg/日) | 副 作 用 | ||
| 镇静作用 | 锥体外系反应 | 降压作用 | ||
| 氯丙嗪 | 300~800 | +++ | ++ | +++(肌注)++口服 |
| 氟奋乃静 | 1~20 | + | +++ | + |
| 三氟拉嗪 | 6~20 | + | +++ | + |
| 奋乃静 | 8~32 | ++ | +++ | + |
| 硫利达嗪 | 200~600 | +++ | + | ++ |
++ +强,+ +次强;+弱
二、硫杂蒽类
硫杂蒽类基本化学结构与吩噻嗪类相似,其代表药物为氯普噻吨(chlorprothixene),又名泰尔登(tardan)。其抗精神分裂症和抗幻觉、妄想作用比氯丙嗪弱,但镇静作用强,而抗肾上腺素作用和抗胆碱作用较弱。因化学结构又与三环类抗抑郁药相似,故有较弱的抗抑郁作用。适用于伴有焦虑或焦虑性抑郁的精神分裂症、焦虑性神经官能症、更年期抑郁症等。副作用为锥体外系反应,与氯丙嗪相似。
三、丁酰苯类
本类药物有氟哌啶醇(haloperidol),其作用及作用机制与吩噻嗪类相似。抗精神病作用及锥体外系反应均很强,镇静、降压作用弱。因抗躁狂、抗幻觉、妄想作用显着,常用于治疗以兴奋躁动、幻觉、妄想为主的精神分裂症及躁狂症。镇吐作用较强,用于多种疾病及药物引起的呕吐,对持续性呃逆也有效。锥体外系反应高达80%,常见急性肌张力障碍和静坐不能。大量长期应用可致心肌损伤。同类药物氟哌利多(droperidol)作用维持时间短,临床常与镇痛药芬太尼合用作安定麻醉术。
四、其他类
五氟利多(penfluridol)为长效抗精神病药。大服后8~16小时血药浓度达峰值,128小时后,血药浓度仍为峰值的30%。一次用药后7天,血中仍可检出。其长效原因与贮存于脂肪组织,并自其中缓慢释放入血及入脑组织有关。每周口服一次即可维持疗效。疗效与氟哌啶醇相似,但无明显镇静作用。副作用中以锥体外系反应常见。适用于急慢性精神分裂症,尤适用于慢性患者维持与巩固疗效。同类药物尚有匹莫齐特(pimozide),其作用维持时间较五氟利多短,每日口服一次,疗效可维持24小时。
舒必利(sulpiride)对急慢性精神分裂症有较好疗效,对长期用其他药物无效的难治病例也有一定疗效。无明显镇静作用,对植物神经系统几无影响,不良反应少,锥体外系反应轻微。本药还有抗抑郁作用,也可用于治疗抑郁症。
氯氮平(clozapine)抗精神病作用较强,对其他药物无效的病例仍可有效,也适用于慢性精神分裂症。几无锥体外系统反应,这可能与氯氮平有较强的抗胆碱作用有关。可引起粒细胞减少,应予警惕。
第二节 抗躁狂抑郁症药
躁狂抑郁症又称情感性精神障碍(affectivedisorders),是一种以情感病态变化为主要症状的精神病。躁狂抑郁症表现为躁狂或抑郁两者之一反复发作(单相型),或两者交替发作(双相型)。其病因可能与脑内单胺类功能失衡有关,但5-HT缺乏是其共同的生化基础。在此基础上,NA功能亢进为躁狂,发作时患者情绪高涨,联想敏捷,活动增多。NA功能不足则为抑郁,表现为情绪低落,言语减少,精神、运动迟缓、常自责自罪,甚至企图自杀。
一、抗抑郁症药
常用抗抑郁药为三环类,包括米帕明(imipramine)、地昔帕明(desipramine)阿米替林(amitriptyline)、多塞平(doxepin)等。它们与吩噻嗪类在化学结构上的主要区别是用-CH2-CH2-代替S。
米帕明
【体内过程】口服吸收良好,但个体差异大。血药浓度于2~8小时达峰值,血浆t1/2为10~20小时。广泛分布于全身各组织,以脑、肝、肾及心肌分布较多。主要在肝代谢,基侧链N脱甲基转化为地昔帕明,后者有显著抗抑郁作用。米帕明及地昔帕明最终被氧化成无效的羟化物或与葡萄糖醛酸结合,自尿排出。
【药理作用】
1.中枢神经系统正常人口服本药后,出现困倦、头晕、口干、视力模糊及血压稍降等。若连续用药数天,以上症状加重,并出现注意力不集中,思维能力下降。相反,抑郁症患者连续服药后,情绪提高,精神振奋,出现明显抗抑郁作用。但米帕明起效缓慢,连续用药2~3周后才见效,故不作应急药物应用。
米帕明抗抑郁作用机制曾经研究,早期发现利血平能引起抑郁症状,而预先给予米帕明则可防止,但若先用利血平耗竭脑内儿茶酚胺后则无效。表明米帕明必须在脑内有儿茶酚胺贮存时,才能发挥抗抑郁作用。因而推测,米帕明可能因抑制突触前膜对NA及(或)5-HT的再摄取,使突触间隙的NA浓度升高,促进突触传递功能而发挥抗抑郁作用。但近年出现的非典型抗抑郁药,并不抑制或仅微弱抑制NA及5-HT的再摄取(如伊普吲哚,iprindole),却仍有较强的抗抑郁作用。此外,米帕明虽可迅速抑制脑内单胺类递质再摄取,但抗抑郁作用的出现却需几周之久,因此增强脑内单胺类递质的作用,只是其复杂作用机制中一个早期环节。
2.植物神经系统 治疗量米帕明能阻断M胆碱受体,引起阿托品样作用。
3.心血管系统米帕明能降低血压,抑制多种心血管反射,易致心律失常,这与它抑制心肌中NA再摄取有关。此外还可以引起体位性低血压及心动过速。心电图中T波倒置可低平。近来证明,米帕明对心肌有奎尼丁样作用,因此心血管疾病患者慎用。
【临床应用】主要用于各型抑郁症的治疗。对内源性、反应性及更年期抑郁症疗效较好,而对精神分裂症的抑郁状态疗效较差。
【不良反应】最常见的副作用为阿托品样作用的口干、便秘、视力模糊、心悸等。因易致尿潴留及升高眼内压,故前列腺肥大及青光眼患者禁用。中枢神经方面表现为乏力、肌肉震颤。某些患者用药后可自抑制状态转为躁狂兴奋状态,剂量大时尤易发生。极少数患者出现皮疹、粒细胞缺乏及黄疸等过敏反应。
【药物相互作用】三环类药物能增强中枢抑制药的作用以及对抗可乐定的降压作用。三环类与安坦等抗帕金森病药或抗精神病药合用,则注意它们的抗胆碱效应可能相互增强。
其他三环类药物的作用比较见表17-3。
其他抗抑郁症药 马普替林(maprotiline)能选择性抑制NA的再摄取。为广谱抗抑郁药,具有奏效快,副作用小的特点。临床用于各型抑郁症,老年抑郁症患者尤为适用。诺米芬新(nomifensine)能显著抑制NA及DA的再摄取,而对5-HT再摄取抑制作用微弱。抗胆碱作用及心血管作用极弱。适用于各型抑郁症,老年患者易于接受,疗效比米帕明略高或相似。此外,本药缓解抑郁患者的严重运动迟缓疗效好,这可能与其抑制DA的再摄取有关。
表17-3 三环类抗抑郁药作用比较
| 药 物 | T1/2(h) | 抑制单胺类递质重摄取 | 镇静作用 | 抗胆碱作用 | |
| 5-HT | NA | ||||
| 米帕明 | 9~24 | ++ | ++ | ++ | ++ |
| 地昔帕明 | 14~76 | +++ | + | + | |
| 阿密替林 | 17~40 | +++ | + | +++ | +++ |
| 多塞平 | 8~24 | 弱 | 弱 | +++ | +++ |
二、抗躁狂症药
抗躁狂症药(antimanicdrugs)氯丙嗪、氟哌啶醇及抗癫痫药卡马西平等对躁狂症也有效。但典型抗躁狂药是锂制剂。
碳酸锂
碳酸锂(lithiumcarbonate)口服吸收快而完全,2~4小时血药浓度达峰值。锂离子先分布于细胞外液,然后逐渐蓄积于细胞内。锂虽吸收快,但通过血脑屏障进入脑组织和神经细胞需要一定时间。因此,锂盐显效较慢。主要自肾排泄,约80%由肾小球滤过的锂在近曲小管与钠竞争重吸收,故增加钠摄入可促进其排泄,而缺钠或肾小球滤出减少时,可导致体内锂潴留,引起中毒。
治疗量锂盐对正常人精神活动几无影响,但对躁狂症发作者则有显着疗效,使言语、行为恢复正常。实验表明锂盐可抑制脑内NA及DA的释放,并促进其再摄取,使突触间隙NA浓度降低,而产生抗躁狂作用。近来发现,锂盐能抑制肌醇磷酸酶,此酶催化磷脂酰肌醇(phosphatidylinositol,PI)系统中三磷酸肌醇(inositol triphosphate,IP3)的脱磷酸化反应,从而阻止肌醇的生成。所以锂盐能抑制脑组织中肌醇的生成,减少PIP2的含量。因而认为锂盐是通过干扰脑内PIP2系统第二信使的代谢,从而发挥其抗躁狂作用的。
临床主要用于治疗躁狂症。对精神分裂症的兴奋躁动也有效,与抗精神病药合用疗效较好,可减少抗精神病药的剂量;同时抗精神病药还可缓解锂盐所致恶心、呕吐等副作用。
锂盐不良反应较多,有个体差异性。用药初期有恶心、呕吐、腹泻、疲乏、肌肉无力、肢体震颤、口干、多尿。常在继续治疗1~2周内逐渐减轻或消失。此外,尚有抗甲状腺作用,可引起甲状腺功能低下或甲状腺肿,一般无明显自觉症状,停药后可恢复。锂盐中毒主要表现为中枢神经症状,如意识障碍、昏迷、肌张力增高、深反射亢进、共济失调、震颤及癫痫发作。静注生理盐水可加速锂的排泄。为确保用药安全,对服用锂盐患者,应每日测定血锂浓度,当血锂高至1.5~2.0mmol/L时,应立即减量或停药。
第三节 抗焦虑药
焦虑是多种精神病的常见症状,焦虑症则是一种以急性焦虑反复发作为特征的神经官能症,并伴有植物神经功能紊乱。发作时,患者多自觉恐惧、紧张、忧虑、心悸、出冷汗、震颤及睡眠障碍等。无论是焦虑症或焦虑状态,临床多用抗焦虑药治疗。常用的为苯二氮![]() 类(详见第十四章)。
类(详见第十四章)。
抗精神失常药 制剂及用法
盐酸氯丙嗪(chlorpromazine hydrochloride)一般口服量12.5~50mg/次,2次/日。肌内注射,25~50mg/次。治疗精神病宜从小剂量开始,轻症300mg/日,重症600~800mg/日,好转后逐渐减用维持量(50~100mg/日)。拒服药者用50~100mg/次,加于25%葡萄糖溶液20ml 内,缓慢静脉注射。
奋乃静(perphenazine)一般2~4mg/次,3次/日。5~10mg/次,肌内注射。治疗精神病:轻症20~30mg/日,重症40~60mg/日,分2次肌内注射。
盐酸三氟拉嗪(trifluperazine hydrochloride)10~30mg/次,分3次服。
盐酸氟奋乃静(fluphenazine hydrochloride)2~20mg/日。
氟奋乃静癸酸酯(fluphenazine decanoate)每2周25mg,肌内注射。
氟普噻吨(chlorprothixene)轻症150mg/日,重症300~600mg/日,口服。
氟哌啶醇(haloperidol)口服2~10mg/次,3次/日,肌内注射,5mg/次。
氟哌利多(droperidol)治疗精神分裂症:10~30mg/日,分1~2次,肌内注射。神经安定镇痛:每次5mg,加入芬太尼0.1mg,在2~3分钟内缓慢静脉注入,5~6分钟内如未达一级浅麻状态,可追加半量至一倍量。麻醉前给药:手术前半小时肌内注射2.5~5mg。
盐酸米帕明(imipramine hydrochloride)25~75mg/次,3次/日。年老体弱者每日自12.5mg开始,逐渐增量。
阿米替林(amitriptyline)75~150mg/日,分3次口服。
碳酸锂(lithium carbonate)由小剂量开始,0.5g/日,递增至0.9~1.8g/日,分3~4次口服。
第十八章 镇痛药
疼痛是多种疾病的症状,它使患者感受痛苦,尤其是剧痛,还可能引起生理功能紊乱,甚至休克。因此,适当地应用药物缓解疼痛,防止可能产生的生理功能紊乱是很必要的。但疼痛发生的原因不同,应区别不同情况选用不同药理作用的药物。另外,疼痛的性质与部位往往是诊断疾病的重要依据,因此,对诊断未明的疼痛不宜先用药物止痛,以免掩盖病情,贻误诊断。
缓解疼痛的药物,按其药理作用及作用机制,可以分为两大类:其一是主要作用于中枢神经系统、选择性地消除或缓解痛觉的药物,在镇痛时,意识清醒,其他感觉不受影响,这类药物称为镇痛药(analgesics),多用于剧痛,属本章叙述范围;其二是具有镇痛、解热、抗炎作用的药物,对各种钝痛(如头痛、牙痛等)有效,详见第二十章。
典型的镇痛药为阿片生物碱类及其合成代用品(或称阿片类药物——opioids),其特点是镇痛作用强大,如反复应用易于成瘾,故又称为成瘾性镇痛药或麻醉性镇痛药。
第一节 阿片生物碱类镇痛药
阿片(opium)为罂粟科植物罂粟(Papaversomniferum)未成熟蒴果浆汁的干燥物,含有20余种生物碱,从化学结构上可分为菲类和异喹啉类两大类型。前者如吗啡(含量约10%)和可待因,具有镇痛作用;后者如罂粟碱,具有平滑肌松弛作用。
吗啡
【化学结构及构效关系】 吗啡(morphine)的分子结构由四部分组成:①保留四个双键的氢化菲核(环A、B、C);②与菲核环B相稠合的N-甲基哌啶环;③连接环A与环C的氧桥;④环A上的一个酚羟基与环C上的醇羟基。酚羟基氢原子被甲基取代,则镇痛作用减弱(如可待因);叔胺氮被烯丙基取代,则不仅镇痛作用减弱,而且成为吗啡的拮抗药,如烯丙吗啡和纳洛酮(表18-1)。
【体内过程】口服后易自胃肠道吸收,但首关消除明显,生物利用度低,故常用注射给药。皮下注射后30分钟已有60%吸收。约1/3与血浆蛋白结合。未结合型吗啡迅速分布于全身,仅有少量通过血脑屏障,但已足以发挥中枢性药理作用。主要在肝内与葡萄糖醛酸结合而失效,其结合物及小量未结合的吗啡于24小时内大部分自肾排泄。血浆t1/22.5~3小时。吗啡有小量经乳腺排泄,也可通过胎盘进入胎儿体内。
【药理作用】吗啡是镇痛药的代表,主要作用于中枢神经系统及胃肠平滑肌。
1.中枢神经系统
(1)镇痛、镇静吗啡有强大选择性镇痛作用,皮下注射5~10mg即能明显减轻或消除疼痛,但意识及其他感觉不受影响。吗啡对各种疼痛都有效,而对持续性慢性钝痛的效力大于间断性锐痛。吗啡还有明显镇静作用;并能消除由疼痛所引起的焦虑、紧张、恐惧等情绪反应,因而显著提高对疼痛的耐受力。随着疼痛的缓解以及对情绪的影响,可出现欣快症(euphoria)。如外界安静,则可使患者入睡。大剂量(15~20mg)时镇痛镇静作用更明显。一次给药,镇痛作用可持续4~5小时。
对吗啡镇痛作用部位曾有研究,我国药理学者于六十年代初期报道微量吗啡注入家兔第三脑室周围能引起镇痛;以后相继证明吗啡注射于第三脑室尾端至第四脑室头端的神经结构均有镇痛作用,最有效的镇痛部位是导水管周围灰质(图)18-1。
(2)抑制呼吸治疗量吗啡即可抑制呼吸,使呼吸频率减慢、潮气量降低;剂量增大,则抑制增强。急性中毒时呼吸频率可减慢至3~4次/分。吗啡可降低呼吸中枢对血液CO2张力的敏感性,同时,对桥脑内呼吸调整中枢也有抑制作用。
(3)镇咳 吗啡抑制咳嗽中枢,有镇咳作用。
(4)其他吗啡可缩瞳,针尖样瞳孔为其中毒特征。吗啡可引起恶心、呕吐。
2.消化道吗啡可止泻及致便秘。其原因主要是吗啡兴奋胃肠平滑肌,提高其张力。甚至
表18-1吗啡及其衍生物的化学结构
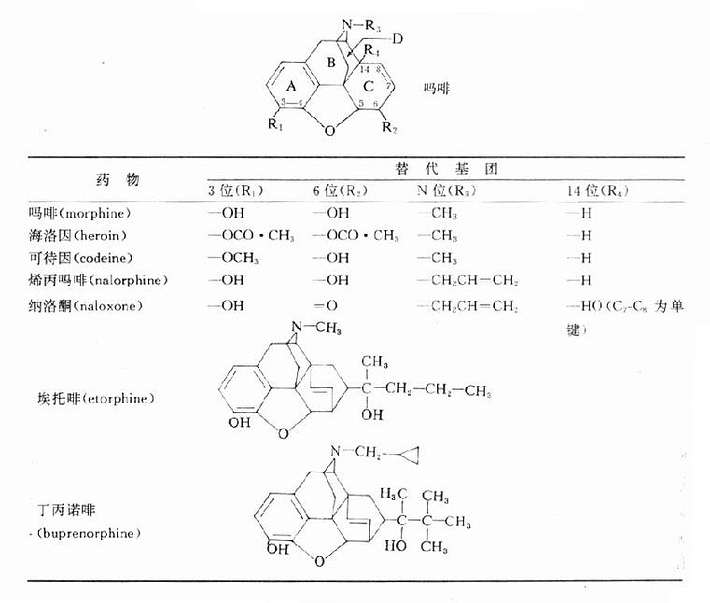
图18-1 吗啡镇痛作用的部位(脑内)箭头表示第三脑室(Ⅲ)尾端、导水管周围灰质及第四脑室(Ⅳ)头端。
C:尾核;T:丘脑;P:壳核;GP:苍白球
图18-2含脑啡肽的神经元与疼痛
疼痛刺激使感觉神经末梢兴奋并释放兴奋性递质(可能为P物质),
该递质与接受神经元上的受体结合,将痛觉冲动传入脑内.感觉神经元末梢上
存在阿片受体,含脑啡肽的神经元释放脑啡肽,后者与阿片受体结合,
减少感觉神经末梢释放P物质,从而防止痛觉冲动传入脑内.E:脑啡肽;SP:P物质
达到痉挛的程度。由于胃窦部及十二指肠上部张力提高,蠕动受抑制,胃排空延迟;小肠及大肠平滑肌张力提高,使推进性蠕动减弱,食糜通过延缓;回盲瓣及肛门括约肌张力提高,肠内容物通过受阻;此外,吗啡抑制消化液的分泌,使食物消化延缓;加上吗啡对中枢的抑制,使患者便意迟钝,因而引起便秘。治疗量吗啡引起胆道奥狄括约肌痉挛性收缩,使胆道排空受阻,胆囊内压力明显提高,可导致上腹不适甚至胆绞痛。阿托品可部分缓解之。
3.心血管系统吗啡扩张阻力血管及容量血管,引起体位性低血压,其降压作用是由于它使中枢交感张力降低,外周小动脉扩张所致。降压作用可部分地被抗组胺药所对抗,因而该作用部分地与吗啡释放组胺有关。吗啡抑制呼吸,使体内CO2蓄积,故致脑血管扩张而颅内压增高。
4.其他治疗量吗啡能提高膀胱括约肌张力,导致尿潴留;还能促进垂体后叶释放抗利尿激素;大剂量吗啡能收缩支气管。
【作用机制】现有资料证明,在体内存在有“抗痛系统”,它由脑啡肽神经元、脑啡肽及阿片受体共同组成。去极化或刺激脑啡肽神经通路可引起脑啡肽释放,并依赖于钙离子。在正常情况下约有20%~30%的阿片受体与脑啡肽结合,起着疼痛感觉的调控作用,维持正常痛阈,发挥生理性止痛机能,镇痛药的作用是激动阿片受体,激活了脑内“抗痛系统”,阻断痛觉传导,产生中枢性镇痛作用。
1.阿片受体及内源性阿片样活性物质七十年代初证实了脑内有阿片受体,而且各种镇痛药与阿片受体的亲和力与它们的镇痛效力之间呈现高度相关性。阿片受体在脑内分布广泛而不均匀,受体密度较高的部位如脊髓胶质区、丘脑内侧、脑室及导水管周围灰质都是和疼痛刺激的传入、痛觉的整合及感受有关的神经结构;而受体密度最高的边缘系统以及蓝斑核,则多是与情绪及精神活动有关的脑区。中脑盖前核的阿片受体可能与缩瞳有关。延脑的孤束核处的阿片受体与药物引起的镇咳、呼吸抑制、中枢交感张力降低有关。脑干极后区、孤束核、迷走神经背核等部位的阿片受体与胃肠活动有关。肠肌本身也有阿片受体。
阿片受体的发现提示脑内可能存在相应的内源性阿片样活性物质,不久即自脑内分离出两种五肽,即甲硫氨酸脑啡肽(M-enkephalin)和亮氨酸脑啡肽(L-enkephalin),它们在脑内的分布与阿片受体的分布近似,并能与阿片受体呈立体特异性结合而产生吗啡样作用,这种作用可被吗啡拮抗药纳洛酮所拮抗。继发现脑啡肽之后,又自垂体中分离出几种较大的肽类,称为内啡肽(endorphins),如β-内啡肽(β-endorphin)及强啡肽(dynorphin A)。迄今已发现近20种作用与阿片生物碱相似的肽类,统称为内阿片样肽(或内阿片肽)。内阿片肽可能是神经递质或神经调质(即调节神经递质释放的物质)或神经激素,在机体内起着痛觉感受的调控或内源性镇痛系统以及调节心血管及胃肠功能的作用,例如在脊髓感觉神经末梢已发现阿片受体,实验资料提示脑啡肽可能通过抑制感觉神经末梢释放一种兴奋性递质(P物质),从而干扰痛觉冲动传入中枢(图18-2)。至于吗啡类药物的作用机制则可能是通过与不同脑区的阿片受体结合,模拟内阿片肽而发挥作用的。
2.阿片受体的多型性、亚型及其效应通过对阿片受体配体结合的实验研究,现认为阿片受体可分为μ、δ、κ、及σ型,μ、δ及σ型各又可分为1和2两种亚型。对各型激动时的效应不同;各种内阿片肽对不同型阿片受体的亲和力不同,现认为亮氨酸脑啡肽及强啡肽分别为δ及κ受体的内源性配体,而μ受体及σ受体的内源性配体则尚未明确。吗啡类药物对不同型的亲和力及内在活性都不完全相同,因此有些药物是激动药(如吗啡、哌替啶);有些是拮抗药(如纳洛酮);还有些药物对一型是拮抗药,而对另一型则是激动药或部分激动药(如喷他佐辛、烯丙吗啡、丁丙诺啡)(表18-2)。近来有研究指出阿片受体之间可能存在变构关系,如与吗啡结合的受体(μ)和与脑啡肽结合的受体(δ)是一个蛋白质分子或一处复合体上的不同结合点;β-内啡肽有两个结合点,即氨基端的脑啡肽结合点和羧基端的吗啡结合点;对其中一个结合点的作用可以变构影响另一结合点的结合性能。
表18-2激动各型阿片受体时的效应及药物作用比较
| 受体型 | ||||
| 激动时效应 | μ | δ | κ | σ |
| 镇痛 | 脊髓以上水平 | 脊髓水平 | 脊髓水平 | - |
| 呼吸抑制 | ++ | ++ | + | - |
| 瞳孔 | 缩小 | 缩小 | - | 散大 |
| 胃肠活动 | 减少 | 减少 | - | - |
| 平滑肌痉挛 | ++ | ++ | - | - |
| 行为、精神活动 | 欣快++ | 欣快++ | 烦躁不安++ | 烦躁不安++ |
| 镇静++ | 镇静++ | 镇静++ | 致幻 | |
| 身体依赖性 | ++ | ++ | + | - |
| 化合物 | ||||
| 阿片肽类 | ||||
| β-内啡肽 | +++ | +++ | +++ | - |
| 亮氨酸脑啡肽 | + | +++ | - | - |
| 甲硫氨酸脑啡肽 | ++ | +++ | - | - |
| 强啡肽 | ++ | + | +++ | - |
| 阿片类药物 | ||||
| 完全激动药 | ||||
| 吗啡 | +++ | + | ++ | - |
| 可待因 | + | + | + | - |
| 哌替啶 | ++ | + | + | - |
| 埃托啡 | +++ | +++ | +++ | - |
| 芬太尼 | +++ | + | - | - |
| 部分/混合激动药 | ||||
| 喷他佐辛 | +* | + | ++ | + |
| 烯丙吗啡 | ++ | (++) | (++) | + |
| 丁丙诺啡 | (+++) | - | - | - |
| 拮抗药 | ||||
| 纳洛酮 | +++ | ++ | ++ | + |
纳曲酮
■[此处缺少一些内容]■
O2的敏感性,使急促浅表的呼吸得以缓解。但对于休克、昏迷及严重肺功能不全者禁用。
3.止泻适用于急、慢性消耗性腹泻以减轻症状。可选用阿片酊或复方樟脑酊;如为细菌感染,应同时服用抗菌药。
【不良反应】
1.治疗量吗啡有时可引起眩晕、恶心、呕吐、便秘、排尿困难、胆绞痛、呼吸抑制、嗜睡等副作用。
2.连续反复多次应用吗啡易产生耐受性及成瘾,一旦停药,即出现戒断症状,表现为兴奋、失眠、流泪、流涕、出汗、震颤、呕吐、腹泻,甚至虚脱、意识丧失等。若给以治疗量吗啡,则症状立即消失。成瘾者为追求吗啡的欣快症及避免停药所致戒断症状的痛苦,常不择手段获取吗啡(称为“强迫性觅药行为”),危害极大。故对吗啡等成瘾性药物应严格控制使用,并按国家颁布的《麻醉药品管理条例》严格管理。吗啡耐受性与成瘾性的产生主要由于神经组织对吗啡的适应性;与吗啡成瘾及戒断症状有直接联系的是蓝斑核,该核由去甲肾上腺素能神经元组成,且有阿片受体密集;吗啡或脑啡肽均可抑制蓝斑核放电,当动物对吗啡耐受或成瘾后,该核放电也出现耐受,一旦停用吗啡,则放电加速,同时出现戒断症状,提示戒断症状与蓝斑核去甲肾上腺素能神经元活动增强有关。据报道,能抑制蓝斑核放电的可乐定可缓解吗啡戒断症状。
【急性中毒】表现为昏迷、瞳孔极度缩小(严重缺氧时则瞳孔散大)、呼吸高度抑制、血压降低甚至休克。呼吸麻痹是致死的主要原因。需用人工呼吸、给氧抢救;吗啡拮抗药纳洛酮对吗啡之呼吸抑制有显著效果,如用药无效,则吗啡中毒的诊断可疑。
【禁忌证】吗啡能通过胎盘或乳汁抑制胎儿或新生儿呼吸,同时能对抗催产素对子宫的兴奋作用而延长产程(原因未明),故禁用于分娩止痛及哺乳妇女止痛。由于抑制呼吸及抑制咳嗽反射以及释放组胺而致支气管收缩,故禁用于支气管哮喘及肺心病患者。颅脑损伤所致颅内压增高的患者、肝功能严重减退患者禁用。
可待因
可待因(codeine)又称甲基吗啡,在阿片中含量约0.5%。口服后易吸收。大部分在肝内代谢,有10%可待因脱甲基后转变为吗啡而发挥作用。
可待因的镇痛作用仅为吗啡的1/12。镇咳作用为其1/4。持续时间则与吗啡相似。镇静作用不明显。欣快症及成瘾性也弱于吗啡。在镇咳剂量时,对呼吸中枢抑制轻微,又无明显便秘、尿潴留及体位性低血压的副作用。
临床上,可待因用于中等程度疼痛止痛,与解热镇痛药合用有协同作用。可待因也是典型的中枢性镇咳药(见第三十章)。{2}第二节 人工合成镇痛药
吗啡易成瘾,是其严重缺点。为了寻找更好的代用品,合成了哌替啶、安那度、芬太尼、美沙酮、喷他佐辛、二氢埃托啡等药,它们的成瘾性均较吗啡轻。较常用的几个药物的化学结构见图18-3
■[此处缺少一些内容]■
~4小时。镇痛效力弱于吗啡,注射80~100mg哌替啶约相当于10mg吗啡的镇痛效力。约10%~20%患者用药后出现欣快。哌替啶与吗啡在等效镇痛剂量时,抑制呼吸的程度相等。对延脑CTZ有兴奋作用,并能增加前庭器官的敏感性,易致眩晕、恶心、呕吐。
2.平滑肌能中度提高胃肠道平滑肌及括约肌张力,减少推进性蠕动,但因作用时间短,故不引起便秘,也无止泻作用。能引起胆道括约肌痉挛,提高胆道内压力,但比吗啡弱。治疗量对支气管平滑肌无影响,大剂量则引起收缩。对妊娠末期子宫,不对抗催产素兴奋子宫的作用,故不延缓产程。
3.心血管系统治疗量可致体位性低血压,原因同吗啡。由于抑制呼吸,也能使体内CO2蓄积,脑血管扩张而致脑脊液压力升高。
【临床应用】
1.镇痛哌替啶对各种剧痛如创伤性疼痛、手术后疼痛、内脏绞痛、晚期癌痛都有止痛效果。但对慢性钝痛则不宜使用,因仍有成瘾性。新生儿对哌替啶抑制呼吸作用极为敏感,故产妇于临产前2~4小时内不宜使用。
2.麻醉前给药及人工冬眠哌替啶的镇静作用可消除患者手术前紧张、恐惧情绪,减少麻醉药用量;与氯丙嗪、异丙嗪合用组成冬眠合剂用于人工冬眠疗法。
【不良反应】治疗量哌替啶与吗啡相似,可致眩晕、出汗、口干?
■[此处缺少一些内容]■
美沙酮
美沙酮(methadone)有左旋体及右旋体。左旋体较右旋体效力强8~50倍。常用其消旋体。药理作用性质与吗啡相似,但它口服与注射同样有效(吗啡口服利用率低)。其镇痛作用强度与持续时间与吗啡相当。耐受性与成瘾性发生较慢,戒断症状略轻,且易于治疗。一次给药后,镇静作用较弱,但多次用药有显着镇静作用。抑制呼吸、缩瞳、引起便秘及升高胆道内压力都较吗啡轻。适用于创伤、手术及晚期癌症等所致剧痛。
喷他佐辛
喷他佐辛(pentazocine,镇痛新)为苯并吗啡烷类衍生物,其哌啶环中N上甲基为异戊烯基取代而成的合成镇痛药。主要激动κ、σ受体;但又可拮抗μ受体(见表18-2)。
【药理作用和临床应用】按等效剂量计算,本药的镇痛效力为吗啡的1/3,一般皮下或肌内注射30mg的镇痛效果与吗啡10mg相当。其呼吸抑制作用约为吗啡的1/2;增加剂量至30mg以上,呼吸抑制作用并不按比例增强;用量达60~90mg,则可产生精神症状,大剂量纳洛酮可对抗之。本药可减慢胃排空并延缓肠管运送肠内容物的时间,但对胆道括约肌的兴奋作用较弱,胆道内压力上升不明显。对心血管系统的作用不同于吗啡,大剂量反而增快心率,升高血压。对冠心病患者,静脉注射能提高平均主动脉压、左室舒张末期压,因而增加心脏作功量。本药能提高血浆中去甲肾上腺素水平,这与它兴奋心血管系统的作用有关。由于本药尚有一定的拮抗μ受体的作用,因而成瘾性很小,在药政管理上已列入非麻醉品。本药能减弱吗啡的镇痛作用;对吗啡已产生耐受性的患者,可促进戒断症状的产生。它拮抗吗啡类抑制呼吸的作用不明显。适用于各种慢性剧痛,口服及注射后吸收均良好,肌内注射后0.25~1小时达血药浓度峰值。口服后,在肝中的首过消除显著,进入全身循环的镇痛新不到20%,故口服后需1~3小时才达血药浓度峰值。本药主要在肝内代谢,代谢速率个体差异较大,这可能是它镇痛效果个体差异大的原因。肌内注射后t1/2约2小时;口服后作用持续5小时以上。
【不良反应】常见镇静、眩晕、恶心、出汗。剂量增大能引起呼吸抑制、血压升高、心率增快;有时可引起焦虑、恶梦、幻觉等。纳洛酮能对抗其呼吸抑制的毒性。
二氢埃托啡
二氢埃托啡(dihydroetorphine)为我国生产的强镇痛药。为吗啡受体激动药。其镇痛作用是吗啡的12000倍。用量小,一次20~40μg 。镇痛作用短暂,仅2小时左右。小剂量间断用药不易产生耐受性而大剂量持续用药则易出现耐受性。它也可成瘾,但较吗啡轻。常用于镇痛或吗啡类毒品成瘾者的戒毒。
第三节 其他镇痛药
曲马朵
曲马朵(tramadol)为阿片受体激动药,其镇痛作用强度与喷他佐辛相似。口服易于吸收,生物利用及度约90%,t1/2约6小时。不良反应和其他镇痛药相似,偶有多汗、头晕、恶心、呕吐、口干、疲劳等。治疗剂量时不抑制呼吸,也不影响心血管功能,不产生便秘等副作用。适用于中度及重度急慢性疼痛及外科手术。不宜作于轻度疼痛,长期应用也可能发生成瘾。
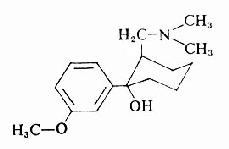
强痛定
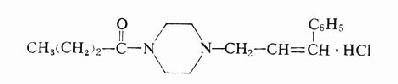
强痛定(fortanodyn,AP-273)的镇痛作用约为吗啡的1/3。临床上多用于偏头痛、三叉神经痛、炎症性及外伤性疼痛、关节痛、痛经及癌疼痛。偶有恶心、头晕、困倦等神经系统反应,停药后即消失。个别例曾出现成瘾性,宜慎用。
延胡索乙素及罗通定
延胡索(Corydalis ambigua)为罂粟科草本博物,药用其块茎。又名玄胡、元胡。能活血散瘀、行气止痛。《本草纲目》中曾记载“治一身上下诸痛,用之中的,妙不可言”。经研究发现所含延胡索乙素有镇痛作用。它是消旋四氢巴马汀(dl-tetrahydropalmatine),有效部分为左旋体,即罗通定(rotundine)。
口服延胡索乙素及罗通定吸收良好,镇痛作用较解热镇痛药强。研究证明其镇痛作用与脑内阿片受体无关。口服延胡索乙素100~200mg,10~30分钟出现镇痛作用,维持2~5小时。对慢性持续性钝痛效果较好,对创伤或手术后疼痛或晚期癌症的止痛效果较差。可用于治疗胃肠及肝胆系统等内科疾病所引起的钝痛、一般性头痛以及脑震荡后头痛等。也可用于痛经及分娩止痛,对产程及胎儿均无不良影响。
第四节 阿片受体拮抗剂——纳洛酮与纳曲酮
纳洛酮(naloxone)化学结构与吗啡极相似,主要区别为叔氮上以烯丙基取代甲基,6位羟基变为酮基(见表18-1)。纳洛酮对4型阿片受体都有拮抗作用(见表18-2)。它本身并无明显药理效应及毒性,给人注射12mg后,不产生任何症状;注射24mg只产生轻微困倦。但对吗啡中毒者,小剂量(0.4~0.8mg)肌内或静脉注射能迅速翻转吗啡的作用,1~2分钟就可消除呼吸抑制现象,增加呼吸频率。对吗啡成瘾者可迅速诱发戒断症状,表明纳洛酮在体内与吗啡竞争同一受体。临床适用于吗啡类镇痛药急性中毒,解救呼吸抑制及其他中枢抑制症状,可使昏迷者迅速复苏。在镇痛药的理论研究中,纳洛酮是重要的工具药。
纳曲酮(naltrexone)的作用与纳洛酮相同,但口服生物利用度较高,作用维持时间较长。
[附]癌痛的镇痛治疗
世界卫生组织提出到2000年达到全世界范围内“使癌症病人不痛”的目标。我国卫生部于1991年4月下达了关于我国开展“癌症病人三级止痛阶梯治疗”工作的指标。
癌痛治疗三阶梯方法就是在对癌痛的性质和原因作出正确的评估后,根据癌症病人的疼痛程度和原因适当选择相应的镇痛药,即对轻度疼痛的患者应主要选用解热镇痛抗炎类药(如阿司匹林、对乙酰氨基酚、布洛芬、吲哚美辛栓剂等);若为中度疼痛者应选用弱阿片类药(如可待因、氨酚待因、强痛定、曲马朵等);若为重度疼痛者应选用强阿片类药(如吗啡、哌替啶、美沙酮、二氢埃托啡等)。在用药过程中要尽量选择口服给药途径;有规律地按时给药而不是按需(只在痛时)给药;药物剂量应个体化;需要时可加用辅助药物,如解痉药(止针刺样痛、浅表性灼痛)、精神治疗药(抗抑郁药或抗焦虑药)等。
镇痛药 制剂及用法
盐酸吗啡(morphine hydrochloride)10mg/次,皮上注射。极量:口服30mg/次,100mg/日。皮下注射20mg/次,60mg/日。
磷酸可待因(codeine phosphate)15~30mg/次,3次/日。极量:0.1mg/次,0.25mg/日,口服。
阿片酊(tincture opium)含吗啡约1%,乙醇3%。0.3~1ml/次,3次/日,口服。极量:2ml/次,6ml/日。
复方樟脑酊(tincture comphor co)每100ml含阿片酊5ml。常用量2~5ml/次(相当于吗啡1~2.5mg),3次/日。用于腹泻、腹痛及镇咳。
盐酸哌替啶(pethidine hydrochloride)100mg/次,肌内注射。极量:150mg/次,600mg/日。
安那度(anadol)10~20mg/次,皮下注射或肌内注射。极量:30mg/次,60mg/日。
盐酸美沙酮(methadone hydrochloride)5~10mg/次,2~3次/日,口服。5~10mg/次,肌内注射。
枸橼酸芬太尼(fentanyl citrate)0.05~0.1mg/次,皮下或肌内注射。
盐酸喷他佐辛(pentazocine hydrochloride)50mg/次,口服。
乳酸喷他佐辛(pentazocine lactate)30mg/次,皮下注射或肌内注射。
盐酸二氢埃托啡(dihydroetorphine hydrochloride)20-40μg/次,180μg/日,舌下含用,10~20μg/次,90μg/日,肌内注射。
盐酸曲马多(tramadol hydrochloride)50mg/次,3次/日,口服。50mg/次。50~200mg/日,缓慢静滴。
强痛定(fortanodyn)60mg/次,3~4次/日,口服。50mg/次,皮下注射。
纳洛酮(naloxone)0.4~0.8mg/次,肌内注射或静脉注射。
硫酸延胡索乙素(tetrahydropalmatine sulfate)100~150mg/次,3次/日,口服。60~100mg/次,皮下注射。
盐酸罗通定(rotundine hydrochloride)60~100mg/次。3次/日,口服。
硫酸罗通定(rotundine sulfate)60mg/次,皮下注射。
第十九章 中枢兴奋药
中枢兴奋药(central stimulants)是能提高中枢神经系统机能活动的一类药物。根据其主要用部位可分为三类:①主要兴奋大脑皮层的药物,如咖啡因等;②主要兴奋延脑呼吸中枢的药物,又称呼吸兴奋药,如尼可刹米等;③主要兴奋脊髓的药物,如土的宁等。这种分类是相对的。随着剂量的增加,其中枢作用部位也随之扩大,过量均可引起中枢各部位广泛兴奋而导致惊厥。脊髓兴奋药因毒性较大,无临床应用价值,故本章不作介绍。
第一节 主要兴奋大脑皮层的药物
咖啡因
咖啡因(caffeine,咖啡碱)为咖啡豆和茶叶的主要生物碱。此外,茶叶还含茶碱(theophyline),均属黄嘌呤类,药理作用相似,但咖啡因的中枢兴奋作用较强,临床主要用作中枢兴奋药;茶碱的舒张平滑肌作用较强,主要用作平喘药。
【药理作用及临床应用】咖啡因对大脑皮层有兴奋作用,人服用小剂量(50~200mg)即可使睡意消失,疲劳减轻,精神振奋,思维敏捷,工作效率提高,因此咖啡和茶叶早就成为世界性的兴奋性饮料成分。在动物实验,咖啡因可引起觉醒型脑电波,损伤其间脑与中脑后,此作用仍存在,这提示作用部位在大脑皮层。较大剂量时则要直接兴奋延脑呼吸中枢和血管运动中枢,使呼吸加深加快,血压升高;在呼吸中枢受抑制时,尤为明显。中毒剂量时则尚兴奋脊髓,动物发生阵挛性惊厥。咖啡因可直接兴奋心脏、扩张血管(冠状血管、肾血管等),但此外周作用常被兴奋迷走中枢及血管运动中枢的作用所掩盖,故无治疗意义。此外,咖啡因还可舒张支气管平滑肌、利尿及刺激胃酸分泌。近报道称治疗量咖啡因和茶碱能在体内竞争性拮抗腺苷受体,又知腺苷有镇静、抗惊厥及收缩支气管平滑肌等作用。这提示咖啡因的中枢兴奋及舒张支气管平滑肌的作用与其阻断腺苷受体之效有关。
咖啡因主要用于对抗中枢抑制状态,如严重传染病、镇静催眠药过量引起的昏睡及呼吸循环抑制等,可肌内注射苯甲酸钠咖啡因。此外,咖啡因还常配伍麦角胺治疗偏头痛;配伍解热镇痛药治疗一般性头痛。此时,它由于收缩脑血管,减少脑血管搏动的幅度而加强以上药物止头痛的作用。
【不良反应】一般少见,但剂量较大时可致激动、不安、失眠、心悸、头痛;剂量过大也可引起惊厥。乳婴高热时易致惊厥,应选用无咖啡因的复方解热药。
哌醋甲酯
哌醋甲酯(methylphenidate)又名利他林(ritalin),化学结构与具有中枢兴奋作用的感胺——苯丙胺相似。作用性质也相类似,但交感作用很弱,中枢兴奋作用较温和,能改善精神活动,解除轻度抑制及疲乏感。大剂量也能引起惊厥。临床用于轻度抑郁及小儿遗尿症,因它可兴奋大脑皮层使之易被尿意唤醒。此外,它对儿童多动综合征有效,该病是由于脑干网状结构上行激活系统内去甲肾上腺素、多巴胺、5-羟色胺等递质中某一种缺乏所致,它能促进这类递质的释放。本药在治疗量时不良反应较少,偶有失眠、心悸、焦虑、厌食、口干。大剂量时可使血压升高而致眩晕、头痛等。癫痫、高血压患者禁用。久用可产生耐受性,并可抑制儿童生长发育。
匹莫林(pemoline)其作用及用途与哌醋甲酯相似,但作用维持时间长,只需一日用药一次。常见副作用为失眠,心血管副作用极少。
甲氯芬酯
甲氯芬酯(meclofenoxate,氯酯醒)能促进脑细胞代谢,增加糖类的利用。对中枢抑制状态的患者有兴奋作用。临床用于颅脑外伤后昏迷、脑动脉硬化及中毒所致意识障碍、儿童精神迟钝、小儿遗尿等。作用出现缓慢,需反复用药。尚未发现不良发应。
吡拉西坦
吡拉西坦(piracetam,吡乙酰胺,脑复康)能促进大脑皮层细胞代谢,增进线粒体内ATP的合成,提高脑组织对葡萄糖的利用率,保护脑缺氧所致的脑损伤,促进正处于发育的儿童大脑及智力的发展。用于脑外伤后遗症,慢性酒精中毒,老年人脑机能不全综合征,脑血管意外及儿童的行为障碍。
第二节 主要兴奋延脑呼吸中枢的药物
尼可刹米
尼可刹米(nikethamide)又名可拉明(coramin),主要直接兴奋延脑呼吸中枢,也可刺激颈动脉体化学感受器而反射性兴奋呼吸中枢,能提高呼吸中枢对CO2的敏感性,使呼吸加深加快。安全性大,但一次静脉注射作用仅维持数分钟。过量可致血压上升、心动过速、肌震颤及僵直、咳嗽、呕吐、出汗。因作用温和,安全范围大,临床常用于各种原因所致中枢性呼吸抑制。一般间歇静脉注射给药效果较好。
二甲弗林
二甲弗林(dimefline,回苏灵)直接兴奋呼吸中枢,作用强于尼可刹米、贝美格,使肺换气量及动脉PO2提高,PCO2降低。临床用于中枢性呼吸抑制。过量可致惊厥。静脉给药需稀释后缓慢注射,并严密观察患者反应。
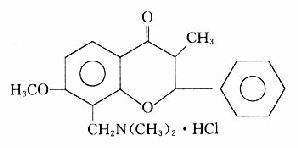
山梗菜碱
山梗菜碱(lobeline,洛贝林)是从山梗菜提取的生物碱。它不直接兴奋延脑,而是通过刺激颈动脉体和主动脉体的化学感受器,反射性地兴奋延脑呼吸中枢。其作用短暂,仅数分钟,但安全范围大,不易致惊厥。临床常用于治疗新生儿窒息、小儿感染性疾病引起的呼吸衰竭以及一氧化碳中毒。剂量较大可兴奋迷走中枢而致心动过缓、传导阻滞。过量时可因兴奋交感神经节及肾上腺髓质而致心动过速。
贝美格
贝美格(bemegride)又名美解眠(megimide),中枢兴奋作用迅速,维持时间短,用量过大或注射太快也可引起惊厥。可用作巴比妥类中毒解救的辅助用药。
以上中枢兴奋药主要用于对抗中枢抑制药中毒或某些传染病引起的中枢性呼吸衰竭。它们的选择性一般都不高,安全范围小,兴奋呼吸中枢的剂量与致惊厥剂量之间的距离小。对深度中枢抑制的患者,大多数中枢兴奋药在不产生惊厥的剂量时往往无效;而且它们的作用时间都很短,需要反复用药才能长时间维持患者呼吸,因而很难避免惊厥的发生。所以除严格掌握剂量外,这类药物的应用宜限于短时就能纠正的呼吸衰竭患者。临床主要采用人工呼吸机维持呼吸,因为它远比呼吸兴奋药有效而且安全可靠。
中枢兴奋药 制剂及用法
苯甲酸钠咖啡因(caffeine andsodium benzoate)0.25~0.5g/次,皮下或肌内注射。极量:0.8g/次,3g/日。
哌醋甲酯(methylphenidate)10mg/次,2~3次/日,口服。10~20mg/次,1~3次/日,肌内或静脉注射。
甲氯芬酯(meclofenoxate)100~200mg/次,3次/日,至少服一周。成人昏迷状态,250mg/次,每2小时肌内注射一次。
吡拉西坦(piracetam)0.4~0.8g/日,2~3次分服。
尼可刹米(nikethamide)皮下、肌内或静脉注射0.25~0.5g/次。必要时,每1~2小时重复一次,或与其他中枢兴奋药交替使用,直到可以“唤醒”患者而无肌震颤或抽搐。极量:皮下、肌内或静脉注射,1.25g/次。
二甲弗林(dimefline)肌内注射8mg/次;静脉注射8~16mg/次,以葡萄糖溶液稀释后缓慢注射;重症患者16~32mg,用生理盐水稀释后,静脉滴注。
盐酸山梗菜碱(lobeinehydrochloride)3~10mg/次,皮下或肌内注射。极量:20mg/次。
贝美格(bemegride)静脉滴注,用5%葡萄糖液稀释后,每3~5分钟静脉滴注50mg,至病情改善或出现毒性症状为止。
第二十章 解热镇痛抗炎药
解热镇痛抗炎药(antipyretic-analgesicand antiinflammatory drugs)是一类具有解热、镇痛,而且大多数还有抗炎、抗风湿作用的药物。它们在化学结构上虽属不同类别,但都可抑制体内前列腺素(prostaglandin,PG)的生物合成,目前认为这是它们共同作用的基础(图20-1)。由于其特殊的抗炎作用,故本类药物又称为非甾体抗炎药(non-steroidalanti-inflammatory drugs,NSAID)。乙酰水杨酸是这类药物的代表,因此有人将这类药物称为乙酰水杨酸类药物。它们有以下三项共同作用。
图20-1 自膜磷脂生成的各种物质及其作用以及抗炎药的作用部位示意图
PLA2-磷脂酶A2;NSAIDS-非甾体抗炎药;PAF-血小板活化因子;
5-HPETE-5-氢过氧化二十碳四烯酸;12HETE-12-羟二十碳四
烯酸;Lipoxin-脂氧素;PGI2-前列环素;PG-前列腺素;TXA2-
血栓素A2;LT-白三烯
1.解热作用 解热镇痛抗炎药能降低发热者的体温,而对体温正常者几无影响。这和氯丙嗪对体温的影响不同,在物理降温配合下,氯丙嗪能使正常人体温降低。
下丘脑体温调节中枢通过对产热及散热两个过程的精细调节,使体温维持于相对恒定水平(正常人为37℃左右)。传染病之所以发热,是由于病原体及其毒素刺激中性粒细胞,产生与释放内热原,可能为白介素-1(IL-1),后者进入中枢神经系统,作用于体温调节中枢,将调定点提高至37℃以上,这时产热增加,散热减少,因此体温升高。其他能引起内热原释放的各种因素也都可引起发热。内热原并非直接作用于体温调节中枢,因为实验证明,全身组织的多种PG都有致热作用,微量PG注入动物脑室内,可引起发热,其中PGE2致热作用最强;其他致热物质引起发热时,脑脊液中PG样物质含量增高数倍。这说明内热原可能使中枢合成与释放PG增多,PG再作用于体温调节中枢而引起发热。解热镇痛药对内热原引起的发热有解热作用,但对直接注射PG引起的发热则无效。因此认为它们是通过抑制中枢PG合成而发挥解热作用的。治疗浓度的解热镇痛药可抑制PG合成酶(环加氧酶),减少PG的合成,而且它们对该酶活性抑制程度的大小与它们的药理作用强弱相一致。这类药物只能使发热者体温下降,而对正常体温没有影响,也支持这一观点。
发热是机体的一种防御反应,而且热型也是诊断疾病的重要依据。故对一般发热患者可不必急于使用解热药;但热度过高和持久发热消耗体力,引起头痛、失眠、谵妄、昏迷、小儿高热易发生惊厥,严重者可危及生命,这时应用解热药可降低体温,缓解高热引起的并发症。但解热药只是对症治疗,因此仍应着重病因治疗。
2.镇痛作用解热镇痛药仅有中等程度镇痛作用,对各种严重创伤性剧痛及内脏平滑肌绞痛无效;对临床常见的慢性钝痛如头痛、牙痛、神经痛、肌肉或关节痛、痛经等则有良好镇痛效果;不产生欣快感与成瘾性,故临床广泛应用。
本类药物镇痛作用部位主要在外周。在组织损伤或发炎时,局部产生与释放某些致痛化学物质(也是致炎物质)如缓激肽等,同时产生与释放PG。缓激肽作用于痛觉感受器引起疼痛;PG则可使痛觉感受器对缓激肽等致痛物质的敏感性提高。因此,在炎症过程中,PG的释放对炎性疼痛起到了放大作用,而PG(E1、E2及F2a)本身也有致痛作用。解热镇痛药可防止炎症时PG的合成,因而有镇痛作用。这说明为何这类药物对尖锐的一过性刺痛(由直接刺激感觉神经末梢引起)无效,而对持续性钝痛(多为炎性疼痛)有效。但它们部分地通过中枢神经系统而发挥镇痛作用的可能性也不能排除。
3.抗炎作用大多数解热镇痛药都有抗炎作用,对控制风湿性及类风湿性关节炎的症状有肯定疗效,但不能根治,也不能防止疾病发展及合并症的发生。PG还是参与炎症反应的活性物质,将极微量(ng水平)PGE2皮内或静脉或动脉内注射,均能引起炎症反应;而发炎组织(如类风湿性关节炎)中也有大量PG存在;PG与缓激肽等致炎物质有协同作用。解热镇痛药抑制炎症反应时PG的合成,从而缓解炎症。
常用的解热镇痛抗炎药按化学结构可分为水杨酸类、苯胺类、吡唑酮类及其他有机酸等四类。各类药物均具有镇痛作用,但在抗炎作用方面则各具特点,如乙酰水杨酸和吲哚美辛的抗炎作用较强,某些有机酸的抗炎作用中等,而苯胺类几无抗炎作用。
第一节 水杨酸类
水杨酸类(salicylates)药物包括乙酰水杨酸(acetylsalicylic acid)和水杨酸钠(sodiumsalicylate)。水杨酸本身因刺激性大,仅作外用,有抗真菌及溶解角质的作用。本类药物中最常用的是乙酰水杨酸。
乙酰水杨酸
乙酰水杨酸又称阿司匹林(aspirin)。
【体内过程】口服后,小部分在胃、大部分在小肠吸收。0.5~2小时血药浓度达峰值。在吸收过程中与吸收后,迅速被胃粘膜、血浆、红细胞及肝中的酯酶水解为水杨酸。因此,乙酰水杨酸血浆浓度低,血浆t1/2短(约15分钟)。水解后以水杨酸盐的形式迅速分布至全身组织。也可进入关节腔及脑脊液,并可通过胎盘。水杨酸与血浆蛋白结合率高,可达80%~90%。水杨酸经肝药本酶代谢,大部分代谢物与甘氨酸结合,少部分与葡萄糖醛酸结合后,自肾排泄。
肝对水杨酸的代谢能力有限。口服小剂量乙酰水杨酸(1g以下)时,水解生成的水杨酸量较少,其代谢按一级动力学进行,水杨酸血浆t1/2约2~3小时;但当乙酰水杨酸剂量≥1g时,水杨酸生成量增多,甘氨酸、葡萄糖醛酸的结合反应已达到饱和,水杨酸的代谢即从一级动力学转变为零级动力学进行,水杨酸血浆t1/2延长为15~30小时,如剂量再增大,血中游离水杨酸浓度将急剧上升,可突然出现中毒症状。
长期大量用药治疗风湿性及类风湿性关节炎时,为保证用药的有效性与安全性,剂量应渐增,并应根据患者用药后的反应及血药浓度监测,据此以确定给药剂量及间隔时间,并在治疗过程中经常调整剂量。
服用剂量较小时,尿中排泄的主要是与甘氨酸或葡萄糖醛酸结合物,也有小部分以水杨酸盐排出。但当剂量大时,结合反应已饱和,就有大量水杨酸盐排出,此时,尿液pH的变化对水杨酸盐排泄量的影响很大,在碱性尿时可排出85%;而在酸性尿时则仅5%。这是由于碱性尿中,水杨酸盐解离增多,再吸收减少而排出增多;尿呈酸性时则相反。故同时服用碳酸氢钠可促进其排泄,降低其血浓度。
【药理作用及临床应用】
1.解热镇痛及抗风湿有较强的解热、镇痛作用,常与其他解热镇痛药配成复方,用于头痛、牙痛、肌肉痛、神经痛、痛经及感冒发热等;抗炎抗风湿作用也较强,可使急性风湿热患者于24~48小时内退热,关节红、肿及剧痛缓解,血沉下降,患者主观感觉好转。由于控制急性风湿热的疗效迅速而确实,故也可用于鉴别诊断。对类风湿性关节炎也可迅速镇痛,消退关节炎症,减轻关节损伤,目前仍是首选药。用于抗风湿最好用至最大耐受剂量,一般成人每日3~5g,分4次于饭后服。
2.影响血栓形成 血栓素(TXA2)是强大的血小板释放ADP及聚集的诱导剂,乙酰水杨酸能使PG合成酶(环加氧酶)活性中心的丝氨酸乙酰化而失活,因而减少血小板中TXA2的生成而抗血小板聚集及抗血栓形成。但在高浓度时,乙酰水杨酸也能抑制血管壁中PG合成酶,减少了前列环素(prostacyclin,PGI2)合成。PGI2是TXA2的生理对抗剂,它的合成减少可能促进血栓形成。实验证明,血小板中PG合成酶对乙酰水杨酸的敏感性远较血管中PG合成酶为高,因而建议采用小剂量(每日口服75mg)用于防止血栓形成。治疗缺血性心脏病、包括稳定型、不稳定型心绞痛及进展性心肌梗塞患者能降低病死率及再梗塞率。此外,应用于血管形成术及旁路移植术也有效。对一过性脑缺血发作者,服用小剂量乙酰水杨酸(30~50mg),可防止脑血栓形成。
【不良反应】短期服用副作用少;长期大量抗风湿则有不良反应。
1.胃肠道反应最为常见。口取可直接刺激胃粘膜,引起上腹不适、恶心、呕吐。血浓度高则刺激延脑催吐化学感应区(CTZ),也可致恶心及呕吐。较大剂量口服(抗风湿治疗)可引起胃溃疡及不易察觉的胃出血(无痛性出血);原有溃疡病者,症状加重。饭后服药,将药片嚼碎,同服抗酸药如碳酸钙,或服用肠溶片可减轻或避免以上反应。内源性PG对胃粘膜有保护作用,如将PGE2与乙酰水杨酸同服,可减少后者引起的胃出血,其疗效与PGE2的剂量成比例,提示乙酰水杨酸致溃疡可能与它抑制胃粘膜合成PG有关。胃溃疡患者禁用。
2.凝血障碍一般剂量乙酰水杨酸就可抑制血小板聚集,延长出血时间。大剂量(5g/日以上)或长期服用,还能抑制凝血酶元形成,延长凝血酶元时间,维生素K可以预防。严重肝损害、低凝血酶元血症、维生素K缺乏等均应避免服用乙酰水杨酸。手术前一周应停用。
3.过敏反应少数患者可出现荨麻疹、血管神经性水肿、过敏性休克。某些哮喘患者服乙酰水杨酸或其他解热镇痛药后可诱发哮喘,称为“阿司匹林哮喘”,它不是以抗原-抗体反应为基础的过敏反应,而与它们抑制PG生物合成有关。因PG合成受阻,而由花生四烯酸生成的白三烯以及其他脂氧酶代谢产物增多,内源性支气管收缩物质居于优势,导致支气管痉挛,诱发哮喘。肾上腺素治疗“阿司匹林哮喘”无效。哮喘、鼻息肉及慢性荨麻疹患者禁用乙酰水杨酸。
4.水杨酸反应 乙酰水杨酸剂量过大(5g/日)时,可出现头痛、眩晕、恶心、呕吐、耳鸣、视、听力减退,总称为水杨酸反应,是水杨酸类中毒的表现。严重者可出现过度呼吸、酸碱平衡失调,甚至精神错乱。严重中毒者应立即停药,静脉滴入碳酸氢钠溶液以碱化尿液,加速水杨酸盐自尿排泄。
5.瑞夷(Reye)综合征据报道患病毒性感染伴有发热的儿童或青年服用乙酰水杨酸后有发生瑞夷综合征的危险,表现为严重肝功能不良合并脑病,虽少见,但可致死,宜慎用。
【药物相互作用】本药与双香豆素合用时,因从血浆蛋白结合部位置换后者,提高游离型双香豆素血浓度,增强其抗凝作用,易致出血。本药也可置换甲磺丁脲,增强其降血糖作用,易致低血糖反应。与肾上腺皮质激素合用,也因蛋白置换而使激素抗炎作用增强,但诱发溃疡的作用也增强。本药妨碍甲氨蝶呤从肾小管分泌而增强其毒性。与呋塞米合用,因竞争肾小管分泌系统而使水杨酸排泄减少,造成蓄积中毒。
第二节 苯胺类
对乙酰氨基酚及非那西丁
对乙酰氨基酚(acetaminophen)又名扑热息痛(paracetamol),是非那西丁(phenacetin)的体内代谢产物,二者都是苯胺衍生物,具有相同的药理作用。
【体内过程】口服对乙酰氨基酚和非那西丁均易吸收,血药浓度0.5~1小时达高峰;约70%~80%非那西丁在肝内迅速去乙基,成为对乙酰氨基酚;其余部分则去乙酰基,成为对氨基苯乙醚;约有60%对乙酰氨基酚与葡萄糖醛酸结合;35%与硫酸结合失效后经肾排泄;有极少部分对乙酰氨基酚进一步代谢为对肝有毒性的羟化物。而对氨苯乙醚也通过羟化,产生某种可使血红蛋白氧化为高铁血红蛋白以及引起溶血的毒性代谢物(图20-2)。
图20-2 非那西丁及对乙酰氨基酚的体内代谢
【药理作用及临床应用】对乙酰氨基酚和非那西丁的解热镇痛作用缓和持久,强度类似乙酰水杨酸,但其抗炎作用很弱,无实际疗效。非那西丁的作用是其本身及其主要代谢物对乙酰氨基酚作用的总和。对乙酰氨基酚抑制中枢PG合成酶的作用强度与乙酰水杨酸相似;但在外周,对此酶的抑制则远比乙酰水杨酸为弱,这可能是两种同功酶的敏感性不同所致。这也可说明它们几无抗炎作用的原因。非那西丁常配成复方应用,但由于它对肾脏及血红蛋白的毒性,近年来已为对乙酰氨基酚所取代。
【不良反应及应用注意】治疗量的对乙酰氨基酚及非那西丁不良反应少,偶见过敏反应,如皮疹,严重者伴有药热及粘膜损害。对乙酰氨基酚过量(成人10~15g)急性中毒可致肝坏死;而非那西丁过量则产生高铁血红蛋白血症,出现紫绀及其它缺氧症状,还可引起溶血性贫血。这类药物长期应用还能导致对药物的依赖及肾损害。
第三节 吡唑酮类
本类药物包括保泰松(phenylbutazone;布他酮,butazolidin)及其化谢产物羟基保泰松(oxyphenbutazone)。
保泰松及羟基保泰松
【体内过程】口服保泰松吸收迅速完全,2小时达峰血药浓度值,吸收后98%与血浆蛋白结合,再缓慢释出,故作用持久,血浆t1/2为50~65小时。保泰松可穿透滑液膜,在滑液膜间隙内的浓度可达血浓度的50%,停药后,关节组织中保持较高浓度可达3周之久。本药主要由肝药酶代谢为羟化物及其葡萄糖醛酸结合物。其苯环化物为羟基保泰松,为活性代谢物;羟基保泰松的血浆蛋白结合率也很高,血浆t1/2长达几天,长期服用保泰松时,羟基保泰松可在体内蓄积,造成毒性。保泰松可诱导肝药酶,加速自身代谢。保泰松及其代谢物由肾缓慢排泄。
【药理作用及临床应用】保泰松抗炎抗风湿作用强而解热镇痛作用较弱;其抗炎作用也是通过抑制PG生物合成而实现。临床主要用于风湿性及类风湿性关节炎、强直性脊柱炎。本药对以上疾病的急性进展期疗效很好;较大剂量可减少肾小管对尿酸盐的再吸收,故可促进尿酸排泄,可用于急性痛风。偶也用于某些高热如恶性肿瘤及寄生虫病(急性丝虫病、急性血吸虫病)引起的发热。
【不良反应】10%~45%患者有不良反应,其中10%~15%患者必须停药,故不宜大量长期用药。
1.胃肠反应最常见为恶心、上腹不适、呕吐、腹泻。饭后服药可减轻。大剂量可引起胃、十二指肠出血、溃疡,与本药抑制PG合成有关。溃疡病者禁用。
2.水钠潴留保泰松能直接促进肾小管对氯化钠及水的再吸收,引起水肿。使心功能不全者出现心衰、肺水肿。故用本药时应忌盐。高血压、心功能不全患者禁用。
3.过敏反应有皮疹。偶致剥脱性皮炎、粒细胞缺乏、血小板减少及再生障碍性贫血,可能致死,应高度警惕。如见粒细胞减少,应立即停药并用抗菌药防治感染。
4.肝、肾损害 偶致肝炎及肾炎。肝、肾功能不良者禁用。
5.甲状腺肿大及粘液性水肿是保泰松抑制甲状腺摄取碘所致。
羟基保泰松除无排尿酸作用及胃肠反应较轻外,作用、用途及不良反应同保泰松。
【药物相互作用】保泰松诱导肝药酶,加速自身代谢,也加速强心甙代谢;还可通过血浆蛋白结合部位的置换,加强口服抗凝药、口服降糖药、苯妥英钠及肾上腺皮质激素的作用及毒性,当保泰松与这些药物合用时,应予注意。
第四节 其他抗炎有机酸类
吲哚美辛
吲哚美辛(indomethacin,消炎痛)为人工合成的吲哚衍生物。口服吸收迅速而完全,3小时血药浓度达峰值。吸收后90%与血浆蛋白结合。主要在肝代谢;代谢物从尿、胆汁、粪便排泄;10%~20%以原形排泄于尿中。血浆t1/2为2~3小时。
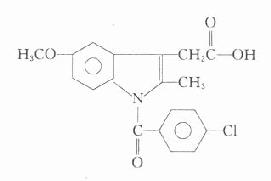
【药理作用及临床应用】吲哚美辛是最强的PG合成酶抑制药之一,有显着抗炎及解热作用,对炎性疼痛有明显镇痛效果。但不良反应多,故仅用于其他药物不能耐受或疗效不显着的病例。对急性风湿性及类风湿性关节炎的疗效与保泰松相似,约2/3患者可得到明显改善。如果连用2~4周仍不见效者,应改用它药。对关节强直性脊椎炎、骨关节炎也有效;对癌性发热及其它不易控制的发热常能见效。
【不良反应】30%~50%患者用治疗量吲哚美辛后发生不良反应;约20%患者必须停药。大多数反应与剂量过大有关。
1.胃肠反应有食欲减退、恶心、腹痛;上消化道溃疡,偶可穿孔、出血;腹泻(有时因溃疡引起);还可引起急性胰腺炎。
2.中枢神经系统 25%~50%患者有前额头痛、眩晕,偶有精神失常。
3.造血系统可引起粒细胞减少、血小板减少、再生障碍性贫血等。
4.过敏反应 常见为皮疹,严重者哮喘。本药抑制PG合成酶作用强大。“阿司匹林哮喘”者禁用本药,因此可发生哮喘。
本药禁用于孕妇、儿童、机械操作人员、精神失常、溃疡病、癫痫、帕金森病及肾病患者。
舒林酸
舒林酸(sulindac,苏林大)的作用及应用均似吲哚美辛,但强度不及后者的一半;其特点是作用较持久,不良反应也较少。
甲芬那酸、氯芬那酸和双氯芬酸
甲芬那酸(mefenamicacid,甲灭酸)、氯芬那酸(clofenamic acid,氯灭酸)和双氯芬酸(diclofenac)均为邻氨苯甲酸(芬那酸)的衍生物。它们都能抑制PG合成酶而具有抗炎、解热及镇痛作用。
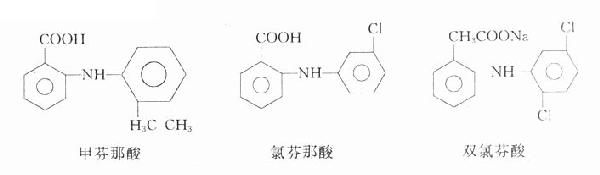
与其他解热镇痛药相比,并无优点。主要用于风湿性及类风湿性关节炎。甲芬那酸常见不良反应有嗜睡、眩晕、头痛、恶心、腹泻,也可发生胃肠溃疡及出血;偶致溶血性贫血及骨髓抑制、暂时性肝功能及肾功能异常。连续用药一般不应超过一周。肝、肾功能损害者及孕妇慎用。氯芬那酸不良反应较少,常见头晕及头痛。双氯芬酸的抗炎作用为芬酸类中最强者,副作用更小,但偶可使肝功能异常、白细胞减少。
布洛芬
布洛芬(ibuprofen,异丁苯丙酸)是苯丙酸的衍生物。口服吸收迅速,1~2小时血浆浓度达峰值,血浆t1/22小时,99%与血浆蛋白结合,可缓慢进入滑膜腔,并在此保持高浓度。口服剂量的90%以代谢物形式自尿排泄。本药是有效的PG合成酶抑制药,具有抗炎、解热及镇痛作用,主要用于治疗风湿性及类风湿性关节炎,也可用于一般解热镇痛,疗效并不优于乙酰水杨酸,但主要特点是胃肠反应较轻,易耐受。
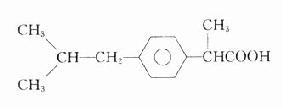
不良反应有轻度消化不良、皮疹;胃肠出血不常见,但长期服用者仍应注意;偶见视力模糊及中毒性弱视,出现视力障碍者应立即停药。
本类药物中的萘普生(naproxen)及酮洛芬(ketoprofen)的作用及用途均相似,但t1/2分别为12~15小时和2小时。
吡罗昔康
吡罗昔康(piroxicam,炎痛喜康)属苯噻嗪类。口服吸收完全,2~4小时血药浓度达峰值。在体外抑制PG合成酶的效力与吲哚美辛相等。对风湿性及类风湿性关节炎的疗效与乙酰水杨酸、吲哚美辛及萘普生相同而不良反应少,患者耐受良好。其主要优点是血浆t1/2长(36~45小时),用药剂量小,每日服1次(20mg)即可有效。由于本药为强效抗炎镇痛药,对胃肠道有刺激作用,剂量过大或长期服用可致消化道出血、溃疡,应予注意。
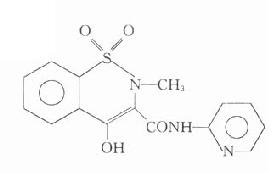
一些常用解热镇痛药常相互配伍,或配伍巴比妥类、咖啡因或抗组胺药(如氯苯那敏)以期提高疗效和减少不良反应。但据一些对照观察,复方并不优于单用,且复方中大多含有非那西丁(苯胺类),久用可致肾乳头坏死,并可能引起肾盂癌;非那西丁还可能与某些复方久用引起依赖性有关。此外,不少复方都含氨基比林(吡唑酮类),少数患者服用后出现粒细胞缺乏。因此,对这些复方需重新评价;对含氨基比林的复方应慎用。
表20-1 常用复方解热镇痛药成分
| 名 称 | 成分与含量(g/片) | 用 法 | |||||||
| 乙酰 水杨酸 |
非那 西丁 |
氨基* 比林 |
安替* 比林 |
咖啡 因 |
苯巴比妥 | 巴比妥 | 氯苯那敏 | ||
| 复方阿司匹林片(APC) | 0.22 | 0.15 | 0.035 | 1~2片/次,3次/日 | |||||
| 复方氯苯那敏片 | 0.2268 | 0.162 | 0.0324 | 0.002 | 1~2片/次,3次/日 | ||||
| 氨啡咖片 | 0.15 | 0.1 | 0.03 | 1~2片/次,3次/日 | |||||
| 去痛片 | 0.15 | 0.15 | 0.05 | 0.015 | 1~2片/次,3次/日 | ||||
| 安痛定注射液(2ml) | 0.1 | 0.04 | 0.18 | 皮下或肌注,一次2ml | |||||
*为吡唑酮类解热镇痛药。
[附]抗痛风药
痛风是体内嘌呤代谢紊乱所引起的一种疾病,表现为高尿酸血症,尿酸盐在关节、肾及结缔组织中析出结晶。急性发作时,尿酸盐微结晶沉积于关节而引起局部粒细胞浸润及炎症反应,治疗痛风的药物有别嘌醇等。
别嘌醇
别嘌醇(allopurinol,别嘌呤醇)为次黄嘌呤的异构体。次黄嘌呤及黄嘌呤可被黄嘌呤氧化酶催化而生成尿酸。别嘌醇也被黄嘌呤氧化酶催化而转变成别黄嘌呤;它及别黄嘌呤都可抑制黄嘌呤氧化酶(图20-3)。因此在别嘌醇作用下,尿酸生成及排泄都减少,避免尿酸盐微结晶的沉积,防止发展为慢性痛风性关节炎或肾病变。别嘌醇不良反应少,偶见皮疹、胃肠反应及转氨酶升高、白细胞减少等。
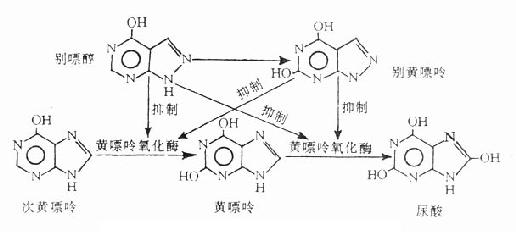
图20-3 别嘌醇对黄嘌呤氧化酶的抑制
丙磺舒
丙磺舒(probenecid)又名羧苯磺胺(benemid),口服吸收完全,血浆蛋白结合率85%~95%;大部分通过肾近曲小管主动分泌而排泄,因脂溶性大,易被再吸收,故排泄较慢。本药竞争性抑制肾小管对有机酸的转运,抑制肾小管对尿酸的再吸收,增加尿酸排泄,可用于治疗慢性痛风。因无镇痛及消炎作用,故不适用于急性痛风。
苯溴马隆
苯溴马隆(benzbromarone)作用似丙碘舒,减少肾小管对尿酸的再吸收而促其排泄。每日用量为20~100mg,宜从20mg/日开始,逐渐递增,不良反应有头痛、恶心、腹泻。
秋水仙碱
秋水仙碱(colchicine)对急性痛风性关节炎有选择性消炎作用,用药后数小时关节红、肿、热、痛即行消退,对一般性疼痛及其他类型关节炎并无作用。它对血中尿酸浓度及尿酸的排泄没有影响,其作用是抑制急性发作时的粒细胞浸润。
本药不良反应较多。常见消化道反应。中毒时出现水样腹泻及血便,脱水,休克;对肾及骨髓也有损害作用。
解热镇痛抗炎药 制剂及用法
乙酰水杨酸(acetylsalicylicacid)解热镇痛 0.3~0.6g/次,3次/日,饭后服。抗风湿:3~5g/日,分4次服,症状控制后逐渐减量。
水杨酸钠(sodiumsalicylate)抗风湿:4~8g/日,分4~6次服,症状控制后逐渐减量。
对乙酰氨基酚(paracetamol)0.5g/次,3次/日。
保泰松(phenylbutazone) 0.1~0.2g/次,3次/日。症状改善后改为1次/日。
羟基保泰松(oxyphenbutazone)0.1g/次,3次/日。餐中服,一周后递减,0.1~0.2g/日。
吲哚美辛(indomethacin)25mg/次,2~3次/日。餐中服,以后每周可递增25mg至每日总量为100~150mg。
舒林酸(sulindac)150~200mg/次,2次/日。每日最大剂量400mg。
甲芬那酸(mefenamicacid)首次0.5g,以后0.25g/次。用药不宜超过一周。
氯芬那酸(clofenamicacid)0.2g/次,3次/日。
双氯芬酸(diclofenac)25mg/次,3次/日。75mg/次,1次/日,深臀部肌注。
布洛芬(ibuprofen)0.2~0.4g/次,3次/日,餐中服。
酮布芬(ketoprofen)50mg/次,3~4次/日。
萘普生(naproxen)口服,0.25g/次,2次/日。
吡罗昔康(piroxicam)口服,20mg/日,分1~2次服。
秋水仙碱(colchicine)0.5mg/次,1~2小时1次,口服,1日总量不得超过4mg。
丙磺舒(probenecid,羧苯磺胺)治疗痛风:开始0.25g/次,2次/日,一周后增至0.5g/次;
别嘌醇(allopurinol)第一周0.1g/日,第二周0.2g/日,第三周以后为0.3g/日,分2~3次服。
第二十一章 钙拮抗药
钙拮抗药(calcium antagonists)是一类阻滞Ca2+从细胞外液经电压依赖性钙通道流入细胞内的药物,又称钙通道阻滞药。第一个钙拮抗药维拉帕米对心有负性肌力和负性频率作用,早期曾被认为是β受体阻断药。1967年A.Fleckenstein发现维拉帕米在降低心肌收缩性时并不影响膜电位的变化和振幅,其作用与脱钙的情况相同,认为其机制是阻滞或减少Ca2+向细胞的内流,并首次提出钙拮抗药名称。
钙拮抗药是发展迅速的一类药物,包含许多化学结构各异的化合物,已广泛用于治疗心律失常、高血压及心绞痛等疾病。
第一节 钙、钙通道与钙拮抗药的分类
一、钙离子的生理意义
钙是体内重要的阳离子之一,具有多项生理作用,早年知钙是心搏动、血液凝固所必需,现知钙还参与许多生理、生化反应,包括神经细胞兴奋性,递质释放,肌肉收缩,腺体分泌,细胞运动等(表21-1)。
表21-1 Ca2+调节的细胞过程与钙拮抗药的效应
| 组织细胞 | 细胞过程 | 钙拮抗药的效应 |
| 窦房结、房室结 | 除极 | 抑窦房结起搏、抑房室结传导 |
| 心脏工作肌 | 除极、收缩 | 抑制动作电位2相、降低收缩性 |
| 动脉(冠状、肺、外周) | 收缩 | 降低外周阻力、解冠脉痉缩、降肺动脉阻力 |
| 静脉 | 收缩 | 减少静脉回流 |
| 支气管、胃肠道、泌尿道及子宫平滑肌 | 收缩 | 缓解哮喘、食管痉缩、胆绞痛、痛经、解输尿管、膀胱疼痛 |
| 胰腺、脑垂体、肾上腺髓质 | 分泌 | 减少胰岛素、垂体激素、儿茶酚胺的分泌 |
| 唾液腺、泪腺、胃粘膜 | 分泌 | 减少唾液、泪、胃泌素的分泌 |
| 肥大细胞 | 组胺释放 | 抑制脱颗粒 |
| 多形核白细胞 | 运动、释溶酶体酶 | 抑制中性白细胞的激活 |
| 血小板 | 聚集、收缩、胞排 | 抑制血小板的激活 |
| 神经细胞 | 递质释放 | 减少递质释放 |
二、钙通道的类型与分子结构
钙通道是细胞膜中蛋白质小孔,Ca2+及Ba2+能通透进入细胞内,其他离子通透率甚低。当膜电位接近—40mV时,钙通道开放率明显增加,单个钙通道每秒钟可通过3×106个Ca2+。
1.钙拮抗药对前者的阻滞作用比对后者为强。电压门控性钙通道中,根据电导值、动力学特性等的不同,又分为几种亚型,现知有L、T、N、P型。L型(long-lasting)开放时间久,约10~20ms,表现为持续长时钙内流,电导值25pS,激活电位-10mV,失活电位-60~-10mV ,衰变时间>500ms。T型(transient)的开放时间短暂,引起瞬间短小Ca2+电流,电导值9pS,激活电位-70mV,失活电位-100~-60mV,衰变时间20~50ms。N型(neither L nor T)见于神经元中,调节神经递质释放,电导值13pS,激活电位-10mV,失活电位-100~-40mV,衰变时间50~80 ms。P型最初在哺乳动物小脑浦肯野细胞中发现,故名,电导值随条件不同变动在9~19pS之间,激活电位-50mV,失活极慢,t1/2约1s。随着研究深入,可能会有新的亚型被发现。
2.对钙通道的分子结构研究最多的是骨骼肌横管中的L型通道,现知它由5个亚单位所组成,即α1(175kD),α2(143kD),β(54kD),γ(30kD),δ(27kD)。其中,α1与β亚单位胞内侧有磷酸化部位,α2·γ·δ亚单位胞外侧有糖基化部位。各亚单位排列如图21-1。
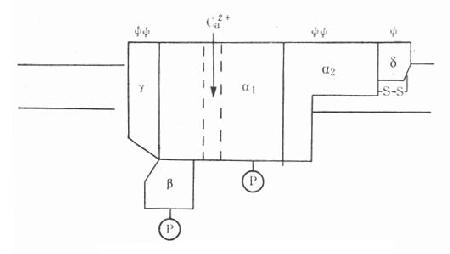
图21-1 钙通道各亚单位位置示意图
P磷酸化部位 Ψ糖基化部位
α1亚单位是主要功能单位,它能单独发挥钙通道的作用。几种钙拮抗药硝苯地平、维拉帕米的受体(结合位点)也在α1上。α1亚单位有4个重复结构域,每域含6个跨膜α-螺旋片段,分别称S1,S2,…S5,S6,都是疏水性的。S4含5~6个带正电荷精氨酸,对膜电位的变化极为敏感,是钙通道的电压传感器。S5与S6之间较长的小袢陷入膜内形成小孔供Ca2+通透,其邻近部位常是钙拮抗药的结合部位。硝苯地平等二氢吡啶类药的结合位点主要在Ⅲ域S6及其细胞外侧,另也结合于Ⅳ域S6及其胞外侧,所以认为二氢吡啶类作用于细胞膜外侧,维拉帕米的结合位点在Ⅳ域S6及其邻近的胞内侧片段上,所以认为它作用于细胞膜内侧。这些钙拮抗药与钙通道结合后,引起通道蛋白质构象变化,终而阻滞Ca2+向细胞的内流。
三、钙拮抗药的分类
钙拮抗药品种繁多,为了便于临床选用,世界卫生组织(WHO)曾于1987年公布钙拮抗药的分类,先按药对钙通道的选择性分为两类;再按药对心血管系的作用,将药分为6类。
(一)选择性钙拮抗药
1.苯烷胺类,维拉帕米、加洛帕米。
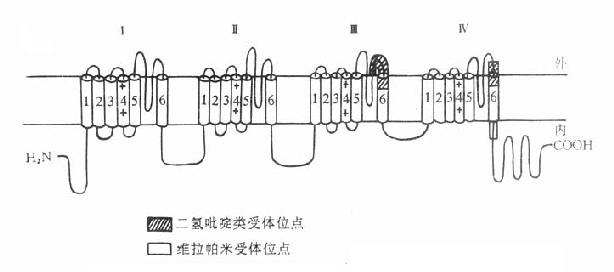
图21-2 钙通道α亚单位的分子结构与二类受体的结合位点
2.二氢吡啶类,硝苯地平、尼莫地平、尼群地平、氨氯地平等。
3.![]() 类,地尔硫
类,地尔硫![]() 。
。
(二)非选择性钙拮抗药
1.氟桂嗪类,氟桂嗪、桂利嗪等。
2.普尼拉明类,普尼拉明等。
3.其他类,哌克昔林等。
第二节 钙拮抗药的作用与临床应用
一.药理作用
鉴于Ca2+对细胞多种生化、生理反应的影响,理论上预计钙拮抗药应有广泛的药理作用。然现有的钙拮抗药却主要作用于心血管系统而对其他组织细胞影响较小,这可能与心血管系统细胞膜上L型钙信道密度较高有关。钙拮抗药由于阻滞Ca2+的内流,使细胞内Ca2+量减少,从而引起各种作用。
1.对心肌的作用
(1)负性肌力作用:钙拮抗药使心肌细胞内Ca2+量减少,因而呈现负性肌力作用。它可在不影响兴奋除极的情况下,明显降低心肌收缩性,这就是兴奋-收缩脱偶联(excitation-contraction decoupling)。
钙拮抗药还能舒张血管降低血压,继而使整体动物中交感神经活性反射性增高,抵消部分负性肌力作用。硝苯地平的这一作用明显,可能超过其负性肌力作用而表现为轻微的正性肌力作用。
收缩性减弱可使心氧耗量相应减少,又由于血管舒张,使心后负荷降低,耗氧量也将进一步减少。
(2)负性频率和负性传导作用:窦房结和房室结等慢反应细胞的0相除极和4相缓慢除极都是Ca2+内流所引起的,所以它们的传导速度和自律性就由Ca2+内流所决定,因此钙拮抗药能减慢房室结的传导速度,延长其有效不应期,可使折返激动消失,用于治疗阵发性室上性心动过速。钙拮抗药对窦房结则能降低自律性,从而减慢心率。这种负性频率作用在整体动物中也可被交感神经活性的反射性增高所部分抵消,所以钙拮抗药治疗窦性心动过速的疗效欠佳,硝苯地平更差。
(3)心肌缺血时的保护作用:缺血时,心细胞的能量代谢出现障碍,渐趋耗竭,使心细胞各项功能衰退。由于钠泵、钙泵抑制及钙的被动转运加强,乃使细胞内钙积储,形成“钙超负荷”,最终引起细胞坏死。钙拮抗药可减少细胞内钙量,避免细胞坏死,起到保护作用。
2.对血管的作用
血管平滑肌细胞的收缩也受细胞内Ca2+量的调控,其细胞内Ca2+量主要来自经钙通道而内流者。细胞内Ca2+量多,将通过钙调蛋白激活肌凝蛋白轻链激酶(MLCK),后者催化肌凝蛋白轻链的磷酸化,继而触发肌纤、肌凝蛋白的相互作用而引起收缩。
钙拮抗药阻滞Ca2+的内流,能明显舒张血管,主要舒张动脉,对静脉影响较小。动脉中又以冠状血管较为敏感,能舒张大的输送血管和小的阻力血管,增加冠脉流量及侧枝循环量,治疗心绞痛有效。
脑血管也较敏感,尼莫地平和氟桂嗪舒张脑血管作用较强,能增加脑血流量。
钙拮抗药也舒张外周血管,解其痉挛,可用于治疗外周血管痉挛性疾病如雷诺病。
表21-2 三种钙拮抗药心血管效应的比较
| 效 应 | 维拉帕米 | 硝苯地平 | 地尔硫 |
| 负性肌力作用 | 4 | 1 | 2 |
| 负性频率作用 | 5 | 1 | 5 |
| 负性传导作用 | 5 | 4 | |
| 舒张血管作用 | 4 | 5 | 3 |
0-5指作用强度由弱到强的程度
3.对其他平滑肌的作用
在其他平滑肌中,钙拮抗药对支气管平滑肌的松弛作用较为明显,较大剂量也能松弛胃肠道、输尿管及子宫平滑肌。钙拮抗药治疗或防止哮喘有效,此时,除松弛支气管平滑肌外,还能减少组胺的释放和白三烯D4的合成,又有减少粘液分泌的作用。
4.改善组织血流的作用
钙拮抗药通过对血小板和红细胞的影响而改善组织血流。
(1)抑制血小板聚集:Ca2+促使血小板第一时相的可逆性聚集和第二时相的不可逆性聚集。因此钙拮抗药能抑制血小板聚集。
(2)增加红细胞变形能力,降低血液滞度:正常时红细胞有良好的变形能力,能缩短其直径而顺利通过毛细血管,保持正常血液粘滞度。当红细胞内Ca2+增多,其变形能力降低,血粘滞度增高,易引起组织血流障碍。钙拮抗药减少红细胞内Ca2+量,即能降低血粘滞度。
5.其他作用
(1)抗动脉粥样硬化作用:钙拮抗药能防止实验性动脉粥样硬化的发生,这一作用与多种效应有关,如减少细胞内Ca2+的超负荷;抑制血小板聚集;减少血管痉挛收缩或舒张血管;抑制血管壁肥厚增殖,特别对血管平滑肌细胞的增殖。
(2)抑制内分泌腺的作用:较大剂量的钙拮抗药能抑制多种内分泌功能,如脑垂体后叶分泌催产素、加压素;垂体前叶分泌促肾上腺皮质激素、促性腺激素、促甲状腺激素;胰素及醛固酮的分泌。此外,还能抑制交感神经未梢对去甲肾上腺素的释放,表现出微弱的非特异性抗交感作用。
二.作用方式
各类钙拮抗药作用略有差异,各有侧重面,不能用统一的构效关系或一种机制来解释其作用。兹介绍与作用方式有关的几个问题。
1.钙信道的三种功能状态电压门控性钙通道受电压调控,在不同电压影响下,通道发生构象变化而表现出不同功能状态。一般设想它有双重门控系统即激活门与失活门,并有三种功能状态即静息态,开放态和失活态(图21-3)。静息态时通道关闭,Ca2+不能通过,通道的激活门关闭而失活门打开,此时,通道小孔被激活门所闭。细胞兴奋除极时通道转为开放态,Ca2+内流,此时,激活门开放,失活门由开放而缓慢趋于关闭,通道小孔开放。随后是失活态,通道关闭,Ca2+不能通过,此时,失活门关闭,激活门由开放而趋于关闭,小孔为失活门所闭。失活是恢复过程,经静息态为下次除极后的开放作好准备。一般,钙拮抗药与静息态的亲和性较低,而对其他状态作用较显著,如维拉帕米作用于开放态,地尔硫![]() 作用于失活态,硝苯地平则主要作用于失活态。
作用于失活态,硝苯地平则主要作用于失活态。
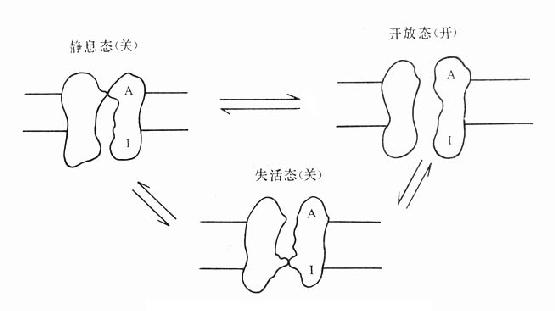
图21-3 通道三种功能状态
A 激活门 I失活门
2.频率依赖性电压门控钙通道的功能和药物对它的作用深受电压的影响,这称“电压依赖性”。此外,药物的作用还呈“频率或使用依赖性”,即通道开放愈频繁,药物的阻滞作用愈强。这样,药物对高频除极的细胞更为有效,治疗频率较高的室上性心动过速就远比治疗频率较低的室性心动过速更为有效。各类钙拮抗药的“频率依赖性”程度不一,维拉帕米和地尔硫![]() 有明显的“频率依赖性”,而硝苯地平则无。
有明显的“频率依赖性”,而硝苯地平则无。
3.受体间的相互影响三类钙拮抗药的受体,即它们在钙通道α1亚单位的结合部位已如前述。这几类受体在钙通道中又相互作用而影响各自对钙拮抗药的亲和力,例如,二氢吡啶受体或地尔硫![]() 受体各被药物占领后,都会提高另一方对药物的亲和力。又如维拉帕米受体被占领后,就会减弱另二类受体对药物的亲和力。反之,另二类受体被占领,也将减弱维帕米受体对药的亲和力。
受体各被药物占领后,都会提高另一方对药物的亲和力。又如维拉帕米受体被占领后,就会减弱另二类受体对药物的亲和力。反之,另二类受体被占领,也将减弱维帕米受体对药的亲和力。
钙通道中药物受体的发现可提示钙拮抗药的作用不是简单的药物分子阻塞通道,而是经通道蛋白构象改变而发生的。又提示体内可能有内源性激动物或阻滞物存在,调控着钙通道。
三.药物体内过程
钙拮抗药化学结构各异,其体内过程既有差异也有共性。三种重要药物口服吸收都较完全,蛋白结合率高,代谢较多。硝苯地平生物利用度高,维拉帕米利用度低,因其首关消除效应较强。维拉帕米在肝中先经N-脱甲基成去甲维拉帕米,仍有拮抗钙作用,后经0-脱甲基及链解裂而失去活性。地尔硫![]() 先脱乙酰基成有活性的代谢产物,后继续代谢成失效物。兹将三种重要钙拮抗药的药代动力学参数列表于下。
先脱乙酰基成有活性的代谢产物,后继续代谢成失效物。兹将三种重要钙拮抗药的药代动力学参数列表于下。
表21-3 三种钙拮抗药的药物代谢动力学参数
| 维拉帕米 | 硝苯地平 | 地尔硫 |
|
| 口服吸收率(%) | >90 | >90 | >90 |
| 生物利用度(%) | 10~20 | 60~70 | 20 |
| 起效时间(分) | iv 1 | 舌下3 | 舌下3 |
| 口服(分) | <30 | <20 | <30 |
| 达峰效时间(分) | iv 5 | 舌下20 | — |
| 口服(小时) | 3~5 | 1~2 | 0.5 |
| 治疗血药浓度(ng/ml) | 30~300 | 25~100 | 50~200 |
| 蛋白结合率(%) | 90 | 90 | 85 |
| 消除t1/2(小时) | 8 | 5 | 5 |
| 代谢部位及产物 | 肝.有活性代谢物 | 肝.无效代谢物 | 肝.有活性代谢物 |
| 首关消除效应(%) | 85 | 20~30 | 90 |
| 分布溶积(L/kg) | 6.1 | 1.3 | 5.3 |
| 排泄(%)消化道 | 15 | 10 | 60 |
| 肾 | 70 | 90 | 30 |
四.临床应用
钙拮抗药的临床应用主要是防治心血管系统疾病,近年也试用于其他系统疾病。
1.心绞痛钙拮抗药对各型心绞痛都有不同程度的疗效。
(1)变异型心绞痛:常在休息时如夜间或早晨发作,由冠状动脉痉挛所引起。钙拮抗药是治疗的首选药物,三种主要药物都能收到良好效果,三者疗效基本相等。
(2)稳定型(劳累型)心绞痛:常见于冠状动脉粥样硬化患者,休息时并无症状,此时心血液供求关系是平衡的。劳累时心作功增加,血液供不应求,导致心绞痛发作。钙拮抗药通过舒张冠脉,减慢心率,降低血压及心收缩性而发挥治疗效果。维拉帕米的负性肌力、频率作用较显,地尔硫![]() 降低血压、减慢心率较强,二药都可应用。硝苯地平降低后负荷较明显,其反射性加快心率的作用可能诱发心绞痛,幸长期给药时未见此不良反应。
降低血压、减慢心率较强,二药都可应用。硝苯地平降低后负荷较明显,其反射性加快心率的作用可能诱发心绞痛,幸长期给药时未见此不良反应。
(3)不稳定型心绞痛:较为严重,昼夜都可发作,由动脉粥样硬化斑块形成或破裂及冠脉张力增高所引起。维拉帕米和地尔硫![]() 疗效较好,硝苯地平宜与β-受体阻断药合用。
疗效较好,硝苯地平宜与β-受体阻断药合用。
2.心律失常钙拮抗药治疗室上性心动过速及后除极触发活动所致的心律失常有良好效果。
三类钙拮抗药减慢心率的作用程度有差异。维拉帕米和地尔硫![]() 减慢心率作用较明显。硝苯地平较差,甚至反射性加速心率,因此它不用于治疗心律失常。
减慢心率作用较明显。硝苯地平较差,甚至反射性加速心率,因此它不用于治疗心律失常。
对阵发性室上性心动过速,静脉注射维拉帕米或地尔硫![]() 可迅速中止发作,口服则可预防发作。二药对房室结的减慢传导和延长不应期的作用可以取消折返激动,使由折返所引起的阵发性室上性心动过速有90%以上转复为窦性节律,合用β-受体阻断药还可维持此效。
可迅速中止发作,口服则可预防发作。二药对房室结的减慢传导和延长不应期的作用可以取消折返激动,使由折返所引起的阵发性室上性心动过速有90%以上转复为窦性节律,合用β-受体阻断药还可维持此效。
房颤时静脉注射二药能抑制房室传导而减少冲动下达心室,控制心室频率;并使少数新发病者转复为窦性节律。地尔硫![]() 合用地高辛控制房颤时的心室频率效果最好。
合用地高辛控制房颤时的心室频率效果最好。
3.高血压高血压时血管平滑肌细胞的Ca2+内流有所增加,因此钙拮抗药治疗有效。三类钙拮抗药都可应用。
硝苯地平控制严重高血压效果较好,用药中并不伴发显著的反射性心动过速,也不引起体位性低血压。长期应用后,全身外周阻力下降30%~40%,肺循环阻力也下降。后一作用特别适合于并发心性哮喘的高血压危象患者。
维拉帕米和地尔硫![]() 治疗轻、中度高血压有效,可以单用,也可与其他抗高血压药合用。单用时可使40%~45%原发性高血压患者的血压得到控制,对老年人疗效较好。二药常能增加心、脑、肾血流量,改善其功能,也适于治疗并发外周动脉阻塞性疾病的高血压。静脉注射可治疗高血压危象。
治疗轻、中度高血压有效,可以单用,也可与其他抗高血压药合用。单用时可使40%~45%原发性高血压患者的血压得到控制,对老年人疗效较好。二药常能增加心、脑、肾血流量,改善其功能,也适于治疗并发外周动脉阻塞性疾病的高血压。静脉注射可治疗高血压危象。
4.肥厚性心肌病肥厚性心肌病时,心肌细胞内Ca2+量超负荷,因此钙拮抗药治疗有效。它能改进舒张功能。维拉帕米疗效较好,还能减轻左心室流出道狭窄。
表21-4 钙拮抗药的治疗应用比较表
| 疾 病 | 维拉帕米 | 硝苯地平 | 地尔硫 |
| 心绞痛 | |||
| 稳定型 | +++ | +++ | +++ |
| 变异型 | +++ | +++ | +++ |
| 不稳定型 | +++ | +++ | +++ |
| 心律失常 | |||
| 阵发性室上性心动过速 | +++ | - | ++ |
| 心房颤动、扑动 | ++ | - | ++ |
| 高血压 | ++ | +++ | + |
| 肥厚性心肌病 | + | - | - |
| 雷诺病 | ++ | ++ | + |
| 脑血管痉挛(出血后) | - | + | - |
注:+++很常用,++常用,+可用,-不用。
5. 脑血管疾病尼莫地平、氟桂嗪等钙拮抗药能较显著舒张脑血管,增加脑血流量。治疗短暂性脑缺血发作、脑血栓形成及脑栓塞等有效。治疗或预防蛛网膜下腔出血所致的脑血管痉挛有效,可减少神经后遗症及病死率。
维拉帕米、氟桂嗪等还能有效地预防偏头痛,长期用药三个月以上也可用作治疗,能减轻症状,减少发作频率及发作时间。
6. 其他雷诺病时由寒冷及情绪激动引起的血管痉挛可被钙拮抗药所解除,常用尼莫地平、硝苯地平。另外,支气管哮喘,食管贲门失弛缓症,急性胃肠痉挛性腹痛,早产,痛经等用钙拮抗药治疗也有效。
第三节 常用钙拮抗药
维拉帕米
维拉帕米(verapamil)又名异搏定(isoptin)。
【药理作用】维拉帕米在离体实验能降低窦房结起搏细胞自律性,减慢窦性频率,但在整体实验中,此作用被反射性的交感神经兴奋所部分抵消,人体用药后窦性频率减慢约10%,对房室结的传导性也能抑制,主要对房室结的上部、中部有作用,对下部作用较差。过大剂量可使窦房结、房室结电活动消失。口服120mg使心电图P-R间期延长。以上作用使维拉帕米成为治疗阵发性室上性心动过速的首选药物。
维拉帕米能舒张冠状血管及外周血管,增加缺血心肌冠脉流量,增加侧枝循环流量,也降低外周阻力,降低血压。
【不良反应】约10%患者出现不良反庆,有1%患者需停用药物。口服易致胃肠道症状,静脉注射可致血压下降,偶见房室传导阻滞及心收缩性下降,故禁用于严重心衰及中、重度房室传导阻滞。
同类药物有加洛帕米(gallopamil)即D600,有较强的心抑制作用,应用价值同维拉帕米。
地尔硫
地尔硫
![]() (diltiazem)
(diltiazem)
【药理作用】地尔硫![]() 对心的电生理作用和维拉帕米相似。能明显抑制窦房结自律性而减慢心率。也减慢房室结传导性,适于治疗阵发性室上性心动过速。它对血管的作用接近硝苯地平,能增加冠脉流量。对心肌还有非竞争性拮抗β-受体的作用,可防治心绞痛。雷诺病及偏头痛等。
对心的电生理作用和维拉帕米相似。能明显抑制窦房结自律性而减慢心率。也减慢房室结传导性,适于治疗阵发性室上性心动过速。它对血管的作用接近硝苯地平,能增加冠脉流量。对心肌还有非竞争性拮抗β-受体的作用,可防治心绞痛。雷诺病及偏头痛等。
【不良反应】较少,约2%~5%患者可能出现,注射给药可引起房室传导阻滞及低血压。其他副作用有皮疹、头痛、面部潮红等。
硝苯地平
硝苯地平(nifedipine),又名心痛定。
【药理作用】硝苯地平的作用与维拉帕米不同,它对窦房结房室结及心收缩性的抑制作用较弱,对血管的舒张作用明显。给药量略大反能加速房室传导,是交感神经活性反射性增高之故。与此同理,在整体中它对心肌也不降低收缩性,因此可与β-受体阻断药合用。
硝苯地平舒张冠脉特别是已痉挛收缩的狭窄冠脉,故能增加缺血区流量,可治心绞痛。也能舒张外周小动脉,降低血压,可治高血压,硝苯地平还降低肺血管阻力及肺动脉压,可治肺动脉高压症。
【不良反应】发生率达20%,一般较轻,主要是低血压。长期用药约有5%患者出现头痛。少数患者偶见心肌缺血症状加重,可能是严重冠脉阻塞、心率加快、血压过低所致。
硝苯地平在化学结构上属二氢吡啶类,这类钙拮抗药发展较快,在作用和药代动力学方面都有所改进,主要有以下几种:
尼卡地平(nicardipine)舒张血管较硝苯地平强,对脑血管有一定选择作用。尼索地平(nisoldipine)舒张血管作用更强,对冠状血管选择性较强,治疗心绞痛及高血压良效。尼莫地平(nimodipine)对脑血管有选择作用而降压作用较弱,用于治疗脑血管痉挛收缩及脑供血不足等疾病,也可防治偏头痛。尼群地平(nitrendipine)血管舒张作用强大,用于治疗高血压。非洛地平(felodipine)对冠状血管及外周血管作用明显,持效较久,用于治疗心绞痛及高血压。伊拉地平(isradipine)对冠脉、脑及外周血管作用较强较久,用于治疗高血压及稳定型心绞痛。氨氯地平(amlodipine)舒张冠脉及外周血管,用于治疗中、轻度高血压作用缓慢持久,降低血压10%~18%,无反射性心动过速。治疗稳定型心绞痛效果明显。它的消除t1/2较久,达35~45小时,日服一次即可。拉西地平(lacidipine)也为长效药物,日服一次,因它有较好的对细胞膜的亲和力及穿透性。作用逐渐开始,不引发心悸及头痛,也不致反射性增高交感神经活性。有抗动脉粥样化的作用,还能减少自由基的形成。适于治疗高血压及冠心病心绞痛。
除上述三类药物外,还有其他钙拮抗药:
苄普地尔(bepridil),作用似维拉帕米,能降低窦房结自律性和房室结传导性。还能阻滞钠通道而降低快反应细胞的自律性和传导性,也阻滞钾通道而延长不应期。临床用于治疗室上性及室性心动过速。对冠脉的舒张作用较维拉帕米为弱,但仍可用于治疗心绞痛。严重的不良反应是致心律失常作用,引起尖端扭转型室性心动过速,值得注意。
氟桂嗪(flunarizine)对脑血管有选择性舒张作用,解除其痉挛,能防止缺血缺氧后神经细胞内Ca2+积储所致的细胞损害。还能促进红细胞的变形能力,改善微循环,保护大脑功能。
氟桂嗪主要用于治疗脑血管功能障碍,如脑血管性痴呆及脑供血障碍,能增加智力,改善记忆。也用于治疗偏头痛或各种眩晕。不良反应较少,偶见嗜眠、皮疹。
哌克昔林(perhexiline)的钙拮抗作用较弱,其舒张冠脉、抑制心肌收缩性等作用也弱。主要用于治疗稳定型心绞痛。不良反应有眩晕、无力,偶见周围神经炎。
普尼拉明(prenylamine),又名心可定(segontin),有非特异性钙拮抗作用,可治疗心绞痛及室性早搏、室性心动过速等。不良反应有食欲不振、乏力、皮疹等。
钙拮抗药 制剂及用法
维拉帕米(verapamil)口服40~80mg/次,3次/日。维持量为40mg/次,3次/日。静脉注射5~10mg/次,隔15分钟可重复1-2次,若无效即停用。
硝苯地平(nifedipine) 口服或舌下含化10~20mg/次,3次/日。
地尔硫![]() (diltiazem)口服30~60mg/次,3次/日。
(diltiazem)口服30~60mg/次,3次/日。
氟桂嗪(flunarizine)口服10mg/日,晚上顿服。开始时早、晚各一次。
普尼拉明(prenylamine)口服15~30mg/次,3次/日。
哌克昔林(perhexiline)口服100mg/次,2次/日,可递增至300~400mg/日。
苄普地尔(bepridil) 口服100mg/次,3次/日。
尼卡地平(nicardipine)口服10~20mg/次,3次/日。
尼莫地平(nimodipine) 静滴治脑血管痉挛,开始1mg/小时,2小时后2mg/小时。
尼群地平(nitrendipine)口服开始10mg/日,可递增至30mg/日。
第二十二章 抗心律失常药
心律失常是心动规律和频率的异常,此时心房心室正常激活和运动顺序发生障碍,是严重的心脏疾病。它有缓慢型和快速型之分,前者常用异丙肾上腺素或阿托品治疗。后者的药物治疗比较复杂,本章讨论的是治疗快速型心律失常的药物。
第一节 心律失常的电生理学基础
一、正常心肌电生理
(一)心肌细胞膜电位
心肌细胞的静息膜电位,膜内负于膜外约-90mV,处于极化状态。心肌细胞兴奋时,发生除极和复极,形成动作电位。它分为5个时相,0相为除极,是Na+快速内流所致。1相为快速复极初期,由K+短暂外流所致。2相平台期,缓慢复极,由Ca2+及少量Na+经慢通道内流与K+外流所致。3相为快速复极末期,由K+外流所致。0相至3相的时程合称为动作电位时间(action potential duration,APD)。4相为静息期,非自律细胞中膜电位维持在静息水平,在自律细胞则为自发性舒张期除极,是特殊Na+内流所致,其通道在—50mV开始开放,它除极达到阈电位就重新激发动作电位。
(二)快反应和慢反应电活动
心工作肌和传导系统细胞的膜电位大(负值较大),除极速率快,传导速度也快,呈快反应电活动,其除极由Na+内流所促成;窦房结和房室结细胞膜电位小(负值较小),除极慢,传导也慢,呈慢反应电活动,除极由Ca2+内流促成。心肌病变时,由于缺氧缺血使膜电位减小,快反应细胞也表现出慢反应电活动。
(三)膜反应性和传导速度
膜反应性是指膜电位水平与其所激发的0相上升最大速率之间的关系。一般膜电位大,0相上升快,振幅大,传导速度就快;反之,则传导减慢。可见膜反应性是决定传导速度的重要因素,其典型曲线呈S状,多种因素(包括药物)可以增高或降低之。
(四)有效不应期
复极过程中膜电位恢复到-60~—50mV时,细胞才对刺激发生可扩布的动作电位。从除极开始到这以前的一段时间即为有效不应期(effectiverefractory period,ERP),它反映快钠通道恢复有效开放所需的最短时间。其时间长短一般与APD的长短变化相应,但程度可有不同。一个APD中,ERP数值大,就意味着心肌不起反应的时间延长,不易发生快速型心律失常。
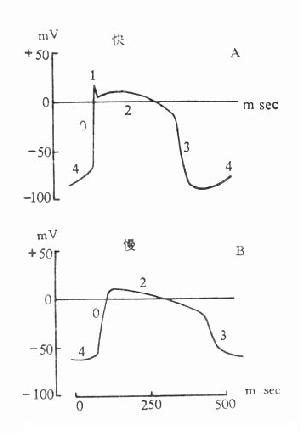
图22-1 浦肯野纤维的快反应与慢反应电活动
A快反应 B慢反应
二、心律失常发生的电生理学机制
心律失常可由冲动形成障碍和冲动传导障碍或二者兼有所引起。
(一)冲动形成障碍
1.自律性增高 自律细胞4相自发除极速率加快或最大舒张电位减小都会使冲动形成增多,引起快速型心律失常。此外,自律和非自律细胞膜电位减小到-60mV或更小时,就引起4相自发除极而发放冲动,即异常自律性。
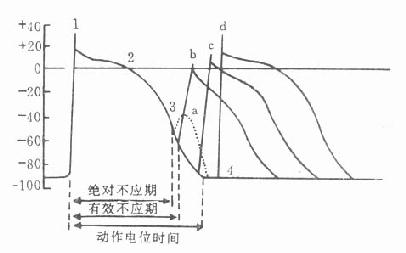
图22-2 不应期与动作电位时间
……局部去极化(局部性兴奋)
──全面去极化(扩布性兴奋)
2.后除极与触发活动 后除极是在一个动作电位中继0相除极后所发生的除极,其频率较快,振幅较小,呈振荡性波动,膜电位不稳定,容易引起异常冲动发放,这称为触发活动(triggeredactivity)。后除极分早后除极与迟后除极两种。前者发生在完全复极之前的2或3相中,主要由Ca2+内流增多所引起;后者发生在完全复极之后的4相中,是细胞内Ca2+过多诱发Na+短暂内流所引起。
图图22-3 A 早后除极与触发活动 b 迟后除极与触发活动
(二)冲动传导障碍
1.单纯性传导障碍包括传导减慢,传导阻滞,单向传导阻滞等。后者的发生可能与邻近细胞不应期长短不一或病变引起的传导递减有关。
Ⅱ常冲动传导 单向阻滞和折返
图22-4 浦肯野纤维末梢正常冲动传导,单向阻滞和折返
2.折返激动 指冲动经传导通路折回原处而反复运行的现象(reentry)。如图所示,正常时浦肯野纤维AB与AC两支同时传导冲动到达心室肌BC,激发除极与收缩,而后冲动在BC段内各自消失在对方的不应期中。在病变条件下,如AC支发生单向传导阻滞,冲动不能下传,只能沿AB支经BC段而逆行至AC支,在此得以逆行通过单向阻滞区而折回至AB支,然后冲动继续沿上述通路运行,形成折返。这样,一个冲动就会反复多次激活心肌,引起快速型心律失常。
邻近细胞ERP长短不一也会引起折返。如图所示,设AC支ERP延长,冲动到达落在ERP中而消失,但可经邻近的AB支下传而后逆行的冲动可因AC支的ERP已过而折回至AB处继续运行,形成折返。
第二节 抗心律失常的基本电生理作用及药物分类
一、抗心律失常的基本电生理作用
药物的基本电生理作用是影响心肌细胞膜的离子通道,通过改变离子流而改变细胞是电生理特性,针对心律失常发生的机制,可将药物的基本电生理作用概括如下:
(一)降低自律性
药物抑制快反应细胞4相Na+内流或抑制慢反应细胞4相Ca2+内流就能降低自律性。药物促进K+外流而增大最大舒张电位,使其较远离阈电位,也将降低自律性。
(二)减少后除极与触发活动
早后除极的发生与Ca2+内流增多有关,因此钙拮抗药对之有效。迟后除极所致的触发活动与细胞内Ca2+过多和短暂Na+内流有关,因此钙拮抗药和钠通道阻滞药对之有效。
(三)改变膜反应性而改变传导性
增强膜反应性改善传导或减弱膜反应性而减慢传导都能取消折返激动,前者因改善传导而取消单向阻滞,因此停止折返激动,某些促K+外流加大最大舒张电位的药如苯妥英钠有此作用;后者因减慢传导而使单向传导阻滞发展成双向阻滞,从而停止折返激动,某些抑制Na+内流的药如奎尼丁有此作用。
(四)改变ERP及APD而减少折返
药物对此约有三种可能的影响:
1.延长APD、ERP 但延长ERP更为显著,奎尼丁类药物能抑制Na+通道,使其恢复重新开放的时间延长,即延长ERP,这称绝对延长ERP。
一般认为ERP对APD的比值(ERP/APD)在抗心律失常作用中有一定意义,比值较正常为大,即说明在一个APD中ERP占时增多,冲动将有更多机会落入ERP中,折返易被取消。
2.缩短APD、ERP 但缩短APD更较显著,利多卡因类药物有此作用。因缩短APD更明显,所以ERP/APD比值仍较正常为大,这称相对延长ERP,同样能取消折返。
3.促使邻近细胞ERP的不均一(长短不一)趋向均一也可防止折返的发生。一般延长ERP的药物,使ERP较长的细胞延长较少,ERP较短者延长较多,从而使长短不一的ERP较为接近。反之亦然,缩短ERP的药物,使ERP短者,缩短少些;ERP长者,缩短多些。所以在不同条件下,这些药物都能发挥促使ERP均一的效应。
二、抗心律失常药物的分类
根据浦肯野纤维离体实验所得的药物电生理效应及作用机制,可将抗心律失常药分为四类,其中Ⅰ类药又分为A、B、C三个亚类。
(一)Ⅰ类——钠通道阻滞药
1.ⅠA类 适度阻滞钠通道,属此类的有奎尼丁等药。
2.ⅠB类 轻度阻滞钠通道,属此类的有利多卡因等药。
3.ⅠC类 明显阻滞钠通道,属此类的有氟卡尼等药。
(二)Ⅱ类——β肾上腺素受体阻断药
它们因阻断β受体而有效,代表性药物为普萘洛尔。
(三)Ⅲ类——选择地延长复极过程的药
它们延长APD及ERP,属此类的有胺碘酮。
(四)Ⅳ类——钙拮抗药
它们阻滞钙通道而抑制Ca2+内流,代表性药有维拉帕米。
上述分类法,既有助于进一步研究基础理论,也有助于临床学家了解药物对各种心律失常的效应和不良反应。
第三节 常用抗心律失常药
一、Ⅰ类药——钠通道阻滞药
(一)ⅠA类药物
它们能适度减少除极时Na+内流,降低0相上升最大速率,降低动作电位振幅,减慢传导速度。也能减少异位起搏细胞4相Na+内流而降低自律性。也延长钠通道失活后恢复开放所需的时间,即延长ERP及APD,且以延长ERP为显。这类药还能不同程度地抑制K+和Ca2+通道。
奎尼丁
奎尼丁(quinidine)是茜草科植物金鸡钠(Cinchonaledgeriana)树皮所含的一种生物碱,是奎宁的右旋体,它对心脏的作用比奎宁强5~10倍。金鸡钠制剂用于治疟,历史已久,1918年又发现一疟疾患者兼患的心房颤动也被治愈。以后的研究证明金鸡钠生物碱确有抗心律失常的作用,其中以奎尼丁为最强。
【药理作用】基本作用是与钠信道蛋白质相结合而阻滞之,适度抑制Na+内流,除这种对钠通道的直接作用外,奎尼丁还通过植物神经而发挥间接作用。
1.降低自律性治疗浓度奎尼丁能降低浦肯野纤维的自律性,对正常窦房结则影响微弱。对病窦综合征者则明显降其自律性。在植物神经完整无损的条件下,通过间接作用可使窦率增加。
2.减慢传导速度 奎尼丁能降低心房、心室、浦肯野纤维等的0相上升最大速率和膜反应性,因而减慢传导速度。这种作用可使病理情况下的单向传导阻滞变为双向阻滞,从而取消折返。
3.延长不应期 奎尼丁延长心房、心室、浦肯野纤维的ERP和APD(图22-5)。延长APD是其减慢减少K+外流所致,在心电图上表现为Q-T间期延长;ERP的延长更为明显,因而可以取消折返。此外,在心脏局部病变时,常因某些浦肯野纤维末梢部位ERP缩短,造成邻近细胞复极不均一而形成折返,此时奎尼丁使这些末梢部位ERP延长而趋向均一化,从而减少折返的形成。
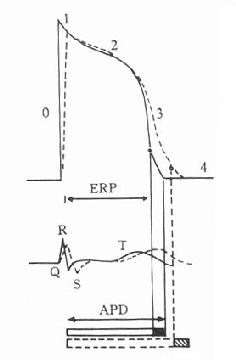
图22-5 奎尼丁对心室肌动作电位、单极电图(中)及ERP、APD影响的模式图
──为正常情况
……给奎尼丁后情况
4.对植物神经的影响动物实验见奎尼丁有明显的抗胆碱作用,阻抑迷走神经的效应。同时,奎尼丁还有阻断肾上腺素α受体的作用使血管舒张,血压下降而反射性兴奋交感神经。这两种作用相合,乃使窦性频率增加。
【体内过程】口服后吸收良好,经2小时可达血浆峰浓度。生物利用度为72%~87%。治疗血药浓度为3~6μg/ml,超过6~8μg/ml即为中毒浓度。在血浆中约有80%~90%与蛋白相结合,心肌中浓度可达血浆浓度的10倍。表现分布容积为2~4L/kg。在肝中代谢成羟化物,仍有一定活性,终经肾排泄。原形排泄约10%~20%,尿pH自7增至8,肾脏排泄下降50%,但因尿中原形者少,故尿碱化不会影响血药浓度。
【临床应用】奎尼丁是广谱抗心律失常药,适用于治疗房性、室性及房室结性心律失常。对心房纤颤及心房扑动,目前虽多采用电转律术,但奎尼丁仍有应用价值,转律前合用强心甙和奎尼丁可以减慢心室频率,转律后用奎尼丁维持窦性节律。预激综合征时,用奎尼丁可以中止室性心动过速或用以抑制反复发作的室性心动过速。
【不良反应】奎尼丁应用过程中约有1/3病员出现各种不良反应,使其应用受到限制。常见的有胃肠道反应,多见于用药早期,久用后,有耳鸣失听等金鸡纳反应及药热、血小板减少等过敏反应。
心脏毒性较为严重,治疗浓度可致心室内传导减慢(Q-Tc延长),延长超过50%表明是中毒症状,必须减量。高浓度可致窦房阻滞,房室阻滞,室性心动过速等,后者是传导阻滞而浦肯野纤维出现异常自律性所致。
奎尼丁治疗心房纤颤或心房扑动时,应先用强心甙抑制房室传导,否则可引起心室频率加快,因奎尼丁可使房性冲动减少而加强,反而容易通过房室结而下传至心室。
奎尼丁晕厥或猝死是偶见而严重的毒性反应。发作时患者意识丧失,四肢抽搐,呼吸停止,出现阵发性室上性心动过速,甚至心室颤动而死。认为这是过量时心室内弥漫性传导障碍及Q-Tc过度延长所致。也有治疗量对个别敏感者及过长Q-T综合征者所引起的尖端扭转型心律失常(室颤前室性心动过速torsadesde pointes)。后者是一种早后除极,发生机制可能与APD延长及复极的不均一有关。发作时宜立即进行人工呼吸,胸外心脏挤压,电除颤等抢救措施。药物抢救可用异丙肾上腺素及乳酸钠,后者提高血液pH值,能促K+进入细胞内,降低血钾浓度,减少K+对心肌的不利影响。同时,血液偏于碱性可增加奎尼丁与血浆蛋白的结合而减少游离奎尼丁的浓度,从而减低毒性。
【药物相互作用】药物代谢酶诱导剂苯巴比妥能减弱奎尼丁的作用。奎尼丁有α受体阻断作用,与其他血管舒张药有相加作用。合用硝酸
■[此处缺少一些内容]■
、粒细胞减少等。大量可致窦性停搏,房室阻滞。久用数月或一年,有10%~20%患者出现红斑性狼疮样综合征,其发生与肝中乙酰化反应的快慢有关,慢者容易发生。
丙吡胺
丙吡胺(disopyramide)的作用与奎尼丁相似。主要用于治疗室性早搏、室性心动过速,心房颤动和扑动。
主要不良反应是由较强的抗胆碱作用所引起,有口干、便秘、尿潴留、视觉障碍及中枢神经兴奋等。久用可引起急性心功能不全,宜慎用。禁用于青光眼及前列腺增生患者。
(二)ⅠB类药物
这类药物能轻度降低0相上升最大速率,略能减慢传导速度,在特定条件下且能促进传导;也能抑制4相Na+内流,降低自律性。由于它们还有促进K+外流的作用,因而缩短复极过程,且以缩短APD更较显着。
利多卡因
利多卡因(lidocaine)是局部麻醉药。现广用于静脉药治疗危及生命的室性心律失常。
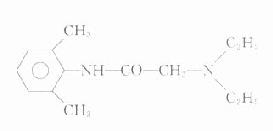
【药理作用】利多卡因对心脏的直接作用是抑制Na+内流,促进K+外流,但仅对希-浦系统发生影响,对其他部位心组织及植物神经并无作用。
1.降低自律性 治疗浓度(2~5μg /ml)能降低浦肯野纤维的自律性,对窦房结没有影响,仅在其功能失常时才有抑制作用。由于4相除极速率下降而提高阈电位,又能减少复极的不均一性,故能提高致颤阈。
2.传导速度利多卡因对传导速度的影响比较复杂。治疗浓度对希-浦系统的传导速度没有影响,但在细胞外K+浓度较高时则能减慢传导。血液趋于酸性时将增强其减慢传导的作用。心肌缺血部位细胞外K+浓度升高而血液偏于酸性,所以利多卡因对之有明显的减慢传导作用,这可能是其防止急性心肌梗塞后心室纤颤的原因之一。对血K+降低或部分(牵张)除极者,则因促K+外流使浦肯野纤维超极化而加速传导速度。大量高浓度(10μg/ml)的利多卡因则明显抑制0相上升速率而减慢传导。
3.缩短不应期 利多卡因缩短浦肯野纤维及心室肌的APD、ERP,且缩短APD更为显著,故为相对延长ERP(图22-6)。这些作用是阻止2相小量Na+内流的结果。
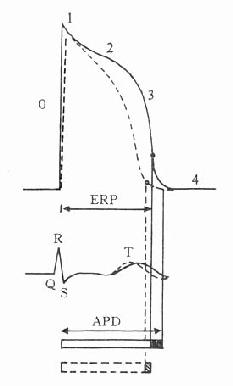
图22-6利多卡因对心室肌动作电位、单极电图(中)及ERP、APD影响的模式图
——为正常情况
……为给利多卡因后情况
【体内过程】口服吸收良好,但肝首关消除明显,仅1/3量进入血液循环,且口服易致恶心呕吐,因此常静脉给药。血浆蛋白结合率约70%,在体内分布广泛,表现分布容积为1L/kg,心肌中浓度为血药浓度的3倍。在肝中经脱乙基化而代谢。仅10%以原形经肾排泄,t1/2约2小时,作用时间较短,常用静脉滴注法。
【临床应用】利多卡因是一窄谱抗心律失常药,仅用于室性心律失常,特别适用于危急病例。治疗急性心肌梗塞及强心甙所致的室性早搏,室性心动过速及心室纤颤有效。也可用于心肌梗塞急性期以防止心室纤颤的发生。
【不良反应】较少也较轻微。主要是中枢神经系统症状,有嗜睡、眩晕,大剂量引起语言障碍、惊厥,甚至呼吸抑制,偶见窦性过缓、房室阻滞等心脏毒性。
苯妥英钠
苯妥英钠(phenytoinsodium)原为抗癫痫药。50年代初发现其有抗心律失常作用,1958年以其治疗耐奎尼丁的室性心动过速获得成功。
【药理作用】与利多卡因相似,也仅作用于希-浦系统。
1.降低自律性抑制浦表野纤维自律性,也能抑制强心甙中毒时迟后除极所引起的触发活动,大剂理才抑制窦房结自律性。
2.传导速度 作用也较复杂,随用药剂量、细胞外K+等因素而异。正常血K+时,小量苯妥英钠对传导速度无明显影响,大剂量则减慢之;低血K+时小量苯妥英钠能加快传导速度,当静息膜电位较小时(强心甙中毒、机械损伤之心肌),加快传导更为明显。
3.缩短不应期 此作用与利多卡因相似。
【体内过程和不良反应】参见第十五章抗癫痫药。
【临床应用】用于治疗室性心律失常,对强心甙中毒者更为有效,其特点是改善被强心甙所抑制的房室传导。对心肌梗塞、心脏手术、麻醉、电转律术、心导管术等所引发的室性心律失常也有效。
美西律
美西律(mexiletine)化学结构与利多卡因相似。对心肌电生理特性的影响也与利多卡因相似。可供口服,持效较久达6~8小时以上,用于治疗室性心律失常,特别对心肌梗塞急性期者有效。不良反应有恶心、呕吐,久用后可见神经症状,震颤、眩晕、共剂失调等。
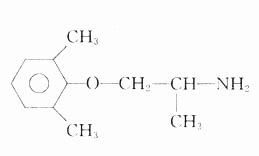
妥卡尼
妥卡尼(tocainide)是利多卡因脱去二个乙基加一个甲基而成。作用及应用与利多卡因相似,然口服有效,也较持久。不良反应与美西律相似。
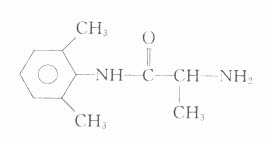
(三)ⅠC类药物
这类药物阻滞钠通道作用明显,能较强降低0相上升最大速率而减慢传导速度,主要影响希-浦系统;也抑制4相Na+内流而降低自律性;对复极过程影响很少。近年报道这类药有致心律失常作用,增高病死率,应予注意。
氟卡尼
氟卡尼(flecainide)抑制希-浦系统的传导速度,降低自律性。能缩短其APD、对ERP则低浓度时缩短,增加浓度又恢复正常。能减慢心室肌的传导,延长其ERP、APD。这种对浦肯野纤维和心肌ERP、APD作用的不同可能是据氟卡尼的致心律失常作用的基础。
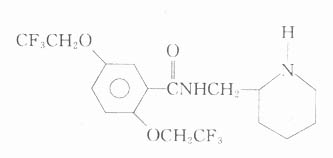
氟卡尼吸收迅速完全,生物利用度约90%,t1/2约20小时(12~27小时)。
氟卡尼用于治疗室性早搏、室性心动过速收到良好效果。但已有治疗心肌梗塞后心律失常的报道称:氟卡尼引起病死率为对照组的2倍。故现认为氟卡尼及恩卡尼应保留用于危及生命的室性心动过速者,不宜用于其他心律失常。
同类药物恩卡尼(encainide)与氟卡尼作用、应用相似。t1/2约4小时。
普罗帕酮
普罗帕酮(propafenone)也主要作用于希-浦系统,降低自律性,减慢传导速度,延长APD、ERP,且减慢传导的程度超过延长ERP的程度,故易引起折返而有致心律失常的作用。也宜限用于危及生命的心律失常。
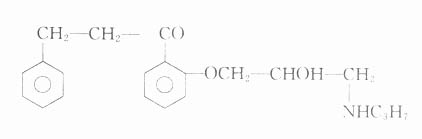
普罗帕酮还有β受体阻断作用,能在治疗上发挥一定的效果。
普罗帕酮口服吸收完全,达100%,但生物利用度却低于20%,首关消除效应明显,t1/2约2.4~11.8小时,肝中氧化甚多,原形经肾排泄小于1%。
不良反应有胃肠道症状,偶见粒细胞缺乏,红斑性狼疮样综合征。心电图QRS波加宽超过20%以上或Q-T间期明显延长者宜减量或停药,否则可致心律失常。
二、Ⅱ类药——β肾上腺素受体阻断药
这类药物主要阻断β受体而对心脏发生影响,同时还有阻滞钠信道、促进钾信道、缩短复极过程的效应。表现为减慢窦房结、房室结的4相除极而降低自律性;也能减慢0相上升最大速率而减慢传导速度;某些β受体阻断药能缩短APD和ERP,且以缩短APD为显着;某些药在高浓度时还有膜稳定作用。这类药物的一般药理内容见第十一章,本节只述及其抗心律失常方面的内容,鉴于普萘洛尔是这类药的典型药,就以它为代表介绍如下。
普萘洛尔
【药理作用】交感神经兴奋或儿茶酚胺释放增多时,心肌自律性增高,传导速度增快,不应期缩短,易引起快速性心律失常。普萘洛尔则能阻止这些反应。
1.降低自律性对窦房结、心房传导纤维及浦肯野纤维都能降低自律性。在运动及情绪激动时作用明显。也能降低儿茶酚胺所致的迟后除极幅度而防止触发活动。
2.传导速度阻断β受体的浓度并不影响传导速度。超过此浓度使血药浓度达100ng/mg以上,则有膜稳定作用,能明显减慢房室结及浦肯野纤维的传导速度,对某些必须应用大量才能见效的病例,这种膜稳定作用是参与治疗的。
3. 不应期 治疗浓度缩短浦肯野纤维APD和ERP,高浓度则延长之。对房室结ERP有明显的延长作用,这和减慢传导作用一起,是普萘洛尔抗室上性心律失常的作用基础。
【临床应用】这类药物适于治疗与交感神经兴奋有关的各种心律失常。
1.室上性心律失常包括心房颤动、扑动及阵发性室上性心动过速,此时常与强心甙合用以控制心室频率,二者对房室结传导有协同作用。也用于治疗由焦虑或甲状腺功能亢进等引发的窦性心动过速。
2.室性心律失常对室性早搏有效,能改善症状。对由运动或情绪变动所引发的室性心律失常效果良好。较大剂量(0.5~1.0g/日)对缺血性心脏病患者的室性心律失常也有效。
其他对心脏具有相对选择性或有膜稳作用的β受体阻断药如醋丁洛尔(acebutolol)治疗室性早搏,室性心动速效果良好。
三、Ⅲ类药——延长APD的药物
这类药物能选择性地延长APD,主要是延长心房肌、心室肌和浦肯野纤维细胞的APD和ERP,而少影响传导速度。
胺碘酮
胺碘酮(amiodarone)分子中含2个碘原子,占其分子量的37.2%。
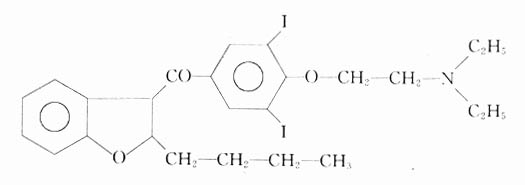
【药理作用】胺碘酮较明显地抑制复极过程,即延长APD和ERP。它能阻滞钠、钙及钾通道,还有一定的α和β受体阻断作用。
1.自律性主要降低窦房结和浦肯野纤维的自律性,可能与其阻滞钠和钙通道及拮抗β受体的作用有关。
2.传导速度减慢浦肯野纤维和房室结的传导速度,也与阻滞钠、钙通道有关。临床还见其略能减慢心室内传导。对心房肌的传导速度少有影响。
3. 不应期 长期口服数周后,心房肌、心室肌和浦肯野纤维的APD、ERP都显著延长,这一作用比其他类抗心律失常药为强,与阻滞钾通道及失活态钠通道有关。
【体内过程】口服吸收缓慢,生物利用度约40%~50%,血浆蛋白结合率为95%,广泛分布于组织中,尤以脂肪组织及血流量较高的器官为多,表现分布容积高达66L/kg。
胺碘酮几乎全部在肝中代谢,初步代谢成脱乙基物,仍属有效,且其阻滞钠通道的作用较强。而胺碘酮则阻滞钙通道的作用较强。长期口服后t1/2平均约40天,全部清除需时4个月。主要经胆汁由肠道排泄,经肾排泄者仅1%,故肾功能减退者不需减量应用。
【临床应用】是广谱抗心律失常药,可用于各种室上性和室性心律失常,如用于心房颤动可恢复及维持窦性节律,治疗阵发性室上性心动过速也有效。对危及生命的室性心动过速及心室颤动可静脉给药,约对40%患者有效。
长期口服能防止室性心动过速和心室颤动的复发,持效较久。对伴有器质性心脏病者,还能降低猝死率。
【不良反应】表现多方面,可引起甲状腺功能亢进或低下,见于约9%的用药者,且能竞争心内甲状腺素受体,这与其抗心律失常作用有一定关系。胺碘酮也影响肝功能,引起肝炎;因少量自泪腺排出,故在角膜可有黄色微型沉着,一般并不影响视力,停药后可自行恢复;胃肠道反应有食欲减退,恶心呕吐、便秘;另有震颤及皮肤对光敏感,局部呈灰蓝色;最为严重的是引起间质性肺炎,形成肺纤维化。静脉注射可致心律失常或加重心功能不全。
与Ⅰ、Ⅱ、Ⅳ类抗心律失常药合用可能互相增强作用,引起窦性心动过缓,甚至停博。
自从明确氟卡尼等ⅠC类药物治疗室性心律失常引起病死率较对照组为高后,胺碘酮的应用重受重视。
索他洛尔(sotalol)原为β受体阻断药,后因明显延长APD而用作Ⅲ类抗心律失常药。它能降低自律性,是其阻断β受体的作用所致。减慢房室结传导。明显延长ERP,使折返激动停止。也延长APD,是阻滞K+通道所致。
索他洛尔口服吸收快,生物利用度达100%,t1/2约10~15小时,几乎全部以原形经肾排出,肾功能不良者宜减量应用。
临床用于各种严重程度的室性心律失常。也治疗阵发性室上性心动过速及心房颤动。
不良反应较少,但有因出现心功能不全(1%)、心律失常(2.5%)、心动过缓(3%)而停药者。少数Q-T间期延长者偶可出现尖端扭转型室性心动过速。
四、Ⅳ类药——钙拮抗药
这类药通过阻滞钙通道而发挥抗心律失常效应,其电生理效应主要是抑制依赖于钙的动作电位与减慢房室结的传导速度。这类药的其他药理学内容详见第二十一章。
维拉帕米
【抗心律失常作用】
1.自律性离体实验中,维拉帕米能降低窦房结起搏细胞的自律性。整体中此效应被反射性的交感神经兴奋所部分抵消,人体窦性频率减慢约10%~15%。对因病变而膜电位减为-60~-40mV的心房肌、心室肌及浦肯野纤维的异常自律性也能降低。此外,也能减少或取消后除极所引发的触发活动。
2.传导速度减慢窦房结和房室结的传导速度。在窦房结中对主导起搏细胞的作用强于对潜在起搏细胞。在房室结中对上部、中部的作用强于对下部。
3,不应期 延长慢反应动作电位的ERP,因维拉帕米阻滞钙通道而延长其恢复开放所需的时间。由于Ca2+内流也参与快反应电活动的复极过程,所以维拉帕米较高浓度也能延长浦肯野纤维的APD和ERP。
【临床应用】维拉帕米治疗房室结折返所致的阵发性室上性心动过速奏效较快较佳,能使80%以上患者转为窦性节律,可作首选药物应用。治疗心房颤动或扑动则能减少室性频率。对房性心动过速也有良好效果。对室性心律失常虽也有效,但与其他药物相比并无特别优越性,因而少用。对缺血复灌后所发生的心律失常也有防止及取消的效果,这是通过其钙拮抗作用和α受体阻断作用所取得的。
维拉帕米一般不与β受体阻断药合用。对窦房结疾病、房室阻滞及严重心功能不全者应慎用或禁用。
其他用于抗心律失常的钙拮抗药还有:
地尔硫![]() ,其电生理作用与维拉帕米相似,以房室传导有明显抑制作用。口服起效较快,可用于阵发性室上性心动过速。治心房颤动可使心室频率减少。
,其电生理作用与维拉帕米相似,以房室传导有明显抑制作用。口服起效较快,可用于阵发性室上性心动过速。治心房颤动可使心室频率减少。
苄普地尔,作用类似维拉帕米,能延长心房肌、心室肌的ERP,延长心电图中Q-Tc间期,用于治疗房室结性折返型心动过速等。
第四节 快速型心律失常的药物选用
选用抗心律失常药物应考虑多种因素,包括心律失常的类别,病情的紧迫性,患者的心功能及医师对各个药物的了解及应用经验等。药物治疗最满意的效果是恢复并维持窦性节律,其次是减少或取消异位节律,再次是控制心室频率,维持一定的循环功能。
各种快速型心律失常的选药如下:
1.窦性心动过速 应针对病因进行治疗,需要时选用β受体阻断药,也可选用维拉帕米。
2.心房纤颤或扑动转律用奎尼丁(宜先给强心甙),或与普萘洛尔合用,预防复发可加用或单用胺碘酮,控制心室频率用强心甙或加用维拉帕米或普萘洛尔。
3.房性早搏必要时选用普萘洛尔,维拉帕米,胺碘酮,次选奎尼丁,普鲁卡因胺,丙吡胺。
4.阵发性室上性心动过速除先用兴奋迷走神经的方法外,可选用维拉帕米、普萘洛尔、胺碘酮、奎尼丁、普罗帕酮。
5.室性早搏必要时首选普鲁卡因胺、丙吡胺、美西律、妥卡尼、胺碘酮,急性心肌梗塞时宜用利多卡因,强心甙中毒者用苯妥英钠。
6.阵发室性心动过速选用利多卡因、普鲁卡因胺、丙吡胺、美西律、妥卡尼等。
7.室纤颤 选利多卡因、普鲁卡因胺(可心腔内注射)。
表22-1 常用抗心律失常药应用比较
| 心律失 常 | 奎尼丁 | 丙吡胺 | 利多卡因 | 苯妥英钠 | 氟卡尼 | 普萘洛尔 | 胺碘酮 | 维拉帕米 |
| 房颤,转律 | 2 | 1 | 1 | 2 | 1 | |||
| 预防 | 3 | 3 | 2 | 2 | ||||
| 控制室率 | 2 | 3 | ||||||
| 阵发室上性心动过速 | 2 | 2 | 1 | 3 | 2 | 4 | ||
| 房性早搏 | 3 | 3 | 1 | 3 | 2 | 2 | ||
| 室性早搏 | 3 | 3 | 4 | 2 | 2 | 1 | 2 | 2 |
| 室性心动过速 | 3 | 2 | 3 | 2 | 2 | 1 | 1 | 1 |
| 强心甙中毒时的 | ||||||||
| 各种心律失常 | 1 | 1 | 3 | 3 | 2 |
*根据各药的效价,不良反应及应用方便等作出比较
0=不用;1=差;2=可;3=良;4=优
[附]抗心律失常药的致心律失常作用
抗心律失常药既能抗心律失常也能导致心律失常,后者即为药物的“致心律失常(proarrhythmia)作用”,它包含引起新的心律失常和加重原有的心律失常二种情况。近年报道IC类氟卡尼等药有致心律失常作用并增加病死率后,这一问题更受到广泛重视。基础与临床研究正在开展,以下介绍有关其发生机制的某些内容。
冲动形成障碍和冲动传导障碍也是药致心律失常的电生理机制。在前者,主要有早后除极的触发活动,如IA类药物延长APD与复极,容易引起2.3相中电位振荡。又如尖端扭转型室性心动过速是一种早后除极,伴有复极高度延长,IA类和Ⅲ类药可能引发。在冲动传导障碍方面,药物可因减慢或抑制传导而引起折返,加Ⅰ、Ⅲ类药可能引发之,其中ⅠB类因与结合部位解离迅速而较少引起。在慢反应细胞中,则Ⅳ类药能抑制或减慢传导。
由于药物的致心律失常作用,近年临床应用抗心律失常药已更趋慎重。对急性心肌梗塞而无严重心律失常者,建议不用Ⅰ类药物,必要时选用Ⅱ类药。对一般心律失常也以少用ⅠA、ⅠC类为宜。
抗心律失常药 制剂与用法
硫酸奎尼丁(quinidine sulfate)心房纤颤或心房扑动:先口服0.1g,观察一天。如无不良反应,次日每隔2~4小时一次,0.2g/次,连续5次。如第一日心律未转为窦性,且无明显毒性反应,第二日用0.3g/次,每2小时1次,连续5次,仍未转为窦性可再服一日,然后改为0.4g/次。每日剂量不宜超过2g。转为窦性后,可用维持量,每6小时一次。0.2g/次,2~3次/日。频率发作过早搏动:0.2g/次,3~4次/日。极量:0.6g/次,3g/日,口服。
盐酸普鲁卡因胺(procainamide hydrochloride)口服首剂0.5~1g/次,以后0.25~0.5g/次,4次/日,心律正常后渐减至0.25g/次,2~3次/日。肌内注射:0.5g/次。静脉滴注:0.5~1g/次,用5%葡萄糖液200ml稀释。每分钟1~2ml,应注意心电图变化。无效可重复一次。极量:1g/次,3g/日,口服。
丙吡胺(disopyramid1e)100mg/次,3次/日,口服。
盐酸利多卡因(lidocaine hydrochloride)先以50~100mg/次或1~2mg/kg/次,静脉注射,见效后可改为100mg以5%葡萄糖液100~200ml稀释后静脉滴注,每分钟1~2ml。
苯妥英钠(phenytoin sodium)口服0.1~0.2g/次,2~3次/日。0.25g/次,以注射用水20~40ml稀释,于6~10分钟注完,静脉注射速度小于25~50mg/分钟为宜,必要时5~10分钟后再静脉注射0.1g,直至心律纠正或总量达0.5g为止。3~5mg/kg/次,隔4~6小时一次,肌内注射。
美西律(mexiletine)口服首剂400~600mg,维持量150~300mg,6~8小时给药1次。静脉注射:开始量30~250mg或75~300mg/5~30分钟内注完,然后静脉滴注1.5~3g/36~48小时。
盐酸妥卡尼(tocainide hydrochloride)0.4~0.6g/次,3次/日,口服。
氟卡尼(flecainide)口服200mg/次,2~3次/日。静脉注射1~2mg/kg,缓慢注入。
普罗帕酮(propafenone)口服150mg/次,2~4次/日,维持量150mg/次,2次/日。必要时静脉注射,每8小时1次,1~2mg/kg。
盐酸普萘洛尔(propranolol hydrochloride)抗心律失常:口服,10~20mg/次,3次/日。1~3mg/次,以5%葡萄糖液100ml稀释,静脉滴注,按需要调整滴注速度。
胺碘酮(amiodarone)口服开始时200mg/次,3次/日。维持量为100mg/次,3次/日。静脉注射,300~450mg/日。或静脉滴注,300mg加至250ml等渗盐水中,于30分钟内滴完。
索他洛尔(sotalol)口服80~320mg/次,2次/日。
维拉帕米(verapamil)口服40~80mg/次,3次/日。静脉注射5~10mg/次,5~10分钟内缓慢注入,2~3次/日。
地尔硫卓(diltiazem)口服30mg/次,3~4次/日,不可嚼碎。静脉注射0.1~0.3mg/kg.
第二十三章 抗慢性心功能不全药
慢性心功能不全又称充血性心力衰竭(congestive heart failure,CHF),是一种多原因多表现的“超负荷心肌病”。在血流动力学方面表现为心脏不能射出足量血液以满足全身组织的需要。心功能受几种生理因素的影响,如心收缩性,心率、前、后负荷及心肌氧耗量等。CHF时收缩性减弱,心率加快,前后负荷增高,氧耗量增加。
近年发现CHF时既有心调节机制的变化,也有心β-肾上腺素受体信息转导系统的变化。
1.交感神经系统激活这是CHF发病过程中早期的代偿机制,是一种快速调节。患者交感神经活性增高,血中去甲肾上腺素浓度升高,从而使心肌收缩性增高,心率加快,血管收缩以维持血压,这都起到代偿作用。久后心肌氧耗量增加,后负荷增加,心工作加重,反使病情恶化,形成恶性循环。
2.肾素-血管紧张素-醛固酮系统(RAAS)激活这一系统对循环的调节较为缓慢。症状明显的患者血浆肾素活性升高,血中血管紧张素Ⅱ(ATⅡ)含量升高。RAAS的激活将强烈收缩血管,久之也将造成恶性循环。醛固酮增多促进水肿,ATⅡ还能促进去甲肾上腺素的释放,加重发病过程。
3.精氨酸加压素分泌增加轻症患者血中精氨酸加压素浓度已有升高,能促使外周血管收缩,既有利地维持血压,又不利地恶化病情,可能参与了CHF晚期的发病过程。
4.其他内源性调节的变化心房排钠因子(ANF)有排钠利尿、扩张血管、拮抗RAAS活性等作用。轻度、重度患者血中ANF含量增多,可能有缓解病情的功效。前列腺素E2、I2也是重要的内源性血管扩张物质,在CHF患者血中其浓度增高,也起到缓解发病过程的作用。内皮依赖性松弛因子(EDRF),即一氧化氮(NO),能明显扩张血管。实验性心衰犬体内EDRF减少。内皮素在CHF患者体内含量增多,可能参与血管收缩过程,但二者确切的发病学意义,尚待研究。
5.心细胞β1-受体的密度下降。CHF患者心肌细胞的β1-受体由占心肌肾上腺素受体的70%~80%降为50%,即β1-受体下调,这是受体长期与较高浓度去甲肾上腺素相接触的结果,也是使心免受过量Ca2+负荷之害的一种保护机制。CHF时β1-受体与G蛋白脱偶联,兴奋性Gs量减少,抑制性Gi量增多,同时腺苷酸环化酶活性下降,细胞内cAMP含量减少,但G蛋白和腺苷酸环化酶的变化是原发还是继发也待研究。
从上述多种调节机制和β1-受体信息转导系统的变化来看,现较重视CHF发病中的神经内分泌因素。治疗上除用正性肌力药加强收缩性,用扩管药及利尿药降低前、后负荷外,现也注意用血管紧张素Ⅰ转化酶抑制药(ACEI)以纠正RAAS的激活,取得较好的治疗效果。
本章将分别介绍强心甙,非甙类正性肌力作用药,血管舒张药及ACEI在CHF中的应用,利尿药则详见第二十七章。
第一节 强心甙
强心甙(cardiacglycosides)是一类有强心作用的甙类化合物,它能选择性地作用于心肌。临床上用于治疗CHF及某些心律失常。
【来源及化学】强心甙来源于植物如紫花洋地黄和毛花洋地黄,所以又称洋地黄类(digitalis)药物。常用的有地高辛(digoxin)和洋地黄毒甙(digitoxin)。
强心甙由糖和甙元结合而成。甙元由甾核与不饱和内酯环构成,糖的部分除葡萄糖外,都是稀有的糖如洋地黄毒糖等。甾核上C3、C14、C17位都有重要取代基。C3位β构型的羟基是甾核与糖相结合的位点,脱糖后此羟基转为α构型,甙元即失去作用;C14必有一个β构型羟基,缺此则甙元失效;C17联结β构型的不饱和内酯环,此环若是饱和或被打开,就会减弱或取消甙元作用。
强心甙加强心肌收缩性的作用来自甙元,糖则能增强甙元的水溶性,延长其作用,一般以三糖甙作用最强。
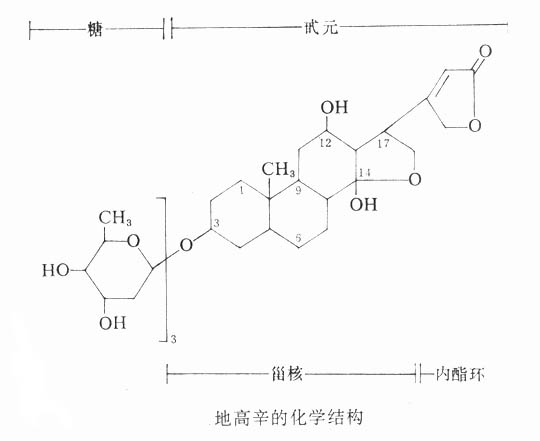
【药理作用】
1.正性肌力作用(positiveinotropic action)即加强心肌收缩性,这是选择性对心肌细胞的作用,可见于离体乳头状肌及体外培养细胞的实验中,这一作用是剂量依赖性的,对心房和心室,对正常心和已衰心都有效。
正性肌力作用表现为心肌收缩最高张力和最大缩短速率的提高,使心肌收缩有力而敏捷。这样,在前后负荷不变的条件下,心每搏作功增加,搏出量增加。
从心动周期中左心室压力与容积的关系看,也能证实这一作用,衰竭心的压力容积环明显右移上移,说明其收缩末和舒张末容积都增大,等容收缩时压力发展较慢,搏出量减少。给予强心甙后则见压力积环左移下移,舒张期压力与容积都下降,搏出量增加。
强心甙对正常人和CHF患者心都有正性肌力作用,但它只增加患者心的搏出量而不增加正常心的搏出量。因为强心甙对正常人还有收缩血管提高外周阻力的作用,由此限制了心搏出量的增加。然而在CHF患者中,通过反射作用,强心甙已降低了交感神经活性,因而这一收缩血管作用难以发挥,使搏出量得以增加。
强心甙对心肌氧耗量的影响也随心功能状态而异。对正常心因加强收缩性而增加氧耗量,对CHF患者,因心脏原已肥厚,室壁张力也已提高,需有较多氧耗以维持较高的室壁张力。强心甙的正性肌力作用能使心体积缩小,室壁张力下降,乃使这部分氧耗降低,降低部分常超过收缩性增加所致的氧耗增加部分,因此总的氧耗有所降低。
2.负性频率作用(negativechronotropic action)即减慢窦性频率,对CHF而窦律较快者尤为明显。这一作用由强心甙增强迷走神经传出冲动所引起,也有交感神经活性反射性降低的因素参与。这主要是增敏窦弓压力感受器的结果。因CHF时感受器细胞Na+-K+-ATP酶活性增高,使胞内多K+,呈超极化,细胞敏感性降低,窦弓反射失灵,乃使交感神经及RAAS功能提高。强心甙直接抑制感受器Na+-K+-ATP酶,敏化感受器,恢复窦弓反射。得以增强迷走神经活性,并降低交感神经活性。
减慢窦性频率对CHF患者是有利的,它使心有较好休息、获得较多的冠状动脉血液供应,又使静脉回心血量更充分而能搏出更多血液。但减慢窦性频率并非强心甙取得疗效的必要条件,临床上常在心率减慢之前或心率并不减慢的情况下,见到强心甙的治疗效果,如水肿减轻及呼吸急促的缓解等。
图23-1 哇巴因对猫心乳头肌的正性肌力作用
图23-2 强心甙对压力容积环的影响
N 正常心;F衰心;D给强心甙后;
1 舒张期; 2心房收缩;3等容收缩;
4 射血时;5等容舒张
3.对电生理特性的影响这些影响比较复杂,它有直接对心肌细胞和间接通过迷走神经等作用之分,还随剂量高低、不同心组织及病变情况而有不同,兹将其主要电生理作用的总效应列表如下:
表23-1 强心甙对心肌的电生理作用
| 电生理特性 | 窦房结 | 心 房 | 房室结 | 浦肯野纤维 |
| 自律性 | ↓ | ↑ | ||
| 传导性 | ↓ | |||
| 有效不应期 | ↓ | ↓ |
治疗量强心甙加强迷走神经活性而降低窦房结自律性,因迷走神经加速K+外流,能增加最大舒张电位(负值更大),与阈电位距离加大,从而降低自律性。与此相反,强心甙能提高浦肯野纤维的自律性,在此迷走神经影响很小,强心甙直接抑制Na+-K+-ARP酶的作用发挥主要影响,结果是细胞内失K+,最大舒张电位减弱(负值减少),与阈电位距离缩短,从而提高自律性。
强心甙减慢房室结传导性是加强迷走神经活性减慢Ca2+内流的结果,慢反应电活动的房室结的除极是Ca2+内流所介导的。
强心甙缩短心房不应期也由迷走神经促K+外流所介导。缩短浦肯野纤维有效不应期是抑制Na+-K+-ATP酶,使细胞内失K+,最大舒张电位减弱,除极发生在较小膜电位的结果。
4.对心电图的影响 治疗量强心甙最早引起T波变化,其幅度减小,波形压低甚至倒置,S-T段降低呈鱼钩状,随后还见P-R间期延长,反映房室传导减慢,Q-T间期缩短,反映浦肯野纤维和心室肌ERP和APD缩短。P-P间期延长则是窦性频率减慢的反映。
中毒量强心甙会引起各种心律失常,心电图也会出现相应变化。
5.对其他系统的作用①对血管:强心甙能使动脉压升高,外周阻力上升,此作用与交感神经、肾上腺及输出量的变化无关,说明是直接收缩血管平滑肌所致。已证明强心甙能收缩下肢、肠系膜血管及冠状血管等。正常人用药后血管阻力升高约23%,局部组织血流减少。CHF患者用药后,因交感神经活性降低,其影响超过直接收缩血管的效应,因此血管阻力下降,心输出量及组织灌流增加,动脉压不变或略升。②对肾:CHF患者用强心甙后利尿明显,是正性肌力作用使肾血流增加所继发的。对正常人或非心性水肿患者也有轻度利尿作用,是抑制肾小管细胞Na+-K+-ATP酶,减少肾小管对Na+的再呼吸的结果。③对神经系统:中毒量可兴奋延脑极后区催吐化学感受区而引起呕吐。严重中毒时还引起中枢神经兴奋症状,如行为失常、精神失常、谵安甚至惊厥。中毒量强心甙还明显增强交感神经的活性,有中枢和外周两方面影响。这也参与了中毒量所致的心律失常的发病过程。
【正性肌力作用机制】三方面因素决定着心肌收缩过程,它们是收缩蛋白及其调节蛋白;物质化谢与能量供应;兴奋-收缩偶联的关键物质Ca2+。已证明强心甙对前二方面并无直接影响,却能增加兴奋时心肌细胞内Ca2+量,并认为这是强心甙正性肌力作用的基本机制。
从原发作用部位的亚细胞或分子结构看,强心甙只与细胞膜上Na+-K+-ATP酶相结合并抑制之。已认为Na+-K+-ATP酶就是强心甙的受体,它是一个二聚体,由α和β亚单位组成。α亚单位是催化亚单位,贯穿膜内外两侧,分子量112000D,约含1021个氨基酸残基。β亚单位是一糖蛋白,分子量约35 000D,可能与α亚单位的稳定性有关。
现知α亚单位有8个疏水性跨膜α-螺旋段,H1~H8,分属于N端和C端1/3,所余中央1/3则折叠成巨大的胞溶部结构域,其中包含ATP结合水解部位501位赖氨酸,ATP水解成的磷酸则结合于369位天冬氨酸。
强心甙与酶的结合位点,曾认为在N端H1-H2间的胞外袢上,但未能最后确定,仅知此胞外袢能影响结合过程中的构象变化,使酶活性下降。体内条件下,治疗量强心甙抑制Na+-K+-ATP酶活性约20%,使钠泵失灵,结果是细胞内Na+量增多,K+量减少。胞内Na+量增多后,再通过Na+-Ca2+双向交换机制,或使Na+内流减少,Ca2+外流减少,或使Na+外流增加,Ca2+内流增加。对Ca2+而言,结果是细胞内Ca2+量增加,肌浆网摄取Ca2+也增加,储存增多。另也证实,细胞内Ca2+少量增加时,还能增强Ca2+离子流,使每一动作电位2相内流的Ca2+增多,此Ca2+又能促使肌浆网释放出Ca2+,即“以钙释钙”的过程。这样,在强心甙作用下,心肌细胞内可利用的Ca2+量增加,使收缩加强。
在多种条件下,强心甙的正性肌力与Na+-K+-ATP酶的抑制之间显示了平行关系:如细胞内Na+增加,能使两种作用的发生速率都加快;细胞外K+增加则降低两作用的发生速率;减少细胞外K+使两种作用都能延长;另见强心甙对不同种类动物的这两种作用在强度上也有差异,然二种作用的差异也是相符的。这些平行关系为上述作用机制提供了有力气的支持。
中毒量强心甙严重抑制Na+-K+-ATP酶,使细胞内Na+、Ca2+大量增加,也使细胞内K+量明显减少,后者导致心细胞自律性增高,传导减慢,容易引起心律失常。
【体内过程】常用的地高辛和洋地黄毒甙的作用性质基本相同,但因药代动力学性状有别,使作用程度上有快慢、久暂之分。洋地黄毒甙仅在C14位有一极性基团羟基,其极性低而脂溶性高,所以口服吸收率较高,原形经肾排泄较少。地高辛在C12、C14位各有一羟基,极性略高,所以口服吸收率略差,原形经肾排泄略多。
1.吸收洋地黄毒甙口服吸收稳定完全,其生物利用度高达100%,地高辛生物利用度约60%~80%,个体差异显著。不同片剂产品的吸收率差异更大,变动在20%~80%之间,这与地高辛原料颗粒大小有关。颗粒小溶出度高,吸收率高,反之则低。经改进制备工艺中颗粒体积后,其生物利用度已经提高,差异缩小,中国药典规定地高辛片剂一小时的溶出度不得低于65%。强心甙口服吸收后,部分经肝与胆管排入肠道而被再吸收,形成肝肠循环。洋地黄毒甙肝肠循环较多,与其作用持久有一定关系。
图23-3 Na+-K+-ATP酶结构模式图 α与β亚单位
注:1-8 H1-H8螺旋段;Oua 哇巴因结合位;ATP ATP结合位;
P磷酸化位; Na+胞内Na+结合位; K+胞外K+结合位。
2.分布强心甙进入血液后可与血浆蛋白发生可逆性结合而分布全身。洋地黄毒甙结合较多,在肾、心、骨骼肌与血清中的浓度比为8.7:5.4:2.9:1。地高辛结合较少,分布于各组织中,以肾内浓度最高,心、骨骼肌中次之。
图23-4 强心甙作用机制示意图
3.代谢转化泮地黄毒甙脂溶性较高,易进入肝细胞,代谢较多。它可经P450氧化脱糖成甙元,再在C3位转为α构型而失效;部分在C12位被羟基化转化成地高辛仍属有效,在人体中此转化约占总代谢量的8%;又有部分甙元的不饱和内酯环被氢化成饱和环而降低效应;代谢产物最终与葡萄糖醛酸或硫酸结合而经肾排泄。地高辛的代谢转化较少,主要被氢化成二氢地高辛,继而再被脱糖,内酯环氢化,与葡萄糖醛酸结合而经肾外排。二氢地高辛的生成有赖于肠道细菌Eubact.lentum的存在,红霉素、四环素等能抑制肠菌,减少二氢地高辛的生成,具有提高地高辛血药浓度的效应。
4.排泄洋地黄毒甙排泄缓慢,是它作用持久的主要原因。它的代谢产物多数经肾,少量经肠道排出。少量原形物也经肾排泄。地高辛经肾小球过滤,部分也经肾小管分泌排出,每日可排出体内量的1/3。
5.影响药代动力学的因素强心甙的小儿用量,按体重计,较成人高。地高辛维持量2岁以下儿童为0.015~0.02mg/kg,2岁以上为0.01~0.015mg/kg,因儿童排泄较多,血浆蛋白结合率较低,分布容积较大。老年人用量以少于成年人20%~30%为宜。地高辛维持量为0.125~0.2mg,因老年人肾排泄少,分布容积小,血浓较高。
表23-2 两种强心甙的药代动力学参数与用量
| 洋地黄毒甙 | 地高辛 | 洋地黄毒甙 | 地高辛 | ||
| 口服吸收% | 90~100 | 60~85 | 给药法 | 口服 | 口服 |
| 蛋白结合% | 97 | 25 | 作用开始(h) | 2 | 1~2 |
| 肝肠循环% | 26 | 7 | 达峰浓度(h) | 8~12 | 4~8 |
| 原形肾排泄% | 10 | 60~90 | 毒性消失(天) | 3~7 | 1~2 |
| 代谢转化% | 70 | 20 | 作用完全消失 | 2~3周 | 3~6天 |
| 分布容积(L/kg) | 0.6 | 5.1~8.1 | 全效量(mg) | 0.8~1.2 | 0.75~1.25 |
| 消除半衰期 | 5~7天 | 36h | 维持量(mg) | 0.05~0.3 | 0.125~0.5 |
| 治疗血浆浓度(ng/ml) | 10~35 | 0.5~2.0 |
肝疾患严重时会影响药的代谢和血浆蛋白结合率。但一般肝病时,洋地黄毒甙的消除并未减慢反而加快。因此时血浆蛋白减少,游离强心甙增多反多被代谢。
肾疾病时,地高辛排泄减少,其用量应根据肌酐清除率计算。洋地黄毒甙的消除则与肾功能无明显关系。
毒毛花甙K(strophantin K)含较多羟基,极性高,口服吸收仅5%,作静脉注射用。几无代谢,以原形经肾排泄。半衰期约19h,是短效药,现已少用。
【临床应用】强心甙主要用于治疗CHF和某些心律失常。
1.CHf 各种原因如心肌缺血、瓣膜病、高血压、先天性心脏病、心肌炎(风湿性、病毒性)、甲状腺机能亢进及严重贫血等所引起的CHF,都可应用强心甙。通过正性肌力作用,增加搏出量及回心血量,可以缓解动脉系统缺血和静脉系统淤血,取得对症治疗效果。但强心甙对不同原因引起的CHF,在对症治疗的效果上却有很大差别。它对瓣膜病、高血压、先天性心脏病等所引起者疗效良好。对继发于严重贫血、甲亢及维生素B1缺乏症的CHF则疗效较差,因这些情况下,心肌能量生产已有障碍,而强心甙又不能改进能量的生产。对肺原性心脏病、严重心肌损伤或活动性心肌炎如风湿活动期的CHF,强心甙疗效也差,因为此时心肌缺氧,既有能量生产障碍,又易发生强心甙中毒,使药量也受到限制,难以发挥疗效。对心肌外机械因素引起的CHF,包括严重二尖瓣狭窄及缩窄性心包炎,强心甙疗效更差甚至无效,因为此时左室舒张充盈受限,搏出量受限,难以缓解症状。
2.强心甙对CHF的治疗价值近年有大规模、随机、对照、双盲的临床研究证实地高辛确能缓解或消除症状,改善血流动力学变化,提高运动耐力,加强左心室功能,效果可靠,但未能降低病死率。
对窦性节律的中、轻度CHF患者,现已肯定地高辛能增加射血分数,改善左心室功能,防止病情恶化。
现认为对有症状的心室收缩功能不全的CHF患者,地高辛疗效明确,仍是常用药物。地高辛合用利尿药是CHF的基础用药。
与其他治疗CHF的药物相比,强心甙有以下优点:它应用方便,每日口服一次即可;长期久用疗效不减;一般有效剂量毒副反应并不严重。强心甙的主要缺点是没有正性松弛作用,不能纠正舒张功能障碍。
3.心律失常强心甙常用于治疗心房纤颤、心房扑动及阵发性室上性心动过速。
心房纤颤时,心房的过多冲动可能下传到达心室,引起心室频率过快,妨碍心排血,导致严重循环障碍,这是心房纤颤的危害所在。此时,强心甙是主要药物。用药目的不在于停止房颤而在于保护心室免受来自心房的过多冲动的影响,减少心室频率。用药后多数患者的心房纤颤并未停止,而循环障碍得以纠正。这是强心甙抑制房室传导的结果,使较多冲动不能穿透房室结下达心室而隐匿在房室结中。
心房扑动时,源于心房的冲动与房颤时相比较少较强,易于传入心室,使室率过快而难以控制,强心甙的治疗功能在于它能不均一地缩短心房不应期,引起折返激动,使心房扑动转为心房纤颤,然后再发挥治疗心房纤颤的作用。某些患者在转为房颤后,停用强心甙,有可能恢复窦性节律。因为停用强心甙就是取消它的缩短心房不应期的作用,就相对地延长不应期,可使折返冲动落入较长的不应期而停止折返,于是窦性节律得以恢复。
阵发性室上性心动过速,强心甙通过兴奋迷走神经减慢房室传导的作用,可有疗效。
【不良反应及其防治】以往用量偏高,中毒发生率接近20%,现用量减少,又常采用逐日给恒量地高辛法,故中毒率明显下降,已低于12%。
1.毒性作用的表现较常见的有胃肠道反应,发厌食、恶心、呕吐、腹泻,应注意与强心甙用量不足心衰未受控制所致的胃肠道症状相鉴别。后者由胃肠道淤血所引起。神经系统反应有眩晕、头痛、疲倦、失眠、谵妄等。还有黄视症、绿视症等。最严重的是心毒性反应,可出现各种心律失常,多见早见的是室性早搏,约占心反应的33%;次为房室阻滞约为18%;房室结性心动过速17%;房室结代节律12%;房性过速兼房室阻滞10%;室性过速8%;窦性停搏2%。这些心律失常由三方面毒性作用所引起:由浦肯野纤维自律性增高及迟后除极触发活动所致的异位节律的出现;房室结传导性的抑制;窦房结自律性的降低。
2.毒性作用的防治 先要明确中毒诊断,可根据心电图的变化与临床症状作出初步判断。测定强心甙的血药浓度则有重要意义。地高辛浓度在3.0ng/ml,洋地黄毒甙在45ng/ml以上可确诊为中毒。预防上应注意诱发因素如低血钾、高血钙、低血镁、心肌缺氧等。还应警惕中毒先兆的出现,如一定次数的室性早搏、窦性心律过缓低于60次/分及色视障碍等。
解救上,对过速性心律失常者可用钾盐静脉滴注,轻者可口服。细胞外K+可阻止强心甙与Na+-K+-ATP酶的结合,能阻止毒性发展。苯妥英钠能控制室性早搏及心动过速而不抑制房室传导,它能与强心甙竞争性争夺Na+-K+-ATP酶而有解毒效应。利多卡因也有效。对中毒时的心动过缓或房室阻滞宜用阿托品解救。地高辛抗体的Fab片断对强心甙有强大选择性亲和力,能使强心甙自Na+-K+-ATP酶的结合中解离出来,解救致死性中毒有明确效果。它与地高辛的结合物可经肾排泄。每毫克地高辛需用80mgFab拮抗之。
【给药方法】强心甙的传统用法分为两步,既先获足够效应而后维持之。用药先给全效量即“洋地黄化”,而后逐日给予维持量。全效量可口服地高辛首次0.5mg,4小时后再给0.5mg,对危急病例可在5分钟内缓慢静脉注射地高辛1.0mg。维持量应每日补充体内消除量,地高辛每日消除体内储存量35%,约为0.125~0.5mg.
现知逐日给恒定剂量的药物,经4~5个t1/2后就能在血中达到稳态浓度。据此,对病情不急的CHF患者,现多采用地高辛(t1/2为36小时)逐日给予0.25~0.375mg,经6~7天就能达到稳定的有效浓度,从而取得稳定疗效。这种给药法可明显降低中毒发生率。
强心甙的用量应做到个体化,同一患者在不同病情下,用量也应增减。当体内失钾或肾功能减退时,为避免中毒应减少用量。当感染而增加心工作负荷时,为了保持疗效,宜酌情加大用量。
【药物相互作用】许多药物干预地高辛的药代动力学变化而影响其血药浓度。消胆胺、新霉素在肠中与地高辛结合,妨碍其吸收,降低血药浓度;奎尼丁能使90%患者的地高辛血浓提高一倍,是奎尼丁自组织中置换出地高辛的结果,合并用药时宜酌减地高辛用量约30%~50%;胺碘酮、维拉帕米等也能升高地高辛的血药浓度。
第二节 非强心甙类的正性肌力作用药
现已合成一些非甙类正性肌力作用药,临床试用有效,兹择要介绍如下:
1.β-受体药β受体部分激动剂扎莫特罗(xamoterol)有双向作用:在轻度CHF或休息时,交感神经活性较低,它发挥激动药作用;在重症或劳累激动时,交感神经活性较高,它发挥阻断药作用。临床称其能增加中、轻度CHF患者休息时的心输出量及血压,对重症患者也能缓解症状。其应用价值仍在研究中。近年用β受体阻断药美托洛尔(metoprolol)治疗特发性扩张型及冠心病CHF而收到较好效果。它能上调β受体数量,又能拮抗交感神经活性增高对CHF发病的不利影响,但并非对每一病例都有效,现仍有研究性试用。异波帕胺(ibopamine)属多巴胺类药物,部分作用是激动β受体,而增加心肌收缩性,能增加心输出量,降低外周阻力,促进利尿。治疗CHF能缓解症状,提高运动耐力,疗效与地高辛相似,有应用价值。
2.磷酸二酯酶抑制药磷酸二酯酶PDE- Ⅲ是cAMP降解酶,抑制此酶活性将增加细胞内cAMP的含量,发挥正性肌力作用和血管舒张作用,临床应用已证明PDE-Ⅲ抑制药能增加心输出量,减轻心负荷,降低心肌氧耗量,缓解CHF症状。
最先应用的PDE- Ⅲ抑制药是氨力农(amrinone),临床有效,但长期口服后约15%患者出现血小板减少,可致死亡。另有心律失常、肝功能减退。现仅供短期静脉滴注用。其代替品米力农(milrinone)抑酶作用较前者强20倍,临床应用有效,能缓解症状、提高运动耐力,不良反应较少,未见引起血小板减少。惜近称久用后疗效并不优于地高辛,反更多引起心律失常,故病死率较高,也仅供短期静脉给药用。依诺昔酮(enoximone)治疗中、重度CHF疗效与米力农相似,近也称其病死率较对照组为高,不作长期口服用。其他有待临床观察的药还有匹罗昔酮(piroximone),匹莫苯(pimobendan),维司力农(Vesnarinone)等。后者作用机制多样,除抑制PDE-Ⅲ外,也增加细胞内Na+量,抑制K+外流,临床试用有效受到重视。
上述多种PDE- Ⅲ抑制药多数还兼有增强心肌收缩成分对Ca2+敏感性的作用,即不用增加细胞内Ca2+量也能加强收缩性。这就可避免因细胞内Ca2+过多而继发的心律失常、细胞损伤甚至坏死。目前正待研制具有选择性的“钙增敏药”。
第三节 血管扩张药治疗慢性心功能不全
一、应用根据
治疗心绞痛和高血压的一些血管扩张药(详见第二十四章及第二十六章),自70年代以来也用于治疗CHF而收到效果。它们能缓解症状,改善血流动力学变化提高运动耐力,但多数扩血管药并不能降低病死率。药物舒张静脉(容量血管)就可减少静脉回心血量、降低前负荷,进而降低左室舒张末压、肺楔压,缓解肺充血症状。药物舒张小动脉(阻力血管)就可降低外周阻力,降低后负荷,进而改善心功能,增加心输出量,增加动脉供血,从而弥补或抵消因小动脉舒张而可能发生的血压下降与冠状动脉供血不足的不利影响。
二、常用药物
1.硝酸酯类主要作用于静脉,降低前负荷,用药后明显减轻呼吸急促和呼吸困难。也略舒张小动脉,略降后负荷。硝酸甘油静脉滴注10μg/min,可每5~10分钟增加5~10μg/min,二硝酸异山梨醇酯也可用。
2.硝普钠能舒张静脉和小动脉,静脉注射给药后2~5分钟即见效,停药后2~15分钟即消退。用药后前、后负荷下降,对急性心肌梗塞及高血压所致CHF效果较好。静脉滴注开始12.5μg/min每5~10分钟增加5~10μg 。
3.肼屈嗪主要舒张小动脉,降低后负荷,用药后心输出量增加,血压不变或略降,不引起反射性心率加快,一般口服每日4次,每次50~75mg。
4.哌唑嗪能舒张静脉和动脉,用药后后负荷下降,心输出量增加,肺楔压也下降。对缺血性心脏病的CHF效果较好。口服首剂0.5mg,以后每6小时1mg。
5.硝苯地平舒张动脉较强,降低后负荷较为显著,能增加心输出量。由于它对受损心肌可能发生的抑制作用,一般不作CHF的常用药。
6.近年报道对Ⅱ、 Ⅲ级CHF患者以地高辛、利尿药为基础,加用肼屈嗪300mg/日和二硝酸异山梨醇酯160mg/日,发现加用二药能降低病死率,3年降低36%,左心室射血分数也增高。
三、药物选用
众多的血管扩张治疗CHF,应根据患者血流动力学变化分别选用。例如对前负荷升高为主,肺淤血症状明显者,宜用舒张静脉的硝酸酯类。对后负荷升高为主、心输出量明显减少者,宜用舒张小动脉的肼屈嗪。多数患者,前、后负荷都有不同程度增高,则宜兼顾。
应用血管扩张药的剂量,可参考血压而定。一般以采用维持血压在12.0~13.3/6.7~8.0kPa(90~100/50~60mmHg)肺楔压2.0~2.4kPa(15~18mmHg)的剂量为宜
第四节 血管紧张素Ⅰ转化酶抑制药(ACEI)
ACEI也属血管扩张药,因其治疗CHF效果突出,作用及机制也有特点,已不限于血管舒张作用,故另作介绍如下。
一、ACEI治疗慢性心功能不全的临床效果
ACEI能缓解或消除CHF患者的症状,改善血流动力学变化及左室功能,提高运动耐力,逆转左室肥厚,更为突出的是ACEI确能降低病死率,有关的临床研究(多中心、随机、双盲对照)有:
1.北欧依那普利生存率研究(CONSENSUS.1987),253例Ⅳ级CHF患者,以地高辛和利尿药作阳性对照,给药组用依那普利开始2.5~5.0mg/次,2次/日,一周后增为10mg/次,2次/日。结果证明依那普利在12个月内降低病死率30%(对照组病死52%,依那普利组病死36%)。
2.左室功能障碍研究(SOLVD.1991)。观察依那普利对射血分数≤35%的CHF患者病死率的影响。治疗试验2569例,用药同上。结果表明在48月内依那普利降低病死率约16%;预防试验无症状者4228例,证实依那普利降低病死率约29%。
3.生存率与心室扩大试验(SAVE 1992)观察卡托普利对2231例急性心肌梗塞3~16日后、射血分数≤40%的患者的疗效,随访42个月。结果发现对照组与卡托普利组的病死率分别为25%与20%,用药组降低约20%。
二、心室肥厚与构形重建
ACEI治疗CHF除发挥一般血管扩张药作用外,还能防止或减轻CFH发病恶性循环中的危险因素即心室肥厚的发生。后者包括肌细胞与非肌细胞的肥大增生,伴有左室形态结构的改变和机械效能的减退。现称这一复杂过程为“构形重建(remodeling)”。此时不仅心肌损伤部位有病变,且有非损伤部位的重建,还有非肌细胞如成纤维细胞、胶原细胞、血管细胞等的增殖生成。这些肌细胞外的间质对收缩力的机机械转导与舒张后的回缩及张力的发展都很重要。间质中胶原增多,重新排列与断裂,间质纤维化,室壁硬化及偏心肥厚都将加剧收缩与舒张功能障碍。
三、血管紧张素Ⅱ的促生长作用
实验发现ATⅡ能引起心肌肥厚,它能增加细胞内DNA、RNA的含量及代谢转化,也增加蛋白质合成。与此相符又发现ACEI能防止或逆转实验动物和人体由高血压所引起的心室肥厚。近知不影响血压的小量ACEI也能防止或逆转由结扎主动脉所引起的大鼠心室肥厚,说明ATⅡ引起肥厚及ACEI阻抑肥厚与它们对血压的影响无关,ATⅡ引起肥厚是一种独立的促生长作用。心肌还有局部RAAS,ATⅡ可发挥自分泌和旁分泌作用,促进细胞生长和心室肥厚。
现已明确ATⅡ能进入细胞核与染色质相互作用,随后促进DNA转录而使细胞生长,发挥生长因子样作用,这种作用又与它激活细胞核内原癌基因c-fos,c-myc有关。ATⅡ通过其受体、G蛋白、磷脂酶C及第二信使IP3、DAG系统的介导,调控胞浆Ca2+浓度与蛋白激酶PKC,从而激活c-fos,c-myc。有实验证明,自心肌培养液中加入ATⅡ,可促进c-fos,c-myc的转录表达。
四、原癌基因与构形重建
原癌基因是正常细胞也包括心肌细胞生长所必需的基因,它能编码细胞生长的许多调节蛋白质如多肽类生长因子及基受体与细胞信息转导系统各组分。c-fos,c-myc较为重要,能表达出“转录因子”,再促进其他基因的转录与表达,引起细胞生长增殖,包括心肌的构形重建。
心肌超负荷时,例如部分结扎大鼠主动脉后,即见心细胞中c-fos,c-myc的转录迅速升高,随后心室开始肥厚,结扎二周后,心室重量增加40%,细胞体积增加。另在离体心灌流实验中,增加灌流压也能增加c-fos,c-myc的转录与表达,随之出现心室肥厚。这说明超负荷时原癌基因的激活是心室肥厚构形重建的始动因素。
以上资料提示ATⅡ能促进心细胞生长,从而引起心室肥厚与构形重建。这样,就不难理解ACEI为何能防止和逆转CHF时的心室肥厚并能降低CHF病死率。
当然,ACEI也非完美无缺,它缓解症状较为缓慢;不良反应有低血压、肾功能下降、呛咳等,因此而停药者达17%~22%。此外应用的适应证,与其他药物联合应用等问题,也有待解决。
归纳看,CHF是多病因、多病理变化、多症状的慢性综合征,很难用一种药物治疗方案统一治疗之。当前临床应用强心甙,利尿药,新型正性肌力作用药,血管扩张药及ACEI等和它们的联合用药,已取得不同程度的疗效。今后将继续研究发病过程中体液、循环和神经内分泌及基因等方面的变化,指导探索合理的、更针对病因的药物治疗,达到进一步提高治疗效果的目的。
抗慢性心功能不全药 制剂及用法
洋地黄毒甙(digitoxin)口服0.05~0.2mg/次。极量:0.4mg/次,1mg/日。
地高辛(digoxin)一般首剂0.25~0.75mg,以后每隔6小时0.25~0.5mg直至洋地黄化,再改用维持量(0.25~0.5mg/日)。轻型慢性病例:0.5mg/日。
毒毛花甙K(strophantin K)静注0.25mg/次,0.5~1.0mg/日,极量0.5mg/次,1.0mg/日。
第二十四章 抗心绞痛药
心绞痛是冠状动脉粥样硬化性心脏病(冠心病)的常见症状,是冠状动脉供血不足,心肌急剧的、暂时的缺血和缺氧所引起的临床综合征。发作时胸骨后部及心前区出现阵发性绞痛或闷痛,并可放射至左上肢,疼痛是由缺血、缺氧的代谢产物乳酸、丙酮酸或类似激肽的多肽类物质等所引起。
参照世界卫生组织有关意见,将心绞痛分型如下:
1.
2.不稳定型心绞痛 包括初发型、恶化型及自发性心绞痛,有可能发展为心肌梗塞或猝死,也可逐渐恢复为稳定型心绞痛。
3.变异型心绞痛 为冠状动脉痉挛所诱发。属于自发性心绞痛,休息时也可发病。
心肌暂时性缺血缺氧是由于血和氧的供需失去平衡所致。现已明确心肌对氧的需求增加和冠状动脉痉挛二方面是心绞痛发生的重要病理生理机制。
心肌的氧供应决定于动、静脉的氧张力差及冠状动脉的血流量。正常情况下心肌细胞摄取血液氧含量65%~75%,已接近于最大量,如再需增加氧的供应,已难以从血中更多地摄取氧,只能通过增加冠状动脉血流量来提供,而后者又取决于冠状动脉阻力、灌流压、侧枝循环及舒张时间等因素。正常情况下冠状动脉系统的小动脉阻力对冠状动脉流量起着重要作用,出现粥样硬化后,狭窄区以下的小动脉因缺氧而舒张,此时较大的心外膜血管则对冠状动脉流量起主要作用。
心肌对氧的需求约为8~10ml/100g心肌/分,决定氧耗的主要因素是心肌的基本代谢,心室壁肌张力,分钟射血时间,心率和收缩性。其中,基本代谢的氧耗用于细胞膜转运功能及合成蛋白质,它较为稳定,少受药物影响;室壁肌张力则影响较大,它与心室容积和心室腔内压力成正比,张力愈高耗氧愈多;分钟射血时间是每搏射血时间与心率的乘积,射血时室壁肌张力最高,所以,射血时间愈久,耗氧愈多;收缩性的强弱也明显影响氧耗,强时耗氧多,反之耗量少。由于测定心肌实际耗氧量较为困难,临床上将影响耗氧量的主要因素简化为“三项乘积”(收缩压×心率×左心室射血时间)或“二项乘积”(收缩压×心率)作为粗略估计心肌耗氧量的指标。
由上可见,药物可通过舒张冠状动脉、解除冠状动脉痉挛或促进侧枝循环的形成而增加冠状动脉供血。药物也可通过舒张静脉,减少回心血量、降低前负荷;舒张外周小动脉、降低血压,减轻后负荷;降低室壁肌张力;减慢心率及降低收缩性等作用而降低心肌对氧的需求。实际上,常用的抗心绞痛药正是通过对这两方面的影响,恢复氧的供需平衡而发挥治疗作用的。
一、硝酸酯类及亚硝酸酯类
硝酸酯类药物有:硝酸甘油,硝酸异山梨酯,单硝酸异山梨酯,其中硝酸甘油最常用,戊四硝酯及吸入用的亚硝酸异戊酯已少用。
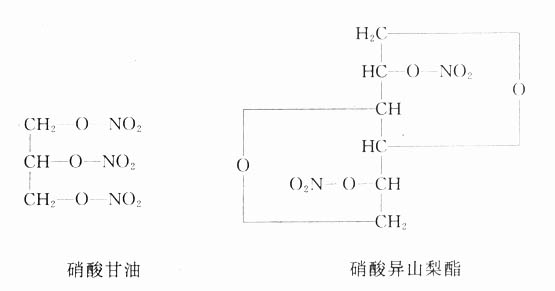
可见,所有硝酸酯类化合物均为硝酸多元酯结构,具有高脂溶性,它们结构中的O-NO2是发挥疗效的关键部分。
硝酸甘油
【药理作用】硝酸甘油(nitroglycerin)的基本作用是松弛平滑肌,但以松弛血管平滑肌的作用最为明显,现分述如下:
1.对血管的作用 能舒张全身静脉和动脉,但舒张毛细血管后静脉(容量血管)远较舒张小动脉的作用为强。对较大的冠状动脉也有明显舒张作用,对毛细血管括约肌则作用较弱。对血管作用的总结果是:血液贮积于静脉及下肢血管,使静脉回心血量减少,降低前负荷、心室充盈度与室壁肌张力。治疗量的硝酸甘油使动脉收缩压约降1.3~2.0kPa(10~15mmHg),舒张压不变,后负荷略降。它也能舒张头、面、颈、皮肤血管及肺血管。
2.对心脏的作用 硝酸甘油对心脏无明显作用。对正常人及无心功能衰竭的冠心病患者,却使每搏及分钟输出量减少,心率不变或轻度加快;剂量加大,可致降压而反射性加快心率。心绞痛患者舌下含用硝酸甘油数分钟后,心脏负荷迅速减轻,表现为心室舒张末压下降,心室内径减少,外周血管阻力下降,使左心室功能改善,心骨耗氧量明显减少。
【抗心绞痛作用的机制】硝酸甘油治疗心绞痛已有100多年历史,对其作用机制的认识曾有反复,现看法如下:
1.硝酸甘油使容量血管扩张而降低前负荷,心室舒张末压力及容量也降低。在较大剂量时也扩张小动脉而降低后负荷,从而降低室壁肌张力及氧耗。
2.硝酸甘油能明显舒张较大的心外膜血管及狭窄的冠状血管以及侧枝血管,此作用在冠状动脉痉挛时更为明显。它对阻力血管的舒张作用微弱。当冠状动脉因粥样硬化或痉挛而发生狭窄时,缺血区的阻力血管已因缺氧而处于舒张状态。这样,非缺血区阻力就比缺血区为大,用药后将迫使血液从输送血管经侧枝血管流向缺血区,而改善缺血区的血流供应。
3.硝酸甘油能使冠状动脉血流量重新分配。已知心内膜下血管是由心外膜血管垂直穿过心肌延伸而来的,因此内膜下血流易受心室壁肌张力及室内压力的影响,张力与压力增高时,内膜层血流量就减少。在心绞痛急性发作时,左心室舒张末压力增高,所以心内膜下区域缺血最为严重。硝酸甘油能降低左心室舒张末压,舒张心外膜血管及侧枝血管,使血液易从心外膜区域向心内膜下缺血区流动,从而增加缺血区的血流量,放射微球法已证明硝酸甘油能增加心内膜下区的血液灌流量。用微型氧电极也测得给硝酸甘油后,心内膜层/心外膜层氧分压比值上升。
图24-1 硝酸酯类(N)及β-受体阻断药(B)对缺血心脏及外周循环的作用
注:↑=增加,↓=降低,→=不变,↓↑=不定
图24-2 硝酸甘油对冠状动脉的作用部位示意图
【舒张血管的作用机制】最近研究证明,血管内皮细胞能释放扩血管物质EDRF(endothelium-derived relaxing factor血管内皮舒张因子,即一氧化氮NO),它是由内皮细胞中的L-精氨酸-NO合成途径产生的,并从内皮细胞弥散到血管平滑肌细胞,在其中它激活鸟苷酸环化酶(GC)增加细胞内cGMP的含量,从而激活依赖于cGMP的蛋白激酶。促使肌球蛋白轻链去磷酸化,而松弛血管平滑肌。硝基扩管药能在平滑肌细胞及血管内皮细胞中产生NO而舒张血管。硝酸酯类在平滑肌细胞能与硝酸酯受体结合,并被硝酸酯受体的巯基还原成NO或-SNO(亚硝巯基)。此外,释出的NO还能抑制血小板聚集和粘附,有利于冠心病的治疗。
【体内过程】硝酸甘油舌下含服易经口腔粘膜迅速吸收,2~5分钟出现作用,3~10分钟作用达峰值,维持20~30分钟,血浆t1/2约为3分钟,舌下含化的生物利用度为80%,也可经皮肤吸收而达到治疗效果。分布容积为0.35L/kg,在肝经有机硝酸酯还原酶脱硝酸而形成二硝酸或单硝酸盐而失效,最后与葡萄糖醛酸结合,从尿排出。
【临床应用】对各型心绞痛均有效,用药后能中止发作,也可预防发作。对急性心肌梗塞不仅能减少耗氧量,尚有抗血小板聚集和粘附作用,使坏死的心肌得以存活或使梗塞面积缩小,但应限制用量,以免过度降压。
【不良反应及耐受性】多数不良反应是其血管舒张作用所继发。如短时的面颊部皮肤发红;而搏动性头痛则是脑膜血管舒张所引起;有进出现体位性低血压及晕厥;眼内血管扩张则可升高眼内压。剂量过大可使血压过度下降,冠状动脉灌注压过低,并可反射性兴奋交感神经、增加心率、加强心肌收缩性反使耗氧量增加而加重心绞痛发作。超剂量时还会引起高铁血红蛋白症。
连续用药后可出现耐受性,停药1~2周后,耐受性可消失。耐受性的发生可能与“硝酸酯受体”中的巯基被耗竭有关。为克服耐受可采用下列措施:调整给药次数和剂量,不宜频繁给药;采用最小剂量;采用间歇给药法,无论采用何种给药途径,如口服、舌下、静注或经皮肤,每天不用药的间歇期必须在8小时以上;补充含巯基的药物,如加用卡托普利,甲硫氨酸等,可能阻止耐受性。
硝酸异山梨酯
硝酸异山梨酯(isosorbidedinitrate)的作用及作用机制与硝酸甘油相似而作用较弱,与硝酸甘油相比作用出现较慢、维持时间较久,经肝代谢后可得二个活性代谢产物,仍具有扩管及抗心绞痛作用。但剂量范围个体差异较大,不良反应较多。见表24-1。
表24-1 硝酸酯类药物的比较
| 药物 | 给药方法 | 一次用量(mg) | 起效(min) | 持效(h) | t1/2(min) | 给药次数(次/日) |
| 硝酸 甘油 |
舌下 口服 |
0.3~0.6 2.5 |
1~2 30 |
20~40分 10 |
4.4 | - 2 |
| 硝酸异山梨酯 | 舌下 口服 |
5~10 10 |
15~30 15~30 |
2~4 2~4 |
45 | 3 3 |
5-单硝酸异山梨酯(isosorbide-5-mononitrate)国内也开始生产。此外,尚有硝酸甘油贴剂(含5~10mg),用时贴在胸前或上臂皮肤缓慢吸收,宜夜间贴,贴皮肤的时间不超过8小时。
二、肾上腺素β受体阻断药
β受体阻断药如普萘洛尔、吲哚洛尔、噻马洛尔及选择性β1受体阻断药如阿替洛尔、美托洛尔、醋丁洛尔等均可用于心绞痛,能使多数患者心绞痛发作次数减少,硝酸甘油用量减少,并增加运动耐量,改善缺血性心电图的变化。兹以普萘洛尔为例介绍如下:
【药理作用】心绞痛时,交感神经活性增强,心肌局部和血中儿茶酚胺含量增高,更大程度地激动β受体,使心肌收缩性加强,心率加快,心肌耗氧量明显增加,因而加重了心肌缺血缺氧。普萘洛尔等β受体阻断药则能明显降低心肌耗氧量,也降低后负荷而缓解心绞痛。临床观察表明,用普萘洛尔后,对心率减慢和收缩性减弱较明显的患者,所获疗效最好。
普萘洛尔还能改善缺血区的供血,因用药后心肌耗氧量减少,非缺血区的血管阻力增高。促使血液向缺血区已舒张的阻力血管流动,从而增加缺血区的供血。其次,β受体阻断药能减慢心率,使舒张期延长,从而冠脉的灌流时间延长,这有利于血液从心外膜血管流向易缺血的心内膜区。普萘洛尔还能促进氧自血红蛋白的解离而增加全身组织包括心肌的供氧。
普萘洛尔抑制心肌收缩性而增大心室容积(增加前负荷),延长射血时间,而相对增加心肌耗氧量、部分抵消其降低氧耗量的有利作用,但多数患者用药后心肌总耗氧量的降低的,见表24-2及图24-2。
【临床应用】治疗稳定及不稳定型心绞痛,可减少发作次数,对兼患高血压或心律失常者更为适用。对心肌梗塞也有效,能缩小梗塞范围。普萘洛尔不宜用于与冠状动脉痉挛有关的变异型心绞痛,因冠脉上的β受体被阻断后,α受体占优势,易致冠状动脉收缩。
普萘洛尔有效剂量的个体差异较大,一般宜从小量开始,以后每隔数日增加10~20mg,多数患者用量可达80~240mg/日。久用停药时,应逐渐减量,否则会加剧心绞痛的发作,引起心肌梗塞或突然死亡,可能是长期用药后β受体数量增加(向上调节),而突然停药时对内源性儿茶酚胺的反应有所增强所致。长期应用后对血脂也有影响,本类药物禁用于血脂异常的患者。合用普萘洛尔和硝酸甘油可相互取长补短,如普萘洛尔可取消硝酸甘油所引起的反射性心率加快;硝酸甘油却可缩小普萘洛尔所扩大的心室容积,而两药对耗氧量的降低却有协同作用,还可减少不良反应的发生。见表24-2。
表24-2 硝酸酯类、β受体阻断药及钙拮抗剂对心脏
氧供及氧需诸因素的影响
| 决定因素 | 硝酸酯类 | β受体阻断药 | 钙拮抗药 |
| 室壁张力 | ↓ | ± | ↓ |
| 心室容量 | ↓ | ↑ | ± |
| 心室压力 | ↓ | ↓ | ↓ |
| 心脏体积 | ↓ | ↑ | ± |
| 心 率 | ↑ | ↓ | ± |
| 收缩性 | ↑ | ↓ | ± |
| 心内膜/心外膜血流比率 | ↑ | ↑ | ↑ |
| 侧枝血流 | ↑ | → | ↑ |
三、钙拮抗药
抗心绞痛常用的钙拮抗药有硝苯地平,维拉帕米,地尔硫卓,哌克昔林及普尼拉明(详见第二十一章)。
【抗心绞痛作用及临床应用】
钙拮抗药通过阻断血管平滑肌电压依赖性钙通道,降低Ca2+内流而扩张冠状动脉和外周动脉,并能使心肌收缩性下降、心率减慢,减轻心脏负荷,从而降低心肌耗氧量。它们也因舒张冠状血管,增加冠状动脉流量而改善缺血区的供血供氧等。上述作用使它们具有“节能”效,且有保护心肌细胞免受缺血的伤害。
钙拮抗药对冠状动脉痉挛及变异型心绞痛是为有效,也可用于稳定型及不稳定型心绞痛,但硝苯地平对不稳定型心绞痛的治疗有一定的局限性,因其有能引起心率加快而增加心肌缺血的危险。但维拉帕米和地尔硫![]() 则不同,可直接作用于心脏,引起心率轻度减慢。钙拮抗药对急性心肌梗塞能促进侧枝循环,缩小梗塞面积。
则不同,可直接作用于心脏,引起心率轻度减慢。钙拮抗药对急性心肌梗塞能促进侧枝循环,缩小梗塞面积。
β受体阻滞药与硝苯地平合用较为理想,与维拉帕米合用时应注意对心脏的抑制和致血压下降的作用。
四、其他抗心绞痛药
吗多明(molsidomine)的作用与硝酸甘油相似,主要能降低心脏前、后负荷,降低心室壁肌张力,因而降低心肌耗氧量,也能舒张冠状动脉,改善心内膜下心肌的供血。临床用于各型心绞痛,作用时间较硝酸甘油为久,一次口服或舌下含化2mg,可维持疗效6~8小时,且不易产生耐受性,与硝酸甘油交替应用可克服耐受性的产生。
抗心绞痛药 制剂及用法
硝酸甘油(nitroglycerin)0.3~0.6mg/次,舌下含化。
硝酸甘油贴剂(Transderm-Nitro5 及10,在24h内分别可吸收5及10mg硝酸甘油)1次/日,贴皮肤时间不超过8h。
硝酸异山梨酯(isosorbidedinitrate,消心痛)5~10mg/次,舌下含化。
5单硝酸异山梨酯(isosorbide-5-mononitrate)20mg/次,2~3次/日,口服。
盐酸普萘洛尔(propranololhydrochloride)抗心绞痛:10mg/次,3次/日,可根据病情增减剂量。
硝苯地平(nifedipine,心痛定)10~20mg/次,3次/日,口服。
硝苯地平缓释片(nifedipineretard)20mg/次,1~2次/日。
第二十五章 抗动脉粥样硬化药
动脉粥样硬化(atherosclerosis)是缺血性心脑血管病的病理基础。在我国,心脑血管病发病率与死亡率近年也明显增加。因而,抗动脉粥样硬化药(antiatherosclerotic drugs)的研究日益受到重视。动脉粥样硬化病因、病理复杂,本类药物涉及面较广。本章主要介绍调血脂药、抗氧化药、多烯脂肪酸类及保护动脉内皮药等。
第一节 调血脂药
血脂以胆固醇酯(CE)和甘油三酯(TG)为核心,外包胆固醇(Ch)和磷脂(PL)构成球形颗粒。再与载脂蛋白(apo)相结合,形成脂蛋白溶于血浆进行转运与代谢。脂蛋白可分为乳糜微粒(CM)、极低密度脂蛋白(VLDL)、中间密度脂蛋白(IDL)、低密度脂蛋白(LDL)和高密度脂蛋白(HDL)等。凡血浆中VLDL、IDL、LDL及apo B浓度高出正常为高脂蛋白血症,易致动脉粥样硬化。近年来证明HDL、apo A浓度低于正常,也为动脉粥样硬化危险因子。
高脂蛋白血症可分为6型(见表25-1)
表25-1 高脂蛋白血症的分型
| 分型 | 脂蛋白变化 | 血脂变化 | ||
| Ⅰ | CM | ↑ | TG↑↑↑ | TC↑ |
| Ⅱa | LDL | ↑ | TC↑↑ | |
| Ⅱb | VLDL及LDL | ↑ | TG↑↑ | TC↑↑ |
| Ⅲ | IDL | ↑ | TG↑↑ | TC↑↑ |
| Ⅳ | VLDL | ↑ | TG↑↑ | |
| Ⅴ | CM及VLDL | ↑ | TG↑↑ | TC↑ |
对血浆脂质代谢紊乱,首先要调节饮食,食用低热卡、低脂肪、低胆固醇类食品,加强体育锻炼及克服吸烟等不良习惯。如血脂仍不正常,再用药物治疗。凡能使LDL、VLDL、TC(总胆固醇)、TG、apo B降低,或使HDL、apo A升高的药物,都有抗动脉粥样硬化作用。
胆汗酸结合树脂
考来烯胺(cholestyramine,消胆胺)和考来替泊(降胆宁,colestipol)都为碱性阴离子交换树脂,不溶于水,不易被消化酶破坏。
【药理作用】能明显降低血浆TC和LDL-c(LDL-胆固醇)浓度,轻度增高HDL浓度。本类药物口服不被消化道吸收,在肠道与胆汁酸形成络合物随粪排出,故能阻断胆汁酸的重吸收。由于肝中胆汁酸减少,使胆固醇向胆汁酸转化的限速酶——7-α羟化酶更多地处于激活状态,肝中胆固醇向胆汁酸转化加强。胆汁酸也是肠道吸收胆固醇所必需,树脂与胆汁酸络合,也影响胆固醇吸收。以上作用使肝中胆固醇水平下降,肝脏产生代偿性改变:一是肝细胞表面LDL受体数量增加,促进血浆中LDL向肝中转移,导致血浆LDL-c和TC浓度下降。另一改变是羟甲基戊二酰辅酶A(HMG-Co A)还原酶(肝脏合成胆固醇限速酶)活性增加,使肝脏胆固醇合成增多。因此,本类药物与HMG-CoA还原酶抑制剂合用,降脂作用增强。
【临床应用】用于Ⅱa型高脂血症,4~7天生效,2周内达最大效应,使血浆LDL、胆固醇浓度明显降低。对纯合子(homozygous)家族性高血脂症,因患者肝细胞表面缺乏LDL受体功能,本类药物无效。
【不良反应】常致恶心、腹胀、便秘等。长期应用,可引起脂溶性维生素缺乏。考来烯胺因以氯化物形式应用,可引起高氯性酸血症。也可妨碍噻嗪类、香豆素类、洋地黄类药物吸收,它们应在本类药用前1小时或用后4小时服用。
烟酸
烟酸(nicotinicacid)是一广谱调血脂药,对多种高脂血症有效。
【药理作用】大剂量烟酸能使VLDL和TG浓度下降,1~4天生效,血浆TG浓度可下降20%~50%,作用程度与原VLDL水平有关。5~7天后,LDL-c也下降。与考来烯胺合用,降LDL-c作用加强。降脂作用可能与抑制脂肪组织中脂肪分解,抑制肝脏TG酯化等因素有关。本品能使细胞cAMP浓度升高,有抑制血小板和扩张血管作用,也可使HDL-c浓度增高。
【体内过程】口服后吸收迅速,服用1g,经30~60分可达血药浓度高峰。血浆t1/2为45分。用量超过3g,以原形自尿中排出增加。
【临床应用】对Ⅱ、 Ⅲ、Ⅳ、Ⅴ型高脂血症均有效。也可用于心肌梗塞。
【不良反应】有皮肤潮红、瘙痒等不良反应,是前列腺素中介的皮肤血管扩张所引起,服药前30分服用阿司匹林325mg可以减轻。胃肠刺激症状如恶心、呕吐、腹泻与较常见。大剂量可引起血糖升高。尿酸增加,肝功异常。
苯氧酸类
氯贝特(氯贝丁酯,clofibrate)又名安妥明(atromid-s)是最早应用的苯氧酸(fibric acid)衍化物,降脂作用明显,但不良反应多而严重。新的苯氧酸类药效强毒性低,有吉非贝齐(gemfibrozil),苯扎贝特(bezafibrate)、非诺贝特(fenofibrate)、环丙贝特(ciprofibrate)等。
【药理作用】口服后,能明显降低病人血浆TG、VLDL、IDL含量,而使HDL升高。对LDL作用与患者血浆中TG水平有关。对单纯高甘油三酯血症患者的LDL无影响,但对单纯高胆固醇血症患者的LDL可下降15%。此外,本类药物也有抗血小板聚集、抗凝血和降低血浆粘度,增加纤溶酶活性等作用。
降低血浆TG、VLDL、IDL作用与增加脂蛋白脂酶活性,促进TG代谢有关,也与减少VLDL在肝脏中合成与分泌有关。升高HDL作用是降低VLDL的结果。正常时VLDL中的甘油三酯与HDL中的胆固醇酯有相互交换作用。VLDL减少,使交换减弱,胆固醇酯留于HDL中,使HDL升高。
【体内过程】口服吸收迅速而完全,数小时即达血药浓度高峰,水解后放出有活性的酸基,能与血浆蛋白结合。部分有肝肠循环,主要以葡萄糖醛酸结合物形式从肾脏排出。
【临床应用】本类药物以降TG、VLDL及IDL为主,所以临床应用于Ⅱb、Ⅲ、Ⅳ型高血脂症。尤其对家族性Ⅲ型高血脂症效果更好。也可用于消退黄色瘤。对HDL-c下降的轻度高胆固醇血症也有较好疗效。
【不良反应】苯氧酸类药物不良反应较轻。有轻度腹痛、腹泻、恶心等胃肠道反应。偶有皮疹、脱发、视物模糊、血象异常等。
HMG-CoA还原酶抑制剂
HMG-CoA(3-羟基-3-甲基戊二酰辅酶A,3-hydroxy-3-methylglutaryl-coenzyme A)还原酶抑制剂最早是从霉菌培养液中提取,有美伐他汀(mevastatin)、乐伐他汀(lovastatin)、以后又有美伐他汀的羟基化、甲基化衍生物普伐他汀(pravastatin)、塞伐他汀(simvastatin)。美伐他汀药效弱而不良反应多,未用于临床。
【药理作用】能明显降低血浆TC和LDL-c.患者每天服用本类药物10~40mg,血浆TC与LDL-c可下降20%~40%。如与胆汁酸结合树脂合用,作用更强,也使VLDL明显下降,对TG作用较弱,可使HDL-c上升。能抑制肝脏合成胆固醇的限速酶HMG-CoA还原酶活性,从而阻断HMG-CoA向甲基二羟戊酸转化,使肝内胆固醇合成减少。由于肝内胆固醇含量下降,可解除对LDL受体基因抑制,使LDL受体合成增加,从而使血浆中LDL、IDL大量被摄入肝脏,使血浆LDL-c、IDL-c降低,由于肝脏胆固醇减少,使VLDL合成减少。降LDL-c作用以乐伐他汀最强,普伐他汀最弱。
【体内过程】乐伐他汀和塞伐他汀口服后在肝脏将内酯环打开才转化成活性物质。用药后1.3~2.4小时血药浓度达到高峰。原药和代谢活性物质与血浆蛋白结合率为95%左右。大部分药物分布于肝脏,随胆汁排出。
【临床应用】对原发性高胆固醇血症、杂合子家族性高胆固醇血症、Ⅲ型高脂蛋白血症,以及糖尿病性、肾性高脂血症均为首选药物。对纯合子家族性高胆固醇血症无降低LDL-c功效,但可使VLDL下降。
【不良反应】本类药物不良反应轻。约10%患者有轻度胃肠症状、头痛或皮疹。少数患者有血清转氨酶、碱性磷酸酶、肌磷酸激酶升高和肌肉触痛。超大剂量乐伐他汀可引起狗白内障,临床应用尚未发现,但应注意。
表25-2 常用调血脂药对血脂水平的影响
| 药 物 | 剂量(日) | 效应 | |||
| TC | LDL-c | TG | HDL-c | ||
| 胆汁酸结合树脂 | 24~30g | ↓20% | ↓20%~25% | 无或↑ | ↑3%~5% |
| 烟酸 | 4g | ↓25% | ↓25% | ↓20%~50% | ↑15%~30% |
| 吉非贝齐 | 1200mg | ↓15% | ↓10%~15% | ↓20%~50% | ↑20% |
| HMC-CoA还原酶抑制剂 | 10~40mg | ↓15%~30% | ↓20%~40% | ↓10%~25% | ↑2%~12% |
第二节 抗氧化剂
氧自由基可使血管内皮损伤,对LDL进行氧化修饰,可促进动脉粥样硬化形成与发展。维生素C、维生素E有抗氧化作用,部分动物实验表明的抗动脉粥样硬化形成的作用。近年发现普罗布考降脂作用较弱,而抗氧化作用较强,对动脉粥样硬化呈现良好防治效应。
普罗布考
普罗布考(probucol,丙丁酚)是70年代创制的降血脂药。
【药理作用】口服能使病人血浆TC下降25%,LDL-c下降10%~15%,HDL-c降低30%,对VLDL、TG影响较少。细胞培养法证明普罗布考有高脂溶性,能结合到脂蛋白之中,从而抑制细胞对LDL的氧化修饰。现知氧化修饰的LDL有细胞毒性,能损伤血管内皮,进而促进血小板,白细胞粘附并分泌生长因子等物质,造成平滑肌细胞移行和过度生长。普罗布考能抑制动脉粥样硬化形成,并使病变消退。可缓解心绞痛,改善缺血性心电图,还能使纯合子家族性高胆固醇血症患者皮肤及肌腱的黄色瘤明显缩小。
【体内过程】口服吸收差。用药后24小时达血药浓度高峰,1~3天出现最大效应。主要在分布于脂肪组织。血浆中以脂蛋白中最多。消除半衰期为23~47天。大部分经粪排出。
【临床应用】用于杂合子及纯合子家族性高胆固醇血症,非家族性高胆固醇血症及糖尿病、肾病所致高胆固醇血症。与考来烯胺、烟酸、HMG-CoA还原酶抑制剂合用作用加强。
【不良反应】仅约10%病人有腹泻、腹胀、腹痛、恶心。偶有嗜酸白细胞增多、感觉异常、血管神经性水肿。个别患者心电图Q-T延长,对心肌损伤、心室应激增强病人应避免使用。
第三节 多烯脂肪酸类
多烯脂肪酸是指有2个或2个以上不饱和键结构的脂肪酸,也称多不饱和脂肪酸(polyunsaturated fatty acids,PUFAs)。根据第一个不饱和键位置不同,可分n-6、n-3二大类。n-6PUFAs包括亚油酸(linoleic acid)、γ-亚麻油酸(γ-linolenic acid)主要含于玉米油、葵花子油、红花油、亚麻子油等植物油中,降脂作用较弱,临床应用疗效可疑。n-3PUFAs 除α-亚麻油酸外,主要有二十碳五烯酸(eicosapentaenoic acid, EPA)和二十二碳六烯酸(docosahexaenoic acid,DHA)等长链PUFAs。含于海洋生物藻、鱼及贝壳类中。
人摄取长链PUFAs后,易结合到血浆磷脂、血细胞、血管壁及其他组织中,改变体内脂肪酸代谢。实验表明,口服EPA,DHA或富含EPA与DHA的鱼油,可使血浆TG、VLDL明显下降,TC和LDL也下降,HDL有所升高。并能抑制血小板聚集,全血粘度下降,红细胞可变性增加,出血时间略有延长。长期服用n-3PUFAs,能预防动脉粥样硬化斑块形成,并使斑块消退。n-3 PUFAs也可使白细胞表面白三烯含量减少,血小板与血管内皮反应减弱,并能抑制血小板活化因子、血小板衍化生长因子的产生,可抑制移植血管增厚,有预防血管再造术后再梗阻作用。目前,国内外已有鱼油或纯EPA,DHA制品。
第四节 保护动脉内皮药
在动脉粥硬化的发病过程中,血管内皮损伤有重要意义。机械、化学、细菌毒素因素都可损伤血管内皮,改变其通透性,引起白细胞和血小板粘附,并释放各种活性因子,导致内皮进一步损伤,最终促使动脉粥样?
■[此处缺少一些内容]■
/日,饭后服。
吉非贝齐(gemfibrozil)胶囊剂,片剂,600mg/次,2次/日。
苯扎贝特(benzafibrate,bezalip)片剂,200mg/次,3次/日。
非诺贝特(fenofibrate)片剂,100mg/次,3次/日,饭后服。
环丙贝特(ciprofibrate)片剂,开始100mg/次,可增至200mg/次,1次/日。
乐伐他汀(lovastatin)片剂,20~40mg/次,1次/日,晚餐时服,必要时4周内可增至80mg/次,1次/日。
普伐他汀(pravastatin)片剂,5~10mg/次,2次/日。
塞伐他汀(simvastatin)片剂,10mg/次,1次/日。
普罗布考(probucol)片剂,250~500mg/次,2次/日。
多烯康 胶丸(300mg,含EPA、DHA 210mg),5~7丸/次,3次/日。
硫酸软骨素A(chondroitinesulfate A) 片剂,600mg/次,3次/日。注射剂,40mg/次,2次/日,肌内注射。
第二十六章 抗高血压药
抗高血压药(antihypertensive)用于治疗高血压。世界卫生组织建议高血压诊断标准为成人血压超过21.3/12.6kPa(160/95 mmHg)者。按发病它又分为原发性高血压及继发性高血压。原发性高血压,约占90%,继发性高血压,约占5%~10%,其血压的升高是某些疾病的一种表现,如继发于肾动脉狭窄、肾实质病变,嗜铬细胞瘤、妊娠,或因药物所致等。原发性高血压是在各种因素影响下,血压调节功能失调所致,其病因因虽未阐明,但它的药物治疗在近几十年中有显着进展,合理应用抗高血压药物,确能控制血压并减少或防止心、脑、肾等并发症,包括心衰、猝死等,从而降低发病率及死亡率,延长寿命。多数高血压患者最终需长期服药以控制症状。若能配合非药物治疗,如低盐饮食,减少饮酒,控制体重,改变生活方式等,当可取得更好的效果。
第一节 抗高血压药物的分类
血压的生理调节极为复杂,在众多的神经体液调节机制中,交感神经系统、肾素-血管紧张素-醛固酮系统及内皮素系统起着重要作用,许多抗高血压药物往往通过影响这些系统而发挥降压效应。
根据药物在血压调节系统中的主要影响及部位,可将抗高血压药物分成以下几类:
(一)主要影响血容量的抗高血压药,利尿药。
(二)β受体阻断药,普萘洛尔等。
(三)钙拮抗药,硝苯地平等。
(四)血管紧张素Ⅰ转化酶抑制剂,即影响血管紧张素Ⅱ形成的抗高血压药、卡托普利等。
(五)交感神经抑制药
1.主要作用于中枢咪唑啉受体部位的抗高血压药,可乐定等。
2.神经节阻断药,美加明等。
3.抗去甲肾上腺素能神经末梢药,利血平,胍乙啶等。
4.肾上腺素能受体阻断药,α受体阻断药,哌唑嗪;α和β受体阻断药,拉贝洛尔。
(六)作用于血管平滑肌的抗高血压药,肼屈嗪等。
第二节 常用抗高血压药
一、主要影响血容量的抗高血压药
利尿药是治疗高血压的常用药,要单独治疗轻度高血压,也常与其他降压药合用以治疗中、重度高血压。一般认为利尿药初期降压机制是排钠利尿、造成体内Na+、水负平衡,使细胞外液和血容量减少之故。长期应用利尿药,当血容量及心输出量已逐渐恢复至正常时,血压仍可持续降低,其可能机制如下:①因排钠而降低动脉壁细胞内Na+的含量,并通过Na+-Ca2+交换机制,使胞内Ca2+量减少。②降低血管平滑肌对血管收缩剂如去甲肾上腺素的反应性。③诱导动脉壁产生扩血管物质,如激肽,前列腺素等。
摄入大量NaCl能对抗利尿药的降压作用,限制NaCl摄入则能增强其降压作用,说明排Na+是利尿药降压的重要原因。
临床治疗高血压以噻嗪类利尿药为主,但长期应用常致不良反应如:降低血钾、钠、镁,增加血中总胆固醇,甘油三酯及低密度脂蛋白胆固醇含量,增加尿酸及血浆肾素活性。大剂量噻嗪类利尿药还可加剧脂血症,降低糖耐量等。但使用低剂量的双氢氯噻嗪,则可避免代谢方面的副作用,其他如呋噻米、氨苯蝶啶等也可应用。
一般情况下,高效利尿药不作为轻症高血压的一线药,而用于高血压危象及伴有慢性肾功能不良的高血压患者,因其不降低肾血流,并有较强的利Na+作用。
二、β受体阻断药
β受体阻断药均有良好的抗高血压作用,现以普萘洛尔为例介绍如下:
【抗高血压作用】用普萘洛尔数天后,收缩压可下降15%~20%,舒张压下降10%~15%,合用利尿药降压作用更显着。静脉注射普萘洛尔后可使心率减慢,心输出量减少,但血压仅略降或不降,这是压力感受器反射使外周阻力增高的结果。
有少数人,使用β阻断药后,总外周阻力增高,推测是激活了血管的α受体,故外周血管有病者,禁用本药。
【作用机制】普萘洛尔降低血压是其β受体阻断作用所继发的,对其进一步机制有以下几种看法。
1.减少心输出量 普萘洛尔阻断心β1受体,抑制心肌收缩性并减慢心率,使心输出量减少,因而降低血压。给药后这一作用出现迅速,而降压作用出现较慢。
2.抑制肾素分泌 肾交感神经通过β1受体促使邻球器分泌并释放肾素,普萘洛尔能抑制之,从而降低血压。具有强内在活性的吲哚洛尔在降压时,并不影响血浆肾素活性。
3.降低外周交感神经活性 普萘洛尔也能阻断某些支配血管的去甲肾上腺素能神经突触前膜的β2受体,抑制其正反馈作用而减少去甲肾上腺素的释放。
4.中枢降压作用 已知下丘脑、延髓等部位有β受体,中枢给予微量普萘洛尔能降低血压,同量静脉注射却无效。与之相反的证据是,不能进入中枢的β阻断药,却有降压作用。因此中枢β受体在血压调节中的意义,尚待阐明。
β受体阻断药的作用机制较为复杂,可能在某种病情发展中某一机制起主导作用,而在另种病情过程中,另一机制占主要地位。
【临床应用】β受体阻断药已广泛用于治疗高血压,对轻、中度高血压有效,对高血压伴心绞痛者还可减少发作。此外,对伴有心输出量及肾素活性偏高者,对伴脑血管病变者疗效也较好。普萘洛尔的用量个体差异较大,一般宜从小量开始,以后逐渐递增,但每日用量以不超过300mg为宜。在β受体阻断药中,选择性β1受体阻断药美托洛尔(metoprolol),阿替洛尔(atenolol)的作用优于普萘洛尔,它们在低剂量时主要作用于心脏,而对支气管的影响小,对伴有阻塞性肺疾患者相对安全些。
三、钙拮抗药
钙拮抗药能抑制细胞外Ca2+的内流,能松弛平滑肌、舒张血管,使血压下降。降血压时并不降低重要器官的血流量,不引起脂质代谢及葡萄糖耐受性的改变(详见二十一章)。
硝苯地平
硝苯地平(nifedipine)对轻、中、重度高血压者均有降压作用,但对正常血压者则无降压效。口服30~60分钟见效,持效3小时,t1/2约3~4小时。在离体血管实验中,它能明显抑制高钾去极化所致的收缩反应,对去甲肾上腺素所致的收缩反应则抑制较弱,但对自发性高血压大鼠的血管标本,由去甲肾上腺素所引起的收缩反应却有明显的抑制作用,这似能说明硝苯地平对血压正常者无降压作用的理由,此外,也可抑制内皮素诱导的肾血管的收缩。
硝苯地平降压时伴有反射性心率加快和心搏出量增加,也增高血浆肾素活性,合用β受体阻断药可免此反应而增其降压作用。
临床用于治疗轻、中、重度高血压,可单用或与利尿药、β阻断药合用。
常见不良反应有头痛、脸部潮红、眩晕、心悸、踝部水肿等。其引起踝部水肿为毛细血管前血管扩张而不是水钠潴留所致。
同类药物维拉帕米、地尔硫![]() 、尼群地平、尼莫地平等也用于治疗高血压,并取得良好的效果。其中有钙拮抗药尚有“利尿作用”,能抑制肾小管细胞对Na+的再吸收,能选择性扩张肾入球小动脉,增加肾小球滤过率。
、尼群地平、尼莫地平等也用于治疗高血压,并取得良好的效果。其中有钙拮抗药尚有“利尿作用”,能抑制肾小管细胞对Na+的再吸收,能选择性扩张肾入球小动脉,增加肾小球滤过率。
粉防已碱
粉防已碱(tetrandrine)是中药粉防已根中所含的生物碱,对自发性高血压大鼠及高血压患者均有明显的降压作用,为其钙拮抗作用所致,已证明它能抑制T及L型钙通道。一般口服给药,对重度高血压及高血压危象者可静脉注射。
本药无严重不良反应,少数患者有轻度恶心及上腹部不适等。
四、影响血管紧张素Ⅱ形成的抗高血压药——血管紧张素Ⅰ转化酶抑制剂
肾素-血管紧张素-醛固酮系统(RAAS)在血压调节及高血压发病中都有重要影响。
近几年来合成了一系列血管紧张素转化酶抑制剂(angiotensinconverting enzyme inhibitors,ACEI),如卡托普利(captopril),依那普利(enalapril),雷米普利(ramipril),赖诺普利(lysinopril)及培哚普利(perindopril)等。它们能有效地降低血压,对心功能不全及缺血性心脏病等也有良效。
现代分子生物学研究证明,在心血管、脑、肾等组织中存在着肾素、血管紧张素原的mRNA,局部有相关基因表达,故提出在组织中存在独立的RAAS(系由局部合成),该系统以旁分泌及自分泌方式对心血管及神经系统功能,甚至结构起调节作用。血管中局部产生的血管紧张素Ⅱ(ATⅡ)可增加血管的收缩性能,并促进去甲肾上腺素的释放,而导致血管收缩,血压上升,实验见ATⅡ能促进培养的血管平滑肌细胞生长、增殖,增加蛋白质合成及细胞体积。
ATⅡ促进血管平滑肌生长的作用可引发血管增生及血管壁中层增厚等。
【药理作用及作用机制】ACEI能使血管舒张,血压下降,其作用机制如下:
1.抑制循环中RAAS
ACEI主要通过抑制ATⅡ的形成而起作用,对血管、肾有直接影响。并通过并感神经系统及醛固酮分泌而发生间接作用。这是改变血流动力学的主要因素,也是用药初期外周血管阻力降低的原因。
2.抑制局部组织中RAAS
组织RAAS对心血管系统的稳定有重要作用,组织中的血管紧张素Ⅰ转化酶(ACE)与药物的结合较持久,因此对酶的抑制时间更长,进而降低去甲肾上腺素释放,降低交感神经对心血管系统的作用,有助于降压和改善心功能。此与药物的长期降压疗效有关,药物与ACE的结合方式见图26-1,以卡托普利为例,卡托普利的三个基团可与酶的三个活性部位相结合,一是脯氨酸羧基与酶的正电荷部位(精氨酸)呈离子键结合;二是肽链的羰基与酶的供氢部位呈氢键结合;三是巯基与酶的Zn2+结合,终使酶失去活性。
3.减少缓激肽的降解 当ACE(即激肽酶Ⅱ)受到药物抑制时,组织内缓激肽(bradykinin,BK)降解减少,局部血管BK浓度增高。BK是血管内皮L-精氨酸-NO途径的重要激活剂,它作用于内皮的β2-受体而引起EDHF(血管内皮超极化因子)及NO的释放,因而发挥强有力的扩血管效应及抑制血小板功能。此外,BK可刺激细胞膜磷脂游离出花生四烯酸(AA),促进前列腺素的合成而增加扩血管效应,见图26-2。
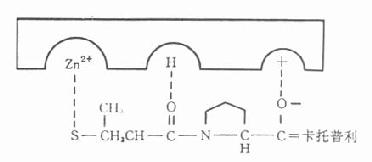
图26-1 卡托普利与酶的活性部位结合图
图26-2 ACEI对RAAS及激肽释放酶-激肽-前列腺素系统的影响
ACEI与其他降压药相比,具有以下特点:
1.适用于各型高血压,在降压的同时,不伴有反射性心率加快,可能是取消了ATⅡ对交感神经传递的易化作用所致。
2.长期应用,不易引起电解质紊乱和脂质代谢障碍,可降低糖尿病、肾病和其他肾实质性损害患者肾小球损伤的可能性,如卡托普利既能有效降压,又能增加机体对胰岛素的敏感性。
3.可防止的和逆转高血压患者血管壁的增厚和心肌细胞增生肥大,可发挥直接及间接的心脏保护作用。
4.能改善高血压患者的生活质量,降低死亡率。
【体内过程】见表26-1
表26-1
| 参数 | 卡托普利 | 依那普利 |
| 前体药 | 否 | 是 |
| 活性代谢产物 | / | 依那普利拉 |
| 生物利用度(%) | 70 | 40 |
| 血浆蛋白结合率(%) | 30 | 50 |
| t1/2(h) | 2 | 30 |
| 维持时间(h) | 3~4 | 12~24 |
| 消除途径 | 肾 | 肾 |
| 12.5-50mg | 10-40 | |
| 剂量 | 2~3次/日 | 1~2次/日 |
【临床应用】治疗原发性及肾性高血压能使血压降低15%~25%,对中、重度高血压合用利尿药、可加强降压效、降低不良反应。
【不良反应】虽不良反应发生率较低,但也不是绝对安全的,主要不良反应有低血压(2%),见于开始剂量过大时,应小量开始试用。高血钾、血管神经性水肿。肾功能受损,对肾血管狭窄者更甚。咳嗽,为刺激性干咳,可能与肺血管床内的激肽及前列腺素等物质的聚积有关。久用可致血锌降低而引起皮疹、味觉、嗅觉缺损、脱发等。补充Zn2+可望克服。
【药物相互作用】合用利尿药可增强降压效,并减少Zn2+的排泄;吲哚美辛可减弱卡托普利的降压效,此与吲哚美辛抑制前列腺素的合成有关;与地高辛合用,可增高地高辛的血浆浓度等。
五、交感神经抑制药
(一)主要作用于中枢部位的抗高血压药
可乐定
可乐定(clonidine,可乐宁)为咪唑类衍化物。
【药理作用】麻醉动物静脉注射可乐定后,先见血压短暂升高,随见血压持久下降,伴有心率减慢、心输出量减少。升压是可乐定激动外周血管α受体所致,随后的降压则与中枢作用有关。口服给药仅见降压而无升压效,继续服用后,外周血管阻力逐渐降低,肾血管阻力也降低,但并不显着影响肾血流量及肾小球滤过率。
可乐定的降压作用中等偏强。它还能抑制胃肠道的分泌和运动,因此适用于兼患溃疡病的高血压患者。
可乐定对中枢神经系统还有镇静作用,减少自发性活动,并显著延长巴比妥类的催眠时间。
【作用机制】动物实验证明微量可乐定注入椎动脉或小脑延髓池均可引起降压,但同等量作静脉注射却并不降压,据此推测,引起降压作用的部位在中枢。通过分层切除脑组织,提示可乐定作用于延髓并降低外周交感神经功能而降压。晚近已证明可乐定引起血压下降的机制是激动了延髓腹外侧嘴部(rostralportion of the ventrolateral medulla)的Ⅰ1-咪唑啉受体(Ⅰ1-imidazoline receptor),降低外周交感张力致血压下降。而其激动中枢α2受体则是其引起镇静等副作用的原因。
此外,从动物脑中已提得内源性可乐定样物质,该物质作用于延髓腹外侧发挥类似可乐定样的作用。另有研究证明,可乐定降压涉及到内源性阿片肽的释放。且可乐定具有镇痛效,此效可被阿片拮抗剂-纳洛酮所拮抗。可乐定也激动外周交感神经突触前膜的α2受体及其相邻的咪唑啉受体,引起负反馈而减少去甲肾上腺素的释放。可见其降压机制复杂。
【体内过程】可乐定口服吸收良好,生物利用度约75%,口服半小时后起效,2~4小时作用达高峰,持续6~8小时。在体内分布均匀,也易透过血脑屏障。t1/2为7.4~13小时。约50%在肝代谢,使结构中的咪唑环裂解,苯环被羟化。余者以原形随尿排出。
【临床应用】可乐定可治疗中度高血压,常于其他药无效时应用。此外,可作为吗啡类镇痛药成瘾者的戒毒药。
【不良反应】常见不良反应有口干,为其作用于胆碱能神经末梢上的α2受体,减少Ach的释放和过量唾液的分泌所致。久用使水、钠潴留,是降压后肾小球滤过率减少的结果。合用利尿药可克服。此外还有镇静、嗜睡、头痛、便秘、腮腺痛、阳萎等,停药后能自行消失,少数患者在突然停药后可出现短时的交感神经功能亢进现象。如心悸、出汗、血压突然升高等,可能是久用后突触前膜α2受体敏感性降低,负反馈减弱,去甲肾上腺素释放过多所致。再用可乐定或用α受体阻断药酚妥拉明能取消之。
甲基多巴
甲基多巴(methyldopa)的降压作用与可乐定相似,属中等偏强,降压时也伴有心率减慢,心输出量减少,外周血管阻力明显降低。治疗中度高血压,适用于肾功能不良的高血压患者。
(二)抗去甲肾上腺素能神经末梢药
利血平是印度萝芙木所含的一种生物碱,国产萝芙木所含总生物碱的制剂称降压灵。该药降压作用弱,不良反应较多,现已少用。作用较强的胍乙啶也因不良反应多而少用。
(三)肾上腺素受体阻断药
α受体阻断药哌唑嗪
哌唑嗪(prazosin)是人工合成的喹唑啉类衍生物。
【药理作用】哌唑嗪能选择性地阻断突触后膜α1受体,能竞争性拮抗激动剂苯福林收缩血管升高血压的作用。能舒张静脉及小动脉,发挥中等偏强的降压作用。它与酚妥拉明不同,降压时并不加快心率,也少增加收缩力及血浆肾素活性,也能增加血中高密度脂蛋白(HDL)是浓度,减轻冠脉病变。
该药口服易吸收,2小时内血浓达峰值,生物利用度为60%,t1/2为2.5~4小时。但口服后降压作用可持续10小时,与血浆蛋白结合率达97%,在肝中广泛代谢,首关消除显著。
【临床应用及不良反应】适用于各型高血压,单用治疗轻、中度高血压,重度高血压合用β受体阻断药及利尿药可增强降压效。
不良反应有眩晕、疲乏、虚弱等,首次给药可致严重的体位性低血压,晕厥、心悸等,称“首剂现象”,在直立体位,饥饿、低盐时较易发生。将首次用减为0.5mg,并在临睡前服用,使可避免发生。
其他α1受体阻断药酮色林(凯他舍林ketanserin)兼有抗5-羟色胺S1受体的作用,也可有效地治疗高血压
α、β受体阻断药
拉贝洛尔
拉贝洛尔(labetalol)对α、β受体均有竞争性拮抗作用,其中,阻断β1、β2受体的作用程度相似,对α1受体作用较弱,对α2受体则无效,故负反馈调节仍然存在,用药后不引起心率加快作用。
本药降压作用温和,适用于治疗各型高血压,无严重不良反应,对梗塞早期,通过其降低心肌壁张力而产生有益的作用。静脉注射可治疗高血压危象。
六、作用于血管平滑肌的抗高血压药
直接作用于血管平滑肌的抗高血压药肼屈嗪等,能直接松弛血管平滑肌,降低外周阻力,纠正血压上升所致的血流动力学异常。与其他类降压药不同的是,本类药物不抑制交感神经活性,不引起直立性低血压及阳萎等。久用后,其神经内分泌及植物神经的反射作用能抵消药物的降压作用(图26-3),从图可见最重要的反射变化是:①激活交感神经,致心输出量和心率增加而抵消降压作用,其增加心肌耗氧量的作用,对有严重冠状动脉功能不全或心脏储备力差者则易诱发心绞痛。②增强血浆肾素活性,肾素血症可增强交感活性导致循环中血管紧张素量增加而使血压上升,以上缺点必须合用利尿药及β受体阻断药加以纠正。
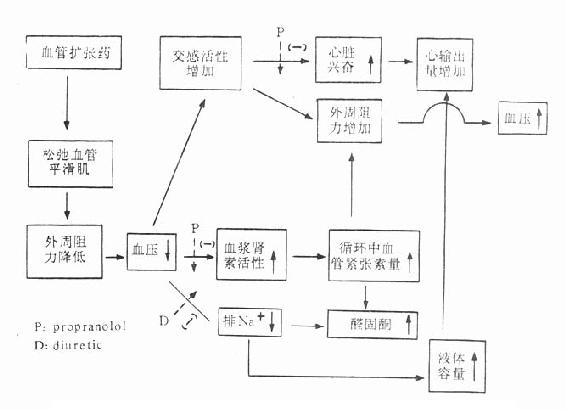
图26-3 久用扩血管药后其神经内分泌及植物神经的反射作用
直接作用于血管平滑肌的扩血管药可能作用于血管平滑肌细胞的兴奋——收缩偶联过程的不同部位,干予Ca2+的内流及Ca2+自胞内储库的释放,降低胞内游离Ca2+及其与平滑肌收缩蛋白的相互作用等。现知某些扩血管药可增加血管平滑肌的cGMP浓度,有的则通过开放钾通道使胞膜超极化而发挥作用。
肼屈嗪
肼屈嗪(hydralazine,肼苯哒嗪),为扩张小动脉的口服有效的降压药,对肾、冠状动脉及内脏血管的扩张作用大于对骨骼肌血管。适用于中度高血压,常与其他降压药合用。
口服吸收好,约65%~90%,给药1小时作用达峰值,维持约6小时。其不良反应有头痛、鼻充血、心悸、腹泻等。较严重时表现为心肌缺血和心衰。高剂量使用时可引起全身性红斑性狼疮样综合征,用量400mg/日,或更大,其发生率可达10%~20%,可见与剂量有关。将剂量降至200mg/日,上述反应少见。本经极少单用。
米诺地尔
米诺地尔(minoxidil,长压定),为作用强大的小动脉扩张药,口服吸收完全,能较持久地贮存于小动脉平滑肌中。其不良反应有水、钠潴留,心悸及多毛症,促进毛发生长可能与皮肤及毛发滤泡的血流增多有关。近证明本药激活了调节毛发杆蛋白的特殊基因而促进毛发杆的生长和成熟,故此药可作为治疗(男性)脱发药。
二氮嗪
二氮嗪(diazoxide,氯苯甲噻嗪),直接舒张血管平滑肌而降压,和米诺地尔一样,其降压机制部分是通过激活平滑肌细胞的IK(ATP)(ATP)所中介的钾通道,促进钾外流,使胞膜超极化,Ca2+通道失活,Ca2+内流减少。
临床上主要作静脉注射用,用于高血压危象及高血压脑病。不作长期用药。因此不良反应少见。如连用几天后,就应检测血糖水平。因本药可至高血糖症,此为药物激活了胰岛β细胞膜的IK(ATP),降低胰岛素释放所致。
钾通道开放剂吡那地尔(pinacidil),莱马卡林(lemakalim)为一类新型抗高血压药物,它们激活血管平滑肌细胞膜的IK(ATP)而发挥降压作用。
硝普钠
硝普钠(sodiumnitroprusside)又称亚硝基铁氰化钠(Na2Fe(CN)6NO·2H2O),属硝基扩张血管药,口服不吸收,需静脉滴注给药,起效快,约1分钟,停药5分钟内血压回升。其作用机制相似于硝酸酯类,能增加血管平滑肌细胞内cGMP水平而扩张血管。
用于高血压危象,特别对伴有急性心肌梗塞者或左室功能衰竭的严重高血压患者,治疗高血压危象一般按3μg/kg/分滴注,通过调整滴注速度来维持血压于所需水平。
由于该药能扩张动、静脉、降低前、后负荷而改善心功能,用于难治性心衰。
该药遇光易破坏,故滴注的药液应新鲜配制和避光。
不良反应有呕吐、出汗、头痛、心悸,均是过度降压所引起。本药毒性较你,在体内产生的CN-,在肝中被转化成SCN-,后者基本无毒,经肾排泄。但连用数日后,SCN-在体内蓄积,其浓度超过20mg/100ml时,可致中毒,因此宜监护SCN-的浓度。
第三节 抗高血压药物的应用原则
目前主张采用个体化治疗方案,其理论根据是:高血压的治疗目的不仅限于控制血压于正常水平,且应扩延为减少致死性及非致死性并发症,即药物也应能防止或逆转其他病理生理过程以延缓病程发展,最终延长患者生命。而高血压的生理病理过程却有很大个体差异。此外,个体化治疗还可照顾到相关的副作用,现分述如下:
1.
根据高血压程度的不同,可参考图26-4。选用药物。
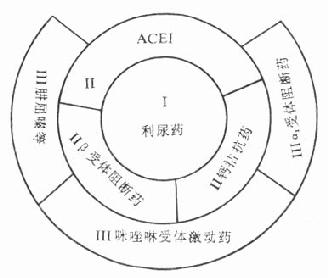
图26-4 高血压药物治疗
由图可见,对于轻、中度高血压患者,首选单药治疗,用Ⅰ或Ⅱ均可。用药后如血压仍大于18.7/12.0kPa(140/90mmHg)者,则二联用药,一般选Ⅰ+Ⅱ,或Ⅱ+Ⅲ,常用利尿药以抗水钠潴留;β受体阻断药与Ⅲ类药合用,可阻反射性肾素释放;ACEI可阻利尿药对RAAS的激活。若仍无效,则三联用药:如Ⅰ+Ⅱ+Ⅱ,或Ⅰ+Ⅱ+Ⅲ,即利尿药加β受体阻断药加扩管药(肼屈嗪、α1受体阻断药、钙拮抗药);或利尿药加钙拮抗药加咪唑啉受体激动药;或ACEI加袢利尿药加钙拮抗药;或利尿药加米诺地尔加β-受体阻断药均可。
必须指出现有抗高血压药物长期单独使用后常会失效,如加大剂量又易引起不良反应而难以继续应用,所以临床实践中常采用联合用药,以增强疗效及减少不良反应的发生。
2.高血压危象及脑病时药物的选用
宜静脉给药以迅速降低血压,可选用硝普钠、二氮嗪、粉防已碱,也可用高效以利尿药如呋噻米等,但应注意不可降压过快,以免造成重要器官灌流不足等。
3.根据并发症选用药物
高血压合并心功能不全、心扩大者,宜用利尿药、卡托普利、哌唑嗪等,不宜用β受体阻断药。
高血压合并肾功能不良者,宜用卡托普利、硝苯地平、甲基多巴。
高血压合并窦性心动过速,年龄在50岁以下者,宜用β受体阻断药。
高血压合并消化性溃疡者,宜用可乐定,不用利血平。高血压合并支气管哮喘、慢性阻塞性肺部疾患者,不用β受体阻断药。高血压伴有潜在性糖尿病或痛风者,不宜用噻嗪类利尿药。高血压伴有精神抑郁者,不宜用利血平或甲基多巴。有关各种并发症的选药原则见表。
表26-2 高血压并发其他病症时的选药
| 利尿剂 | β受体阻断药 | α受体阻断药 | 钙拮抗药 | ACEI | |
| 老年人 | ++ | +/- | + | + | + |
| 冠心病 | +/- | ++ | + | ++ | + |
| 心衰 | ++ | - | + | - | ++ |
| 脑血管病 | + | + | + | ++ | + |
| 肾功能不全 | ++ | +/- | + | ++ | ++※ |
| 糖尿病 | - | - | ++ | + | ++ |
| 血脂异常 | - | - | ++ | + | + |
| 哮喘 | + | - | + | + | + |
| 外周血管病 | + | - | + | ++ | + |
注:+ 适宜;+/- 一般不用;- 禁忌;※隐匿性肾血管病慎用
4.个体化治疗是90年代治疗高血压的特点,主要应根据患者的年龄、性别、种族、同时患有的疾病和接受的治疗等,使治疗个体化,要使患者得到最佳的抗高血压治疗,要防止动脉粥样梗化,控制其他危险因子(如高脂血症、糖尿病、吸烟等),逆转靶器官的损伤,维持和改善患者的生活质量,降低心血管的发病率及死亡率等。
药物治疗时的剂量个体化也是比较重要的,因不同患者或同一患者在不同病程时期,所需剂量不同。如可乐定、普萘洛尔,肼屈嗪等药物的治疗量可相差数倍,所以也应根据“最好疗效最少不良反应”的原则,选择每一患者的最佳剂量。
抗高血压药 制剂及用法
盐酸可乐定(clonidinehydrochloride)0.075~0.15mg/次,3次/日,口服,根据病情可适当逐渐增加剂量。0.15~0.3mg/次,肌内注射或静脉注射,必要时6小时重复一次。
盐酸哌唑嗪(prazosinhydrochloride)1mg/次,3次/日,口服。
盐酸普萘洛尔(propranololhydrochloride)10~20mg/次,3~4次/日,口服,以后每周增加剂量10~20mg,每日剂量有用至120mg者。
盐酸肼屈嗪(hydralazinehydrochloride)10~25mg/次,3次/日,口服。
二氮嗪(diazoxide)300mg快速静脉注射,溶液碱性极强,避免漏至血管外。
米诺地尔(minoxidil)2.5mg/次,2次/日,口服,逐渐增至5~10mg/次,2次/日。
硝普钠(sodiumnitroprusside)50mg临用时以5%葡萄糖溶液2~3ml溶解后再用同一溶液500ml稀释缓慢静脉滴注(容器避光),速度每分钟不超过3μg/kg。配制时间超过4小时的溶液不宜使用。
氢氯噻嗪(hydrochlorothiazide)12.5~25mg/次,2次/日,口服,见效后酌减,给维持量。
粉防已碱(tetrandrine)100mg/次,3次/日,口服,120~150mg/次,2次/日,静脉注射。
硝苯地平(nifedipine)5~10mg/次,3次/日,舌下含化。
卡托普利(captopril)开始12.5~25mg/次,口服,渐增至50mg/次,2~3次/日,每日最大剂量为450mg。
依那普利(enalapril)开始2.5~5mg,口服,渐增至5~40mg,分1~2次服用。
第二十七章 利尿药及脱水药
第一节 利尿药
利尿药(diuretics)是作用于肾,增加电解质及水排泄、使尿量增多的药物。临床应用很广。常用的利尿药按它们的效应力分类如下:
1.高效利尿药有呋塞米、依他尼酸及布美他尼等。
2.中效利尿药包括噻嗪类利尿药及氯酞酮等。
3.低效利尿药包括留钾利尿药如螺内酯,氨苯蝶啶、阿米洛利。碳酸酐酶抑制剂乙酰唑胺。
为了正确理解各类利尿药的作用及其机制,合理使用利尿药,将先介绍与利尿药有关的肾泌尿生理并分析各类利尿药的作用部位。
一、肾脏泌尿生理及利尿药作用部位
尿液的生成是通过肾小球滤过、肾小管再吸收及分泌而实现的,现分述如下:
(一)肾小球
血液流经肾小球、除蛋白质和血细胞外,其他成份均可滤过而形成原尿。原尿量的多少决定于有效滤过压。凡能增加有效滤过压的药物当可利尿。如氨茶碱,通过增加心肌收缩性,增加肾血流量及小球滤过率而利尿。但其利尿作用极弱,一般不作利尿用。正常人每日能形成180升原尿,但进入输尿管的终尿每日仅1~2升,可见约99%的原尿在肾小管被再吸收,它是影响终尿量的主要因素。目前常用的利尿药多数是通过减少肾小管对电解质及水的再吸收而发挥利尿作用的。
(二)肾小管
1.近曲小管 此段再吸收Na+约占原尿Na+量的60%~65%,原尿中约有90%的NaHCO3及部分NaCl在此段被再吸收。
Na+在近曲小管的转运可分成二相,Na+通过腔膜侧进入胞内;再通过基底膜离开细胞,后者由钠泵(K+、Na+、-ATPase)所驱动,此外,Na+在近端管可通过Na+-H+反向转运系统(antiporter)与H+按1:1进行交换而进入细胞内。H+由小管细胞分泌到小管液中,并将小管液中的Na+换回到细胞内。H+的产生来自H2O与CO2所生成的H2CO3,这一反应需上皮细胞内碳酸酐酶的催化,然后H2CO3再解离成H+和HCO3-,H+将Na+换入细胞内,然后由Na+泵将Na+送至组织间液。
![]()
若H+的生成减少,则Na+-H+交换减少,致使Na+的再吸收减少而引起利尿。碳酸酐酶抑制剂乙酰唑胺(acetazolamide,diamox)能使H+的生成减少而发挥利尿作用。但作用弱,易致代谢性酸血症,故现少用。
目前尚无高效的作用于近曲小管的利尿药,原因是药物抑制了近曲小管Na+的再吸收后,使近曲小管腔内原尿增多,小管有所扩张,原尿吸收面积增大,尿流速度减慢而停留时间延长,从而近曲小管本身出现代偿性再吸收,同时近曲小管以下各段肾小管也出现代偿性再吸收增多现象。
2. 髓袢升枝粗段的髓质和皮质部髓袢升枝的功能与利尿药作用关系密切。也是高效利尿药的重要作用部位,此段再吸收原尿中30%~35%的Na+,而不伴有水的再吸收。髓袢升枝粗段NaCl的再吸收受腔膜侧K+-Na+-2Cl-共同转运(co-transport)系统所控。该转运系统可将2个Cl-,一个Na+和一个K+同向转运到细胞内,其驱动力来自间液侧K+、Na+-ATP酶对胞内Na+的泵出作用,即共同转运的能量来自Na+浓度差的势能,进入胞内的Cl-,通过间液侧离开细胞,K+则沿着腔膜侧的钾通道进入小管腔内,形成K+的再循环。
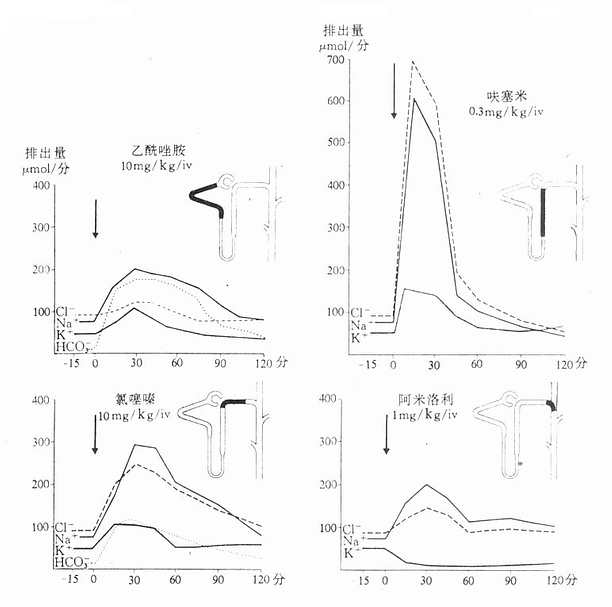
图27-1 比较三类(四个药)重要利尿药的作用部位(在肾单位用黑色标记)、作用强度、作用持续时间以及不同电解质自尿中的排出量
当原尿流经髓袢升枝时,随着NaCl的再吸收,小管液由肾乳头部流向肾皮质时,也逐渐由高渗变为低渗,进而形成无溶质的净水(free water,CH2O),这就是肾对尿液的稀释功能。同时NaCl被再吸收到髓质间质后,由于髓袢的逆流倍增作用,以及在尿素的共同参与下,使髓袢所在的髓质组织间液的渗透压逐步提高,最后形成呈渗透压梯度的髓质高渗区。这样,当尿液流经开口于髓质乳头的集合管时,由于管腔内液体与高渗髓质间存在着渗透压差。并经抗利尿激素的影响,水被再吸收,即水由管内扩散出集合管,大量的水被再吸收回去、称净水的再吸收,这就是肾对尿液的浓缩功能。
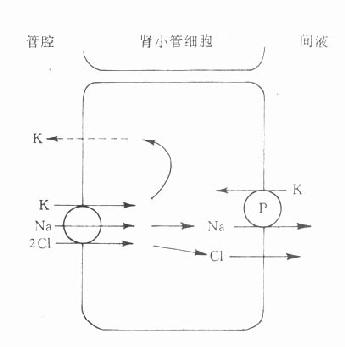
图27-2 髓袢升枝粗段细胞的Na+、K+、2Cl-共同转运系统
P=Na+-K+-ATP酶
实线──代表主动转运或次级主动转运
虚线┄┄代表被动弥散
综上所述,如当髓袢升枝粗段髓质和皮质部对NaCl的再吸收被抑制时,一方面肾的稀释功能降低(净水,即非渗透压所吸引的水生成减少);另一方面肾的浓缩功能也降低(净水再吸收减少),排出大量渗透压较正常尿为低的尿液,就能引起强大的利尿作用。高效利尿药呋塞米等,可抑制升枝粗段髓质和皮质部对氯化钠的再吸收,使肾的稀释功能降低,净水生成减少,同时又使肾的浓缩功能降低。中效噻嗪类利尿药等,抑制髓袢升枝粗段皮质部(远曲小管开始部分)对NaCl的再吸收,使肾的稀释功能降低,但不影响肾的浓缩功能。
3. 远曲小管及集合管 此段再吸收原尿Na+约5%~10%,其再吸收方式除继续进行Na+-H+交换外,同时也有Na+-K+交换过程,这是在醛固酮调节下进行的。醛固酮有三个作用:增加渗透酶蛋白(permeaseproteins)的合成而增强腔膜侧Na+的内流;兴奋间液侧的K+-Na+-ATP酶;促进细胞的生物氧化过程以提供ATP,为Na+泵活动供能,通过这些作用增加远曲小管、集合管对Na+的再吸收并分泌K+。如能抗醛固酮的调节功能或直接抑制K+-Na+交换,就会造成排Na+留K+而致利尿。螺内酯、氨苯蝶啶等药作用于此部位,它们又称留钾利尿药。
了解利尿药的作用部位,有助于理解各类利尿药效应力的高低,因后者是作用部位对Na+的再吸收能力所决定的。当然,实际用药效果还受药物用量、肾血流及血容量等因素的限制。
某种利尿药对电解质的排泄也与该药的作用部位有关,人体主要电解质是Na+、K+、Cl-、HCO3-,现有的各种利尿药都是排钠利尿药,用药后Na+和Cl-的排泄都是增加的,其排泄量的多少与利尿效应力一致,对钾的排泄,除留钾利尿药外,其他利尿药都能促进钾排泄。因它们不抑制远曲小管的K+-Na+交换,而且由于它们在远曲小管以上各段减少了Na+的再吸收,使到达远曲小管的尿液中含有较多的Na+,因而K+-Na+交换有所增加,另一方面由于利尿药降低血容量而激活肾压力感受器及肾交感神经而促进肾素的释放,其结果是使醛固酮分泌增加,因而促进K+-Na+交换而导致K+外排增多。故应用这些利尿药时应注意补K+。排HCO3-最多的利尿药为乙酰唑胺。噻嗪类利尿药也可使HCO3-的排泄略有增加,见表27-1。
表27-1 常用利尿药对电解质排泄及排钠力比较
| 药物 | 尿电解质的排泄 | 排钠力(滤过Na+量%) | 主 要 作 用 部 位 |
机 制 | |||
| Na+ | K+ | Cl- | HCO3- | ||||
| 呋塞米 依他尼酸 布美他尼 |
+++ | + | ++++ | ~23 | 髓袢升枝粗段髓质和皮质部 | 抑制Na+、K+、 2Cl-共同转运 系统 |
|
| 噻嗪类 氯酞酮 |
++ | + | ++ | + | ~8 | 髓袢升粗段皮质部(远曲小管开始部位) | 抑制NaCl再吸收 |
| 螺内酯 氨苯蝶啶 阿米洛利 乙酰唑胺 |
+ + |
- ++ |
+ |
+++ |
~2 ~4 |
远曲小管及集合管 近曲小管 |
竞争Ald受体阻 Na+通道,抑制 NaCl再吸收 ↓胞内H+形成 |
二、常用的利尿药
(一)高效利尿药
有呋塞米(furosemide,呋喃苯胺酸),依他尼酸(etacrynic acid,利尿酸),布美他尼(bumetanide)。它们的化学结构如下:
以上三药的药理特性相似,它们作用于髓袢升枝粗段,能特异性地与Cl-竞争Na+-K+-2Cl-共同转运系统的Cl-结合部位,抑制NaCl再吸收而发挥强大的利尿作用。
【药理作用】三药使肾小管对Na+的再吸收由原来的99.4%下降为70%~80%。在水与电解质平衡保持于正常水平时,持续给予大剂量呋塞米可使成人24小时内排尿50~60升。静注呋塞米可增加肾血流30%,并见前列腺素E(PGE)量增加,它在内源性肾功能受损的情况下可发挥保护作用,对急性肾功能衰竭有利。
高效利尿药可使小管液中Na+、Cl-浓度,尤其是Cl-的浓度显著增高,因而降低肾的稀释与浓缩功能,排出大量近于等渗的尿液。同时也增加Ca2+、Mg2+的排泄。Cl-的排出量往往超过Na+,故可出现低氯碱血症。
布美他尼也有磺胺基团,但效价较呋塞米高,量效曲线提示,0.125mg的布美他尼相当于11.5mg的呋塞米,通常单次口服0.5mg布美他尼相当于28.7mg呋塞米。
【体内过程】高效利尿药口服30分钟内、静注5分钟后生效,2小时作用达峰值,维持6~8小时。布美他尼与呋塞米,二者的表现分布容积约为0.15L/kg,血浆蛋白结合率约95%,然布美他尼的生物利用度是呋塞米的二倍。可能是由于所需剂量较低而吸收更完全之故。它们都经肾小球滤过,并通过近曲小管有机酸转运机制分泌而从尿中排出。约1/3经胆汁排泄,反复给药不易在体内蓄积。
【不良反应】
1.水与电解质紊乱常为过度利尿所引起,表现为低血容量、低血钾、低血钠、低氯碱血症等。低血钾症的症状为恶心、呕吐、腹胀、肌无力及心律失常等,严重时可引起心肌、骨骼肌及肾小管的器质性损害及肝昏迷,故应注意及时补充钾盐,加服留钾利尿药可避免或减少低血钾的发生。长期应用还可引起低血镁、K+和Mg2+之间的内在关系已明确,当低血K+与低血Mg2+同时存在时,如不纠正低血Mg2+,即使补充K+也不易纠正低血钾。
2.高尿酸血症和高氮质血症前者主要由利尿后血容量降低、胞外液浓缩,使尿酸经近曲小管的再吸收增加所致。另一原因是利尿药和尿酸经有机酸分泌途径排出时相互竞争,长期用药时多数病人可出现高尿酸血症,但临床痛风的发生率较低。
3.胃肠道反应表现为恶心、呕吐、上腹部不适,大剂量时尚可出现胃肠出血。
4.耳毒性呈剂量依赖性,表现为眩晕、耳鸣、听力减退或暂时性耳聋,依他尼酸最易引起,且可发生永久性耳聋。组织学检查发现耳蜗管基底膜毛细胞受损伤,内淋巴电解质成分改变,如Na+、Cl-浓度的升高等可能与耳毒有关,耳毒性主要发生在肾衰者使用高剂量利尿药时。布美他尼的耳毒性最小,为呋塞米的1/6,对听力有缺陷及急性肾衰者宜选用布美他尼。
【药物相互作用】氨基苷甙类抗生素及第一、二代头孢菌素等可增强高效利尿药的耳毒作用,应避免合用。非甾体抗炎药如吲朵美辛可减弱或抑制它们的排Na+作用,尤其在血容量降低时。华法林、氯贝特等可与它们竞争血浆蛋白的结合部位,而增加药物的毒性。
(二)中效利尿药
噻嗪类利尿药
噻嗪类(thiazides)利尿药有共同的基本结构,是由杂环苯并噻二嗪与一个磺酰胺基(-SO2NH2)组成。其一系列的衍生物是在2、3、6位代入不同基团而得。
化学结构上的微小改变就能改善药物的吸收,增强利尿强度,同时减轻对碳酸酐酶的抑制等,按等效剂量比,本类药物中各个利尿药的效价强度可相差达千倍,从弱到强的顺序依次为:氯噻嗪(chlorothiazide)<氢氯噻嗪(hydrochlorothiazide)<氢氟噻嗪(hydroflumethiazide)<苄氟噻嗪(bendroflumethiazide)<环戊噻嗪(cyclopenthiazide)。但噻嗪类药物的效能相同,所以有效剂量的大小在各药的实际应用中并无重要意义。氯酞酮(chlortalidon)无噻嗪环结构,但其药理作用相似,故在此一并介绍。
表27-2 噻嗪类利尿药及氯酞酮的化学结构
【药理作用】
1.利尿作用噻嗪类药物作用于髓袢升枝粗段皮质部(远曲小管开始部位)抑制NaCl的再吸收,此段排Na+量达原尿Na+的10%~15%,尿中除含较多的Cl-及Na+外,还含K+。长期服用可致低血钾、低血镁。本类药物具有碘酰胺基的结构,对碳酸酐酶有轻度抑制作用,所以也略增加HCO3-的排泄。
2.抗尿崩症作用噻嗪类利尿药能明显减少尿崩症患者的尿量,主要用于肾性尿崩症及加压素无效的垂体性尿崩症。其机制与噻嗪类对磷酸二酯酶的抑制作用有关,因此增加远曲小管及集合管细胞内cAMP的含量,后者能提高远曲小管对水的通透性。同时因增加NaCl的排出、造成负盐平衡,导致血浆渗透压的降低,减轻口渴感和减少饮水量,也使胞外容量减少和导致尿量减少。
3.降压作用它们是重要的抗高血压药物,详见抗高血压药章。
【体内过程】口服吸收良好,除氯噻嗪吸收率只有30%~35%外,其他噻嗪类药因脂溶性高,吸收率都在80%以上。它们在体内不被代谢,主要通过肾小球滤过及近曲小管分泌而排泄,少量由胆汁排泄。其中氢氯噻嗪的生物利用度约为70%,有较大的Vd,t1/2为8~10小时。本类药物可通过胎盘进入乳汁。
【不良反应】
1.电解质紊乱 如低血钾、低血镁、低氯碱血症等。
2.潴留现象如高尿酸血症、高钙血症,主要是药物减少细胞外液容量,增加近曲小管对尿酸的再吸收所致,痛风者慎用。
3.代谢性变化与剂量有关,可致高血糖、高脂血症。致肾素、醛固酮的过度分泌。对血脂可使血清甘油三酯及低密度脂蛋白-胆固醇(LDL-cho)量增加,同时伴有高密度脂蛋白(HDL)的减少,有报道同时应用β受体阻断药可防止利尿药引起的LDL-cho的升高。还可降低糖耐量,使血糖升高,可能是抑制了胰岛素的分泌或抑制肝PDE,因此使cAMP中介的糖原分解作用加强,糖尿病者慎用。
4.高敏反应 如发热、皮疹、过敏反应。
5.其他 可增高血尿素氮,加重肾功能不良。无尿及对磺胺过敏者禁用本类药物。
(三)低效利尿药
包括留钾利尿药及乙酰唑胺。
螺内酯
螺内酯(spironolactone)又名安体舒通(antisterone),化构与醛固酮相似,具有抗醛固酮作用,已知醛固酮从肾上腺皮质释放后,能进入远曲小管细胞,并与胞浆同受体结合成醛固酮-受体复合物,然后移入胞核,并诱导特异mRNA的形成,后者促进一种能调控管腔膜对Na+、K+通透性的蛋白质的合成等。螺内酯可与醛固酮竞争醛固酮受体,最终阻碍蛋白质的合成,因此螺内酯能抑制Na+-K+交换,减少Na+的再吸收和钾的分泌,表现出排Na+留K+作用。
螺内酯的利尿作用不强,起效慢而维时久,其利尿作用与体内醛固酮的浓度有关。仅当体内有醛固酮存在时,它才发挥作用。对切除肾上腺的动物则无利尿作用。由于其利尿作用较弱,抑制Na+再吸收量还不到3%,因此较少单用。常与噻嗪类利尿药或高效利尿药合用以增强利尿效果并减少K+的丧失。
【不良反应】久用可引起高血钾,尤当肾功能不良时,故肾功能不良者禁用。还有性激素样副作用,可引起男子乳房女性化和性功能障碍,致妇女多毛症等。
氨苯蝶啶及阿米洛利
氨苯蝶啶(triamterene,三氨蝶啶)及阿米洛利(amiloride,氨氯吡脒)虽化构不同,却有相同的药理作用,它们的化学结构如下:
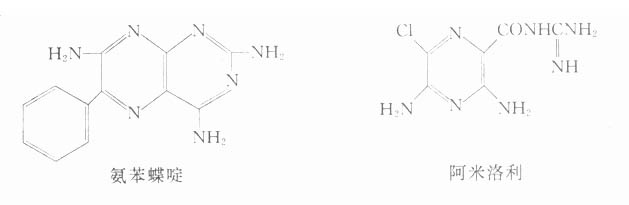
二药作用于远曲小管及集合管,阻滞Na+通道而减少Na+的再吸收,因而可发挥较弱的利尿作用。同时K+的排泄减少。当醛固酮分泌量过多时,或与其他利尿药合用时,其留钾作用更为明显,二药作用并非竞争性拮抗醛固酮所致。因它们对切除肾上腺的动物仍有留钾利尿作用。现认为是药物抑制远曲小管及集合管对K+的分泌所致。但原始作用仍是减少Na+的再吸收,由于Na+的再吸收减少,使管腔的负电位减小,管腔内外电位差下降,乃使钾分泌的驱动力减小。在远曲小管阿米洛利还抑制钙的排泄,这一作用也是与抑制Na+再吸收相偶联的。
【不良反应】二药长期服用均可引起高血钾症。肾功能不良者、糖尿病者、老人较易发生。其中氨苯蝶啶还抑制二氢叶酸还原酶,引起叶酸缺乏。肝硬化病人服用此药可发生巨幼红细胞性贫血,偶可引起高敏反应及形成肾结石。
三、利尿药的临床应用
1.消除水肿这是利尿药的主要适应证,水肿常见于心、肝、肾性疾病,其病因及病理变化虽不相同,但基本表现均是细胞间液增加,Na+潴留是细胞间液增加的主要因素,利尿通过排Na+、排水而治疗水肿。在应用利尿药前要注意下列几点:①对基本疾病作病因治疗;②动员组织间水肿液或体腔中积液进入血液循环,便于利尿消肿。这就要求患者卧床休息并进行支持疗法等;③用低盐饮食以减少体内Na+量;④注意治疗失败的可能,如观察肾小球滤过率是否下降,醛固酮分泌是否继发性增多等。
(1)心性水肿治疗心性水肿主要依靠改善心功能,利尿药能减少或消除水肿而降低心负荷、改善心功能。用噻嗪类利尿药,宜加用钾盐。对中度水肿可用氢氯噻嗪加留钾利尿药。对一般利尿药无效的严重水肿,可合用高效利尿药和留钾利尿药,要定期检查血钾含量。此外,利尿药的用量不宜过大,否则尿液排泄速度超过水肿液进入血浆的速度,会引起继发性醛固酮增多而降低利尿作用。
(2)肾性水肿急性肾炎时,一般不用利尿药,必要时用氢氯噻嗪。主要采用无盐膳食和卧床休息以消退水肿。肾病综合征时,水肿的形成与大量蛋白尿引起血浆白蛋白减少及血浆胶体渗透压下降有关。又因循环血量减少可继发性地致醛固酮分泌增加,导致NaCl及水再吸收的增加而加重水肿。故应限水限NaCl,并给予白蛋白以提高胶体渗透压。对高度水肿者可用噻嗪类药物加留钾利尿药。效果不明显时可用高效利尿药加留钾利尿药。
(30)肝性水肿肝性水肿多伴有继发性醛固酮增多症,所以开始治疗时不宜采用高效利尿药,否则将会引起严重的电解质紊乱,甚至因严重低血钾症而诱发肝昏迷。一般宜先用留钾利尿药,或留钾利尿药加噻嗪类利尿药,如疗效不显著,可合用留钾及高效利尿药。
(4)急、慢性肾功能衰竭高效利尿药可预防急性肾功能衰竭和治疗急性肾衰早期的少尿,能增加尿量及尿流速度,防止肾小管萎缩、坏死及急性肾衰时的无尿。呋塞米还可治疗慢性肾功能衰竭,但需大量。用药后可使尿量明显增加。当肾小球滤过率降至2ml/min时,或当其他利尿药无效时,仍可有效。
(5)急性肺水肿及脑水肿静脉注射呋塞米等高效利尿药可对此发挥良好效果。它们能使血容量及细胞外液明显减少,进而降低回心血量,减少左室充盈压及降低肺楔压。还可通过舒张血管、增加静脉容量、降低左室舒张末压而消除肺水肿,已证明呋塞米等促进血管扩张物前列腺素的释放,也可中介上述效应。对脑水肿合并左心室功能不全者也有良效。
2.慢性心功能不全的治疗 详见二十五章
3.高血压的治疗 详见二十六章
4.尿崩症肾性尿崩症,最常用噻嗪类利尿药,用药二天排出大量Na+后见效。垂体性尿崩症,常用加压素治疗,而利尿药可作为辅助治疗,通过降低胞外容量,促进远曲小管再吸收而使尿量减少。
5.特发性高尿钙症和钙结石治疗量噻嗪类利尿药可使正常人、原发性甲状旁腺功能亢进及高尿钙患者尿钙的排出显著降低,用于防止钙结石的形成,是因本类药物能增强远曲小管对Ca++的再吸收。
6.高血钙症强效利尿药抑制髓袢升枝粗段对钙的再吸收,增加钙排出而降低血钙。
7.加速某些毒物的排泄当某些药物或毒物急性中毒时,可选用高效利尿药强迫利尿,同进配合输液,使尿量在一天内达5升以上,可加速毒物排出,但这一作用仅对以原型自尿排出的药物或毒物有效。
8.青光眼乙酰唑胺已少作利尿药使用,但可用于治疗青光眼,因它可使眼房水生成减少而降低眼内压。
9.选用药物可参考表27-3。
表27-3 常用利尿药利尿作用的比较
| 药 物 | 利尿作用 | ||
| 开始时间 | 峰值时间(h) | 维持时间 | |
| 袢利尿药 | |||
| 呋塞米 | 口服 60分 静注5~10分 |
1~2 15~20分 |
6~8 1~3 |
| 依他尼酸 | 口服 30分 静注5~10分 |
1~2 15~20分 |
6~8 1~3 |
| 布美他尼 | 口服 30分 静注5~10分 |
1~2 15~20分 |
6~8 1~3 |
| 噻嗪类利尿药 | |||
| 氯噻嗪 | 2 | 4 | 6~12 |
| 氢氯噻嗪 | 2 | 4 | 6~12 |
| 氢氟噻嗪 | 1~2 | 3~4 | 18~24 |
| 苄氟噻嗪 | 1~2 | 6~12 | 18~24 |
| 环戊噻嗪 | 6 | 7~12 | 18~24 |
| 氯酞酮 | 2 | 6 | 48~72 |
| 留钾利尿药 | |||
| 螺内酯 | 24 | 48~72 | 72~96 |
| 氨苯蝶啶 | 2~4 | 6 | 7~9 |
| 阿米洛利 | 2 | 6 | 12~24 |
第二节 脱水药
脱水药又称渗透性利尿药(osmotic diuretics)是指能使组织脱水的药物。包括甘露醇、山梨醇、高渗葡萄糖等。它们的药物作用完全决定于溶液中药物分子本身所发挥的渗透压作用。它们应具备如下特点:①易经肾小球滤过;②不易被肾小管再吸收;③在体内不被代谢;④不易从血管透入组织液中。根据上述特性,这类药物在大量静脉给药时,可升高血浆渗透压及肾小管腔液的渗透压而产生脱不及利尿作用。
甘露醇
甘露醇(mannitol)为一已六醇结构,临床用其20%的高渗溶液。
【药理作用】
1.静脉注射后,该药不易从毛细血管渗入组织,能迅速提高血浆渗透压,使组织间液水分向血浆转移而产生组织脱水作用。
2.利尿作用 静注高渗甘露醇后,一般在10分钟左右起效,能迅速增加尿量及排出Na+、K+。经2~3小时利尿作用达高峰。
甘露醇产生排钠利尿作用的原因是通过稀释血液而增加循环血容量及肾小球滤过率,并间接抑制Na+、K+、2Cl共同转运系统,减少髓袢升枝对NaCl的再吸收,降低髓质高渗区的渗透压,使集合管中水的再吸收减少。甘露醇还能扩张肾血管、增加肾髓质血流量,使髓质间液Na+和尿素易随血流移走,这也有助于降低髓质高渗区的渗透压而利尿。
【临床应用】
1.脑水肿及青光眼该药不易进入脑组织或眼前房等有屏障的特殊组织,静脉滴入甘露醇的高渗溶液使这些组织特别容易脱水,对多种原因引起的脑水肿(如脑瘤、颅脑外伤外缺氧等情况时)是首选药。甘露醇也降低青光眼患者的房水量及眼内压,短期用于急性青光眼,或术前使用以降低眼内压。
2.预防急性肾功能衰竭肾功能衰竭时应用甘露醇,能在肾小管液中发生渗透效应,阻止水分再吸收,维持足够的尿流量,且使肾小管内有害物质稀释,从而保护肾小管,使其免于坏死。还能改善急性肾衰早期的血流动力学变化,对肾功能衰竭伴有低血压者,该药维持肾小球滤过率的效果也远比盐水为佳。
【不良反应】注射过快时可引起一过性头痛、眩晕和视力模糊。禁用于慢性心功能不全者,因可增加循环血量而增加心脏负荷。
山梨醇
山梨醇(sorbitol)是甘露醇的同分异构体,作用与临床应用同甘露醇,但其水溶性较高,一般可制成25%的高渗液使用,进入体内后可在肝内部分转化为果糖,故作用较弱。
葡萄糖
50%的高渗葡萄糖也有脱水及渗透性利尿作用,但高渗作用维持不久,因易被代谢,并能部分地从血管弥散到组织中,常与甘露醇合用以治疗脑水肿。
利尿药及脱水药 制剂及用法
呋塞米(furosemide)口服,20mg/次,1~3次/日,为避免发生电解质紊乱,应从小量开始,间歇给药,即服药1~3日,停药2~4日。注射,20mg/次,每日或隔日一次,肌内注射或稀释后缓慢静脉注射。
布美他尼(bumetanide) 口服,0.5~2mg/日,单次,注射,0.5~1mg/次,肌内注射或静脉注射。
依他尼酸(etacrynic acid) 口服,25mg/次,1~3次/日,小量开始,可增加剂量至有效为止。注射,25mg/次,每日一次,静脉注射每次25mg加于25%葡萄糖液20ml中,注射部位需经常更换,以免发生局部血栓性脉管炎。
氢氯噻嗪(hydrochlorothiazide) 口服,25~50mg/次,2~3次/日。
环戊噻嗪(cyclopenthiazide) 口服,0.25g/次,1~2次/日。
苄氟噻嗪(bendroflumethiazide) 口服,5~15mg/次,每日一次。
氯酞酮(chlortalidone) 口服,100mg/次,每日或隔日一次。
螺内酯(antisterone,aldactone) 口服,20mg/次,3~4次/日。
氨苯蝶啶(triamterene) 口服,50~100mg/次,2~3次/日。
乙酰唑胺(acetazolamide) 口服,0.25g/次。治疗青光眼,2~3次/日。利尿1次/日或隔日。
甘露醇(mannitol)注射,1~2g/kg/次,静脉滴注,10ml/分必要时4~6小时重复使用,使在血液中迅速达到所需浓度。
山梨醇(sorbitol)注射,1~2g/kg/次,静脉滴注,必要时可重复注射。
葡萄糖(glucose)注射,50%溶液20ml,40~60ml/次,静脉注射。
第二十八章 作用于血液及造血器官的药物
第一节 抗凝血药
血液凝固是一个复杂的蛋白质水解活化的连锁反应,最终使可溶性的纤维蛋白原变成稳定、难溶的纤维蛋白,网罗血细胞而成血凝块。参与的凝血因子包括以罗马数字编号的12个凝血因子和前激肽释放酶(prekallikrein,Pre-K)、激肽释放酶(kallikrein,Ka)、高分子激肽原(high molecular weight kininogen, HMWK)、血小板磷脂(PL或PF3)等,反应过程见图28-1。抗凝血药(anticoagulants)是一类干扰凝血因子,阻止血液凝固的药物,主要用于血栓栓塞性疾病的预防与治疗。
图28-1 凝血机制图示
HMWK:高分子激肽原 Ka:激肽释放酶
TPL:组织凝血活素 PL:血小板磷脂
肝素
【来源与化学】 肝素(heparin)含有长短不一的酸性粘多糖。主要由硫酸-D-葡萄糖胺、硫酸-L-艾杜糖醛酸、硫酸-D-葡萄糖胺及D-葡萄糖醛酸中两种双糖单位交替连接而成,是一分子量为5000~30000的混合物。含有大量硫酸基和羧基,带大量阴电荷呈强酸性。药用肝素是从猪小肠和牛肺中提取而得。
【药理作用】肝素在体内、体外均有强大抗凝作用。静脉注射后,抗凝作用立即发生,这与其带大量负电荷有关,可使多种凝血因子灭活。这一作用依赖于抗凝血酶Ⅲ(antithrombin Ⅲ,AT Ⅲ)。At Ⅲ是凝血酶及因子Ⅻα、Ⅺα、Ⅸα、Ⅹα等含丝氨酸的蛋白酶的抑制剂。它与凝血酶通过精氨酸-丝氨酸肽键相结合。形成At Ⅲ凝血酶复合物而使酶灭活,肝素可加速这一反应达千倍以上。肝素与At Ⅲ所含的赖氨酸结合后引起AT Ⅲ构象改变,使At Ⅲ所含的精氨酸残基更易与凝血酶的丝氨酸残基结合。一旦肝素-At Ⅲ凝血酶复合物形成,肝素就从复合物上解离,再次与另一分子At Ⅲ结合而被反复利用。At Ⅲ-凝血酶复合物则被网状内皮系统所消除。抑制凝血酶活性的作用与肝素分子长度有关。分子越长则酶抑制作用越大。
肝素也有降脂作用,因它能使血管内皮释放脂蛋白脂酶,水解乳糜微粒及VLDL。但停药后会引起“反跳”,使血脂回升。
【体内过程】肝素是带大量阴电荷的大分子,口服不被吸收。常静脉给药,60%集中于血管内皮,大部分经网状内皮系统破坏,极少以原形从尿排出。肝素抗凝活性t1/2与给药剂量有关,静脉注射100,400,800U/kg,抗凝活性t1/2分别为1,2.5和5小时。肺栓塞、肝硬化患者t1/2延长。
【临床应用】
1.血栓栓塞性疾病,防止血栓形成与扩大,如深静脉血栓、肺栓塞、脑栓塞以及急性心肌梗塞。
2.弥漫性血管内凝血(DIC),应早期应用,防止因纤维蛋白原及其他凝血因子耗竭而发生继发性出血。
3.心血管手术、心导管、血液透析等抗凝。
【不良反应】应用过量易引起自发性出血。一旦发生,停用肝素,注射带有阳电荷的鱼精蛋白(protamine),每1mg鱼精蛋白可中和100U肝素。部分病人应用肝素2~14天期间可出现血小板缺乏,与肝素引起血小板聚集作用有关。
肝素不易通过胎盘屏障,但妊娠妇女应用可引起早产及胎儿死亡。
连续应用肝素3~6月,可引起骨质疏松,产生自发性骨折。肝素也可引起皮疹、药热等过敏反应。肝、肾功能不全,有出血素质、消化性溃疡、严重高血压患者、孕妇都禁用。
香豆素类
是一类含有4-羟基香豆素基本结构的物质,口服参与体内代谢才发挥抗凝作用,故称口服抗凝药。有双香豆素(dicoumarol)、华法林(warfarin,苄丙酮香豆素)和醋硝香豆素(acenocoumarol,新抗凝)等。它们的药理作用相同。
【药理作用】香豆素类是维生素K拮抗剂,在肝脏抑制维生素K由环氧化物向氢醌型转化,从而阻止维生素K的反复利用,影响含有谷氨酸残基的凝血因子Ⅱ、Ⅶ、Ⅸ、Ⅹ的羧化作用,使这些因子停留于无凝血活性的前体阶段,从而影响凝血过程。对已形成的上述因子无抑制作用,因此抗凝作用出现时间较慢。一般需8~12小时后发挥作用,1~3天达到高峰,停药后抗凝作用尚可维持数天。双香豆素抗凝作用慢而持久,持续4~7天。华法林作用出现较快,持续2~5天。
【体内过程】 华法林口服吸收完全,1小时后血浆中即能测到,2~8小时达高峰。与血浆蛋白结合率为90%~99%。t1/2为10~60小时。主要在肝及肾中代谢。双香豆素吸收不规则。与血浆蛋白结合率为90%~99%。t1/2为10~30小时。醋硝香豆素t1/2为8小时,还原型代谢产物仍有抗凝作用,t1/2为20小时。
【临床应用】用途与肝素同,可防止血栓形成与发展。也可作为心肌梗塞辅助用药。口服有效,作用时间较长。但作用出现缓慢,剂量不易控制。也用于风湿性心脏病、髋关节固定术、人工置换心脏瓣膜等手术后防止静脉血栓发生。
【不良反应】剂量应根据凝血酶原时间控制在25~30秒(正常值12秒)进行调节。过量易发生出血,可用维生素K对抗,必要时输新鲜血浆或全血。禁忌证同肝素。其他不良反应有胃肠反应、过敏等。
【药物相互作用】 ①食物中维生素K缺乏或应用广谱抗生素抑制肠道细菌,使体内维生素K含量降低,可使本类药物作用加强。②阿司匹林等血小板抑制剂可与本类药物发生协同作用。③水合氯醛、羟基保泰松、甲磺丁脲、奎尼丁等可因置换血浆蛋白,水杨酸盐、丙咪嗪、甲硝唑、西咪替丁等因抑制肝药酶均使本类药物作用加强。④巴比妥类、苯妥英钠因诱导肝药酶,口服避孕药因增加凝血作用可使本类药物作用减弱。
第二节 抗血小板药
血小板在止血、血栓形成、动脉粥样硬化等过程中起着重要作用。药物主要通过抑制花生四烯酸代谢,增加血小板内cAMP浓度等机制而抑制血小板粘附、聚集和分泌功能。
阿司匹林能与环加氧酶活性部分丝氨酸发生不可逆的乙酰化反应,使酶失活,抑制花生四烯酸代谢,减少对血小板有强大促聚集作用的血栓烷A2(TXA2)的产生,使血小板功能抑制。环加氧酶的抑制,也抑制血管内皮产生前列环素(PGI2),后者对血小板也有抑制作用。然而阿司匹林对血小板中环加氧酶的抑制是不可逆的,只有当新的血小板进入血液循环才能恢复。而血管内皮细胞中环加氧酶因DNA合成而较快恢复。因此,每天口服75mg的阿司匹林就能引起最大抗血小板作用。现已明确,阿司匹林对血小板功能亢进而引起血栓栓塞性疾病效果肯定。对急性心肌梗塞或不稳定性心绞痛患者,可降低再梗塞率及死亡率;对一过性脑缺血也可减少发生率及死亡率。
双嘧达莫
双嘧达莫(dipyridamole)又名潘生丁(persantin),对血小板有抑制作用。能抑制磷酸二酯酶,使cAMP增高,也能抑制腺苷摄取,进而激活血小板腺苷环化酶使cAMP浓度增高。单独应用作用较弱。与华法林合用防止心脏瓣膜置换术术后血栓形成。
前列环素
前列环素(prostacyclin,PGI2)能激活腺苷环化酶而使cAMP浓度增高。既能抑制多种诱导剂引起的血小板聚集与分泌,又能扩张血管,有抗血栓形成作用。PGI2极不稳定,t1/2仅2~3分。采用静脉滴注,用于急性心肌梗塞,外周闭塞性血管疾病等。
噻氯匹啶
噻氯匹啶(ticlopidine)为一强效血小板抑制剂,能抑制ADP、AA、胶原、凝血酶和血小板活化因子等所引起的血小板聚集。口服吸收良好,临床应用24~48小时出现作用,3~5天达高峰,t1/2为24~33小时。用于预防急性心肌再梗塞,一过性脑缺血及中风和治疗间歇性跛行,有稳定型心绞痛等。
第三节 纤维蛋白溶解药
凝血中形成的纤维蛋白,可经纤溶酶作用从精氨酸-赖氨酸键上分解成可溶性产物,使血栓溶解。纤维蛋白溶解药(fibrinolytic drugs)激活纤溶酶而促进纤溶,也称溶栓药(thrombolytic drugs),用于治疗急性血栓栓塞性疾病。对形成已久并已机化的血栓难以发挥作用。目前应用的纤溶药主要缺点是对纤维蛋白无特异性,诱发血栓溶解同时伴有严重出血。较新纤溶药t-PA,scu-PA有一定程度的特异性,但人体应用仍有出血并发症,半衰期又短。为加强特异性以减少出血并发症,并延长半衰期,采用生物工程学方法研制开发高效而特异的新纤溶药的工作正在进行。
链激酶
链激酶(streptokinase,SK)是C组β溶血性链球菌产生的一种蛋白质,能与纤溶酶原结合,形成SK-纤溶酶原复合物后,促使游离的纤溶酶原转变成纤溶酶,溶解纤维蛋白。因此,需选合适剂量以发挥最大效应。合适量应使SK-纤溶酶原复合物与纤溶酶之比为1:10。静脉或冠脉内注射可使急性心肌梗塞面积缩小,梗塞血管重建血流。对深静脉血栓、肺栓塞、眼底血管栓塞均有疗效。但须早期用药,血栓形成不超过6小时疗效最佳。严重不良反应为出血,因为被激活的纤溶酶不但溶解病理性,也溶解生理性的纤维蛋白。SK有抗原性,体内若有SK抗体可中和SK,还可引起过敏反应。活动性出血三个月内,有脑出血或近期手术史者禁用。有出血倾向、胃、十二指肠溃疡,分娩未满四周,严重高血压、癌症患者禁用。
尿激酶
尿激酶(urokinase,UK)由人肾细胞合成,自尿中分离而得,无抗原性。能直接激活纤溶酶原,使纤溶酶原从精氨酸-缬氨酸处断裂成纤溶酶。UK在肝、肾灭活。t1/2为11~16分。临床应用同SK,用于脑栓塞疗效明显。因价格昂贵,仅用于SK过敏或耐受者。不良反应为出血及发热,较SK少。禁忌证同SK。
组织纤溶酶原激活因子
组织纤溶酶原激活因子(tissue plasminogen activator,t-PA)为较新的纤溶药。内源性t-PA由血管内皮产生,已能用DNA重组技术制备。t-PA对循环血液中纤溶酶原作用很弱,对与纤维蛋白结合的纤溶酶原作用则强数百倍,所以对血栓部位有一定选择性。t1/2为3分,静脉滴注用于急性心肌梗塞。剂量过大也引起出血。
茴酰化纤溶酶原链激酶激活剂复合物
茴酰化纤溶酶原链激酶激活剂复合物(anisoylated plasminogen streptokinaseactivator complex,APSAC)是一种新型纤溶酶原激活剂。其特点是通过茴酰化使纤溶酶原的活性部位得到保护,这样可避免注射时非特异地激活,进入体内缓慢脱茴酰而生效。血浆t1/2为105~120分,一次静脉注射30mg就能产生较好溶栓效果。对发病6小时内的急性心肌梗塞患者,血管再通率与冠脉注射SK相近。不良反应与等剂量SK相等。
第四节 促凝血药
维生素K
维生素K(vitamin K)的基本结构为甲萘醌。存在于植物中的为K1,由肠道细菌合成或得自腐败鱼粉者为K2,均为脂溶性。人工合成的K3为亚硫酸氢钠甲萘醌(menadionesodium bisulfate),K4为乙酰甲萘醌(menadione diacetate),均为水溶性。
【药理作用】维生素K作为羧化酶的辅酶参与凝血因子Ⅱ、Ⅶ、Ⅸ、Ⅹ的合成。这些因子上的谷氨酸残基必须在肝微粒体酶系统羧化酶的作用下形成9~12个γ-羧谷氨酸,才能使这些因子具有与Ca2+结合的能力,并连接磷脂表面和调节蛋白,从而使这些因子具有凝血活性。在羧化反应中,氨醌型维生素K被转为环氧型维生素K,后者在NADH作用下还原为氢醌型,继续参与羧化反应。维生素K缺乏或环氧化物还原反应受阻(被香豆素类),因子Ⅱ、Ⅶ、Ⅸ、Ⅹ合成停留于前体状态,凝血酶原时间延长,引起出血。
图28-2 维生素K作用示意图
KH2氢醌型维生素K,KO环氧型维生素K
【临床应用】用于维生素K缺乏引起的出血,如梗阻性黄疸、胆瘘,慢性腹泻所致出血,新生儿出血,香豆素类、水杨酸钠等所致出血。长期应用广谱抗生素应作适当补充,以免维生素K缺乏。
【不良反应】维生素K1静脉注射太快可产生潮红、呼吸困难、胸痛、虚脱。较大剂量维生素K3对新生儿、早产儿可发生溶血及高铁血红蛋白症。葡萄糖-6-磷酸脱氢酶缺乏病人也可诱发溶血。
抗纤维蛋白溶解剂
抗纤溶剂(antifibrinolysin)是一类竞争性对抗纤溶酶原激活因子,高浓度也抑制纤溶酶活性的物质。用于纤溶亢进所致出血,如肺、肝、脾、前列腺、甲状腺、肾上腺等手术时的异常出血。口服吸收良好,也可注射给药。临床常用的有氨甲苯酸(P-aminomethylbenzoicacid,PAMBA)、氨甲环酸(tranexamicacid,AMCHA)等。用量过大可致血栓形成,诱发心肌梗塞。
第五节 抗贫血药
循环血液中红细胞数或血红蛋白量低于正常称为贫血。临床常见贫血为缺铁性贫血,也有巨幼红细胞性贫血和再生障碍性贫血。后者是骨髓造血功能抑制所致,治疗比较困难。缺铁性贫血可用铁剂,巨幼细胞性贫血可用叶酸和维生素B12治疗。
铁剂
常用的有硫酸亚铁(ferrous sulfate)、枸橼酸铁铵(ferric ammonium citrate)和右旋糖酐铁(iron dextran)等。
【体内过程】口服铁剂或食物中外源性铁都以亚铁形式在十二指肠和空肠上段吸收。胃酸、维生素C、食物中果糖、半胱氨酸等有助于铁的还原,可促进吸收。胃酸缺乏以及食物中高磷、高钙、鞣酸等物质使铁沉淀,有碍吸收。四环素等与铁络合,也不利于吸收。食物中肉类的血红素中铁吸收最佳。蔬菜中铁吸收较差。一般食物中铁吸收率为10%,成人每天需补充铁1mg ,所以食物中铁为10~15mg就能满足需要。铁的吸收与体内贮存铁多少有关。吸收进入肠粘膜的铁根据机体需要或直接进入骨髓供造血使用,或与肠粘膜去铁蛋白结合以铁蛋白(ferritin)形式贮存其中。
体内铁的转运需要转铁蛋白(transferrin)。它是分子量为76000的β1糖蛋白,有2个铁结合位。胞浆膜上有转铁蛋白受体,铁-转铁蛋白复合物与受体结合,通过受体调节的胞饮作用进入细胞,铁分离后,去铁的转铁蛋白被释出细胞外继续发挥作用。
铁的排泄主要通过肠粘膜细胞脱落以及胆汁、尿液、汗液而排除体外,每日约1mg。
【临床应用】治疗缺铁性贫血,疗效基佳。口服铁剂一周,血液中网织红细胞即可上升,10~14天达高峰,2~4周后血红蛋白明显增加。但达正常值常需1~3月。为使体内铁贮存恢复正常,待血红蛋白正常后尚需减半量继续服药2~3月。
硫酸亚铁吸收良好,价格也低,最常用。枸橼酸铁铵为三价铁,吸收差,但可制成糖浆供小儿应用。右旋糖酐铁供注射应用,仅限于少数严重贫血而又不能口服者应用
【不良反应】口服铁剂对胃肠道有刺激性,可引起恶心、腹痛、腹泻。饭后服用可以减轻。也可引起便秘,因铁与肠腔中硫化氢结合,减少了硫化氢对肠壁的刺激作用。小儿误服1g以上铁剂可引起急性中毒,表现为坏死性胃肠炎、呕吐、腹痛、血性腹泻、休克、呼吸困难、死亡。急救措施为以磷酸盐或碳酸盐溶液洗胃,并以特殊解毒剂去铁胺(deferoxamine)注入胃内以结合残存的铁。
叶酸类
叶酸(folic acid)是由喋啶核、对氨苯甲酸及谷氨酸三部分组成。广泛存在于动、植物性食品中。
【药理作用】食物中叶酸和叶酸制剂进入体内被还原和甲基化为具有活性的5-甲基四氢叶酸(5-CH3H4PteGlu)。进入细胞后5-CH3H4PteGlu作为甲基供给体使维生素B12转成甲基B12,而自身变为H4PteGlu,后者能与多种一碳单位结合成四氢叶酸类辅酶,传递一碳单位,参与体内多种生化代谢,包括①嘌呤核苷酸的从头合成;②从尿嘧啶脱氧核苷酸(dUMP)合成胸嘧啶脱氧核苷酸(dTMP);③促进某些氨基酸的互变(图28-3)。当叶酸缺乏时,上述代谢障碍,其中最为明显的是dTMP合成受阻,导致DNA合成障碍,细胞有丝分裂减少。由于对RNA和蛋白质合成影响较少,使血细胞RNA:DNA比率增高,出现巨幼红细胞性贫血。消化道上皮增殖受抑制,出现舌炎、腹泻。
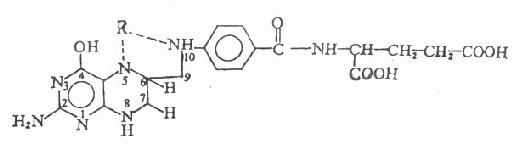
| 位置 | R | 四氢叶酸及其类似物名称 |
| N5 | -H | H4PteGlu(四氢叶酸) |
| N5 | -CH3 | 5-CH3H4PteGlu(5-甲基四氢叶酸) |
| N5 | -CHO | 5-CHOH4PteGlu(5-甲酰四氢叶酸) |
| N10 | -CHO | 10-CHOH4PteGlu(10-甲酰四氢叶酸) |
| N5,10 | =CH- | 5,10-CHH4PteGlu(5,10-甲炔四氢叶酸) |
| N5,10 | -CH2- | 5,10-CH2H4PteGlu(5,10-甲烯四氢叶酸) |
四氢叶酸及其一碳单位取代物的结构和名称
图28-3 叶酸和维生素B12的代谢及它们的相互关系
【体内过程】正常机体每日最低需要叶酸50μg,食物中每天有50~200μg叶酸在十二指肠和空肠上段吸收,妊娠妇女可增至300~400μg。食物中叶酸多为聚谷氨酸形式,吸收前必须在肠粘膜经α-L-谷胺酰转移酶(α-L-glutamyl transferase)水解成单谷氨酸形式,并经还原和移甲基作用形成5-CH3H4PteGlu后才吸收入肝及血液,广泛分布于体内。经尿和胆汁排出。
【临床应用】作为补充治疗用于各种原因所致巨幼红细胞性贫血。与维生素B12合用效果更好。对叶酸对抗剂甲氨蝶呤、乙胺嘧啶、甲氧苄氨嘧啶等所致巨幼红细胞性贫血,由于二氢叶酸还原酶抑制,应用叶酸无效,需用甲酰四氢叶酸钙(calciumleucovorin)治疗。对维生素B12缺乏所致“恶性贫血”,大剂量叶酸治疗可纠正血象,但不能改善神经症状。
维生素B12
维生素B12(vitamin B12)为含钴复合物,广泛存在于动物内脏、牛奶、蛋黄中。钴原子带有各种配体如-CN,-OH,-CH3和5′-脱氧腺苷基,因而有氰钴胺、羟钴胺、甲钴胺和5′-脱氧腺苷钴胺等维生素B12同类物。药用维生素B12为氰钴胺、羟钴胺,性质稳定。体内具有辅酶活性的维生素B12为甲钴胺和5′-脱氧腺苷钴胺。
【药物作用】维生素B12为细胞分裂和维持神经组织髓鞘完整所必需。体内维生素B12主要参与下列两种代谢过程。
1.同型半胱氨酸甲基化成甲硫氨酸需有甲基B12参与。该甲基是维生素B12自5-CH3H4PteGlu得来,然后转给同型半胱氨酸,5-CH3H4PteGlu则转变成H4PteGlu,促进四氢叶酸循环利用。故维生素B12缺乏会引起叶酸缺乏症状。
2.甲基丙二酰辅酶A变为琥珀酰辅酶A而进入三羧酸循环,需有5′-脱氧腺苷B12参与。维生素B12缺乏,甲基丙二酰辅酶A积聚,导致异常脂肪酸合成,影响正常神经髓鞘脂质合成,出现神经症状。
从以上代谢过程可知,对巨幼红细胞性贫血二药可以互相纠正,但神经症状必须用维生素B12治疗。
【体内过程】维生素B12必须与胃壁细胞分泌的糖蛋白即“内因子”结合才能免受胃液消化而进入空肠吸收。胃粘膜萎缩致“内因子”缺乏可影响维生素B12吸收,引起“恶性贫血”。吸收后有90%贮存于肝。正常人每天需要维生素B121μg,每天从食物中提供2~3μg,即可满足需要。由于肝有大量贮存,食物中即使无维生素B12,也不易造成缺乏。
【临床应用】主要用于恶性贫血及巨幼红细胞性贫血。
红细胞生成素
红细胞生成素(erythropoietin)是由肾脏近典小管管周细胞(peritubular cells)产生的糖蛋白激素,分子量约34000。现用基因工程人工合成。能刺激红系干细胞生成,促成红细胞成熟,使网织细胞从骨髓中释出。贫血、缺氧时红细胞生成素在肾脏的合成与分泌大量增加。但肾脏疾病、骨髓损伤或缺铁时,此合成增加机制即被破坏。已发现红系干细胞表面有红细胞生成素受体,结合后引起细胞内磷酸化及Ca2+浓度增加。注射2000~10000U,可用于慢性肾功能不全、肿瘤化疗及爱滋病药物治疗等引起的贫血。不良反应有血压上升、注射部位血栓形成以及流感样症状。
第六节 促进白细胞增生药
维生素B4、鲨肝醇等作为升白细胞药应用多年,但疗效较差。基因重组及克隆技术则为集落刺激因子的生产和应用创造了条件。
粒细胞集落刺激因子
粒细胞集落刺激因子(granulocyte colony-sitmulating factor,G-CSF)是血管内皮细胞、单核细胞和成纤维细胞合成的糖蛋白。能促进中性粒细胞成熟;刺激成熟的粒细胞从骨髓释出;增强中性粒细胞趋化及吞噬功能。对巨噬细胞、巨核细胞影响很小。现用的G-CSF为基因重组产品。皮下注射5~10μg/kg,4~6小时达血浓高峰,高于10μg/kg,血浓可维持16小时。静静滴注(60μg/kg,20~30分滴完),t1/2为0.75~7.2小时。1987年起用于肿瘤化疗、放疗引起骨髓抑制,也用于自体骨髓移值。对再生障碍性贫血、骨髓再生不良和爱滋病也有应用。可升高中性粒细胞,减少感染发生率。患者耐受良好,略有轻度骨骼疼痛,长期静脉滴注可引起静脉炎。应在化疗药物应用前或后24小时应用。
粒细胞/巨噬细胞集落刺激因子
粒细胞/巨噬细胞集落刺激因子(生日能,granulocyte-macrophage colony-stimulatingfactor,GM-CSF)在T-淋巴细胞、单核细胞、成纤维细胞、血管内皮细胞均有合成。它与白细胞介素3(interleukin 3)共同作用于多向干细胞和多向祖细胞等细胞分化较原始部位,因此可刺激粒细胞、单核细胞、巨噬细胞和巨核细胞等多种细胞的集落形成和增生。对红细胞增生也有间接影响。对成熟中性粒细胞可增加其吞噬功能和细胞毒性作用,但降低其能动性。临床应用的也为基因重组产品。静脉注射,t1/2α为5~15分,t1/2β为1~9小时。皮下注射10μg/kg,t1/2为2.9小时。临床应用同G-CSF,也可用于血小板减少症。不良反应有皮疹、发热、骨及肌肉疼痛、皮下注射部位红斑。首次静脉滴注时可出现潮红、低血压、呼吸急促、呕吐等症状,应以吸氧及输液处理。
第七节 血容量扩充剂
大量失血或失血浆(如烧伤)可引起血容量降低,导致休克。迅速补足以至扩充血容量是抗休克的基本疗法。除全血和血浆外,也可应用人工合成的血容量扩充剂。对血容量扩充剂的基本要求是能维持血液胶体渗透压;排泄较慢;无毒、无抗原性。目前最常用的是右旋糖酐。
右旋糖酐
右旋糖酐(dextran)是葡萄糖的聚合物,由于聚合的葡萄糖分子数目不同,可得不同分子量的产品。临床应用的有中分子量(平均分子量为70000),低分子量(平均分子量为40 000)和小分子量(平均分子量为10000)右旋糖酐。分别称右旋糖酐70,右旋糖酐40和右旋糖酐10。
【药理作用】右旋糖酐分子量较大,不易渗出血管,可提高血浆胶体渗透压,从而扩充血容量,维持血压。作用强度与维持时间依中、低、小分子量而逐渐缩小。低分子和小分子右旋糖酐能抑制血小板和红细胞聚集,降低血液粘滞性,并对凝血因子Ⅱ有抑制作用,因而能防止血栓形成和改善微循环。它们还有渗透性利尿作用。
【体内过程】右旋糖酐70在血液中存留时间较久,24小时约排出50%,作用维持12小时。右旋糖酐10则仅维持3小时。
【临床应用】各类右旋糖酐主要用于低血容量休克,包括急性失血、创伤和烧伤性休克。低分子右旋糖酐由于能改善微循环,抗休克效应更好。低、小分子右旋糖酐酐也用于DIC,血栓形成性疾病,如脑血栓形成、心肌梗塞、心绞痛、血管闭塞性脉管炎、视网膜动静脉血栓等。
【不良反应】少数患者用药后出现皮肤过敏反应,极少数人可出现过敏性休克。故首次用药应严密观察5~10分,发现症状,立即停药,及时抢救。用量过大可出现凝血障碍。禁用于血小板减少症及出血性疾病。心功能不全病人慎用。
作用于血液及造血器官的药物 制剂与用法
肝素钠(heparin sodium)静脉注射或静脉滴注,500~10 000U/次,稀释后用,1次/3~4小时,总量为25000U/日,过敏体质者先试用1 000U,如无反应,可用至足量。
依诺肝素(enoxaparin)注射剂,20~40mg/次,1次/日,皮下注射。用于血液透析,1mg/kg,从动脉导管中注入。
华法令(warfarin sodium)片剂,首次6~20mg,以后每日2~8mg。
双香豆素(dicoumarol)片剂,0.1g/次,第一天2~3次/日,第二天1~2次/日,后0.05~0.1g/日。
新抗凝(acenocoumarol)片剂,第一天16~28mg/次,第二天起2~10mg/次,1次/日。
阿司匹林(asparin)片剂,75mg/次,1次/日。
盐酸噻氯匹定(ticlopidine hydrochloride)片剂,250~500mg/次,1次/日,进餐时服。
链激酶(streptokinase)粉针剂,初导剂量,50万单位溶于生理盐水或5%葡萄糖液中,静脉滴注,30分滴完。维持剂量,60万单位/小时静脉滴注。疗程一般24~72小时。为防止过敏反应可给糖皮质激素。
尿激酶(urokinase)粉针剂,以注射用水3~5ml溶解后加于10%葡萄糖液20~40ml静注,1.5万~2万单位/次,2次/日,第4天起改为1万~2万单位/次,1次/日,一般7~10天。静脉滴注则先以负荷剂量2000~4000单位/30分,继以2000~4000单位/小时,维持12小时。
组织纤溶酶原激活因子(t-PA)粉针剂,首剂10mg,静脉注射。以后第1小时50mg,第2、3小时各20mg静脉滴注。
维生素K1(vitamin K1)肌内或静脉注射,10mg/次,2~3次/日。
维生素K3(vitamin K3)肌内注射,4mg/次,2~3次/日。
维生素K4(vitamin K4)片剂,2~4mg/次,3次/日。
硫酸亚铁(ferrous sulfate)片剂,0.3~0.6g/次,3次/日。
枸橼酸铁铵(ferric ammonium citrate)糖浆,1~2ml/kg/日,分3次服。
右旋糖酐铁(iron dextran)深部肌内注射,25~50mg/次,1次/日。总剂量(mg)=(正常Hb-患者Hb/100)×血容量×3.5×1.5
叶酸(folic acid)口服,5~10mg/次,3次/日。肌内注射,15~30mg/次,1次/日。
甲酰四氢叶酸钙(calcium leucovorin)肌内注射,3~6mg/次,1次/日。
维生素B12(vitamin B12)肌内注射,50~500μg/次,1~2次/日。
红细胞生成素(erythropoietin)注射剂,开始50~100单位/kg,皮下或静脉注射,3次/周。2周后视血细胞比容增减剂量。
生白能(leucomax,GM-CSF)干粉注射剂(300μg/支,合3.33×106单位),5~10μg/kg,每日一次皮下注射,于化疗停止一天后使用,连用7~10天。
右旋糖酐(dextran)6%,10%,12%溶液,视病情选用,静脉滴注。
第二十九章 组胺受体阻断药
组胺(histamine)是广泛存在于人体组织的自身活性物质(autacoids)。组织中的组胺主要含于肥大细胞及嗜碱细胞中。因此,含有较多肥大细胞的皮肤、支气管粘膜和肠粘膜中组胺浓度较高,脑脊液中也有较高浓度。肥大细胞颗粒中的组胺常与蛋白质结合,物理或化学等刺激能使肥大细胞脱颗粒,导致组胺释放。组胺与靶细胞上特异受体结合,产生生物效应;如小动脉、小静脉和毛细血管舒张,引起血压下降甚至休克;增加心率和心肌收缩力,抑制房室传导;兴奋平滑肌,引起支气管痉挛,胃肠绞痛;刺激胃壁细胞,引起胃酸分泌。组胺受体有H1、H2、H3亚型。各亚型受体功能见表29-1。组胺的临床应用已逐渐减少,但其受体阻断药在临床上却有重大价值。
表29-1 组胺受体分布及效应表
| 受体类型 | 所在组织 | 效 应 | 阻断药 |
| H1 | 支气管,胃肠, 子宫等平滑肌 皮肤血管 心房,房室结 |
收缩 扩张 收缩增强,传导减慢 |
苯海拉明 异丙嗪及 氯苯那敏等 |
| H2 | 胃壁细胞 血管 心室,窦房结 |
分泌增多 扩张 收缩加强,心率加快 |
西米替丁 雷尼替丁等 |
| H3 | 中枢与外周 神经末梢 |
负反馈性调节组胺 合成与释放 |
thioperamide |
第一节 H1受体阻断药
人工合成的H1受体阻断药多具有乙基胺的共同结构,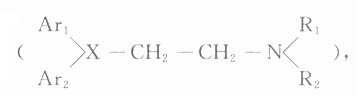
Ar1和Ar2可为苯环或杂环,X可为氮、氧或碳,并与主核相联。乙基胺与组胺的侧链相似,对H1受体有较大亲和力,但无内在活性,故能竞争性阻断之。
【药理作用】
1.抗外周组胺H1受体效应 H1受体被激动后即能通过G蛋白而激活磷脂酶C,产生三磷酸肌醇(IP3)与二酰基甘油(DG),使细胞内Ca2+增加,蛋白激酶C活化,从而使胃、肠、气管、支气管平滑肌收缩。又释放血管内皮松弛因子(EDRF)和PGI2,使小血管扩张,通透性增加。H1受体阻断药可拮抗这些作用。如先给H1受体阻断药,可使豚鼠接受百倍致死量的组胺而不死亡。对组胺引起的血管扩张和血压下降,H1受体阻断药仅有部分拮抗作用,因H2受体也参与心血管功能的调节。
2.中枢作用 治疗量H1受体阻断药有镇静与嗜唾作用。作用强度因个体敏感性和药物品种而异,以苯海拉明、异丙嗪作用最强;阿司咪唑、特非那丁因不易通过血脑屏障,几无中枢抑制作用。苯茚胺略有中枢兴奋作用。它们引起中枢抑制可能与阻断中枢H1受体有关。个别患者也出现烦躁失眠。它们还有抗晕、镇吐作用,可能与其中枢抗胆碱作用有关。
3.其他作用 多数H1受体阻断药有抗乙酰胆碱、局部麻醉和奎尼丁样作用。各种H1受体阻断药的作用特点见表29-2。
表29-2 常用H1-受体阻断药作用的比较
| 药 物 | 镇静程度 | 止吐作用 | 抗胆碱作用 | 作用时间(小时) |
| 苯海拉明 | +++ | ++ | +++ | 4~6 |
| 异丙嗪 | +++ | ++ | +++ | 4~6 |
| 吡苄明 | ++ | / | / | 4~6 |
| 氯苯那敏 | + | - | ++ | 4~6 |
| 布可立嗪 | + | +++ | + | 16~18 |
| 美克洛嗪 | + | +++ | + | 12~24 |
| 阿司咪唑 | - | - | - | 10(天) |
| 特非那定 | - | - | - | 12~24 |
| 苯茚胺 | 略兴奋 | - | ++ | 6~8 |
(+++ 作用强;++ 作用中等;+ 作用弱;- 无作用)
【体内过程】多数H1受体阻断药口服吸收良好,2~3小时达血浓高峰,作用持续4~6小时。药物在肝内代谢后,经尿排出。肝病可使药物作用时间延长。特非那丁口服后1~2小时达血浓高峰,t1/2为4~5小时,然而作用持续12~24小时以上,因其代谢产物尚有活性。阿司咪唑口服后2~4小时达血药浓度高峰,t1/2约20小时。在肝脏代谢成去甲基阿司咪唑,仍具活性,t1/2为10天,数星期后才达稳态血浓。
【临床应用】
1.变态反应性疾病本类药物对由组胺释放所引起的荨麻疹,枯草热和过敏性鼻炎等皮肤粘膜变态反应效果良好。对昆虫咬伤引起的皮肤瘙痒和水肿也有良效。对药疹和接触性皮炎有止痒效果。对慢性过敏性荨麻疹与H2受体阻断药合用效果比单用好。本类药物能对抗豚鼠由组胺引起的支气管痉挛,但对支气管哮喘患者几乎无效。因引起人类哮喘的活性物质复杂,药物不能对抗其他活性物质的作用。对过敏性休克也无效。
2.晕动病及呕吐苯海拉明、异丙嗪、布可立嗪、美克洛嗪对晕动病、妊娠呕吐以及放射病呕吐有镇吐作用。防晕动病应在乘车、船前15~30分服用。
3.失眠对中枢有明显抑制作用的导丙嗪、苯海拉明可用于失眠。
【不良反应】常见镇静、嗜唾、乏力等,故服药期间应避免驾驶车、船和高空作业。少数患者则有烦躁、失眠。此外尚有消化道反应及头痛、口干等。美克洛嗪可致动物畸胎,妊娠早期禁用。局部外敷可致皮肤过敏。阿司咪唑过量可致晕厥、心跳停止。
第二节 H2受体阻断药
以含有甲硫乙胍的侧链代替H1受体阻断药的乙基胺链,获得有选择作用的H2受体阻断药,它拮抗组胺引起的胃酸分泌,对H1受体无作用。H2受体阻断药是治疗消化性溃疡很有价值的新药。当前临床应用的有西米替丁(cimetidine)、雷尼替丁(ranitidine)、法莫替丁(famotidine)和尼扎替丁(nizatidine)。
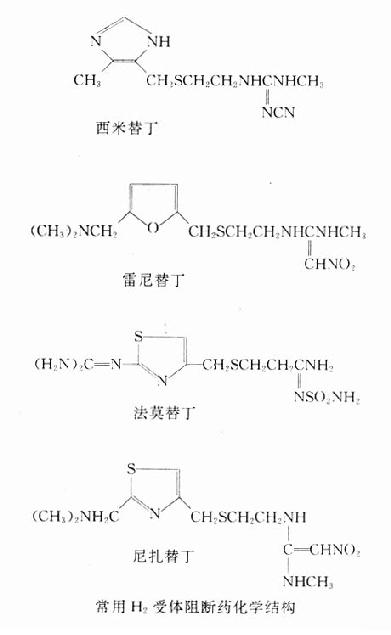
常用H2受体阻断药化学结构
【药理作用】本类药物竞争性拮抗H2受体,能抑制组胺、五肽胃泌素、M胆碱受体激动剂所引起的胃酸分泌。能明显抑制基础胃酸及食物和其他因素所引起的夜间胃酸分泌。用药后胃液量及氢离子浓度下降。用药4周,在内窥镜检查下,十二指肠溃疡愈合率为77%~92%。晚饭时1次给药疗效与一日多次给药的疗效相仿或更佳。对胃溃疡疗效发挥较慢,用药8周愈合率为75%~88%。雷尼替丁尼扎替丁抑制胃酸分泌作用比西米替丁强4~10倍,法莫丁比西米替丁强20~50倍。
【体内过程】本类药物口服吸收良好,但首关消除使生物利用度降为50%~60%。消除t1/2尼扎替丁为1.3小时,其他三药为2~3小时。大部分药物以原形经肾排出,但肝功能不良者雷尼替丁半衰期明显延长。
【临床应用】用于十二指肠溃疡,胃溃疡,应用6~8周,愈合率较高,延长用药可减少复发。卓-艾(Zollinger-Ellison)综合征需用较大剂量。其他胃酸分泌过多的疾病如胃肠吻合溃疡,反流性食道炎等及消化性溃疡和急性胃炎引起的出血也可用。
【不良反应】发生较少,尤其是雷尼替丁、法莫替丁和尼扎替丁,长期服用耐受良好。偶有便秘、腹泻、腹胀及头痛、头晕、皮疹、瘙痒等。静脉滴注速度过快,可使心率减慢,心收缩力减弱。长期服用西米替丁的男性青年,可引起阳萎、性欲消失及乳房发育。可能与其抑制二氢睾丸素与雄性素受体相结合及增加血液雌二醇浓度有关。
西米替丁能抑制细胞色素P-450肝药酶活性,抑制华法林、苯妥英钠、茶碱、苯巴比妥、安定、普萘洛尔等代谢。合用时,应调整这些药物剂量。雷尼替丁这一作用很弱,法莫替丁、尼扎替丁对其无影响。
组胺受体阻断药 制剂及用法
盐酸苯海拉明(diphenhydraminehydrochloride,benadryl)片剂,25~50mg/次,3次/日。注射剂,20mg/次,肌内注射,1~2次/日。
茶苯海明(dimenhydrinate;晕海宁,theohydramine)片剂,为苯海拉明与氨茶碱复合物,预防晕动病,行前半小时服50mg。
盐酸异丙嗪(promethazinehydrochloride;非那根,phenergan)片剂,1.25~25mg/次,2~3次/日。注射剂,25~50mg/次,肌内或静脉注射。
盐酸吡苄明(pyribenzaminehydrochloride去敏灵,扑敏宁)片剂,25~50mg/次,3次/日。
马来酸氯苯那敏(chlorpheniraminemaleate,扑尔敏)片剂,4mg/次,3次/日,小儿0.35mg/kg·日,分3~4次/日。注射剂,5~20mg/次,皮下或肌内注射。
盐酸布可立嗪(buclizinehydrochloride,安其敏)片剂,25~50mg/次,2次/日。
盐酸美克洛嗪(meclizinehydrochloride,敏可静)片剂,25mg/次,2次/日。
酒石酸苯茚胺(phenindaminetartrate)片剂,25mg/次,2~3次/日。
阿司咪唑(astemizole,息斯敏)片剂,10mg/次,1次/日。
特非那定(terfenadine)片剂,60mg/次,2次/日。
西米替丁(cimetidine)片剂,400mg/次,3次/日,或800mg,晚饭后服,1次/日。注射剂,200mg/次,静脉滴注,1~2次/日。
盐酸雷尼替丁(ranitidinehydrochloride)片剂,150mg/次,2次/日,或300mg,晚饭后服,1次/日,4~8周为一疗程。注射剂,50mg/次,每6~8小时肌内注射或静脉注射。
法莫替丁(famotidine)片剂,20mg/次,2次/日,或40mg,晚饭后服,1次/日。注射剂,20mg/次,2次/日,静脉点滴。
尼扎替丁(nizatidine)胶囊,150mg/次,2次/日,或300mg,晚饭后服,1次/日,4~8周为一疗程。
第三十章 作用于呼吸系统的药物
咳、痰、喘为呼吸系统疾病的常见症状。镇咳药(antitussives)、祛痰药(expectorants)和平喘药(antiasthmatic drugs)是呼吸系统疾病对症治疗的常用药物。
第一节 平喘药
喘息是支气管哮喘和喘息性支气管炎的主要症状。其基本病理变化是炎症细胞浸润,释放炎症介质,引起气道粘膜下组织水肿,微血管通透性增加,纤毛上皮剥离,气管分泌物增多,支气管平滑肌痉挛。细胞浸润包括肥大细胞、巨噬细胞、嗜酸细胞、淋巴细胞和中性白细胞等。释放的炎症介质有组胺、前列腺素D2、F2α、血栓素(TXA2)、白三烯(LT)及氧自由基等。喘息时气道反应性亢进。因此,除抗原能致变态反应性喘息外,寒冷、烟尘等非特异性刺激也可引起喘息。上述资料提示,抑制气道炎症及炎症介质是喘息的根本治疗。
一、肾上腺素受体激动药
本类药物因激动β受体,激活腺苷环化酶而增加平滑肌细胞内cAMP浓度,从而使平滑肌松弛。对各种刺激引起支气管平滑肌痉挛有强大的舒张作用。也能抑制肥大细胞释放过敏介质,可预防过敏性哮喘的发作。对炎症过程并无影响。长期应用可使支气管对各种刺激反应性增高,发作加重。目前主要发展对β2受体有高度选择性的药物,并以吸入给药,用于哮喘急性发作治疗和发作前预防用药。
肾上腺素
对α和β受体都有强大激动作用。其舒张支气管主要靠激动β受体。α受体激动可使支气管粘膜血管收缩,减轻水肿,有利气道畅通。但α受体激动也收缩气道平滑肌,并使肥大细胞释放活性物质,有不利影响。现在本药仅作皮下注射,以缓解支气管哮喘急性发作。
麻黄碱
作用与肾上腺素相似,但缓慢、温和、持久。口服有效。用于轻症和预防哮喘发作。
异丙肾上腺素
选择作用于β受体,对β1和β2受体无选择性。平喘作用强大,可吸入给药。但心率增快、心悸、肌震颤等不良反应较多。哮喘患者如有严重缺氧或剂量太大易致心律失常,甚至心室颤动、突然死亡。
β2受体激动剂
本类药物对β2受体有较强选择性,对α受体无作用。口服有效,作用维持4~6小时。采用吸入给药法几无心血管系统不良反应。但剂量过大,仍可引起心悸、头晕、手指震颤(激动骨骼肌β2受体)等。临床常用的有:
沙丁胺醇(salbutamol,舒喘灵)对β2受体作用强于β1受体,兴奋心脏作用仅为异丙肾上腺素1/10。口服30分起效,维持4~6小时。气雾吸入5分起效,维持3~4小时。近年来有缓释和控释剂型,可使作用时间延长,适用于夜间发作。
克伦特罗(clenbuterol)为强效选择性β2受体激动剂,松弛支气管平滑肌效为沙丁胺醇的100倍。口服30μg,10~20分起效,持效4~6小时。气雾吸入5~10分起效,持效2~4小时。心血管系统不良反应较少。
特布他林(terbutaline)作用与沙丁胺醇相似,即可口服,又可注射,是选择作用于β2受体药中唯一能作皮下注射的药。虽肾上素也作皮下注射用,但本品作用持久。皮下注射5~15分生效,30~60分达高峰,持续1.5~5小时。重复用药易致蓄积作用。
二、茶碱
茶碱(theophylline),能松弛平滑肌,兴奋心肌,兴奋中枢,并有利尿作用。其松弛平滑肌的作用对处于痉挛状态的支气管更为突出,对急、慢性哮喘,不论口服、注射或直肠给药,均有疗效,对喘息性慢性支气管炎,由于它能兴奋骨骼肌,可增强呼吸肌收缩力和减轻患者呼吸肌疲劳的感觉。茶碱短期应用能促进儿茶酚胺类物质释放。近年证明腺苷可引起哮喘病人支气管平滑肌收缩,而茶碱有阻断腺苷受体的作用。
茶碱难溶于水,为提高水溶性,与乙二胺或胆碱制成复盐为氨茶碱(aminophylline)、胆茶碱(choline theophylline)等供临床应用。茶碱的安全范围较小,尤其是静脉注射太快易引起心律失常、血压骤降、兴奋不安甚至惊厥。
三、M胆碱受体阻断药
各种刺激引起内源性乙酰胆碱的释放在诱发哮喘中有重要作用。M胆碱受体阻断剂能阻断乙酰胆碱作用,可用于治疗哮喘。例如异丙基阿托品(ipratropium)。吸入给药很少吸收。因此有明显扩张支气管作用,增加第一秒钟最大呼气量,而不影响痰液分泌,也无明显全身性不良反应。主要用于喘息型慢性支气管炎。
四、肾上腺皮质激素
糖皮质激素是目前治疗哮喘最有效的抗炎药物。这一作用与其抗炎和抗过敏作用有关(详见第三十四章)。它能抑制前列腺素和白三烯生成;减少炎症介质的产生和反应;能使小血管收缩,渗出减少。糖皮质激素是哮喘持续状态或危重发作的重要抢救药物。近年应用吸入治疗法,充分发挥了糖皮质激素对气道的抗炎作用,也避免了全身性不良反应。
倍氯米松
倍氯米松(beclomethasone)为地塞米松衍化物。局部抗炎作用比地塞米松强500倍。气雾吸入,直接作用于气道发生抗炎平喘作用,能取得满意疗效,且无全身不良反应,长期应用也不抑制肾上腺皮质功能。可以长期低剂量或短期高剂量应用于中度或重度哮喘患者,对皮质激素依赖者,可代替泼尼松的全身给药,并使肾上腺皮质功能得到必得。本品起效较慢,故不能用于急性发作的抢救。长期吸入,可发生口腔霉菌感染,宜多漱口。
氟尼缩松(flunisolide)作用与倍氯米松相似,但作用时间较长,一天用药2次。
五、肥大细胞膜稳定药
色甘酸钠
色甘酸钠(sodium cromoglycate) 又名咽泰(intal)。
【药理作用】色甘酸钠无松弛支气管及其他平滑肌的作用,也没有对抗组胺、白三烯等过敏介质的作用。但在接触抗原前用药,可预防Ⅰ型变态反应所致的哮喘,也能预防运动或其他刺激所致的哮喘。它能抑制肺肥大细胞对各种刺激,包括IgE与抗原结合所引起脱颗粒作用,抑制组胺及颗粒中其他内容物的释放。这一作用有种属及器官选择性,人支气管肺泡洗液中的肥大细胞最为敏感。作用的发生与受刺激肥大细胞内Ca2+浓度的降低有关。它还能逆转哮喘患者白细胞功能改变。
【体内过程】 口服吸收仅1%,治疗支气管哮喘主要用其微粒粉末(直径约6μm)吸入给药。约10%达肺深部组织并吸收入血,15分达血药浓度峰值。血浆蛋白结合率约60%~75%。t1/245~100分。以原形从胆汁和尿排出。
【临床应用】主要用于气管哮喘的预防性治疗,能防止变态反应或运动引起的速发和迟发性哮喘反应。应用2~3天,能降低支气管的较高反应性。也可用于过敏性鼻炎、溃疡性结肠炎及其他胃肠道过敏性疾病。
【不良反应】毒性很低。少数患者困粉末的刺激可引起呛咳、气急、甚至诱发哮喘,与少量异丙肾上腺素合用可以预防。
奈多罗米
奈多罗米(nedocromil)能抑制支气管粘膜炎症细胞释放多种炎症介质,作用比色甘酸钠强。吸入给药能降低哮喘患者的气道反应,改善症状和肺功能。可预防性治疗哮喘、喘息性支气管炎。偶有头痛。儿童、妊娠期妇女慎用。
平喘药的临床应用
1.哮喘急性发作需用β2受体激动剂吸入作抢救治疗,无效则口服或注射。β2受体激动剂与异丙基阿托品联合吸入,可起协同作用。对中、重度急性发作或β2受体激动剂无效者,全身应用糖皮质激素,常可缓解病情。
2.对慢性哮喘的处理。目的在于控制症状,减少复发,恢复日常生活。用药随病情而定,轻度者应选短效β2受体激动剂间歇吸入,接触已知抗原前呼入色甘酸钠。对中度者的基本治疗是每天吸入小剂量糖皮质激素或色甘酸钠。有症状时加用β2受体激动剂,但每天不应超过4次。无效时可增加吸入糖皮质激素量。也可加用长效支气管扩张药(包括茶碱类)。对严重慢性哮喘,在吸入高剂量糖皮质激素和口服长效支气管扩张剂同时,吸入长效β2受体激动剂。
第二节 镇咳药
镇咳药可作用于中枢,抑制延脑咳嗽中枢;也可作用于外周,抑制咳嗽反射弧中的感受器和传入神经纤维的末梢。
可待因
可待因(codeine)为阿片生物碱之一。与吗啡相似,有镇咳、镇痛作用,对咳嗽中枢的作用为吗啡的1/4,镇痛作用为吗啡的1/7~1/10。镇咳剂量不抑制呼吸,成瘾性也较吗啡弱。临床主要用于剧烈的刺激性干咳,也用于中等强度的疼痛。作用持续4~6小时。久用也能成瘾,应控制使用。少数患者能发生恶心、呕吐,大剂量可致中枢兴奋、烦躁不安。
右美沙芬
右美沙芬(dextromethorphan,右甲吗南)为中枢性镇咳药,强度与可待因相等,但无成瘾性,无镇痛作用。用于干咳。偶有头晕、嗳气。中毒量时才有中枢抑制作用。
喷托维林
喷托维林(pentoxyverine,咳必清)为人工合成的非成瘾性中枢镇咳药。选择性抑制咳嗽中枢,强度为可待因的1/3。并有阿托品样作用和局部麻醉作用,能松弛支气管平滑肌和抑制呼吸道感受器。适用于上呼吸道感染引起的急性咳嗽。偶有轻度头痛、头昏、口干、便秘等。有阿托品样作用,青光眼患者禁用。
苯丙哌林
苯丙哌林(benproperine)为非成瘾性镇咳药。能抑制咳嗽中枢,也能抑制肺及胸膜牵张感受器引起的肺-迷走神经反射,且有平滑肌解痉作用。是中枢性和末梢性双重作用的强效镇咳药,其镇咳作用比可待因强。口服后1~-20分生效,镇咳作用维持4~7小时,可用于各种原因引起的刺激性干咳。有轻度口干、头晕、胃部烧灼感和皮疹等不良反应。
苯佐那酯
苯佐那酯(benzonatate)又名退嗽露(tessalon)为丁卡因的衍生物。有较强的局部麻醉作用,抑制肺牵张感受器及感觉神经末梢。止咳剂量不抑制呼吸,反能增加肺每分钟通气量。用药后20分钟左右出现作用,维持3~4小时。对干咳、阵咳效果良好,也可用于支气管镜等检查前预防咳嗽。有轻度嗜睡、头晕、鼻塞等不良反应,偶见过敏性皮炎。服用时勿将药丸咬碎,以免引起口腔麻木。
第三节 祛痰药
能使痰液易于排出的药物称祛痰药。气道上的痰液刺激气管粘膜而引起咳嗽。粘痰积于小气道内可使气道狭窄而致喘息。因此,祛痰药还能起到镇咳、平喘作用。
氯化铵
氯化铵(ammonium chloride)口服对胃粘膜产生局部刺激作用,反射性地引起呼吸道的分泌,使痰液变稀,易于咳出。本品很少单独应用,常与其他药物配伍制成复方。应用于急、慢性呼吸道炎症而痰多不易咳出的患者。氯化铵吸收可使体液及尿呈酸性,可用于酸化尿液及某些碱血症。溃疡病与肝、肾功能不良者慎用。
乙酰半胱氨酸
乙酰半胱氨酸(acetylcysteine)性质不稳定,能使粘痰中连接粘蛋白肽链的二硫键断裂,变成小分子的肽链,从而降低痰的粘滞性,易于咳出。雾化吸入用于治疗粘稠痰阻塞气道,咳嗽困难者。紧急时气管内滴入,可迅速使痰变稀,便于吸引排痰。有特殊臭味,可引起恶心、呕吐。对呼吸道有刺激性,可致支气管痉挛,加用异丙肾上腺素可以避免。支气管哮喘患者慎用。滴入气管可产生大量分泌液,故应及时吸引排痰。雾化吸入不宜与铁、铜、橡胶和氧化剂接触,应以玻璃或塑料制品作喷雾器。也不宜与青霉素、头孢菌素、四环素混合,以免降低抗生素活性。
溴已新
溴已新(bromhexine,溴已铵)可裂解粘痰中的粘多糖,并抑制其合成,使痰液变稀。也有镇咳作用。适用于慢性支气管炎,哮喘及支气管扩张症痰液粘稠不易咳出患者。少数患者可感胃部不适、偶见转氨酶升高。消化性溃疡、肝功不良者慎用。
作用于呼吸系统的药物 制剂及用法
硫酸沙丁胺醇(salbutamol sulfate)片剂,2~4mg/次,3次/日。长效喘乐宁片(缓释):8mg/次,早、晚各1次。喘特宁片(控释):8mg /次,早、晚各1次。气雾剂(0.2%),1~2揿/次,1次/4小时。
硫酸特布他林(terbutaline sulfate)片剂,2.5mg/次,2~3次/日。注射剂,0.25mg /次,皮下注射,15~30分无效,可重复注射1次。
盐酸克仑特罗(clenbuterol hydrochloride)片剂,20~40μg/次,3次/日。气雾剂,10~20μg/次吸入,3~4次/日。
氨茶碱(amminophylline)片剂,0.1~0.2g/次,3次/日。氨茶碱控释片,300mg/12小时或400mg/24小时。注射剂,0.25~0.5g,以25%~50%葡萄糖溶液稀释后缓慢静脉推注。
胆茶碱(choline theophylline)片剂,0.2g/次,3次/日。
溴化异丙基阿托品(ipratropium bromide)气雾剂,40~80μg/次,3~6次/日。
色甘酸钠(sodium cromoglycate)粉雾剂,吸入20mg/次,4次/日。气雾剂,吸入2~4mg/次,4次/日。软膏(5%~10%),滴眼剂(2%)外用。
二丙酸倍氯米松(beclomethasone dipropionate)气雾剂,1~3揿/次,2~3次/日。
磷酸可待因(codeine phosphate)片剂,15~30mg/次,3次/日。注射剂,15~30mg/次,皮下注射。
氢溴酸右美沙芬(dextromethorphan hydrobromide)片剂,15~30mg/次,3~4次/日。
枸橼酸喷托维林(pentoxyverine citrate)片剂,25mg/次,3次/日。复方咳必清糖浆每100ml内含喷托维林0.2g,氯化铵3.0g。10ml/次,3~4次/日。
苯丙哌林(benproperine)糖衣片,20mg/次,3次/日。
苯佐那酯(benzonatate)糖衣片,25~50mg/次,3次/日。
氯化铵(ammonium chloride)片剂,0.3~0.6g/次,用水稀释或配成合剂,3次/日。
喷雾用乙酰半胱氨酸(acetylcysteinum pro nebula)每瓶0.5g,1g。喷雾,以10%溶液喷雾吸入,1~3ml/次,2~3次/日。
盐酸溴已新(bromhexine hydrochloride)片剂,8mg/次,3次/日。喷雾剂,2mg/次,3次/日。
第三十一章 作用于消化系统的药物
消化系统包括抗消化性溃疡药、助消化药、止吐药、泻药、止泻药和胆药等。
第一节 抗消化性溃疡药
消化性溃疡(peptic ulcer)的发病与粘膜局部损伤和保护机制之间的平衡失调有关。损伤因素(胃酸、胃蛋白酶和幽门螺旋菌)增强或保护因素(粘液/HCO3-屏障和粘膜修复)减弱,均可引起消化性溃疡。当今的治疗主要着眼于减少胃酸和增强胃粘膜的保护作用。
一、抗酸药
抗酸药(antacids)是一类弱碱性物质。口服后能降低胃内容物酸度,从而解除胃酸对胃、十二指肠粘膜的侵蚀和对溃疡面的刺激,并降低胃蛋白酶活性,发挥缓解疼痛和促进愈合的作用。餐后服药可延长药物作用时间。合理用药应在餐后1、3小时及临睡前各服一次,一天7次。理想的抗酸药应该是作用迅速持久、不吸收、不产气、不引起腹泻或便秘,对粘膜及溃疡面有保护收敛作用。单一药物很难达到这些要求,故常用复方制剂。
氢氧化镁(magnesium hydroxide)抗酸作用较强、较快。镁离子有导泻作用,少量吸收经肾排出,如肾功能不良可引起血镁过高。
三硅酸镁(magnesium trisilicate)抗酸作用较弱而慢,但持久。在胃内生成胶状二氧化硅对溃疡面有保护作用。
氢氧化铝(aluminum hydroxide)抗酸作用较强,缓慢。作用后产生氧化铝有收敛、止血和引起便秘作用。还可影响磷酸盐、四环素、地高辛、异烟肼、强的松等的吸收。
碳酸钙(calcium carbonate)抗酸作用较强、快而持久。可产生CO2气体。进入小肠的Ca2+可促进胃泌素分泌,引起反跳性胃酸分泌增多。
碳酸氢钠(sodium bicarbonate)又称小苏打。作用强、快而短暂。可产生CO2气体。未被中和的碳酸氢钠几乎全部吸收,能引起碱血症。
二、H2受体阻断药
H2受体阻断药西米替丁等详见第二十九章。
三、M胆碱受体阻断药
M胆碱受体阻断药如阿托品及其合成代用品可减少胃酸分泌、解除胃肠痉挛。但在一般治疗剂量下对胃酸分泌抑制作用较弱,增大剂量则不良反应较多,已很少单独应用。而哌仑西平(pirenzepine)对引起胃酸分泌的M1胆碱受体亲和力较高,而对唾液腺、平滑肌、心房的M胆碱受体亲和力低。治疗效果与西米替丁相仿,而不良反应轻微。
四、胃壁细胞H+泵抑制药
壁细胞通过受体(M1、H2受体、胃泌素受体)、第二信使和H+,K+-ATP酶三个环节来分泌胃酸。H+,K+-ATP酶(H+泵)位于壁细胞的管状囊泡和分泌管上。它能将H+从壁细胞内转运到胃腔中,将K+从胃腔中转运到壁细胞内,进行H+-K+交换。抑制H+,K+-ATP酶,就能抑制胃酸形成的最后环节,发挥治疗作用。
奥美拉唑
奥美拉唑(omeprazole)又名洛赛克(losec),由一个砜根联接苯咪唑环和吡啶环所成。1982年试用于临床治疗消化性溃疡收到明显效果。
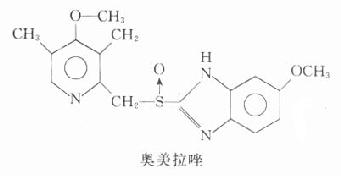
【药理作用】奥美拉唑口服后,可浓集于壁细胞分泌小管周围,并转变为有活性的次磺酰胺衍生物。它的硫原子与H+,K+-ATP酶上的巯基结合,形成酶-抑制剂复合物,从而抑制H+泵功能,抑制基础胃酸与最大胃酸分泌量。给狗口服2µmol/kg,连续用药一周,胃酸分泌抑制82%。十二指肠溃疡患者每日口服10mg,30mg,连续服药一周。24小时胃内H+活性抑制率分别为37%和97%。本品缓解疼痛迅速,服药1~3天即效。经4~6周,胃镜观察溃疡愈合率达97%。其他药物无效者用药4周,愈合率也高达90%左右。还使贲门、胃体、胃窦处粘膜血流量增加。也使幽门螺旋菌数量下降,约有83%~88%患者的幽门螺旋菌转阴。
胃酸分泌的抑制,可使胃窦G细胞分泌胃泌素增加。用药4~6周,血浆中胃泌素增加2~4倍。由于其促进胃酸分泌作用已被阻断,可发挥胃酸分泌以外的其他作用,如促进血流量的作用,对溃疡愈合有利。
【体内过程】本品口服生物利用度为35%。重复给药,可能因胃内pH降低,使生物利用度增为60%。1~3小时达血浓高峰。其活性代谢产物不易透过壁细胞膜,增高了药物选择性和特异性。t1/2为0.5~1小时,但因抑制H+泵为非可逆性,故作用持久。80%代谢产物由尿排出,其余随粪排出。
【临床应用】对其他药,包括H2受体阻断药无效的消化性溃疡患者,能收到较好效果。对反流性食道炎,有效率达75%~85%,优于雷尼替丁。卓-艾(Zollinger-Ellison)综合征给药第一天胃酸度降低,症状改善。
【不良反应】不良反应率为1.1%~2.8%。主要有头痛、头昏、口干、恶心、腹胀、失眠。偶有皮疹、外周神经炎、男性乳房女性化等。长期持续抑制胃酸分泌,可致胃内细菌过度滋长,亚硝酸类物质升高,是否会引起胃嗜铬细胞增生与胃类癌形成,尚无定论。
五、胃泌素受体阻断药
丙谷胺
丙谷胺(proglumide)由于化学结构与胃泌素相似,可竞争性阻断胃泌素受体,减少胃酸分泌。并对胃粘膜有保护和促进愈合作用。可用于胃溃疡,十二指肠溃疡和胃炎。也可用于急性上消化道出血。
六、粘膜保护药
前列腺素衍生物
胃粘膜能合成前列腺素E2(PGE2)及前列环素(PGI2),它们能防止有害因子损伤胃粘膜。实验证明它们能预防化学刺激引起的胃粘膜出血、糜烂与坏死,发挥细胞或粘膜保护作用。临床应用比较稳定的作用较强的衍生物。
米索前列醇(misoprostol),性质稳定,口服吸收良好,t1/2为1.6~1.8小时。口服后能抑制基础胃酸和组胺、胃泌素、食物刺激所致的胃酸分泌,胃蛋白酶分泌也减少。给动物应用小于抑制胃酸分泌的剂量,也能预防乙酰水杨酸等引起胃出血、溃疡或坏死,表明有强大的细胞保护作用。治疗十二指肠溃疡4周和8周的愈合率分别为61%和71%。临床应用于胃、十二指肠溃疡及急性胃炎引起消化道出血。其主要不良反应为稀便或腹泻。因能引起子宫收缩,孕妇禁用。
恩前列醇(enprostil)可使基础胃酸下降71%,也可明显抑制组胺、胃泌素和假餐所引起的胃酸分泌。也有细胞保护作用。用于胃溃疡,6周和8周愈合率分别为80%和86%。口服后t1/2α和t1/2β为1.75和34.3小时。主要从尿排出。用途及不良反应同米索前列醇。
硫糖铝
硫糖铝(sucralfate,ulcerlmine)是蔗糖硫酸酯的碱式铝盐,在pH<4时,可聚合成胶冻,牢固地粘附于上皮细胞和溃疡基底,抵御胃酸和消化酶的侵蚀;能减少胃酸和胆汁酸对胃粘膜的损伤;能促进胃粘液和碳酸氢盐分泌,从而发挥细胞保护效应。治疗消化性溃疡、慢性糜烂性胃炎、返流性食道炎有较好疗效。硫糖铝在酸性环境中才发挥作用,所以不能与抗酸药、抑制胃酸分泌药同用。不良反应较轻,约有2%患者可有便秘。小于1%病人发生口干。偶有恶心、胃部不适、腹泻、皮疹、瘙痒及头晕。
胶体碱式枸橼酸铋
又名三钾二枸橼酸铋(tripotassium dicitrate bismuthate,枸橼酸铋钾),溶于水形成胶体溶液。本品不抑制胃酸,在胃液pH条件下能形成氧化铋胶体沉着于溃疡表面或基底肉芽组织,形成保护膜而抵御胃酸、胃蛋白酶、酸性食物对溃疡面刺激。也能与胃蛋白酶结合而降低其活性。还能促进粘液分泌。用于胃、十二指肠溃疡、疗效与H2受体阻断剂相似,但复发率较低。牛奶、抗酸药可干扰其作用。服药期间可使舌、粪染黑。偶见恶心等消化道症状。肾功不良者禁用,以免引起血铋过高。
七、抗幽门螺旋菌药
幽门螺旋菌是慢性胃窦炎的主要病因,它能产生有害物质,分解粘液,引起组织炎症。虽然它与消化性溃疡发病的关系有待阐明,但已知消除幽门螺旋菌中明显减少十二指肠溃疡的复发率。因此,根治幽门螺旋菌具有重要意义。幽门螺旋菌在体外对多种抗菌药非常敏感,但体内单用一种药物,几全无效。临床常以甲硝唑、四环素、氨苄青霉素、羟氨苄青霉素等的2~3药作联合应用。
第二节 助消化药
助消化药多为消化液中成分或促进消化液分泌的药物。能促进食物的消化,用于消化道分泌机能减弱,消化不良。有些药物能阻止肠道的过度发酵,也用于消化不良的治疗。
稀盐酸(dilute hydrochloric acid)为10%的盐酸溶液,服后使胃内酸度增加,胃蛋白酶活性增强。适用于慢性胃炎、胃癌、发酵性消化不良等。服后可消除胃部不适、腹胀、嗳气等症状。
胃蛋白酶(pepsin)得自牛、猪、羊等胃粘膜。常与稀盐酸同服用于胃蛋白酶缺乏症。
胰酶(pancreatin)得自牛、猪、羊等动物的胰腺。含胰蛋白酶、胰淀粉酶及胰脂肪酶。在酸性溶液中易被破坏,一般制成肠衣片吞服。
乳酶生(biofermin,表飞呜)为干燥活乳酸杆菌制剂,能分解糖类产生乳酸,使肠内酸性增高,从而抑制肠内腐败菌的繁殖,减少发酵和产气。常用于消化不良,腹胀及小儿消化不良性腹泻。不宜与抗菌药或吸附剂同时服用,以免抗菌而降低疗效。
第三节 止吐药
延脑的呕吐中枢,可接受来自催吐化学感受区(CTZ)、前庭器官、内脏等传入冲动而引发呕吐。已知CTZ含有丰富的多巴胺、组胺、胆碱受体,前庭器官有胆碱能、组胺能神经纤维与呕吐中枢相联。5-羟色胺的5-HT3亚型受体通过外周、中枢部位如孤束核也与呕吐有关。M胆碱受体阻断药东莨菪碱、组胺H1受体阻断药苯海拉明等抗晕动病呕吐已在有关章节中叙述。本节主要介绍某些多巴胺受体阻断药和5-HT3受体阻断药的止吐作用。
甲氧氯普胺
甲氧氯普胺(metoclopramide,胃复安)对多巴胺D2受体有阻断作用,阻断CTZ的D2受体,发挥止吐作用。阻断胃肠多巴胺受体,可引起从食道至近段小肠平滑肌运动,加速胃的正向排空(多巴胺使胃体平滑肌松弛,幽门肌收缩)和加速肠内容物从十二指肠向回盲部推进,发挥胃肠促动药(prokinetics)作用。口服生物利用度为75%,易通过血脑屏障和胎盘屏障。t1/2为4~6小时。常用于包括肿瘤化疗、放疗所引起的各种呕吐,对胃肠的促动作用可治疗慢性功能性消化不良引起的胃肠运动障碍包括恶心、呕吐等症。大剂量静脉注射或长期应用,可引起锥体外系反应,如肌震颤、震颤麻痹(又名帕金森病),坐立不安等。也可引起高泌乳素血症,引起男子乳房发育、溢乳等。对胎儿影响尚待深入观察,孕妇慎用。
多潘立酮
多潘立酮(domperidone)又名马丁林(motilium)阻断多巴胺受体而止吐。不易通透血脑屏障。外周作用能阻断多巴胺对胃肠肌层神经丛突触后胆碱能神经元的抑制作用,加强胃肠蠕动,促进胃的排空与协调胃肠运动,防止食物返流,发挥胃肠促动药的作用。生物利用度较低,t1/2为7小时,主要经肝代谢。对偏头痛、颅外伤,放射治疗引起恶心、呕吐有效,对胃肠运动障碍性疾病也有效。不良反应较轻,遇有轻度腹部痉挛,注射给药引起过敏。
胃肠促动药还有西沙必利(cisapride),它能促进食管、胃、小肠直至结肠的运动。无锥体外系、催乳素释放及胃酸分泌等不良反应。能促使肠壁肌层神经丛释放乙酰胆碱。t1/2为10小时。用于治疗胃肠运动障碍性疾病,包括胃食管返流、慢性功能性和非溃疡性消化不良,胃轻瘫及便秘等有良好效果,每日3次,每次10mg。
奥丹西隆
奥丹西隆(ondansetron)能选择性阻断中枢及迷走神经传入纤维5-HT3受体,产生强大止吐作用。对抗肿瘤药顺铂、环磷酰胺、阿霉素等引起呕吐的止吐作用迅速强大。对顺铂引起的呕吐,完全或满意控制者达60%~73%,环磷酰胺引起呕吐控制率达92%,明显优于甲氧氯普胺。但对晕动病及多巴胺激动剂去水吗啡引起呕吐无效。生物利用度为60%。t1/2为3~4小时,代谢产物大多经肾排泄。临床用于化疗、放疗引起的恶心、呕吐。不良反应较轻,可有头痛、疲劳或便秘、腹泻。
同类新药格雷西隆(granisetron)、托比西隆(tropisetron)作用更强,目前正在临床试用中。
第四节 泻药
泻药(laxatives, catharitics)是能增加肠内水分,促进蠕动,软化粪便或润滑肠道促进排便的药物。临床主要用于功能性便秘。分为容积性、刺激性和润滑性泻药三类。
一、容积性泻药
为非吸收的盐类和食物性纤维素等物质。
1.硫酸镁(magnesium sulfate, MgSO4·7H2O)和硫酸钠(sodium sulfate, Na2SO4·10H2O)也称盐类泻药。在肠道难以吸收,大量口服形成高渗压而阻止肠内水分的吸收,扩张肠道,刺激肠壁,促进肠道蠕动。此外镁盐还能引起十二指肠分泌缩胆囊素(cholecystokinin),此激素能刺激肠液分泌和蠕动。一般空腹应用,并大量饮水,1~3小时即发生下泻作用,排出液体性粪便。导泻作用剧烈,故临床主要用于排除肠内毒物及某些驱肠虫药服后连虫带药一起排出。
口服高浓度硫酸镁或用导管直接注入十二指肠,因反射性引起总胆管括约肌松弛,胆囊收缩,发生利胆作用。可用于阻塞性黄疸、慢性肿囊炎。
硫酸镁、硫酸钠下泻作用较剧,可引起反射性盆腔充血和失水。月经期、妊娠妇女及老人慎用。
2.乳果糖(lactulose)为半乳糖和果糖的双糖。它在小肠内不被消化吸收,故能导泻。未被吸收部分进入结肠后被细菌代谢成乳酸等,进一步提高肠内渗透压,发生轻泻作用。
乳果糖还能降低结肠内容物的pH值,降低肠内氨的形成;H+又可与已生成的氨形成铵离子(NH4+)而不被吸收,从而降低血氨。可用于慢性门脉高压及肝性脑病。应注意因腹泻而造成水、电解质丢失,可使肝性脑病恶化。
3.食物纤维素包括蔬菜、水果中天然和半合成的多糖及纤维素衍生物如甲基纤维素、羧甲基纤维素等不被肠道吸收,增加肠内容积并保持粪便湿软,有良好通便作用。可防治功能性便秘。
二、接触性泻药
1.酚酞(phenolphthalein)口服后在肠道内与碱性肠液相遇形成可溶性钠盐,能促进结肠蠕动。服药后6~8小时排出软便,作用温和,适用于慢性便秘。口服酚酞约有15%被吸收。从尿排出,如尿液为碱性则呈红色。部分由胆汁排泄,并有肝肠循环而延长其作用时间,故一次服药作用可维持3~4天。遇有过敏性反应,发生肠炎、皮炎及出血倾向等。同类药物吡沙可啶(bisacodyl,双醋苯啶)用于便秘或X线,内窥镜检查或术前排空肠内容。
2.蒽醌类(anthroquinones)大黄、番泻叶和芦荟等植物,含有蒽醌甙类,口服后被大肠内细菌分解为蒽醌,能增加结肠推进性蠕动。用药后6~8小时排便,常用于急、慢性便秘。
三、滑润性泻药
滑润性泻药是通过局部滑润并软化粪便而发挥作用。适用于老人及痔疮、肛门手术患者。
1.液体石蜡(liquid paraffin)为矿物油,不被肠道消化吸收,产生滑润肠壁和软化粪便的作用,使粪便易于排出。
2.甘油(glycerin)以50%浓度的液体注入肛门,由于高渗压刺激肠壁引起排便反应,并有局部润滑作用,数分内引起排便。适用于儿童及老人。
泻药应用注意事项
1.治疗便秘,尤其是习惯性便秘,首先应从调节饮食、养成定时排便习惯着手。多吃蔬菜、水果等常能收到良好效果。
2.应根据不同情况选择不同类型泻药。如排除毒物,应选硫酸镁、硫酸钠等盐类泻药。一般便秘,以接触性泻药较常用。老人、动脉瘤、肛门手术等,以润滑性泻药较好。
3.腹痛患者在诊断不明情况下不能应用泻药。年老体弱、妊娠或月经期妇女不能用作用强烈的泻药。
第五节 止泻药
腹泻是多种疾病的症状,治疗时应采取对因疗法。例如肠道细菌感染引起的腹泻,应当首先用抗菌药物。但剧烈而持久的腹泻,可引起脱水和电解质紊乱,可在对因治疗的同时,适当给予止泻药。常用的药物如下:
1.阿片制剂 多用于较严重的非细菌感染性腹泻(参见第十八章)。
2.不良反应轻而少见。大剂量长期服用可产生成瘾性,一般则少见。
3.洛哌丁胺(loperamide,苯丁哌胺)结构类似地芬诺酯,除直接抑制肠道蠕动外,还可减少肠壁神经末梢释放乙酰胆碱。作用强而迅速。用于急、慢性腹泻。不良反应轻微。
4.收敛剂(astringents)和吸附药(adsorbants)口服鞣酸蛋白(tannalbin)在肠中释出鞣酸能与肠粘膜表面的蛋白质形成沉淀,附着在肠粘膜上,减轻刺激,降低炎性渗出物,起收敛止泻作用。次碳酸铋(bismuth subcarbonate)也有相同作用。药用炭(medicinal activated charcoal)是不溶性粉末,因其颗粒很小,总面积很大,能吸附大量气体、毒物,起保护、止泻和阻止毒物吸收的作用。
第六节 利胆药
利胆药为促进胆汁分泌或促进胆囊排空的药物。
去氢胆酸
去氢胆酸(dehydrocholic acid)可增加胆汁的分泌,使胆汁变稀。对脂肪的消化吸收也有促进作用。临床用于胆囊及胆道功能失调,胆汁淤滞,阻止胆道止行性感染,也可用于排除胆结石。对胆道完全梗阻及严重肝肾功能减退者禁用。
熊脱氧胆酸
熊脱氧胆酸(ursodeoxycholic acid)可减少普通胆酸和胆固醇吸收,抑制胆固醇合成与分泌,从而降低胆汁中胆固醇含量,不仅可阻止胆石形成,长期应用还可促胆石溶解。对胆色素结石、混合性结石无效。对胆囊炎、胆道炎也有治疗作用。
作用于消化系统的药物 制剂及用法
镁乳(milk of magnesia)8%氢氧化镁混悬液。口服,5ml/次,7次/日。
三硅酸镁(magnesium trisilicate)为氧化镁及二氧化硅的复合物。口服,0.3~0.9g/次,7次/日。
氢氧化铝凝胶(aluminum hydroxide gel)为白色混悬液。含4%氢氧化铝。口服,4~8ml/次,7次/日。
碳酸钙(calcium carbonate)口服,0.5~2.0g/次,7次/日。
碳酸氢钠(sodium bicarbonate)口服,0.3~1.0g/次,7次/日。纠正酸中毒:轻者可口服,较重者可用4%~5%碳酸氢钠静脉滴注,0.25g/kg。
哌仑西平(pirenzepine)片剂,50mg/次,2次/日。早、晚饭前1.5小时服,疗程4~6周。严重者,可50mg/次,3次/日。
奥美拉唑(omeprazole)片剂,20mg/次,1次/日,疗程2~4周。治疗反流性食道炎,20~60mg/次,1次/日。卓-艾氏综合征,60mg/次,1次/日。
丙谷胺(proglumide)片剂,0.4g/次,3次/日,4~6周一疗程。注射剂,静脉注射,0.4g/次,6小时一次,用于急性胃粘膜病变及急性上消化道出血。
米索前列腺(misoprostol)片剂,口服,200µg/次,1次/日。
恩前列醇(enprostil)片剂,35~70µg/次,2次/日。
硫糖铝(sucralfate)片剂,1g/次,2次/日。
胶体碱式枸橼酸铋(colloidal bismuth subcitrate;德诺,denol)片剂,120mg/次,4次/日,餐前,睡前各一次,4~8周一疗程。
稀盐酸(dilute hydrochloric acid)0.5~2ml/次,用水稀释饭前服。
胃蛋白酶(pepsin)粉剂,0.2~0.6g/次,3次/日,饭前或饭时服。合剂,每10ml含胃蛋白酶0.2~0.3g,稀盐酸0.1ml,10ml/次,3次/日?
■[此处缺少一些内容]■
ctulose)糖浆剂(60%),30~40ml/次,2~3次/日。
酚酞(phenolphthalein)片剂,0.05~0.2g/次,睡前服。
甘油(glycerin)栓剂,纳入肛门,成人2.67g/次,儿童1.33g/次。
开塞露(10ml,20ml)为50%甘油,或含适量的山梨醇制剂,10ml一支,供小儿用;20ml一支,供成人用。每次用一支,注入直肠内。
复方地芬诺酯(lomitil)片剂,每片含盐酸苯乙哌啶2.5mg ,硫酸阿托品0.025mg),口服,1~2片/次,3次/日。
洛哌丁胺(loperamide)胶囊,2mg/次,3次/日,首剂加倍。
鞣酸蛋白(tannalbin)片剂,1~2g/次,3次/日。
次碳酸铋(bimuth subcarbonate)片剂,0.3~1.0g/次,3次/日。
药用炭(medicinal activated charcoal)片剂,1g/次,3次/日。粉剂,1~3g/次,3次/日。
去氢胆酸(dehydrocholic acid(片剂,0.25g/次,3次/日。
熊去氧胆酸(ursodeoxycholic acid,UDCA)片剂,150mg/次,3次/日,或300mg/次,2次/日,饭后服用,持续6个月。
第三十二章 子宫平滑肌兴奋药和抑制药
第一节 子宫平滑肌兴奋药
子宫平滑肌兴奋药(oxytocics)是一类选择性直接兴奋子宫平滑肌的药物,它们的作用可因子宫生理状态及剂量的不同而有差异,或使子宫产生节律性收缩,或产生强直性收缩。如用于催产或引产,则希望发挥近似生理分娩的节律性收缩作用;如用于产后止血或子宫复原,则希望引起强直性收缩。如使用不当,可能造成子宫破裂与胎儿窒息的严重后果。因此,必须慎重使用和适当掌握剂量。
缩宫素
【来源与化学】缩宫素(oxytocin;催产素pitocin)和加压素(vasopressin;抗利尿素,antidiuretic hormone,ADU)是垂体后叶激素的两种主要成分,都先在下丘脑的视上核与室旁核的神经元内合成大分子的前激素,然后与载体,后叶激素运载蛋白(neurophysin)结合成为复合体。此复合体含于分泌颗粒中,沿神经轴突(下丘脑-垂体束)以每日约3mm速度转运至垂体;在转运途中,前激素转化为九肽的缩宫素或加压素,然后贮存在神经末梢,当神经冲动到达时即被释放。这一过程类似神经递质;不同之处是作为激素的缩宫素和加压素不是释放入神经突触间隙中,而是通过毛细血管进入血液循环到达远离器官而发挥作用。
缩宫素和加压素都是含有二硫键的9肽,只是3位和8位的氨基酸不同。因此它们的作用既有各自的特点,又有一定的交叉,即缩宫素有较弱的抗利尿和加压活性,而加压素也有轻微的兴奋子宫作用。
垂体后叶注射液是从牛或猪垂体后叶提得的,主要含缩宫素也含加压素,现在常用的催产素可从牛、猪垂体后叶提取,也可人工合成。我国药典规定缩宫素的效价以单位计算,一个单位相当于2µg纯缩宫素。
S────────────S
||
缩宫素半-酪-异-谷 · NH2-门 · NH2-半-脯-亮-甘· NH2
12345 6789
S────────────S
||
加压素半-酪-苯-谷 · NH2-门 · NH2-半-脯-精-甘· NH2
123456789
缩宫素和加压素的氨基酸序列
异=异亮氨酸
苯=苯丙氨酸
【药理作用】
1.兴奋子宫 缩宫素直接兴奋子宫平滑肌,加强其收缩。小剂量缩宫素加强子宫(特别是妊娠末期的子宫)的节律性收缩,使收缩振幅加大,张力稍增加(图32-1),其收缩的性质与正常分娩相似,即使子宫底部肌肉发生节律性收缩,又使子宫颈平滑肌松弛,以促进胎儿娩出。随着剂量加大,将引起肌张力持续增高,最后可致强直性收缩,这对胎儿和母体都是不利的。子宫平滑肌对缩宫素的敏感性与体内雌激素和孕激素水平有密切关系。雌激素可提高敏感性,孕激素则降低此敏感性;在妊娠早期,孕激素水平高,敏感性低,妊娠后期雌激素水平高,敏感性高。在妊娠20周至39周之间,敏感性可增加8倍。临产时子宫最为敏感,分娩后子宫的敏感性又逐渐降低。临床资料证明:晚期妊娠应用缩宫素引产者,其中反应快者,血中孕酮值低于反应慢者。
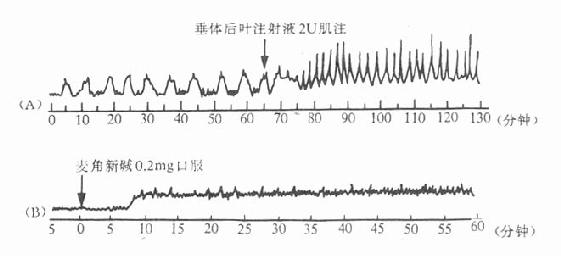
图32-1 垂体后叶剂与麦角新碱对人子宫的作用
A.分娩开始数小时后子宫的活动及肌注垂体后叶2U对子
宫活动的促进作用;
B.分娩七天后子宫的活动及口服麦角新碱0.2mg对子宫
活动的促进作用。
妊娠子宫对缩宫素的敏感性有个体差异,对63例引产成功的初产妇监测资料提示,有效缩宫素滴注速度最低为0.4mU/分,最大可达180mU/分。
已证明在人子宫平滑肌有缩宫素受体,故认为缩宫素通过与受体结合而发挥作用。妊娠期间缩宫素受体数量增加。钙通道的开放引起Ca2+的内流也参与缩宫素的作用机制。也有认为缩宫素作用于蜕膜的受体,促进PGF2α及PGF2α的代谢物,13,14二氢15-酮PGF2α(PGFM)的合成。前列腺素,尤其是PGFM能兴奋子宫并使子宫颈变软、展平及扩张。也发现在缩宫素引产成功的孕妇血浆中PGF和PGFM含量明显升高。
2.其他作用 缩宫素能使乳腺泡周围的肌上皮细胞(属平滑肌)收缩,促进排乳。大剂量还能短暂地松弛血管平滑肌,引起血压下降,并有抗利尿作用。
【体内过程】口服后在消化道易被破坏,故无效。能经鼻腔及口腔粘膜吸收。肌内注射吸收良好,3~5分钟内生效。可透过胎盘。大部分经肝及肾破坏,效果维持20~30分钟。
【临床作用】
1.催产和引产 对于无产道障碍而宫缩无力的难产,可用小剂量缩宫素加强子宫的收缩性能,促进分娩。对于死胎、过期妊娠、或因患严重心脏病等病的孕妇,需提前中断妊娠者,可用缩宫素引产。用法:一般每次2~5U,用5%葡萄糖液500ml稀释后,先以8~10滴/分的速度静脉滴注,必须密切观察,以后根据子宫收缩和胎心情况调整滴注速度,最快不超过40滴/分。
2.产后止血 产后出血时立即皮下或肌内注射较大剂量缩宫素(5~10U),迅速引起子宫强直性收缩,压迫子宫肌层内血管而止血。但缩宫素作用不持久,应加用麦角制剂使子宫维持收缩状态。
【不良反应】缩宫素过量引起子宫高频率甚至持续性强直收缩,可致胎儿窒息或子宫破裂,因此作催产或引产时,必须注意下列两点:①严格掌握剂量,避免发生子宫强直性收缩。②严格掌握禁忌证,凡产道异常、胎位不正、头盆不称、前置胎盘,以及三次妊娠以上的经产妇或有剖腹产史者禁用,以防引起子宫破裂或胎儿窒息。
垂体后叶素
垂体后叶素(pituitrin)是从牛、猪的垂体后叶中提取的粗制品,内含缩宫素和加压素,故对子宫平滑肌的选择性不高,在作为子宫兴奋药的应用上,已逐渐被缩宫素所代替。它所含的加压素能与肾脏集合管的受体相结合,增加集合管对水分的再吸收,使尿量明显减少;可用于治疗尿崩症。加压素对未孕子宫有兴奋作用,但对妊娠子宫反而作用不强。加压素还能收缩血管(特别是毛细血管和小动脉),在肺出血时可用来收缩小动脉而止血。它也能收缩冠状血管,故冠心病者禁用。此外,加压素尚有升高血压和兴奋胃肠道平滑肌的作用。
本品不良反应有面色苍白、心悸、胸闷、恶心、腹痛及过敏反应等。
表32-1垂体后叶素、缩宫素、加压素作用的比较
| 作 用 | 垂体后叶素(含加压素10U/ml;缩宫素10U/ml | 缩宫素(含缩宫素10U/ml;加压素<1U/ml | 加压素(含加压素10U/ml;缩宫素<1U/ml |
| 子宫收缩作用(人) | |||
| 未孕 | ++ | ++ | |
| 妊娠初期 | |||
| 妊娠末期 | ++++ | ++++ | +(偶而++) |
| 排乳作用 | ++++ | ++++ | + |
| 抗利尿作用 | +++ | ++++ | |
| 血压(人) | ++ | -- | ++ |
| 冠状血管收缩作用 | + | -- | ++++ |
| 肠环状肌收缩作用 | +++ | +++ |
+:增加;0:基本无作用;-:减少。
前列腺素
前列腺素(prostaglandins,PGs)是一类广泛存在于体内的不饱和脂肪酸,早期是从羊精囊提取,现可用生物合成法或全合成法制成。对心血管、呼吸、消化以及生殖系统等有广泛的生理和药理作用。目前研究较多并与生殖系统有关的前列腺素有前列腺素E2(PGE2)、前列腺素F2α(PGF2α)和15-甲基前列腺素F2α等。
前列腺素类药物的化学结构
与缩宫素不同,上述几种前列腺素对各期妊娠的人子宫都有显著的兴奋作用,仅对分娩前的子宫更敏感些。故除用于足月引产外,对早期或中期妊娠子宫也能引起足以导致流产的高频率和大幅度的收缩。除静脉滴注外,阴道内、宫腔内或羊膜腔内给药,也能有效。前列腺素也可能发展成为一种用于月经过期不久的早孕妇女的催经抗早孕药物。目前正从剂型、给药途径以及提高选择性等方面进行研究。
不良反应主要为恶心、呕吐、腹痛等胃肠道兴奋现象。
麦角生物碱
麦角(ergot)是寄生在黑麦中的一种麦角菌的干燥菌核,在麦穗上突出如角,故名。目前已用人工培养方法生产。
麦角中含多种作用强大的成分,主要是麦角碱类,此外尚有组胺、酪胺、胆碱和乙酰胆碱等。麦角碱类在化学结构上都是麦角酸的衍生物,可分为两类。
1.氨基酸麦角碱类 包括麦角胺(ergotamine)和麦角毒(ergotoxine),后者是三种麦角碱(ergocristine,ergokryptine,ergocornine)的混合物。口服吸收不良,且不规则,作用缓慢而持久。
2.氨基麦角碱类 以麦角新碱(ergometrine,ergonovine)为代表,口服吸收容易而规则,作用迅速而短暂。
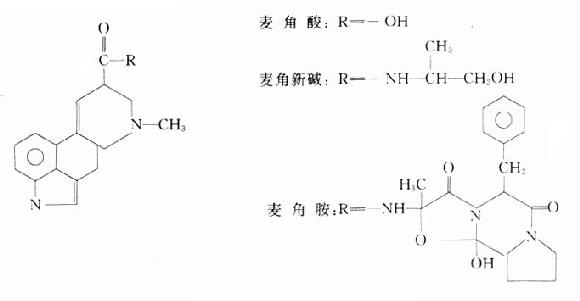
麦角酸和麦角新碱的化学结构
【药理作用】
1.兴奋子宫 麦角碱类能选择性地兴奋子宫平滑肌,其作用也取决于子宫的机能状态,妊娠子宫对麦角碱类比未妊娠子宫敏感。在临产时或新产后则最敏感。与缩宫素的不同,它们的作用比较强而持久,剂量稍大即引起子宫强直性收缩,对子宫体和子宫颈的兴奋作用无明显差别,因此,不宜用于催产和引产。麦角新碱的作用最快最强。
2.收缩血管 氨基酸麦角碱类,特别是麦角胺,能直接作用于动静脉血管使其收缩;大剂量还会伤害血管内皮细胞,长期服用可导致肢端干性坏疽。
3.阻断α受体,氨基酸麦角碱类尚有阻断α肾上腺素受体的作用,使肾上腺素的升压作用翻转。但在临床上,此剂量已能引起很多副作用,故无应用价值。麦角新碱则无此作用。
【临床作用】
1.治疗子宫出血 产后或其他原因引起的子宫出血都可用麦角新碱止血,它能使子宫平滑肌强直性收缩,机械地压迫血管而止血。
2.产后子宫复原 产后的最初十天子宫复原过程进行很快,如进行缓慢就易发生出血或感染,因此,须服用麦角制剂等子宫兴奋药以加速子宫复原。常用麦角流浸膏。
3.治疗偏头痛 偏头痛可能为脑动脉舒张和搏动幅度加大之结果,麦角胺与咖啡因都能收缩脑血管,减少动脉搏动的幅度。合用咖啡因还可使麦角胺的吸收速率和血药峰浓度提高到两倍。
4.中枢抑制作用 麦角毒的氢化物称氢麦角毒(海得琴,dihydroergotoxine,hydergine)具有抑制中枢、舒张血管(主要由于抑制血管运动中枢)和降低血压的作用。可与异丙嗪、哌替啶配成冬眠合剂。
【不良反应】注射麦角新碱可致呕吐,血压升高等,因此对妊娠毒血症产妇的产后应用须慎重。麦角流浸膏中含有麦角毒和麦角胺,长期应用可损害血管内皮细胞,特别是肝脏病或外周血管有病者更为敏感。此外,麦角新碱偶致过敏反应。
麦角制剂禁用于催产和引产,血管硬化及冠状动脉疾病患者。
中草药
益母草(Leonurus heterophyllus)为唇形科植物,又名益母蒿,药用全草,全国各地都有分布。有效成分为生物碱(如益母草碱leonurine等)。动物实验显示益母草能兴奋子宫平滑肌,增加子宫收缩频率,也能提高其张力,但作用较脑垂体后叶制剂为弱。临床用于产后止血和促使产后子宫复原。
当归(Angelica sinensis)为伞形科植物,药用其根,为中医妇科常用药物,应用历史悠久。动物实验证明,当归对子宫的作用与其化学成分及子宫机能状态有关。当归所含挥发油成分对子宫有抑制作用;其水溶性非挥发性碱性成分则对子宫具有兴奋作用。当子宫内加压时,当归使其收缩力加强;不加压时,则有轻微抑制作用。用于治疗痛经和月经不调。
第二节 子宫平滑肌抑制药
人的子宫平滑肌含有β肾上腺素受体,且以β2受体占优势。许多常见β2受体激动药如沙丁醇胺(舒喘灵)等都有松弛子宫平滑肌作用,并试用于防治早产。其中利托君(nitodrine)是专作为子宫松弛药而设计发展的。其化学结构与异丙肾上腺素相似,对非妊娠和妊娠子宫都有抑制作用。可用于防治早产。除β2受体激动药外,钙通道阻滞药也有良好的子宫平滑肌松弛作用,具有开发的前途。这一类子宫平滑肌抑制药被称为抗分娩药(tocolytic drugs)。
子宫平滑肌兴奋药和抑制药 制剂及用法
缩宫素(oxytocin)皮下或肌内注射,2~5U/次,用5%葡萄糖液500ml稀释后缓慢静脉滴注,具体用法见缩宫素“临床应用”项。极量:肌注20U/次。
垂体后叶素(pituitrin)皮下或肌内注射,5~10U/次;静脉滴注5~10U/次,可用5%葡萄糖液500ml稀释后缓慢滴入。
马来酸麦角新碱(ergometrine maleate)口服,0.2~0.5mg/次。肌内注射,0.2~0.5mg/次,必要时半小时后重复一次。静脉滴注0.2mg以5%葡萄糖溶液稀释后应用。极量:肌内或静脉注射,0.5mg/次,1mg/日。
酒石酸麦角胺(ergotamine tartrate)口服1mg/次。皮下或肌内注射,0.25mg/次。
麦角胺咖啡因片 每片含酒石酸麦角胺1mg,咖啡因100mg。偏头痛发作时即口服半片至1片半;如无效,可于间隔1小时后重复同剂量。
麦角流浸膏 2ml/次,3次/日,连续口服2~3天。极量:12ml/日。
乙磺酸二氢麦角碱(dihydroergotoxine aethanosulfonate)将盐酸哌替啶100mg、盐酸异丙嗪25mg、乙磺酸二氢麦角碱0.6~0.9mg加入5%葡萄糖液250ml中,配成冬眠合剂进行静脉滴注。
益母草流浸膏 2~5ml/次,3次/日,连续口服2~3天。
当归浸膏片 0.5/片。4~6片/次,2~3次/日。
第三十三章 性激素类药及避孕药
性激素(sex hormones)为性腺分泌的激素,包括雌激素、孕激素和雄激素。目前临床应用的是人工合成品及其衍生物。常用的避孕药(contraceptives)大多属于性激素制剂。
【化学】性激素属甾体(steroids)激素,其基本结构是甾体核。
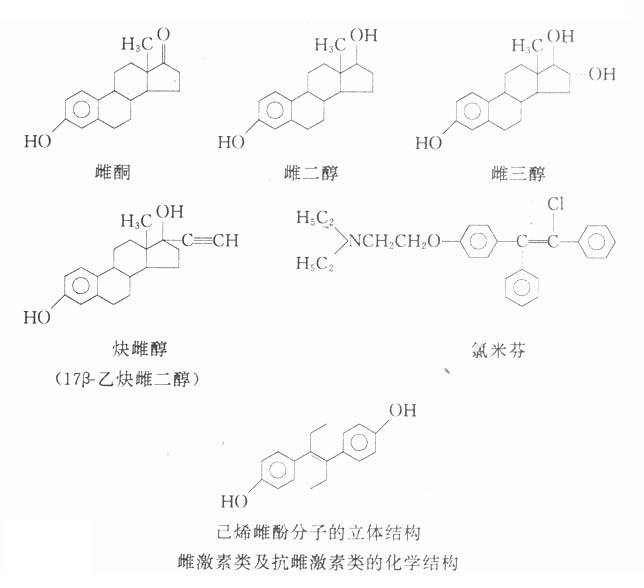
【性激素分泌的调节】雌激素和孕激素的分泌受下丘脑-垂体前叶的调节。下丘脑分泌促性腺激素释放激素(gonadotropin-releasing hormone,GnRH),它促进垂体前叶分泌促卵泡素(follicle stimulating hormone,FSH)和黄体生成素(luteinizing hormone,LH)。FSH促进卵巢的卵泡生长发育,而在FSH和LH共同作用下,使成熟的卵泡分泌雌激素和孕激素。
性激素对垂体前叶的分泌功能具有正反馈和负反馈两方面的调节作用,这取决于药物剂量和机体性周期。例如在排卵前,雌激素水平较高可直接或通过下丘脑促进垂体分泌LH,导致排卵(正反馈)。在月经周期的黄体期,由于血中雌激素、孕激素都高,从而减少GnRH的分泌,抑制排卵(负反馈)。常用的甾体避孕药就是根据这一负反馈而设计的。以上的反馈途径称“长反馈”。垂体促性腺激素的水平也能影响下丘脑GnRH的释放,这种反馈途径称“短反馈”(见图33-1)。
图33-1 女性激素的分泌(A)与调节(B)示意图
(A)促性腺激素分泌,女性激素分泌皆与卵泡发育过程和子宫
内膜变化相应。以28日为一周期,月经开始作为第1日。
(B)各激素曲线仅为相对数值。从图中可见FSH与LH的分泌高峰
在第14日左右,并在其后出现排卵。黄体酮的高峰与子宫内膜的
分泌期相应,雌激素则先后有两个高峰。GF,囊状卵泡;CL,黄体
峰在成年男性,垂体前叶所释放的LH可促进睾丸间质细胞分泌雄激素。雄激素也有抑制促性腺激素释放的作用。
【性激素的作用机制】性激素受体都位于细胞内,属第4类受体(可参阅图34-2)。如雌激素受体位于靶细胞的胞质液中,雌激素进入细胞后,首先与受体结合成复合物,然后复合物进入细胞核,从而诱导功能不同的蛋白质的合成,产生不同效应;例如雌激素诱导的蛋白质可使子宫肥大、代谢增加。但近年用单克隆抗体免疫组织化学方法发现雌激素受体都位于细胞核内,故认为是雌激素直接进入细胞核内而发挥作用。
第一节 雌激素类药及抗雌激素类药
一、雌激素类药
【来源】卵巢分泌的雌激素(estrogens)主要是雌二醇(estradiol)。从孕妇尿提出的雌酮(estrone)和雌三醇(estriol)等,多为雌二醇的代谢产物。雌二醇是传统的雌激素类药物,近年来以雌二醇为母体,人工合成许多高效的衍生物,如炔雌醇(ethinyl estradiol)、炔雌醚(quinestrol)及戊酸雌二醇(estradiol valerate)等。此外,也曾合成一些结构较简单的具有雌激素样作用的制剂,如已烯雌酚(diaethylstilbestrol ;乙菧酚,stilbestrol),它虽非甾体,但据其立体结构也可将它看作为断裂的甾体结构。
【体内过程】天然雌激素如雌二醇可经消化道吸收,但易在肝破坏,故口服效果远较注射为差。在血液中大部分与性激素结合球蛋白结合,也可与白蛋白非特异性地结合。部分以葡萄糖醛酸及硫酸结合的形式从肾脏排出,也有部分从胆道排泄并形成肝肠循环。人工合成的炔雌醇、炔雌醚或已烯雌酚等在肝内破坏较慢,口服效果好,作用较持久。油溶液制剂或与脂肪酸化合成酯,作肌内注射,可以延缓吸收,延长其作用时间。炔雌醚在体内可贮存于脂肪组织中,口服一剂作用可维持7~10天。
【生理及药理作用】
1.对未成年女性,雌激素能促使女性第二性征和性器官发育成熟。如子宫发育、乳腺腺管增生及脂肪分布变化等。
2.对成年妇女,除保持女性性征外,并参与形成月经周期。它使子宫内膜增殖变厚(增殖期变化),并在黄体酮的协同作用下,使子宫内膜进而转变为分泌期(见图33-1)状态,提高子宫平滑肌对缩宫素的敏感性。同时使阴道上皮增生,浅表层细胞发生角化。
3.较大剂量时,可作用于下丘脑-垂体系统,抑制GnRH的分泌,发挥抗排卵作用。并能抑制乳汁分泌,是在乳腺水平干扰催乳素的作用所致。此外还有对抗雄激素的作用。
4.在代谢方面,有轻度水、钠潴留作用。能增加骨骼钙盐沉积,加速骨骺闭合。大剂量可使甘油三酯和磷脂升高而胆固醇降低,也使糖耐量降低。尚有促进凝血作用。
【临床应用】
1.绝经期综合征绝经期综合征是更年期妇女因雌激素分泌减少,而垂体促性腺激素分泌增多,造成内分泌平衡失调的现象。雌激素可抑制垂体促性腺激素的分泌从而减轻各种症状。绝经期和老年性骨质疏松症可用雌激素与雄激素合并治疗。
除绝经期综合征外,老年性阴道炎及女阴干枯症等,局部用药也能奏效。
2.卵巢功能不全和闭经原发性或继发性卵巢功能低下患者以雌激素替代治疗,可促进外生殖器、子宫及第二性征的发育。与孕激素类合用,可产生人工月经周期。
3.功能性子宫出血可用雌激素促进子宫内膜增生,修复出血创面,也可适当配伍孕激素,以调整月经周期。
4.乳房胀痛部分妇女停止授乳后可发生乳房胀痛,可用大剂量雌激素制剂抑制乳汁分泌,克服胀痛,俗称回奶。由于此时垂体分泌的催乳素并不减少,故认为大剂量雌激素类抑制泌乳主要是在乳腺水平干扰催乳素的作用。
5.晚期乳腺癌绝经五年以上的乳腺癌可用雌激素制剂治疗,缓解率可达40%左右。但绝经期以前的患者禁用,因这时反可能促进肿瘤的生长。
6.前列腺癌大剂量雌激素类可使症状改善,肿瘤病灶退化。这是其抑制垂体促性腺激素分泌,使睾丸萎缩而抑制雄激素的产生所致,也有抗雄激素的作用参与。
7.痤疮青春期痤疮是由于雄激素分泌过多所致,故可用雌激素类治疗。
8.避孕(见本章第四节)
【不良反应及应用注意】
1.常见恶心、食欲不振,早晨较多见。如从小剂量开始,逐渐增加剂量可减轻反应;反应发生后减少剂量也可减轻反应;注射剂此种反应较轻。
2.长期大量应用可引起子宫内膜过度增生及子宫出血,故有子宫出血倾向者及子宫内膜炎患者慎用。
3.肿瘤患者(前列腺癌和绝经期后乳腺癌除外)不用。本药在肝灭活,并可能引起胆汁郁积性黄疸,故肝功能不良者慎用。
二、抗雌激素类药
氯米芬
氯米芬(clomiphene,氯酜酚胺)为三苯乙烯衍生物,与已烯雌酚的化学结构相似。
本品有较弱的雌激素活性,能与雌激素受体结合,发挥竞争性拮抗雌激素的作用。它能促进人的垂体前叶分泌促性腺激素,从而诱使排卵;这可能是因阻断下丘脑的雌激素受体,从而消除雌二醇的负反馈性抑制。用于不孕症和闭经,乳房纤维囊性疾病和晚期乳癌等。连续服用大剂量可引起卵巢肥大,故卵巢囊肿患者禁用。
第二节 孕激素类药
【来源】孕激素(progestogens)主要由卵巢黄体分泌,妊娠3~4个月后,黄体逐渐萎缩而由胎盘分泌代之,直至分娩。在近排卵期的卵巢及肾上腺皮质中也有一定量的孕激素产生。自黄体分离出的天然孕激素为黄体酮(孕酮,progesterone)含量很低。临床应用的是人工合成品及其衍生物。
【分类】孕激素类按化学结构可分为两大类:
1.17α-羟孕酮类从黄体酮衍生而得。如醋酸甲羟孕酮(醋酸甲孕酮,安宫黄体酮,medroxyprogesterone acetate)、甲地孕酮(megestrol)、氯地孕酮(chlormadinone)和羟孕酮已酸酯(17α-hydroxyprogesterone caproate)。
2.19-去甲睾丸酮类从妊娠素衍生而得。如炔诺酮(norethisterone,norethindrone,norlutin)、双醋炔诺醇(etynodiol diacetate)、炔诺孕酮(18甲基炔诺酮,甲基炔诺酮,norgestrel)等。
【体内过程】黄体酮口服后在胃肠及肝迅速破坏,效果差,故采用注射给药。血浆中的黄体酮大部分与蛋白结合,游离的仅占3%。其代谢产物主要与葡萄糖醛酸结合,从肾排出,人工合成的炔诺酮、甲地孕酮等作用较强,在肝破坏较慢,可以口服,是避孕药的主要成分。油溶液肌内注射可发挥长效作用。
【生理及药理作用】
1.生殖系统
(1)当月经后期,在雌激素作用的基础上,使子宫内膜继续增厚、充血、腺体增生并分支,由增殖期转为分泌期,有利于孕卵的着床和胚胎发育。
(2)抑制子宫的收缩,并降低子宫对缩宫素的敏感性。
(3)一定剂量可抑制垂体前叶LH的分泌,从而抑制卵巢的排卵过程。
(4)可促使乳腺腺泡发育,为哺乳作准备。
2.代谢 竞争性地对抗醛固酮,从而促进Na+和Cl-的排泄并利尿。
3.升温作用有轻度升高体温作用,使月经周期的黄体相基础体温较高。
【临床应用】
1.功能性子宫出血因黄体功能不足。所致子宫内膜不规则的成熟与脱落而引起子宫出血时,应用孕激素类可使子宫内膜协调一致地转为分泌期,故可维持正常的月经。
2.痛经和子宫内膜异位症可抑制排卵并减轻子宫痉挛性收缩从而止痛,也可使异位的子宫内膜退化。与雌激素制剂合用,疗效更好。
3.先兆流产与习惯性流产由于黄体功能不足所致的先兆流产与习惯性流产,孕激素类有时可以安胎,但对习惯性流产,疗效不确实。19-去甲睾酮类具雄激素作用,可使女性胎儿男性化,故不宜采用,黄体酮有时也可能引起生殖性畸形,须注意。
4.子宫内膜腺癌、前列腺肥大或癌症(见第四十九章)。
【不良反应】不良反应较少,偶见头晕、恶心及乳房胀痛等。长期应用可引起子宫内膜萎缩,月经量减少,并易发阴道真菌感染。19-去甲睾酮类大剂量时可致肝功能障碍。
第三节 雄激素类药和同化激素类药
一、雄激素类药
【来源】天然雄激素(androgens)主要是睾丸间质细胞分泌的睾酮(睾丸素,testosteron0一些新衍生物,临床常用的为甲睾酮(android;甲基睾丸素,methyltestosterone)、丙酸睾酮(andronate;丙酸睾丸素,testosterone propionate)和苯乙酸睾酮(苯乙酸睾丸素,testosterone phenylacetate)。
睾酮 甲睾酮
雄激素类的化学结构
【体内过程】睾酮口服易吸收,但在肝被迅速破坏,因此口服无效。大部分与蛋白结合。代谢物与葡萄糖醛酸或硫酸结合失去活性,经尿排泄。也可作成片剂植于皮下,吸收缓慢,作用可长达6周。睾酮的酯类化合物极性较低,溶于油液中肌内注射后,吸收缓慢,持续时间也较长,例如丙酸睾酮一次肌内注射可维持2~4天。甲睾酮不易被肝脏破坏,口服有效。也可舌下给药。
【生理及药理作用】
1.生殖系统促进男性性征和生殖器官发育,并保持其成熟状态。睾酮还可抑制垂体前叶分泌促性腺激素(负反馈),对女性可减少雌激素分泌。尚有抗雌激素作用。
2.同化作用雄激素能明显地促进蛋白质合成(同化作用),减少氨基酸分解(异化作用),使肌肉增长,体重增加,降低氮质血症,同时出现水、钠、钙、磷潴留现象。
3.骨髓造血功能在骨髓功能低下时,大剂量雄激素可促进细胞生长。是促进肾脏分泌促红细胞生成素所致,也可能是直接刺激骨髓造血功能。
【临床应用】
1.睾丸功能不全无睾症或类无睾症(睾丸功能不全)时,作替代疗法。
2.功能性子宫出血利用其抗雌激素作用使子宫平滑肌及其血管收缩,内膜萎缩而止血。对严重出血病例,可用已烯雌酚、黄体酮和丙酸睾酮等三种混合物作注射,以收止血之效,停药后则出现撤退性出血。
3.晚期乳腺癌对晚期乳腺癌或乳腺癌转移者,采用雄激素治疗可使部分病例的病情得到缓解。这可能与其抗雌激素作用有关,也可能通过抑制垂体促性腺激素的分泌,减少卵巢分泌雌激素。此外,雄激素尚有抗催乳素刺激乳腺癌的作用。治疗效果与癌细胞中雌激素受体含量有关,受体浓度高者,疗效较好。
4.再生障碍性贫血及其他贫血用丙酸睾酮或甲睾酮可使骨髓机能改善。
【不良反应】
1.如长期应用于女性病人可能引起痤疮、多毛、声音变粗、闭经、乳腺退化、性欲改变等男性化现象。发现此现象应立即停药。
2.多数雄激素均能干扰肝内毛细胆管的排泄功能,引起胆汁郁积性黄疸。应用时若发现黄疸或肝功能障碍时,则应停药。
【禁忌证及应用注意】对孕妇及前列腺癌病人禁用。因有水、钠潴留作用,对肾炎、肾病综合征、肝功能不良、高血压及心力衰竭病人也应慎用。
二、同化激素类药
临床应用雄性激素虽有较强的同化作用,但用于女性或非性腺功能不全的男性,常可出现雄激素作用,从而限制了它的临床应用;因此,合成了同化作用较好,而雄激素样作用较弱的睾酮的衍生物,即同化激素(anabolic steroids),如南诺龙(苯丙酸诺龙,nandrolone phenylpropionate)、司坦唑(stanozolol,康力龙)及美雄酮(methandienone,去氢甲基睾丸素)等。
本类药物主要用于蛋白质同化或吸收不足,以及蛋白质分解亢进或损失过多等情况;如严重烧伤、手术后慢性消耗性疾病、老年骨质疏松和肿瘤恶液质等病人。服用时应同时增加食物中的蛋白质成分。是体育竞赛的一类违禁药。
长期应用可引起水钠潴留及女性轻微男性化现象。有时引起肝内毛细胆管胆汁郁积而发生黄疸。肾炎、心力衰竭和肝功能不良者慎用,孕妇及前列腺癌病人禁用。
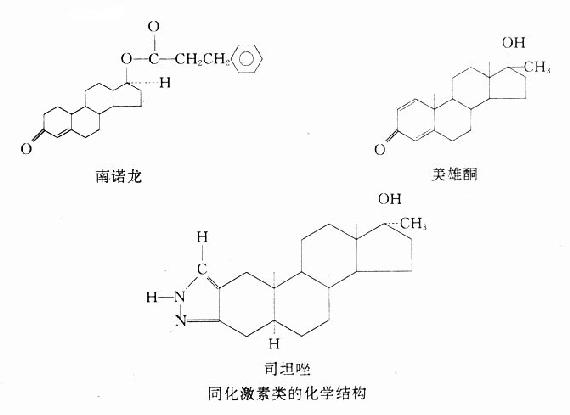
第四节 避孕药
使用避孕药是计划生育的一项重要措施。
生殖过程是一个复杂的生理过程,包括精子和卵子的形成与成熟、排卵、受精、着床以及胚胎发育等多个环节,阻断其中任何一个环节都可以达到避孕和终止妊娠的目的。这些环节多发生在女性体内,这使女性避孕药较男性避孕药发展为快。
与其他药物比较,避孕药有下列几个特点:①应用广,例如女用口服避孕药在目前全世界有数千万人使用;②服药时间长,可达10年以上;③对于安全度要求特别高;④一般药物的疗效达到70%~80%已算不错,避孕药则远非如此,目前应用的避孕药虽尚未能达到100%的疗效,但总要接近99%才算满意。
一、主要抑制排卵的避孕药
【药理作用】现应用的女性避孕药以此类为主。它们由不同类型的雌激素和孕激素类组成,主要避孕作用是抑制排卵。一般认为雌激素通过负反馈机制抑制下丘脑GnRH的释放,从而减少FSH分泌,使卵泡的生长成熟过程受到抑制,同时孕激素又抑制LH释放,两者协同作用而抑制排卵。动物实验证明,甾体避孕药的抗排卵作用可被外源性促性腺激素所防止,此结果支持上述说法。如按规定用药,用药期间避孕效果可达99%以上。停药后,垂体前叶产生和释放FSH和LH以及卵巢排卵功能都可以很快恢复。
除以上作用外,此类药物还可干扰生殖过程的其他环节,例如:可能使子宫内膜的正常增殖受到抑制,腺体少而内膜萎缩,因此不适宜受精卵的着床;还可能影响子宫和输卵管制的正常活动,改变受精卵在输卵管的运行速度,以致受精卵不能适时地到达子宫。此外,宫颈粘液变得更粘稠,使精子不易进入子宫腔等。
【分类及用途】
1.短效口服避孕药如复方炔诺酮片、复方甲地孕酮片及复方炔诺孕酮片等(其成分见表33-1)。从月经周期第5天开始,每晚服药1片,连服22天,不能间断。一般于停药后2~4天就可以发生撤退性出血,形成人工月经周期。下次服药仍从月经来潮第5天开始。如停药7天仍未来月经,则应立即开始服下一周期的药物。偶尔漏服时,应于24小时内补服一片。
2.长效口服避孕药是以长效雌激素类药物炔雌醚与不同孕激素类如炔诺孕酮或氯地孕酮等配伍而成的复方片剂。每月服一次,成功率为98.3%。服法是从月经来潮当天算起,第5天服一片,最初两次间隔20天,以后每月服一次,每次一片。
3.长效注射避孕药 如复方已酸孕酮注射液(即避孕针1号),第一次于月经周期的第5日深部肌内注射2支,以后每隔28日或于每次月经周期的第11~12天注射一次,每次1支。注射后一般于14天左右月经来潮。如发生闭经,仍应按期给药,不能间断。
4.埋植剂 以已内酮小管(约φ2mm×30mm)装入炔诺孕酮70mg,形成棒状物,植入臂内侧或左肩胛部皮下。
5.多相片剂为了使服用者的激素水平近似月经周期水平并减少月经期间出血的发生率,可将避孕药制成多相片剂,如炔诺酮双相片、三相片和炔诺孕酮三相片。双相片是开始10天每日服一片含炔诺酮0.5mg和炔雌醇0.035mg的片剂,后11天每日服一片含炔诺酮1mg和炔雌醇0.035mg的片剂,这种服用法,很少发生突破性出血,是其优点。三相片则分为开始7天每日服一片含炔诺酮0.5mg和炔雌醇0.035mg的片剂,中期7天,每日服用一片含炔诺酮0.75mg和炔雌醇0.035mg的片剂,最后7天每日服用一片含炔诺酮1mg和炔雌醇0.035mg的片剂,其效果较双相片更佳。炔诺孕酮三相片则为开始6天每日服用一片含炔诺孕酮0.05mg和炔雌醇0.03mg的片剂,中期5天每日服用一片含炔诺孕酮0.075mg和炔雌醇0.04mg的片剂,后10天每日服用一片含炔诺孕酮0.125mg和炔雌醇0.03mg的片剂,这种服法更符合人体内源性激素的变化规律,临床效果更好。
【不良反应】
表33-1 几种甾体避孕制剂的成分
| 制剂名称 | 孕激素 成分(mg) | 雌激素 |
| 短效口服避孕药 | ||
| 复方炔诺酮片(口服避孕药片Ⅰ号) | 炔诺酮0.6 | 炔雌醇0.035 |
| 复方甲地孕酮片(口服避孕药片Ⅱ号) | 甲地孕酮1.0 | 炔雌醇0.035 |
| 复方炔诺孕酮甲片 | 炔诺孕酮0.3 | 炔雌醇0.03 |
| 长效口服避孕药 | ||
| 复方炔诺孕酮乙片(长效避孕片) | 炔诺孕酮12.0 | 炔雌醚3.0 |
| 复方氯地孕酮片 | 氯地孕酮12.0 | 炔雌醚3.0 |
| 复方次甲氯地孕酮片 | 16-次甲氯地孕酮12.0 | 炔雌醚3.0 |
| 长效注射避孕药 | ||
| 复方已酸孕酮注射液(避孕针1号) | 已酸孕酮250.0 | 戊酸雌二醇5.0 |
| 复方甲地孕酮注射液 | 甲地孕酮25.0 | 雌二醇3.5 |
| 探亲避孕药 | ||
| 甲地孕酮片(探亲避孕1号片) | 甲地孕酮2.0 | |
| 炔诺酮片(探亲避孕片) | 炔诺酮5.0 | |
| 双炔失碳酯片(53号避孕片) | 双炔失碳酯7.5 |
1.类早孕反应少数妇女在用药初期可出现轻微的类早孕反应,如恶心、呕吐及择食等。一般坚持用药2~3个月后可减轻或消失。
2.子宫不规则出血较常见于用药后最初几个周期中,如出现不规则出血,可加服炔雌醇。
3.闭经 约有1%~2%服药妇女发生闭经,有不正常月经史者较易发生。如连续两个月闭经,应予停药。
4.乳汁减少少数哺乳妇女乳汁减少。长效口服避孕药可通过乳汁影响乳儿,使其乳房肿大。
5.凝血功能亢进国外报道本类药物可诱发血栓性静脉炎、肺栓塞或脑血管栓塞等。国内虽尚未见报道,但仍应注意。
6.其他 可能出现座疮、皮肤色素沉着,个别人可能血压升高。
【禁忌证及应用注意】充血性心力衰竭或有其他水肿倾向者慎用。急慢性肝病及糖尿病需用胰岛素治疗者不宜用。对长期用药是否会增加肿瘤发病率的问题,各家报道不一,但仍应注意,如长时用药过程中出现乳房肿块,应立即停药。宫颈癌患者禁用。
二、抗着床避孕药
此类药物也称探亲避孕药,主要使子宫内膜发生各种功能和形态变化,使之不利于孕卵着床。我国多用大剂量炔诺酮(5mg/次)或甲地孕酮(2mg/片);此外还研制成一种新型抗着床药双炔失碳酯(anorethidrane dipropionate,53抗孕片)。本类药物主要优点是应用不受月经周期的限制,无论在排卵前、排卵期或排卵后服用,都可影响孕卵着床。一般于同居当晚或事后服用。同居14日以内必须连服14片,如超过14日,应接服Ⅰ号或Ⅱ号口服避孕药。
三、男性避孕药
棉酚(gossypol)是棉花根、茎和种子中所含的一种黄色酚类物质。其作用部位在睾丸细精管的生精上皮,可使精子数量减少,直至无精子。停药后可逐渐恢复。经健康男子试用,每天20mg,连服两个月即可达节育标准,有效率达90%以上。
不良反应有乏力、食欲减退、恶心、呕吐、心悸及肝功能改变等。服药者如发生低血钾肌无力症状,应加处理。
性激素类药及避孕药 制剂及用法
苯甲酸雌二醇(estradiol benzoate)肌内注射,1~2mg/次,2~3次/周。
已烯雌酚(diethylstilbestrol)用于卵巢功能不全、垂体功能异常的闭经或绝经期综合征:一日量不超过0.25mg;用于人工周期,口服0.25mg/日,连服20日,待月经后再服,用法同前,共3周;或先用已烯雌酚1mg/次,每晚1次,连用22天,于服药后第16日开始肌内注射黄体酮10mg,共5日。阴道栓剂:0.1~0.5mg/粒。
炔雌醇(乙炔雌二醇,ethinylestradiol)作用比已烯雌酚强,用量为后者的1/20。
黄体酮(progesterone) 肌内注射。先兆流产或习惯性流产:10~20mg/日。检查闭经的原因:10mg/日,共3~5日,停药后2~3日若见子宫出血,说明闭经并非由于妊娠。
醋酸甲羟孕酮(medroxyprogesterone acetate)口服,2~10mg/日。
枸橼酸氯米芬(clomifene citrate) 促排卵,口服,50mg/次,1次/日,连服5日。
甲地孕酮醋酸酯(megestrol acetate) 口服,2~4mg/次,1次/日。
炔诺酮(norethisterone) 口服,1.25~5mg/次,1次/日。
丙酸睾酮(testosterone propionate) 肌内注射,10~50mg/日,1~3次/周。
甲睾酮(methyltestosterone) 舌下给药或口服,5~10mg/次,1~2次/日。
苯乙酸睾酮(testosteronephenylacetate)肌内注射。效力较丙酸睾酮强而持久,故称长效睾酮。10~25mg/次,2~3次/周。
睾酮小片 75mg/片,每6周植入皮下1片。用于无睾症等作补充(代替)疗法。
南诺龙(nandrolone phenylpropionate)肌内注射,25mg/次,1~2次/周。
美雄酮(methandienone)口服,5~10mg/次,2~3次/日。
司坦唑(stanozolol) 口服,2mg/次,2~3次/日。
避孕药见表33-1。
第三十四章 肾上腺皮质激素类药物
肾上腺皮质激素(adrenocortical hormones)是肾上腺皮质所分泌的激素的总称,属甾体类化合物。可分为三类:①盐皮质激素(mineralocorticoids),由球状带分泌,有醛固酮(aldosterone)和脱氧皮质酮(desoxycortone,desoxycorticosterone)等。②糖皮质激素(glucocorticoids),由束状带合成和分泌,有氢化可的松(hydrocortisone)和可的松(cortisone)等,其分泌和生成受促皮质素(ACTH)调节。③性激素,由网状带所分泌,通常所指肾上腺皮质激素,不包括后者。临床常用的皮质激素是指糖皮质激素。
【化学结构及构效关系】肾上腺皮质激素的基本结构为甾核(图34-1),构效关系非常密切:①C3的酮基、C20的羰基及C4-5的双键是保持生理功能所必需;②糖皮质激素的C17上有-OH;C11上有=O或-OH;③盐皮质激素的C17上无-OH;C11上无=O或有O与C18相联;④C1~2为双键以及C6引入-CH3则抗炎作用增强、水盐代谢作用减弱;⑤C9引入-F,C16引入-CH3或-OH则抗炎作用更强、水盐代谢作用更弱。
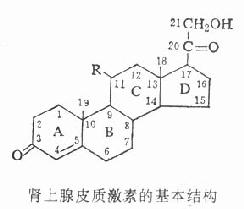
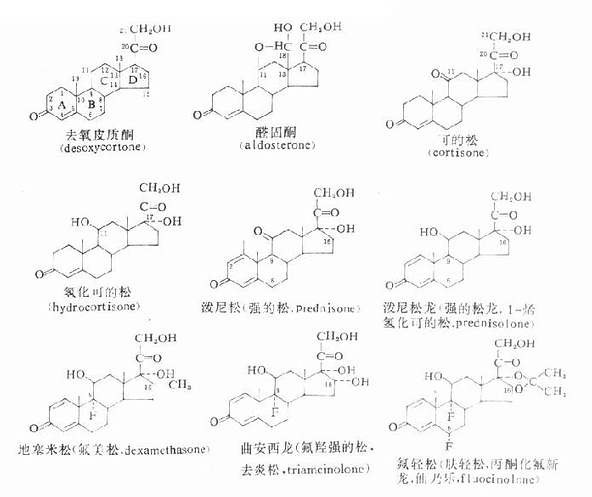
为了提高皮质激素的临床疗效,曾对它们的结构进行改造并获得多种新型药物。
第一节 糖皮质激素
糖皮质激素作用广泛而复杂,且随剂量不同而异。生理情况下所分泌的糖皮质激素主要影响物质代谢过程,超生理剂量的糖皮质激素则还有抗炎、抗免疫等药理作用。
【生理效应】
1.糖代谢 糖皮质激素能增加肝糖原、肌糖原含量并升高血糖,其机制为:促进糖原异生;减慢葡萄糖分解为CO2的氧化过程;减少机体组织对葡萄糖的利用。
2.蛋白质代谢 促进淋巴和皮肤等的蛋白质分解,抑制蛋白质的合成,久用可致生长减慢、肌肉消瘦、皮肤变薄、骨质疏松、淋巴组织萎缩和伤口愈合延缓等。
3.脂肪代谢 促进脂肪分解,抑制其合成。久用能增高血胆固醇含量,并激活四肢皮下的脂酶,使四肢脂肪减少,还使脂肪重新分布于面部、胸、背及臀部,形成满月脸和向心性肥胖。
4.水和电解质代谢 也有较弱的盐皮质激素的作用,能潴钠排钾。增加肾小球滤过率和拮抗抗利尿素,故可利尿。过多时还可引起低血钙,长期应用可致骨质脱钙。
【药理作用】
1.抗炎作用 糖皮质激素有强大的抗炎作用,能对抗各种原因如物理、化学、生理、免疫等所引起的炎症。在炎症早期可减轻渗出、水肿、毛细血管扩张、白细胞浸润及吞噬反应,从而改善红、肿、热、痛等症状;在后期可抑制毛细血管和纤维母细胞的增生,延缓肉芽组织生成,防止糖连及瘢痕形成,减轻后遗症。但必须注意,炎症反应是机体的一种防御功能,炎症后期的反应更是组织修复的重要过程。因此,糖皮质激素在抑制炎症、减轻症状的同时,也降低机体的防御功能,可致感染扩散、阻碍创口愈合。
皮质激素抗炎作用的基本机制在于糖皮质激素(GCS)与靶细胞浆内的糖皮质激素受体(G-R)相结合后影响了参与炎症的一些基因转录而产生抗炎效应。糖皮质激素的靶细胞广泛分布于肝、肺、脑、骨、胃肠平滑肌、骨骼肌、淋巴组织、成纤维细胞、胸腺等处。各类细胞中受体的密度也各不相同。
G-R由约800个氨基酸构成。其C端与GCS结合;其中央有两个锌指(zinc finger),各结合4个半胱氨酸;其N端的功能区τ1涉及与DNA结合后的转录性基因转移活化以及与其他转录因子的结合,人的G-R的结合功能区中还有τ2,它对受体进入核内有重要作用。(图34-1)。
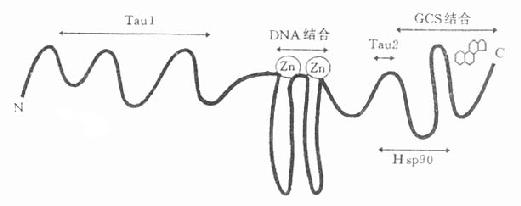
图34-1 皮质激素受体功能区示意图
未活化的G-R与一大分子(约30kDa)蛋白质复合物相结合,该复合物含有两个热休克蛋白90(heat shock protein 90,Hsp 90),其C端与受体相结合;该复合物可能还结合有其他蛋白,如抑制性蛋白等。一旦GCS与G-R结合,Hsp 90被解离,则被活化的GCS-G-R复合物迅速进入核内,进而与靶的基因的启动子(promoter)序列的糖皮质激素反应成分(glucocorticoid response element,GRE)或负性糖皮质激素反应成分(negativeglucocorticoid respones element,nGRE)相结合,相应地引起转录增加或减少,继而通过mR-NA影响介质蛋白合成(图34-2)。
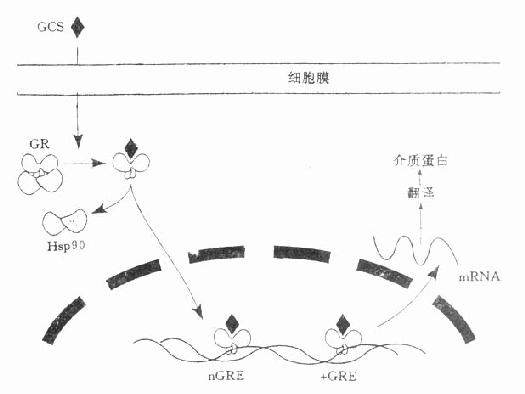
图34-2 皮质激素作用机制示意图
GCS可通过增加或减少基因转录而抑制炎症过程的某些环节,如对细胞因子、炎症介质及一氧化氮合成酶等的影响等。
细胞因子(cytokine)在慢性炎症起到重要作用。它们能促进血管内皮细胞粘附白细胞,进而使其从血液渗出到炎性部位,并能使内皮细胞、嗜中性白细胞及区噬细胞活化,还能使血管通透性增加、刺激成纤维细胞增生以及刺激淋巴细胞增殖与分化。GCS通过与G-R结合-nGRE的相互作用而抑制了一些与慢性炎症有关的细胞因子白介素1(IL-1)、肿瘤坏死因子α(TNFα)、巨噬细胞集落刺激因子(GM-CSF)、白介素3(IL-3)、白介素4(IL-4)、白介素5(IL-5)、白介素6(IL-6)及白介素8(IL-8)等的转录,而强烈地抑制细胞因子介导的炎症。也有证据表明GCS可增加mRNA的断裂而使IL-1、IL-3及GM-CSF减少。GCS也可通过抑制IL-2受体的合成,或通过将活化转录因子活化蛋白-1(activator protein-1,AP-1)等的活化调节逆转,或通过直接与AP-1相互作用,而对抗细胞因子的效应。
炎症介质,如白三烯(LT)、前列腺素(PG)等,前者有较强的白细胞趋化作用和增加血管通透性的作用,后者可引起红、肿、热、痛等炎症反应。GCS可通过增加脂皮素(lipocortin-1)的合成及释放而抑制脂质介质白三烯(LT)、前列腺素(PG)及血小板活化因子(PAF)的生成,因为脂皮素(37kDa)可抑制脂质生成所必需的磷脂酶A2(PLA2)。皮质激素还可以诱导血管紧张素转化酶(ACE)而降解缓激肽(可引起血管舒张和致痛),产生抗炎作用。
GCS可抑制巨噬细胞中一氧化氮合酶(NO synthase,NOS)而发挥抗炎作用,因为各种细胞因子均可诱导NOS,使NO生成增多而增加炎性部位的血浆渗出,水肿形成及组织损伤,加重炎症症状。
2.免疫抑制作用(详见第五十章)对免疫过程的许多环节均有抑制作用。首先抑制巨噬细胞对抗原的吞噬和处理。其次,对敏感动物由于淋巴细胞的破坏和解体,使血中淋巴细胞迅速减少;糖皮质激素对人也引起暂时性淋巴细胞减少,其原因可能与淋巴细胞移行至血液以外的组织有关,而不是淋巴细胞溶解所致。动物实验指出,小剂量主要抑制细胞免疫;大剂量则能抑制由B细胞转化成浆细胞的过程,使抗体生成减少,干扰体液免疫,原因可能与其选择性地作用于T细胞亚群,特别是增强了T。抑制B细胞的作用有关。但在人体迄未证实糖皮质激素在治疗剂量时能抑制抗体产生。
3.抗休克超大剂量的皮质激素类药物已广泛用于各种严重休克,特别是中毒性休克的治疗,对其评价虽尚有争论,但一般认为其作用与下列因素有关:①扩张痉挛收缩的血管和加强心脏收缩;②降低血管对某些缩血管活性物质的敏感性,使微循环血流动力学恢复正常,改善休克状态;③稳定溶酶体膜,减少心肌抑制因子(myocardio-depressantfactor,MDF)的形成。④提高机体对细菌内毒素的耐受力。保护动物耐受脑膜炎双球菌、大肠杆菌等内毒素致死量数倍至数十倍。
4.其他作用
(1)血液与造血系统 皮质激素能刺激骨髓造血机能,使红细胞和血红蛋白含量增加,大剂量可使血小板增多并提高纤维蛋白原浓度,缩短凝血时间;促使中性白细胞数增多,但却降低其游走、吞噬、消化及糖酵解等功能,因而减弱对炎症区的浸润与吞噬活动。对淋巴组织也有明显影响,在肾上腺皮质功能减退者,淋巴组织增生,淋巴细胞增多;而在肾上腺皮质功能亢进者,淋巴细胞减少,淋巴组织萎缩。
(2)中枢神经系统 能提高中枢神经系统的兴奋性,出现欣快、激动、失眠等,偶可诱发精神失常。大剂量对儿童能致惊厥。
(3)消化系统 糖皮质激素能使胃酸和胃蛋白酶分泌增多,提高食欲,促进消化,但大剂量应用可诱发或加重溃疡病。
【体内过程】口服、注射均可吸收。口服可的松或氢化可的松后1~2小时血药浓度可达高峰。一次给药作用持续8~12小时。
氢化可的松在血浆中(浓度小于25µg%时)约有90%以上与血浆蛋白结合,其中77%与皮质激素转运蛋白(transcortin,corticosteroid binding globulin,CBG)结合,CBG在血浆中含量少,虽亲和力大(3×10-7mol/L),但结合容量仍小;另有15%与白蛋白结合,其血浆含量高,结合量大。CBG在肝中合成,雌激素可促进其合成,妊娠期间或雌激素治疗时,血中CBG浓度增高而游离的氢化可的松减少,但通过反馈调节,可使游离型者恢复正常水平。肝、肾病时CBG合成减少,可使游离型增多。
吸收后,在肝分布较多。主要在肝中代谢,与葡萄糖醛酸或硫酸结合,与未结合部分一起由尿排出。
氢化可的松的血浆t1/2为80~144分钟,但在2~8小时后仍具有生物活性,剂量大或肝、肾功能不全者可使t1/2延长;甲状腺功能亢进时,肝灭活皮质激素加速,使t1/2缩短。泼尼松龙因不易被灭活,t1/2可达200分钟。
可的松和泼尼松在肝内分别转化为氢化可的松和泼尼松龙而生效,故严重肝功能不全的病人只宜应用氢化可的松或泼尼松龙。与肝微粒体酶诱导剂如苯巴比妥、苯妥英钠等合用时需加大皮质激素的用量。
表34-1 常用糖皮质激素类药物的比较
| 类别 | 药物 | 对受体的亲和力* | 水盐代谢(比值) | 糖代谢(比值) | 抗炎作用(比值) | 等效剂量(mg) | 半衰期(分) | 半效期(小时) | 一次口服常用量(mg) |
| 短效 | 氢化可的松 | 1 | 1.0 | 1.0 | 1.0 | 20 | 90 | 8~12 | 10~20 |
| 可的松 | 0.01 | 0.8 | 0.8 | 0.8 | 25 | 90 | 8~12 | 12.5~25 | |
| 中效 | 泼尼松 | 0.05 | 0.6 | 3.5 | 3.5 | 5 | >200 | 12~36 | 2.5~10 |
| 泼尼松龙 | 2.2 | 0.6 | 4.0 | 4.0 | 5 | >200 | 12~36 | 2.5~10 | |
| 甲泼尼龙 | 11.9 | 0.5 | 5.0 | 5.0 | 4 | >200 | 12~36 | 2.0~8 | |
| 曲安西龙(去炎松) | 1.9 | 5.0 | 5.0 | 4 | >200 | 12~36 | 2.0~8 | ||
| 长效 | 地塞米松 | 7.1 | 30 | 30 | 0.75 | >300 | 36~54 | 0.75~1.5 | |
| 倍他米松 | 5.4 | 30~35 | 25~35 | 0.60 | >300 | 36~54 | 0.6~1.2 | ||
| 外用 | 氟氢可的松 | 3.5 | 125 | 12 | |||||
| 氟轻松 | 1 | 40 |
*胎儿肺细胞
【临床应用】
1.替代疗法 用于急、慢性肾上腺皮质功能减退症(包括肾上腺危象)、脑垂体前叶功能减退及肾上腺次全切除术后作替代疗法。
2.严重感染或炎症
(1)严重急性感染,如中毒性菌痢、暴发型流行性脑膜炎、中毒性肺炎、重症伤寒、急性粟粒性肺结核、猩红热及败血症等,在应用有效的抗菌药物治疗感染的同时,可用皮质激素作辅助治疗,但对其疗效尚有不同看法。病毒性感染一般不用激素,因用后可减低机体的防御能力反使感染扩散而加剧。但对严重传染性肝炎、流行性腮腺炎、麻疹和乙型脑炎等,也有缓解症状的作用。
(2)防止某些炎症后遗症,如结核性脑膜炎、脑炎、心包炎、风湿性心瓣膜炎、损伤性关节炎、睾丸炎以及烧伤后疤痕挛缩等,早期应用皮质激素可防止后遗症发生。对虹膜炎、角膜炎、视网膜炎和视神经炎等非特异性眼炎,应用后也可迅速消炎止痛、防止角膜混浊和疤痕粘连的发生。
3.自身免疫性疾病及过敏性疾病
(1)自身免疫性疾病风湿热、风湿性心肌炎、风湿性及类风湿性关节炎、全身性红斑狼疮、结节性动脉周围炎、皮肌炎、自身免疫性贫血和肾病综合征等应用皮质激素后可缓解症状。一般采用综合疗法,不宜单用,以免引起不良反应。异体器官移植手术后所产生的排异反应也可应用皮质激素。
(2)过敏性疾病荨麻疹、枯草热、血清热、血管神经性水肿、过敏性鼻炎、支气管哮喘和过敏性休克等,应以肾上腺受体激动药和抗组胺药治疗,病情严重或无效时,也可应用皮质激素辅助治疗,使能抑制原-杭体反应反致的组织损害和炎症过程。
4.抗休克治疗 感染中毒性休克时,在有效的抗菌药物治疗下,可及早、短时间突击使用大剂量皮质激素,见效后即停药;对过敏性休克,皮质激素为次选药,可与首选药肾上腺素合用;对心原性休克,须结合病因治疗;对低血容量性休克,在补液补电解质或输血后效果不佳者,可合用超大剂量的皮质激素。
5.血液病 可用于急性淋巴细胞性白血病、再生障碍性贫血、粒细胞减少症、血小板减少症和过敏性紫癜等的治疗,但停药后易复发。
6.局部应用 对接触性皮炎、湿疹、肛门搔痒、牛皮癣等都有疗效。宜用氢化可的松、泼尼松龙或氟轻松。对天疱疮及剥脱性皮炎等严重病例仍需全身用药。
【不良反应】
1.长期大量应用引起的不良反应
(1)类肾上腺皮质功能亢进综合征 因物质代谢和水盐代谢紊乱所致,如满月脸、水牛背、向心性肥胖、皮肤变薄、痤疮、多毛、浮肿、低血钾、高血压、糖尿等(图34-3)。停药后可自行消退,必要时采取对症治疗,如应用降压药、降糖药、氯化钾、低盐、低糖、高蛋白饮食等。
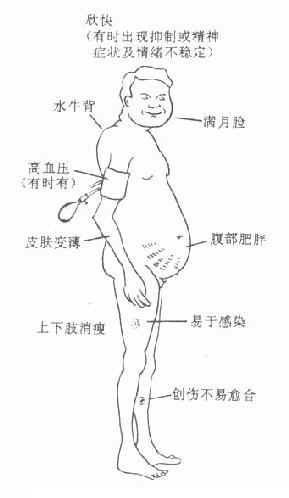
还有:负氮平衡、骨质疏松、食欲增加、低血钾、高血糖倾向、消化性溃疡
图34-3 长期服用糖皮质激素后的不良反应示意图
(2)诱发或加重感染 因皮质激素抑制机体防御功能所致。长期应用常可诱发感染或使体内潜在病灶扩散,特别是在原有疾病已使抵抗力降低如肾病综合征者更易产生。还可使原来静止的结核病灶扩散、恶化。故结核病患者必要时应并用抗结核药。
(3)消化系统并发症 使胃酸、胃蛋白酶分泌增加,抑制胃粘液分泌,降低胃肠粘膜的抵抗力,故可诱发或加剧胃、十二指肠溃疡,甚至造成消化道出血或穿孔。对少数患者可诱发胰腺炎或脂肪肝。
(4)心血管系统并发症 长期应用可引起高血压和动脉粥样硬化。
(5)骨质疏松、肌肉萎缩、伤口愈合迟缓等与激素促进蛋白质分解、抑制其合成及增加钙、磷排泄有关。骨质疏松多见于儿童、老人和绝经妇女,严重者可有自发性骨折。因抑制生长素分泌和造成负氮平衡,还可影响生长发育。对孕妇偶可引起畸胎。
(6)其他 精神失常。有精神病或癫痫病史者禁用或慎用。
2.停药反应
(1)长期应用尤其是连日给药的病人,减量过快或突然停药时,由于皮质激素的反馈性抑制脑垂体前叶对ACTH的分泌,可引起肾上腺皮质萎缩和机能不全。多数病人可无表现。肾上腺皮质功能恢复的时间与剂量、用药期限和个体差异有关。停用激素后垂体分泌ACTH的功能需经3~5个月才恢复;肾上腺皮质对ACTH起反应机能的恢复约需6~9个月或更久。因此不可骤然停药。停药后也有少数患者遇到严重应激情况如感染、创伤、手术时可发生肾上腺危象,如恶心、呕吐、乏力、低血压、休克等,需及时抢救。这种皮质功能不全需半年甚至1~2年才能恢复。
(2)反跳现象 因病人对激素产生了依赖性或病情尚未完全控制,突然停药或减量过快而致原病复发或恶化。常需加大剂量再行治疗,待症状缓解后再逐渐减量、停药。
【禁忌证】曾患或现患严重精神病和癫痫,活动性消化性溃疡病,新近胃肠吻合术,骨折,创伤修复期,角膜溃疡,肾上腺皮质功能亢进症,严重高血压,糖尿病、孕妇,抗菌药不能控制的感染如水痘、霉菌感染等都是皮质激素的禁忌证。当适应证与禁忌证同时并存时,应全面分析,权衡利弊,慎重决定。一般说,病情危重的适应证,虽有禁忌证存在,仍不得不用,待危急情况过去后,尽早停药或减量。
【用法及疗程】宜根据病人、病情、药物的作用和不良反应特点确定制剂、剂量、用药方法及疗程:
1.大剂量突击疗法 用于严重中毒性感染及各种休克。氢化可的松首次剂量可静脉滴注200~300mg,一日量可达1g以上,疗程不超过3天。对于休克有人主张用超大剂量,每次静脉注射1g,一日4~6次。
2.一般剂量长期疗法 用于结缔组织病、肾病综合征、顽固性支气管哮喘、中心性视网膜炎、各种恶性淋巴瘤、淋巴细胞性白血病等。一般开始时用泼尼松口服10~20mg或相应剂量的其他皮质激素制剂,每日3次,产生临床疗效后,逐渐减量至最小维持量,持续数月。
3.小剂量替代疗法 用于垂体前叶功能减退、阿狄森病及肾上腺皮质次全切除术后。一般维持量,可的松每日12.5~25mg,或氢化可的松每日10~20mg。
4.隔日疗法 皮质激素的分泌具有昼夜节律性,每日上午8~10时为分泌高潮(约450nmol/L),随后逐渐下降(下午4时约110nmol/L),午夜12时为低潮,这是由ACTH昼夜节律所引起。临床用药可随这种节律进行,即长期疗法中对某些慢性病采用隔日一次给药法,将一日或两日的总药量在隔日早晨一次给予,此时正值激素正常分泌高峰,对肾上腺皮质功能的抑制较小。实践证明,外源性皮质激素类药物对垂体-肾上腺皮质轴的抑制性影响,在早晨最小,午夜抑制最大,隔日服药以用泼尼松、泼尼松龙等中效制剂较好。
第二节 促皮质素及皮质激素抑制药
一、促皮质素
促皮质素(corticotrophin,adreno-cortico-tropic-hormone,ACTH)是维持肾上腺正常形态和功能的重要激素。它的合成和分泌是垂体前叶在下丘脑促皮质素释放激素(CRH)的作用下,在腺垂体嗜碱细胞内进行的。糖皮质激素对下丘脑及垂体前叶起着长负反馈作用,抑制CRH及ACTH的分泌。在生理情况下,下丘脑、垂体和肾上腺三者处于相对的动态平衡中,ACTH缺乏,将引起肾上腺皮质萎缩、分泌功能减退。ACTH还有控制本身释放的短负反馈调节(图33-4)。
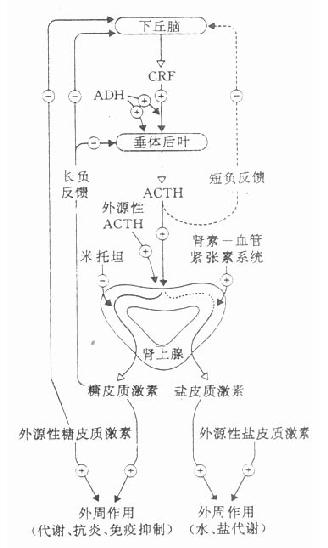
图34-4 下丘脑-垂体前叶-肾上腺皮质的调节系统示意图
抑制兴奋
ADH抗利尿激素 CRH促皮质素释放激素
ACTH口服后在胃内被胃蛋白酶破坏而失效,只能注射应用。血浆t1/2为15分钟。它在正常人的血浆浓度,晨8时为22pg/ml,晚10时为9.6pg/ml。其主要作用是促进糖皮质激素分泌,但只有在皮质功能完好时方能发挥治疗作用。一般在给药后2小时,皮质才开始分泌氢化可的松。临床用于诊断脑垂体前叶-肾上腺皮质功能水平及长期使用皮质激素的停药前后,以防止发生皮质功能不全。由于ACTH易引起过敏反应(因临床应用制剂来自牛、羊、猪垂体),现已少用。
二、皮质激素抑制药
皮质激素抑制剂可代替外科的肾上腺皮质切除术,临床常用的有米托坦和美替拉酮。
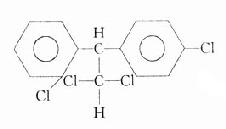
米托坦(mitotan;双氯苯二氯乙烷0,p'-DDD)为杀虫剂滴滴涕(DDT)一类化合物。它能选择性地使肾上腺皮质束状带及网状带细胞萎缩、坏死,但不影响球状带,故醛固酮分泌不受影响。用药后血、尿中氢化可的松及其代谢物迅速减少。主要用于不可切除的皮质癌、切除后复发癌以及皮质癌术后辅助治疗。可有厌食、恶心、腹泻、皮疹、嗜眠、头痛、眩晕、乏力、中枢抑制及运动失调等反应。
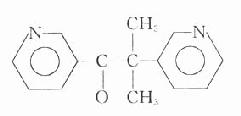
美替拉酮(metyrapone,甲吡酮)能抑制11β-羟化反应,干扰11-去氧皮质酮转化为皮质酮及11-去氧氢化可的松转化为氢化的松,而降低它们的血浆水平,但通过反馈性地促进ACTH分泌导致11-去氧皮质酮和11-去氧氢化可的松代偿性增加,故尿中17-羟类固醇排泄也相应增加。临床用于治疗肾上腺皮质肿瘤和产生ACTH的肿瘤所引起的氢化可的松过多症和皮质癌。还可用于垂体释放ACTH功能试验。不良反应较少,可有眩晕、消化道反应等。
第三节 盐皮质激素
肾上腺皮质激素类药物 制剂及用法
醋酸可的松(cortisone acetate)替代(补充)疗法:口服,12.5~37.5mg/日,分两次;药理治疗:口服,开始75~300mg/日,分3~4次,维持量25~50mg/日。肌内注射25~125mg/次,2~3次/日,用前摇匀。
氢化可的松(hydrocortisone,cortisol)替代(补充:疗法)口服,20~30mg/日,分两次;药理治疗:口服,开始60~120mg/日,分3~4次。维持量20~40mg/日。静脉滴注,100~200mg/次或更多,1~2次/日,临用时以等渗氯化钠溶液或5%葡萄糖溶液500ml稀释。0.5%~2.5%软膏外用。
氢化可的松琥珀酸钠酯(hydrocortisone sodium succinate) 肌内或静脉注射。135mg相当于氢化可的松100mg。
泼尼松(prednisone)开始一般剂量5~15mg/次,3~4次/日,维持量5~10mg。
泼尼松龙(prednisolone) 口服,开始20~40mg/日,分3~4次。维持量5mg/日。静脉滴注,10~20mg/次,加入5%葡萄糖液50~500ml中。
甲泼尼龙(methylprednisolone) 口服,开始16~40mg/日,分4次;维持量4~8mg/日。注射用其琥珀酸钠酯,53mg相当于甲泼尼龙40mg。
氟氢可的松(fludrocortisone) 口服,0.1~0.3mg/日,用于替代疗法。
地塞米松(dexamethasone) 口服,开始0.75~1.5mg/次,3~4次/日,维持量0.5~0.75mg/日。皮下、肌内或静脉注射,5~10mg/次,2次/日。
曲安西龙(triamcinolone) 开始8~40mg/日,分1~3次,维持量4~8mg/日。肌内注射,40~80mg/次,1次/周。关节腔内或皮损部位注射,10~25mg/次。
倍他米松(betametasone,celestone) 口服,开始1.5~2mg/日,分3~4次,维持量0.5~1mg/日。
氟轻松(fluocinolone acetonide) 0.01%~0.025%软膏剂、洗剂、霜剂,外用,3~4次/日。
促皮质素(corticotrophin) 静脉滴注,5~25U/次,溶于生理盐水内,于8小时内滴入,1次/日。肌内注射,25~50U/次。
美替拉酮(metyrapone) 在两天对照观察期后,口服,每4小时750mg,共6次。
第三十五章 甲状腺激素及抗甲状腺药
第一节 甲状腺激素
甲状腺激素为碘化酪氨酸的衍化物,包括甲状腺素(thyroxin,T4)和三碘甲状腺原氨酸(triiodothyronine,T3)。正常人每日释放T4与T3量分别为75及25µg。它们的结构如下:
【甲状腺激素的合成、贮存、分泌与调节】
T3、T4在体内的合成与贮存部分是在甲状腺球蛋白上(TG)进行的,过程如下:①血液循环中的碘化物被甲状腺细胞通过碘泵主动摄取;②碘化物在过氧化物酶的作用下被氧化成活性碘或氧化碘中间产物(I+)。活性碘与TG上的酪氨酸残基结合,生成一碘酪氨酸(MIT)和二碘酪氨酸(DIT);③在过氧化物酶作用下,一分子MIT和一分子DIT偶联生成T3,二分子DIT偶联成T4。合成的T3、T4贮存于滤泡腔内的胶质中;④在蛋白水解酶作用下,TG分解并释出T3、T4进入血液。
垂体、心、肝、肾、骨骼肌、肺、肠组织的细胞都含有甲状腺素受体。在胞膜、线粒体、核内也有该受体分布。T3可与膜上受体结合,也可被动进入胞内,与胞浆结合蛋白(cytosol binding protein,CBP)结合并与游离的T3形成平衡状态。甲状腺激素通过调控由核内T3受体所中介的基因表达而发挥作用。近来证明甲状腺激素受体是具有结合DNA能力的非组蛋白,分子量为52kD,可能是原癌基因的产物。T3也可与胞核染色质上的受体结合,使RNA多聚酶活性增加,启动、调控转录,增加mRNA及蛋白质合成。结果多种酶和细胞活性加强。
【药理作用】
1.维持生长发育 甲状腺激素为人体正常生长发育所必需,其分泌不足或过量都可引起疾病。甲状腺功能不足时,躯体与智力发育均受影响,可致呆小病(克汀病),成人甲状腺功能不全时,则可引起粘液性水肿。
2.促进代谢 甲状腺激素能促进物质氧化,增加氧耗,提高基础代谢率,使产热增多,而又不能很好利用。甲状腺功能亢进时有怕热、多汗等症状。
3.神经系统及心血管效应 呆小病患者的中枢神经系统的发育发生障碍。甲状腺功能亢进时出现神经过敏、急躁、震颤、心率加快、心输出量增加等现象。因甲状腺激素可增强心脏对儿茶酚胺的敏感性。
【体内过程】口服易吸收,T3及T4的生物利用度分别为50%~75%及90%~95%,与血浆蛋白结合率均高达99%以上。但T3与蛋白质的亲和力低于T4,其游离量可为T4的10倍,T3作用快而强,维持时间短,而T4则作用慢而弱、维持时间长。t1/2较长,T4为5天,T3为2天,主要在肝、肾线粒体内脱碘,并与葡萄糖醛酸或硫酸结合而经肾排泄。甲状腺激素可通过胎盘和进入乳汁、妊娠和哺乳期应注意。
【临床应用】甲状腺激素主要用于甲状腺功能低下的替代补充疗法。
1.呆小病 功能减退始于胎儿或新生儿,若尽早诊治,则发育仍可正常。若治疗过晚,则智力仍然低下,应终身治疗。
2.粘液性水肿 一般服用甲状腺片,从小量开始,逐渐增大至足量。剂量不宜过大,以免增加心脏负担而加重心脏疾患。垂体功能低下的病人宜先用皮质激素再给予甲状腺激素,因易发生急性肾上腺皮质功能不全。粘液性水肿昏迷者必须立即静注大量L-T4(左旋甲状腺素),以后每日给50µg,待患者苏醒后改为口服。
3.单纯性甲状腺肿 其治疗取决于病因。由于缺碘所致者应补碘。临床上无明显原因发现可给予适量甲状腺激素,以补充内源性激素的不足,并可抑制甲状腺激素过多分泌,以缓解甲状腺组织代偿性增生肥大。
【不良反应】过量可引起甲状腺功能亢进的临床表现,在老人和心脏病患者中,可发生心绞痛和心肌梗塞,宜用β受体阻断药对抗,并应停用甲状腺激素。
第二节 抗甲状腺药
可用于治疗甲状腺功能亢进(甲亢)的药物有硫脲类、碘化物、放射性碘及β受体阻断药。
一、硫脲类
硫脲类可分为二类:(1)硫氧嘧啶类,包括甲硫氧嘧啶(methylthiouracil),丙硫氧嘧啶(propylthiouracil);(2)咪唑类,包括甲巯咪唑(thiamazole,他巴唑),卡比马唑(carbimazole,甲亢平)它们的化学结构如下:
【药理作用及作用机制】硫脲类的基本作用是抑制甲状腺过氧化物酶所中介的酪氨酸的碘化及偶联,而药物本身则作为过氧化物酶的底物而被碘化,使氧化碘不能结合到甲状腺球蛋白上,从而抑制甲状腺激素的生物合成。硫脲类药物对已合成的甲状腺激素无效,须待已合成的激素被消耗后才能完全生效。一般用药2~3周甲亢症状开始减轻,1~3个月基础代谢率才恢复正常。本类药物长期应用后,可使血清甲状腺激素水平显著下降,反馈性增加TSH分泌而引起腺体代偿性增生,腺体增大、充血,重者可产生压迫症状。
丙硫氧嘧啶还能抑制外周组织的T4转化为T3,能迅速控制血清中生物活性较强的T3水平,故在重症甲亢、甲亢危象时该药可列为首选。此外,硫脲类药物尚有免疫抑制作用,能轻度抑制免疫球蛋白的生成,使血循环中甲状腺刺激性免疫球蛋白(thyroid stimulating immunoglobulin TSI)下降,因此对甲亢患者除能控制高代谢症状外,对病因也有一定的治疗作用,因认为甲亢的发病与自体免疫机制异常有关。
【临床应用】 主要用于甲状腺功能亢进。
1.内科药物治疗 适用于轻症和不宜手术或131I治疗者,如儿童、青少年及术后复发而不适于131I治疗者可用。开始治疗给大剂量以对甲状腺激素合成产生最大抑制作用。经1~3个月后症状明显减轻,当基础代谢率接近正常时,药量即可递减,直至维持量,疗程1~2年。
2.手术前准备 为减少甲状腺次全切除手术病人在麻醉和手术后的合并症,防止术后发生甲状腺危象。在手术前应先服用硫脲类药物,使甲状腺功能恢复或接近正常。然后于术前两周加服碘剂,以利手术进行及减少出血。
3.甲状腺危象的治疗 甲状腺危象的患者可因高热、虚脱、心力衰竭、肺水肿、电解质紊乱而死亡。此时除主要应用大剂量碘剂和采取其他综合措施外,大剂量硫脲类可作为辅助治疗,以阻断甲状腺激素的合成。
普萘洛尔等也是甲亢及甲状腺危象时有价值的辅助治疗药,用于不宜用抗甲状腺药,不宜手术及131I治疗的甲亢患者。主要通过其阻断β受体的作用而改善甲亢的症状。此外还能抑制外周T4脱碘成为T3,因T3是主要的外周激素,故这一作用有助于控制甲亢。
β受体阻断药不干扰硫脲类药物对甲状腺的作用,且作用迅速,对甲亢所致的心率加快,心收缩力增强等交感神经活动增强的表现很有效。但单用时其控制症状的作用有限。若与硫脲类药物合用则疗效迅速而显著。
【体内过程】硫氧嘧啶类药物口服后吸收迅速,生物利用度约为80%。血浆蛋白结合率约为75%,在体内分布较广、易进入乳汁和通过胎盘。主要在肝内代谢。t1/2为2小时。
甲巯咪唑的血浆t1/2约为4.7小时,但在甲状腺组织中药物浓度可维持16~24小时,其疗效与甲状腺内药物浓度有关,而后者的高低又与每日给药量呈正相关。每日给药一次(30mg)与每日给药三次(每次10mg)一样,都可发挥较好的疗效。
卡比马唑为甲巯咪唑的衍化物,在体内转化成甲巯咪唑而发挥作用。
【不良反应】常见的不良反应有搔痒、药疹等过敏反应,多数情况下不需停药也可消失。严重不良反应有粒细胞缺乏症。一般发生在治疗后的2~3个月内,故应定期检查血象,若用药后出现咽痛或发热,立即停药则可恢复。特别要注意与甲亢本身所引起的白细胞总数偏低相区别。
二、碘及碘化物
【药理作用及临床应用】碘(iodine)和碘化物(iodide)是治疗甲状腺病最古老的药物,不同剂量的碘化物对甲状腺功能可产生不同的作用。小剂量的碘用于治疗单纯性甲状腺肿,可在食盐中按1/104~1/105的比例加入碘化钾或碘化钠可有效地防止发病。
大剂量碘产生抗甲状腺作用,主要是抑制甲状腺素的释放,可能是抑制了蛋白水解酶,使T3、T4不能和甲状腺球蛋白解离所致。此外大剂量碘还可抑制甲状腺激素的合成。大剂量碘的抗甲状腺作用快而强。用药1~2天起效,10~15天达最大效应。此时若继续用药,反使碘的摄取受抑制、胞内碘离子浓度下降,因此失去抑制激素合成的效应,甲亢的症状又可复发。这就是碘化物不能单独用于甲亢内科治疗的原因。大剂量碘的应用只限于以下情况:①甲状腺机能亢进的手术前准备,一般在术前二周给予复方碘溶液(卢戈液Lugol's solution)以使甲状腺组织退化、血管减少,腺体缩小变韧、利于手术进行及减少出血;②甲状腺危象的治疗,可将碘化物加到10%葡萄糖溶液中静脉滴注,也可服用复方碘溶液,并在二周内逐渐停服,需同时配合服用硫脲类药物。
【不良反应】
1.急性反应 可于用药后立即或几小时后发生,主要表现为血管神经性水肿,上呼吸道水肿及严重喉头水肿。
2.慢性碘中毒表现为口腔及咽喉烧灼感、唾液分泌增多,眼刺激症状等。
3.诱发甲状腺功能紊乱长期服用碘化物可诱发甲亢。碘还可进入乳汁并通过胎盘引起新生儿甲状腺肿,故孕妇及乳母应慎用。
三、放射性碘
临床应用的放射性碘是131I,其t1/2为8天。
【药理作用】利用甲状腺高度摄碘能力,131I可被甲状腺摄取,并可产生β射线(占99%),在组织内的射程仅约2mm,因此其辐射作用只限于甲状腺内,破坏甲状腺实质,而很少波及周围组织。131I还产生γ射线(占1%),可在体外测得,故可用作甲状腺摄碘功能的测定。
【临床应用】
1.13131I能使腺泡上皮破坏,萎缩、减少分泌。同时可降低腺泡内淋巴细胞从而减少抗体产生。一般用药后一个月见效,3~4个月后甲状腺功能恢复正常。
2.甲状腺功能检查 小量131I可用于检查甲状腺功能。甲状腺功能亢进时,摄碘率高,摄碘高峰时间前移。反之,摄碘率低,摄碘高峰时间后延。
【不良反应】易致甲状腺功能低下,故应严格掌握剂量和密切观察有无不良反应,一旦发生甲状腺功能低下可补充甲状腺激素对抗之。
甲状腺激素及抗甲状腺药 制剂及用法
甲状腺(thyroid)是家畜甲状腺的干燥微黄色粉末,不溶于水,片剂含碘量为0.17%~0.23%,治疗粘液性水肿,开始不超过15~30mg/日,渐增至90~180mg/日,分三次服。基础代谢恢复到正常(成人在-5%左右,儿童应在+5%左右)后,改用维持量(成人一般为60~120mg/日)。单纯性甲状腺肿,开始每日60mg,渐增至120~180mg/日,疗程一般为3~6个月。
三碘甲状腺原氨酸钠(sodium triiodothyronine,甲碘安)成人开始10~20µg/日,以后渐增至80~100µg/日,2~3次服。儿童体重在7公斤以下者开始2.5µg/日,7公斤以上者5µg/日,以后每隔一周增加5µg/日,维持量15~20µg/日,分2~3次服。
甲状腺素钠(sodium thyroxine)本品0.1mg相当于甲状腺片60mg,口服0.1~0.2mg/日,静脉注射0.3~0.5mg/日。
丙硫氧嘧啶(propylthiouracil)开始剂量300~600mg/日,分3~4次;维持量25~100mg/日,分1~2次服。
甲硫氧嘧啶(methylthiouracil)剂量基本同上。
甲巯咪唑(thiamazole,他巴唑,tapazole)开始剂量20~60mg/日,分三次服,维持量5~10mg/日,服药最短不能少于1年。
卡比马唑(carbimazole)15~30mg/日,分3次服。服用4~6周后如症状改善,改用维持量,2.5~5mg/日,分次服。
碘化钾(potassium iodide)治疗单纯性甲状腺肿开始剂量宜小,10mg/日,20日为一疗程,连用2疗程,疗程间隔30~40日,约1~2月后,剂量可渐增大至20~25mg/日,总疗程约3~6个月。
复方碘溶液(卢戈液,Lugol's solution)每1000ml含碘50g、碘化钾100g,治疗单纯性甲状腺肿:0.1~0.5ml/次,1次/日,2周为一疗程,疗程间隔30~40日。用于甲亢术前准备:3~10滴/次,3次/日,用水稀释后服用,约服2周。用于甲状腺危象:首次服2~4ml,以后每4小时1~2ml。或静脉滴注,3~5ml加于10%葡萄糖液500ml中。
第三十六章 胰岛素及口服降血糖药
第一节 胰岛素
胰岛素(insulin)是一个分子量为56kD的酸性蛋白质,由两条多肽链组成(A、B链),其间通过两个二硫链以共价相联。药用胰岛素一般多由猪、牛胰腺提得。目前可通过重组DNA技术利用大肠杆菌合成胰岛素,还可将猪胰岛素β链第30位的丙氨酸用苏氨酸代替而获得人胰岛素。
【体内过程】口服无效,因易被消化酶破坏,因此所有胰岛素制剂都必须注射,皮下注射吸收快。代谢快,t1/2为9~10分钟,但作用可维持数小时。因其分布于组织后,与组织结合而在其中发挥作用。主要在肝、肾灭活,经谷胱甘肽转氨酶还原二硫键,再由蛋白水解酶水解成短肽或氨基酸,也可被肾胰岛素酶直接水解。严重肝肾功能不良者能影响其灭活。为延长胰岛素的作用时间,可制成中效及长效制剂。用碱性蛋白质与之结合,使等电点提高到7.3,接近体液pH值,再加入微量锌使之稳定,这类制剂经皮下及肌内注射后,在注射部位发生沉淀,再缓慢释放、吸收。所有中、长效制剂均为混悬剂,不可静注。见表36-1。
表36-1 胰岛素制剂及其作用时间
| 分类 | 药 物 | 注射途径 | 作用时间(小时) | 给药时间 | ||
| 开始 | 高峰 | 维持 | ||||
| 短效 | 正规胰岛素(Regular Iusulin) | 静脉 | 立即 | 0.5 | 2 | 急救 |
| 皮下 | 0.5~1 | 2~3 | 6~8 | 餐前0.5小时,3~4次/日。 | ||
| 中效 | 低精蛋白锌胰岛素(Isophane Insulin) | 皮下 | 2~4 | 8~12 | 18~24 | 早餐或晚餐前1小时,一日1~2次 |
| 珠蛋白锌胰岛素(Globin Zinc Insulin) | 皮下 | 2~4 | 6~10 | 12~18 | ||
| 长效 | 精蛋白锌胰岛素(Protamine Zinc Insulin) | 皮下 | 3~6 | 16~18 | 24~36 | 早餐或晚餐前1小时,一日1次 |
【药理作用】胰岛素对代谢过程具有广泛的影响。
1.糖代谢 胰岛素可增加葡萄糖的转运,加速葡萄糖的氧化和酵解,促进糖原的合成和贮存,抑制糖原分解和异生而降低血糖。
2.脂肪代谢 胰岛素能增加脂肪酸的转运,促进脂肪合成并抑制其分解,减少游离脂肪酸和酮体的生成。
3.蛋白质代谢 胰岛素可增加氨基酸的转运和蛋白质的合成(包括mRNA的转录及翻译),同时又抑制蛋白质的分解。
【作用机制】已知胰岛素受体为一糖蛋白,是由两个13kD的α-亚单位及两个90kD的β-亚单位组成的大分子蛋白复合物。α-亚单位在胞外,含胰岛素结合部位,β-亚单位为跨膜蛋白,其胞内部分含酪氨酸蛋白激酶,胰岛素需与靶细胞膜受体结合后,才能产生一系列的生物效应,对产生效应的机制有以下假说,一认为胰岛素可诱导第二信使的形成,它们模拟或具有胰岛素样的活性。二认为胰岛素与α-亚单位结合,移入胞内后可激活酪氨酸蛋白激酶,继而催化受体蛋白自身及胞内其他蛋白的酪氨酸残基磷酸化,因而启动了磷酸化的连锁反应(phosphorylation cascade)。三认为胰岛素可使葡萄糖载体蛋白(glucose transporter)和其他蛋白质从胞内重新分布到胞膜,从而加速葡萄糖的转运。
图36-1 胰岛素受体的基本结构
图注:α、β=亚单位 -S-S-=双硫键 =糖基化部位
胰岛素与α-亚单位相结合;β-亚单位胞内部分含酪氨酸蛋白激酶
【临床应用】胰岛素仍是治疗胰岛素依赖型糖尿病(insulin-dependent diabetis mellitus,IDDM)的唯一药物,对胰岛素缺乏的各型糖尿病均有效。主要用于下列情况:①重症糖尿病(IDDM,I型);②非胰岛素依赖型糖尿病(noninsulin dependent diabetis mellitus,NIDDM)经饮食控制或用口服降血糖药未能控制者;③糖尿病发生各种急性或严重并发症者,如酮症酸中毒及非酮症高血糖高渗性昏迷(要建立和维持电解质的平衡)。④合并重度感染、消耗性疾病、高热、妊娠、创伤以及手术的各型糖尿病。
【不良反应】
1.过敏反应 多数为使用牛胰岛素所致,它作为异体蛋白进入人体后可产生相应抗体如IgE并引起过敏反应。一般反应轻微而短暂,偶可引起过敏休克。可用猪胰岛素代替,因其与人胰岛素较为接近。
2.低血糖症 为胰岛素过量所致,正规胰岛素能迅速降低血糖,出现饥饿感、出汗、心跳加快、焦虑、震颤等症状,严重者引起昏迷、惊厥及休克,甚至脑损伤及死亡。长效胰岛素降血糖作用较慢,不出现上述症状,而以头痛和精神情绪、运动障碍为主要表现。
为防止低血糖症的严重后果,应教会病人熟知反应,以便及早发现和摄食,或饮用糖水等。严重者应立即静脉注射50%葡萄糖。必须在糖尿病患者中鉴别低血糖昏迷和酮症酸中毒性昏迷及非酮症性糖尿病昏迷。
3.胰岛素耐受性 产生急性耐受常由于并发感染、创伤、手术、情绪激动等应激状态所致。此时血中抗胰岛素物质增多,或因酮症酸中毒时,血中大量游离脂肪酸和酮体的存在妨碍了葡萄糖的摄取和利用。出现急性耐受时,需短时间内增加胰岛素剂量达数千单位。产生慢性耐受的原因较为复杂(系指每日需用200U以上的胰岛素并且无并发症者)。可能是体内产生了抗胰岛素受体抗体(AIRA),对此可用免疫抑制剂控制症状,能使患者对胰岛素的敏感性恢复正常;也可能是胰岛素受体数量的变化,如高胰岛素血症时,靶细胞膜上胰岛素受体数目减少;还可能是靶细胞膜上葡萄糖转运系统失常。此时换用其他动物胰岛素或改用高纯度胰岛素,并适当调整剂量常可有效。
第二节 口服降血糖药
常用的口服降血糖药:包括磺酰脲类及双胍类。
一、磺酰脲类
常用的有甲苯磺丁脲(tolbutamid,D860,甲糖宁)、氯磺丙脲(chlorpropamide),格列本脲(glyburide,glibenclamide,优降糖),格列吡嗪(glipizide,吡磺环已脲),格列齐特(gliclazipe,达美康)等,化学结构如下:
表36-2磺酰脲类药物的化学结构
![]()
【药理作用及作用机制】胰岛β细胞膜含有磺酰脲受体及与之相偶联的ATP敏感的钾通道[Ik(ATP)],以及电压依赖性的钙通道。当磺酰脲类药物与其受体相结合后,可阻滞Ik(ATP)而阻钾外流,致使细胞膜去极化,增强电压依赖性钙通道开放,胞外钙内流。胞内游离钙浓度增加后,触发胞吐作用及胰岛素的释放。长期服用且胰岛素已恢复至给药前水平的情况下,其降血糖作用仍然存在,这可能与抑制胰高血糖素的分泌,提高靶细胞对胰岛素的敏感性有关。也可能与增加靶细胞膜上胰岛素受体的数目和亲和力有关。
【体内过程】磺酰脲类药物在胃肠道吸收迅速而完全,与血浆蛋白结合率很高。其中多数药物在肝内氧化成羟基化合物,并迅速从尿中排出。磺酰脲类药物的药代动力学见表36-3。
甲苯磺丁脲作用最弱、维持时间最短,而氯磺丙脲t1/2最长,且排泄慢、每日只需给药一次。新型磺酰脲类作用较强,可维持24小时,每日只需给药1~2次。
表36-3 磺酰脲类药物的药代动力学参数
| 药 物 | 给药途径 | 效强 | 血浆蛋白结 合 | 作用持续时间(h) | t1/2 | 代谢途径 | 排泄(经肝、肾) |
| 甲苯磺丁脲 | 口服 | + | >90% | 4~6 | 3~5 | 氧化 | 95% |
| 氯磺丙脲 | 口服 | +++ | >90% | 60 | 24~48 | 不代谢 | 90% |
| 格列本脲 | 口服 | ++++ | >90% | 24 | 10~16 | 氧化 | 50% |
| 格列吡嗪 | 口服 | ++++ | >90% | 24 | 3~7 | 氧化 | 90% |
【临床应用】
1.糖尿病 用于胰岛功能尚存的非胰岛素依赖型糖尿病且单用饮食控制无效者。对胰岛素产生耐受的患者用后可刺激内源性胰岛素的分泌而减少胰岛素的用量。
2.氯磺丙脲能促进抗利尿素的分泌,可治疗尿崩症。
【不良反应】常见不良反应为胃肠不适、恶心、腹痛、腹泻。大剂量氯磺丙脲还可引起中枢神经系统症状,如精神错乱、嗜睡、眩晕、共济失调。也可引起粒细胞减少和胆汁郁积性黄疸及肝损害,一般在服药后1~2个月内发生。因此需定期检查肝功能和血象。较严重的不良反应为持久性的低血糖症,常因药物过量所致,尤以氯磺丙脲为甚。老人及肝、肾功能不良者较易发生,故老年糖尿病人不宜用氯磺丙脲。新型磺酰脲类较少引起低血糖。
【药物相互作用】由于磺酰脲类有较高的血浆蛋白结合率,因此在蛋白结合上能与其他药物(如保泰松、水杨酸钠、吲哚美辛、青霉素、双香豆素等)发生竞争,使游离药物浓度上升而引起低血糖反应。此外,氯丙嗪、糖皮质激素、噻嗪类利尿药、口服避孕药均可降低磺酰脲类药物的降血糖作用。
二、双胍类
国内应用的有甲福明(metformin,二甲双胍)、苯乙福明(phenformine,苯乙双胍)。
甲福明作用短,在体内不与蛋白结合,不被代谢,从尿中排出。其作用机制可能是降低食物吸收及糖原异生、促进组织摄取葡萄糖等。主要用于轻症糖尿病患者,尤适用于肥胖者,单用饮食控制无效者。不良反应为食欲下降、恶心、腹部不适、腹泻等,危及生命的不良反应为乳酸血症,尤以苯乙福明的发生率高。与苯乙福明相比,甲福明一般不引起乳酸血症,应用较广。
三、α-葡萄糖甙酶抑制药
α-葡萄糖甙酶抑制剂是一类新型口服降血糖药,其中阿卡波糖(acarbose)已用于临床,其降血糖的机制是:在小肠上皮刷状缘与碳水化合物竞争水解碳水化合物的酶,从而减慢水解及产生葡萄糖的速度并延缓葡萄糖的吸收。血糖峰值降低。主要副作用为胃肠道反应。服药期间应增加碳水化合物的比例,并限制单糖的摄入量,以提高药物的疗效。
胰岛素及口服降血糖药 制剂及用法
胰岛素(insulin,正规胰岛素,regular insulin)剂量和给药次数按病情而定,通常24小时内所排尿糖每2~4g者,给胰岛素1U,中型糖尿病人每日需给5~10U,重型者每日用量在40U以上。一般饭前半小时皮下注射,3~4次/日,必要时可作静脉注射或肌内注射。
低精蛋白锌胰岛素(isophane insulin)剂量视病情而定,早饭前(或加晚饭前)30~60分钟给药,仅作皮下注射。
珠蛋白锌胰岛素(globin zinc insulin)剂量视病情而定,早饭前(或加晚饭前)30分钟给药,1~2次/日,皮下注射。
精蛋白锌胰岛素(protamine zinc insulin)剂量视病情而定,早饭前30~60分钟给药,1次/日,皮下注射。
甲苯磺丁脲(tolbutamide,D860,甲糖宁)口服,第一天服1g/次,3次/日;第2天起0.5g/次,3次/日,饭前服、待血糖正常或尿糖少于每日5g时,改为维持量,0.5g/次,2次/日。
氯磺丙脲(chlorpropamide,P-607)口服,治糖尿病:0.1~0.3g/次,1次/日,待血糖降到正常时,剂量酌减至0.1~0.2g/日,早饭前一次服。治疗尿崩症:0.125~0.25g/日。
格列本脲(glibenclamide,HB-419)口服,开始每日早饭后服2.5mg,以后逐渐增量,但每日不得超过15mg,待增至每日10mg时,应分早、晚二次服,至出现疗效后,逐渐减量至2.5~5mg/日。
甲福明(metformin,Diabex,DMBG,二甲双胍,降糖片)0.25~0.5g/次,3次/日,饭后服。以后根据尿糖(或血糖)情况增减。
第三十七章 抗菌药物概论
抗菌药物对病原菌具有抑制或杀灭作用,是防治细菌感染性疾病的一类药物。细菌和其他微生物、寄生虫及癌细胞所致疾病的药物治疗统称为化学治疗学(chemotherapy,简称化疗)。化学治疗学的目的是研究、应用对病原体有选择毒性(即强大杀灭作用),而对宿主无害或少害的药物以防治病原体所引起的疾病。
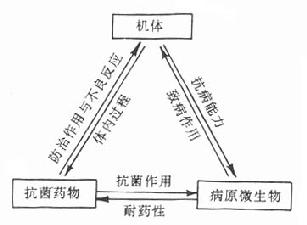
图37-1 机体、抗菌药物及病原微生物的相互作用关系
在应用化疗药物治疗感染性疾病过程中,应注意机体、病原体与药物三者的相互关系(图37-1)。感染性疾病的罹患与康复是微生物与机体相互斗争的过程。病原微生物在疾病的发生上无疑起着重要作用。但病原体不能决定疾病的全过程,人体的反应性、免疫状态和防御功能对疾病的发生、发展与转归也有重要作用。当机体防御功能占主导地位时,就能战胜致病微生物,使它不能致病,或发病后迅速康复。抗菌药物的抑菌或杀菌作用是制止疾病发展与促进康复的外来因素,为机体彻底消灭病原体和导致疾病痊愈创造有利条件。事物总是有两面性的,矛盾是不断转化的。在某种条件下微生物可产生耐药性,而使药物失去抗菌效果;在治疗中药物的治疗作用是主要的,但使用不当时,药物可产生不良反应,影响患者健康,甚至使治疗失败。
第一节 常用术语
抗菌谱每种抗菌药物都有一定的抗菌范围,称为抗菌谱。某些抗菌药物仅作用于单一菌种或局限于一属细菌,其抗菌谱窄,如异烟肼只对抗酸分支杆菌有效。另一些药物抗菌范围广泛称之为广谱抗菌药,如四环素和氯霉素,它们不仅对革兰阳性细菌和革兰阴性细菌有抗菌作用,且对衣原体、肺炎支原体、立克次体及某些原虫等也有抑制作用。近年新发展的青霉素类和头孢菌素类抗生素也有广谱抗菌作用。
抗菌活性抗菌活性是指药物抑制或杀灭微生物的能力。一般可用体外与体内(化学实验治疗)两种方法来测定。体外抗菌试验对临床用药具有重要意义。能够抑制培养基内细菌生长的最低浓度称之为最低抑菌浓度(MIC);能够杀灭培养基内细菌的最低浓度称之为最低杀菌浓度(MBC)。
抑菌药是指仅有抑制微生物生长繁殖而无杀灭作用的药物,如四环素等。
杀菌药这类药不仅能抑制微生物生长繁殖,而且能杀灭之,如青霉素类、氨基甙类等。
化疗指数理想的化疗药物一般必须具有对宿主体内病原微生物有高度选择性的毒性,而对宿主无毒性或毒性很低,最好还能促进机体防御功能并能与其他抗菌药物联合应用消灭病原体。化疗药物的价值一般以动物半数致死量(LD50)和治疗感染动物的半数有效量(ED50)之比,或5%致死量(LD5)与95%有效量(ED95)的比来衡量。这一比例关系称为化疗指数。化疗指数愈大,表明药物的毒性愈小,疗效愈大,临床应用的价值也可能愈高。但化疗指数高者并不是绝对安全,如几无毒性的青霉素仍有引起过敏休克的可能。
第二节 抗菌药作用机制
抗菌药物的作用机制,现多以干扰细菌的生化代谢过程来解释。兹将几种主要方式(图37-2)简介如下:
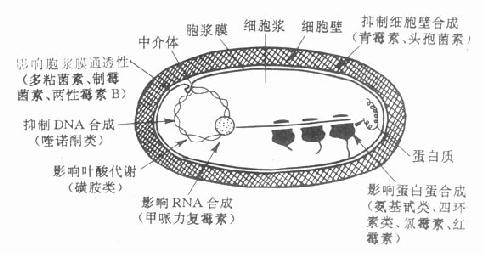
图37-2 细菌结构与抗菌药作用部位示意图
一、抗叶酸代谢
磺胺类与甲氧苄啶(TMP)可分别抑制二氢叶酸合成酶与二氢叶酸还原酶,妨碍叶酸代谢,最终影响核酸合成,从而抑制细菌的生长和繁殖(见42章)。
二、抑制细菌细胞壁合成
细菌细胞膜外是一层坚韧的细胞壁,能抗御菌体内强大的渗透压,具有保护和维持细菌正常形态的功能。细菌细胞壁主要结构成分是胞壁粘肽,由N-乙酰葡萄糖胺(GNAc)和与五肽相连的N-乙酰胞壁酸(MNAc)重复交替联结而成。胞壁粘肽的生物合成可分为胞浆内、胞浆膜与胞浆外三个阶段。胞浆内粘肽前体的形成可被磷霉素与环丝氨酸所阻碍。磷霉素抑制有关酶系阻碍N-乙酰胞壁酸的形成;环丝氨酸通过抑制D-丙氨酸的消旋酶和合成酶阻碍了N-乙酰胞壁酸五肽的形成。胞浆膜阶段的粘肽合成可被万古霉素和杆菌肽所破坏,它们能分别抑制MNAc-五肽与脂载体结合并形成直链十肽二糖聚合物和聚合物转运至膜外受体的过程及脱磷酸反应。青霉素与头孢菌素类抗生素则能阻碍直链十肽二糖聚合物在胞浆外的交叉联接过程。青霉素等的作用靶位是胞浆膜上的青霉素结合蛋白(PBPs),表现为抑制转肽酶的转肽作用,从而阻碍了交叉联接。能阻碍细胞壁合成的抗生素可导致细菌细胞壁缺损。由于菌体内的高渗透压,在等渗环境中水分不断渗入。致使细菌膨胀、变形,在自溶酶影响下,细菌破裂溶解而死亡。
三、影响胞浆膜的通透性
细菌胞浆膜主要是由类脂质和蛋白质分子构成的一种半透膜,具有渗透屏障和运输物质的功能。多粘菌素类抗生素具有表面活性物质,能选择性地与细菌胞浆膜中的磷酯结合;而制霉菌素和二性霉素等多烯类抗生素则仅能与真菌胞浆膜中固醇类物质结合。它们均能使胞浆膜通透性增加,导致菌体内的蛋白质、核苷酸、氨基酸、糖和盐类等外漏,从而使细菌死亡。
四、抑制蛋白质合成
细菌为原核细胞,其核蛋白体为70S,由30S和50S亚基组成,哺乳动物是真核细胞,其核蛋白体为80S,由40S与60S亚基构成,因而它们的生理、生化与功能不同,抗菌药物对细菌的核蛋白体有高度的选择性毒性,而不影响哺乳动物的核蛋白体和蛋白质合成。多种抗生素能抑制细菌的蛋白质合成,但它们的作用点有所不同。①能与细菌核蛋白体50S亚基结合,使蛋白质合成呈可逆性抑制的有氯霉素、林可霉素和大环内酯类抗生素(红霉素等)。②能与核蛋白体30S亚基结合而抑菌的抗生素如四环素能阻止氨基酰tRNA向30S亚基的A位结合,从而抑制蛋白质合成。③能与30S亚基结合的杀菌药有氨基甙类抗生素(链霉素等)。它们的作用是多环节的。影响蛋白质合成的全过程,因而具有杀菌作用。
五、抑制核酸代谢
喹诺酮类药物能抑制DNA的合成,利福平能抑制以DNA为模板的RNA多聚酶。
第三节 细菌的耐药性
细菌的耐药性又称抗药性,一般是指细菌与药物多次接触后,对药物的敏感性下降甚至消失,致使药物对耐药菌的疗效降低或无效。
一、耐药性产生机制
1.产生灭活酶灭活酶有两种,一是水解酶,如β-内酰胺酶可水解青霉素或头孢菌素。该酶可由染色体或质粒介导,某些酶的产生为体质性(组构酶);某些则可经诱导产生(诱导酶)。二是钝化酶又称合成酶,可催化某些基团结合到抗生素的OH基或NH2基上,使抗生素失活。多数对氨基甙类抗生素耐药的革兰阴性杆菌能产生质粒介导的钝化酶,如乙酰转移酶作用于NH2基上,磷酸转移酶及核苷转移酶作用于OH基上。上述酶位于胞浆膜外间隙,氨基甙类被上述酶钝化后不易与细菌体内的核蛋白体结合,从而引起耐药性。
2.胞浆膜通透性细菌可通过各种途径使抗菌药物不易进入菌体,如革兰阴性杆菌的细胞外膜对青霉素G等有天然屏障作用;绿脓杆菌和其他革兰阴性杆菌细胞壁水孔或外膜非特异性通道功能改变引起细菌对一些广谱青霉素类、头孢菌素类包括某些第三代头孢菌素的耐药;细菌对四环素耐药主要由于所带的耐药质粒可诱导产生三种新的蛋白,阻塞了细胞壁水孔,使药物无法进入。革兰阴性杆菌对氨基甙类耐药除前述产生钝化酶外,也可由于细胞壁水孔改变,使药物不易渗透至细菌体内。
3.细菌体内靶位结构的改变链霉素耐药株的细菌核蛋白体30S亚基上链霉素作用靶位P10蛋白质发生改变;利福平的耐药性是细菌RNA多聚酶的β'亚基发生改变,使其与药物的结合力降低而耐药。由质粒介导的对林可霉素和红霉素的耐药性,系细菌核蛋白体23S亚基的腺嘌呤甲基化,使药物不能与细菌结合所致。某些肺炎球菌、淋球菌对青霉素G耐药,以及金葡菌对甲氧苯青霉素耐药,乃因经突变引起青霉素结合蛋白(PBPs)改变,使药物不易与之结合。这种耐药株往往对其他青霉素(如苯唑或邻氯青霉素)和头孢菌素类也都耐药。
4.其他细菌对磺胺类的耐药,可由对药物具拮抗作用的底物PABA的产生增多所致;也可能通过改变对代谢物的需要等途径。
二、避孕细菌耐药性的措施
为了克服细菌对药物产生耐药性,临床医生要注意抗菌药物的合理应用,给予足够的剂量与疗程,必要的联合用药和有计划的轮换供药。此外,医药学专家还应努力开发新的抗菌药物,改造化学结构,使其具有耐酶特性或易于透入菌体。
第四节 抗菌药的合理使用
由于抗菌药的使用,过去许多致死性的疾病已得到控制。但随着抗菌药物的广泛使用,特别是滥用,也给治疗带来许多新问题,如毒性反应、过敏反应、二重感染、细菌产生耐药性等。因此,合理使用抗菌药物日益受到重视。
一、抗菌药临床应用的基本原则
(一)严格按照适应证选药
每一种抗菌药物各有不同抗菌谱与适应证。临床诊断、细菌学诊断和体外药敏试验可作为选药的重要参考。表37-1供选药时参考。此外,还应根据病人全身情况,肝、肾功能,感染部位,药物代谢动力学特点,细菌产生耐药性的可能性、不良反应和价格等方面因素综合考虑。
表37-1 药敏试验中的抗菌药物选择
| 肠杆菌科 | 假单胞菌属 | 金葡菌 | 肠球菌属 | 流感杆菌 | |
| 第一线 | 氨苄西林 | 哌拉西林 | 头孢噻吩 | 青霉素 | 氨苄西林 |
| 氨苄西林-舒巴坦 | 羧苄西林 | 氨苄西林-舒巴坦 | (或氨苄西林) | 氨苄西林-舒巴坦 | |
| 头孢噻吩 | 庆大霉素 | 红霉素 | 头孢呋新 | ||
| 庆大霉素 | 阿米卡星 | 苯唑西林 | 氯霉素 | ||
| 阿米卡星 | 青霉素 | 复方SMZ-TMP | |||
| 万古霉素 | |||||
| 第二线 | 头孢呋新 | 诺氟沙星 | 庆大霉素 | 万古霉素 | |
| 氯霉素 | 复方SMZ-TMP | 阿米卡星 | |||
| 哌拉西林 | 头孢他啶 | 诺氟沙星 | |||
| 复方SMZ-TMP | (或头孢哌酮) | 利福平 | |||
| 第三代头孢菌素 | 复方SMZ-TMP | ||||
| 诺氟沙星 | |||||
| 尿液 | 呋喃妥因 | 诺氟沙星 | 复方SMZ-TMP | 诺氟沙星 | |
| 诺氟沙星 | 复方SMZ-TMP | 呋喃妥因 | 红霉素 | ||
| 复方SMZ-TMP | 诺氟沙星 |
(二)病毒性感染和发热原因不明者
感冒、上呼吸道感染等病毒性疾病,发病原因不明者(除病情严重并怀疑为细菌感染外)不宜用抗菌药,否则可使临床症状不典型和病原菌不易被检出,以致延误正确诊断与治疗。
(三)抗菌药剂量
剂量要适当,疗程应足够。剂量过小,不但无治疗作用,反易使细菌产生耐药性;剂量过大,不仅造成浪费,还会带来严重的毒副作用。疗程过短易使疾病复发或转为慢性。
(四)皮肤粘膜等局部感染
应尽量避免局部应用抗菌药,因其易发生过敏反应和耐药菌的产生。
(五)预防应用及联合应用
对此均应严格掌握适应证,抗菌药物的预防应用仅限于少数情况,如经临床实践证明确有效果者;联合用药,也必须谨慎掌握指征、权衡利弊。
二、抗菌药的联合应用
(一)抗菌药联合应用的意义
联合用药的主要优点是:①发挥药物的协同抗菌作用以提高疗效;②延迟或减少耐药菌的出现;③对混合感染或不能作细菌学诊断的病例,联合用药可扩大抗菌范围;④联合用药可减少个别药剂量,从而减少毒副反应。
滥用抗菌药物的联合应用,可能产生不利后果:如增加不良反应发生率;容易出现二重感染;耐药菌株更加增多;浪费药物;给人一种虚伪的安全感染,延误正确治疗。
(二)联合用药的指征
联合用药的指征有:①病原菌未明的严重感染;②单一抗菌药物不能控制的严重混合感染,如肠穿孔后腹膜炎的致病菌常有多种需氧菌和厌氧菌等;③单一抗菌药物不能有效控制的感染性心内膜炎或败血症;④长期用药细菌有可能产生耐药者,如结核、慢性尿路感染、慢性骨髓炎等;⑤用以减少药物毒性反应,如两性霉素B和氟胞嘧啶合用治疗深部真菌,前者用量可减少,从而减少毒性反应;⑥临床感染一般用二药联用即可,常不必要三药联用或四药联用。
(三)联合用药可能产生结果
两种抗菌药联合应用在体外或动物实验中可获得无关、相加、协同(增强)和拮抗等四种效果。抗菌药物依其作用性质可分为四大类:一类为繁殖期杀菌,如青霉素类、头孢菌素类等;二类为静止期杀菌,如氨基甙类、多粘菌素等,它们对静止期、繁殖期细菌均有杀灭作用;三类为速效抑菌,如四环素类、氯霉素类与大环内酯类抗生素等、四类为慢效抑菌剂,如磺胺类等。第一类和第二类合用常可获得协同(增强)作用,例如青霉素与链霉素或庆大霉素合用治疗肠球菌心内膜炎;青霉素破坏细菌细胞壁的完整性,有利于氨基甙类抗生素进入细胞内发挥作用。第一类与第三类合用可能出现拮抗作用。例如青霉素类与氯霉素或四环素类合用。由于后二药使蛋白质合成迅速被抑制,细菌处于静止状态,致使繁殖期杀菌的青霉素干扰细胞壁合成的作用不能充分发挥,使其抗菌活性减弱。第二类和第三类合用可获得增强或相加作用。第四类慢效抑菌药与第一类可以合用,例如,治疗流行性脑膜炎时,青霉素可以和磺胺嘧啶合用而提高疗效。
应该指出上述资料多来自体外与动物试验在特定条件下的观察,与临床实际不尽相同,仅供参考。联合用药产生的作用也可因不同菌种和菌株而异,药物剂量和给药顺序也会影响效果。
三、肝肾功能损害时抗菌药的应用
(一)肾功能损害
肾功能减退时,应用主要经肾排泄的药物宜减量或延长给药时间。对肾有毒的药物,如两性霉素B、万古霉素及氨基甙类等,宜避免使用。对肾功能无损害或损害不大的药物在一般情况下,可按常规给药,但要求肝功能必须正常。
肾功能轻、中和重度减退的给药量分别为正常剂量的2/3~1/2,1/2~1/5和1/5~1/10。
(二)肝功能障碍的影响
肝功能减退者,应避免使用或慎用氯霉素、林可霉素、红霉素、利福平、四环素类等。早产和新生儿的肝脏对氯霉素的解毒功能较低,氯霉素列为禁用。
第三十八章 β-内酰胺类抗生素
β-内酰胺类抗生素(β-lactams)系指化学结构中具有β-内酰胺环的一大类抗生素,包括临床最常用的青霉素与头孢菌素,以及新发展的头霉素类、硫霉素类、单环β-内酰胺类等其他非典型β-内酰胺类抗生素。此类抗生素具有杀菌活性强、毒性低、适应症广及临床疗效好的优点。本类药化学结构,特别是侧链的改变形成了许多不同抗菌谱和抗菌作用以及各种临床药理学特性的抗生素。
一、抗菌机制、影响抗菌作用因素及细菌耐药性
(一)抗菌作用机制
各种β-内酰胺类抗生素的作用机制均相似,都能抑制胞壁粘肽合成酶,即青霉素结合蛋白(penicillin binding proteins,PBPs),从而阻碍细胞壁粘肽合成,使细菌胞壁缺损,菌体膨胀裂解(胞壁粘肽合成过程见三十七章)。除此之外,对细菌的致死效应还应包括触发细菌的自溶酶活性,缺乏自溶酶的突变株则表现出耐药性。哺乳动物无细胞壁,不受β-内酰胺类药物的影响,因而本类药具有对细菌的选择性杀菌作用,对宿主毒性小。近十多年来已证实细菌胞浆膜上特殊蛋白PBPs是β-内酰胺类药的作用靶位,PBPs的功能及与抗生素结合情况归纳于图38-1。各种细菌细胞膜上的PBPs数目、分子量、对β-内酰胺类抗生素的敏感性不同,但分类学上相近的细菌,其PBPs类型及生理功能则相似。例如大肠杆菌有7种PBPs,PBP1A,PBP1B与细菌延长有关,青霉素、氨苄西林、头孢噻吩等与PBP1A、PBP1B有高度亲和力,可使细菌生长繁殖和延伸受抑制,并溶解死亡,PBP2与细管形状有关,美西林、棒酸与硫霉素(亚胺培南)能选择性地与其结合,使细菌形成大圆形细胞,对渗透压稳定,可继续生几代后才溶解死亡。PBP3功能与PBP1A相同,但量少,与中隔形成,细菌分裂有关,多数青霉素类或头孢菌素类抗生素主要与PBP1和(或)PBP3结合,形成丝状体和球形体,使细菌发生变形萎缩,逐渐溶解死亡。PBP1,2,3是细菌存活、生长繁殖所必需,PBP4,5,6;与羧肽酶活性有关,对细菌生存繁殖无重要性,抗生素与之结合后,对细菌无影响。

图38-1 大肠杆菌PBPs的酶功能及与其结合的抗生素的作用
(二)影响β-内酰胺类抗菌作用素
革兰阳性菌与阴性菌的结构差异甚大,β-内酰胺类各药与母核相联接的侧链不同可影响其亲脂性或亲水性。有效药物必需能进入菌体作用于细胞膜上的靶位PBPs。影响抗菌作用的主要因素:①药物透过革兰阳性菌细胞壁或阴性菌脂蛋白外膜(即第一道穿透屏障)的难易;②对β-内酰胺酶(第二道酶水解屏障)的稳定性;③对抗菌作用靶位PBPs的亲和性。根据这些因素,目前临床应用的β-内酰胺类对革兰阳性与阴性菌的作用大致有6种类型(见图38-2)。
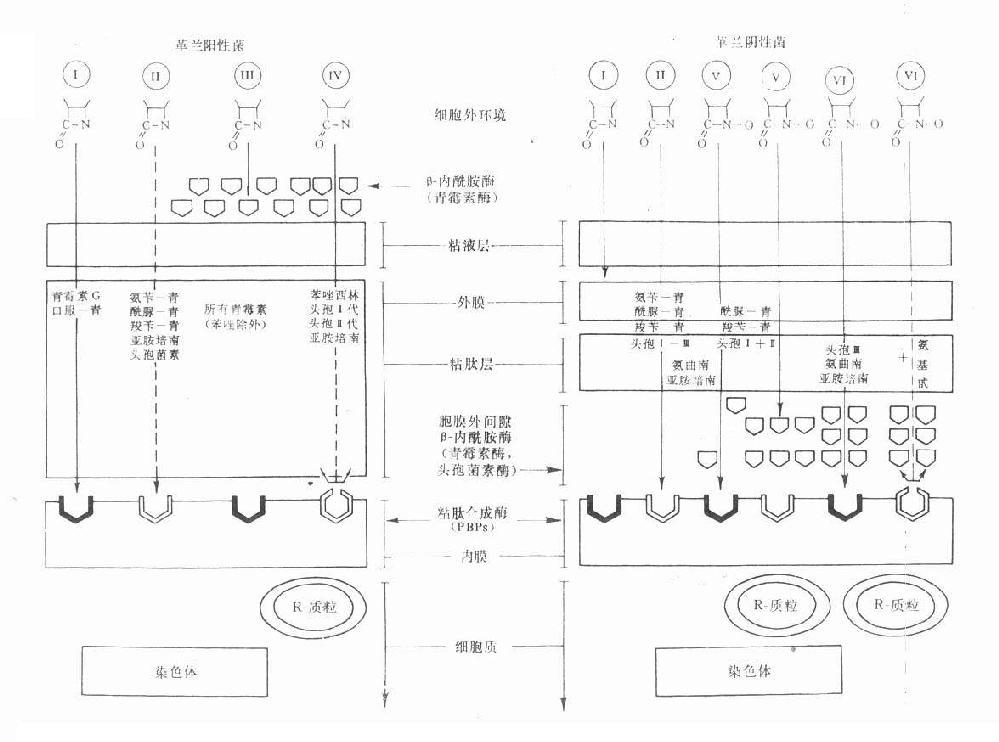
图38-2 革兰阳性与阴性菌的结构及β内酰胺类药的穿透情况及其对β-内酰胺酶与胞壁合成酶(PBPs)的关系示意图
Ⅰ类为青霉素及口服青霉素V易透过革兰阳性菌胞壁粘肽层,但它们不能透过革兰阴性菌糖蛋白磷脂外膜,因而属窄谱的仅对革兰阳性菌有效。Ⅱ类包括有氨苄西林、羧苄西林、酰脲类青霉素、亚胺培南及若干头孢菌素,能适度透过革兰阳性菌的胞壁粘肽层,对革兰阴性菌的外膜透过性则很好,因而是广谱抗菌药物。Ⅲ类为青霉素等容易被革兰阳性菌的胞外β-内酰胺酶即青霉素酶破坏灭活的青霉素类,对产酶菌往往表现明显的耐药性。Ⅳ类为异噁唑类青霉素、头孢菌素一、二代及亚胺培南等对青霉素酶稳定,对革兰阳性的产酶菌有效,但对染色体突变而改变的PBPs结构,可使药物与PBPs的亲和力下降或消失,因而无效。Ⅴ类包括酰脲类青霉素(阿洛西林与美洛西林等)、羧苄青霉素及头孢菌素一、二代,当胞膜外间隙的β-内酰胺酶少量存在时有抗菌效果,大量酶存在时,则被破坏而无效。Ⅵ类包括第三代头孢菌素、氨曲南、亚胺培南等对β-内酰胺酶十分稳定,即使大量β-内酰胺酶存在时仍然有效,但对因染色体突变而改变了的PBPs则无效,加用氨基甙类抗生素也仍然无效。
(三)细菌耐药机制
细菌对β-内酰胺类抗生素耐药机制可概括为:①细菌产生β-内酰胺酶(青霉素酶、头孢菌素酶等)使易感抗生素水解而灭活;②对革兰阴性菌产生的β-内酰胺酶稳定的广谱青霉素和第二、三代头孢菌素,其耐药发生机制不是由于抗生素被β-内酰胺酶水解,而是由于抗生素与大量的β-内酰胺酶迅速、牢固结合,使其停留于胞膜外间隙中,因而不能进入靶位(PBPs)发生抗菌作用。此种β-内酰胺酶的非水解机制又称为“牵制机制”(trappingmechanism);③PBPs靶蛋白与抗生素亲和力降低、PBPs增多或产生新的PBPs均可使抗生素失去抗菌作用。例如MRSA(methicillin resistant Staphylococcusaureus)具有多重耐药性,其产生机制是PBPs改变的结果,高度耐药性系由于原有的PBP2与PBP3之间产生一种新的PBP2'(即PBP2a),低、中度耐药系由于PBPs的产量增多或与甲氧西林等的亲和力下降所致;④细菌的细胞壁或外膜的通透性改变,使抗生素不能或很少进入细菌体内到达作用靶位。革兰阴性菌的外膜是限制β-内酰胺类抗生素透入菌体的第一道屏障。近年研究已证实抗生素透入外膜有非特异性通道与特异性通道两种。大肠杆菌K-12外膜有亲水性的非特异性孔道蛋白(porin)为三聚体结构,有二个孔道蛋白,即OmpF与OmpC,其合成由OmpB3基因调控。OmpF的直径为1nm,许多重要的β-内酰胺类抗生素大多经过此通道扩散入菌体内。鼠伤寒杆菌OmpF与OmpC缺陷突变株对头孢噻啶的通透性要比野生株小10倍,因而耐药。仅含微量OmpF与OmpC的大肠杆菌突变株,对头孢唑啉、头孢噻吩的透入也较野生株成倍降低,其MIC明显增高,也出现耐药。绿脓杆菌对β-内酰胺类抗生素耐药性的产生已证明是由于外膜非特异性孔道蛋白OprF缺陷而引起的。革兰阴性外膜的特异性通道,在绿脓杆菌耐亚胺培南的突变株已证明系由于外膜缺失一种分子量为45~46kD蛋白OprD。如将此OprD重组于缺陷OprD的突变株外膜蛋白脂质体中,又可使亚胺培南透过性增加5倍以上,其MIC也相应地降低,于是细菌的耐药性消除。⑤由于细菌缺少自溶酶而出现细菌对抗生素的耐药性,即抗生素具有正常的抑菌作用,但杀菌作用差。
二、青霉素类
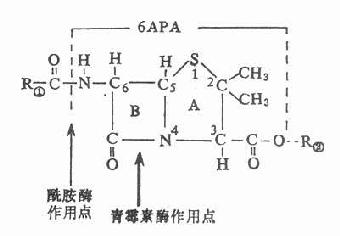
青霉素的基本结构
青霉素G是最早应用于临床的抗生素,由于它具有杀菌力强、毒性低、价格低廉、使用方便等优点,迄今仍是处理敏感菌所致各种感染的首选药物。但是青霉素有不耐酸、不耐青霉素酶、抗菌谱窄和容易引起过敏反应等缺点,在临床应用受到一定限制。1959年以来人们利用青霉素的母核6-氨基青霉烷酸(6-APA),进行化学改造,接上不同侧链,合成了几百种“半合成青霉素”,有许多已用于临床,常用青霉素的化学结构和药理特性。见表38-1,38-2。
表38-1 常用青霉素类抗生素的化学结构
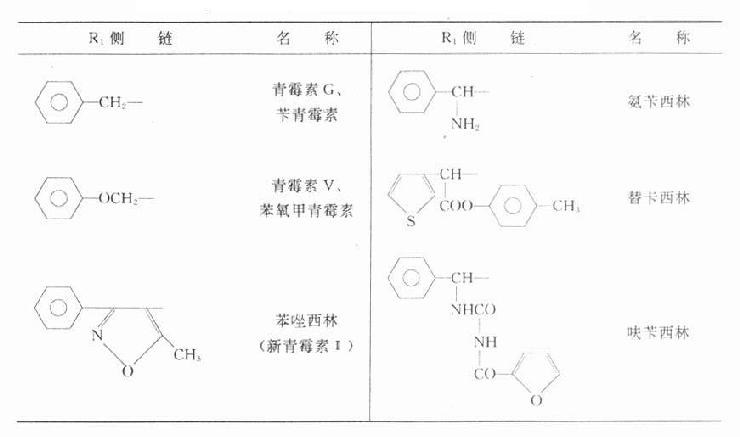
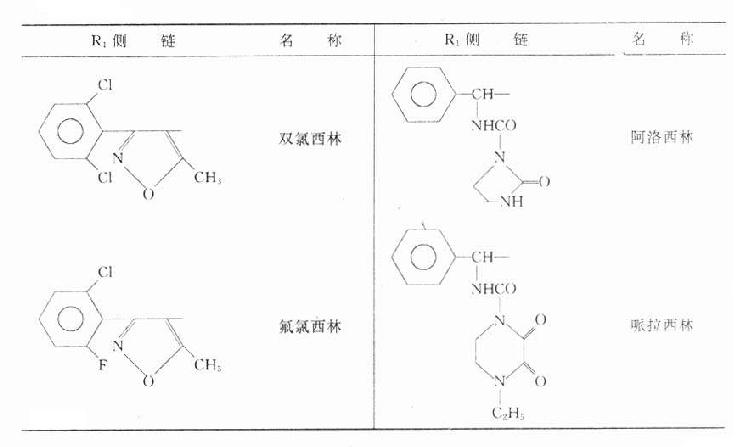
表38-2 青霉素类各种重要药理特性
| 耐酸 | 耐酶 | 广谱 | 抗绿脓杆菌 | 蛋白质结合率(%) | 尿中排出量(%) | t1/2(小时) | 常用剂量(g/日) | ||
| 正常 | 无尿 | ||||||||
| 青霉素G | - | - | - | - | 46~67 | 60~80 | 0.5 | 6~12 | 变化的 |
| 青霉素V | + | - | - | - | 80 | 20~40 | 1~2 | 6~10 | 1~2 |
| 甲氧西林 | - | + | - | - | 40 | 50~70 | 0.5 | 4~6 | 8~12 |
| 苯唑西林 | + | + | - | - | 90 | 40~55 | 0.4~0.7 | 1~2 | 8~12 |
| 氯唑西林 | + | + | - | - | 95 | 55~62 | 0.6 | 0.8~3 | 1~2 |
| 双氯西林 | + | + | - | - | 96 | 60 | 0.8 | 1~2 | 1~2 |
| 氨苄西林 | + | - | + | - | 20 | 60~80 | 1~1.3 | 8~12 | 1~4 |
| 阿莫西林 | + | - | + | - | 20 | 45~68 | 1~1.3 | 5~7 | 1~2 |
| 羧苄西林 | - | - | + | + | 50 | 76~90 | 1.0 | 12~16 | 20~30 |
| 呋苄西林 | - | - | + | + | 90 | 25 | 1.2 | 4~8 | |
| 替卡西林 | - | + | + | 45 | 92 | 1.3 | 15~20 | ||
| 阿洛西林 | - | - | + | + | 16~42 | 8~22 | 1.3 | 变化<24 | |
| 哌拉西林 | - | - | + | + | 16 | 50~70 | 1.0 | 3.3 | 变化<24 |
(一)青霉素
青霉素(penicillin G)又名苄青霉素(benzyl penicillin),是天然青霉素,侧链为苄基。常用其钠盐或钾盐,其晶粉在室温中稳定,易溶于水,水溶液在室温中不稳定,20℃放置24小时,抗菌活性迅速下降,且可生成有抗原性的降解产物,故青霉素应在临用前配成水溶液。
【抗菌作用】青霉素主要作用于革兰阳性菌、革兰阴性球菌、嗜血杆菌属以及各种致病螺旋体等。
青霉素对溶血性链球菌、草绿色链球菌、肺炎球菌等作用强,肠球菌敏感性较差。不产生青霉素酶的金葡菌及多数表葡菌对青霉素敏感,但产生青霉素酶的金葡菌对之高度耐药。革兰阳性杆菌,白喉杆菌、炭疽杆菌及革兰阳性厌氧杆菌如产气荚膜杆菌、破伤风杆菌、难辨梭菌、丙酸杆菌、真杆菌、乳酸杆菌等皆对青霉素敏感。革兰阴性菌中脑膜炎球菌对青霉素高度敏感,耐药者罕见。对青霉素敏感的淋球菌日益少见。百日咳杆菌对青霉素敏感。致病螺旋体,如梅毒螺旋体、钩端螺旋体对之高度敏感。
【体内过程】青霉素遇酸易被分解,口服吸收差,肌注100万单位后吸收快且甚完全,0.5小时达血药浓度峰值,约为20U/ml,消除半衰期为1/2小时。6小时内静滴500万单位青霉素钠,2小时后能获得20~30U/ml的血药浓度。青霉素的血清蛋白结合率为46%~58%。青霉素主要分布于细胞外液,并能广泛分布于各种关节腔、浆膜腔、间质液、淋巴液、胎盘、肝、肾、肺、横纹肌、中耳液等。青霉素的脂溶性低,进入细胞量减少;房水与脑脊液含量也较低,但炎症时青霉素透入脑脊液和眼的量可略提高,能达有效浓度。青霉素几乎全部以原形迅速经尿排泄,约10%经肾小球过滤。90%经肾小管分泌。无尿患者青霉素t1/2可延长达10小时。丙磺舒可与青霉素竞争肾小管分泌,两药合用时能提高青霉素血药浓度,延长其半衰期。
为了延长青霉素的作用时间,还可采用难溶制剂普鲁卡因青霉素(procacaine penicillin)和苄星青霉素(benzathine penicillin;长效西林,bicillin)。它们的水悬剂或油制剂肌内注射后,在注射部位缓慢溶解吸收。普鲁卡因青霉素一次注射40万单位,可维持24小时,苄星青霉素溶解度极小,一次注射120万单位,可维持15天,这两种制剂的血药浓度很低,只用于轻症病人或用于预防感染。
【临床应用】青霉素为治疗A组和B组溶血性链球菌感染、敏感葡萄球菌感染、气性坏疽、梅毒、鼠咬热等的首选药。肺炎球菌感染和脑膜炎时也可采用,当病原菌比较耐药时,可改用万古霉素或利福平。青霉素也是治疗草绿色链球菌心内膜炎的首选药。还可作为放线菌病、钩端螺旋体病、梅毒、回归热等及预防感染性心内膜炎发生的首选药。破伤风、白喉病人采用青霉素时应与抗毒素合用。
【不良反应】青霉素的毒性很低,除其钾盐大量静注易引起高血钾症、肌内注射疼痛外,最常见的为过敏反应,有过敏休克、药疹、血清病型反应、溶血性贫血及粒细胞减少等。毒霉素制剂中的青霉噻唑蛋白、青霉烯酸等降解物、青霉素或6-APA高分子聚合物均可成为致敏原。为防止各种过敏反应,应详细询问病史,包括用药史,药物过敏史,家属过敏史,并进行青霉素皮肤过敏试验。应用青霉素及皮试时应作好急救准备,如肾上腺素、氢化可的松等药物和注射器材,以便一旦发生过敏休克,能及时治疗。
在青霉素治疗梅毒或钩端螺旋体病时可有症状加剧现象,称为赫氏反应(Herxheimerreaction),或治疗矛盾,此反应一般发生于青霉素开始治疗后6~8小时,于12~24小时消失,表现为全身不适、寒战、发热、咽痛、胁痛、心跳加快等;同时可有病变加重现象,甚至危及生命。此反应可能为螺旋体抗原与相应抗体形成免疫复合物的结果,或与螺旋体释放非内毒素致热原有关。
【用药注意】肌内注射局部可发生周围神经炎,鞘内注射和全身大剂量应用可引起青霉素脑痛。严重感染宜静脉滴注给药,大剂量静注应监测血清离子浓度,以防发生高血钠、高血钾症。
(二)半合成青霉素
1.耐酸青霉素苯氧青霉素包括青霉素V和苯氧乙基青霉素。抗菌谱与青霉素相同,抗菌活性不及青霉素,耐酸、口服吸收好,但不耐酶,不宜用于严重感染。
2.耐酶青霉素化学结构特点是通过酰基侧链(R1)的空间位障作用保护了β-内酰胺环,使其不易被酶水解,主要用于耐青霉素的金葡菌感染。
异恶唑类青霉素 侧链为苯基异恶唑,耐酸、耐酶、可口服。常用的有:苯唑西林(oxacillin,新青霉素Ⅱ),氯唑西林(cloxacillin),双氯西林(dicloxacillin)与氟氯西林(flucloxacillin)。
【抗菌作用】本类药的抗菌谱及对耐药性金葡菌的作用均基本相似,对甲型链球菌和肺炎球菌效果最好,但不及青霉素,对耐药金葡菌的效力以双氯西林最强,随后依次为氟氯西林、氯唑西林与苯唑西林,对革兰阴性的肠道杆菌或肠球菌无明显作用。
【体内过程】胃肠道吸收较好,食物残渣会影响其吸收,因此,应在饭前一小时,空腹一次服药,大约1~1.5小时血药浓度达峰值,有效浓度可维持2~3小时。各药的吸收以苯唑西林最差,氯唑西林次之,双氯西林最好。血浆蛋白结合率均很高(95%以上)。主要以原型从尿排泄,速度较青霉素慢。
【不良反应】在胃肠道反应,个别有皮疹或荨麻疹。
【临床应用】用于耐药金葡菌的各种感染,或需长期用药的慢性感染等。对严重金葡菌感染,宜注射给药。
3.广谱青霉素对革兰阳性及阴性菌都有杀菌作用,还耐酸可口服,但不耐酶。
⑴氨苄西林(ampicillin) 对青霉素敏感的金葡菌等的效力不及青霉素,但对肠球菌作用优于青霉素。对革兰阴性菌有较强的作用,与氯霉素,四环素等相似或略强,但不如庆大霉素与多粘菌素,对绿脓杆菌无效。
【体内过程】口服后2小时达血药浓度峰值,经肾排泄,丙磺舒可延缓其排泄。体液中可达有效抗菌浓度,脑膜炎时脑脊液浓度较高。
【临床应用】主要用于伤寒、副伤寒、革兰阴性杆菌败血症、肺部、尿路及胆道感染等,严重者应与氨基甙类抗生素合用。
【不良反应】有轻微胃肠反应。
⑵阿莫西林(amoxycillin) 为对位羟基氨苄西林,抗菌谱与抗菌活性与氨苄西林相似,但对肺炎双球菌与变形杆菌的杀菌作用比氨苄西林强。经胃肠道吸收良好,血中浓度约为口服同量氨苄西林的2.5倍。阿莫西林用于治疗下呼吸道感染(尤其是肺炎球菌所致)效果超过氨苄西林。
⑶匹氨西林(pivampicillin) 为氨苄西林的双酯,口服吸收比氨苄西林好,能迅速水解为氨苄西林而发挥抗菌作用。正常人口服250mg,其血、尿浓度较相当剂量的氨苄西林分别高3与2倍。
4.抗绿脓杆菌广谱青霉素
⑴羧苄西林(carbenicillin) 其抗菌谱与氨苄西林相似。特点是对绿脓杆菌及变形杆菌作用较强。口服吸收差,需注射给药,肾功能损害时作用延长,主要用于绿脓杆菌及大肠杆菌所引起的各种感染。单用时细菌易产生耐药性,常与庆大霉素合用,但不能混合静脉注射。毒性低,偶也引起粒细胞缺乏及出血。
⑵磺苄西林(sulbenicillin) 抗菌谱和羧苄西林相似,抗菌活性较强。口服无效,胆汁中药物浓度为血药浓度的3倍,尿中浓度尤高,主要用于治疗泌尿生殖道及呼吸道感染。副作用为胃肠道反应,偶有皮疹、发热等。
⑶替卡西林(ticarcillin) 抗菌谱与羧苄西林相似,抗绿脓杆菌活性较其强2~4倍。对革兰阳性球菌活性不及青霉素,口服不吸收,肌内注射后0.5~1.0小时达血药浓度峰值。分布广泛,胆汁中药物浓度高,大部分经肾排泄,主要用于绿脓杆菌所致各种感染。
⑷呋苄西林(furbenicillin) 抗绿脓杆菌较羧苄西林强6~10倍,对金葡菌、链球菌、痢疾杆菌等也有强大抗菌作用。副作用同羧苄西林。
⑸阿洛西林(azlocillin) 抗菌谱和羧苄西林相似,抗菌活性与哌拉西林相近,强于羧苄西林。对多数肠杆菌科细菌和肠球菌以及绿脓杆菌均有较强作用。对耐羧苄西林和庆大霉素的绿脓杆菌也有较好作用。主要用于治疗绿脓杆菌、大肠杆菌及其他肠杆菌科细菌所致的感染。
⑹哌拉西林(piperacillin) 抗菌谱广与羧苄西林相似,而抗菌作用较强,对各种厌氧菌均有一定作用。与氨基甙类合用对绿脓杆菌和某些脆弱拟杆菌及肠杆菌科细菌有协同作用。除产青霉素酶的金葡菌外,对其他革兰阴性球菌和炭疽杆菌等均甚敏感。不良反应较少,可供肌注及静脉给药。目前在临床已广泛应用。
三、头孢菌素类
【化学与分类】头孢菌素类抗生素是从头孢菌素的母核7-氨基头孢烷酸(7-ACA)接上不同侧链而制成的半合成抗生素。本类抗生素具有抗菌谱广、杀菌力强、对胃酸及对β-内酰胺酶稳定,过敏反应少,(与青霉素仅有部分交叉过敏现象)等优点。根据其抗菌作用特点及临床应用不同,可分为三代头孢菌素(表38-3)。
表38-3 常用头孢菌素类抗生素的化学结构、特点与分类
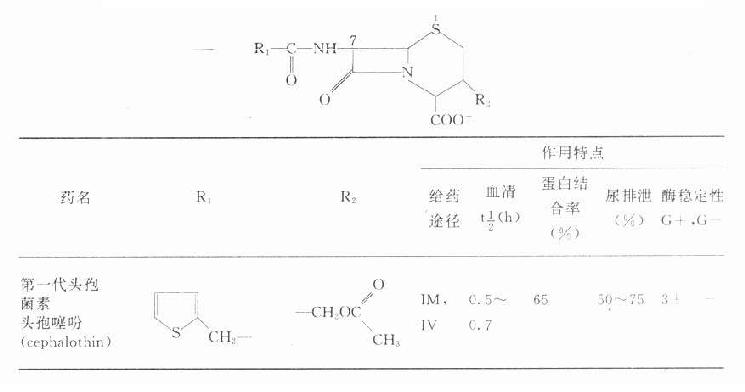
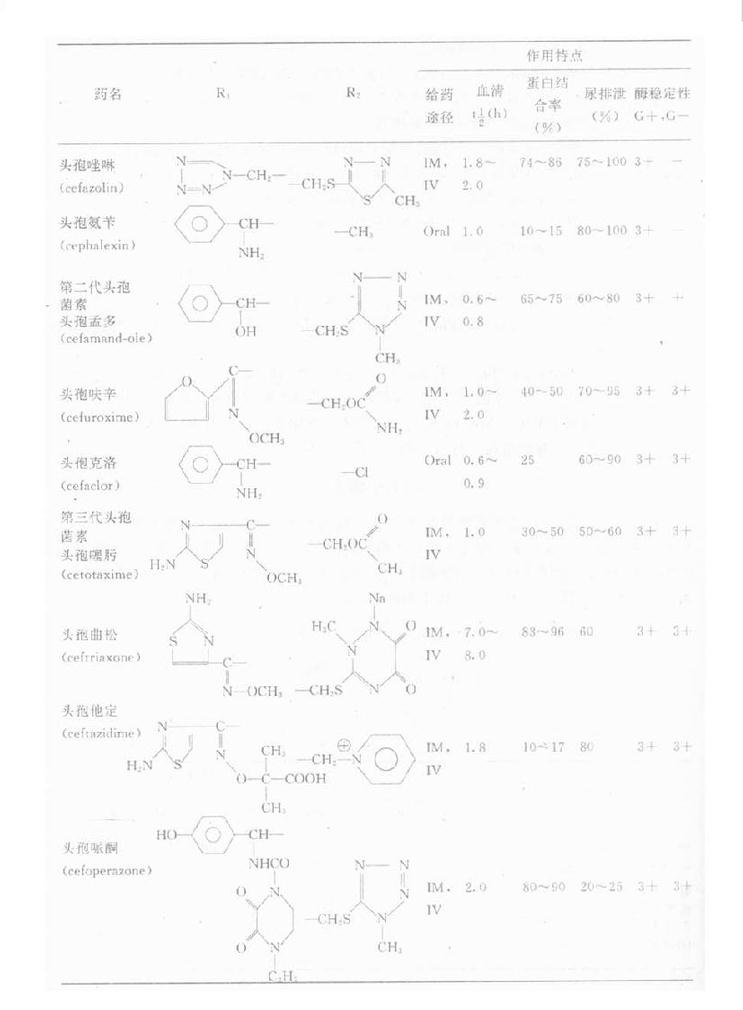
第一代头孢菌素的特点有:①对革兰阳性菌(包括对青霉素敏感或耐药的金葡菌)的抗菌作用较第二、三代强,对革兰氏阴性菌的作用较差;②对青霉素酶稳定,但仍可为革兰阴性菌的β-内酰胺酶所破坏;③对肾脏有一定毒性。
第二代头孢菌素的特点有:①对革兰阳性菌作用与第一代头孢菌素相仿或略差,对多数革兰阴性菌作用明显增强,部分对厌氧菌有高效,但对绿脓杆菌无效;②对多种β-内酰胺酶比较稳定;③对肾脏的毒性较第一代有所降低。
第三代头孢菌素的特点有:①对革兰阳性菌有相当抗菌活性,但不及第一、二代头孢菌素,对革兰阴性菌包括肠杆菌属和绿脓杆菌及厌氧菌如脆弱类杆菌均有较强的作用;②其血浆t1/2较长,体内分布广,组织穿透力强,有一定量渗入脑脊液中;③对β-内酰胺酶有较高稳定性;④对肾脏基本无毒性。
【抗菌作用及作用机制】抗菌谱广,多数革兰阳性菌对之敏感,但肠球菌常耐药;多数革兰阴性菌极敏感,除个别头孢菌素外,绿脓杆菌及厌氧菌常耐药。本类药与青霉素类,氨基甙类抗生素之间有协同抗菌作用。
头孢菌素类为杀菌药,抗菌作用机制与青霉素类相似,也能与细胞壁上的不同的青霉素结合蛋白(PBPs)结合(见图38-1,38-2)。
细菌对头孢菌素类与青霉素类之间有部分交叉耐药现象。
【体内过程】多需注射给药。但头孢氨苄、头孢羟氨苄和头孢克洛能耐酸,胃肠吸收好,可口服。
头孢菌素吸收后,分布良好,能透入各种组织中,且易透过胎盘。在滑囊液、心包积液中均可获得高浓度。头孢呋辛和第三代头孢菌素多能分布于前列腺。第三代头孢菌素还可透入眼部眼房水。胆汁中浓度也较高。其中以头孢哌酮为最高,其次为头孢曲松。头孢呋辛、头孢曲松、头孢噻肟、头孢他定、头孢哌酮等可透过血脑屏障,并在脑脊液中达到有效浓度。多数头孢菌素的血浆t1/2均较短(0.5~2.0小时),但头孢曲松的t1/2最长,可达8小时。
【不良反应】常见者为过敏反应,偶可见过敏性休克,哮喘及速发型皮疹等,青霉素过敏者约有5%~10%对头孢菌素有交叉过敏反应;静脉给药可发生静脉炎;第一代的头孢噻吩、头孢噻啶和头孢氨苄大剂量时可出现肾脏毒性,这与近曲小管细胞损害有关。由于头孢菌素钠盐含钠量可达2.0~3.5Eq/g,大量静注时应注意高钠血症的发生。
头孢孟多、头孢哌酮等可出现双硫仑(disulfiram)样反应,第三代头孢菌素偶见二重感染或肠球菌、绿脓杆菌和念珠菌的增殖现象。头孢孟多、头孢哌酮高剂量可出现低凝血酶原血症。
【临床应用】
第一代头孢菌素,主要用于耐药金葡菌感染,常用头孢噻吩、头孢拉定、及头孢唑啉,后者肌注血浓度为头孢菌素类中最高,是一代中最广用的品种。口服头孢菌素主要用于轻、中度呼吸道和尿路感染。
第二代头孢菌素用以治疗大肠杆菌、克雷伯菌、肠杆菌、吲哚阳性变形杆菌等敏感菌所致的肺炎、胆道感染、菌血症、尿路感染和其他组织器官感染。应用较多的有头孢呋辛及头孢孟多等。
第三代头孢菌素治疗尿路感染以及危及生命的败血症、脑膜炎、肺炎等严重感染可获满意效果。第三代头孢菌素治疗脑膜炎球菌肺炎也可选用。头孢他定为目前临床应用的抗绿脓杆菌最强的抗生素,此外头孢哌酮也可选用。对肠杆菌科细菌头孢曲松和头孢噻肟相仿,头孢哌酮稍差。新生儿脑膜炎和肠杆菌科细菌所致的成人脑膜炎须选用第三代头孢菌素。
四、非典型β-内酰胺类抗生素
(一)头霉素类
头霉素(cephamycin)自链霉菌获得的β-内酰胺抗生素,有A、B、C三型,C型最强。抗菌谱广,对革兰阴性菌作用较强,对多种β-内酰胺酶稳定。头霉素化学结构与头孢菌素相仿,但其头孢烯母核的7位碳上有甲氧基。目前广泛应用者为头孢西丁(cefoxitin),抗菌谱与抗菌活性与第二代头孢菌素相同,对厌氧菌包括脆弱拟杆菌有良好作用,适用于盆腔感染、妇科感染及腹腔等需氧与厌氧菌混合感染。
(二)拉氧头孢
拉氧头孢(latamoxef)又名羟羧氧酰胺菌素(moxalactam),化学结构属氧头孢烯,1位硫为氧取代,7位碳上也有甲氧基,抗菌谱广,抗菌活性与头孢噻肟相仿,对革兰阳性和阴性菌及厌氧菌,尤其脆弱拟杆菌的作用强,对β-内酰胺酶极稳定,血药浓度维持较久。
(三)硫霉素类
硫霉素(thienamycin)化学结构属碳青霉烯类,噻唑环有饱和链,1位硫为碳取代,抗菌谱广,抗菌作用强,毒性低,但稳定性极差,无实用意义,亚胺培南(imipenem,亚胺硫霉素)具有高效、抗菌谱广、耐酶等特点。在体内易被去氢肽酶水解失活。所用者为本品与肽酶抑制剂西司他丁(cilastatin)的合剂,称为泰宁(tienam),稳定性好,供静脉滴注。
(四)β-内酰胺酶抑制剂
1.克拉维酸(clavulanic acid,棒酸) 为氧青霉烷类广谱β-内酰胺酶抑制剂,抗菌谱广,但抗菌活性低。与多种β-内酰胺类抗菌素合用时,抗菌作用明显增强。临床使用奥格门汀(augmentin,氨菌灵)与泰门汀(timentin),为克拉维酸分别和阿莫西林与替卡西林配伍的制剂。
2.舒巴坦(sulbactam,青霉烷砜) 为半合成β-内酰胺酶抑制剂,对金葡菌与革兰阴性杆菌产生的β-内酰胺酶有很强且不可逆抑制作用,抗菌作用略强于克拉维酸,但需要与其他β-内酰胺类抗生素合用,有明显抗菌协同作用。优立新(unasyn)为舒巴坦和氨苄西林(1:2)的混合物,可供肌肉或静脉注射。舒巴哌酮(sulperazone)为舒巴坦和头孢哌酮(1:1)混合物,可供在静脉滴注。
(五)单环β-内酰胺类抗生素
氨曲南(aztreonam)是第一个成功用于临床的单环β-内酰胺类抗生素,对需氧革兰阴性菌具有强大杀菌作用,并具有耐酶、低毒、对青霉素等无交叉过敏等优点,可用于青霉素过敏患者并常作为氨基甙类的替代品使用。
β-内酰胺类抗生素 制剂及用法
青霉素钾盐或钠盐(penicillin G potassium,penicillin G sodium,苄青霉素钾或钠)临用前配成溶液,一般40~80万单位/次,肌内注射,普通感染2次/日,严重感染4次/日,必要时每日总量可再增大。严重感染时可用作静脉滴注,但钾盐忌静脉推注,滴注时亦要计算含钾量(每60万单位青霉素钾盐含钾离子39mg),并注意滴注速度,以防血钾过高。用量较大或病人肾功能不全时,则应改用钠盐滴注。
普鲁卡因青霉素(procaine benzylpenicillin)40万单位/次,1次/日,肌内注射,可产生速效及长效作用。
苄星青霉素(benzathine benzylpenicillin)成人每月1~2次,儿童每月1次,60~120万单位/次,肌内注射。
苯唑西林钠(oxacillin sodium,新青霉素Ⅱ)成人0.5~1.0g/次,4~6次/日,儿童50~100mg/kg/日,分4~6次用。宜在饭前1小时或饭后2小时服用,以免食物干扰吸收。肌内注射剂量同口服,静脉滴注,成人4~6g/日,儿童50~100mg/kg/日。
氯唑西林钠(cloxacillin sodium)成人250~500mg/次,2~4次/日;儿童30~60mg/kg/日,分2~4次口服。肌内注射剂量同口服。
双氯西林(dicloxacillin)成人1~3g/日,儿童30~50mg/kg/日,分4次服用。
氟氯西林(flucloxacillin)成人0.125~0.25g/次,每日4次或0.5~1.0g,每日3次口服。
氨苄西林(ampicillin)成人0.25~1g/次,4次/日;儿童20~80mg/kg/日,分4次服。肌内注射剂量同口服。静脉注射或静脉滴注,成人2~6g/日,儿童50~150mg/kg/日。
阿莫西林(amoxycillin)成人0.3~0.6g/次,每日3~4次口服,儿童10岁以下,病情轻者0.15g/次,3次/日,口服。
匹氨西林(pivampicillin)轻、中度感染,成人1.5~2.0g/日,严重感染3~4g/日,儿童40~80mg/kg/日,3~4次分服。
羧苄西林(carbenicillin)肌内注射,成人4g/日,儿童100mg/kg/日,分4次。静脉注射或静脉滴注用于绿脓杆菌感染,成人10~20g/日,儿童100~400mg/kg/日。
呋苄西林(furbenicillin)成人4~8g/日,儿童50~150mg/kg/日,分4次静脉注射或静脉滴注。
磺苄西林(sulbenicillin)成人2~4g/日,严重者可用8~13g/日,分次肌内注射、静脉注射或静脉滴注,儿童40~160mg/kg/日。
替卡西林(ticarcillin)肌内注射或静脉注射,剂量同羧苄青霉素或磺苄青霉素。
哌拉西林(piperacillin)成人4~8g/日,儿童100~150mg/kg/日;静脉注射,成人8~16g/日,儿童100~300mg/kg/日,皆分4次注射。
美西林(mecillinam)成人1.6~2.4g/日,儿童30~50mg/kg/日,分4次静脉或肌内注射。
头孢噻吩钠(cephalothin sodium,头孢菌素I)成人0.5g/次,4次/日,肌内注射;严重感染时每日2~4g,静脉推注或静脉滴注。
头孢噻啶(cephaloridine)成人0.5~1.0g/次,2~3次/日,肌内注射,每日量不超过4g。
头孢氨苄(cephalexin)成人1~4g/日,分3~4次服。
头孢唑啉(cefazolin)成人500mg/次,2~4次/日,肌内注射或静脉注射,病情严重或耐药菌株,剂量可增大为3~5g/日。儿童剂量为20~100mg/日。
头孢拉定(cefradine)成人1~4g/日,分4次服,对重症者可静脉注射,每日不超8g,儿童50~100mg/kg/日,分4次服。
头孢羟氨苄(cefadroxil)成人2g/日,分2次服;儿童30~60mg/kg/日,分2~3次服。
头孢孟多(cefamandole)成人2~4g/日,儿童50~100mg/kg/日,分3~4次肌内注射。静脉注射成人8~12g/日,儿童100~200mg/kg/日,分2~4次。
头孢呋辛(cefuroxime)肌内注射,成人2~2.5g/日,儿童30~60mg/kg/日,分3~4次。静脉注射,成人4.5~6g/日,儿童50~100mg/kg/日,分2~4次。
头孢克洛(cefaclor)成人2~4g/日,分4次口服。
头孢噻肟(cefotaxime)肌内注射,成人2~6g/日,儿童50~100mg/kg/日,分3~4次;静脉注射,成人2~8g/日,儿童50~150mg/kg/日,分2~4次。
头孢曲松(ceftriaxone)肌内注射,1g/日,溶于利多卡因注射液3.5ml中,深部注入。静脉滴注,成人0.5~2g/日,一次溶于生理盐水或5%葡萄糖液中,30分钟滴完。
头孢他定(ceftazidime)成人1.5~6g/日,儿童50~100mg/kg/日,分3次静脉注射,快速静脉滴注或肌内注射,后者一般溶于1%利多卡因0.5ml,深部注入。
头孢哌酮(cefoperazone)成人2~4g/日,儿童50~150mg/kg/日,分2~3次静脉滴注,推注或肌内注射。
头孢西丁(cefoxitin)成人3~8g/日,分3~4次,儿童45~120mg/kg/日,分4~6次静脉滴注,也可肌内注射。
拉氧头孢(latamoxef)成人1~2g/日,分2次静脉注射,静脉滴注或肌内注射,重症者4g/日或更高剂量。儿童40~80mg/kg/日,严重者可增至150mg/kg/日,分2~4次注射。
亚胺培南(imipenem)成人1~2g/日,分4次静脉注射,应与去氢肽酶抑制剂合用,如泰宁。
氨曲南(aztreonam) 成人1.5~6g/日,分3次,肌内注射,静脉注射或静脉滴注(药物加入100ml生理盐水中,于30分钟内滴完)。
第三十九章 大环内酯类、林可霉素类及其他抗生素
一、大环内酯类抗生素
大环内酯类抗生素是一类具有12~16碳内酯环共同化学结构的抗菌药。目前使用的有红霉素、麦迪霉素、麦白霉素、乙基螺旋霉素、交沙霉素及吉他霉素等。本类药的共同特点为:①抗菌谱窄,比青霉素略广,主要作用于需氧革兰阳性菌和阴性球菌、厌氧菌,以及军团菌、胎儿弯曲菌、衣原体和支原体等;②细菌对本类各药间有不完全交叉耐药性;③在碱性环境中抗菌活性较强,治疗尿路感染时常需碱化尿液;④口服后不耐酸,酯化衍生物可增加口服吸收;⑤血药浓度低,组织中浓度相对较高,痰、皮下组织及胆汁中明显超过血药浓度;⑥不易透过血脑屏障;⑦主要经胆汁排泄,进行肝肠循环;⑧毒性低微。口服后的主要副作用为胃肠道反应,静脉注射易引起血栓性静脉炎。
红霉素
红霉素(erythromycin)从链丝菌(S. erythreus)分离而得。
【抗菌作用】红霉素对革兰阳性细菌有强大抗菌作用,革兰阴性菌如脑膜炎球菌、淋球菌、流感杆菌、百日咳杆菌、布氏杆菌等及军团菌(Legionella)对红霉素也都高度敏感。红霉素对某些螺旋体、肺炎支原体及螺杆菌也有抑制作用。金葡菌对红霉素可产生耐药性,大环内酯类抗生素之间有部分交叉耐药性。
红霉素的抗菌机制是它能与细菌核蛋白体的50S亚基结合,抑制转肽作用及(或)信使核糖核酸(mRNA)移位,而抑制蛋白质合成。
【体内过程】红霉素不耐酸,口服用糖衣片。无味红霉素是其丙酸酯的十二烷酸盐,能耐酸、无味,适于儿童患者服用。红霉素口服吸收快,2小时血药浓度达到高峰,可维持6~12小时,t1/2约2小时。乙琥红霉素为酯化红霉素在体内释出红霉素。红霉素吸收后可迅速分布于组织、各种腺体并易透过胎盘和滑膜囊腔等。药物在体内大部分经肝破坏,胆汁中浓度高,约为血浆浓度的10倍,仅少量药物(12%)由尿排泄。
【临床应用】红霉素主要用于治疗耐青霉素的金葡菌感染和青霉素过敏患者。它效力不及青霉素,且易产生耐药性,但停药数月后,又可恢复其敏感性。红霉素是白喉带菌者、支原体肺炎、沙眼衣原体所致婴儿肺炎及结肠炎、弯曲杆菌所致败血症或肠炎及军团病的首选药。
【不良反应】口服大剂量可出现胃肠道反应。无味红霉素或乙琥红霉素(后者低些)可引起肝损害,如转氨酶升高、肝肿大及胆汁郁积性黄疸等,一般于停药后数日可恢复。口服红霉素也可出现伪膜性肠炎、静脉滴注其乳糖酸盐可引起血栓性静脉炎。
乙酰螺旋霉素
乙酰螺旋霉素(acetylspiramycin)抗菌谱和其他大环内酯类抗生素相似,但其抗菌活性较弱。本品耐酸,口服吸收后,脱乙酰基转为螺旋霉素,体外抗菌作用低于红霉素,但其体内作用较强,组织浓度较高,维持时间也较长。主要用于防治革兰阳性菌引起的呼吸道和软组织感染。
吉他霉素
吉他霉素(kitasamycin),柱晶白霉素(leucomycin)是由链丝菌(S.kitasatoensis)所产生的,抗菌谱与红霉素相似,但其抗菌活性不如红霉素,葡萄球菌也可产生耐药性,但比红霉素慢。吉他霉素可以口服、也可以注射,其体内过程与红霉素相似。临床应用与红霉素相同,优点是对大多数耐红霉素或耐青霉素的金葡菌仍有效,还可用于治疗百日咳、白喉、猩红热、胆道感染及支原体肺炎等,不良反应较少。
麦迪霉素与麦白霉素
麦迪霉素(medecamycin)由链丝菌(S. mycarofaciens)产生,含有麦迪霉素A1、A2和少量A3、A4等组分。国内生产菌所得产品也为多组分,含较多量的白霉素A6,因而称为麦白霉素(meleumycin)。它们的抗菌性能与红霉素相似或稍弱。口服吸收后分布于各组织,以肝、肺、脾、肾较高,胆汁浓度也高。本品主要在体内代谢,仅少量经尿排出,不能透过正常脑膜。主要作为红霉素替代品应用于敏感菌所致的咽部、呼吸道、皮肤和软组织、胆道等部位感染。米欧卡霉素(miocamycin)为麦迪霉素的二醋酸酯,口服吸收较麦迪霉素好,血药浓度高,作用时间长,且味不苦,适合于儿童使用。
交沙霉素
交沙霉素(jossamycin)是单一组分的14碳大环内酯抗生素,抗菌谱与抗菌作用与红霉素相同,对革兰阳性菌和厌氧菌具有较好抗菌作用;对部分耐红霉素的金葡菌仍有效。体内分布较广,在痰、胆汁和组织中浓度较高,不能透过政正常血脑屏障。适应证同麦迪霉素,胃肠反应小。
阿齐霉素及其他
阿齐霉素(azithromycin)与罗红霉素(roxithromycin)是近年用于临床的大环内酯类新品种,其抗菌谱和抗菌作用与红霉素相近或稍差,但具有良好的药动学特性,如罗红霉素血及组织浓度高,并衰期长(12~14小时),从而可降低用量,减少给药次数(每日1~2次),减少不良反应。
二、林可霉素及克林霉素
林可霉素(lincomycin)由链丝菌(S. -lincolensis)产生,克林霉素(clindamycin )是林可霉素7位OH为Cl取代而成。两者具有相同的抗菌谱。由于克林霉素抗菌作用更强、口服吸收好且毒性较小,故临床较为常用。
【抗菌作用】两药对金葡菌(包括耐青霉素者)、溶血性链球菌、草绿色链球菌、肺炎球菌及大多数厌氧菌都有良好抗菌作用。对革兰阴性菌大都无效。两药的抗菌机制相同,能与核蛋白体50S亚基结合,抑制肽酰基转移酶,使蛋白质肽链的延伸受阻。红霉素与林可霉素能互相竞争结合部位,而呈拮抗作用,故不宜合用。
【体内过程】克林霉素较林可霉素的口服吸收为好,且不受食物影响。两药都能渗入骨及其他组织,前者的血药浓度约为后者的2倍,但不透过血脑屏障,其t1/2为2~2.5小时,药物主要在肝代谢灭活,约90%经尿排出。
【临床应用】主要用于急、慢性敏感菌引起的骨及关节感染。用于治疗厌氧菌也有较好疗效。两药中可林霉素尤为常用。
【不良反应】两药口服或注射均可引起胃肠道反应,一般反应轻微,表现为胃纳差,恶心、呕吐、胃部不适和腹泻,但也有出现严重的假膜性肠炎者,多见于林可霉素。
三、万古霉素及去甲万古霉素
万古霉素(vancomycin )和去甲万古霉素(demethylvancomycin)属多肽类化合物,化学结构相近,作用相似,后者略强,仅对革兰阳性菌有强大杀菌作用。抗菌机制为阻碍细菌细胞壁合成(见第三十七章)。细菌对本品不产生耐药性,且与其他抗生素无交叉耐药性。
口服不吸收,粪便中浓度高。药物广泛分布于各组织,主要经肾排泄。静脉滴注正常人血浆t1/2为5~11小时,肾功能不全者t1/2可延长(2~9)天。
万古霉素主要用于治疗耐青霉素金葡萄菌引起的严重感染,如败血症、肺炎、心内膜炎、结肠炎及其他抗生素,尤其是克林霉素引起的假膜性肠炎。静脉滴注时偶可发生恶心、寒战、药热、皮疹及皮肤瘙痒等。较大剂量,严重者可致耳聋、耳鸣及听力损害。
大环内酯类、林可霉素类及其他抗生素 制剂与用法
红霉素(erythromycin)口服0.2~0.5g/次,4次/日。注射用其乳糖酸盐0.3~0.6g/次,3~4次/日,一般用5%葡萄糖液稀释后静脉滴注。
乙酰螺旋霉素(acetylspiramycin)成人2g/日,分2~4次口服;儿童50~100mg/kg/日,分4次口服。
吉他霉素(kitasamycin,leucomycin)口服0.8~1.2g/日,分4~6次用。静脉注射0.4~0.8g/日,分2次,注射速度宜慢,加入静脉滴注液中应用更好。
麦迪霉素(medecamycin),麦白霉素(meleumycin)口服0.8~1.2g/日,分3~4次口服。
交沙霉素(jossamycin)片剂,每片0.1g,成人0.8~1.2g,分3~4次口服。
阿齐霉素(azithromycin)口服:成人500mg/日,每日一次连续3日,或第1日500mg,2~5日,每日250mg,儿童10mg/kg,每日一次,连用3日。
罗红霉素(roxithromycin)成人口服300~600mg/日,分二次服。
林可霉素(lincomycin,洁霉素)口服:成人1.5~2.0g/日,分3~4次服。儿童30~60mg/kg/日,分3~4次服。肌内注射或静脉注射,成人300~600mg/日,3次/日,儿童15~40mg/kg/日。
克林霉素(clindamycin,氯洁霉素)口服:成人0.6~1.2g/日,分3~4次服;儿童8~16mg/kg/日,分3~4次服。肌内注射或静脉注射0.6~1.8g/日,2~4次/日。
万古霉素(vancomycin)口服:2.0g/日,分4次服。静脉滴注:成人1~2g/日,儿童20~40mg/kg/日,分2~4次用,一般应稀释后缓慢滴注。
去甲万古霉素(demethylvancomycin)为国产品,其效价比万古霉素高10%,成人每剂量0.8~1.6g,分2次静滴。
第四十章 氨基甙类抗生素及多粘菌素
第一节 氨基甙类抗生素
氨基甙类抗生素(aminoglycosides)都由氨基糖分子和非糖部分的甙元结合而成,它包括链霉素、庆大霉素、卡那霉素、西索米星以及人工半合成的妥布霉素、阿米卡星、奈替米星等。
一、氨基甙类抗生素的共性
氨基甙类抗生素的化学结构基本相似,因此具有共同特点,如水溶性好,性质稳定;此外,在抗菌谱,抗菌机制,血清蛋白结合率,胃肠吸收,经肾排泄,及不良反应等方面也有共性。
【抗菌作用】氨基甙类对各种需氧革兰阴性菌如大肠杆菌、克雷伯菌属、肠杆菌属、变形杆菌属等具高度抗菌活性。此外,对沙雷菌属、产碱杆菌属、布氏杆菌、沙门菌、痢疾杆菌、嗜血杆菌及分枝杆菌也具有抗菌作用。氨基甙类对革兰阴性球菌如淋球菌、脑膜炎球菌的作用较差。流感杆菌及肺炎支原体呈中度敏感,但临床疗效不显着。绿脓杆菌只对庆大霉素、阿米卡星、妥布霉素敏感,其中以妥布霉素为最强。对各型链球菌的作用微弱,肠球菌对之多属耐药,但金葡菌包括耐青霉素菌株对之甚为敏感。结核杆菌对链霉素、卡那霉素、阿米卡星和庆大霉素均敏感,但后者在治疗剂量时不能达到有效抑菌浓度。
按相同重量比较,庆大霉素和西索米星的抗菌活性较卡那霉素、妥布霉素、奈替米星和阿米卡星稍强,但临床用量中它们的抗菌作用并无明显差别。
【抗菌作用机制】氨基甙类的抗菌作用机制是阻碍细菌蛋白质的合成。许多基本成分如活化氨基酸,转运核糖核酸(tRNA),信息核糖核酸(mRNA),酶Mg2+,始动因子(F1,F2,F3),ATP,GTP等都参与了蛋白质合成(图40-1)。
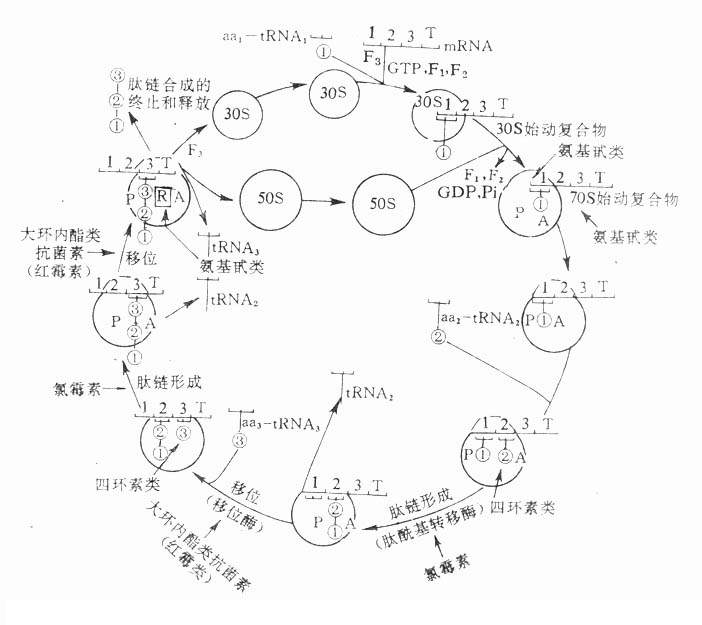
图40-1核蛋白体循环及有关抗生素作用部位图解
30s、50s表示组成核蛋白体的两个亚基;A、P分别表示A位、P位,F1,F2,F3,为始动因子;R表示终止因子;aa1、aa2、aa3-tRNA表示tRNA1、2、3携带三种不同的活化氨基酸;┗┷┛表示tRNa上面三个小点表示反密码,它能翻译mRNA上相应的密码;mRNA上的1、2、3、T表示四个密码,T为终止密码,即停止合成的密码,T前可能有许多密码,为图示简便,仅顺序标出在T前的三个密码。每个密码包括三个核苷酸,称为三联密码。
细菌蛋白质合成分为三个阶段:①起始阶段 30S亚基与新生成的mRNA结合成mRNA-30S复合物,然后接上第一个氨基酰-tRNA(即甲酰蛋氨酰-tRNA),接在相当于50S的P位,称为30S起始复合物,后者很快与50S亚基结合成70S起始复合物。②肽链延长阶段 新的氨基酰-tRNA按mRNA的密码要求进入核蛋白体50S亚基的A位上,此时P位上的甲酰蛋氨酰或以后合成的肽链经肽酰转移酶的作用,基羧基与A位新接上的氨基酸的氨基结合而形成肽链。此时,在P位上的tRNA被释放回到细胞质内转运其他相应的氨基酸,核蛋白体30S亚基上的mRNA发生移位,把带有肽链的tRNA从A位移至P位。空出的A位又接受新的氨基酰-tRNA,如此反复使肽链不断延长。③终止阶段mRNA上出现终止信号时,表示蛋白质合成已结束,此时释放因子(R)进入A位,使肽链释放,tRNA及mRNA与核蛋白体分离,核蛋白体70S又解离为30S与50S亚基,重新参与蛋白质合成。
氨基甙类能影响蛋白质合成的许多环节:①起始阶段,抑制70S始动复合物的形成,②选择性地与30S亚基上靶蛋白结合(如P10),使mRNA上的密码错译,导致异常的、无功能的蛋白质合成;③阻碍终止因子(R)与核蛋白体A位结合,使已合成的肽链不能释放并阻止70S核蛋白体的解离,最终造成菌体内核蛋白体的耗竭。此外,氨基甙类通过离子吸附作用附着于细菌体表面造成胞膜缺损致使胞膜通透性增加,细胞内钾离子、腺嘌呤核苷酸、酶等重要物质外漏,从而导致细菌死亡。氨基甙类与β内酰胺类都是杀菌药,但它与后者不同,对静止期细菌有较强的作用。
【体内过程】氨基甙类在胃肠道不吸收或极少吸收(<1%)。口服后血药浓度很低,可用于胃肠道消毒,但在肾功能损害时,多次口服或直肠内给药,血药浓度可蓄积至中毒水平。肌内注射后氨基甙类吸收迅速且完全。30~90分钟达到峰浓度。常用的氨基甙类药代动力学参数见表40-1。氨基甙类静脉内给药,其浓度高低随剂量而异,一般在静脉滴注20~30分钟后,血浆中浓度与肌内注射者相同,本类药物中除链霉素外,与血浆蛋白很少结合。药物主要分布于细胞外液,组织与细胞内药物含量较低,分布容积大致与细胞外液容积相当,成人为15L(0.56L/kg)。肾脏皮质内药物浓度可超过血药浓度10~50倍。消除t1/2平均可达112~693小时。肾脏皮质内药物蓄积浓度越高,对肾毒性越大。氨基甙类可进入内耳外淋巴液,浓度与用药量成正比,其t1/2较血浆t1/2长5~6倍(图40-2)。当肾功能减退(无尿)时其浓度与t1/2均明显增加。氨基甙类在体内不被代谢,约90%以原形经肾小球过滤排出,尿药浓度极高,约为血浆峰浓度的25~100倍。
表40-1 常用氨基甙类的药代动力学
| 抗生素 | IM. 血药浓度达峰时间(小时) | t1/2(小时) | 24小时尿排出(%) | 蛋白结合率(%) | |
| 正常 | 无尿 | ||||
| 链霉素 | 0.5~1.5 | 2.0~3.0 | 50~110 | 80 | 35 |
| 庆大霉素 | 0.75~1.0 | 1.7~2.3 | 48~72 | 70~80 | <10 |
| 妥布霉素 | 0.33~0.75 | 2.0~2.8 | 56~60 | 80~90 | <10 |
| 卡那霉素 | 0.75~1.0 | 2.1~2.4 | 60~90 | 84~90 | |
| 阿米卡星 | 0.75~2.0 | 2.2~2.5 | 56~150 | 81~98 | 4.0 |
| 西索米星 | 0.75~1.0 | 2.0~2.3 | 35~37 | 85~87 | |
| 奈替米星 | 0.5~1.0 | 2.2 | 33 | 80~90 | <10 |
图40-2正常和无尿豚鼠庆大霉素皮下注射50mg/kg后
血清(A)和外淋巴液(B)的药动学
庆大霉素浓度用生物测定法,每点为6只动物的均值,
无尿动物于给药前3小时切除肾脏
【不良反应】
1.耳毒性 临床反应可分为二类:一为前庭功能损害,有眩晕、恶心、呕吐、眼球震颤和平衡障碍,其发生率依次为:新霉素(已少用)>卡那霉素>链霉素>西索米星>庆大霉素>妥布霉素>奈替米星。另一为耳蜗神经损害,表现为听力减退或耳聋,其发生率依次为:新霉素>卡那霉素>阿米卡星>西索米星>庆大霉素>妥布霉素>链霉素。必须指出耳聋性的许多自觉症状并不明显,但经仪器监测显示有前庭功能或听力损害的“亚临床耳毒性”反应的发生率则可达10%~20%,最先影响为高频听力,随后逐渐波及低频部分。耳毒性发生机制可能是内耳淋巴液中药物浓度过高,损害内耳柯蒂氏器内、外毛细胞的糖代谢和能量利用,导致内耳毛细胞膜上钾钠离子泵发生障碍,终使毛细胞的功能受损。
为防止和减少耳毒性反应,在治疗过程中应注意观察耳鸣、眩晕等早期症状的出现,进行听力监测,并根据患者的肾功能(肌酐清除率等)及血药浓度来调整用药剂量。除非必要,应避免与高效利尿药或其他耳毒性药物合用。
2.肾毒性 氨基甙类主要经肾排泄并在肾(尤其是皮质部)蓄积,主要损害近曲小管上皮细胞,但不影响肾小球,临床化验可见蛋白尿、管形尿、尿中红细胞、肾小球过滤减少,严重者可发生氮质血症及无尿等。年老、剂量过高
■[此处缺少一些内容]■
其硫酸盐,性质稳定,水溶液在室温可保持一周。口服不吸收,肌肉注射吸收快,30~60分钟达峰浓度,t1/2为2~3小时,一次注射有效浓度可达6~8小时,年龄超过40岁t1/2可延长至9小时,主要分布于细胞外液,大部分经肾排泄,肾功不全时,排泄减慢。
链霉素对多数革兰阴性菌有强大抗菌作用,但因毒性与耐药性问题,限制了它的临床应用。目前临床主要用于:①鼠疫与兔热病,对此链霉素是首选药;②布氏杆菌病,链霉素与四环素合用也有满意的效果;③感染性心内膜炎,对草绿色链球菌引起者,以青霉素合并链霉素为首选;对肠球菌引起者,也需青、链合用治疗,但部分菌株对链霉素耐药,可改用庆大霉素或妥布霉素等;④结核病,链霉素为最早的抗结核药,现仍有应用,但必须与其他抗结核药联合应用,以延缓耐药性的发生;⑤链霉素与青霉素或氨苄西林合用,可用于预防常发的细菌性心内膜炎及呼吸、胃肠道及泌尿系统手术后感染。
链霉素治疗时常可出现头痛、头晕、呕吐、耳鸣、平衡失调和眼球震颤。多是可逆的。严重者可致永久性耳聋。对肾脏的毒性为氨基甙类中最轻者,但肾功能不全者仍应慎用。
2.庆大霉素(gentamicin)是目前临床最为常用的广谱氨基甙类。庆大霉素水溶液稳定,水针剂常作肌内或静脉滴注给药。体内过程与链霉素相仿(表40~1)。但其有效与安全的血药浓度较低(4~8mg/L)。药物主要经肾排泄,部分经胆汁入肠,胆汁药物浓度可达血药浓度的60%~80%,t1/2约3小时。
庆大霉素广泛用于治疗敏感菌的感染:①严重革兰阴性杆菌的感染如败血症、骨髓炎、肺炎、腹膜感染、脑膜炎等、庆大霉素是首选药;②绿脓杆菌感染,庆大霉素常与羧苄西林合用可获协同作用,但两药不可同时混合滴注,因后者可使本药的活力降低;③病因未明的革兰阴性杆菌混合感染,庆大霉素与广谱半合成青霉素类(羧苄西林或哌拉西林等)或头孢菌素联合应用可以提高疗效;④与青霉素联合治疗肠球菌心内膜炎;与羧苄西林、氯霉素联合治疗革兰阴性杆菌心内膜炎;⑤庆大霉素口服可用于肠道感染或肠道术前准备;⑥庆大霉素局部用于皮肤、粘膜表面感染、眼、耳、鼻部感染,但因可致光敏感反应,大面积应用易致吸收毒性,故少作局部应用。
不良反应有前庭神经功能损害,但较链霉素少见,对肾脏毒性则较多见。
3.卡那霉素(kanamycin)由链丝菌培养液获得。卡那霉素体内过程与链霉素、庆大霉素基本相同。其抗菌谱与链霉素相似,但稍强,对多数常见的革兰阴性菌及结核菌有效,但对绿脓杆菌无效。体内抗菌有效的血药浓度范围为8~16µg/ml。卡那霉素由于毒性及耐药菌较多见,其在临床应用已为庆大霉素等其他氨基甙类药所取代。
4.妥布霉素(tobramycin)由链丝菌培养液中提得,也可由卡那霉素B脱氧而成,其水溶液非常稳定。
抗菌作用与庆大霉素相似,对绝大多数肠杆菌科细菌、绿脓杆菌及葡萄球菌具良好的抗菌作用。最突出的是对绿脓杆菌作用较庆大霉素强2~4倍,并且对庆大霉素耐药者仍有效,对肠球菌及除绿脓杆菌外的假单孢菌属及厌氧菌无效,对肺炎杆菌、肠杆菌属与变形杆菌属的作用较庆大霉素略强;但对沙雷菌和沙门菌的作用略差。
妥布霉素与庆大霉素相同,主要用于各种严重的革兰阴性杆菌感染,但一般不作为首选药。对绿脓杆菌感染或需较长时间用药者,如感染性心内膜炎,以选用妥布霉素为宜。
妥布霉素的耳毒性较庆大霉素略低,但仍应警惕。一般每日剂量不宜超过5mg/kg,血药浓度不宜超过12mg/L。在肾功能减退时还应根据血清肌酐清除率,调整剂量与给药间隔。
5.阿米卡星(amikacin,丁胺卡那霉素)是卡那霉素的半合成衍生物,其抗菌谱为本类药物中最宽的。其突出优点是对许多肠道革兰阴性菌和绿脓杆菌?
■[此处缺少一些内容]■
、沙雷杆菌、各型变形杆菌和绿脓杆菌都具有较强抗菌活性,对流感嗜血杆菌、沙门菌、志贺菌和奈瑟菌也有效。对某些耐其他氨基甙类的革兰阴性杆菌及耐青霉素类的金葡菌也有效。适用于尿路、肠道、呼吸道、皮肤软组织、骨和关节、腹腔及创口部分的感染。奈替米星的耳、肾毒性是氨基甙类抗生素中最低者。但仍宜注意。
8.新霉素(neomycin)抗菌谱似卡那霉素。肌内注射吸收快,毒性比卡那霉素大,易引起永久性耳聋和肾损害,不宜注射给药。口服很少吸收,毒性较小,可用于肠道感染和肠道消毒。局部外用治疗皮肤粘膜浅表感染。
【附】:大观霉素(spectinomycin)由链霉菌所产生的一种氨基环醇类(aminocyclitols)抗生素,主要对淋球菌有高度抗菌活性,6.3mg/L可抑制大多数淋球菌。肌注2g,1小时血药浓度达峰100mg/L,t1/2约2.5小时。药物主要经尿排泄。临床的唯一适应证是无并发症的淋病,限于对青霉素、四环素等耐药的淋病或患者对青霉素过敏者。
三、药物相互作用
氨基甙类与两性霉素、杆菌肽、头孢噻吩、多粘菌素或万古霉素合用能增加肾脏毒性。呋塞米(速尿)、利尿酸及甘露醇等能增加氨基甙类的耳毒性。苯海拉明、敏可静、安其敏等抗组胺药可掩盖氨基甙类的耳毒性。氨基甙类能增强骨骼肌松弛药及全身麻醉药引起的肌肉松弛作用,可导致呼吸抑制。
第二节 多粘菌素类
多粘菌素包括多粘菌素B(polymyxin B)及多粘菌素E(polymyxin E;粘菌素,colistin),二者具有相似的药理作用。是多肽类抗生素,由于静脉给药可致严重肾毒性现已少用。
【体内过程】多粘菌素口服不易吸收。肌内注射50mg后2小时血药浓度达峰值(2~8mg/L),有效血药浓度可维持8~12小时,t1/2约6小时。肾功能不全者清除慢,t1/2可达2~3天。它分布于全身组织,以肝、肾为最高,并保持较长时间。多粘菌素不易弥散进入胸、腹腔、关节腔,即使在脑膜炎症时也不易透入脑脊液中,胆汁中浓度也较低。药物经肾缓慢排泄。
【抗菌作用及临床应用】对多数革兰阴性杆菌有杀灭作用。多肽类抗生素具有表面活性,含有带阳电荷的游离氨基,能与革兰阴性菌细胞膜的磷脂中带阴电荷的磷酸根结合,使细菌细胞膜面积扩大,通透性增加,细胞内的磷酸盐、核苷酸等成份外漏,导致细菌死亡。多粘菌素对生长繁殖期和静止期的细菌都有效,过去曾用于对其他抗生素耐药的绿脓杆菌和革兰阴性杆菌所致感染如败血症、脑膜炎、心内膜炎、烧伤后感染等。但现在已被疗效好、毒性低的其他抗生素所取代。仍可局部用于敏感菌的眼、耳、皮肤、粘膜感染及烧伤绿脓杆菌感染。多粘菌素口服用于肠道手术前准备。
【不良反应】毒性较大。主要表现在肾脏及神经系统两方面,其中多粘菌素B较E尤为多见,症状为蛋白尿、血尿等。大剂量、快速静脉滴注时,由于神经肌肉的阻滞可导致呼吸抑制。
氨基甙类抗生素及多粘菌素 制剂及用法
硫酸链霉素(streptomycin sulfate)成人0.75~1.0g/日,儿童15~30mg/kg/日,分1~2次肌内注射。成人1~3g/日,儿童1mg/kg/日,分4次服。
硫酸庆大霉素(gentamicin sulfate)成人16~24万单位/日,儿童3,000~5,000u/kg/日,分3~4次肌内注射。静脉滴注剂量同上,忌与青霉素等混合滴注。鞘内注射,成人每次5,000~10,000单位。口服,成人24万~64万单位/日,儿童1万~1.5万单位/kg/日,分四次服。
硫酸卡那霉素(kanamycin sulfate)成人1.0~1.5g/日,儿童20~30mg/kg/日,分2~3次肌内注射。静脉滴注用,剂量同肌内注射。疗程一般不超过10~14天。
硫酸妥布霉素(tobramycin sulfate)成人或儿童每次1.5mg/kg/日,每8小时一次,肌内或静脉注射,总量不超过5mg/kg/日,疗程一般不超过10~14天。
硫酸阿米卡星(amikacin sulfate)成人1.0~1.5g/日,分2~3次肌内注射。
硫酸西索米星(sisomicin sulfate)全身性感染用3mg/kg/日,分3次肌内注射,尿道感染可按2mg/kg/日,分2次肌内注射。
硫酸奈替米星(netilimicin sulfate)成人用4~6mg/kg/日,严重感染7.5mg/kg/日,分2~3次肌内注射;儿童按6~7.5mg/kg/日,分3次给予。
硫酸新霉素(neomycin sulfate)成人1~4g/日,儿童25~50mg/kg/日,分4次口服。
大观霉素(spectinomycin)2g溶于3.2ml特殊稀释液(0.9%苯甲醇溶液)深部肌注,每日1~2次。
硫酸多粘菌素B(polymyxin B sulfate)成人50~100万单位/日,儿童1.0~2.0万单位/日,分2~3次肌内注射或静脉滴注,疗程一般不超过7~14天,鞘内注射,成人1万单位/次,儿童5,000单位/次。
硫酸多粘菌素E(polymyxin E sulfate,colistin sulfate)肌内注射,成人100~150万单位/日,儿童1.5~2.5万单位/kg/日,分2~3次。静脉滴注,成人50~100万单位/日,分2次;儿童1.5~2.5万单位/kg/日,分1~2次。疗程一般不超7天。口服,100~200万单位/日,分3~4次服。
第四十一章 四环素类及氯霉素
四环素类和氯霉素的抗菌谱极广,包括革兰氏阳性和阴性菌、立克次体、衣原体、支原体和螺旋体,故常称为广谱抗生素。
第一节 四环素类
四环素类抗生素具有共同的基本母核(氢化骈四苯),仅取代基有所不同。它们是两性物质,可与碱或酸结合成盐,在碱性水溶液中易降解,在酸性水溶液中则较稳定,故临床一般用其盐酸盐。
四环素类可分为天然品与半合成品两类。天然品有金霉素、土霉素、四环素和去甲金霉素等。金霉素已被淘汰,去甲金霉素我国不生产。四环素和土霉素较常用。半合成品有多西环素和米诺环素,前者在我国较为常用。
四环素与土霉素
四环素(tetracycline)和土霉素(terramycin;氧四环素,oxytetracycline),由于抗菌谱广,口服有效,应用方便,故曾长期广用于临床。近年来由于耐药菌株日益增多,疗效不够理想,且副作用较多,其临床应用已明显减少。
【抗菌作用】抗菌谱广,对革兰阳性的肺炎球菌、溶血性链球菌、草绿色链球菌及部分葡萄球菌、破伤风杆菌和炭疽杆菌等;对革兰阴性细菌中的脑膜炎球菌、痢疾杆菌、大肠杆菌、流感杆菌、巴氏杆菌属、布氏杆菌等及某些厌氧菌(如拟杆菌、梭形杆菌、放线菌)都有效。此外,对肺炎支原体、立克次体、螺旋体、放线菌也有抑制作用,还能间接抑制阿米巴原虫。对绿脓杆菌、病毒与真菌无效。
四环素类属快速抑菌剂,在高浓度时也有杀菌作用。其抗菌机制主要为与细菌核蛋白体30S亚单位在A位特异性结合,阻止aa-tRNA在该位置上的联结,从而阻止肽链延伸和细菌蛋白质合成。其次四环素类还可引起细胞膜通透性改变,使胞内的核苷酸和其他重要成分外漏,从而抑制DNA复制。
细菌对四环素类的耐药性在体外发展较慢,然本类药物之间有交叉耐药性。大肠杆菌和其他肠杆菌科细菌的耐药性主要通过耐药质粒介导,并可传递、诱导其他敏感细菌转成耐药,带耐药质粒细菌的细胞膜对四环素类药物摄入减少或泵出增加。
【体内过程】口服易吸收,但不完全,四环素吸收较土霉素好,2~4小时血药浓度可达高峰,t1/2约为8.5小时,土霉素血药浓度较低,t1/2为9.6小时,由于四环素类能与多价阳离子如Mg2+、Ca2+、Al3+及Fe2+等起络合作用,因而含这些离子的药物和食物均可妨碍其吸收。饭后服盐酸四环素较空腹服用时血药浓度低50%左右;铁剂可使四环素的吸收率下降约40%~90%,如需要两药合用,服药时间应相隔3小时。胃液中酸度高时,药物溶解完全,吸收较好。此外,口服四环素与土霉素吸收量有一定限度。服药量超过0.5g以上,血药浓度并不随剂量增加而提高,只增加粪便中的排泄量。
吸收后广泛分布于各组织中,并能沉积于骨及牙组织内。它们与血浆蛋白结合率约为20%~30%,因此四环素容易渗入胸腔、腹腔、胎儿循环及乳汁中,但不易透过血脑屏障,脑脊液中的药物浓度一般仅为血药浓度的1/10。四环素、土霉素主要以原形经肾小球过滤排出,故尿药浓度较高,有利于治疗尿路感染。土霉素口服排泄快,且较完全,排泄量可达60%~70%。四环素排泄量较少,约20%~30%。本类药物经肝浓缩排入胆汁,形成肝肠循环。胆汁中药物浓度为血药浓度的10~20倍。
【临床应用】四环素类临床应用范围比较广泛。对立克次体感染和斑疹伤寒、恙虫病以及支原体引起的肺炎有良效,为首选药物。对革兰阳性菌和阴性菌感染,百日咳、痢疾、肺炎杆菌所致的尿道、呼吸道与胆道感染,可用新四环素类作次选药。
【不良反应】
1.胃肠道反应 本药口服后直接刺激而引起恶心、呕吐、上腹不适、腹胀、腹泻等症状,尤以土霉素多见,与食物同服可以减轻。
2.二重感染 正常人的口腔、鼻咽、肠道等都有微生物寄生,菌群间维持平衡的共生状态。广谱抗生素长期应用,使敏感菌受到抑制,而不敏感菌乘机在体内繁殖生长,造成二重感染,又称菌群交替症。多见于老幼和体质衰弱、抵抗力低的患者。此外,合并应用肾上腺皮质激素、抗代谢或抗肿瘤药物也更容易诱发二重感染。常见的二重感染有:①真菌病,致病菌以白色念珠菌最多见。表现为口腔鹅口疮、肠炎、可用抗真菌药治疗。②葡萄球菌引起的假膜性肠炎,此时葡萄球菌产生强烈的外毒素,引起肠壁坏死,体液渗出、剧烈腹泻、导致失水或休克等症状,有死亡危险。此种情况必须停药并口服万古霉素。
3.对骨、牙生长的影响 四环素类能与新形成的骨、牙中所沉积的钙相结合。妊娠五个月以上的妇女服用这类抗生素,可使出生的幼儿乳牙釉质发育不全并出现黄色沉积,引起畸形或生长抑制。
4.其他 长期大量口服或静脉给予(每日超过1~2g)可造成严重肝脏损害。也能加剧原有的肾功能不全,影响氨基酸代谢而增加氮质血症。此外,四环素类抗生素还可引起药热和皮疹等过敏反应。
多西环素
多西环素(doxycycline,强力霉素)是土霉素的脱氧物。易溶,遇光不稳定。
【抗菌作用】抗菌谱和四环素相似。但抗菌作用强2~10倍,且对土霉素、四环素的耐药金葡菌有效。
【体内过程】脂溶性较大,因此口服吸收快而完全,分布于全身,脑脊液中浓度也较高。强力霉素的吸收不受食物的影响。药物大部经胆汁排入肠道又可再吸收,经肾小管时也可再吸收,因此t1/2长达20小时,可维持有效血药浓度24小时以上。一般细菌性感染每日服药一次即可。药物小部分从肾排泄。大部分以结合或络合的无活性代谢产物由粪便排泄,故对肠道菌群无影响,肾功能不全时仍可使用。
【临床应用】同四环素,用于呼吸道感染如老年慢性气管炎、肺炎、麻疹肺炎,也用于泌尿道感染及胆道感染等。对肾功能不良患者的肾外感染也可使用。对产肠毒素大肠杆菌所致的腹泻也有效,但宜慎用。
【不良反应】常见胃肠道刺激性反应,如恶心、呕吐、腹泻、舌炎、口腔炎及肛门炎等,宜饭后服药。皮疹及二重感染少见。在静脉注射过程中可出现舌头麻木及口内特殊气味,个别可有呕吐。
【相互作用】多西环素与肝药酶诱导剂苯巴比妥、苯妥英钠等同服,可使其t1/2缩短为7小时左右,并使血药浓度降低而影响疗效。
米诺环素
米诺环素(minocycline,二甲胺四环素)是长效高效的半合成四环素,其抗菌谱和四环素相近,抗菌作用为四环素类中最强,对四环素耐药的金葡菌、链球菌和大肠杆菌对本品仍敏感。
口服吸收迅速,2~3小时后血药浓度可达高峰,经尿与粪排泄量为本类药中最低者,t1/2约为13(10~20)小时。药物在体内长时间存留于脂肪组织,给药后10天尿中仍可测出。
临床用于尿路、胃肠道、呼吸道感染、脓皮病、骨髓炎,眼耳鼻喉部感染等。此外对疟疾也有一定效果。
不良反应与其他四环类基本相同,但能引起可逆性前庭反应,包括恶心、呕吐、头昏、眼花及运动失调等,常在开始服药时出现,停药后24~48小时可消失。
第二节 氯霉素
氯霉素(chloramphenicol,chloromycetin)是由委内瑞拉链丝菌产生的抗生素。分子中含有氯。
【抗菌作用】氯霉素对革兰阳性、阴性细菌均有抑制作用,且对后者的作用较强。其中对伤寒杆菌、流感杆菌、副流感杆菌和百日咳杆菌的作用比其他抗生素强,对立克次体感染如斑疹伤寒也有效,但对革兰阳性球菌的作用不及青霉素和四环素。抗菌作用机制是与核蛋白体50S亚基结合,抑制肽酰基转移酶,从而抑制蛋白质合成。
各种细菌都能对氯霉素发生耐药性,其中以大肠杆菌、痢疾杆菌、变形杆菌等较为多见,伤寒杆菌及葡萄球菌较少见。细菌对氯霉素产生耐药性比较慢,可能是通过基因的逐步突变而产生的,但可自动消失。细菌也可以通过R因子的转移而获得耐药性,获得R因子的细菌能产生氯霉素乙酰转移酶(acetyltransferase)使氯霉素灭活。
【体内过程】氯霉素自肠道上部吸收,一次口服1.0g后2小时左右血中药物浓度可达到峰值(约10~13mg/L)。血浆t1/2平均为2.5小时,6~8小时后仍然维持有效血药浓度。氯霉素广泛分布于各组织和体液中,脑脊液中的浓度较其他抗生素为高。氯霉素的溶解和吸收均与制剂的颗粒大小及晶型有关。肌内注射吸收较慢,血浓度较低,仅为口服同剂量的50%~70%,但维持时间较长。注射用氯霉素为琥珀酸钠盐,水中溶解度大,在组织内水解产生氯霉素。
氯霉素在体内代谢大部分是与葡萄糖醛酸相结合,其原形药及代谢物迅速经尿排出,口服量5%~15%的有效原形药经肾小球过滤而排入尿中,并能达到有效抗菌浓度,可用于治疗泌尿系统感染。肾功能不良者使用时应减量。
【临床应用】氯霉素曾广泛用于治疗各种敏感菌感染,后因对造血系统有严重不良反应,故对其临床应用现已做出严格控制。可用于有特效作用的伤寒、副伤寒和立克次体病等及敏感菌所致的严重感染。氯霉素在脑脊液中浓度较高,也常用于治疗其他药物疗效较差的脑膜炎患者。必要时可用静脉滴注给药。
【不良反应】主要不良反应是抑制骨髓造血机能。症状有二:一为可逆的各类血细胞减少,其中粒细胞首先下降,这一反应与剂量和疗程有关。一旦发现,应及时停药,可以恢复;二是不可逆的再生障碍性贫血,虽然少见,但死亡率高。此反应属于变态反应与剂量疗程无直接关系。可能与氯霉素抑制骨髓造血细胞内线粒体中的与细菌相同的70S核蛋白体有关。为了防止造血系统的毒性反应,应避免滥用,应用时应勤查血象,氯霉素也可产生胃肠道反应和二重感染。此外,少数患者可出现皮疹及血管神经性水肿等过敏反应,但都比较轻微。新生儿与早产儿剂量过大可发生循环衰竭(灰婴综合征),这是由于他们的肝发育不全,排泄能力差,使氯霉素的代谢、解毒过程受限制,导致药物在体内蓄积。因此,早产儿及出生两周以下新生儿应避免使用。
【应用注意】
1.开始治疗前应检查血象(白细胞、分类及网织细胞计数),随后每48小时再查一次,治疗结束还要定期检查血象,一旦出现异常,应立即停药。
2.氯霉素治疗时,对用口服降血糖药的糖尿病患者或服抗凝血药者,尤其是老年人,应分别检测血糖及凝血酶原时间,以防药效及毒性增强。
3.对肝肾功能不良,G-6PDH缺陷者、婴儿、孕妇、乳妇应慎用。
4.用药时间不宜过长,一般不超过二个月,能达到防止感染复发即可,避免重复疗程。
四环素类及氯霉素 制剂与用法
盐酸四环素(tetracycline hydrochloride)口服:0.25~0.5g/次,3~4次/日。
盐酸土霉素(oxytetracycline hydrochloride)口服一次0.5g,3~4次/日。8岁以下小儿30~40mg/kg/日,分3~4次服用。
多西环素(doxycycline)成人首剂0.2g,以后0.1~0.2g/次,每日1次。儿童首剂4mg/kg,以后每日2~4mg/kg,每日1次。
米诺环素(minocycline)首剂0.2g,以后0.1g,每12小时1次。
氯霉素(chloramphenicol,chloromycetin)口服1.5g/次,每日4次,氯霉素肌注、静注或静滴0.5或1g,每12小时1次。
琥珀氯霉素(chlamphenicol sodium succinate)注射剂:0.69(相当氯霉素0.5g),成人1~2g/日,分2~4次肌注或静滴,儿童每日25~50mg/kg,分二次静滴。
第四十二章 人工合成抗菌药
第一节 喹诺酮类药物
喹诺酮类quinolones是人工合成的含4-喹诺酮基本结构,对细菌DNA螺旋酶(DNA gyrase)具有选择性抑制作用的抗菌药物。目前发展迅速,临床广为使用。
一、喹诺酮类药物概述
(一)简史
萘啶酸(nalidixicacid)是1962年用于临床的第一个喹诺酮类药(实是萘啶酮),抗菌谱窄,口服吸收差,副作用多,现已不用。吡哌酸(pipemidicacid)抗菌活性强于萘啶酸,口服少量吸收,不良反应较萘啶酸少,可用于敏感菌的尿路感染与肠道感染。1979年合成诺氟沙星(norfloxacin),随又合成一系列含氟的新喹诺酮类药,通称为氟喹诺酮类(fluoroquinolones)。
(二)化学结构与作用关系
本类药物的构效关系研究表明:4-喹诺酮母核的3位均有羧酸基,6位引入氟原子可增强抗菌作用并对金葡菌有抗菌活性;7位引进哌嗪环可提高对金葡菌及绿脓杆菌的抗菌作用(如诺氟沙星),哌嗪环被甲基哌嗪环取代(如培氟沙星),则脂溶性增加,肠道吸收增强,细胞的穿透性提高,半衰期延长。在8位引进第二个氟原子,可进一步提高肠道吸收,延长半衰期(如洛美沙星等),N-1修饰以环丙基团(环丙沙星)或噁嗪基团(氧氟沙星)可扩大抗菌谱,增强对衣原体、支原体及分支杆菌(结核杆菌与麻风杆菌等)的抗菌活性,噁嗪环还可提高水溶性,使药物在体内不被代谢,以原形经尿排泄。
(三)抗菌作用机制
喹诺酮类通过抑制DNA螺旋酶作用,阻碍DNA合成而导致细菌死亡。大肠杆菌的DNA螺旋酶是四叠体结构的蛋白,由2个A亚单位与2个B亚单位组成,分子量分别为105kD与95kD(见图42-1)。细菌在合成DNA过程中,DNA螺旋酶的A亚单位将染色体DNA正超螺旋的一条单链(后链)切开,接着B亚单位使DNA的前链后移,A亚单位再将切口封住,形成了负超螺旋。根据实验研究,氟喹诺酮类药并不是直接与DNA螺旋酶结合,而是与DNA双链中非配对碱基结合,抑制DNA抑螺旋酶的A亚单位,使DNA超螺旋结构不能封口,这样DNA单链暴露,导致mRNA与蛋白合成失控,最后细菌死亡。
本类药体外对DNA螺旋酶的半抑制浓度(IC50)与其对细菌的MIC呈一定的平行关系。哺乳动物的细胞内也含有生物活性与细菌DNA螺旋酶相似的酶,称为拓朴异构酶Ⅱ(topoisomerase Ⅱ)。氟喹诺酮类药对人体细胞拓朴异构酶Ⅱ影响较小(见表42-1)。从该表可见氧氟沙星与环丙沙星对动物细胞内拓朴异构酶Ⅱ的作用明显比依诺沙星与萘啶酸小,IC50很高,选择指数很大。这可能是氧氟沙星与环丙沙星不良反应较少的原因。
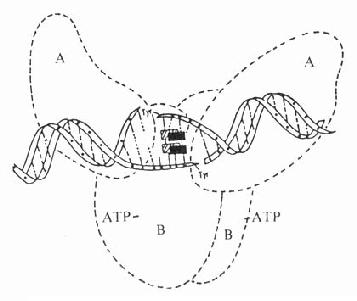
图42-1 喹诺酮类药物-DNA结合抑制DNA螺旋酶活性的示意图
图中实心和斜线长方形示喹诺酮类药物分子,A、B为DNA螺旋酶的A、B亚单位。在DNA螺旋酶作用下,DNA双链打开,而药物分子嵌入双链。与非配对碱基结合,阻碍DNA双链封口
(四)细菌耐药机制
氟喹诺酮类药物广泛应用后,已出现细菌耐药性。耐药机理研究证实主要是染色体突变,不存在质粒介导的耐药性。耐药机制有二:①细菌DNA螺旋酶的改变,与细菌高浓度耐药有关;②细菌细胞膜孔蛋白通道的改变或缺失与低浓度耐药有关。耐药菌株DNA螺旋酶的活性改变主要由于gyrA基因突变所致。
(五)氟喹诺酮类药理学共同特性
①抗菌谱广,尤其对革兰阴性杆菌包括绿脓杆菌在内有强大的杀菌作用,对金葡菌及产酶金葡菌也有良好抗菌作用;某些品种对结核杆菌,支原体,衣原体及厌氧菌也有作用;②细菌对本类药与其他抗菌药物间无交叉耐药性;③口服吸收良好,部分品种可静脉给药;体内分布广,组织体液浓度高,可达有效抑菌或杀菌水平;血浆半衰期相对较长,大多为3~7小时以上。血浆蛋白结合率低(14%~30%),多数经尿排泄,尿中浓度高;④适用于敏感病原菌所致的呼吸道感染、尿路感染、前列腺炎,淋病及革兰阴性杆菌所致各种感染,骨、关节、皮肤软组织感染;⑤不良反应少(5%~10%),大多轻微,常见的有恶心、呕吐、食欲减退、皮疹、头痛、眩晕。偶有抽搐精神症状,停药可消退。
表42-1 喹诺酮类药物对大肠杆菌和哺乳动物细胞DNA旋转酶的选择作用
| 药物 | IC50(mg/L) | 选择指数B/A | |
| A大肠杆菌KL-16DNA螺旋酶 | B胎牛胸腺局部拓朴异构酶Ⅱ | ||
| 氧氟沙星 | 0.76 | 1870 | 2461 |
| 环丙沙星 | 0.13 | 155 | 1192 |
| 依诺沙星 | 1.72 | 93 | 54 |
| 萘啶酸 | 23.0 | 385 | 17 |
二、各种喹诺酮类药特点
吡哌酸(pipemidic acid,PPA)对革兰阴性菌的抗菌作用较萘啶酸强,对革兰阳性和部分绿脓杆菌有一定作用。口服400mg后血浓度达不到治疗效果,但尿中浓度高,可达900mg/L以上,主要用于治疗尿路和肠道感染。
诺氟沙星(norfloxacin)又名氟哌酸,是第一个氟喹诺酮类药,抗菌谱广,抗菌作用强,对革兰阳性和阴性菌包括绿脓杆菌均有良好抗菌活性,明显优于吡哌酸。口服吸收约35%~45%;易受食物影响,空腹比饭后服药的血浓度高2~3倍,血浆蛋白结合率为14%,体内分布广,组织浓度高,药物消除半衰期为3~4小时。主要用于尿路及肠道感染。
氧氟沙星(ofloxacin)又名氟嗪酸,抗菌活性强,对革兰阳性菌(包括甲氧西林耐药金葡菌,MRSA)革兰阴性菌包括绿脓杆菌均有较强作用;对肺炎支原体,奈瑟菌病,厌氧菌及结核杆菌也有一定活性。对感染小鼠的保护效果明显强于诺氟沙星、依诺沙星。口服吸收快而完全,血药浓度高而持久,血浆消除半衰期为5~7小时,药物体内分布广,尤以痰中浓度较高,70%~90%药物经肾排泄,48小时尿中药物浓度仍可达到对敏感菌的杀菌水平,胆汁中药物浓度约为血药浓度的7倍左右。
依诺沙星(enoxacin)又名氟啶酸,抗菌谱和抗菌活性和诺氟沙星相似,对厌氧菌作用较差。口服吸收好,不受食物影响,血药浓度介于诺氟沙星与氧氟沙星之间,口服后约50%~65%经肾排泄,消除半衰期为3.3~5.8小时。副作用以消化道反应为主,偶有中枢神经系统毒性反应。
培氟沙星(pefloxacin)又名甲氟哌酸,抗菌谱广与诺氟沙星相似,抗菌活性略逊于诺氟沙星,对军团菌及MRSA有效,对绿脓杆菌的作用不及环丙沙星。口服吸收好,生物利用度为90%~100%。血药浓度高而持久,半衰期可达10小时以上,体内分布广泛,尚可通过炎症脑膜进入脑脊液。
环丙沙星(ciprofloxacin)又名环丙氟哌酸,抗菌谱广,体外抗菌活性为目前在临床应用喹诺酮类中最强,对耐药绿脓杆菌,MRSA,产青霉素酶淋球菌、产酶流感杆菌等均有良效,对肺炎军团菌及弯曲菌亦有效,一些对氨基甙类、第三代头孢菌素等耐药的革兰阴性和阳性菌对本品仍然敏感。口服后本品生物利用度为38%~60%,血浓度较低,静脉滴注可弥补此缺点。半衰期为3.3~5.8小时,药物吸收后体内分布广泛。
洛美沙星(lomefloxacin)抗菌谱广,体外抗菌作用与诺氟沙星、氧氟沙星、氟罗沙星相似,但比环丙沙星弱;体内抗菌活性比诺氟沙星与氧氟沙星强,但不及氟罗沙星。本品口服吸收好,生物利用度为85%,血药浓度高而持久,半衰期约7小时,体内分布广,药物经肾排泄。
氟罗沙星(fleroxacin)又名多氟沙星,抗菌谱广,体外抗菌活性略逊于环丙沙星,但其体内抗菌活性强于现有各喹诺酮药。口服吸收好,生物利用度达99%。口服同剂量(400mg)的血药浓度比环丙沙星高2~3倍,半衰期为9小时。体内分布广,药物经肾排泄,约为给药量50%~60%。
表42-2 几种常用氟喹诺酮类药的药代动力学参数
| 药物 | 单次口服剂量(mg) | Cmax(mg/L) | t1/2kel(h) | 绝对生物利用度(%) | Vd(L) | 总清除率(L/h) | 原药尿中排泄率(%) | 粪便排泄率(%) |
| 诺氟沙星 | 400 | 1.58 | 3~4 | 35~45 | >100 | 51.6 | 25~30 | 28 |
| 培氟沙星 | 400 | 3.80 | 7.5~11 | 90~100 | 139 | 8.94 | 11 | |
| 依诺沙星 | 400 | 3.70 | 3.3~5.8 | 80~89 | 175 | 21.0 | 52 | 18 |
| 氧氟沙星 | 400 | 5.60 | 5.0~7.0 | 85~95 | 120 | 12.84 | 70~80 | 4 |
| 环丙沙星 | 500 | 2.56 | 3.3~4.9 | 38~60 | 307 | 39.12 | 29~44 | 15 |
三、应用注意事项
1.对幼年动物可引起软骨组织损害,故不宜用于妊娠期妇女和骨骼系统未发育完全的小儿。药物可分泌于乳汁,乳妇应用时应停止哺乳。
2.可引起中枢神经系统不良反应,不宜用于有中枢神经系统病史者,尤其是有癫痫病史的患者。
3.可抑制茶碱类、咖啡因和口服抗凝血药在肝中代谢,使上述药物浓度升高引起不良反应。产生上述相互作用最显著者为依诺沙星、其次为环丙沙星与培氟沙星,氧氟沙星无明显影响。因此应避免与有相互作用的药物合用,如有指征合用时,应对有关药物进行必要的监测。
4.吸收,宜避免合用。
5.肾功能减退者应用主要经肾排的药物如氧氟沙星和依诺沙星应减量。
第二节 磺胺类药
磺胺类药是三十年代发现的能有效防治全身性细菌性感染的第一类化疗药物。在临床上现已大部被抗生素及喹诺酮类药取代,但由于磺胺药有对某些感染性疾病(如流脑、鼠疫)、具有疗效良好,使用方便、性质稳定、价格低廉等优点,故在抗感染的药物中仍占一定地位。磺胺类药与磺胺增效剂甲氧苄啶合用,使疗效明显增强,抗菌范围增大。
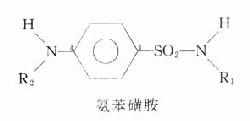
【结构和分类】磺胺类药是人工合成的氨苯磺胺衍生物。氨苯磺胺分子中的磺酰胺基上一个氢原子(R1)被杂环取代可得到口服易吸收的,用于全身性感染的磺胺药如磺胺嘧啶,磺胺异噁唑,磺胺甲噁唑等。如将氨苯碘胺分子中的对位氨基上一个氢原子(R2)取代则可得到口服难吸收的,用于肠道感染的磺胺药如柳氮磺胺吡啶等等。此外,还有外用磺胺药如磺胺嘧啶银等。
【药理学特点】①抗菌谱广,对金葡菌、溶血性链球菌、脑膜炎球菌,志贺菌属,大肠杆菌、伤寒杆菌,产气杆菌及变形杆菌等有良好抗菌活性,此外对少数真菌,衣原体、原虫(疟原虫和弓形体也有效);②细菌对各种磺胺药间有交叉耐药性;③磺胺药中有可供局部应用,肠道不易吸收及口服易吸收者,后者吸收完全血药浓度高,组织分布广;④磺胺嘧啶(SD)、磺胺甲噁唑(SMZ)脑膜通透性好,脑脊液内药物浓度高;⑤主要经肝代谢灭活,形成乙酰化物后溶解度低,易引起血尿,结晶尿及肾脏损害;⑥不良反应较多,常见有恶心、呕吐、皮疹、发热、溶血性贫血,粒细胞减少,肝脏损害、肾损害等。
【作用机制】磺胺药是抑菌药,它通过干扰细菌的叶酸代谢而抑制细菌的生长繁殖。与人和哺乳动物细胞不同,对磺胺药敏感的细菌不能直接利用周围环境中的叶酸,只能利用对氨苯甲酸(PABA)和二氢蝶啶,在细菌体内经二氢叶酸合成酶的催化合成二氢叶酸,再经二氢叶酸还原酶的作用形成四氢叶酸。四氢叶酸的活化型是一碳单位的传递体,在嘌呤和嘧啶核苷酸形成过程中起着重要的传递作用。磺胺药的结构和PABA相似,因而可与PABA竞争二氢叶酸合成酶,障碍二氢叶酸的合成,从而影响核酸的生成,抑制细菌生长繁殖。
【各种磺胺药特点】
1.用于全身性感染的磺胺药 口服易吸收的磺胺药,可用于治疗全身感染,根据血浆t1/2长短将药物分为三类:短效类(<10小时)、中效类(10~24小时)和长效类(>24小时)。短效和中效磺胺药抗菌力强,血中或其他体液中浓度高,临床最为常用;长效磺胺药抗菌力弱,血药浓度低,且过敏反应多见,许多国家已淘汰不用。
磺胺异噁唑(sulfafurazole,sulfisoxazole,SIZ)又名菌得清,是短效磺胺药,血浆t1/2为5~7小时,乙酰化率较低。尿中浓度最高,可达1000~2000mg/L,适于治疗尿路感染。在尿中不易析出结晶。每日需服药4次,消化道反应多见。
磺胺嘧啶(sulfadiazine,SD)中效磺胺药,口服易吸收,给药后3~4小时,血药浓度达峰值,血浆t1/2为10~13小时。抗菌力强,血浆蛋白结合率最低约25%,易透过血脑屏障,脑脊液浓度可达血浆浓度的40%~80%。是治疗流行性脑脊髓膜炎的首选药物,也适用于治疗尿路感染。但在尿中易析出结晶,需注意对肾的损害。
磺胺甲噁唑(sulfamethoxazole,sinomin,SMZ)又名新诺明,是中效磺胺药,血浆t1/2为10~12小时。抗菌作用与SIZ相似。蛋白结合率较高(60%~80%),脑脊液浓度不及SD,尿中浓度虽低于SIZ但与SD接近,故也适用于治疗尿路感染。在酸性尿液中可析出结晶而损害肾,需注意碱化尿液。
磺胺甲氧嘧啶(sulfamethoxydiazine,SMD)是长效磺胺药,血浆t1/2为30~40小时。抗菌力较弱。在体内维持时间较长,可每日服药一次。乙酰化率低,尿中溶解度高,不易析出结晶。
磺胺多辛(sulfadoxine,SDM′)又名周效磺胺;是长效磺胺药,血浆t1/2为150~200小时。在体内维持时间最长,可每3~7日服药一次。抗菌力较弱,适于轻症感染及预防链球菌感染,对疟疾等也有效。
2.用于肠道感染的磺胺药
柳氮磺吡啶(sulfasalazine,salicylazosulfapyridine)口服吸收较少,对结缔组织有特殊的亲和力并从肠壁结缔组织中释放出磺胺吡啶而起抗菌、抗炎和免疫抑制作用。适于治疗非特异性结肠炎,长期服用可防止发作。由于疗程长,易发生恶心,呕吐皮疹及药热等反应。
3.外用磺胺药
磺胺嘧啶银(sulfadiazinesilver)能发挥SD及硝酸银两者的抗菌作用,抗菌谱广,对绿脓杆菌抑制作用强大,尚有收敛作用,能促进创面的愈合,适用于二度或三度烧伤。
磺胺米隆(sulfamylon,SML)又名甲磺灭脓,是对位氨甲基磺胺药物,因此其抗菌作用不受脓液和坏死组织的影响。对绿脓杆菌、金葡菌及破伤风杆菌有效。能迅速渗入创面及焦痂中,并能促进创面上皮生长愈合及提高植皮成活率。适用于烧伤和大面积创伤后感染。
磺胺醋酰(sulfacetamide,SA)其钠盐水溶液(15%~30%)接近中性,局部应用几乎无刺激性,穿透力强。用于治疗沙眼,结膜炎和角膜炎等。
第三节 其他合成抗菌药
一、甲氧苄啶
甲氧苄啶(trimethoprim,TMP)又名磺胺增效剂,抗菌谱和磺胺药相似,但抗菌作用较强,对多种革兰阳性和阴性细菌有效。最低抑菌浓度常低于10mg/L。单用易引起细菌耐药性。
TMP的抗菌作用机制是抑制细菌二氢叶酸还原酶,使二氢叶酸不能还原成氢叶酸,阻止细菌核酸的合成。因此,它与磺胺药合用,可使细菌的叶酸代谢遭到双重阻断,增强磺胺药的抗菌作用达数倍至数十倍,甚至出现杀菌作用,而且可减少耐药菌株的产生,对磺胺药已耐药的菌株也可被抑制。TMP还可增强多种抗生素(如四环素、庆大霉素等)的抗菌作用。
口服吸收迅速而完全,血浆浓度高峰常在服药后1~2小时内达到。迅速分布全身组织及体液,肺、肾和痰液中。大部分以原形由肾排泄,尿中浓度约高出血浆浓度100倍,血浆t1/2约为10小时,和SMZ相近。
TMP常与SMZ或SD合用,治疗呼吸道感染、尿路感染、肠道感染和脑膜炎、败血症等。对伤寒、副伤寒疗效不低于氨苄西林,也可与长效磺胺药合用于耐药恶性疟的防治。
TMP毒性较小,不致引起叶酸缺乏症。大剂量(0.5g/日以上)长期用药可致轻度可逆性血象变化如白细胞减少、巨幼红细胞性贫血,必要时可注射四氢叶酸治疗。
二、硝基呋喃类药
本类药物抗菌谱广,且不易产生耐药性,对多种细菌的抑菌浓度为5~10mg/L,主要用于治疗尿路感染。
呋喃妥因(nitrofurantoin)又名呋喃坦啶(furadantin),对大肠杆菌、金葡萄、表葡萄、腐生葡萄球菌和肠球菌属均具抗菌作用。口服吸收迅速而完全。在体内约50%很快被组织破坏,其余以原形迅速自肾排出。血浆t1/2约20分钟。血药浓度很低,不适用于全身感染的治疗。但尿中浓度高,一般剂量下可达50~250mg/L以上。主要用于敏感菌所致铁急性肾炎、肾盂肾炎、膀胱炎、前列腺炎、尿道炎等尿路感染。酸化尿液可增强其抗菌活性。消化道反应较常见。剂量过大或肾功能不全者可引起严重的周围神经炎。偶见过敏反应。
呋喃唑酮(furazolidone)又名痢特灵,体外对沙门菌属、志贺菌属、大肠杆菌、肠杆菌属、金葡菌、粪肠球菌、霍乱弧菌和弯曲菌属均有抗菌作用。口服吸收少(5%),肠内浓度高,主要用于肠炎和菌痢。也可用于尿路感染、伤寒、副伤寒和霍乱。国内也曾试治溃疡病。不良反应同呋喃妥因。
人工合成抗菌药 制剂与用法
吡哌酸(pipemidicacid,PPA)成人1.5~2g/日,分2~4次服;儿童30~40mg/kg/日,分3次服。
■[此处缺少一些内容]■
lfamethoxazole,sinomin,SMZ,新诺明)成人1g/次,首剂加倍,2次/日,儿童25mg/kg/次,2次/日。复方新诺明片,每片含SMZ0.1g,TMP0.02g。成人2片/次,2次/日。
磺胺多辛(sulfadoxine,sulfadimoxine,SDM′,周效磺胺)0.5~1.0g/次,首剂1g,每3~7日服药1次。
柳氮磺吡啶(salicylazosulfapyridine,SASP)1~1.5g/次,3~4次/日,症状好转后减为0.5g/次。
磺胺醋酰钠(sulfacetamidesodium,SA)用10%~30%水溶液,滴眼用。
磺胺米隆(sulfamylone,SML,甲磺灭脓)5%~10%溶液湿敷或5~10%软膏涂敷,或用其散剂撒布。
磺胺嘧啶银(sulfadiazinesiliver,SD-Ag)用1%~2%软膏或乳膏涂敷创面,也可用乳膏油纱布包扎创面。
甲氧苄啶(trimethoprim,TMP,磺胺增效剂)0.1~0.2g/次,2次/日,小儿5~10mg/kg/日,分2次服用。
呋喃妥因(nitrofurantoin)成人0.05~0.1g/次,4次/日;儿童5~10mg/kg/日,分4次服,连续服用不宜超过两周。
呋喃唑酮(furazolidone,痢特灵)成人0.1g/次,3~4次/日;儿童5~10mg/kg/日,分4次服,5~7天为一疗程。
第四十三章 抗真菌药及抗病毒药
第一节 抗真菌药
真菌感染可分为浅部和深部感染两类。前者常由各种癣菌引起,主要侵犯皮肤、毛发、指(趾)甲等,发病率高,治疗药物有灰黄霉素、制霉菌素或局部应用的咪康唑和克霉唑。深部感染常由白色念珠菌和新型隐球菌引起,主要侵犯内脏器官和深部组织,发病率虽低,但危害性大,常可危及生命,治疗药物有两性霉素B及咪唑类抗真菌药等。
一、灰黄霉素
灰黄霉素(griseofulvin)为抗浅表真菌抗生素。
【抗菌作用】对各种皮肤癣菌(表皮癣菌属、小孢子菌属和毛癣菌属)有较强的抑制作用,但对深部真菌和细菌无效。其化学结构类似鸟嘌呤,故能竞争性抑制鸟嘌呤进入DNA分子中,从而干扰真菌核酸合成,抑制其生长。
【体内过程】口服易吸收,吸收量与颗粒大小有关,极微颗粒(2.7μm)比细颗粒(10μm)吸收完全,t1/2约14小时.油脂食物能促进药物吸收。吸收后,分布全身,以脂肪、皮肤、毛发等组织含量较高,能掺入并贮存在皮肤角质层和新生的毛发、指(趾)甲角质部分。大部分在肝代谢为6-去甲基灰黄霉素而灭活。原形药自尿排泄不足1%,约16%-36%随粪便排泄。
【临床应用】主要用于治疗上述真菌所致的头癣、体癣、股癣、甲癣等。与巴比妥类药(酶诱导剂)合用可加速在肝灭活,减弱药效;本品可促进抗凝药代谢,使后者的作用降低,故不宜与上述药物合用。
【不良反应】常见有恶心、腹泻、皮疹、头痛、白细胞减少等。
二、两性霉素B
两性霉素B(amphotericin B)是多烯类抗深部真菌药。国产庐山霉素与本药是同一物质。
【抗菌作用】对多种深部真菌如新型隐球菌、白色念珠菌、皮炎芽生菌及组织胞浆菌等,有强大抑制作用,高浓度有杀菌作用。它能选择性地与真菌细胞膜的麦角固醇相结合形成孔道,从而增加膜的通透性,导致细胞内重要物质外漏而致死。细菌的细胞膜不含固醇类物质,故本品对细菌无效。
【体内过程】口服、肌内注射均难吸收,一次静脉滴注,有效浓度可维持24小时以上。它不易透过血脑屏障,体内消除缓慢,每日约2%~5%以原形从尿排出,停药2周后仍可从尿中检出。血浆t1/2约24小时。
【临床应用】主要用于治疗全身性深部真菌感染。治疗真菌性脑膜炎时,需加用小剂量鞘内注射,其疗效良好。
【不良反应】静脉滴注不良反应较多,最常见的是滴注开始或滴注后数小时可发生寒战、高热、头痛、恶心和呕吐。其肾毒性呈剂量依赖性,约80%患者发生氮血症,与氨基甙类、环孢素合用肾毒性增加。应用时应注意:①静脉滴注液应新鲜配制,滴注前常需给病人服用解热镇痛药和抗组织胺药,滴注液中加生理量的氢化可的松或地塞米松可以减轻反应。②定期作用血钾、血尿常规、肝肾功能和心电图检查。
三、制霉菌素
制霉菌素(nystatin)也属多烯抗真菌药,其体内过程和抗菌作用与两性霉素B基本相同,但毒性更大,不作注射用。口服用于防治消化道念珠菌病,局部用药对口腔、皮肤、阴道念珠菌病有效。较大剂量口服可致恶心、呕吐、腹泻。局部用药刺激性小,个别阴道用药可见白带增多。
四、咪唑类抗真菌药
咪唑类(imidazoles)为合成的抗真菌药。抗菌作用与两性霉素相似,它能选择性抑制真菌细胞色素P-450依赖性的14-α-去甲基酶,使14-α-甲基固醇蓄积,细胞膜麦角固醇不能合成,使细胞膜通透性改变,导致胞内重要物质丢失而使真菌死亡。本类药物在肝脏代谢,主要经胆汁排出,在肾功能不全时不需改变剂量。主要毒性为贫血、胃肠道反应、皮疹等。
1.克霉唑(clotrimazole)对大多数真菌具有抗菌作用,对深部真菌作用不及两性霉素B。口服吸收差,一次服3g的血药峰浓度仅1.29mg/L,t1/2为3.5~5.5小时。连续给药由于肝药酶诱导作用可使血药浓度降低。不良反应多见,目前仅局部用于治疗浅部真菌病或皮肤粘膜的念珠菌感染。
2.咪康唑(miconazole)抗菌谱和抗菌力与克霉唑基本相同。口服吸收差,生物利用度约25%~30%,且不易透过血脑屏障,t1/2约24小时。静脉给药用于治疗多种深部真菌病。在两性霉素B不能耐受时,作为替代药。局部用药治疗皮肤粘膜真菌感染,疗效优于克霉唑和制霉菌素。静脉给药可致血栓静脉炎,此外,还有恶心、呕吐、过敏反应等。临床应用的具体剂量应随病原真菌而异。
3.酮康唑(ketoconazole)是广谱抗真菌药。对念珠菌和表浅癣菌有强大抗菌力。口服易吸收,血浆蛋白结合率达80%以上,不易透过血脑屏障,血浆t1/2为7~8小时。口服治疗多种浅部真菌病的疗效至少相当于或优于灰黄霉素、两性霉素B和咪康唑。酮康唑在酸性溶液中溶解吸收,因此不能与抗酸药、胆碱受体阻断药及H2受体阻断药同服,必要时至少相隔2小时。老年人胃酸缺乏,应将药片溶于4ml的稀盐酸中服下。不良反应有胃肠道反应,血清转氨酶升同,偶有严重肝毒性及过敏反应等。
4.氟康唑(fluconazole)广谱抗真菌药,抗菌谱与酮康唑相近似,体外抗真菌作用不及酮康唑,但其体内抗真菌作用比酮康唑强10-20倍。口服吸收后,生物利用度达90%,口服150mg于1.5~2.0小时达峰浓度3.8mg/L,蛋白结合率低,体内分布广,可渗入脑脊液,体内代谢甚少,约63%以原形由尿排出,血浆t1/2约30小时。本品可供口服及注射用。主要用于念珠菌病与隐球菌病。不良反应在本类药中最低,有轻度消化系统反应,过敏反应,头痛、头晕、失眠。
五、氟胞嘧啶
氟胞嘧啶(flurocytosine)对隐球菌、念珠菌和拟酵母菌等具有较高的抗菌活性,对着色真菌、少数曲菌有一定抗菌活性,对其他真菌和细菌作用均差。本品为抑菌剂,高浓度时具有杀菌作用,作用机制为药物通过真菌细胞的渗透系统进入细胞内,转换为氟尿嘧啶,替代尿嘧啶进入真菌的DNA中,从而阻断核酸合成。本品口服吸收良好,3~4小时血浓度达峰值,血中t1/2为8~12小时,可透过血脑屏障。临床上用于念珠菌和隐球菌感染,单用效果不如两性霉素B,且易产生耐药性,与两性霉素合用本品进入真菌细胞增多发挥协同作用。不良反应有胃肠道反应,一过性转氨酶升高,碱性磷酸酶升高,白细胞、血小板减少。
第二节 抗病毒药
病毒寄生于宿主细胞内,依赖宿主细胞代谢系统进行增殖复制。在病毒基因提供的遗传信息调控下合成病毒核酸和蛋白质,然后在胞浆内装配为成熟的感染性病毒体,以各种方式自细胞释出而感染其他细胞。
抗病毒感染的途径很多,如直接抑制或杀灭病毒、干扰病毒吸附、阻止病毒穿入细胞、抑制病毒生物合成、抑制病毒释放或增强宿主抗病毒能力等。常用抗病毒药有:
一、金刚烷胺
金刚烷胺(amantadine)能特异性地抑制甲型流感病毒,干扰RNA病毒穿入宿主细胞,它还能抑制病毒脱壳及核酸的释放,可用于甲型流感的防治,但对乙型流感病毒、麻疹病毒、腮腺炎病毒和单纯疱疹病毒(HSV)无效。它还能抗震颤麻痹(见第十六章)。口服易吸收,在体内不被代谢,约90%以原形自尿排出,血浆t1/2约12~17小时。不良反应有厌食、恶心、头痛、眩晕、失眠、共济失调等。
二、碘苷
碘苷(idoxuridine)又名疱疹净,竞争性抑制胸苷酸合成酶,使DNA合成受阻,故能抑制DNA病毒,如HSV和牛痘病毒的生长,对RNA病毒无效。本品全身应用毒性大,临床仅限于局部用药,以治疗眼部或皮肤疱疹病毒和牛痘病毒的感染,对急性上皮型疱疹性角膜炎疗效最好,对慢性溃疡性实质层疱疹性角膜炎疗效很差,对疱疹性角膜虹膜炎无效。局部反应有痛、痒,结膜炎和水肿等。
三、阿昔洛韦
阿昔洛韦(acyclovir)又名无环鸟苷,是核苷类抗DNA病毒药。抗疱疹病毒作用比碘苷强10倍,比阿糖腺苷强160倍。对乙型肝炎病毒也有一定作用。对牛痘病毒和RNA病毒无效。它在感染细胞内经病毒胸苷激酶和细胞激酶催化,生成三磷酸无环鸟苷,抑制病毒DNA多聚酶。口服吸收差,生物利用度为15%~30%,血浆t1/2约3小时。血浆蛋白结合率很低,易透过生物膜。药物部分经肝代谢,主要以原型自肾排出。本品适用于HSV所致的各种感染,带状疱疹,EB病毒,艾滋病患者并发水痘带状疱疹等,局部滴眼治疗单纯性疱疹性角膜炎或用霜剂治疗带状疱疹等疗效均佳,不良反应较少。
四、阿糖腺苷
阿糖腺苷(adenine arabinoside,Ara~A)为核苷类抗DNA病毒药,能抑制DNA复制,对疱疹病毒与痘病毒均有作用。静脉滴注t1/2为3~4小时,脑脊液中药物浓度约为血药浓度的35%,主要经肾排出。临床用于治疗HSV脑炎、角膜炎、新生儿单纯疱疹,艾滋病患者合并带状疱疹等。静滴可出现消化道反应及血栓静脉炎。偶见血清转氨酶升高。
五、利巴韦林
利巴韦林(ribavirin)又名病毒唑(virazole),为核苷、次黄嘌呤核苷类似物,能抑制病毒核酸的合成,具广谱抗病毒性能,对RNA和DNA病毒均有抑制作用。对甲、乙型流感病毒、腺病毒肺炎、甲型肝炎、疱疹、麻疹等均有防治作用。口服吸收良好,1~1.5小时后血药浓度达到峰值,t1/2为20小时,主要由肾排出。
抗真菌药及抗病毒药 制剂及用法
灰黄霉素(griseofulvin)成人500~600mg/日,儿童10~15mg/kg/日,分2~4次口服。滴丸(固体分散物),剂量减半。疗程10~14日。
两性霉素B(amphotericin B)静脉滴注时溶于5%葡萄糖液中,稀释为0.1mg/ml,必要时可在滴注液中加入地塞米松。成人与儿童剂量均按体重计算.从0.1mg/kg/日开始,逐渐增至1mg/kg/日为止,可每日或隔日给药1次,药液宜避光缓慢滴入。鞘内注射:首次0.1~0.2mg,渐增至0.5~1.0mg/次,浓度不超过0.3mg/ml,应与地塞米松合用。
制霉菌素(nystatin)成人50万~100万单位/次,4次/日,儿童酌减。此外,尚有软膏、阴道栓剂、混悬剂供局部用。
克霉唑(clotrimazole)成人0.5~1.0g/次,3次/日;儿童20~60mg/kg/日,分3次服。软膏、栓剂、霜剂可供外用。
咪康唑(miconazole)成人静脉滴注200~400mg,每8小时1次,最大剂量不宜超过每日30mg/kg或2g。药物稀释于生理盐水或5%葡萄糖液200ml中,于30~60分钟内静脉滴注。鞘内注射成人最大量每次为20mg。
酮康唑(ketoconazole)成人200mg/次,1次/日,必要时剂量可加大至600mg/次/日。疗程根据真菌感染的性质而定,可达5~6月以上。儿童15kg以下20mg/次,3次/日;15~30kg为100mg/次,1次/日。
氟康唑(fluconazole)胶囊剂(或片剂)50、100、150mg/粒,每日1次,每次50或100mg,必要时150或300mg/日。注射剂100mg/50ml静滴,100~200mg/日。
氟胞嘧啶(flurocytosine)片剂,每片250mg、500mg,每日100~150mg/kg,分3~4次服,疗程自数周至数月。
盐酸金刚烷胺(amantadine hydrochloride)成人早、晚各服1次,0.1g/次。儿童酌减,可连用3~5日,至多不超过10日。
碘苷(idoxuridine,疱疹净)治疗疱疹性角膜炎:白天每小时滴眼1次,夜间2小时1次,症状显著改善后,改为白天每2小时1次,夜间4小时一次。
阿昔洛韦(acyclovir,无环鸟苷)成人口服每次200mg,每4小时1次,静滴,每次5mg/kg,加入输液中,1小时内滴完,每8小时1次,疗程7天。另有眼膏、霜剂供外用。
利巴韦林(ribavirin,病毒唑)口服0.8~1.0g/日,分3~4次服用,肌注或静脉滴注每支100mg/kg/日,分三次给予,静滴宜缓慢。滴眼液为0.1%,滴鼻液为0.5%。
第四十四章 抗结核病药及抗麻风病药
第一节 抗结核病药
抗结核药(antituberculous drugs)中疗效高、不良反应少、患者较易接受的如异烟肼、利福平、乙胺丁醇、吡嗪酰胺、链霉素等,列为“一线药”。其余为“二线药”,如对氨基水杨酸、丙硫异烟胺、卡那霉素等,抗菌作用弱,毒性较大,仅用于细菌对“一线药”耐药时。
一、各类抗结核病药
异烟肼
异烟肼(isoniazid,INH)又名雷米封(rimifon)。性质稳定,易溶于水。具有疗效高、毒性小、口服方便、价廉等优点
【抗菌作用】异烟肼对结核杆菌有高度选择性,抗菌力强,在试管中0.025~0.05mg/L的浓度即可抑菌,较高浓度对繁殖期细菌有杀菌作用。单用时结核杆菌易产生耐药性,但与其他抗结核药无交叉耐药性,如与其他抗结核病联用,则能延缓耐药性的发生并增强疗效。抗菌机制可能是抑制分枝菌酸(mycolic acid)的合成,使细菌丧失耐酸性、疏水性和增殖力而死亡。此酸是结核杆菌细胞所特有的重要成分,因此异烟肼对其他细菌无作用。
【体内过程】口服吸收快而完全,1~2小时后血药浓度达高峰。吸收后广泛分布于全身体液和组织中,当脑膜发炎时,脑脊液中的浓度可与血浆浓度相近。穿透力强,可渗入关节腔,胸、腹水以及纤维化或干酪化的结核病灶中,也易透入细胞内,作用于已被吞噬的结核杆菌。异烟肼大部分在肝中被代谢为乙酰异烟肼、异烟酸等,最后与少量原形药一起由肾排出,异烟肼乙酰化的速度有明显的人种和个体差异。分为快代谢和慢代谢型,前者尿中乙酰化异烟肼较多,后者尿中游离异烟肼较多。慢型者在白种人中占50%~60%,在中国人中慢代谢型约占25.6%,快代谢型者约占49.3%。慢代谢型者肝中缺少乙酰化酶,服药后异烟肼血药浓度较高,t1/2延长,显效较快。快、慢代谢型的t1/2分别为0.5~1.5小时与2~3小时。
【临床应用】适用于各种类型的结核病,除早期轻症肺结核或预防应用外,均宜与其他第一线药联合应用。对急性粟粒性结核和结核性脑膜炎应增大剂量,必要时采用静脉滴注。
【不良反应】发生率与剂量有关,治疗量时不良反应少而轻。
1.神经系统毒性 周围神经炎系继发于维生素B6缺乏,多见于营养不良及慢乙酰化型患者,表现为手、脚震颤、麻木,同服维生素B6可治疗及预防此反应。中枢神经系统毒性反应常因用药过量所致,出现昏迷、惊厥、神经错乱,偶见有中毒性脑病或中毒性精神病。因而有癫痫、嗜酒、精神病史者慎用。其发生可能与维生素B6的利用降低有关,因此时抑制性递质GABA生成减少。
2.肝毒性 以35岁以上及快代谢型患者较多见,可有暂时性转氨酶值升高。用药时应定期检查肝功,肝病患者慎用。
利福平
利福平(rifampicin)又名甲哌力复霉素(rifampin),简称RFP,是人工半合成的力复霉素类衍生物,为砖红色结晶性粉末。具有高效低毒、口服方便等优点。
【抗菌作用】利福平有广谱抗菌作用,对结核杆菌、麻风杆菌和革兰阳性球菌特别是耐药性金葡菌都有很强的抗菌作用,对革兰阴性菌、某些病毒和沙眼衣原体也有抑制作用。对结核杆菌的最低抑菌浓度平均为0.018mg/L,口服治疗量后血药浓度为此浓度的100倍,故可发挥杀菌作用。抗结核作用与异烟肼相近,而较链霉素强。结核杆菌对利福平易产生耐药性,故不宜单用。与异烟肼、乙胺丁醇等合用有协同作用,并能延缓耐药性的产生。利福平的抗菌机制是特异性地抑制细菌依赖于DNA的RNA多聚酶,阻碍mRNA合成,对动物细胞的RNA多聚酶则无影响。
【体内过程】 中服吸收迅速而完全,1~2小时血药浓度达峰值,但个体差异很大。食物可减少吸收,故应空腹服药。t1/2约为4小时,有效血药浓度可维持8~12小时。吸收后分布于全身各组织,穿透力强,能进入细胞、结核空洞、痰液及胎儿体内。脑膜炎时,脑脊液中浓度可达血浓度的20%。主要在肝内代谢成去乙酰基利福平,其抑菌作用约为利福平的1/8~1/10。重复口服利福平可透导肝药酶,加快自身及其他药物的代谢。主要从胆汁排泄,形成肝肠循环,约60%经粪与尿排泄,患者的尿、粪、泪液、痰等均可染成桔红色。
【临床应用】主要与其他结核病药合用,治疗各种结核病及重症患者。对耐药性金葡菌及其他细菌所致的感染也有效。还用于治疗麻风病。
【不良反应】较常见的为胃肠道刺激症状;少数病人可见肝脏损害而出现黄疸,有肝病或与异烟肼合用时较易发生。过敏反应如皮疹、药热、血小板和白细胞减少等多见于间歇疗法,出现过敏反应时应停药。利福平可激活肝微粒体酶,加速皮质激素和雌激素等的代谢,因而它能降低肾上腺皮质激素、口服避孕药、双香豆素和甲苯磺丁脲等的作用。对动物有致畸胎作用;妊娠早期的妇女和肝功能不良者慎用。
利福喷汀与利福定
利福喷汀(rifapentine)和利福定(rifandine)均为利福霉素衍生物。它们的抗菌谱和利福平相同,抗菌效力分别比利福平强8倍与3倍以上,与其他抗结核药,如异烟肼、乙胺丁醇等有协同抗菌作用。此外,它们对革兰阳性与阴性菌也有强大的抗菌活性。利福喷汀(微晶)与利福定的t1/2分别为30与5小时。利福定的治疗剂量为利福平的1/2~1/3,利福喷汀剂量与利福平相同,每周用药1~2次。
乙胺丁醇
乙胺丁醇(ethambutol)现作为一线药应用。
【抗菌作用】乙胺丁醇过去列为抑菌药,近年发现对细菌内、外结核杆菌有较强杀菌作用。对链霉素或异烟肼等有耐药性的结核杆菌,本药仍有效。主要与利福平或异烟肼等合用。单用也可产生耐药性,但较缓慢。抗菌机制可能是与二价金属离子如Mg2+结合,干扰菌体RNA的合成。
【体内过程】 口服吸收良好,迅速分布于组织与体液,2小时血药浓度达峰值,t1/2为8小时,排泄缓慢,24小时内尿排口服量的50%,肾功能不全时可引起蓄积中毒,宜禁用。
【不良反应】视神经炎是最重要的毒性反应,多发生在服药后2~6月内,表现为视力下降、视野缩小,出现中央及周围盲点。反应发生率与剂量、疗程有关,早日发现及时停药,数周至数月可自行消失。此外有胃肠道不适,恶心、呕吐及肝功能损害等。
吡嗪酰胺
吡嗪酰胺(pyrazinamide)口服迅速吸收,分布于各组织与体液,2小时血药浓度达峰值,t1/2为6小时,经肝代谢为吡嗪酸,约70%经尿排泄。酸性环境中抗菌作用增强,故能在细胞内有效杀灭结核杆菌。结核菌对吡嗪酰胺易产生耐药性,但与其他抗结核药无交叉耐药。它已列为抗结核病基本药在短程化疗中应用。过去高剂量、长疗程应用常见肝毒性与关节痛等不良反应,现用低剂量、短程疗法,不良反应已明显减少。
对氨水杨酸
对氨水杨酸(para-aminosalicylic acid,PAS)其钠盐和钙盐,口服吸收快而完全。分布于全身组织、体液及干酪样病灶中,但不易透入脑脊液及细胞内。对结核杆菌只有抑菌作用,引起耐药性缓慢,与其他抗结核病药合用,可以延缓耐药性的发生。最常见的不良反应为恶心、呕吐、厌食、腹痛及腹泄。饭后服药或加服抗酸药可以减轻反应。
二、抗结核药的治疗原则
1.早期用药 早期病灶内结核菌生长旺盛,对药物敏感,同时病灶部位血液供应丰富,药物易于渗入病灶内,达到高浓度,可获良好疗效。
2.联合用药 联合用药可提高疗效、降低毒性、延缓耐药性,并可交叉消灭对其他药物耐药的菌株,使不致成为优势菌造成治疗失败或复发。联合用药二联、三联或四联则取决于疾病的严重程度,以往用药情况以及结核杆菌对药物的敏感性。
3.短期疗法 短期疗法(6~9个月)是一种强化疗法,疗效好,目前已广泛采用。主要是利福平和异烟肼联合,大多用于单纯性结核的初治。如病灶广泛,病情严重则应采用三联甚至四联。目前常用的有:最初两个月每日给于异烟肼,利福平与吡嗪酰胺,以后四个月每日给于异烟肼和利福平(即2HRZ/4Hr 方案)。异烟肼耐药地区在上述三联与二联的基础上分别增加链霉素与乙胺丁醇(即2SHRZ/4HRe 方案)。对营养不良、恶性病而免疫功能低下者,宜用12个月疗程,对选药不当,不规则治疗或细菌产生耐药,可选用或增加二线药联合,复发而有合并症者,宜用18~24个月治疗方案。
通过新的药物治疗方案现已能做到治愈率高,复发率低(约1%),耐药菌少,疗程短(六个月),安全性高,使防治工作取得满意成效。
第二节 抗麻风病药
防治麻风病的药物主要为氨苯砜、利福平和氯法齐明等。目前多采用联合疗法。
一、砜类
本类药最常用的是氨苯砜(dapsone,DDS),此外,还有苯丙砜(phenprofen)醋氨苯砜(acedapsone),它们须在体内转化为氨苯砜或乙酰氨苯砜而显效。
【体内过程】氨苯砜口服吸收较慢,但吸收完全,口服100mg约4~8小时达到峰浓度,血中t1/2为28小时,有效抑菌浓度可持续约10天左右,蛋白结合率为50%,它分布全身,皮肤病变部位的浓度远高于正常部位。经肝乙酰化,并有肝肠循环,消除缓慢,70%~80%经尿排泄,故易蓄积,宜周期性地作短暂停药。苯丙砜较难吸收,用量较大。
【临床应用】砜类的抗菌机制和磺脑类相似,但对革兰阳性菌和阴性菌无抗菌活性,对麻风杆菌有较强的直接抑制作用。患者服用3~6月后,症状即可改善,粘膜病变好转,细菌逐渐消失,皮肤及神经损害的恢复,瘤型患者细菌消失则需要较长时间。麻风杆菌对砜类可产生耐药性,因而须采用联合疗法以减少或延缓耐药性的发生,减少复发和较快消除其传染性。对多菌型患者的联合疗法采用WHO推荐的方案为氨苯砜100mg/日自服,利福平及氯法齐明(见后)每月一次分别为600与300mg监服,疗程二年或查菌阴转后,再继续治疗一年并随访观察。少菌型麻风治疗方案为氨苯砜100mg/日自服,利福平600mg每月一次监服,疗程为六个月。
【不良反应】较常见为贫血,偶可引起急性溶血性贫血,G-6-PDH缺乏者尤易发生。有时出现胃肠刺激症状、头痛、失眠、中毒性精神病及过敏反应。剂量过大还可引起肝损害及剥脱性皮炎。治疗早期或增量过快,患者可发生麻风症状加剧的反应(麻风反应),一般认为是机体对菌体裂解产生的磷脂类颗粒的过敏反应,多认为是预后良好的现象。麻风反应可用沙利度胺(thalidomide,反应停)防治。其他处理方法是减量停药或暂改用另一些抗麻风药,并用肾上腺皮质激素进行治疗。
二、其他药物
1. 利福平 对麻风杆菌包括对氨苯砜耐药菌株有快速杀菌作用,用药数日至数周,菌体即碎裂呈粒变现象。临床应用600或1200mg后,在4天内即可杀灭99.9%的活菌,但仍需坚持长期治疗,单独使用易致耐药性。利福平是治疗麻风联合疗法中的必要组成药。利福霉素类均有类似的抗麻风作用,以利福平为最常用。
2. 氯法齐明(clofazimine)又名氯苯吩嗪,对麻风杆菌有抑制作用,其作用机制为干扰核酸代谢,抑制菌体蛋白合成,作用较氨苯砜缓慢。本品还能抑制麻风结节红斑反应。口服微粒晶体后吸收率为50%~70%,迅速分布于体内各组织中;组织药物浓度高于血药浓度;其消除半衰期为70天,本品为联合疗法药物之一,或作为抗麻风反应治疗药物。主要副作用为皮肤色素沉着等。
抗结核病药及抗麻风病药 制剂及用法
异烟肼(isoniazid,INH)300~400mg/日,分1~3次服。粟粒性结核、结核性脑膜炎、干酪性肺炎等重症应增加剂量至200mg/次,3次/日。儿童一般10~20mg/kg/日。注射剂量视病情而定,一般与口服量相同。可作肌内注射、腔内注射或用50%葡萄糖或等渗盐水稀释至0.1%静脉滴注(如用于结核性脑膜炎等)。
利福平(rifampicin;甲哌力复霉素rifampin)450~600mg/日,清晨空腹顿服。儿童20mg/kg/日。用于其他急性细菌感染,600mg/日,分2次早晚空腹服用。
利福喷汀(rifapentine)胶囊剂,每粒300mg,成人每次600mg,每周1~2次空腹服用。
利福定(rifandine,异丁基哌嗪力复霉素)成人每日150~200mg,晨一次空腹口服,儿童3~4mg/kg。
乙胺丁醇(ethambutol,myambutol)初治病例15mg/kg/日,1次或分2~3次服。复治病例25mg/kg/日,2月后减为15mg/日。
对氨水杨酸钠(sodium para-aminosalicylate)2~3g/次,4次/日。重症或口服不能耐受者,用静脉滴注。注射液应新鲜配制,避光条件下2小时内滴完。
链霉素(streptomycin)结核重症:0.75~1.0g/日,分2次肌内注射;轻症:1.0g/次,每周2~3次。小儿20~40mg/kg/日,不应超过1.0g/日。疗程3~6个月。
吡嗪酰胺(pyrazinamide)0.5g/次,3次/日。
氨苯砜(dapsone,DDS)12.5~100mg/日,1次服用,从小量开始,因有轻度蓄积性,每周服药6天,连服三月,停药2周。
苯丙砜(phenprofone)开始0.5g/日,以后渐增至2~3g/日,分次服用,也可每周服药6天,服药2.5~3月,停药半月。肌内注射,每周2次,第1周每次1ml,第2周每次2ml,第三周起每周3ml,10周为一疗程。
醋氨苯砜(acedapsone)每支300mg(2ml),肌注其油注射液,一次1.5~2ml,每60~70天一次,疗程数年。
氯法齐明(clofazimine)胶丸,每粒50mg,剂量与服法随联合疗法而异。对麻风反应开始每日3次,每次100mg,随后根据反应控制并和胃肠道反应逐渐减量至每日100mg。
第四十五章 抗疟药
第一节 概述
抗疟药(antimalarialdrugs)是防治疟疾的重要手段。现有抗疟药中尚无一种能对疟原虫生活史的各个环节都有杀灭作用。因此,必须了解各种抗疟药对疟原虫生活史的不同环节的作用,以便根据不同目的正确选择药物。
一、疟原虫的生活史和抗疟药的作用环节
使人致病的疟原虫主要有三种:恶性疟、间日疟和三日疟。后二者又称良性疟。
疟原虫的生活史可分为雌按蚊体内的有性生殖阶段和人体内的无性生殖阶段(见图45-1)。
图45-1 疟原虫生活史和各类抗疟药的作用部位
1.人体内的无性生殖阶段 可分为以下各期:
(1)原发性红细胞外期:受感染按蚊叮咬人时,将其唾液中的子孢子输入人体。要30分钟内子孢子即侵入肝细胞中开始其红细胞前期发育和裂体增殖。经过10~14天,生成大量裂殖子。此期不发生症状,为疟疾的潜伏期。对此期有杀灭作用的药物,如乙胺嘧啶,可起病因预防作用。
(2)红细胞内期:原发性红细胞外期在肝细胞内生成的大量裂殖子破坏肝细胞而进入血液,侵入红细胞,经滋养体发育成裂殖体,并破坏红细胞,释放出大量裂殖子及其代谢产物,再加上红细胞破坏产生的大量变性蛋白,至一定程度,刺激机体,引起寒战、高热等症状。从红细胞内逸出的裂殖子又复进入红细胞进行发育。如此周而复始,每完成一个无性生殖周期,引起一次症状发作。不同种的疟原虫完成无性生殖周期所需时间不同:恶性疟36~48小时,间日疟48小时,三日疟72小时。对此期疟原虫有杀灭作用的药物,如氯喹、奎宁、青蒿素等,有控制症状发作和症状抑制性预防作用。
(3)继发性红细胞外期:间日疟原虫在进行红细胞内期无性生殖时,在肝细胞内仍有疟原虫生长、发育。此时肝细胞内疟原虫的来源尚无定论。有认为来源于原发性红细胞外期生成的裂殖子。但近又证明,间日疟原虫的子孢子在遗传学上有不同的两个型,即速发型子孢子和迟发型子孢子。它们同时进入肝细胞,速发型子孢子完成原发性红细胞外期后,即全部由肝细胞释放,进入红细胞内期。而迟发型子孢子则在相当长的时间内处于休眠状态(称休眠子),然后才开始并完成其红细胞外期裂体增殖,并向血液释放裂殖子,引起间日疟复发。能杀灭继发性红细胞外期的药物,如伯氨喹,对间日疟有根治(阻止复发)作用。恶性疟和三日疟原虫无继发性红细胞外期,故无须用药进行根治。
2.雌按蚊体内的有性生殖阶段红细胞内期疟原虫一方面不断进行裂体增殖,同时也产生雌、雄配子体。按蚊在吸入血时,雌、雄配子体随血液进入蚊体。二者结合成合子,进一步发育产生子孢子,移行至唾液腺内,成为感染人的直接传染源。能抑制雌、雄配子体在蚊体内发育的药物,如乙胺嘧啶,则有控制疟疾传播和流行的作用。
二、疟原虫的耐药性
当前防治疟疾所遇到的最大困难是恶性疟原虫对抗疟药,特别是对氯喹,其次是对奎宁、乙胺嘧啶等产生耐药性。而且耐氯喹的虫株常对乙胺嘧啶和周效磺胺等有交叉耐药性。恶性疟是流行最广、对人类危害性最大的一种疾病。因此亟需寻找新型抗疟药。近国外报道钙拮抗药(包括粉防己碱)与氯喹合用,能部分恢复疟原虫对氯喹的敏感性。
第二节 常用抗疟药
一、主要用于控制症状的抗疟药
此类药物是主要杀灭红细胞内期疟原虫的药物。
氯喹
氯喹(chlorquine)是人工合成的4-氨喹啉类衍生物。
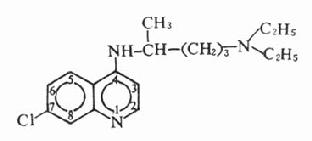
【药理作用和临床应用】
1.抗疟作用氯喹对间日疟和三日疟原虫,以及敏感的恶性疟原虫的红细胞内期的裂殖体有杀灭作用。能迅速治愈恶性疟;有效地控制间日疟的症状发作,也可用于症状抑制性预防。其特点是疗效高、生效快。多数病例在用药后24~48小时内发作停止,48~72小时内血中疟原虫消失。由于此药在体内代谢和排泄都很缓慢,加之在内脏组织中的分布量大,停药后可逐渐释放入血,故作用持久。用于间日疟,症状复发较迟。用于症状抑制性预防,每周服药一次即可。其本身毒性小,与伯氨喹合用时不增加后者的毒性。对红细胞外期无效,既不能作病因性预防,也不能根治间日疟。
氯喹抗疟作用机制复杂。应用氯喹后,疟原虫溶酶体内药物的含量高出宿主溶酶体千倍以上,由此认为疟原虫有浓集氯喹的特异机制。氯喹可插入DNA双螺旋链之间,形成DNA-氯喹复合物,影响DNA复制和RNA转录,并使RNA断裂,从而抑制疟原虫的分裂繁殖。再者,氯喹为弱碱性药物,大量进入疟原虫体内,必然使其细胞液的PH值增大,形成对蛋白质分解酶不利的环境,使疟原虫分解和利用血红蛋白的能力降低,导致必需氨基酸缺乏,也可干扰疟原虫的繁殖。疟原虫对氯喹耐药性的发生,可能与其从体内排出药物增多和代谢加速有关。
2.抗肠道外阿米巴病作用氯喹对阿米巴痢疾无效。但由于它在肝组织内分布的浓度比血药浓度高数百倍,对阿米巴肝脓肿有效。详见第四十六章。
3.其他作用氯喹偶而用于类风湿性关节炎,也常用于碟形红斑狼疮。但对后者的疗效尚无定论,而且用量大,易引起毒性反应。
【不良反应】氯喹用于治疗疟疾时,一般能良好耐受,仅有轻度头晕、头痛、胃肠不适和皮疹等,停药后迅速消失。大剂量、长疗程用药可引起视力障碍,以及对肝脏和肾脏的损害。
奎宁
奎宁(quinine)是从金鸡纳树皮中提得的一种生物碱。金鸡纳树原产南美洲,自古当地居民即用其树皮治疗疟疾。1820年分离出奎宁后,迅即用于临床,曾是治疗疟疾的主要药物。自合成氯喹等药后,奎宁已不作首选抗疟药用。但当今氯喹的耐药性问题日趋严重,因而奎宁又被重视。
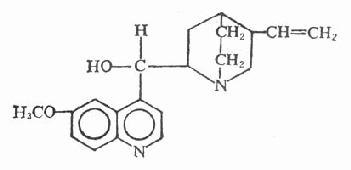
【抗疟作用和临床应用】奎宁对各种疟原虫的红细胞内期滋养体有杀灭作用,能控制临床症状。但疗效不及氯喹而毒性较大。主要用于耐氯喹或耐多药的恶性疟,尤其是严重的脑型疟。奎宁在肝内迅速氧化失活并由肾排出,加之毒性较大,因此不用于症状抑制性预防。对红细胞外期无效,对配子体亦无明显作用。
【不良反应】
1.金鸡纳反应表现为恶心、呕吐、耳鸣、头痛、听力和视力减弱,甚至发生暂时性耳聋。因为奎宁得自金鸡纳树皮,金鸡纳树的其他生物碱也有此反应,故称金鸡纳反应。
2.心肌抑制作用奎宁降低心肌收缩力、减慢传导和延长心肌不应期。但不及其D-异构体奎尼丁的作用明显。静脉注射时可致血压下降和致死性心律失常。用于危急病例时,仅可静脉滴注。
3.特异质反应少数恶性疟患者即使应用很小剂量也能引起急性溶血,发生寒战、高热、背痛、血红蛋白尿(黑尿)和急性肾功能衰竭,甚至死亡。
4.子宫兴奋作用 奎宁对妊娠子宫有兴奋作用,故孕妇忌用。
5.中枢神经抑制有微弱的解热镇痛作用。也可引起头晕、精神不振等症状。
甲氟喹
甲氟喹(mefloquine)和奎宁都属喹啉-甲醇衍生物。鉴于奎宁对耐多药虫株至少还保留部分抗疟作用,通过改变奎宁的结构而获得甲氟喹。
甲氟喹也是一种杀疟原虫红细胞内期滋养体的药物。用于控制症状,生效较慢。在某些地区已发现恶性疟对此药产生耐药性,但与奎宁和氯喹之间并无必然的交叉耐药关系。单独或与长效磺胺和乙胺嘧啶合用,对耐多药恶性疟虫株感染有一定疗效。
甲氟喹的另一特点是血浆半衰期较长(约30天),可能是其在体内有肝肠循环的缘故。用于症状抑制性预防,每2周给药一次。
青蒿素和蒿甲醚
青蒿素(artemisinin,qinhaosu)是从黄花蒿(Artemisia annua L.)及其变种大头黄花蒿中提取的一种倍半萜内酯过氧化物。是根据中医“青蒿截疟”的记载而发掘出的新型抗疟药。应将黄花蒿与同属另一植物青蒿(A.apiacea)相区别,后者不含青蒿素,无抗疟作用。由于对耐氯喹虫株感染有效,青蒿素受到国内、外广泛重视。
【抗疟作用和临床应用】青蒿素对红细胞内期滋养体有杀灭作用,对红细胞外期无效。用于治疗间日疟和恶性疟,即期症状控制率可达100%。与氯喹只有低度交叉耐药性,用于耐氯喹虫株感染仍有良好疗效。青蒿素可透过血脑屏障,对凶险的脑型疟疾有良好抢救效果。
青蒿素也可诱发耐药性,但比氯喹为慢。与周效磺胺或乙胺嘧啶合用,可延缓耐药性的发生。
青蒿素治疗疟疾最大的缺点是复发率高,口服给药时近期复发率可达30%以上。这可能与其在体内消除快,代谢产物无抗疟活性有关。与伯氨喹合用,可使复发率降至10%左右。
蒿甲醚(artemether)为青蒿素的12-β-甲基二氢衍生物。其溶解度较大,可制成澄明的油针剂注射给药。抗疟活性比青蒿素强,近期复发率比青蒿素低(8%),与伯氨喹合用,可进一步降低复发率。
【不良反应】不良反应少见,偶见四肢麻木感和心动过速。未见对重要内脏有损害作用。动物试验中应用大剂量时,曾见骨髓抑制和肝损害,并有胚胎毒性作用。与青蒿素相比,蒿甲醚的不良反应较轻。
二、主要用于控制复发和传播的药物
伯氨喹
伯氨喹(primaquine)是人工合成的8-氨喹啉类衍生物。
【药理作用和临床作用】伯氨喹主要对间日疟红细胞外期(或休眠子)和各种疟原虫的配子体有较强的杀灭作用,是根治间日疟和控制疟疾传播最有效的药物。对红细胞内期无效,不能控制疟疾症状的发作。通常均需与氯喹等合用。疟原虫对此药很少产生耐药性。
【不良反应】毒性较大是此药的一大缺点,但目前尚无适当药物可以取代之。治疗量即可引起头晕、恶心、呕吐、紫绀、腹痛等。停药后可消失。
严重的反应是少数特异质者发生的急性溶血性贫血和高铁血红蛋白血症。现已查明,此特异质的本质是红细胞内缺乏6-磷酸葡萄糖脱氢酶(G6PD),这是一种性联染色体遗传缺陷。因为伯氨喹的氧化代谢产物能引起氧化应激反应(oxidativestress),产生高铁血红蛋白、自由基和过氧化物,以及氧化型谷胱甘肽(GSSG)。正常时,在G6PD催化下,可迅速补充NADPH,后者使GSSG还原为谷胱甘肽(GSH)。GSH对红细胞膜、血红蛋白和红细胞内的某些含巯基的酶有保护作用,使之免受伯氨喹氧化代谢物引起的氧化应激反应的损害。但红细胞内缺乏G6PD的个体不能迅速补充NADPH,因此不能保护红细胞而发生溶血。另一方面,也不能将高铁血红蛋白还原为血红蛋白,引起高铁血红蛋白血症。
三、主要用于病因性预防的抗疟药
乙胺嘧啶
乙胺嘧啶(pyrimethamine)是目前用于病因性预防的首选药。
【药理作用和临床作用】乙胺嘧啶对恶性疟和间日疟某些虫株的原发性红细胞外期有抑制作用,用作病因预防药,作用持久,服药一次,预防作用可维持一周以上。对红细胞内期的未成熟裂殖体也有抑制作用,对已成熟的裂殖体则无效。用于控制耐氯喹株恶性疟的症状发作,生效较慢,常需在用药后第二个无性增殖期才能显效。此药并不能直接杀灭配子体,但含药血液随配子体被按蚊吸入后,能阻止疟原虫在蚊体内的孢子增殖,起控制传播的作用。
【作用机制和联合用药】疟原虫不能利用环境中的叶酸和四氢叶酸,必须自身合成叶酸并转变为四氢叶酸后,才能在合成核酸的过程中被利用。乙胺嘧啶对疟原虫的二氢叶酸还原酶有较大的亲和力,并能抑制其活性,从而阻止四氢叶酸的生成,阻碍核酸的合成。与二氢叶酸合成酶抑制剂磺胺类或砜类合用,在叶酸代谢的两个环节上起双重抑制作用,可收协同作用之效,且可延缓耐药性的发生。因此,此药常与半衰期相近的周效磺胺或氨苯砜合用。但近年已发现耐氯喹恶性疟原虫对乙胺嘧啶–周效磺胺合剂有交叉耐药性。
【不良反应】治疗量时基本上不发生不良反应。此药略带甜味,易被儿童误服而中毒,表现恶心、呕吐、发热、发绀、惊厥,甚至死亡。成人长期大量服用时,可因二氢叶酸还原酶受抑制而引起巨细胞性贫血。偶可引起皮疹。
磺胺类和砜类
磺胺类和砜类与PABA竞争二氢叶酸合成酶,从而抑制疟原虫二氢叶酸的合成。单用时效果较差,仅抑制红细胞内期,主要用于耐氯喹的恶性疟。对红细胞外期无效。与乙胺嘧啶或TMP等二氢叶酸还原酶抑制剂合用,可增强疗效。常用制剂为周效磺胺和氯苯砜。
抗疟药 制剂及用法
磷酸氯喹(chloroquinephosphate)口服,治疗疟疾(3日疗法):第1日先服1.0g,8小时后再服0.5g;第2,3日各0.5g。预防:0.5g/次,1次/周。
硫酸奎宁(quinine sulfate)口服0.3~0.6g/次,3次/日,连续服5~7日。
二盐酸奎宁(quininedihydrochloride)静脉滴注:0.25~0.5g/次,用葡萄糖液稀释成每ml含0.5~1.0mg后,静脉缓慢滴注。切忌静脉推注.
甲氟喹(mefloquine)口服,用于耐多药恶性疟治疗,成人1000~1500mg(盐基)一次;儿童25mg/kg,一次。用于耐多药恶性疟预防,250mg/周,连用4周,以后每周125mg。
青蒿素油混悬剂 肌内注射,间日疟及恶性疟总量为500~800mg,疗程2~3天。
磷酸伯氨喹(primaquinephosphate)口服,4日疗法:52.8mg(4片)/日.连服4日。8日疗法:39.6mg(3片)/日,连服8日。14日疗法:26.4mg(2片)/日,连服14日。以上剂量均以其盐基计算.
乙胺嘧啶(pyrimethamine)病因性预防:口服,25mg/次,1次/周或50mg/次,1次/2周。
防疟片1号 每片含氨苯砜100mg,乙胺嘧啶20mg。每7日服1次,每次1片(首次连服2天,每天1片),可连续服用3个月。
防疟片2号 每片含周效磺胺250mg,乙胺嘧啶17.5mg。每10~15天服1次,每次2片(首次连服2天,每天2片)。连续服用不宜超过3个月。
第四十六章 抗阿米巴病药及抗滴虫病药
第一节 抗阿米巴病药
阿米巴病为溶组织内阿米巴原虫的感染。人经口感染阿米巴包囊,在肠腔内脱囊而出成为小滋养体,在结肠内与肠道菌丛共生。小滋养体在随宿主肠内容下移过程中,逐渐转变成包囊。此时并无症状,称为排包囊者,是重要的传染源。小滋养体在一定条件下侵入肠壁,成为大滋养体,因破坏肠组织而引起阿米巴痢疾。大滋养体不能形成包囊,但可经血流至肝和其他器官引起阿米巴炎症和脓肿,统称为肠外阿米巴病。
抗阿米巴病药的选用主要根据感染部位和类型。急性阿米巴痢疾和肠外阿米巴病首选甲硝唑;而依米丁和氯喹只在甲硝唑无效或禁忌时偶可使用。对于排包囊者肠腔内的小滋养体和阿米巴痢疾急性症状控制后肠腔内残存的小滋养体,则宜选用主要分布于肠腔内的二氯尼特,偶可考虑应用卤化喹啉类、巴龙霉素和四环素等。
甲硝唑
甲硝唑(metronidazole)又称灭滴灵,为咪唑衍生物。
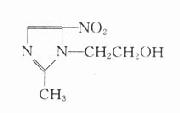
【体内过程】甲硝唑口服吸收迅速而完全。500mg一剂,约1小时后血浆浓度达10μg/ml,远超过平均有效浓度。t1/2约8小时,通常8小时给药一次。在体内各组织和体液中分布均匀。主要在肝中代谢,由肾排出,粪中只含少量。
【药理作用和临床作用】
1.抗阿米巴作用甲硝唑对阿米巴大滋养体有直接杀灭作用。治疗急性阿米巴痢疾和肠外阿米巴病效果最好。但对肠腔内阿米巴原虫则无明显作用。因此,单用甲硝唑治疗阿米巴痢疾时,复发率颇高,须再用肠腔抗阿米巴药继续治疗。同理,甲硝唑不适用于排包囊者。
2.抗滴虫作用甲硝唑对阴道滴虫亦有直接杀灭作用。口服后可出现于阴道分泌物、精液和尿中,故对女性和男性泌尿生殖道滴虫感染都有良好疗效。250mg,每日3次,7日一疗程,对阴道滴虫症的治愈率为90%。2g一剂疗法也有同样疗效。偶有耐药虫株出现。
3.抗贾第鞭毛虫作用甲硝唑是目前治疗贾第鞭毛虫病最有效的药物。成人每次250mg,儿童每次10~15mg/kg,每日3次,5天一疗程,治愈率均在90%以上。
4.抗厌氧菌作用对厌氧性革兰阳性和阴性杆菌和球菌都有较强的抗菌作用。其中尤以对脆弱杆菌的杀菌作用受到重视。至今未发现耐药菌株。长期应用不诱发二重感染。对口腔及盆腔和腹腔内厌氧菌感染及由此引起的败血症,以及气性坏疽等,本品均有良好的防治作用。
【不良反应】甲硝唑不良反应一般较少而轻。最常见者为恶心和口腔金属味,偶见呕吐、腹泻、腹痛、头痛、眩晕、肢体麻木。少数患者可出现白细胞暂时性减少。重复疗程前应作白细胞计数。极少数人可出现脑病、共济失调和惊厥。如发生四肢麻木和感觉异常应立即停药,因为严重的感觉障碍恢复甚慢且不完全。甲硝唑干扰乙醛代谢,如服药期间饮酒,可出现急性乙醛中毒,引起腹部不适、恶心、呕吐、头痛和味觉改变等。
啮齿动物试验证明,长期、大量口服有致癌作用。对细菌有致突变作用。因此,妊娠早期禁用,以防引起胎儿畸形。
替硝唑
替硝唑(tinidazole)也为咪唑衍生物。与甲硝唑相比,其半衰期较长(12~24小时)。口服一次,有效血药浓度可维持72小时。每日50~60mg/kg,3~5天一疗程,对阿米巴痢疾和肠外阿米巴病的疗效与甲硝唑相当而毒性略低。也可用于阴道滴虫症。
二氯尼特
二氯尼特(diloxanide)通常用其糠酸酯(diloxanide furoate),是目前最有效的杀包囊药。口服后主要靠其未吸收部分杀灭阿米巴原虫的囊前期,对于无症状或仅有轻微症状的排包囊者有良好疗效。对于急性阿米巴痢疾,单用二氯尼特疗效不佳;但在甲硝唑控制症状后再用二氯尼特肃清肠腔内的小滋养体,可有效地预防复发。对肠外阿米巴病无效。本品对阿米巴原虫有直接杀灭作用,对脊椎动物无明显作用,不良反应轻微,偶而出现呕吐和皮疹等。很大剂量时可致流产,但无致畸作用。
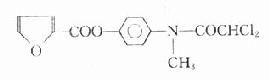
卤化喹啉类
本类药包括喹磺方(chiniofon)、双碘喹啉(diiodhydroxyquinoline)和氯碘羟喹(clioquinol)。
此类药物有直接杀阿米巴作用,口服吸收较少,曾广泛用作肠腔内抗阿米巴药,用于排包囊者,或与甲硝唑合用于急性阿米巴痢疾。此类药物毒性低,但可致腹泻。每日量超过2g,疗程较长时;或为儿童,则危险性较大。在日本曾见引起亚急性脊髓-视神经病,可致视神经萎缩和失明。许多国家已禁止或限制其应用。
依米丁和去氢依米丁
依米丁(emetine)是吐根中提得的一种生物碱,又称吐根碱。其衍生物去氢依米丁(dehydroemetine)抗阿米巴作用更强。
依米丁和去氢依米丁主要对组织中的阿米巴滋养体有直接杀灭作用。由于其刺激性很强,口服可致吐,只能深部肌肉注射。除引起胃肠道反应外,对心肌有严重毒性。仅在急性阿米巴痢疾和肠外阿米巴病病情严重,甲硝唑疗效不满意时才考虑使用。必须住院,在严密监护下给药。
氯喹
氯喹为抗疟药(见第四十五章),也有杀灭阿米巴滋养体的作用。口服后肝中浓度比血浆浓度高数百倍,而肠壁的分布量很少,对肠阿米巴病无效,仅用于甲硝唑无效或禁忌的阿米巴肝炎或肝脓肿病人。
第二节 抗滴虫病药
滴虫病主要指阴道滴虫病,但阴道毛滴虫也可寄生于男性尿道内。甲硝唑是治疗滴虫病最有效的药物。详见第一节。偶遇抗甲硝唑株滴虫感染时,可考虑改用乙酰胂胺局部给药。
乙酰胂胺
乙酰胂胺(acetarsol)为五价胂剂,其复方制剂称滴维净。以其片剂置于阴道穹窿部有直接杀滴虫作用。此药有轻度局部刺激作用,使阴道分泌物增多。
抗阿米巴病药及抗滴虫病药 制剂和用法
甲硝唑(metronidazole,灭滴灵)250和500mg片剂,口服给药。阿米巴痢疾:0.4~0.8g/次,3次/日,共5日。肠外阿米巴病:0.75g/次,3次/日,共10日。阴道滴虫病和男性尿道滴虫感染:250mg/次,3次/日,共7日,或2g顿服。贾第鞭毛虫病:0.25g,3次/日,共5~7日;或2g/日,连用3日。厌氧菌感染:7.5mg/kg,q ·6h,首剂加倍,共7~10日,静脉注射。
二氯尼特糠酸酯(diloxanide furoate,furamide)成人:500mg,3次/日,共10日;儿童:20mg/kg/日,分3次给药,共10日;口服给药。
喹碘方(chiniofon)0.25~0.5g/次,3次/日,共10日;口服给药。
氯碘羟喹(clioquinol)0.25g/次,3次/日,共10日;口服给药。
双碘喹啉(diiodohydroquinoline)0.6g/次,3次/日,共14~21日。
去氢依米丁(dehydro emetine,去氢吐根碱)成人:1~1.5mg/kg/日,极量90mg,深部肌内注射 ,连用5日;儿童也按上述方法按体重计算剂量,每12小时各给半量。重复疗程时宜间隔30日。
磷酸氯喹(chloroquine phosphate)肠外阿米巴病:0.25g/次,3~4次/日,儿童酌减,3~4周一疗程。必要时可适当延长疗程。
滴维净(devegan)每次1~2片塞入阴道穹窿部,1~3次/日,10~14日一疗程。
第四十七章 抗血吸虫病药和抗丝虫病药
第一节 抗血吸虫病药
日本血吸虫病在我国长江流域和长江以南十三个省、直辖市、自治区严重流行,解放初期估计有患者千余万人。是我国危害最严重的寄生虫病。解放后政府开展了大规模的防治工作,流行情况得到基本控制。但文革期间,由于预防工作中断,使血吸虫病又复流行和蔓延。积极开展防治工作仍很有必要。
长期以来,酒石酸锑钾是主要的特效药。但它有毒性大、疗程长、必须静脉注射等缺点。70年代发现吡喹酮高效、低毒、疗程短、口服有效,是血吸虫病防治史上的一个突破,现已完全取代酒石酸锑钾。
吡喹酮
吡喹酮(praziquantel)为吡嗪异喹啉衍生物,为广谱抗吸虫药和驱绦虫药,尤以对血吸虫有杀灭作用而受重视。对线虫和原虫感染无效。
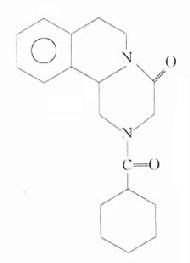
【抗虫作用】吡喹酮除对血吸虫有杀灭作用外,对其他吸虫,如华支睾吸虫、姜片吸虫、肺吸虫,以及各种绦虫感染和其幼虫引起的囊虫症、包虫病都有不同程度的疗效。本章着重讨论其抗血吸虫作用;其他抗肠蠕虫的作用和用途,见第四十八章。
在体外实验中,吡喹酮能为血吸虫迅速摄取。在最低有效浓度(0.2~1.0μg/ml)时,可使虫体兴奋、收缩和痉挛。略高浓度时,则可使血吸虫体被形成空泡和破溃,粒细胞和吞噬细胞浸润,终至虫体死亡。整体实验结果表明,用药后数分钟内,肠系膜静脉内95%的血吸虫向肝转移,并在肝内死亡。
吡喹酮的上述作用可能与其增加体被对Ca2+的通透性,干扰虫体内Ca2+平衡有密切关系。除去培养液中的Ca2+或加入Mg2+,则可取消上述作用。由于虫体发生痉挛性麻痹,使其不能附着于血管壁,被血流冲入肝,即出现肝移。在肝内由于失去完整体被的保护,更易被吞噬细胞所消灭。吡喹酮对哺乳动物细胞膜则无上述作用,由此表现出其作用的高度选择性。
【体内过程】吡喹酮口服吸收迅速而完全,于服药后1~2小时达血药峰浓度。由于首关消除多,限制了其生物利用度。主要在肝内羟化而失活,经肾排出。24小时内排出用药量的90%。以原形药经肾排泄者不超过用药量的0.02.消除t1/2健康人为1~1.5小时,晚期血吸虫病患者则明显处长。
【临床作用】对慢性日本血吸虫病,20mg/kg,一日三次,一日疗法,或10mg/kg,每日三次,连用2日,远期治愈率可达90%以上。对急性血吸虫病,有迅速退热和改善全身症状的作用,远期疗效也可达87%。有心、肝等并发症的晚期患者多能顺利完成疗程。
【不良反应】副作用轻微、短暂。可在服药后短期内发生腹部不适、腹痛、恶心,以及头昏、头痛、肌束颤动等。少数出现心电图改变。
第二节 抗丝虫病药
乙胺嗪
乙胺嗪(diethylcarbamazine)的枸橼酸盐称海群生(hetrazan)。
【抗虫作用和临床应用】服用乙胺嗪后,班氏丝虫和马来丝虫的微丝蚴迅速从患者血液中减少或消失。对淋巴系统中的成虫也有毒杀作用,但需较大剂量或较长疗程。
在体外,乙胺嗪本身或服过乙胺嗪的动物血清,对微丝蚴和成虫并无毒杀作用。其抗虫作用机制可能有两方面:一是其分子中的哌嗪部分使微丝蚴之肌组织发生超极化,失去活动能力,以致不能停留于宿主周围血液中;再一是破坏微丝蚴体被之完整性,使其易于遭受宿主防卫机制的破坏。
【不良反应】乙胺嗪本身毒性较低而短暂,可引起厌食、恶心、呕吐、头痛、无力等。但因丝虫成虫和蚴虫死亡释出大量异体蛋白引起的过敏反应则较明显。表现为皮疹、淋巴结肿大、血管神经性水肿、畏寒、发热、哮喘,以及心率加快、胃肠功能紊乱等。一般于给药之日开始,持续3~7天。
制剂及用法
吡喹酮(praziquantel)片剂,每片0.2mg;口服,血吸虫病:10mg/kg/次,3次/日,连服2日,或20mg/kg/次,3次/日,服1日,治其他蠕虫病的用法和用量见驱肠蠕虫药章。
枸橼酸乙胺嗪 (diethylcarbamazine citrate;海群生,hetrazam)口服,一天疗法:1.5g,一次或分二次服。七天疗法:0.2g/次,3次/日,连服7天。
第四十八章 抗肠蠕虫药
肠道蠕虫包括绦虫、钩虫、蛔虫、蛲虫、鞭虫和姜片虫等。不同蠕虫对不同药物的敏感性不同,因此,必须针对不同的蠕虫感染正确选药。近年来不断有广谱、高效的驱肠蠕虫药问世,使选药更为方便易行,而且有些药物对由肠蠕虫病引起的组织型感染也有效。
甲苯达唑
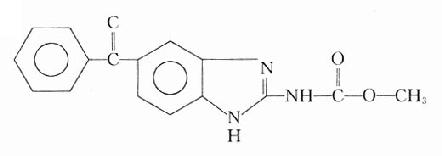
【驱虫作用】甲苯达唑(mebendazole)为一高效、广谱驱肠蠕虫药。它选择性地使线虫的体被和肠细胞中的微管消失,抑制虫体对葡萄糖的摄取减少糖原量,减少ATP生成,妨碍虫体生长发育。对多种线虫的成虫和幼虫有杀灭作用。对蛔虫、蛲虫、鞭虫、钩虫、绦虫感染的疗效常在90%以上,尤其适用于上述蠕虫的混合感染。
甲苯哒唑显效缓慢,给药后数日才能将虫排尽。
本品对钩虫卵、蛔虫卵和鞭虫卵有杀灭作用,有控制传播的重要意义。
【不良反应】本品口服吸收少,首过效应明显,无明显不良反应。少数病例可见短暂腹痛、腹泻。大剂量时偶见过敏反应、脱发、粒细胞减少等。大鼠试验发现有致畸胎作用和胚胎毒作用,故孕妇忌用。对2岁以下儿童和对本品过敏者不宜使用。
阿苯达唑
阿苯达唑(albendazole)是继甲苯达唑之后研制成功的又一同类药,别名肠虫清,具有广谱、高效、低毒的特点。
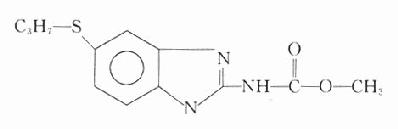
【驱虫作用】阿苯达唑对肠道寄生虫,如线虫类的蛔虫、蛲虫、钩虫、鞭虫和粪类圆线虫,绦虫类的猪肉绦虫、牛肉绦虫、短膜壳绦虫等的驱杀作用及其机制基本同甲苯达唑。但由于它口服后吸收迅速,血药浓度比口服甲苯达唑后高出100倍,肝、肺等组织中均能达到相当高的浓度,并能进入棘球蚴囊内。因此,对肠道外寄生虫病,如棘球蚴病(包虫病)、囊虫症、旋毛虫病,以及华支睾吸虫病、肺吸虫病等也有较好疗效,为甲苯达唑所不及。对于脑囊虫症,也有较缓和的治疗作用,比吡喹酮较少引起颅内压升高和癫痫发作等强烈反应,但仍应住院治疗,随时警惕脑疝等反应的发生。对华支睾吸虫病的疗效则稍逊于吡喹酮,疗程也稍长。
【不良反应】本品副作用轻,一般耐受良好。每日400mg时,20%~30%的病例可出现消化道反应和头晕、思睡、头痛等。多在数小时内自行缓解。每日800mg时,初期有30%出现白细胞减少,5~6个月后可恢复。少数可见肝功能障碍,1~2周内恢复。
治疗囊虫症和包虫病时,所用剂量较大,疗程很长,但也多能耐受。主要反应系由猪囊尾蚴解体后释出异体蛋白所致。可见头痛、发热、皮疹、肌肉酸痛。脑型囊虫症时则可引起癫痫发作、视力障碍、颅内压升高,甚至脑水肿和脑疝。治旋毛虫病时也可发生发热、肌痛和水肿加重等反应。
【临床应用】
1、蛔虫、钩虫、鞭虫感染 400mg顿服。
2、牛肉绦虫感染,每日800mg,连用3天;猪肉绦虫或短膜壳绦虫感染,可参考上述疗法进行。
3、囊虫症 200~300mg,每日三次,10天为一疗程,一般给予2~3个疗程,疗程间隔15~21天。脑型病例应住院治疗。如治疗过程中出现癫痫大发作,应停药2~3周。如有颅内压增高(常在给药后1~3周逐渐明显),应先行降低颅内压。尤须警惕发生脑疝。
4、包虫病(棘球蚴病)5~7mg/kg,每日二次,30日一疗程,重复数疗程,疗程间隔2周。
5、华支睾吸虫病 每日8mg/kg,顿服,共7日。
6、旋毛虫病 对肠内期和肠外期旋毛虫均有驱杀作用。每日24或32mg/kg,5天一疗程,给予1~2个疗程。
7、对肺吸虫病和梨形鞭毛虫病也有效。
吡喹酮
吡喹酮为一广谱抗蠕虫药。其抗血吸虫作用和一般药理作用已详第四十七章。此处仅介绍其抗其他蠕虫的作用。对线虫和原虫感染无效。
【抗蠕虫作用】吡喹酮对牛肉绦虫、猪肉绦虫、阔节裂头绦虫和短膜壳绦虫都有良好的疗效。10或25mg/kg顿服的治愈率高于氯硝柳胺。对于囊虫症,皮下-肌肉型用总量120mg/kg4天疗法;脑型用总量180mg/kg的9天疗法;间隔3~4个月再进行第2疗程,共3个疗程。疗效不低于阿苯达唑,而杀虫作用迅速,但引起的颅内压升高的反应较重。如囊泡在重要中枢附近,则更宜谨慎从事。
对华支睾吸虫病和其他肝吸虫病,总量100mg/kg,一天疗法,或总量120mg/kg,2天疗法均有良好疗效。对肺吸虫病也有效。
姜片虫对吡喹酮甚为敏感,5~15mg/kg一剂疗法即效。
用本品治包虫病不能获得寄生虫学治愈,但可杀灭已生成的原头蚴或使其感染能力明显降低。用于术前准备以防术中棘球蚴扩散。一般每日25~30mg/kg,给药6~10天。这点与阿苯达唑直接杀死棘球蚴不同。不宜于手术者应采用阿苯达唑。
哌嗪
哌嗪(piperazine),其枸橼酸盐称驱蛔灵,对蛔虫和蛲虫有较强的驱除作用。主要改变虫肌细胞膜对离子的通透性。使虫体肌肉超极化,抑制神经-肌肉传递,致虫体发生弛缓性麻痹而随肠蠕动排出。治蛔虫,1~2天疗法的治愈率可达70%~80%。对蛲虫,需用药7~10天,远不如使用阿苯达唑等方便。
本品不易吸收,副作用少见。
噻嘧啶
噻嘧啶(pyrantel),其枸橼酸盐称驱虫灵。为一广谱驱线虫药,对蛔虫、钩虫、蛲虫和毛圆线虫感染均有较好疗效,但对鞭虫无效。它使虫体神经-肌肉去极化,引起痉挛和麻痹。
口服不易吸收。不良反应轻而短暂,主要为胃肠不适,其次为头昏、发热。
氯硝柳胺
氯硝柳胺(niclosamide)原为杀钉螺药,对血吸虫尾蚴和毛蚴也有杀灭作用,用于血吸虫病的预防。后发现对牛肉绦虫、猪肉绦虫、阔节裂头绦虫和短膜壳绦虫感染都有良好疗效,尤以对牛肉绦虫的疗效为佳。
本品主要抑制绦虫线粒体内ADP的无氧磷酸化,阻碍产能过程,也抑制葡萄糖摄取,从而杀死其头节和近端节片,但不能杀死节片中的虫卵。已死头节可被部分消化而在粪中难以辨认。如欲急于考核疗效,应在服药1~3小时内,即在头节未被消化前服泻药一剂。猪肉绦虫死亡节片被消化后,释出的虫卵逆流入胃,有引起囊虫症的危险。因此,有人主张宁可用吡喹酮治猪肉绦虫症。
本品口服不易吸收,也无直接刺激作用,仅偶见消化道反应。
其他驱肠蠕虫药
左旋咪唑和扑蛲灵等也有驱肠蠕虫作用,其适应症见表48-1。
表48-1 抗肠蠕虫药的适应症和合理选用
| 适应症 | 可选用药物 |
| 蛔虫感染 | 甲苯达唑*,阿苯达唑*,噻嘧啶,哌嗪,左旋咪唑 |
| 蛲虫 | 甲苯达唑*,阿苯达唑*,噻嘧啶,扑蛲灵,哌嗪 |
| 钩虫 | 甲苯达唑*,阿苯达唑*,噻嘧啶 |
| 鞭虫 | 甲苯达唑 |
| 绦虫 | 吡喹酮*,氯硝柳胺 |
| 姜片虫 | 吡喹酮 |
| 华支睾吸虫 | 吡喹酮*,阿苯达唑 |
| 囊虫 | 吡喹酮*,阿苯达唑* |
| 包虫 | 阿苯达唑*,吡喹酮,甲苯达唑 |
* 表示首选
抗肠蠕虫药 制剂和用法
甲苯达唑片(mebendazole)成人和2岁以上儿童服用同样剂量。蛲虫:100mg,顿服,2周后再服一剂;蛔虫、钩虫、鞭虫:100mg,早晚各一剂,连服3天;绦虫:300mg,每日3次,连用3天。
阿苯达唑片(albendazole,肠虫清片)剂量和用法详正文。
枸橼酸哌嗪(piperazinecitrate)蛔虫:75mg/kg/日,极量4g/日,顿服;儿童75~150mg/kg/日,极量3g/日,空腹顿服,连用2日。蛲虫:成人1.0~1.2g/次,2次/日;儿童60mg/kg/日,分两次,连用7日。
双羟萘酸噻嘧啶 (pyrantelpamoate)钩虫:5~10mg/kg,顿服,连服2~3日。蛔虫:剂量同上,用药一次;蛲虫:剂量同上,连服一周.
氯硝柳胺(niclosamide)猪肉、牛肉绦虫:晨空服1g,顿服,1小时后再服一剂,1~2小时后服硫酸镁导泻;短膜壳绦虫:清晨空腹嚼服2g,1小时后再服一剂,连服7~8天。
第四十九章 抗恶性肿瘤药
恶性肿瘤是严重威胁人类健康的常见病、多发病,是世界各国医学科学领域中的重大科研课题,目前尚无满意的防治措施。治疗恶性肿瘤的方法仍为手术切除、放射治疗和化学治疗,后者仍为临床治疗的重要方法。抗恶性肿瘤药对癌细胞和人体正常细胞的选择性差别不大,因而应用过程中的不良反应广泛而严重。另外,易产生耐药性也是治疗过程中的问题之一。近年来,在分子生物学、细胞动力学、免疫学的理论指导下以及采用联合用药的方法,恶性肿瘤化学治疗的疗效有显着的提高,并明显减少了不良反应及耐药性的发生。
随着恶性肿瘤分子生物学研究的开展,如对生长因子(血小板衍生的生长因子)、生长抑制因子(干扰素)、原癌基因(C-ras、H-ras、myc、fos)以及癌促进因子(phorbol酯)等的研究,新的抗恶性肿瘤药物及基因疗法已开始出现。近年来,对癌细胞分化诱导剂维甲酸(retinoids)的研究,在实验研究方面也已取得较大进展,在临床初步试用,出现了可喜的苗头。
第一节 抗恶性肿瘤药的作用及分类
抗恶性肿瘤药的主要作用是杀伤癌细胞,阻止其分裂繁殖。兹先介绍其分类与细胞增殖动力学内容。
一、对生物大分子的作用及药物分类
(一)影响核酸(DNA,RNA)生物合成的药物
核酸是一切生物的重要生命物质,它控制着蛋白质的合成。核酸的基本结构单位是核苷酸,而核苷酸的合成需要嘧啶类前体和嘌呤前体及其合成物,所以这一类型作用的药物又可分为①阻止嘧啶类核苷酸形成的抗代谢药,如5-氟尿嘧啶等。②阻止嘌呤类核苷酸形成的抗代谢药,如6-巯嘌呤等。③抑制二氢叶酸还原酶的药,如甲氨蝶呤等。④抑制DNA多聚酶的药,如阿糖胞苷。⑤抑制核苷酸还原酶的药,如羟基脲。
(二)直接破坏DNA并阻止其复制的药物
有烷化剂、丝裂霉素C、博来霉素等。
(三)干扰转录过程阻止RNA合成的药物
有多种抗癌抗生素,如放线菌素D及蒽环类的柔红霉素、阿霉素等。
(四)影响蛋白质合成的药物
可分为①影响纺锤丝的形成纺锤丝是一种微管结构,由微管蛋白的亚单位聚合而成。长春碱类和鬼臼毒素类属本类药物。②干扰核蛋白体功能的药物如三尖杉酯碱。③干扰氨基酸供应的药物 如L-门冬酰胺酶。
(五)影响激素平衡发挥抗癌作用的药物
有肾上腺皮质激素、雄激素 、雌激素等。
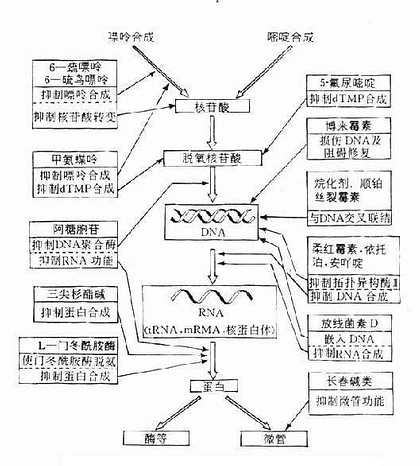
图49-1 抗恶性肿瘤药的作用部位示意图
二、对细胞增殖动力学的影响
肿瘤组织主要由增殖细胞群和非增殖细胞(G)群组成(见图49-2)前者可不断按指数分裂增殖,这部分细胞在肿瘤全部细胞群的比例称为生长比率(growthfraction,GF)。增长迅速的肿瘤(如急性白血病等)GF值较大,接近1,对药物最敏感,药物疗效也好;增长慢的肿瘤(如多数实体瘤),GF值较小,0.5~0.01,对药物敏感性低,疗效较差。同一种肿瘤早期的GF值较大,药物的疗效也较好。
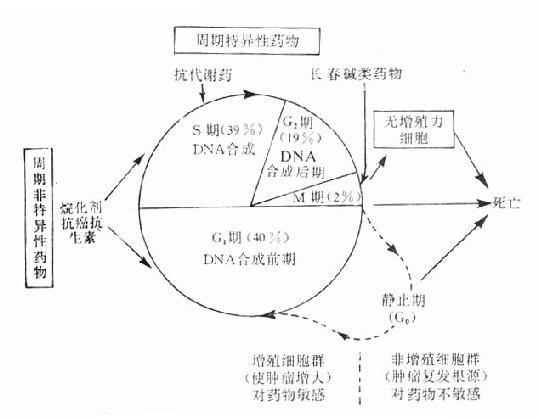
图49-2 细胞增殖周期及药物作用示意图
1.周期非特异性药物(cellcyclenon-specific drugs)主要杀灭增殖细胞群中各期细胞,如烷化剂。它们对小鼠骨髓干细胞和淋巴肿瘤细胞的量效曲线都呈指数性,其中氮芥和丝裂霉素选择性低(杀伤两类细胞的曲线斜率很接近),而大多数其他烷化剂选择性较高(表现于对两类细胞的量效曲线的斜率相差较大,见图49-3A,B)。
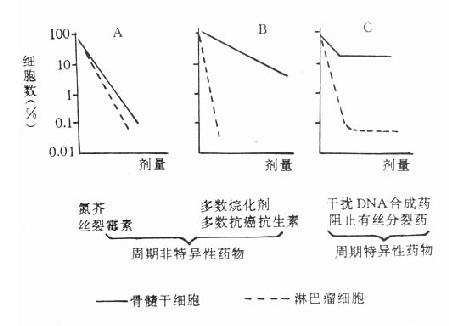
图49-3 各类抗肿瘤药杀灭小鼠骨髓干细胞及淋巴瘤细胞的量效曲线
2.周期特异性药物(cell cyclespecific drugs)仅对增殖周期中的某一期有较强的作用,如抑制核酸合成的药对S期作用显著;长春碱等作用于M期。这类药物对骨髓及瘤细胞的量效曲线也随剂量增大而下降,但达到一定剂量时即向水平方向转折,成为一个坪,即再增加剂量,不再有更多的细胞被杀死(见图49-3C)。
第二节 常用的抗肿瘤药物
一、影响核酸生物合成的药物
又称抗代谢药,是模拟正常代谢物质,如叶酸、嘌呤碱、嘧啶碱等的化学结构所合成的类似物,与有关代谢物质发生特异性的拮抗作用,从而干扰核酸,尤其是DNA的生物合成,阻止瘤细胞的分裂繁殖。它们是细胞周期特异性药物,主要作用于S期。
5-氟尿嘧啶
5-氟尿嘧啶 (5-fluorouracil,5-FU),是尿嘧啶5位的氢被氟取代的衍生物,是抗嘧啶药。
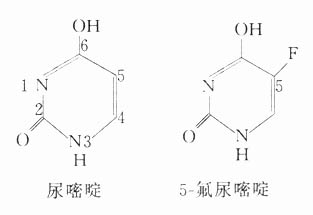
【药理作用】在细胞内转变为5-氟尿嘧啶脱氧核苷酸(5F-dUMP)而抑制脱氧胸苷酸合成酶,阻止脱氧尿苷酸(dUMP)甲基化为脱氧胸苷酸(dTMP),从而影响DNA的合成。另外,5-FU在体内转化为5-氟尿嘧啶核苷(5-FUR)后,也能掺入RNa 中干扰蛋白质合成,故对其他各期细胞也有作用。
【体内过程】口服吸收不规则。常静脉给药。分布于全身体液,肿瘤组织中的浓度较高,易进入脑脊液内。由肝代谢灭活,变为CO2和尿素分别由肺和尿排出。
【不良反应】主要为胃肠道反应,重者血性下泻而死。骨髓抑制、脱发、共济失调等。因刺激性可致静脉炎或动脉内膜炎。偶见肝、肾功能损害。
【临床应用】对多种肿瘤有效,特别是对消化道癌症和乳腺癌疗效较好;对卵巢癌、宫颈癌、绒毛膜上皮癌、膀胱癌等也有效。
图49-4 几种药物阻断DNA合成的作用环节
MTX=甲氨蝶呤;6-MP=6-颈嘌呤;5-FU=5-氟尿嘧啶;
HU=羟基脲;6-TG=6-硫鸟嘌呤;Ara-C=阿糖胞苷
6-颈基嘌呤
6-颈嘌呤(6-mercaptopurine,6-MP)是腺嘌呤6位上的-NH2被-SH所取代的衍生物,为抗嘌呤药。
【药理作用】在体内先经酶催化变成硫代肌苷酸,它阻止肌苷酸转变为腺苷酸和鸟苷酸,干扰嘌呤代谢、阻碍核酸合成,对S期细胞及他期细胞有效。肿瘤细胞对6-MP可产生耐药性,因耐药性细胞中6-MP不易转变成硫代肌苷酸或产生后迅速降解之故。
【体内过程】口服吸收良好。分布到各组织,部分在肝内经黄嘌呤氧化酶催化为无效的硫尿酸(6-thiouricacid)与原形物一起由尿排泄。静脉注射的t1/2约为90分钟。抗痛风药别嘌醇可干扰6-MP变为硫尿酸,故能增强6-MP的抗肿瘤作用及毒性,合用时应注意减量。
【不良反应】多见胃肠道反应和骨髓抑制;少数病人可出现黄疸和肝功能障碍。偶见高尿酸血症。
【临床应用】对儿童急性淋巴性白血病疗效好,因起效慢,多作维持药用。大剂量用于治疗绒毛上皮癌有一定疗效。
甲氨蝶呤
甲氨蝶呤(methotrexate,MTX)又名氨甲蝶呤,(amethopterin),化学结构与叶酸相似,是抗叶酸药。
【药理作用】甲氨蝶呤对二氢叶酸还原酶有强大而持久的抑制作用,使5,10-甲撑四氢叶酸不足,脱氧胸苷酸(dTMP)合成受阻,影响DNA合成(图49-4);MTX也可阻止嘌呤核苷酸的合成,因为嘌呤环上的第2和第8碳原子是由FH4携带的一碳基团(如-CHO-,=C-)所供给,故能干扰RNA和蛋白质的合成。
【体内过程】口服吸收良好。1小时内血中浓度达峰值,3~7小时后已不能测到。与血浆蛋白质结合率为50%;t1/2约2小时。由尿中排出的原形约50%;少量通过胆道从粪排出。MTX不易透过血脑屏障。
【不良反应】较多。可致口腔及胃肠道粘膜损害,如口腔炎、胃炎、腹泻、便血甚至死亡。骨髓抑制可致白细胞、血小板减少以至全血象下降。也有脱发、皮炎等。孕妇可致畸胎、死胎。大剂量长期用药可致肝、肾损害。
【临床应用】用于儿童急性白血病和绒毛膜上皮癌。甲酰四氢叶酸能拮抗MTx 治疗中的毒性反应,现主张先用很大剂量MTX(3~5~25g/m2),以后再用甲酰四氢叶酸作为救援剂,以保护骨髓正常细胞,对成骨肉瘤等有良效。
近年发现癌细胞可对MTX产生耐药性,主要是基因扩增产生更多二氢叶酸还原酶所致,也与MTX进入细胞减少等有关。
阿糖胞苷
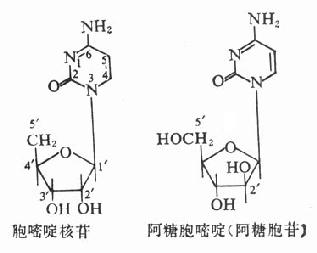
【药理作用】阿糖胞苷(cytarabine,AraC)在体内经脱氧胞苷激酶催化成二或三磷酸胞苷,进而抑制DNA多聚酶的活性而影响DNA合成;也可掺入DNA中干扰其复制,使细胞死亡。S期细胞对之最敏感,属周期特异性药物。
【体内过程】因不稳定,口服易破坏。静脉注射(5~10mg/kg)20分钟后多数患者血中已测不到。主要在肝中被胞苷酸脱氨酶催化为无活性的阿糖尿苷,迅速由尿排出。
【不良反应】对骨髓的抑制可引起白细胞及血小板减少。久用后胃肠道反应明显。对肝功能有一定影响,出现转氨酶升高。
【临床应用】治疗成人急性粒细胞或单核细胞白血病的有效药物。对实体瘤单独应用疗效不满意。
羟基脲
H2N-CO-NHOH
【药理作用】羟基脲(hydroxycarbamide,hydroxyurea,HU)能抑制核苷酸还原酶,阻止胞苷酸转变为脱氧胞苷酸,从而抑制DNA的合成。它能选择性地作用于S期细胞。口服吸收很快,1小时血药浓度达峰值,6小时消失。能透过红细胞膜和血脑屏障。主要由肾排泄。
【不良反应】主要为抑制骨髓。也可有胃肠道反应。可致畸胎,孕妇忌用,肾功能不良者慎用。
【临床应用】对慢性粒细胞白血病有确效,也可用于急性变者。对转移性黑色素瘤也有暂时缓解作用。用药后可使瘤细胞集中于G1期,故常作为同步化药物以提高肿瘤对化疗或放疗的敏感性。
表49-1 其他常用抗代谢药
| 药名 | 作用特点 | 不良反应 |
| 替加氟(tegafur;呋喃氟尿嘧啶,ftorafur,FT-207) | 为5-FU的衍生物,在肝内活化为氟尿嘧啶而起作用。主要用于胃、直肠、结肠、肝、乳癌等 | 与5-FU相似,较轻 |
| 安西他滨(ancitabin;环胞苷,cyclocytidine,cyclo-C) | 为Ara-C脱水衍生物,在体内逐渐水解放出Ara-C而显效。抗瘤谱广。t1/2较长。用于急性白血病、恶性淋巴瘤等。 | 骨髓抑制,较轻;可有体位性低血压,腮腺痛及流涎 |
| 6-硫鸟嘌呤(tioguanine,6-thioguanine,6-TG) | 与6-MP相似。主要用于急性白血病。与Ara-C联合应用对缓解急性粒细胞或单核白血病疗效较好。 | 骨髓抑制和胃肠道反应,较6-MP轻。 |
| 溶癌呤(tisupurine,AT-1438) | 为6-MP的磺酸钠盐,易溶于水,在体内分解为6-MP而显效。作用快,不易通过血脑屏障。用于急性白血病、绒癌和恶性葡萄胎较好 | 骨髓抑制,胃肠道反应。 |
二、直接破坏DNA并阻止其复制的药物
(一)烷化剂
烷化剂(alkylatingagents)又称烃化剂,是一类化学性质很活泼的化合物。它们具有活泼的烷化基团,能与细胞中DNA或蛋白质中的氨基、颈基、羟基和磷酸基等起作用,常可形成交叉联结或引起脱嘌呤作用,使DNA链断裂,在下一次复制时,又可使核碱酸对错码,造成DNA结构和功能的损害,重者可致细胞死亡(图49-5)。
图49-5 氮芥为例的烷化作用和DNA交叉联结示意图
氮芥为一双(氯乙)胺化合物(R=CH3)
(1)经释放Cl、分子间环化,形成不隐定的乙撑亚胺离子(叔胺)
(2)后,即刻打开环链而形成具有活性的碳鎓(caubonium)离子
(3)随即与DNA-鸟嘌呤(4)的N-7反应生成7-烷鸟嘌泠(5)N-7则可再转
变为季胺N。具有双功能基团的烷化剂尚可进行第二个环化(6)再产生一个碳鎓离子
(7)与另一条互补DNA链上的鸟嘌呤形成链间交叉联结(8)
氮芥
氮芥(chlorethamine,nitrogenmustard,mechlorethamine,HN2)是最早应用的烷化剂,选择性低,局部刺激性强,必须静脉注射。作用迅速而短暂(数分钟),但对骨髓等抑制的后果却较久。目前主要利用其速效的特点,作为纵隔压迫症状明显的恶性淋巴瘤的化学治疗,以及区域动脉内给药或半身化疗(压迫主动脉阻断下身循环),治疗头颈部等肿瘤,以提高肿瘤局部的药物浓度和减少毒性反应。可有恶心、呕吐、昡晕、视力减退、脱发、黄疸、月经失调和皮疹等不良反应。
环磷酰胺
环磷酰胺(cyclophosphamide,endoxan,cytoxan,CTX)为氮芥与磷酰胺基结合而成的化合物(见图49-6)。
图49-6 环磷酰胺的体内代谢
【药理作用】环磷酰胺在体外无活性,在体内经肝细胞色素P-450氧化、裂环生成中间产物醛磷酰胺(aldophosphamide),它在肿瘤细胞内,分解出有强效的磷酰胺氮芥(phosphamide mustard),才与DNA发生烷化,形成交叉联结,抑制肿瘤细胞的生长繁殖。环磷酰胺抗瘤谱较广,对恶性淋巴瘤疗效显著。对多发性骨髓瘤、急性淋巴细胞白血病、卵巢癌、乳腺癌等也有效。
【体内过程】口服吸收良好,1小时后血中药物达峰浓度,17%~31%的药物以原形由粪排出。30%以活性型由尿排出,对肾和膀胱有刺激性。静脉注射6~8mg/kg后,血浆t1/2约为6.5小时。在肝及肝癌组织中分布较多。
【不良反应】环磷酰胺可口服或注射;呕吐、恶心反应较轻,静脉注射大剂量时仍多见;脱发发生率较其他烷化剂为高约30%~60%,多发生于服药3~4周后;抑制骨髓,对粒细胞的影响更明显;对膀胱粘膜刺激可致血尿、蛋白尿;偶可影响肝功能,导致黄疸;还致凝血酶原减少;久用可致闭经或精子减少。
噻替派
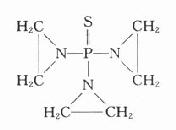
噻替派(thio-tepa,triethylenethiophosphoramide,TSPA)结构中含三个乙撑亚胺基,能形成有活性的碳三离子与细胞内DNA的碱基结合,影响瘤细胞的分裂。其选择性较高,抗瘤谱较广,主要用于乳腺癌、卵巢癌、肝癌和恶性黑色素瘤等,对骨髓有抑制作用,引起白细胞和血小板减少,但较氮芥轻。胃肠道反应少见,局部刺激小,可作静脉注射、肌内注射及动脉内给药与胸(腹)腔内给药。
白消安
白消安(busulfan)又名马利兰(myleran),属磺酸酯类,在体内解离后起烷化作用。小剂量即可明显抑制粒细胞生成,对慢性粒细胞白血病疗效显着(缓解率80%~90%)。剂量提高可抑制全血象。对慢性粒细胞白血病急性病变及急性白血病无效。对其他肿瘤疗效不明显。口服吸收良好。静脉注射后2~3分钟内90%药物自血中消失。绝大部分代谢成甲烷磺酸由尿排出。本药的胃肠道反应少,对骨髓有抑制作用。久用可致闭经或睾丸萎缩,偶见出血、再生障碍性贫血及肺纤维化等严重反应。
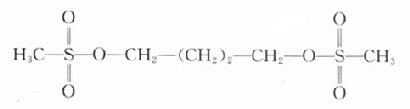
表49-2 其他常用烷化剂
| 药 名 | 作用特点 | 不良反应 |
| 氮甲(N-formylsarcolysin,N-F) | 对精原细胞瘤、多发性骨髓瘤疗效较明显。对恶性淋巴瘤亦有效 | 骨髓抑制及胃肠道反应 |
| 消卡芥(消瘤芥nitrocophac,AT-1258 | 对肺癌、乳腺癌、鼻咽癌、喉癌等均有效 | 同上 |
| 卡莫司汀(卡氮芥,carmustine,BCNU) | 脂溶性大,能透过血脑屏障进入脑组织,用于原发性脑瘤、脑转移瘤、脑膜白血病等 | 同上 |
| 罗莫司汀(环己亚硝脲,lumustine,CCNU)及司莫司汀(甲环亚硝脲,semustine,meCCNU) | 均较BCNU为优,对静止细胞的杀灭作用较强,能延长S期。多用于肺癌、肠癌,对脑瘤和肺癌脑转移、骨转移亦有效 | 同上,肝功不良者慎用 |
| 丙卡巴肼(甲基芐肼,procarbazine,PCZ) | 对何杰金病,淋巴瘤有效 | 同上,肾萎缩,骨髓抑制,胃肠道反应 |
| 达卡巴嗪(甲氮咪唑胺,dacarbazine,DTIC) | 对黑色素瘤,脑瘤,淋巴瘤类有效 | 胃肠道反应 |
| 六甲密胺(altretamine,hexamethylmelamine,HMM) | 抑制核酸合成,对小细胞肺癌、卵巢癌乳癌有效 | 胃肠道反应 |
| 美法仑(melphalen) | 对乳腺癌、卵巢癌、慢性淋巴细胞和粒细胞白血病、恶性淋巴瘤、多发性骨髓瘤有效 | 胃肠道反应,骨髓抑制 |
(二)抗生素类
丝裂霉素C
【药理作用】丝裂霉素C(mitomycin C,MMC)化学结构中有乙撑亚胺及氨甲酰酯基团,具有烷化作用。能与DNA的双链交叉联结。可抑制DNA复制,也能使部分DNA断裂。属周期非特异性药物。注射后迅速由血浆消失,经肾排泄。
【不良反应】骨髓抑制以白细胞和血小板下降最明显,也常有恶心、呕吐、腹泻等症状。注射局部刺激性较大。偶见心脏毒性。
【临床应用】抗瘤谱广,可用于胃、肺、乳癌、慢性粒细胞白血病、恶性淋巴瘤等。
博来霉素
博来霉素(平阳霉素,bleomycin,BLM)为多种糖肽抗生素的混合物。
【药理作用】能与铜或铁离子络合,使氧分子转成氧自由基,从而使DNA单链断裂,阻止DNA复制,干扰细胞分裂繁殖。属周期非特异性药物,作用于G2及M期,并延缓S/G2边界期及G2期时间。
【体内过程】给药后广泛分布到各组织,以肺及鳞癌较多,在该处不易被灭活,而其他组织的水解酶能使之迅速灭活。肉瘤使其灭活较癌瘤者快。主要由肾排泄。
【不良反应】对骨髓和免疫的抑制及胃肠道反应均不严重;约有1/3患者用药后可有发热、脱发等。少数患者可有皮肤色素沉着。最严重是肺纤维化,与剂量有关。
【临床应用】主要用于鳞状上皮癌(头、颈、口腔、食管、阴茎、外阴、宫颈等)。与DDP及VLB合用治疗睾丸癌,可达根治效果。也用于淋巴瘤的联合治疗。
(三)顺铂及卡铂
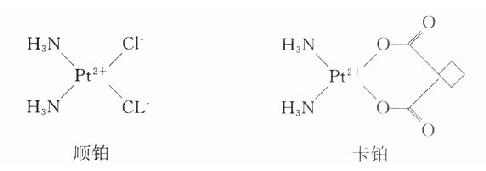
顺铂(顺氯氨铂,cisplatin,DDP)先将所含之氯解离,然后与DNA上的核碱鸟嘌呤、腺嘌呤和胞嘧啶形成DNA单链内两点的交叉联结,也可能形成双链间的交叉联结,从而破坏DNA的结构和功能。对RNA和蛋白质合成的抑制作用较弱。属周期非特异性药物。
它在体内主要聚积于肝、肾及膀胱。在血浆中与蛋白的结合率约90%,1小时后残留在血浆的铂不足10%,主要以原形经肾排泄,排泄较慢。
主要不良反应有肾毒性,呕吐、恶心的发生率较高。还能致听力减退及神经症状。
顺铂抗瘤谱广。对睾丸肿瘤与BLM及VLB联合化疗,可以根治;对卵巢癌、肺癌、鼻咽癌、淋巴瘤、膀胱癌等也有效。
卡铂(carboplatin)的抗癌作用与顺铂相似,但其不良反应不同,主要是骨髓抑制。
新的铂类抗肿瘤药草酸铂(oxaliplatin)具有高效和低毒的特点,目前正在进行临床试用中。
三、干扰转录过程阻止RNA合成的药物
放线菌素D
放线菌素D(dactinomycin,DACT)是多肽抗生素,国产品称更生霉素。
【药理作用】放线菌素D能嵌入到DNA双螺旋链中相邻的鸟嘌呤和胞嘧啶(G-C)碱基对之间,与DNA结合成复合体,阻碍RNA多聚酶的功能,阻止RNA特别是mRNA的合成,从而妨碍蛋白质合成而抑制肿瘤细胞生长。属周期非特异性药物,但对G1期作用较强,且可阻止G1向S期的转变。
【体内过程】口服疗效差。静脉注射后2分钟内迅速分布到组织内。肝、肾中药物浓度较高。24小时内,有10%~20%由尿中排出,50%~90%由胆汁排泄。
【不良反应】恶心、呕吐、口腔炎常见。骨髓抑制先呈血小板减少,后即出现全血细胞减少。有局部刺激作用,可致疼痛和脉管炎。还致脱发、皮炎、畸胎等。
【临床应用】抗瘤谱较窄。对恶性葡萄胎、绒毛膜上皮癌、淋巴瘤、肾母细胞瘤、横纹肌肉瘤及神经母细胞瘤等的疗效较好。
阿霉素
阿霉素(doxorubicin,adriamycin,ADM)能嵌入DNA碱基对之间,阻止转录过程,抑制RNA合成,也阻止DNA复制。属周期非特异性药物。
不良反应有骨髓抑制及口腔炎,尤应注意其心脏毒性,早期可出现各种心律失常,积累量大时可致心肌损害或心力衰竭。应将总量限制在550mg/m2以下。
临床抗瘤谱广,疗效高,可用于多种联合化疗。如非何杰金淋巴瘤、乳癌、卵巢癌、小细胞肺癌、胃癌、肝癌、膀胱癌及肉瘤类。
表49-3 其他抑制转录过程的抗生素
| 药 名 | 作用特点 | 不良反应 |
| 柔红霉素(daunorubicin,DNR) | 能嵌入DNA碱基对中,破坏DNA的模板功能,阻止转录过程而抑制DNA及RNA的合成。主要用于急性淋巴细胞白血病和急性粒细胞白血病 | 骨髓抑制和心脏毒性较大 |
| 普卡霉素(plicamycin;光辉霉素,mithramycin) | 可与DNA以非共价键结合,阻碍RNA合成,干扰转录过程。还能阻断甲状旁腺激素对骨钙的代谢作用,主要用于睾丸胚胎瘤,也用于纠正乳癌等所致血钙过高 | 恶心、呕吐、腹泻、皮肤色素沉着、血小板减少。对肝、肾也有一定损害。 |
四、影响蛋白质合成的药物
长春碱类
主要有长春碱(vinblastin,VLB)及长春新碱(vincristin,VCR),它们为夹竹桃科长春花(Vinca rosea L.)植物所含的生物碱。
【药理作用】可使细胞有丝分裂停止于中期。对有丝分裂的抑制作用,VLB较VCR强,但后者的作用不可逆。作用机制在于药物与纺锤丝微管蛋白结合,使其变性,从而影响微管装配和纺锤丝的形成。是作用于M期的药。
【不良反应】VLB可引起骨髓抑制、白细胞及血小板减少。也有脱发、恶心等。偶有外周神经症状。静脉注射因刺激导致血栓性静脉炎。VCR对骨髓抑制不明显,主要引起神经症状,表现为指、趾麻木、腱反射迟钝或消失、外周神经炎等。
【临床应用】VLB主要用于急性白血病、何杰金病及绒毛膜上皮癌。VCR对小儿急性淋巴细胞白血病疗效较好,起效较快,常与强的松合用作诱导缓解药。对淋巴瘤类也有效,并常与其他类型抗癌药合用于多种癌瘤的治疗。
鬼臼毒素类
鬼臼毒素(podophyllotoxin)是植物西藏鬼臼(Podophyllus emodii Wall)的有效成分,经改造半合成又得依托泊甙(鬼臼乙叉甙,足草乙甙,etoposid,vepesid,VP16)。鬼臼毒素能与微管蛋白相结合而破坏纺锤丝的形成。但VP16则不同,它能干扰DNA拓朴异构酶,阻止DNA复制。
VP16单用虽也有效,但临床上常与顺铂联合用于治疗肺癌及睾丸肿瘤,有良好效果。也用于作淋巴瘤类的二结治疗。同类药鬼臼噻吩甙(VM26)治脑瘤有效。
不良反应有骨髓抑制及胃肠道反应。
三尖杉酯碱
三尖杉酯碱(harringtonine)从三尖杉属植物的枝、叶和树皮中提取而得。其作用机制是抑制蛋白质合成的起步阶段,并使核蛋白体分解,释出新生肽链,但对mRNA或tRNA与核蛋白体的结合并无阻抑作用。
它对急性粒细胞白血病疗效较好,对急性单核细胞白血病也有效。只作缓慢静脉滴注用。不良反应有白细胞减少及胃肠道反应,也有心率加快、心肌缺血等。
L-门冬酰胺酶
L-门冬酰胺是重要氨基酸,某些肿瘤细胞不能自行合成,需从细胞外摄取。L-门冬酰胺酶(L-asparaginase)可将血清门冬酰胺水解而使肿瘤细胞缺乏门冬酰胺供应,生长受抑。正常细胞能合成门冬酰胺,受影响较少。主要用于急性淋巴细胞白血病,缓解率约60%,但不持久。常见的不良反应有胃肠道反应及精神症状。也可有血浆蛋白低下及出血。偶见过敏反应,应作皮试。
五、激素类
人们早已注意到乳腺癌、前列腺癌、甲状腺癌、宫颈癌、卵巢肿瘤及睾丸肿瘤等均与相应的激素失调有关,因此应用某些激素或其拮抗药,改变失调状态,可以抑制这些肿瘤生长,且无骨髓抑制等不良反应。但激素作用广泛,使用不当,也有害。
肾上腺皮质激素
能抑制淋巴组织,使淋巴细胞溶解。对急性淋巴细胞白血病及恶性淋巴瘤的疗效较好,效快但短暂,且易产生耐药性。对慢性淋巴细胞白血病除减低淋巴细胞数目外,还可缓解伴发的自身免疫性贫血。对其他癌无效,且可能因抑制免疫功能而助长癌瘤扩展。仅在癌瘤引起发热不退、毒血症状明显时可少量短期应用以改善症状(应合用抗癌药及抗菌药)。常用的有泼尼松、泼尼松龙、氟美松等。
雌激素
用于前列腺瘤治疗,因可抑制下丘脑及垂体,减低促间质细胞激素的分泌,从而减少睾丸间质细胞分泌睾丸酮;减少肾上腺皮质分泌雄激素;还用于绝经7年以上的乳癌而有内脏或软组织转移者。
雄激素
对晚期乳癌,尤其是骨转移者效佳;可抑制促卵泡激素的分泌,在肿瘤细胞对抗乳腺促进激素(或催乳素)的促进作用,不利于乳癌生长。
他莫西芬
他莫西芬(tamoxifen)为抗雌激素药,它可在靶组织上拮抗雌激素的作用.已证实,某些乳癌细胞的生长有赖于雌激素,且在乳癌组织上已检出雌激素受体,故可用于治疗晚期乳癌。与雄激素的疗效同,但无后者的男性化副作用。
氨鲁米特
氨鲁米特(氨基导眠能,aminoglutethimide)为催眠药格鲁米特(导眠能)的衍生物,具有抑制肾上腺皮质激素合成及阻止雄激素转变为雌激素的作用。可用于绝经后晚期乳腺癌。
第三节 联合应用抗肿瘤药物的原则
根据抗肿瘤药物的作用机制和细胞增殖动力学,设计出联合用药方案,可以提高疗效、延缓耐药性的产生,而毒性增加不多。联合用药有先后使用的序贯疗法,也有同时应用的联合疗法。一般原则如下。
1.继用周期特异性药物杀死之。相反,对生长比率高和肿瘤如急性白血病,则先用杀灭S期或M期的周期特异性药物,以后再用周期非特异性药物杀灭其它各期细胞。待G期细胞进入周期时,可重复上述疗程。此外,瘤细胞群中的细胞往往处于不同时期,若将作用于不同时期的药物联合应用,还可收到各药分别打击各期细胞的效果。
2.从抗肿瘤药物的作用机制考虑不同作用机制的抗肿瘤药合用可能增强疗效,如甲氨蝶呤和巯嘌呤的合用。
3.从药物的毒性考虑多数抗肿瘤药均可抑制骨髓,而泼尼松、长春新碱、博来霉素的骨髓抑制作用较少,可合用以降低毒性并提高疗效。
4.从抗瘤谱考虑胃肠道腺癌宜用氟尿嘧啶、噻替派、环磷酰胺、丝裂霉素等。鳞癌可用博来霉素、消卡芥、甲氨蝶呤等。肉瘤可用环磷酰胺、顺铂、阿霉素等。
5.给药方法一般均采用机体能耐受的最大剂量,特别是对病期较早、健康状况较好的肿瘤病人应用环磷酰胺、阿霉素、卡氮芥、甲氨蝶呤等时,大剂量间歇用药法往往较小剂量连续法的效果好。因为前者杀灭瘤细胞数更多;而且间歇用药也有利于造血系统等正常组织的修复,与补充,有利于提高机体的抗瘤能力及减少耐药性。
抗恶性肿瘤药 制剂及用法
5-氟尿嘧啶(5-fluorouracil)静脉注射,10~12mg/kg/日,连用3~5日后改为5~6mg/kg/隔日,总量5~10g为一疗程。必要时间隔1~2个月开始第2个疗程。
替加氟(tegafur)口服,15~20mg/kg/次,一般0.8~1.0g,总量20~40g为一疗程。
6-巯嘌呤(6-mercaptopurine)白血病:1.5~2.5mg/kg/日,分2~3次口服,疾病缓解后用原量1/3~1/2维持。绒癌:6.0~6.5mg/kg/日,10日为一疗程。
溶癌呤(tisupurine)治疗白血病:静脉滴注或静脉注射,4~5mg/kg/日,将药物溶于5%葡萄糖溶液或生理盐水中,10~14日为一疗程。治疗绒癌,8mg/kg/日,将药物溶于5%葡萄糖溶液中静脉滴注,10日为一疗程。
6-硫鸟嘌呤(tioguanine)口服2~2.5mg/kg/日,一次或分次服用,5~10日为一疗程。
甲氨蝶呤(methotrexate)治疗白血病:口服,成人5~10mg/次,4岁以上5mg/次、4岁以下2.5mg/次,每周2次,总量为50~150mg。绒毛膜上皮癌:静脉滴注,每日10~20mg,5~10次为一疗程。头颈部癌:动脉连续滴注,5~10mg/日,连用5~10日。鞘内注射:5~15mg/次,每周1~2次。
盐酸阿糖胞苷(cytarabine hydrochloride)静脉注射或静脉滴注,1~3mg/kg/日,10~14日为一疗程。鞘内注射,25mg/次,,2~3次/周,连用3次,6周后重复应用。
安西他滨(ancitabin)静脉注射或肌内注射,5~10mg/kg/日。用5~10日为一疗程。口服,剂量同注射。
羟基脲(hydroxycarbamide)20~40mg/kg/日,分次口服,或60~80mg/kg/每3日,4~6周为一疗程。
盐酸氮芥(chlorethamine hydrochloride)静脉注射或动脉插管灌注,0.1mg/kg/次,每1~3日一次,4~6次为一疗程,必要时间隔4周进行第2疗程。
环磷酰胺(cyclophosphamide)静脉注射,4mg/kg/日,每日或隔日一次,总量8~10g为一疗程。大剂量冲击疗法为10~20mg/kg/次,每周一次,8g为一疗程,以口服维持,2~4mg/kg/日,分次服用。
噻替派(thio-tepa)静脉注射、动脉注射或肌内注射,0.2mg/kg/次/日,连用5~7日,以后改为每周2~3次,总量约200~400mg。体腔注射,20~40mg/次,1~2次/周。
白消安(busulfan)口服,2~8mg/日,分3次空腹服用,有效后用维持量,0.5~2mg/日,一次/日。
氮甲(N-formylsarcolysin)口服,3~4mg/kg/日,分3~4次服用或睡前一次服,6~8g为一疗程.
消卡芥(nitrocophan)静脉注射,20~40mg/次,总量200~400mg为一疗程。口服,每次20mg,2~3次/日,1g为一疗程。
卡莫司汀(carmustine)静脉滴注,2.5mg/kg/次/日,溶于5%葡萄糖溶液或生理盐水内,连用3日为一疗程 ,每疗程间隔6~8周。
罗莫司汀(lomustine)口服,100mg/m2/次,每6~8周用一次。
司莫司汀(semustine)口服,130~200mg/m2/次,每6~8周用一次。
博来霉素(bleomycin)静脉或肌内注射,15~30mg/次,每日或隔日一次,总量450mg。
丝裂霉素(mitomycin C)静脉注射,2mg/次/日,或10mg/次,每周一次。总量60mg为一疗程。
顺铂(cisplatin)静脉注射或静脉滴注,30mg/日,连用5日为一疗程,疗程间隔2~4周,可用药4~5个疗程。或以50~100mg/m2,静脉注射或滴注一次,间隔3~4周再用。
放线菌素D(dactinomycin)静脉注射,200μg/日,10~14日为一疗程。
柔红霉素(daunorubicin)静脉注射或静脉滴注,开始0.2mg/kg/日,增至0.4mg/kg/日,每日或隔日一次,3~5次为一疗程,间隔5~7日再给下一个疗程。最大总量600mg/m2。
阿霉素(doxorubicin)静脉注射,30mg/m2,连用2日,间隔3周后可重复应用。60~75mg/m2,每3周应用一次。或30mg/m2,连用3日,间隔4周可再用。积累总量不得超过550mg/m2。
普卡霉素(plicamycin)静脉注射或静脉滴注,2~6mg/次,每日或隔日一次,10~30次为一疗程。
长春碱(vinblastin)静脉注射,0.2mg/kg/次,每周一次,总量60~80mg为一疗程。
长春新碱(vincristin)静脉注射,0.02mg/kg/次,每周一次,总量20~30mg为一疗程。
三尖杉酯碱(harringtonine)静脉滴注0.1~0.2mg/kg/日,7日为一疗程,停2周后再用。
L-门冬酰胺酶(L-asparaginase)肌内或静脉注射,20~200u/kg/次,每日或隔日一次,10~20次为一疗程。用药前皮内注射10~50u作过敏试验,观察3小时。
第五十章 影响免疫功能的药物
免疫系统包括参与免疫反应的各种细胞、组织和器官,如胸腺、淋巴结、脾、扁桃体以及分布在全身体液和组织中的淋巴细胞和浆细胞。这些组分及其正常功能是机体免疫功能的基本保证,任何一方面的缺陷都将导致免疫功能障碍,丧失抵抗感染能力或形成免疫性疾病。
机体免疫系统在抗原刺激下所发生的一系列变化称为免疫反应,可分为以下三期(图50-1):
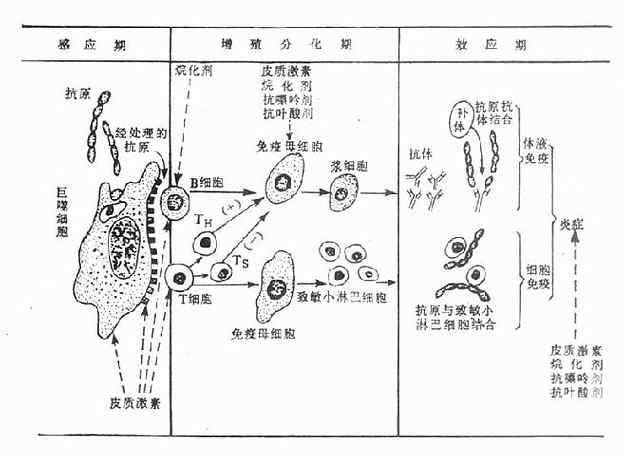
图50-1 免疫反应的基本过程和药物作用环节
T细胞主要有两个亚群;TH:辅助性T细胞(Helper T cell)能促进B细胞
增殖分化;Ts抑制性T细胞(Suppressor T cell)能抑制B细胞分化
1.感应期 为处理和识别抗原的阶段;先由巨噬细胞吞噬和处理,在胞浆内降解、消化之,暴露出活性部位而与巨噬细胞mRNA结合形成复合体,使T、B细胞得以识别。
2.增殖分化期 抗原-mRNA复合体能刺激B或T细胞,使其转化为免疫母细胞并进行增殖。B细胞增殖分化为浆细胞,可合成多种免疫球蛋白IgG、IgM、IgA、IgD、IgE等抗体。T细胞增殖分化为致敏小淋巴细胞,分别对相应抗原起特异作用。
3.效应期 致敏小淋巴细胞或抗体再次与抗原相反应,产生细胞免疫或体液免疫效应。致敏小淋巴细胞再受抗原刺激时,可有直接杀伤作用或释放淋巴毒素、炎症因子等免疫活性物质,使抗原所在细胞破坏或发生异体器官移植的排异反应等。这称为细胞免疫。抗原与抗体结合,直接或在补体协同下破坏抗原的过程称为体液免疫。不论细胞免疫或体液免疫,其最终效果都是消除抗原,保护机体。
在调节免疫和炎症方面,淋巴因子(lymphokine)或单核因子(monokines)等细胞调节蛋白也起到重要的作用。它们可以由淋巴细胞、单核细胞及巨噬细胞产生,如干扰素、白细胞介素、肿瘤坏死因子、克隆刺激因子、巨噬细胞激活和抑制因子等,其中已有多种作用免疫调节剂应用。
影响免疫功能的药物有两类:免疫抑制药(immunosuppressivedrugs)能抑制免疫活性过强者的免疫反应;免疫增强药(immunopotentiating drugs)能扶持免疫功能低下者的免疫功能。
第一节 免疫抑制药
临床常用的免疫抑制药有环孢素、肾上腺皮质激素类、烷化剂和抗代谢药等。
免疫抑制药都缺乏选择性和特异性,对正常和异常的免疫反应均呈抑制作用。故长期应用后,除了各药的特有毒性外,尚易出现降低机体抵抗力而诱发感染、肿瘤发生率增加及影响生殖系统功能等不良反应。
环孢素
环孢素(ciclosporin,cyclosporinA)是霉菌Tolypocladium inflatum生成的一种脂溶性环状十一肽化合物。它可选择地作用于T淋巴细胞活化初期。辅助性T细胞被活化后可生成增殖因子白细胞介素2(interleukin 2,IL-2),环孢素可抑制其生成;但它对抑制性T细胞无影响。它的另一个重要作用是抑制淋巴细胞生成干扰素。它对网状内皮系统吞噬细胞无影响。因而环孢素不同于细胞毒类药物的作用,它仅抑制T细胞介导的细胞免疫而不致显着影响机体的一般防御能力。
口服环孢素可被吸收,但不完全,其生物利用度仅20%~50%。口服后2~4小时血浆浓度达峰值。有40%的药物存在于血浆,50%在红细胞,10%在白细胞。在血浆中与蛋白的结合率为95%。它在体内几乎全部被代谢,从尿中排出的原形物不足服用量的0.1%。其t1/2约16小时。
环孢素在临床上主要用于防止异体器官或骨髓移植时排异等不利的免疫反应,常和糖皮质激素合用。在治疗自体免疫性疾病方面的临床应用尚在探索中。其毒性反应主要在肝与肾,在应用过程中宜监测肝、肾功能。
肾上腺皮质激素类
常用的有泼尼松、泼尼松龙、地塞米松等。它们对免疫反应的许多环节均有影响。主要是抑制巨噬细胞对抗原的吞噬和处理;也阻碍淋巴细胞DNA合成和有丝分裂,破坏淋巴细胞,使外周淋巴细胞数明显减少,并损伤浆细胞,从而抑制细胞免疫反应和体液免疫反应,缓解变态反应对人体的损害。
烷化剂
常用的有环磷酰胺、白消安、噻替派等。它们能选择性地抑制B淋巴细胞,大剂量也能抑制T淋巴细胞。还可抑制免疫母细胞,从而阻断体液免疫和细胞免疫反应。环磷酰胺作用明显,副作用较小,且可口服,故常用。
抗代谢药类
常用6-巯嘌呤与硫唑嘌呤。它们主要抑制DNA、RNA和蛋白质合成。硫唑嘌呤的毒性较小,故较常用。本类药物对T细胞的抑制较明显,并可抑制两类母细胞,故兼能抑制细胞免疫和体液免疫反应,但不抑制巨噬细胞的吞噬功能。用于肾移植的排异反应和自体免疫性疾病如类风湿性关节炎和全身性红斑狼疮等。
抗淋巴细胞球蛋白
抗淋巴细胞球蛋白(antilymphocyteglobulin,ALG)是直接抗淋巴细胞的抗体,现已能用单克隆抗体技术生产,特异性高,安全性好。它可与淋巴细胞结合,在补体的共同作用下,使淋巴细胞裂解。可用于器官移植的排斥反应,多在其他免疫抑制药无效时应用。
第二节 免疫增强药
又称免疫调节药,因大多数免疫增强药可能使过高的或过低的免疫功能调节到正常水平,临床主要用其免疫增强作用,治疗免疫缺陷疾病、慢性感染和作为肿瘤的辅助治疗。近年来也发现人参、黄芪、五味子、枸杞子、党参、冬虫夏草、灵芝和银耳多糖等具有提高免疫功能的作用。
左旋咪唑
左旋咪唑(levamisole,LMS)为四咪唑的左旋体。它有免疫增强作用,能使受抑制的巨噬细胞和T细胞功能恢复正常。这可能与激活环核苷酸磷酸二酯酶,从而降低淋巴细胞和巨噬细胞内cAMP含量有关。其不良反应不严重,可有胃肠道症状、头痛、出汗、全身不适等。少数病人有白细胞及血小板减少,停药后可恢复。主要用于免疫功能低下者,恢复免疫功能后,可增强机体的抗病能力。肺癌手术合用左旋咪唑可延长无瘤期,减低复发率及肿瘤死亡率。对鳞癌较好,可减少远处转移。多种自身免疫性疾病,如类风湿性关节炎、红斑性狼疮等用药后均可得到改善,可能与提高T细胞功能,恢复其调节B细胞的功能有关。
白细胞介素
白细胞介素2又名T细胞生长因子(t cell growth factor,TCGF),由TH细胞产生,为Ts和TC(杀伤)细胞分化增殖所需的调控因子,它对B细胞、自然杀伤(NK)细胞、抗体依赖性杀伤细胞和淋巴因子激活的杀伤(LAK)细胞等均可促进其分化增殖。它在抗恶性肿瘤、免疫缺陷病和自身免疫性疾病的治疗和诊断方面有潜在的重要意义。
白细胞介素3(interleukin-3,IL-3)由激活的T细胞产生,可刺激某些细胞分化为成熟的T细胞,还能刺激骨髓多能造血干细胞和各系统细胞分化、增殖,可促进自然细胞毒细胞(natural cytoxicity cell)的杀瘤活性。
近年来也从激活的T细胞的产物中分离出白细胞介素4、5、6,对于它们的作用和应用前景正在研究中。
干扰素
干扰素(interferon,IFN)是一类糖蛋白,它具有高度的种属特异性,故动物的IFN对人无效。干扰素具有抗病毒、抑制细胞增殖、调节免疫及抗肿瘤作用。
在抗病毒方面,它是一个广谱抗病毒药,其机制可能是作用于蛋白质合成阶段,临床可用于病毒感染性疾病,如疱疹性角膜炎、病毒性眼病、带状疱疹等皮肤疾患、慢性乙型肝炎等。
其免疫调节作用在小剂量时对细胞免疫和体液免疫都有增强作用,大剂量则产生抑制作用。
IFN的抗肿瘤作用在于它既可直接抑制肿瘤细胞的生长,又可通过免疫调节发挥作用。临床试验表明,它对肾细胞癌、卡波济肉瘤、多毛细胞白血病,、某些类型的淋巴瘤、黑色素瘤、乳癌等有效;而对肺癌、胃肠道癌、及某些淋巴瘤无效、
在临床应用时常见的不良反应有发热和白细胞减少等,少数病人快速静注时可出现血压下降。约5%的病人用后可产生IGN抗体。
转移因子
转移因子(transfer factor,TF)是从正常人的淋巴细胞或淋巴组织、脾、扁桃体等制备的一种核酸肽。它可将供体细胞免疫作息转移给受者的淋巴细胞,使之转化、增殖、分化为致敏淋巴细胞,从而获得供体样的免疫力。由此获得的免疫力较持久。其作用机制可能是TF的RNA通过逆转录酶(reverse transcriptase)的作用掺入到受者的淋巴细胞中,形成含这TF密码的特异DNA所致。主要用于原发性或继发性细胞免疫缺陷的补充治疗。还试用于慢性感染、麻风及恶性肿瘤等。
胸腺素
胸腺素(thymosin)又称胸腺多肽,为小分子多肽。可促进T细胞分化成熟,即诱导前T细胞(淋巴干细胞)转变为T细胞,并进一步分化成熟为具有特殊功能的各亚型群T细胞。临床主要用于细胞免疫缺陷的疾病,某些自身免疫和晚期肿瘤。除少数过敏反应外,一般无严重不良反应。
影响免疫功能的药物 制剂及用法
环孢素(ciclosporin,cyclosporin A)口服,一日10~15mg/kg。于器官移植前3小时开始应用并持续1~2周,然后逐渐减至维持量5~10mg/kg。静脉滴注时可将50mg以生理盐水或5%葡萄糖注射液200ml稀释后于2~6小时内缓慢点滴,剂量为口服剂量的1/3。
盐酸左旋咪唑(levamisole hydrochloride)治疗肿瘤:每2周用药3天或每周2天,3次/日,50mg/次。自身免疫性疾病:2-3次/日,50mg/次,连续用药。
转移因子(transfer factor)肌内注射,每次2ml,相当于108个淋巴细胞(或1g扁桃体),1~2次/周。
附录 某些药物代谢动力学数据
| 药 物 | 生物利用度(%) | 尿排泄(%) | 血浆蛋白结合(%) | 清除率(ml·min-1·kg-1) | 分布容积(L/kg) | 半衰期(h) |
| 醋丁洛尔 acebutolol | 37 | 40 | 26 | 6.8 | 1.2 | 2.7 |
| 阿昔洛韦 aciclovir | 15~30 | 75 | 15 | 3.37 | 0.69 | 2.4 |
| 别嘌醇 allopurinol | 80~90 | <10 | <1 | 12.7 | 0.6~1.6 | 1~3 |
| 阿普洛尔 alprenolol | 8.6 | 0.2 | 85 | 15 | 3.3 | 2.5 |
| 金刚烷胺 amantadine | 50~90 | 50~90 | 67 | 4.8 | 6.6 | 16 |
| 两性霉素B amphotericin B | - | 2~5 | >90 | 0.46 | 0.76 | 18 |
| 阿米卡星 amikacin | - | 98 | 4 | 1.3 | 0.27 | 2.3 |
| 胺碘酮 amiodarone | 46 | 100 | 1.9 | 66 | 25d | |
| 阿米替林 amitriptyline | 48 | <2 | 94.8 | 11.5 | 15 | 21.5 |
| 氨苄西林 ampicillin | 62 | 82 | 18 | 1.7 | 0.28 | 1.3 |
| 阿斯匹林 aspirin | 68 | 1.4 | 49 | 9.3 | 0.15 | 0.25 |
| 阿替洛尔 atenolol | 56 | 94 | <5 | 2.4 | 0.95 | 6.1 |
| 阿曲库铵 atracurium | - | <1 | - | 5.5 | 0.16 | 0.33 |
| 阿托品 atropine | 50 | 57 | 14~22 | 5.9 | 1.7 | 4.3 |
| 硫唑嘌呤 azathioprine | 60 | <2 | - | 57 | 0.81 | 0.16 |
| 倍他米松 betamethasone | 72 | 4.8 | 64 | 2.9 | 1.4 | 5.6 |
| 博莱霉素 bleomycin | - | 68 | - | 1.1 | 0.27 | 3.1 |
| 布美他尼 bumetanide | 81 | 62 | 99 | 2.6 | 0.13 | 0.8 |
| 布比卡因 bupivacaine | - | 2 | 95 | 7.1 | 0.9 | 2.4 |
| 咖啡因 caffeine | 100 | 1.1 | 36 | 1.4 | 0.61 | 4.9 |
| 卡托普利 captopril | 65 | 38 | 30 | 12 | 0.81 | 2.2 |
| 卡马西平 carbamazepine | >70 | <1 | 74 | 1.3 | 1.4 | 15 |
| 羧苄西林 carbenicillin | - | 82 | 50 | 0.68 | 0.18 | 1.0 |
| 头孢克洛 cefaclor | - | 52 | 25 | 6.1 | 0.36 | 0.67 |
| 头孢氨苄 cefalexin | 90 | 91 | 14 | 4.3 | 0.26 | 0.9 |
| 头孢孟多 cefamandole | - | 96 | 74 | 2.8 | 0.16 | 0.78 |
| 头孢唑林 cefazolin | - | 80 | 89 | 0.95 | 0.12 | 1.8 |
| 头孢哌酮 cefoperazone | - | 29 | 89~93 | 1.2 | 0.09 | 2.1 |
| 头孢噻肟 cefotaxime | - | 50 | 36 | 3.7 | 0.23 | 1.1 |
| 头孢拉定 cefradine | >90 | 86 | 14 | 5.1 | 0.25 | 0.77 |
| 头孢曲松 ceftriaxone | - | 46 | 90~95 | 0.24 | 0.16 | 7.3 |
| 头孢呋辛 cefuroxime | - | 96 | 33 | 0.94 | 0.19 | 1.7 |
| 苯丁酸氮芥 chlorambucil | 87 | <1 | 99 | 2.6 | 0.29 | 1.3 |
| 氯霉素 chloramphenicol | 75~90 | 25 | 53 | 2.4 | 0.94 | 4.0 |
| 氯喹 chloroquine | 89 | 61 | 61 | 1.8 | 115 | 41 |
| 氯噻嗪 chlorothiazide | 9~56 | 92 | 94.6 | 4.5 | 0.20 | 1.5 |
| 氯丙嗪 chlorpromazine | 32 | 〈1 | 95~98 | 8.6 | 21 | 30 |
| 氯酞酮 chlortalidone | 64 | 65 | 75 | 1.6 | 3.9 | 44 |
| 西米替丁 cimetidine | 62 | 62 | 19 | 8.3 | 1.0 | 2.0 |
| 环丙沙星 ciprofloxacin | 60 | 65 | 40 | 6.0 | 1.8 | 4.1 |
| 顺铂 cisplatin | - | 23 | - | 6.3 | 0.28 | 0.53 |
| 氯贝特 clofibrate | 95 | 5.7 | 96.5 | 0.12 | 0.11 | 13 |
| 氯硝西泮 clonazepam | 98 | <1 | 86 | 1.55 | 3.2 | 23 |
| 可乐定 clonidine | 95 | 62 | 20 | 3.1 | 2.1 | 12 |
| 可卡因 cocaine | 57 | <2 | 91 | 32 | 2.0 | 0.8 |
| 可待因 codeine | 50 | 7 | 11 | 2.6 | 2.9 | |
| 环磷酰胺 cyclophosphamide | 74 | 6.5 | 13 | 1.3 | 0.78 | 7.5 |
| 环孢霉素 cyclosporine | 23 | <1 | 93 | 5.9 | 1.2 | 5.6 |
| 氨苯砜 dapsone | 93 | 15 | 73 | 0.6 | 1.0 | 22 |
| 地塞米松 dexamethasone | 78 | 2.6 | 68 | 3.7 | 0.82 | 3.0 |
| 地西泮 diazepam | 100 | <1 | 98.7 | 0.38 | 1.1 | 43 |
| 洋地黄毒甙 digitoxin | >90 | 32 | 97 | 0.055 | 0.54 | 6.7d |
| 地高辛 digoxin | 70 | 60 | 25 | 0.88 | - | 39 |
| 地尔硫 |
44 | <4 | 78 | 12 | 3.1 | 3.7 |
| 苯海拉明 diphenhydramine | 72 | 1.9 | 78 | 6.2 | 4.5 | 8.5 |
| 丙吡胺 disopyramide | 83 | 55 | - | 1.2 | 0.59 | 6.0 |
| 多巴酚丁胺 dobutamine | - | - | - | 59 | 0.2 | 2.4 |
| 阿霉素 doxorublicin | 5 | <15 | 79~85 | 17 | 25 | 30 |
| 多西环素 doxycycline | 93 | 41 | 88 | 0.53 | 0.75 | 16 |
| 依那普利 enalapril | 41 | 88 | <50 | 4.9 | 1.7 | 11 |
| 依诺沙星 enoxacin | 79 | 60 | - | - | 5 | |
| 红霉素 erythromycin | 35 | 12 | 84 | 9.1 | 0.78 | 1.6 |
| 艾司洛尔 esmolol | - | <1 | 55 | 170 | 1.9 | 0.13 |
| 乙胺丁醇 ethambutol | 77 | 79 | <5 | 8.6 | 1.6 | 3.1 |
| 乙醇 ethanol | 80 | <3 | - | - | 0.54 | 0.24 |
| 炔雌醇 ethinylestradiol | 40 | - | 98 | 6.3 | 1.5~4.3 | 6~20 |
| 法莫替丁 famotidine | 45 | 67 | 17 | 7.1 | 1.3 | 2.6 |
| 芬太尼 fentanyl | - | 8 | 84 | 13 | 4.0 | 3.7 |
| 氟卡尼 flecainide | 70 | 43 | 61 | 5.6 | 4.9 | 11 |
| 氟氯西林 flucloxacillin | 49 | 41 | 96 | - | 0.11 | 0.8 |
| 氟尿嘧啶 fluorouracil | 28 | <10 | 8~12 | 16 | 0.25 | 11 |
| 氟奋乃静 fluphenazine | - | - | - | - | 20 | 14.7~15.3 |
| 呋噻米 furosemide | 61 | 66 | 98.8 | 2.0 | 0.11 | 92 |
| 庆大霉素 gentamicin | - | >90 | <10 | 0.82 | 0.31 | 2~3 |
| 格列本脲 glibenclamide | 90 | 1 | 99 | 1.5 | 0.12 | 1.6 |
| 胍乙啶 guanethidine | 35 | 43 | - | 60 | 120 | |
| 氟哌啶醇 haloperidol | 61 | 1 | 92 | 11.8 | 18 | 18 |
| 肝素 heparin | - | - | - | 0.058 | 12 | |
| 肼屈嗪 hydralazine | 16~35 | 1~15 | 87 | 56 | 1.5 | 0.96 |
| 氢氯噻嗪 hydrochlorothiazide | 71 | >95 | 58 | 4.9 | 0.83 | 2.5 |
| 氢化可的松 hydrocortisone | - | <1 | 90~95 | - | - | 1~2 |
| 布洛芬 ibuprofen | >80 | <1 | >99 | 0.75 | 0.15 | 2 |
| 米帕明 imipramine | 40 | <2 | 90.1 | 15 | 23 | 18 |
| 吲哚美辛 indometacin | 98 | 15 | 90 | 2.0 | 0.26 | 2.4 |
| 胰岛素 insulin | - | <2 | 5 | - | 0.08 | 0.1~0.3 |
| 异烟肼 isoniazid | - | 7~29 | 3.7~7.4 | 0.67 | 1.1~3.1 | |
| 硝酸异山梨酯 isosorbide dinitrate | 22~30 | <1 | 28 | 45 | 1.5 | 0.8 |
| 5-单硝酸异山梨酯 isosorbide-5-mononitrate | 93 | <5 | 1.81 | 0.79 | 4.4 | |
| 卡那霉素 kanamycin | - | 90 | 1.4 | 0.26 | 2.1 | |
| 酮康唑 ketoconazole | - | <1 | 99 | 8.4 | 2.4 | 3.3 |
| 拉贝洛尔 labetalol | 18 | <5 | 50 | 25 | 9.4 | 4.9 |
| 左旋多巴 levodopa | - | - | - | 7.7~13 | 0.8~1.1 | 2.2 |
| 利多卡因 lidocaine | 35 | 2 | 70 | 9.2 | 1.1 | 1.8 |
| 林可霉素 lincomycin | 100 | 20 | 72 | - | 0.44 | 5 |
| 锂盐 lithium | 100 | 95 | 0.35 | 0.79 | 22 | |
| 洛伐他汀 lovastatin | 5 | <10 | 95 | 5~21 | - | 1.1~1.7 |
| 巯嘌呤 mercaptopurine | 12 | 22 | 19 | 11 | 0.56 | 0.9 |
| 美沙酮 methadone | 92 | 24 | 89 | 1.4 | 3.8 | 35 |
| 甲氨蝶呤 methotrexate | 70 | 48 | 34 | 2.1 | 0.55 | 7.2 |
| 甲氧氯普胺 metoclopramide | 76 | 20 | 40 | 6.2 | 3.4 | 5.0 |
| 美托洛尔 metoprolol | 38 | 10 | 11 | 15 | 4.2 | 3.2 |
| 甲硝唑 metronidazole | 99 | 10 | 10 | 1.3 | 0.74 | 8.5 |
| 美西律 mexiletine | 87 | 4~15 | 63 | 6.3 | 4.9 | 9.2 |
| 米诺环素 minocycline | 95~100 | 11 | 76 | 1.0 | 1.3 | 16 |
| 米诺地尔 minoxidil | - | 20 | 24 | 2.7 | 3.1 | |
| 吗啡 morphine | 24 | 6~10 | 35 | 24 | 3.3 | 1.9 |
| 纳多洛尔 nadolol | 34 | 73 | 20 | 2.9 | 1.9 | 16 |
| 纳洛酮 naloxone | 2 | - | 22 | 2.1 | 1.1 | |
| 萘普生 naproxen | 99 | <1 | 99.7 | 0.13 | 0.16 | 14 |
| 新斯的明 neostigmine | - | 67 | - | 8.4 | 0.7 | 1.3 |
| 尼卡地平 nicardipine | 19~38 | <1 | 89~99.5 | 13.4 | 1.1 | 1.3 |
| 硝苯地平 nifedipine | 50 | 96 | 7.0 | 0.78 | 1.8 | |
| 硝西泮 nitrazepam | 78 | <1 | 87 | 0.86 | 1.9 | 26 |
| 硝酸甘油 nitroglycerin | <1 | <1 | - | 230 | 3.3 | 2.3min |
| 炔诺酮 norethisterone | 65 | - | 80 | - | 1.5~4.3 | 5~14 |
| 诺氟沙星 norfloxacin | 30~40 | 26~32 | 15~20 | 7.2 | 3.2 | 5 |
| 奥美拉唑 omeprazole | - | <1 | 95 | - | 0.19~0.48 | 1~2.3 |
| 昂丹司琼 ondansetron | 60 | <10 | 70~76 | 10~12 | 2.3 | 3~3.5 |
| 对乙酰氨基酚 paracetamol | 88 | 3 | 5.0 | 0.95 | 2.0 | |
| 培氟沙星 pefloxacin | 1 | <10 | 20~30 | 2.3 | 1.5~1.9 | 7~14 |
| 青霉素G penicillin G | 22 | 70 | 60 | - | 0.2 | 0.6 |
| 青霉素V penicillin V | 60~73 | 26~65 | 75~89 | - | 0.5 | 0.5 |
| 戊巴比妥 pentobarbital | >90 | <1 | 35~45 | 0.8 | 1 | 35~45 |
| 哌替啶 pethidine | 52 | 1~25 | 58 | 17 | 4.4 | 3.2 |
| 苯巴比妥 phenobarbital | 100 | 24 | 51 | 0.062 | 0.54 | 99 |
| 保泰松 phenybutazone | 80~100 | 1 | 96.1 | 0.023 | 0.097 | 56 |
| 苯妥英 phenytoin | 90 | 2 | 89 | - | 0.64 | 6~24 |
| 吲哚洛尔 pindolol | 75 | 54 | 51 | 8.3 | 2.3 | 3.6 |
| 哌拉西林 piperacillin | - | 71 | 16~48 | 2.6 | 0.18 | 0.93 |
| 吡罗昔康 piroxicam | - | <5 | 99.3 | 0.036 | 0.15 | 48 |
| 哌唑嗪 prazosin | 68 | <1 | 95 | 3.0 | 0.6 | 2.9 |
| 泼尼松龙 prednisolone | 82 | 26 | 90~95 | 8.7 | 1.5 | 2.2 |
| 泼尼松 prednisone | 80 | 3 | 75 | 3.6 | 0.97 | 3.6 |
| 普鲁卡因胺 procainamide | 83 | 67 | 16 | 2.7 | 1.9 | 3.0 |
| 异丙嗪 promethazine | - | - | 70~80 | 18 | 13 | 12 |
| 普罗帕酮 propafenone | 3~40 | <1 | 97 | - | - | 9~25(PM) 3~8(EM) |
| 普萘洛尔 propafenone | 26 | <0.5 | 87 | 16 | 4.3 | 3.9 |
| 乙胺嘧啶 pyrimethamine | - | 65 | 87 | 0.41 | 2.9 | 83 |
| 奎尼丁 quinidine | 80 | 18 | 87 | 4.7 | 2.7 | 6.2 |
| 雷尼替丁 ranitidine | 52 | 69 | 15 | 10.4 | 1.3 | 2.1 |
| 利福平 rifampin | - | 7 | 89 | 3.5 | 0.97 | 3.5 |
| 东莨菪碱 scopolamine | 27 | 6 | - | 16 | 1.4 | 2.9 |
| 索他洛尔 sotalol | 60 | 60 | 54 | - | 0.7 | 9 |
| 链激酶 streptokinase | - | - | 0.15 | 0.016 | 1.4 | |
| 链霉素 streptomycin | - | 50~60 | 48 | 1.2 | 0.25 | 2.6 |
| 磺胺嘧啶 sulfadiazine | 100 | 57 | 54 | 0.55 | 0.29 | 9.9 |
| 磺胺异恶唑 sulfafurazole | 96 | 49 | 91.4 | 0.33 | 0.15 | 6.6 |
| 磺胺甲恶唑 sulfamethoxazol | 100 | 14 | 62 | 0.32 | 0.21 | 10.1 |
| 舒林酸 sulindac | - | 99.4 | 1.5 | 2 | 15 | |
| 特布他林 terbutaline | 14 | 56 | 20 | 3.4 | 1.8 | 14 |
| 硫喷妥 thiopental | - | <1 | 85 | 3.9 | 2.3 | 9.0 |
| 替卡西林 ticarcillin | - | 92 | 65 | 2.0 | 0.21 | 1.3 |
| 噻马洛尔 timolol | 50 | 15 | 60 | 7.3 | 2.1 | 4.1 |
| 妥布霉素 tobramycin | - | 90 | <10 | 0.98 | 0.33 | 2.2 |
| 甲苯磺丁脲 tolbutamide | 93 | 96 | 0.24 | 0.10 | 5.9 | |
| 氨苯蝶啶 triamterene | 54 | 52 | 61 | 63 | 13.4 | 4.2 |
| 筒箭毒碱 tubocurarine | - | 63 | 50 | 1.9 | 0.39 | 2.0 |
| 万古霉素 vancomycin | - | 79 | 30 | 1.4 | 0.39 | 5.6 |
| 维库溴铵 vecuronium bromide | - | 18 | 30 | 3.0 | 2.1 | 1.5 |
| 维拉帕米 verapamil | 22 | <3 | 90 | 15 | 5 | 4.0 |
| 华法林 warfarin | 93 | <2 | 99 | 0.045 | 0.14 | 37 |
本表资简化自①A.G.Gilman et al(ed):Goodman and Gilman’s Thepharmacological basis of therapeuties. 8th ed.Macmillan,1990:1655~1715.
②Melmon K.L.andMorrelli:Clinical pharmacology.3rd ed.Mcgraw-Hill,1992:1029~1072.
③Herfindal E.T.etal:Clinical pharmacy and Therapeutics.3rd ed.1992:364~368.
药名外文索引
| A | B |
| acebutolol 158 | barbiturates 96 |
| acedapsone 319 | beclomethasone 223 |
| acenocumarol 209 | bemegride 129 |
| acetarsol 329 | benactyzine hydrochloride 65 |
| acetazolamide 197 | bendroflumethiazide 201 |
| acetylcholine 50 | benproperine 225 |
| acetycysteine 226 | benserazide 107 |
| acetylsalicylic acid 131 | benzathine penicillin 281 |
| acetylspiramycin 289 | benzbromaron 138 |
| actinomycin 345 | benzhexol 107 |
| acyclovir 313 | benzodiazepines 94 |
| adenine arabinoside Ara-A 314 | benzonatate 225 |
| adrenaline 73 | benzylpenicillin 280 |
| adriamycin 346 | bepridil 148 |
| albendazole 332 | betamethasone 259 |
| aldosterone 251 | bezafibrate 181 |
| allopurinol 137 | biofermin 230 |
| altretamine 344 | bisacodyl 232 |
| aluminum hydroxide 227 | bismuth subcarbonate 233 |
| amantadine 107,313 | bleomycin 345 |
| ambenonium 56 | bromhexine 226 |
| amethopterine 340 | bromocriptine 107 |
| amikacin 296 | buclizine 221 |
| amiloride 203 | bumetanide 200 |
| aminoglutethimide 348 | bupivacaine 87 |
| aminoglycosides 292 | buprenorphine 118 |
| p-Aminomethylbenzoic acid 212 | busulfan 343 |
| aminophylline 223 | butazolidin 134 |
| amiodarone 158 | |
| amitriptyline 113 | C |
| amlodipine 148 | |
| ammonium chloride 225 | caffeine 127 |
| amobarbital 98 | caffeine and sodium benzoate 129 |
| amoxycillin 282 | calcium carbonate 227 |
| amphotericin B 311 | captopril 188 |
| ampicillin 282 | carbachol 50 |
| amrinone 170 | carbamazepine 101 |
| anadol 123 | carbamylcholine 50 |
| ancitabin 349 | carbamylmethylcholine 52 |
| anesthetic ether 91 | carbenicillin 282 |
| anisodamine 63 | carbidopa 107 |
| anorethidrane dipropionate 349 | carbimazole 261 |
| antibiotic 270 | carboplatine 345 |
| antilymphocyte glubulin 353 | cardiac glycosides 163 |
| antisterone 203 | carmustine 344 |
| Ara C 340 | cedilanid 164 |
| aramine 73 | cefadroxil 287 |
| artane 107 | cefamandole 287 |
| artemisia 324 | cefazolin 284 |
| aspirin 131 | cefoperazone 284 |
| astemizole 221 | cefoxitin 284 |
| atenolol 85 | cefradine 287 |
| atracurium 68 | ceftazidime 284 |
| atromid-S 181 | celestone 259 |
| atropine 61 | cephalexin 284 |
| augmetin 286 | cephaloridine 287 |
| azithromycin 290 | cephalosporine 283 |
| azlocillin 283 | cephalothin 283 |
| aztreonam 286 | cephradine 287 |
| chiniofon 328 | |
| D | chloral hydrate 97 |
| chloramphenicol 301 | |
| dacarbazine 344 | chlordiazepoxide 94 |
| dactinomycin 345 | chlorethamine 342 |
| dapsone 319 | chlorethamine hydrochloride 342 |
| daunorubicin 346 | chlorfenamic acid 139 |
| demethylvancomycin 291 | chlormadinone 244 |
| desipramine 113 | chloromycetin 301 |
| desoxycorticosterone 258 | chloroquine 322 |
| desoxycortone 251 | chlorothiazide 201 |
| devegan 329 | chlorpheniramine 221 |
| dexamethasone 251 | chlorpromazine 110 |
| dextran 216 | chlorpropamide 267 |
| dextromethorphan 225 | chlorprothixene 112 |
| diamox 197 | chlortalidone202 |
| diazepam 94 | cholestyramine 180 |
| diazoxide 193 | choline theophylline 223 |
| dibenzyline 81 | chondroidin sulfate A 184 |
| dichlorvos 56 | ciclosporin 352 |
| diclofenac 136 | cilastatin 286 |
| dicloxacillin 282 | cimetidine 219 |
| dicoumarol 209 | ciprofibrate 181 |
| dietary fibers 232 | ciprofloxacin 305 |
| diethylcarbamazine 331 | cisplatine 345 |
| diethylstilbestrol 242 | clavulanic acid 286 |
| digitalis 163 | clenbuterol 223 |
| digitoxin 164 | clindamycin 290 |
| digoxin 164 | clioquinol 328 |
| dihydroergotoxine aethanosulfonate 239 | clofazimine 319 |
| dihydroetorphine 125 | clofenamic acid 136 |
| diloxanide 328 | clofibrate 181 |
| diltiazem 147,178 | clomiphene 243 |
| dilute hydrochloride acid 230 | clomiphene citrate 249 |
| dimefline 128 | clonazepam 102 |
| diphenhydramine 221 | clonidine 190 |
| diphenoxylate 232 | clotrimazole 312 |
| dipyridamole 210 | cloxacillin 282 |
| disodium cromoglycate 224 | clozapine 118 |
| disopyramide 155 | codeine 121,225 |
| dobutamine 77 | codeine phosphate 121 |
| docosahexaenoic acid 184 | colchicine 138 |
| dolantin 122 | colestipol 180 |
| domperidone 230 | colistin 297 |
| dopamine 75 | colloidal bismuth subcitrate 233 |
| doxepin 113 | coramine 128 |
| doxorubicin 346 | corticotrophin 257 |
| doxycycline 300 | cortisol 251 |
| droperidol 113 | cortisone 251 |
| cortisone acetate 259 | |
| E | curare 68 |
| cyclo-C 341 | |
| eicosapentaenoic acid 184 | cyclocytidine 341 |
| emetine 328 | cyclopenthiazide 201 |
| enalapril 188 | cyclophosphamide 343 |
| encainide 157 | cyclosporin A 352 |
| endoxan 343 | cytarabine 340 |
| enflurane 91 | cytarabine hydrochloride 340 |
| enoxacin 303 | cytoxan 340 |
| enoxaparin 217 | |
| enprostil 233 | F |
| ephedrine 75 | |
| epinephrine 73 | famotidine 219 |
| ergometrine 238 | felodipiine 148 |
| ergotamine 238 | fenofibrate 181 |
| ergotoxine 238 | fentanyl 123 |
| erythromycin 289 | fentanyl citrate 126 |
| erythropoietin 215 | ferric ammonium citrate 213 |
| eserine 56 | ferrous sulfate 213 |
| esmolol 48 | flecainide 157 |
| estradiol 242 | fleroxacin 307 |
| estradiol benzoate 249 | flucloxacillin 282 |
| estradiol valerate 242 | fluconazole 312 |
| etacrynic acid 200 | fludrocortisone 259 |
| ethambutol 317 | flunarizine 148 |
| ethinylestradiol 242 | flunisolide 223 |
| ethosuximide 101 | fluocinolone 250 |
| etoposide 347 | fluocinolone acetonide 250 |
| etorphine 124 | fluorocytosine 313 |
| etynodiol diacetate 244 | fluoroquinolones 303 |
| fluorouracil 338 | |
| G | fluphenazine 112 |
| flurazepam 94 | |
| galanthamine 56 | folic acid 213 |
| gallamine triethiodide 68 | fortanodyn 125 |
| gallopamil 147 | ftorafur 341 |
| gemfibrozil 181 | furadantin 309 |
| gentamicin 295 | furazolidone 309 |
| glibenclamide 267 | furbenicillin 283 |
| glipizide 267 | furosemide 200 |
| globin zinc insulin 265 | |
| glutethimide 97 | H |
| glyburide 267 | |
| GM-CSF 216 | haloperidol 112 |
| gossypol 249 | halothane 91 |
| granisetron 231 | harringtonine 347 |
| griseofulvin 311 | heparan sulfate 184 |
| heparin 208 | |
| I | heroine 118 |
| hetrazan 331 | |
| ibuprofen 136 | hexamethonium 66 |
| imidazoles 312 | hexamethylmelamine 344 |
| imipenem 286 | homatropine 64 |
| imipramine 113 | hydralazine 193 |
| indomethacin 135 | hydrochlorothiazide 201 |
| insulin 265 | hydrocortisone 251 |
| intal 224 | hydroflumethiazide 201 |
| interferon 354 | hydroprednisone 251 |
| interleukin 2 353 | hydroxycarbamide 341 |
| iodine 262 | hydroxyurea 341 |
| ipratropium 223 | hyoscyamine 61 |
| iron dextran 213 | |
| isoflurane 91 | J |
| isoflurophate 56 | |
| isoniazid ,INH 316 | jossamycin 290 |
| isophane insulin 265 | |
| isoprenaline 76 | K |
| isoproterenol 76 | |
| isoptin 147 | kanamycin 296 |
| isosorbide –5-mononitrate 177 | kemadrin 107 |
| isosorbide dinitrate 177 | ketamine 92 |
| isradipin 148 | ketanserin 192 |
| ketoconazole 312 | |
| L | ketoprofen 136 |
| kitasamycin 290 | |
| L-dopa 105 | |
| labetalol 85,192 | M |
| lacidipine 148 | |
| lactulose 231 | madopa 107 |
| latamoxef 286 | magnesium hydroxide 227 |
| lemakalim 194 | magnesium sulfate 231 |
| leucomycin 290 | magnesium trisilicate 227 |
| levamisole 353 | malathion 56 |
| levodopa 105 | mannitol 205 |
| lidocaine 87 | maprotilin 114 |
| lincomycin 290 | marcaine 87 |
| liquid paraffin 232 | mebendazole 332 |
| lithium carbonate 115 | mecamylamine 66 |
| lobeline 129 | MeCCNU 344 |
| lomitil 234 | mechlorethamine 342 |
| lomefloxacin 306 | mecillinam 287 |
| loperamide 233 | meclizine 221 |
| losec 228 | meclofenoxate 128 |
| lovastain 182 | medecamycin 290 |
| lumstine 344 | medicinal activated charcoal 233 |
| medroxyprogesterone acetate 244 | |
| N | mefenamic acid 136 |
| mefloquine 323 | |
| nadolol 82 | megestrol 244 |
| nafcillin 282 | megimide 129 |
| nalidixic acid 303 | menadione sodium bisulfate 212 |
| nalorphine 118 | menadione diacetate 212 |
| naloxone 125 | meprobamate 97 |
| naltrexone 125 | mercaptopurine 339 |
| naproxen 136 | metaraminol 73 |
| nedocromil 224 | metformin 268 |
| neomycin 296 | methadone 124 |
| neostigmine 55 | methandienone 246 |
| netilmicin 296 | methaqualone 97 |
| nicardipine 148 | methimazol 261 |
| niclosamide 334 | methotrexate 340 |
| nicotine 53 | methoxamine 73 |
| nicotinic acid 181 | methyldopa 191 |
| nifedipine retard 179 | methyldopahydrazine 107 |
| nifedipine 148,188 | methylnorethindrone 244 |
| nikethamide 128 | methylphenidate 127 |
| nimodipine 148 | methylprednisolone 259 |
| nisoldipine 148 | methyltestosterone 245 |
| nitrazepam 102 | methylthiouracil 261 |
| nitrendipine 148 | metoclopramide 230 |
| nitrocophan 344 | metoprolol 85 |
| nitrofurantoin 309 | metronidazole 327 |
| nitrogen mustard 342 | metyrapone 258 |
| nitroglycerin 175 | mevastatin 182 |
| nitrous oxide 91 | mexiletine 156 |
| nizatidine 219 | miconazole 312 |
| nomifensine 114 | milk of magnesia 233 |
| noradrenaline 70 | minocycline 301 |
| norepinephrine 70 | minoxidil 193 |
| norethindrone 244 | miocamycin 290 |
| norethisterone 244 | misoprostol 229 |
| norfloxacin 303 | mithramycin 346 |
| norlutin 244 | mitomycin C 344 |
| nystatin 312 | mitotan 258 |
| molsidomine 179 | |
| P | morphine 117 |
| morphine hydrochloride 117 | |
| pancreatin 230 | motilium 230 |
| pancuronium 68 | moxalactam 286 |
| para-amino salicylic acid 318 | myleran 343 |
| paracetamol 133 | |
| pefloxacin 305 | O |
| pemoline 128 | |
| penicillin G 280 | o,p-DDD 258 |
| penicillin V 282 | ofloxacin 305 |
| pentaerythritol 174 | omeprazole 228 |
| pentaerythritol tetranitrate 174 | ondansetron 231 |
| pentazocine 124 | oxacillin 282 |
| pentoxyverine 225 | oxaliplatin 345 |
| pepsin 231 | oxazepam 94 |
| perhexiline 148 | oxyphenbutazone 134 |
| perindopril 188 | oxytetracycline 299 |
| persantin 210 | oxytocics 235 |
| pethidine 122 | oxytocin 235 |
| phenacetin 133 | |
| phenergan 221 | Q |
| phenformin 268 | |
| phenindamine 221 | quinestrol 242 |
| phenobarbital 97 | quinidine 153 |
| phenolphthalein 232 | quinine 323 |
| phenoxybenzamine 81 | quinolones 303 |
| phenprofen 319 | |
| phentolamine 79 | R |
| phenylbutazone 134 | |
| phenylephrine 73 | ramipril 188 |
| phenytoin 99 | ranitidine 219 |
| physostigmine 56 | reserpine 186 |
| pilocarpine 51 | ribavirin 314 |
| pimozide 113 | rifampicin 317 |
| pinacidil 194 | rifampin 317 |
| pindolol 85 | rifandine 317 |
| pipemidic acid 303 | rifapentine 317 |
| piperacillin 283 | rimifon 316 |
| piperazine 333 | ritalin 127 |
| piracetam 128 | rogor 56 |
| pirenzepine 64,227 | rotundine 125 |
| piroxicam 139 | roxithromycin 290 |
| pituitrin 237 | |
| pivampicillin 282 | S |
| plicamycin 346 | |
| polymycin B 297 | salbutamol 223 |
| polymyxin E 297 | salicylates 131 |
| pralidoxime iodide 58 | salicylazosulfapyridine 308 |
| pravastatin 182 | scoline 66 |
| praziquantel 330 | scopolamine 64 |
| prazosin 81,192 | seconal 98 |
| prednisolone 251 | semustine 344 |
| prednisone 251 | simvastatin 182 |
| prenylamine 149,178 | sinomin 309 |
| primaquine 324 | sisomicin 296 |
| primidone 101 | sodium bicarbonate 227 |
| probenecid 138 | sodium cromoglycate 224 |
| probucol 183 | sodium nitroprusside 194 |
| procainamide 154 | sodium salicylate 131 |
| procaine 87 | sodium sulfacetamide 308 |
| procaine penicillin 281 | sodium sulfate 231 |
| progesterone 244 | sodium valproate 102 |
| proglumide 228 | sorbitol 206 |
| promethazine 221 | spectinomycin 296 |
| propafenone 157 | spironolactone 203 |
| propantheline bromide 65 | stanozolol 246 |
| propranolol 84,158 | stilbestrol 242 |
| propylthiouracil 261 | streptokinase 211 |
| procarbazine 344 | streptomycin 295 |
| prostacyclin 210 | strophantin K 168 |
| prostigmine 55 | succinylcholine 66 |
| protamine sulfate 209 | sucralfate 229 |
| protamine zinc insulin 265 | sulbactam 286 |
| pyraloxime methylchloride 59 | sulbenicillin 283 |
| pyrantel 334 | sulfacetamide 308 |
| pyrazinamide 318 | sulfadiazine –Ag 308 |
| pyribenzamine 221 | sulfadiazine 307 |
| pyridostigmine 60 | sulfadoxine 308 |
| pyrimethamine 325 | sulfafurazole 307 |
| sulfamethoxazole 309,307 | |
| T | sulfamethoxydiazine 308 |
| sulfamylon 308 | |
| t-PA 211 | sulfasalazine 308 |
| tamoxifen 348 | sulfisoxazole 307 |
| tannalbin 233 | sulindac 136 |
| tapazole 264 | sulperazone 286 |
| tardan 112 | sulpiride 113 |
| tegafur 341 | |
| terbutaline 223 | U |
| terfenadine 221 | |
| terramycin 299 | ulcerlmin 229 |
| tessalon 225 | urokinase 211 |
| testosterone 245 | ursodeoxycholic acid 233 |
| testosterone phenylacetate 245 | |
| testosterone propionate 245 | V |
| tetracaine 87 | |
| tetracycline 299 | valproate 102 |
| tetrandrine 188 | vancomycin 291 |
| theohydramine 221 | vecuronium 68 |
| theophylline 127,223 | verapamil 147,159 |
| thiamazole 261 | vinblastin 346 |
| thienamycin 286 | vincristin 346 |
| thio-tepa 343 | virazole 314 |
| thiopental sodium 93 | vitamin B12215 |
| thioridazine 112 | vitamin K 212 |
| thymosin 354 | vitamin K1212 |
| thyroxine 260 | vitamin K2212 |
| ticarcillin 283 | vitamin K3212 |
| ticlopidine 210 | |
| tienam 286 | W |
| timentin 286 | |
| timolol 84 | warfarin 209 |
| tinidazole 328 | wintermine 110 |
| tioguanine 341 | |
| tisupurine 341 | X |
| tobramycin 296 | |
| tolazoline 81 | xylocaine 87 |
| tolbutamide 267 | |
| tramadol 124 | |
| tranexamic acid 212 | |
| transfer factor 354 | |
| triamcinolone 251 | |
| triamterene 203 | |
| triazolam 94 | |
| triethylene thiophosphoramide 343 | |
| trifluoperazine 112 | |
| trihexyphenidyl 107 | |
| triiodothyronine 260 | |
| trimethaphan 66 | |
| trimethoprim 308 | |
| tropicamide 64 | |
| tropisetron 231 | |
| tubocurarine chloride 68 |