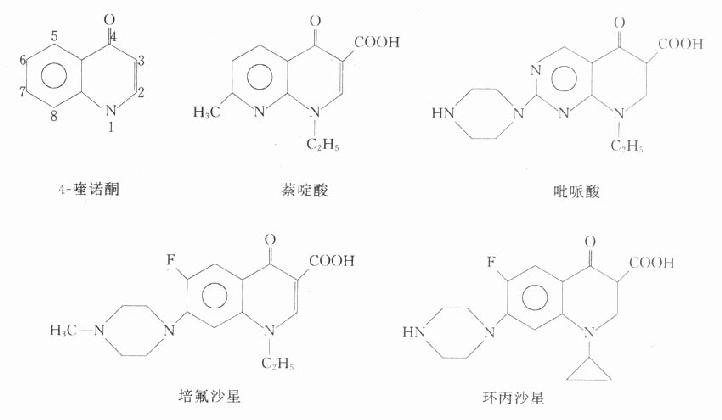
(一)简史
萘啶酸(nalidixicacid)是1962年用于临床的第一个喹诺酮类药(实是萘啶酮),抗菌谱窄,口服吸收差,副作用多,现已不用。吡哌酸(pipemidicacid)抗菌活性强于萘啶酸,口服少量吸收,不良反应较萘啶酸少,可用于敏感菌的尿路感染与肠道感染。1979年合成诺氟沙星(norfloxacin),随又合成一系列含氟的新喹诺酮类药,通称为氟喹诺酮类(fluoroquinolones)。
(二)化学结构与作用关系
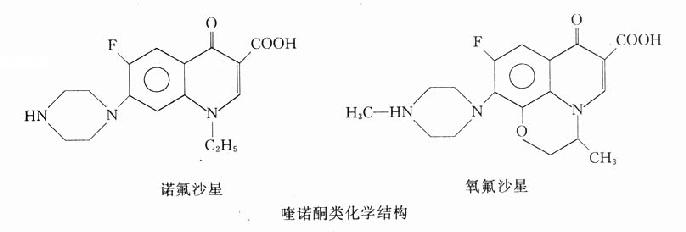
本类药物的构效关系研究表明:4-喹诺酮母核的3位均有羧酸基,6位引入氟原子可增强抗菌作用并对金葡菌有抗菌活性;7位引进哌嗪环可提高对金葡菌及绿脓杆菌的抗菌作用(如诺氟沙星),哌嗪环被甲基哌嗪环取代(如培氟沙星),则脂溶性增加,肠道吸收增强,细胞的穿透性提高,半衰期延长。在8位引进第二个氟原子,可进一步提高肠道吸收,延长半衰期(如洛美沙星等),N-1修饰以环丙基团(环丙沙星)或噁嗪基团(氧氟沙星)可扩大抗菌谱,增强对衣原体、支原体及分支杆菌(结核杆菌与麻风杆菌等)的抗菌活性,噁嗪环还可提高水溶性,使药物在体内不被代谢,以原形经尿排泄。
(三)抗菌作用机制
喹诺酮类通过抑制DNA螺旋酶作用,阻碍DNA合成而导致细菌死亡。大肠杆菌的DNA螺旋酶是四叠体结构的蛋白,由2个A亚单位与2个B亚单位组成,分子量分别为105kD与95kD(见图42-1)。细菌在合成DNA过程中,DNA螺旋酶的A亚单位将染色体DNA正超螺旋的一条单链(后链)切开,接着B亚单位使DNA的前链后移,A亚单位再将切口封住,形成了负超螺旋。根据实验研究,氟喹诺酮类药并不是直接与DNA螺旋酶结合,而是与DNA双链中非配对碱基结合,抑制DNA抑螺旋酶的A亚单位,使DNA超螺旋结构不能封口,这样DNA单链暴露,导致mRNA与蛋白合成失控,最后细菌死亡。
本类药体外对DNA螺旋酶的半抑制浓度(IC50)与其对细菌的MIC呈一定的平行关系。哺乳动物的细胞内也含有生物活性与细菌DNA螺旋酶相似的酶,称为拓朴异构酶Ⅱ(topoisomerase Ⅱ)。氟喹诺酮类药对人体细胞拓朴异构酶Ⅱ影响较小(见表42-1)。从该表可见氧氟沙星与环丙沙星对动物细胞内拓朴异构酶Ⅱ的作用明显比依诺沙星与萘啶酸小,IC50很高,选择指数很大。这可能是氧氟沙星与环丙沙星不良反应较少的原因。
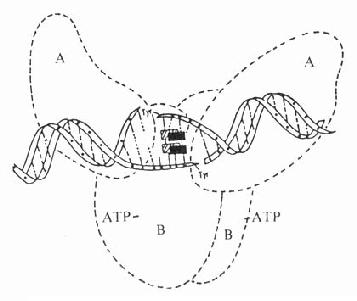
图42-1 喹诺酮类药物-DNA结合抑制DNA螺旋酶活性的示意图
图中实心和斜线长方形示喹诺酮类药物分子,A、B为DNA螺旋酶的A、B亚单位。在DNA螺旋酶作用下,DNA双链打开,而药物分子嵌入双链。与非配对碱基结合,阻碍DNA双链封口
(四)细菌耐药机制
氟喹诺酮类药物广泛应用后,已出现细菌耐药性。耐药机理研究证实主要是染色体突变,不存在质粒介导的耐药性。耐药机制有二:①细菌DNA螺旋酶的改变,与细菌高浓度耐药有关;②细菌细胞膜孔蛋白通道的改变或缺失与低浓度耐药有关。耐药菌株DNA螺旋酶的活性改变主要由于gyrA基因突变所致。
(五)氟喹诺酮类药理学共同特性
①抗菌谱广,尤其对革兰阴性杆菌包括绿脓杆菌在内有强大的杀菌作用,对金葡菌及产酶金葡菌也有良好抗菌作用;某些品种对结核杆菌,支原体,衣原体及厌氧菌也有作用;②细菌对本类药与其他抗菌药物间无交叉耐药性;③口服吸收良好,部分品种可静脉给药;体内分布广,组织体液浓度高,可达有效抑菌或杀菌水平;血浆半衰期相对较长,大多为3~7小时以上。血浆蛋白结合率低(14%~30%),多数经尿排泄,尿中浓度高;④适用于敏感病原菌所致的呼吸道感染、尿路感染、前列腺炎,淋病及革兰阴性杆菌所致各种感染,骨、关节、皮肤软组织感染;⑤不良反应少(5%~10%),大多轻微,常见的有恶心、呕吐、食欲减退、皮疹、头痛、眩晕。偶有抽搐精神症状,停药可消退。
表42-1 喹诺酮类药物对大肠杆菌和哺乳动物细胞DNA旋转酶的选择作用
| 药物 | IC50(mg/L) | 选择指数B/A | |
| A大肠杆菌KL-16DNA螺旋酶 | B胎牛胸腺局部拓朴异构酶Ⅱ | ||
| 氧氟沙星 | 0.76 | 1870 | 2461 |
| 环丙沙星 | 0.13 | 155 | 1192 |
| 依诺沙星 | 1.72 | 93 | 54 |
| 萘啶酸 | 23.0 | 385 | 17 |